

Abstract
A single bout of prolonged endurance exercise stimulates glucose transport in skeletal muscles, leading to post-exercise muscle glycogen supercompensation if sufficient carbohydrate is provided after the cessation of exercise. Although we recently found that short-term sprint interval exercise also stimulates muscle glucose transport, the effect of this type of exercise on glycogen supercompensation is uncertain. Therefore, we compared the extent of muscle glycogen accumulation in response to carbohydrate feeding following sprint interval exercise with that following endurance exercise. In this study, 16-h-fasted rats underwent a bout of high-intensity intermittent swimming (HIS) as a model of sprint interval exercise or low-intensity prolonged swimming (LIS) as a model of endurance exercise. During HIS, the rats swam for eight 20-s sessions while burdened with a weight equal to 18% of their body weight. The LIS rats swam with no load for 3 h. The exercised rats were then refed for 4, 8, 12, or 16 h. Glycogen levels were almost depleted in the epitrochlearis muscles of HIS- or LIS-exercised rats immediately after the cessation of exercise. A rapid increase in muscle glycogen levels occurred during 4 h of refeeding, and glycogen levels had peaked at the end of 8 h of refeeding in each group of exercised refed rats. The peak glycogen levels during refeeding were not different between HIS- and LIS-exercised refed rats. Furthermore, although a large accumulation of muscle glycogen in response to carbohydrate refeeding is known to be associated with decreased insulin responsiveness of glucose transport, and despite the fact that muscle glycogen supercompensation was observed in the muscles of our exercised rats at the end of 4 h of refeeding, insulin responsiveness was not decreased in the muscles of either HIS- or LIS-exercised refed rats compared with non-exercised fasted control rats at this time point. These results suggest that sprint interval exercise enhances muscle glycogen supercompensation in response to carbohydrate refeeding as well as prolonged endurance exercise does. Furthermore, in this study, both HIS and LIS exercise prevented insulin resistance of glucose transport in glycogen supercompensated muscle during the early phase of carbohydrate refeeding. This probably led to the enhanced muscle glycogen supercompensation after exercise.
Keywords: Insulin, Glucose uptake, Akt, Exercise, Epitrochlearis
Introduction
A single bout of prolonged endurance exercise stimulates glucose transport in skeletal muscle independently of the insulin signaling pathway [1]. This effect is evident during and immediately after exercise, but reverses progressively, with little or no residual effect being found at 2–3 h after exercise in rats [1–3]. As the acute increase in glucose transport reverses after the cessation of endurance exercise, there is a substantial increase in insulin-stimulated muscle glucose transport [1–4]. As a result of the exercise-induced increase in glucose transport, glucose floods into the muscles, leading to muscle glycogen supercompensation, if sufficient carbohydrate is provided after exercise. The term “glycogen supercompensation” refers to an increase in the muscle glycogen level far above that found in the sedentary fed state [5].
Our previous studies demonstrated that a bout of high-intensity intermittent swimming (HIS: total exercise time 160 s) as a model of sprint interval exercise increases insulin-independent glucose transport in rat epitrochlearis muscles to a much higher level than that caused by low-intensity prolonged swimming (LIS; total exercise time 3 h) as a model of endurance exercise [6, 7]. Furthermore, we previously reported that HIS exercise can enhance the insulin-stimulated glucose transport in rat epitrochlearis muscles after cessation of exercise as well as LIS exercise does [6, 7]. However, the effect of sprint interval exercise on post-exercise glycogen supercompensation is uncertain.
In our present study, we compared the extent of glycogen accumulation in rat skeletal muscle in response to carbohydrate feeding following sprint interval exercise with that following prolonged endurance exercise. We found that sprint interval exercise enhances muscle glycogen supercompensation in response to carbohydrate refeeding as well as endurance exercise does. Moreover, glycogen supercompensation is known to be associated with insulin resistance of glucose transport [8–10], which prevents the excess accumulation of glycogen in muscle cells. However, we observed that both sprint interval and endurance exercise reduced insulin resistance of glucose transport in glycogen supercompensated muscle during the early phase of carbohydrate refeeding. This probably led to the sustained increase of glucose flux into skeletal muscles and enhanced muscle glycogen supercompensation after exercise.
