

Abstract
As the first step in evaluating the possibility of low-temperature atmospheric plasma for clinical applications in the treatment of rhabdomyosarcoma (RMS), we determined the effects of plasma exposure on C2C12 myoblasts. The low-temperature atmospheric plasma was generated through an electrical discharge in argon gas. One minute of plasma exposure every 24 h inhibited the cell proliferation, whereas myoblast differentiation was not affected. Plasma exposure increased the phosphorylation of ERK and JNK at 30 min after the exposure, but the phosphorylation of both was decreased to less than control levels at 1 and 4 h after the exposure. Plasma exposure increased the percentage of cells in the G2/M phase at 8 h after the exposure. In conclusion, plasma exposure retarded the proliferation of C2C12 myoblasts by G2/M arrest. Therefore, plasma exposure can be a possible treatment for the anti-proliferative effects of malignant tumors, such as RMS, without affecting differentiated skeletal muscle cells.
Keywords: Cell cycle, Electrical discharge, MAP kinases, Plasma, Myoblast
Introduction
In general, plasma of an electrical discharge can be produced only at high temperatures or in a vacuum, but technical innovations have allowed the development of plasma at low-temperatures and atmospheric pressures. Many types of low-temperature atmospheric plasma devices have recently been developed for medical applications such as the surface reformulation of biomaterials [1, 2] and the sterilization of medical instruments [3, 4]. Plasma exposure has also been used directly on living tissues or cells for medical treatment (reviewed by [5–7]).
Plasma consists of many active components including reactive species, ions, electrons, and photons. These components can chemically interact with living cells. In-vitro experiments with fibroblasts [8], endothelial cells, and smooth muscle cells [9] demonstrated that plasma affects cells in a dose-dependent manner. It was also shown that treatment with low-intensity plasma induced cell proliferation in endothelial cells [10]. In contrast, it was reported that plasma exerted anti-proliferative effects on a variety of cancer cells by inducing apoptosis [11–14]. The results of these studies have suggested possible applications of plasma in cancer therapy.
Rhabdomyosarcoma (RMS) is a childhood cancer thought to be derived from skeletal myoblasts or their progenitors, and is the most common soft tissue sarcoma in children [15]. Standard treatment of RMS is systemic chemotherapy with radiation therapy and/or surgery for local control. In addition, intraoperative radiotherapy represents an effective means of improving the therapeutic ratio of radiation therapy in the treatment of patients with soft tissue sarcomas [16]. The purpose of the intraoperative radiation therapy is to destroy remaining cancer cells after surgical excision while causing as little damage as possible to normal cells.
In the present study, we focused on the effect of plasma exposure on C2C12 cells. The C2C12 cell line was derived from the mouse skeletal muscle C2 cell line, which shares similar properties with isolated skeletal muscle cells [17]. The myoblast proliferation and differentiation are highly coordinated processes, and are critical factors for muscle development. As the first step in evaluating the possibility of low-temperature atmospheric plasma for clinical applications in the treatment of soft tissue sarcoma, including RMS, we determined the effects of plasma exposure on the proliferation and differentiation of myoblasts. Here we show that 1 min of plasma exposure every 24 h inhibited the proliferation of C2C12 cells by G2/M phase cell-cycle arrest, whereas the cells’ differentiation was not affected by plasma exposure. Our results indicate that the effect of plasma exposure was apparent in the proliferating cells, and the cell-cycle arrest may be mediated by downregulation of MAP kinase pathways and cell-cycle checkpoint proteins. Therefore, low-temperature atmospheric plasma exposure can be a possible treatment for the anti-proliferative effects of malignant tumors, such as RMS, without affecting differentiated skeletal muscle cells.
Materials and methods
Materials
Culture medium, fetal bovine serum (FBS), horse serum (HS), and antibiotics were purchased from Sigma-Aldrich (St. Louis, MO). Antibodies against phospho-extracellular-regulated kinase (ERK1/2) (Thr202/Tyr204), phospho-c-Jun N-terminal kinase (SAPK/JNK) (Thr183/Tyr185), phospho-cdc2 (Tyr15), cyclin B1, β-actin, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were obtained from Cell Signaling Technology (Beverly, MA). Antibody against fast myosin heavy chain (MHC) was obtained from Novocastra Laboratories (Newcastle, UK). Antibodies against myogenin and p21 Waf-1/Cip-1 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
Low-temperature atmospheric plasma
Low-temperature atmospheric plasma was generated by a microwave-type plasma torch (AD-TEC Plasma Technology Co., Hiroshima, Japan) (Fig. 1). The torch has a stainless steel electrode placed inside an aluminum cylinder. Microwave power at 2.45 GHz frequency was applied to the electrode via a three-stub tuner and coaxial cables. The microwave power can be set at 0–15 W. Argon gas is used to produce plasma at the flow rate of 1 standard liter/min. The distance between the surface of the culture medium and the tip of the plasma was approximately 7 mm.
Fig. 1.

Photograph of the low-temperature atmospheric plasma device. C2C12 cells were cultured in 0.5 mL of growth or differentiation medium and treated with plasma exposure for 1 min
C2C12 culture
C2C12 cells (a mouse myoblast cell line) were obtained from the Riken BRC Cell Bank (Ibaraki, Japan). C2C12 cells were plated in a four-well dish (1.9 cm2/well, Thermo Fisher Scientific, Yokohama, Japan) in 0.5 mL of Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 4.5 g/L glucose, 10 % FBS, 100 units/mL penicillin, and 100 mg/mL streptomycin sulfate (growth medium) in a 5 % CO2-humidified chamber at 37 °C. Growing cells were exposed to plasma for 1 min (10 s × six different areas) every 24 h (Plas group). The growth medium was changed every 24 h prior to plasma exposure. Two different control groups were set; one was an incubator control without any treatment (IC group), and the other was treated with argon gas flow only (Cont group). To evaluate the indirect effect of plasma exposure through culture media, the culture medium of the control cells were replaced with plasma-exposed medium (Med group).
Effect of antioxidant, N-acetyl-l-cysteine (NAC) on plasma-exposure was investigated. One hour prior to plasma exposure, culture medium was changed to media containing 0.2 or 2 mM NAC. The cells were then exposed to plasma for 1 min (10 s × six different areas) every 24 h. The medium containing NAC was changed every 24 h prior to plasma exposure.
To investigate the effect of plasma exposure on cell differentiation, the culture medium was changed to 0.5 mL of differentiation medium (DMEM supplemented with 1.5 g/L glucose, 2 % HS, 100 units/mL penicillin, and 100 mg/mL streptomycin sulfate) when the cells had reached approx. 100 % confluence. The cells were then exposed to plasma for 1 min (10 s × six different areas) every 24 h. The differentiation medium was changed every 24 h prior to plasma exposure.
Cell proliferation assay
Cell proliferation was examined by counting the numbers of cells with trypan blue staining. At the indicated time, plasma-exposed cells and control cells were recovered by 0.25 % trypsin-EDTA solution. Cells were stained with 0.2 % trypan blue/phosphate-buffered saline (PBS) for 10 min. Stained and non-stained cells were counted under a microscope using a hemocytometer.
SDS-PAGE and immunoblotting
Cells were rinsed with cold PBS and lysed in RIPA lysis buffer (Santa Cruz Biotechnology). Equal amounts of protein were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotting. Separated proteins were transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA). Nonspecific binding to the membrane was blocked with Tris-buffered saline (pH 7.4) containing 0.05 % (v/v) Tween-20 and 5 % (w/v) bovine serum albumin (BSA) or 5 % (w/v) skim milk. Following an overnight incubation with the indicated primary antibody, the membranes were washed and then incubated with horseradish peroxidase-conjugated anti-rabbit IgG or anti-mouse IgG as appropriate. Immunoreactive protein bands were visualized by ECL-prime (GE Healthcare UK, Buckinghamshire, UK) with an ECL mini-camera (GE Healthcare UK). The band intensity was quantified by a computer analysis package (NIH ImageJ).
Cell-cycle analysis by flow cytometry
The measurement of the cell-cycle progression was performed using the Cell Cycle Phase Determination Kit (Cayman Chemical Co., Ann Arbor, MI). Briefly, plasma-exposed cells and control cells were recovered by 0.25 % trypsin-EDTA solution and washed twice with assay buffer. Then, cells were resuspended in fixative solution for at least 2 h. Fixed and permeated cells were collected and treated with staining solution containing propidium iodide (PI) and RNase A for 30 min. Stained cells were analyzed using a FACS CantoII Flow Cytometer (BD Biosciences, San Jose, CA).
Statistical analyses
All values are expressed as mean ± SD. Statistical analyses for multiple comparisons were performed using a one- or two-way analysis of variance (ANOVA) followed by Scheffe’s post-hoc test. Comparisons between the control and plasma-exposed groups were made using the unpaired Student’s t test. Differences were considered significant at p < 0.05.
Results
Effect of plasma exposure on cell proliferation
One minute of plasma exposure every 24 h inhibited the proliferation of the C2C12 cells, without cell death. Cell viability was assayed by trypan blue staining, and the numbers of stained cells (0–2 cells/100 cells) were not different among the four groups. The cell numbers in the plasma-exposure (Plas) groups were significantly lower than IC, Cont, and Med groups at 1 and 2 days after the plasma exposure (Fig. 2A). The total protein yield per well was significantly decreased in the Plas groups at 2 days after the plasma exposure (Fig. 2B). An inhibitory effect of plasma exposure on cell proliferation was also observed by bright-field microscopy images (Fig. 3).
Fig. 2.
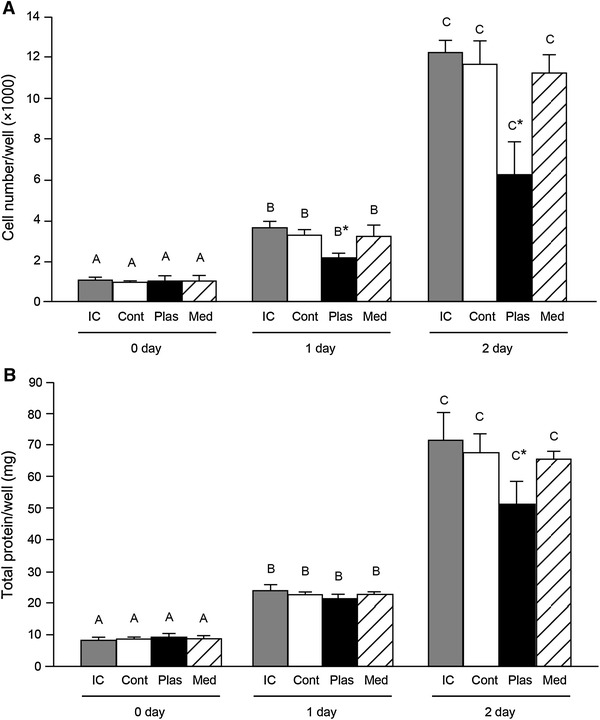
Plasma exposure reduced the cell proliferation. C2C12 cells were treated with plasma for 1 min at 3.5 W every 24 h. A Effect of plasma exposure on cell numbers per well. At the indicated day of 2 h after the plasma exposure, cells were recovered by 0.25 % trypsin-EDTA solution. Cells were stained with 0.2 % trypan blue/PBS for 10 min. Non-stained (viable) cells were counted under a microscope using a hemocytometer (n = 4–6 per group). B Total protein content in recovered cells per well was determined by a BCA protein assay. IC incubator control cells, Cont the cells were treated with argon gas flow without plasma, Plas the plasma-exposed cells, Med the culture medium of control cells were replaced with plasma-exposed medium. *Significantly different from IC, Cont, and Med groups on the same day (p < 0.05). Values with different letters are significantly different (p < 0.05)
Fig. 3.