Materials and methods
Materials
Purified human insulin was purchased from Eli Lilly Japan (Kobe, Japan). Rabbit anti-Akt, anti-phospho-Akt (Ser473), and anti-phospho-Akt (Thr308) were purchased from Cell Signaling Technology (Beverly, MA). Rabbit anti-GLUT4 was from Biogenesis (Poole, UK), and horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG was from Biosource International (Camarillo, CA). Enhanced chemiluminescence (ECL or ECL plus) kits from Amersham were used for immunodetection according to the manufacturer’s instructions. BCA assay kits for determination of total protein were purchased from Pierce (Rockford, IL). All other reagents were obtained from Sigma (St. Louis, MO).
Treatment of animals
This research was approved by the Animal Studies Committee of Niigata University of Health and Welfare. Four-week-old male specific pathogen-free Wistar rats were obtained from Clea Japan, Inc. (Tokyo, Japan). The animals were housed in individual cages, and fed a diet of Purina rodent laboratory chow and water ad libitum. Animals with a body weight 100–120 g were assigned randomly to either a non-exercised fasted control, non-exercised refed, HIS-exercised refed, or LIS-exercised refed group. Rats in the HIS- and LIS-exercised refed groups were accustomed to swimming for 10 min/day, 2 days before the experiment.
The non-exercised refed, HIS-exercised refed, and LIS-exercised refed animals were fasted for 16 h from 6:00 a.m. to 10:00 p.m. After 16 h fasting, the animals had free access to Purina chow (containing 530 g of carbohydrate/kg) and 10% sucrose in their drinking water for either 0 h (non-access to food and water), 4 h (until 2:00 a.m.), 8 h (until 6:00 a.m.), 12 h (until 10:00 a.m.), or 16 h (until 2:00 p.m.). Tissue samples were collected at each of these times. The rationale for adding sucrose to the drinking water was to ensure an adequate intake of carbohydrate. Just before the start of refeeding at 10:00 p.m., rats in the HIS-exercised refed group underwent eight 20-s bouts of swimming carrying a weight equal to 18% of their body weight, with a 40-s rest between bouts [6, 7, 11]. Single rats swam in a barrel filled to a depth of 30 cm with a surface area of 450 cm2. Meanwhile, just before the start of refeeding at 10:00 p.m., rats in the LIS-exercised refed group underwent swimming for 3 h without a weight, with four rats swimming simultaneously in a barrel filled to a depth of 40 cm with an average surface area of 240 cm2/rat [6, 7]. The water temperature was maintained at a constant 35°C during the swimming protocol.
The 4, 8, 12, and 16 h refed groups were time-matched with non-exercised fasted control animals, with the tissues of the control animals being collected at the same times as those of the refed animals. The time-matched non-exercised fasted control animals for the 4, 8, 12, and 16 h refed groups were fasted for 16 h from 10:00 a.m. to 2:00 a.m., from 2:00 p.m. to 6:00 a.m., from 6:00 p.m. to 10:00 a.m., and from 10:00 p.m. to 2:00 p.m., respectively.
Preparation of muscles
The animals were anesthetized with an intraperitoneal injection of pentobarbital (5 mg/100 g of body weight), followed by dissection of the epitrochlearis muscles. The epitrochlearis muscles were either clamp-frozen in liquid nitrogen for measurement of glycogen concentrations or used for subsequent incubation as described below.
Muscle incubation
The epitrochlearis muscles were incubated with shaking for 60 min at 35°C in 3 ml of oxygenated Krebs-Henseleit buffer (KHB) containing 8 mM glucose, 32 mM mannitol, and 0.1% RIA grade bovine serum albumin (BSA), in the presence or absence of 2 mU/ml purified human insulin. This concentration of insulin is considerably greater than that required to stimulate maximum glucose uptake in this preparation. The flasks were then gassed continuously with 95% O2–5% CO2 during the incubations.