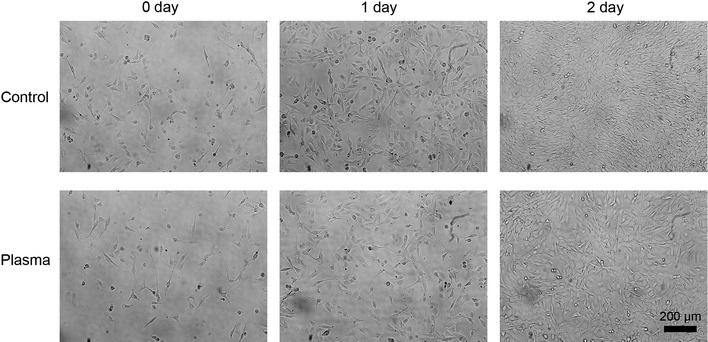
Images of control and plasma-exposed cells in proliferative phase. Bright-field images of control (argon gas flow only) and plasma-exposed C2C12 cells at 0, 1, and 2 days after plasma exposure (×20) in growth medium
Effect of antioxidant NAC on plasma exposure-induced inhibition of cell proliferation
To investigate the mechanisms for the inhibitory effect of cell proliferation by plasma exposure, the cells were pretreated with non-specific antioxidant NAC prior to plasma exposure. Treatment of NAC at 0.2 or 2 mM did not rescue the inhibitory effect of plasma exposure on cell proliferation (Fig. 4A, B).
Fig. 4.
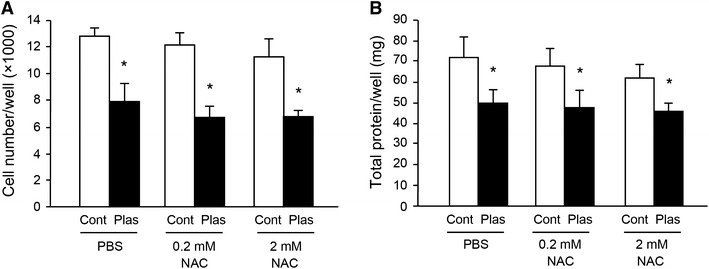
Antioxidant NAC did not rescue the reduction of cell proliferation by plasma exposure. C2C12 cells were pretreated with antioxidant N-acetyl-l-cysteine (NAC) at 0.2 or 2 mM prior to plasma exposure (n = 4–6). Plasma exposure was performed as noted in the legend of Fig. 2 for 2 days. Viable cell number/well was counted under a microscope (A). Total protein content in recovered cells per well was determined by a BCA protein assay (B). *Significantly different from control (Cont; argon gas flow only) groups at same treatment (p < 0.05)
Effect of plasma exposure on cell differentiation
Cell differentiation was induced with differentiation medium containing 2 % HS instead of 10 % FBS. The expressions of the two marker proteins for differentiation, myogenin (Fig. 5A, C) and MHC (Fig. 5A, D), were not different between the Cont and Plas groups at both 1 and 3 days after the plasma exposure. The total protein yield per well was not significantly different between the Cont and Plas groups at 1 and 3 days after the plasma exposure (Fig. 5B). The observation by bright-field images indicated that plasma exposure did not affect the myotube formation (Fig. 6).
Fig. 5.
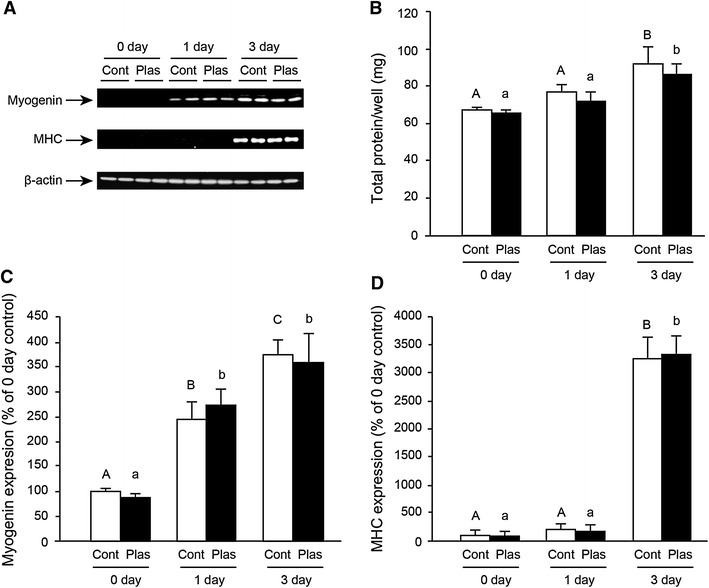
Plasma exposure did not affect cell differentiation. C2C12 cells at approx. 100 % confluence were cultured in differentiation medium, which was changed every 24 h. Cells were treated with plasma for 1 min at 3.5 W every 24 h. Cells were lysed at 0, 1, and 3 days of culture in differentiation medium (n = 4–6). Representative western blots showing myogenin, fast myosin heavy chain (MHC), and β-actin proteins in control (Cont; argon gas flow only) and plasma exposed (Plas) cells are shown in (A). The total protein content in recovered cells was measured by BCA protein assay (B). The expressions of myogenin (C) and MHC (D) were analyzed by densitometry for quantification. Values with different letters are significantly different (p < 0.05)
Fig. 6.