Measurement of 2DG uptake rate
The non-metabolizable glucose analogue 2-deoxyglucose (2DG) is phosphorylated by hexokinase after transport into cells, but is not metabolized further. In this study, 2DG was used to estimate the rate of glucose transport, based on the method described by Ueyama et al. [12]. After the 60-min incubation described above, the muscles were rinsed for 20 min at 30°C in 3 ml of KHB containing 40 mM mannitol, 0.1% BSA, and 10 mU/ml of insulin (if present in the previous incubation) to remove glucose. After this rinse, the muscles were incubated at 30°C for 20 min in 3 ml of KHB containing 8 mM 2DG, 32 mM mannitol, 0.1% BSA, and 10 mU/ml of insulin (if present in the previous incubation). The flasks were gassed continuously with 95% O2–5% CO2 during the incubations. After incubation, the muscles were blotted briefly on filter paper and frozen in liquid N2. The samples were then weighed, homogenized in 0.3 M perchloric acid, and centrifuged at 1,000g. After centrifugation, the supernatant was collected and neutralized by the addition of 2 N KOH, followed by fluorometric measurement of 2-deoxyglucose-6-phosphate (2-DG-6-P) [13].
Under the above incubation conditions, intracellular accumulation of free 2-DG in muscles is negligible, while intracellular accumulation of 2-DG-6-P is linear [14]. The rate of intracellular accumulation of 2-DG-6-P (2-DG uptake rate) therefore reflects the muscle glucose transport activity [14].
Muscle glycogen
Perchloric acid extracts of muscle were assayed for glycogen by the amyloglucosidase method [15].
Immunoblotting
After the first 60-min incubation described above, the epitrochlearis muscles were blotted and then clamp frozen. The muscle samples were homogenized in ice-cold buffer containing 50 mM HEPES (pH 7.4), 150 mM NaCl, 10% glycerol, 1% Triton-X-100, 1.5 mM MgCl2, 1.0 mM EDTA, 10 mM Na4P2O7, 100 mM NaF, 2.0 mM Na3VO4, aprotinin (10 μg/ml), leupeptin (10 μg/ml), pepstatin (0.5 μg/ml), and phenylmethylsulfonyl fluoride (2 mM) [16]. The homogenates were then incubated with end-over-end rotation at 4°C for 60 min and centrifuged at 4,000g for 30 min at 4°C. Aliquots of the supernatants were treated with 2× Laemmli sample buffer containing 100 mM dithiothreitol. All samples (20 μg protein) were subjected to 10% SDS-PAGE. The resolved proteins were transferred to PVDF membranes and then blocked in 5% nonfat dry milk in TBS containing 0.1% Tween 10 (TBST), pH 7.5. After blocking, the membranes were rinsed in TBST and incubated overnight with an appropriate antibody at 4°C, followed by rinsing in TBST and incubation for 120 min with HRP-conjugated goat anti-rabbit IgG. Antibody-bound protein was visualized by enhanced chemiluminescence (ECL or ECL plus; Amersham), with the intensity of the bands being assessed using the NIH images.
Statistics
The results are expressed as the mean ± SE, with statistical significance being defined by P < 0.05. Significant differences between groups were evaluated using one-way analysis of variance (ANOVA), with a subsequent post hoc comparisons with Fisher's least significant difference analysis.
Results
Carbohydrate intake
Total carbohydrate intake, which includes sucrose in the drinking water during the first 4 h of refeeding, averaged 4.3 ± 0.3 g (n = 10) in non-exercised refed rats. Neither HIS nor LIS exercise, each of which was imposed immediately before the start of refeeding, had any effect on carbohydrate intake during the 4 h of refeeding [4.3 ± 0.2 g (n = 10) for HIS and 4.1 ± 0.2 g (n = 9) for LIS]. There was also no significant difference in carbohydrate intake during 4 h of refeeding between HIS-exercised and LIS-exercised refed rats. Furthermore, there was no significant difference in carbohydrate intake during 8, 12, and 16 h of refeeding among non-exercised refed rats [7.2 ± 0.2 g (n = 7) for 8 h refeeding, 9.1 ± 0.4 g (n = 8) for 12 h refeeding, and 10.1 ± 0.6 g (n = 7) for 16 h refeeding], HIS-exercised refed rats [7.2 ± 0.3 g (n = 7) for 8 h refeeding, 9.3 ± 0.3 g (n = 8) for 12 h refeeding, and 10.4 ± 0.5 g (n = 7) for 16 h refeeding], and LIS-exercised refed rats [6.7 ± 0.3 g (n = 7) for 8 h refeeding, 8.7 ± 0.4 g (n = 8) for 12 h refeeding, and 10.3 ± 0.1 g (n = 6) for 16 h refeeding] at any of the tested time points.