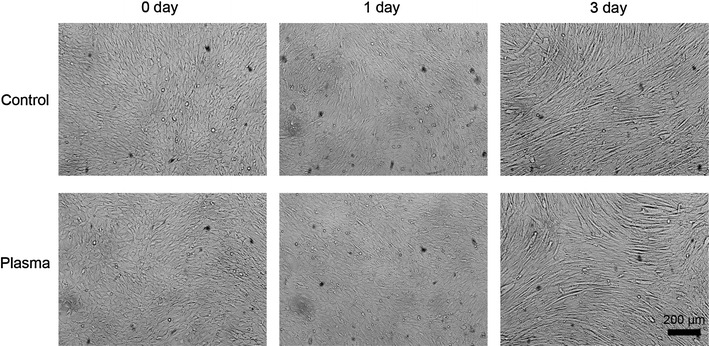
Images of control and plasma-exposed cells in differentiation phase. Bright-field images of control (argon gas flow only) and plasma-exposed C2C12 cells at 0, 1, and 3 days after plasma exposure (×20) in differentiation medium
Effect of plasma exposure on the phosphorylation of ERK and JNK
To investigate the mechanisms involved in plasma exposure, we examined the phosphorylation of ERK and JNK because both enzymes quickly respond to external stimuli and play an important role in signal transduction. Proliferating C2C12 cells were exposed to 1 min of low-temperature atmospheric plasma at 3.5 or 4.5 W. Plasma exposure significantly increased the phosphorylations of ERK (Fig. 7A) and JNK (Fig. 7B) in an electrical power-dependent manner. However, time-course experiments revealed that the phosphorylations of ERK (Fig. 8A) and JNK (Fig. 8B) were decreased markedly to less than the control levels at 1 and 4 h after the plasma exposure, and returned to the control levels at 24 h.
Fig. 7.
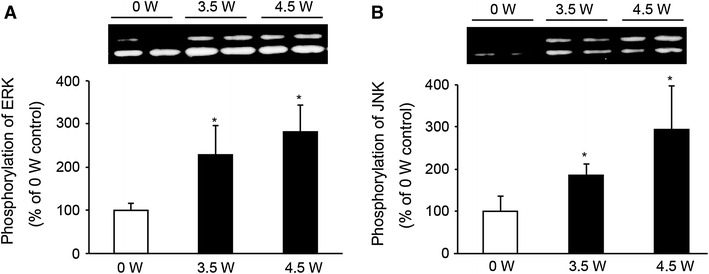
Plasma exposure increased the phosphorylation of ERK and JNK. C2C12 cells were treated with plasma exposure and electrical power at 0, 3.5, or 4.5 W for 1 min, and at 30 min after the exposure, the cells were lysed with RIPA lysis buffer. The phosphorylations of ERK (A) and JNK (B) were analyzed by western blotting (n = 4–6). *Significantly different from 0 W (argon gas flow only) group (p < 0.05)
Fig. 8.
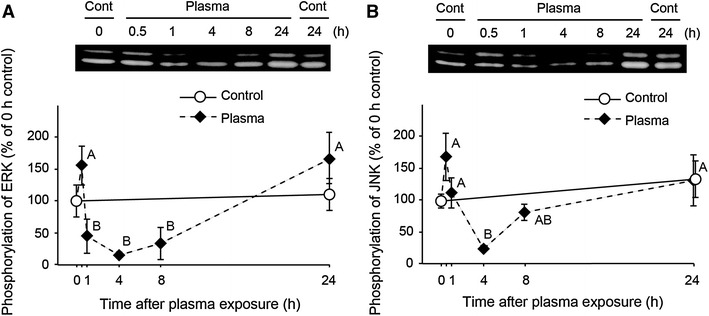
Time-course analysis of the phosphorylation of ERK and JNK in response to plasma exposure. Cells were treated with plasma exposure (3.5 W) for 1 min and returned to the CO2 incubator. Cells were lysed at 0, 0.5, 1, 4, 8, and 24 h after the plasma exposure. The phosphorylations of ERK (A) and JNK (B) were analyzed by western blotting (n = 4–6 for each time points). Values with different letters are significantly different (p < 0.05)
Effect of plasma exposure on cell-cycle progression
Cell-cycle progression was analyzed by flow cytometry. One minute of plasma exposure significantly affected the percentage of cells in cell-cycle phases at 8 h after the exposure (Fig. 9A, B). Plasma exposure decreased the percentage of cells in G0/G1 and S phases and increased those in the G2/M phase at 8 h after the exposure (Fig. 9C). At 24 h after plasma exposure, the percentage of cells in the cell-cycle phase was not different between the control and plasma-exposed cells (Fig. 9D).
Fig. 9.
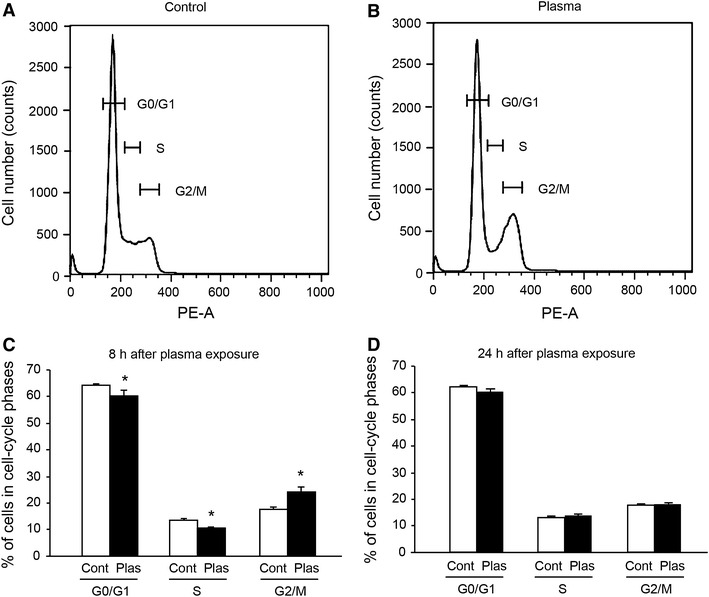
Plasma exposure affected the percentage of cells in cell-cycle phases. Cells were treated with plasma exposure (3.5 W) for 1 min and returned to the CO2 incubator. Cells were lysed at 8 and 24 h after the plasma exposure. Flow cytometry plots of DNA content were measured by PI staining. Representative histograms from flow cytometry for the control (argon gas flow only) (A) and plasma-exposed (Plas) (B) cells at 8 h after exposure are shown. The percentages of cells in each cell-cycle phase at 8 h (C) and at 24 h (D) after plasma exposure (n = 5–6) are also shown. *Significantly different from Cont (argon gas flow only) group (p < 0.05)
To further characterize the plasma exposure-induced G2/M arrest, we measured the expression of cell-cycle checkpoint proteins (Fig. 10). Immunoblots revealed increased levels of phospho-cdc2 (Tyr15) in plasma-exposed cells (Fig. 10B), suggesting that the plasma exposure inhibited the cyclin B1/cdc2 complex by increasing the phosphorylation of cdc2 (inactive), despite the elevated levels of cyclin B1 (Fig. 10C). The expression of p21 Waf-1/Cip-1, which is known to control the entry of cells at the G2/M phase transition checkpoint, was also elevated in the plasma-exposed cells (Fig. 10D).
Fig. 10.
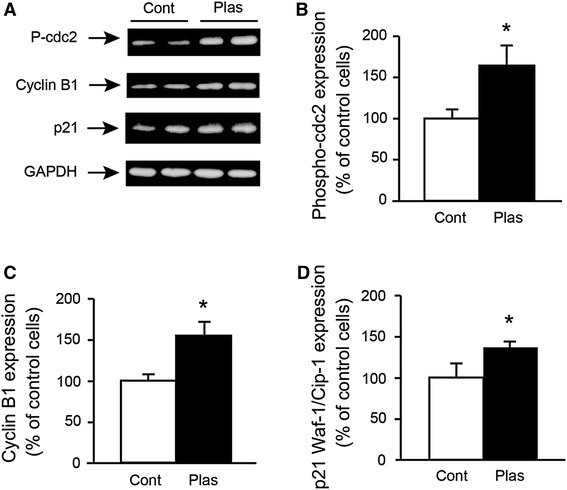
Plasma exposure affected the expression of cell-cycle checkpoint proteins. Cells were treated with plasma exposure (3.5 W) for 1 min, and at 8 h after plasma exposure, the cells were lysed with RIPA lysis buffer and subjected to western blot analysis for phospho (P)-cdc2, cyclin B1, p21 Waf-1/Cip-1 (p21) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (A). Effect of plasma exposure on the expression of phospho-cdc2 (B), cycline B1 (C) and p21 Waf-1/Cip-1 S (D) were analyzed by densitometry for quantification (n = 4–6). *Significantly different from Cont (argon gas flow only) group (p < 0.05)
Discussion
The results of the present study demonstrate that 1 min of low-temperature atmospheric plasma exposure every 24 h retarded the proliferation of C2C12 myoblasts, without cell death. The effect of plasma exposure was apparent only in the proliferative phase of the cells; the differentiation of the cells was not affected by plasma exposure.
We measured the effect of plasma exposure on cell proliferation by counting the cell numbers. The cell numbers in the plasma-exposed group were significantly lower than those of the other groups. The proliferation of the cells treated with argon gas flow without plasma (Cont group) was not different from incubator control (IC group). In addition, the proliferation of the cells was not affected by the plasma-exposed culture medium. These results suggest that retardation of cell proliferation was due to the direct effect of plasma exposure. We assessed the effect of plasma exposure on cell viability by trypan blue straining, and we found no difference among the four groups (0–2 cells/100 cells). These results suggest that the inhibition of cell proliferation by plasma exposure was not caused by cell death. Previous studies demonstrated that non-thermal plasma produces dose-dependent effects that range from increased cell proliferation to apoptosis in epithelia and endothelial cells [18, 19]. In the present study, we applied 1 min of plasma exposure at 3.5 W every 24 h to the cells. This exposure condition might be too severe for the proliferation of C2C12 cells. The effect of shorter and milder conditions of plasma exposure should be examined in future experiments.