Muscle glycogen
We measured glycogen concentrations in epitrochlearis muscles. At the start of refeeding, the glycogen concentrations in the muscles of both HIS- and LIS-exercised rats were lower than those of non-exercised refed rats (Fig. 1). The muscle glycogen concentration measured immediately after HIS exercise was significantly (P < 0.05) lower compared with that after LIS exercise (Fig. 1). After 4, 8, 12, and 16 h of refeeding, the glycogen concentrations in the muscles of non-exercised refed rats were 3.0-, 3.4-, 3.4-, and 2.2-fold higher, respectively (P < 0.05), than in the muscles of non-exercised fasted controls (Fig. 1). On the other hand, at the end of 4, 8, 12, and 16 h of refeeding, the glycogen concentrations in the muscles of HIS-exercised refed rats were 3.6-, 4.5-, 3.8-, and 3.1-fold higher, respectively (P < 0.05), than in the muscles of non-exercised fasted controls (Fig. 1). Furthermore, at the end of 4, 8, 12, and 16 h of refeeding, the glycogen concentrations in the muscles of LIS-exercised refed rats were 3.3-, 4.5-, 4.0-, and 3.2-fold higher, respectively (P < 0.05), than in the muscles of non-exercised fasted controls (Fig. 1). This implied that a rapid increase in muscle glycogen levels occurred during 4 h of refeeding and that glycogen levels had peaked at the end of 8 h of refeeding in each group (Fig. 1). Furthermore, peak glycogen levels in the muscles of either HIS- or LIS-exercised refed rats were significantly higher (P < 0.05) than those in the muscles of non-exercised refed rats at the end of 8 h of refeeding (Fig. 1). On the other hand, there was no significant difference in muscle glycogen levels between HIS- and LIS-exercised refed rats at the end of 8 h of refeeding (Fig. 1).
Fig. 1.
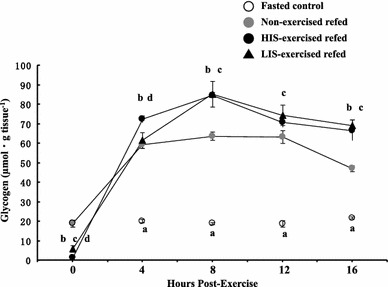
Glycogen concentrations in epitrochlearis muscles at the start of refeeding or after 4, 8, 12, or 16 h of refeeding in time-matched fasted controls, non-exercised refed rats, HIS-exercised refed rats, and LIS-exercised refed rats. Open circles time-matched fasted controls, gray circles non-exercised refed rats, filled circles HIS-exercised refed rats, filled triangles LIS-exercised refed rats. The animals were fasted for 16 h and then allowed free access to standard chow (containing 530 g carbohydrate/kg) and drinking water containing 10% sucrose for 4, 8, 12, or 16 h after the cessation of fasting. Exercised refed rats performed a bout of high-intensity intermittent swimming (HIS) or low-intensity prolonged swimming (LIS) just before the start of refeeding. The time-matched fasted control rats were fasted for 16 h before collection of the muscle samples. Values are expressed as the mean ± SE; n = 6–9 muscles/group. a Time-matched fasted controls versus the other three groups, P < 0.05. b Non-exercised refed rats versus HIS-exercised refed rats, P < 0.05. c Non-exercised refed rats versus LIS-exercised refed rats, P < 0.05. d HIS-exercised refed rats versus LIS-exercised refed rats, P < 0.05
2DG uptake
We measured the basal rate of 2DG uptake and maximum rate of insulin-stimulated 2DG uptake in the epitrochlearis muscles of time-matched non-exercised fasted controls, non-exercised refed, LIS-exercised refed, and HIS-exercised refed rats (Fig. 2).