The effects of plasma exposure were reported to being due to active species—mainly oxygen/hydroxyl radicals and nitric oxide—generated in the plasma [8, 20]. To examine the involvement of reactive oxygen species (ROS), which are generated by plasma exposure, the cells were pretreated with non-specific antioxidant NAC prior to plasma exposure. It has been reported that arsenic trioxide-induced reduction of C2C12 cell viability was attributed to the generation of ROS, and pretreatment of NAC reversed this effect [21]. However, in the present study, plasma exposure-induced retardation of cell proliferation was not attenuated by pretreatment with NAC. This result suggests that ROS are not major contributors for the plasma exposure-induced inhibition of cell proliferation.
To investigate the mechanisms involved in plasma exposure, we first examined the phosphorylation of MAP kinase family members ERK and JNK. MAP kinase family members are essential signal transduction components, and they are involved in various stimulatory and inhibitory signals [22, 23]. ERK and JNK are activated (phosphorylated) by growth factors, cytokines, and environmental stress. The activated state of ERK and JNK plays a role in cell survival and/or cell proliferation [24–27]. Here, 1 min of plasma exposure increased the phosphorylation of ERK and JNK at 30 min after the exposure. However, the phosphorylation of ERK and JNK was markedly decreased to less than control levels at 1 and 4 h after the plasma exposure, respectively, and returned to the control levels at 24 h. These results suggest that the predominant effect of plasma exposure on ERK and JNK was inactivation (dephosphorylation) of the enzymes. The inactivation of ERK and JNK may be involved in the retardation of cell proliferation by plasma exposure. This hypothesis is supported by studies showing that the downmodulation of ERK and JNK activity inhibits the proliferation of various cell types (reviewed in [26]). It was also reported that polyunsaturated fatty acid decreased C2C12 cell proliferation through an MAPK-ERK pathway [28]. Inhibition of JNK and phosphatidylinositol 3-kinase pathways blocked the proliferation of pulmonary artery smooth muscle cells in response to growth factor [29].
Our flow cytometric analysis revealed that at 8 h after the 1-min plasma exposure, the proportion of cells in the G2/M phase was increased, suggesting that plasma exposure inhibited the cell proliferation by cell-cycle arrest at the G2/M phase. On the other hand, at 24 h after the exposure, the proportion of the cells in each cell-cycle phase was not different between the control and plasma-exposed cells. The reduced level of ERK and JNK phosphorylation in the plasma-exposed cells also returned to the control level at 24 h after the exposure. These results suggest that the effect of the plasma exposure applied in this study is temporary and reversible.
The progression of cell-cycle phases is under the control of the cyclins and their partner kinases, the cyclin-dependent kinases (CDKs) [30]. The transition from G2 phase into mitosis is controlled by the activation of the cyclin B1/cdc2 complex [31]. Activation of cdc2 (also known as CDK1) is prevented by its phosphorylation at Thr14/Tyr 15 by the protein kinase Wee1 and Myt1 [32, 33]. In the present study, the phosphorylation of cdc2 at Tyr 14 was significantly increased in the plasma-exposed cells, further suggesting that the plasma exposure induced G2/M arrest by downregulation of the cyclin B1/cdc2 complex. Disruption of cell-cycle progression in the plasma-exposed cells was also confirmed by the upregulation of p21, a member of the Cip/Kip family of cyclin-dependent kinase inhibitors involved in cell-cycle regulation [34, 35].
In contrast to the inhibitory effect of plasma exposure on cell proliferation, the differentiation of these myoblasts to myotubes was not affected. The expression of myogenin, a marker for the entry of myoblasts into the differentiation pathway, was not different between the control and plasma-exposed cells. MHC, another marker of differentiation, was also not affected by the plasma exposure. The differentiation of muscle cells and the subsequent myotube formation depend not only on muscle transcription factors’ expression and activity but also on irreversible G0/G1 cell-cycle arrest [36, 37]. When C2C12 cells were cultured in differentiation medium, the cell cycle was already arrested by the low-mitogen concentration in the medium. This may be one reason why the plasma exposure did not affect the cell differentiation.
From a clinical point of view, low-temperature atmospheric plasma exposure can be applied for the treatment of RMS and other soft tissue sarcomas. Intraoperative plasma exposure is a possible candidate for treatment instead of radiation, because it inhibited growth of the cells in the proliferative phase and did not affect the cells in the differentiation phase. Thus, plasma exposure may specifically act on uncontrolled proliferation of cancer cells. More detailed studies in vitro and animal studies are needed to investigate the potentiality of low-temperature atmospheric plasma exposure against soft tissue sarcoma.
In conclusion, low-temperature atmospheric plasma exposure retarded the proliferation of C2C12 myoblasts, without cell death, but it had no effect on the cells’ differentiation. The inhibition of cell proliferation in the plasma-exposed cells was caused by the induction of G2/M cell-cycle arrest. Downregulation of ERK and JNK activity and modulation of cell-cycle checkpoint proteins may be involved in plasma exposure-induced cell-cycle arrest.
Acknowledgments
We thank S. Yoshida, T. Fukuda, T. Ogawa, S. Yamamoto, T. Uchida, and J. Mano (Kyoto Institute of Technology) for their help with the feasibility studies. This work was supported by a Grant-in-Aid for Challenging Exploratory Research (23650405 to N.N.) from the Japan Society for the Promotion of Science (JSPS).
Conflict of interest
The authors declare that there are no conflicts of interest.
References
- 1.Meyer-Plath AA, Schroder K, Finke B, Ohl A. Current trends in biomaterial surface functionalization-nitrogen-containing plasma assisted processes with enhanced selectivity. Vacuum. 2003;71:391–406. doi: 10.1016/S0042-207X(02)00766-2. [DOI] [Google Scholar]
- 2.Sardella E, Detomaso L, Gristina R, Senesi GS, Agheli H, Sutherland DS, d’Agostino R, Favia P. Nano-structured cell-adhesive and cell-repulsive plasma-deposited coatings: Chemical and topographical effects on keratinocyte adhesion. Plasma Process Polym. 2008;5:540–551. doi: 10.1002/ppap.200800005. [DOI] [Google Scholar]
- 3.Sarrette JP, Cousty S, Clement F, Canal C, Ricard A. Biological decontamination using high and reduced pressure nitrogen afterglows. Plasma Process Polym. 2012;9:576–584. doi: 10.1002/ppap.201100096. [DOI] [Google Scholar]
- 4.Sarrette JP, Cousty S, Merbahi N, Negre-Salvayre A, Clement F. Observation of antibacterial effects obtained at atmospheric and reduced pressures in afterglow conditions. Eur Phys J-Appl Phys. 2010;49:13108–13111. doi: 10.1051/epjap/2009169. [DOI] [Google Scholar]
- 5.Yousfi M, Merbahi N, Pathak A, Eichwald O. Low-temperature plasmas at atmospheric pressure: toward new pharmaceutical treatments in medicine. Fundam Clin Pharmacol. 2013;28(2):123–135. doi: 10.1111/fcp.12018. [DOI] [PubMed] [Google Scholar]
- 6.Fridman G, Friedman G, Gutsol A, Shekhter AB, Vasilets VN, Fridman A. Applied plasma medicine. Plasma Process Polym. 2008;5:503–533. doi: 10.1002/ppap.200700154. [DOI] [Google Scholar]
- 7.Kong MG, Kroesen G, Morfill G, Nosenko T, Shimizu T, van Dijk J, Zimmermann JL (2009) Plasma medicine: an introductory review. New J Phys 11. doi:10.1088/1367-2630/11/11/115012
- 8.Kieft IE, Kurdi M, Stoffels E. Reattachment and apoptosis after plasma-needle treatment of cultured cells. IEEE T Plasma Sci. 2006;34:1331–1336. doi: 10.1109/TPS.2006.876511. [DOI] [Google Scholar]
- 9.Kieft IE, Darios D, Roks AJM, Stoffels E. Plasma treatment of mammalian vascular cells: a quantitative description. IEEE T Plasma Sci. 2005;33:771–775. doi: 10.1109/TPS.2005.844528. [DOI] [Google Scholar]
- 10.Kalghatgi SU, Fridman G, Fridman A, Friedman G, Clyne AM. Non-thermal dielectric barrier discharge plasma treatment of endothelial cells. Annual international conference of the IEEE engineering in medicine and biology society conference. 2008;2008:3578–3581. doi: 10.1109/IEMBS.2008.4649979. [DOI] [PubMed] [Google Scholar]
- 11.Vandamme M, Robert E, Pesnel S, Barbosa E, Dozias S, Sobilo J, Lerondel S, Le Pape A, Pouvesle JM. Antitumor effect of plasma treatment on U87 glioma xenografts: preliminary results. Plasma Process Polym. 2010;7:264–273. doi: 10.1002/ppap.200900080. [DOI] [Google Scholar]
- 12.Utsumi F, Kajiyama H, Nakamura K, Tanaka H, Mizuno M, Ishikawa K, Kondo H, Kano H, Hori M, Kikkawa F. Effect of indirect nonequilibrium atmospheric pressure plasma on anti-proliferative activity against chronic chemo-resistant ovarian cancer cells in vitro and in vivo. PLoS ONE. 2013;8:e81576. doi: 10.1371/journal.pone.0081576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sensenig R, Kalghatgi S, Cerchar E, Fridman G, Shereshevsky A, Torabi B, Arjunan KP, Podolsky E, Fridman A, Friedman G, Azizkhan-Clifford J, Brooks AD. Non-thermal plasma induces apoptosis in melanoma cells via production of intracellular reactive oxygen species. Ann Biomed Eng. 2011;39:674–687. doi: 10.1007/s10439-010-0197-x. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 14.Fridman G, Shereshevsky A, Jost MM, Brooks AD, Fridman A, Gutsol A, Vasilets V, Friedman G. Floating electrode dielectric barrier discharge plasma in air promoting apoptotic behavior in melanoma skin cancer cell lines. Plasma Chem Plasma P. 2007;27:163–176. doi: 10.1007/s11090-007-9048-4. [DOI] [Google Scholar]
- 15.Parham DM, Barr FG. Classification of rhabdomyosarcoma and its molecular basis. Adv Anat Pathol. 2013;20:387–397. doi: 10.1097/PAP.0b013e3182a92d0d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nag S, Shasha D, Janjan N, Petersen I, Zaider M, American Brachytherapy S. The American Brachytherapy Society recommendations for brachytherapy of soft tissue sarcomas. Int J Radiat Oncol Biol Phys. 2001;49:1033–1043. doi: 10.1016/S0360-3016(00)01534-0. [DOI] [PubMed] [Google Scholar]
- 17.Yaffe D, Saxel O. Serial passaging and differentiation of myogenic cells isolated from dystrophic mouse muscle. Nature. 1977;270:725–727. doi: 10.1038/270725a0. [DOI] [PubMed] [Google Scholar]