Fig. 2.
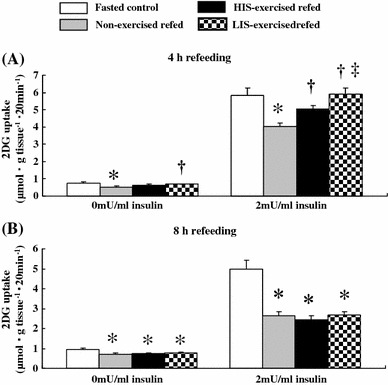
Basal and insulin-stimulated 2DG uptake rates in epitrochlearis muscles after 4 or 8 h of refeeding in time-matched fasted controls, non-exercised refed rats, HIS-exercised refed rats, and LIS-exercised refed rats. Open bars time-matched fasted controls, gray bars non-exercised refed rats, filled bars HIS-exercised refed rats, dark gray bars LIS-exercised refed rats. See the legend of Fig. 1 for a description of the animal treatment. The values are expressed as the mean ± SE; n = 6–7 muscles/group for basal 2DG uptake and n = 7–10 muscles/group for insulin-stimulated 2DG uptake. *Significant differences P < 0.05, non-exercised refed, HIS-exercised refed, or LIS-exercised refed rats versus time-matched fasted controls. †Significant differences P < 0.05, HIS-exercised refed or LIS-exercised refed rats versus non-exercised refed rats. ‡Significant differences P < 0.05, LIS-exercised refed rats versus HIS-exercised refed rats
At the end of 4 h of refeeding, the basal rate of 2DG uptake was 31% lower (P < 0.05) in the muscles of the non-exercised refed rats compared to that in the muscles of the non-exercised fasted controls (Fig. 2), whereas in LIS-exercised refed rats this uptake was 36% higher (P < 0.05) than that in the muscles of non-exercised refed rats (Fig. 2). On the other hand, at this time point, insulin-stimulated 2DG uptake was 31% lower (P < 0.05) in the muscles of the non-exercised refed rats compared to that in the muscles of the non-exercised fasted controls, whereas in both HIS- and LIS-exercised refed rats this uptake was not significantly different from that in the muscles of the non-exercised fasted controls (Fig. 2). Insulin-stimulated 2DG uptake was 24 and 46% higher (P < 0.05) in the muscles of HIS- and LIS-exercised refed rats than in the muscles of non-exercised refed rats at the end of 4 h of refeeding (Fig. 2), respectively. Insulin-stimulated 2DG uptake was 15% lower (P < 0.05) in the muscles of HIS-exercised refed rats than in those of LIS-exercised refed rats (Fig. 2).
At the end of 8 h of refeeding, the basal rate of 2DG uptake was 26, 24, and 20% lower (P < 0.05) in the muscles of non-exercised refed, HIS-exercised refed, and LIS-exercised refed rats, respectively, than in the muscles of non-exercised fasted controls (Fig. 2). After 8 h of refeeding, the insulin-stimulated 2DG uptake was 47, 51, and 46% lower (P < 0.05) in the muscles of non-exercised refed rats, HIS-exercised refed rats, and LIS-exercised refed rats, respectively, than in the muscles of non-exercised fasted controls (Fig. 2).
We also measured the insulin-independent 2DG uptake rate in the epitrochlearis muscles immediately after exercise (Table 1). The insulin-independent 2DG uptake measured immediately after HIS and LIS exercise was increased by 6.3- and 1.8-fold (P < 0.05), respectively, compared with the non-exercised fasted controls. The 2DG uptake rate was higher (P < 0.05) with HIS than with LIS (Table 1).
Table 1.