- 18.Kalghatgi S, Kelly CM, Cerchar E, Torabi B, Alekseev O, Fridman A, Friedman G, Azizkhan-Clifford J. Effects of non-thermal plasma on mammalian cells. PLoS ONE. 2011;6:e16270. doi: 10.1371/journal.pone.0016270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kalghatgi S, Friedman G, Fridman A, Clyne AM. Endothelial cell proliferation is enhanced by low dose non-thermal plasma through fibroblast growth factor-2 release. Ann Biomed Eng. 2010;38:748–757. doi: 10.1007/s10439-009-9868-x. [DOI] [PubMed] [Google Scholar]
- 20.Stoffels E, Roks AJM, Deelmm LE. Delayed effects of cold atmospheric plasma on vascular cells. Plasma Process Polym. 2008;5:599–605. doi: 10.1002/ppap.200800028. [DOI] [Google Scholar]
- 21.Yen YP, Tsai KS, Chen YW, Huang CF, Yang RS, Liu SH. Arsenic induces apoptosis in myoblasts through a reactive oxygen species-induced endoplasmic reticulum stress and mitochondrial dysfunction pathway. Arch Toxicol. 2012;86:923–933. doi: 10.1007/s00204-012-0864-9. [DOI] [PubMed] [Google Scholar]
- 22.Lange-Carter CA, Pleiman CM, Gardner AM, Blumer KJ, Johnson GL. A divergence in the MAP kinase regulatory network defined by MEK kinase and Raf. Science. 1993;260:315–319. doi: 10.1126/science.8385802. [DOI] [PubMed] [Google Scholar]
- 23.Minden A, Lin A, Claret FX, Abo A, Karin M. Selective activation of the JNK signaling cascade and c-Jun transcriptional activity by the small GTPases Rac and Cdc42Hs. Cell. 1995;81:1147–1157. doi: 10.1016/S0092-8674(05)80019-4. [DOI] [PubMed] [Google Scholar]
- 24.Yujiri T, Sather S, Fanger GR, Johnson GL. Role of MEKK1 in cell survival and activation of JNK and ERK pathways defined by targeted gene disruption. Science. 1998;282:1911–1914. doi: 10.1126/science.282.5395.1911. [DOI] [PubMed] [Google Scholar]
- 25.Zhang W, Liu HT. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002;12:9–18. doi: 10.1038/sj.cr.7290105. [DOI] [PubMed] [Google Scholar]
- 26.Wilkinson MG, Millar JB. Control of the eukaryotic cell cycle by MAP kinase signaling pathways. FASEB J. 2000;14:2147–2157. doi: 10.1096/fj.00-0102rev. [DOI] [PubMed] [Google Scholar]
- 27.Grosch S, Tegeder I, Schilling K, Maier TJ, Niederberger E, Geisslinger G. Activation of c-Jun-N-terminal-kinase is crucial for the induction of a cell cycle arrest in human colon carcinoma cells caused by flurbiprofen enantiomers. FASEB J. 2003;17:1316–1318. doi: 10.1096/fj.02-0919fje. [DOI] [PubMed] [Google Scholar]
- 28.Peng Y, Zheng Y, Zhang Y, Zhao J, Chang F, Lu T, Zhang R, Li Q, Hu X, Li N. Different effects of omega-3 fatty acids on the cell cycle in C2C12 myoblast proliferation. Mol Cell Biochem. 2012;367:165–173. doi: 10.1007/s11010-012-1329-4. [DOI] [PubMed] [Google Scholar]
- 29.Garat CV, Fankell D, Erickson PF, Reusch JE, Bauer NN, McMurtry IF, Klemm DJ. Platelet-derived growth factor BB induces nuclear export and proteasomal degradation of CREB via phosphatidylinositol 3-kinase/Akt signaling in pulmonary artery smooth muscle cells. Mol Cell Biol. 2006;26:4934–4948. doi: 10.1128/MCB.02477-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Obaya AJ, Sedivy JM. Regulation of cyclin-Cdk activity in mammalian cells. Cell Mol Life Sci. 2002;59:126–142. doi: 10.1007/s00018-002-8410-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Castedo M, Perfettini JL, Roumier T, Kroemer G. Cyclin-dependent kinase-1: linking apoptosis to cell cycle and mitotic catastrophe. Cell Death Differ. 2002;9:1287–1293. doi: 10.1038/sj.cdd.4401130. [DOI] [PubMed] [Google Scholar]
- 32.Mueller PR, Coleman TR, Kumagai A, Dunphy WG. Myt1: a membrane-associated inhibitory kinase that phosphorylates Cdc2 on both threonine-14 and tyrosine-15. Science. 1995;270:86–90. doi: 10.1126/science.270.5233.86. [DOI] [PubMed] [Google Scholar]
- 33.Russell P, Nurse P. Negative regulation of mitosis by wee1+, a gene encoding a protein kinase homolog. Cell. 1987;49:559–567. doi: 10.1016/0092-8674(87)90458-2. [DOI] [PubMed] [Google Scholar]
- 34.el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-P. [DOI] [PubMed] [Google Scholar]
- 35.Dotto GP. p21(WAF1/Cip1): more than a break to the cell cycle? Biochim Biophys Acta. 2000;1471:M43–56. doi: 10.1016/s0304-419x(00)00019-6. [DOI] [PubMed] [Google Scholar]
- 36.Gu W, Schneider JW, Condorelli G, Kaushal S, Mahdavi V, Nadal-Ginard B. Interaction of myogenic factors and the retinoblastoma protein mediates muscle cell commitment and differentiation. Cell. 1993;72:309–324. doi: 10.1016/0092-8674(93)90110-C. [DOI] [PubMed] [Google Scholar]
- 37.Joulia D, Bernardi H, Garandel V, Rabenoelina F, Vernus B, Cabello G. Mechanisms involved in the inhibition of myoblast proliferation and differentiation by myostatin. Exp Cell Res. 2003;286:263–275. doi: 10.1016/S0014-4827(03)00074-0. [DOI] [PubMed] [Google Scholar]