Glycogen concentrations and insulin-independent 2DG uptake rates in epitrochlearis muscles immediately after HIS or LIS exercise
| Fasted control rats | HIS-exercised rats | LIS-exercised rats | |
|---|---|---|---|
| Glycogen (μmol/g tissue) | 18.9 ± 1.9 | 1.4 ± 0.6* | 5.8 ± 1.6*,‡ |
| 2DG uptake (μmol/g tissue/20 min) | 1.01 ± 0.13 | 6.33 ± 0.22* | 1.78 ± 0.20*,‡ |
Values are mean ± SE for 7–9 muscles/group. Significant differences * P < 0.05, HIS-exercised or LIS-exercised rats versus fasted control rats. Significant differences ‡ P < 0.05, LIS-exercised rats versus HIS-exercised rats
Akt phosphorylation
We wanted to determine whether the decrease in insulin-stimulated Akt phosphorylation and 2DG uptake occurred concurrently during refeeding. At the end of 4 h of refeeding, basal phosphorylation of Ser473 was lower in the muscles of both non-exercised and HIS-exercised refed rats than in the muscles of non-exercised fasted controls (P < 0.05, Fig. 3). At the end of 4 h of refeeding, insulin-stimulated phosphorylation of Ser473 was decreased 30% in the muscles of non-exercised refed rats compared with the muscles of non-exercised fasted controls (P < 0.05, Fig. 3). In contrast, after 4 h of refeeding, insulin-stimulated phosphorylation of Ser473 was not significantly decreased in the muscles of HIS-exercised refed rats compared with the level in the muscles of non-exercised fasted controls (Fig. 3). There was no significant difference in insulin-stimulated Ser473 phosphorylation between non-exercised and HIS-exercised refed groups (P = 0.115, Fig. 3).
Fig. 3.
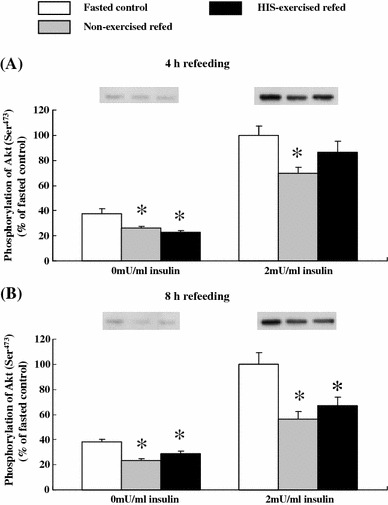
Basal and insulin-stimulated Ser473 phosphorylation of Akt in epitrochlearis muscles after 4 or 8 h of refeeding in time-matched fasted controls, non-exercised refed rats, HIS-exercised refed rats, and LIS-exercised refed rats. Open bars time-matched fasted controls, gray bars non-exercised refed rats, filled bars HIS-exercised refed rats. The mean values for insulin-stimulated phosphorylation of time-matched fasted controls were set at 100.0 arbitrary OD units. Representative Western blots of Ser473 phosphorylation of Akt are shown at the top of the figure. See the legend of Fig. 1 for a description of the animal treatment. The values are expressed as the mean ± SE; n = 6 muscles/group for basal phosphorylation and n = 6–8 muscles/group for insulin-stimulated phosphorylation. *Significant differences P < 0.05, non-exercised refed or HIS-exercised refed rats versus time-matched fasted controls
At the end of 8 h of refeeding, basal phosphorylation of Ser473 was lower in the muscles of both non-exercised and HIS-exercised refed rats compared with the level in the muscles of non-exercised fasted controls (P < 0.05, Fig. 3). At the end of 8 h of refeeding, insulin-stimulated Ser473 phosphorylation was 45% lower (P < 0.01) in the muscles of non-exercised refed rats than in the muscles of non-exercised fasted controls (Fig. 3). At this time point, insulin-stimulated phosphorylation was 44% lower for Ser473 (P < 0.05) in muscles of the HIS-exercised refed rats compared to the level in the muscles of non-exercised fasted controls (Fig. 3). The changes in insulin-stimulated Thr308 phosphorylation of Akt after 4 and 8 h of refeeding were identical to those observed for Ser473 phosphorylation (Fig. 4).
Fig. 4.
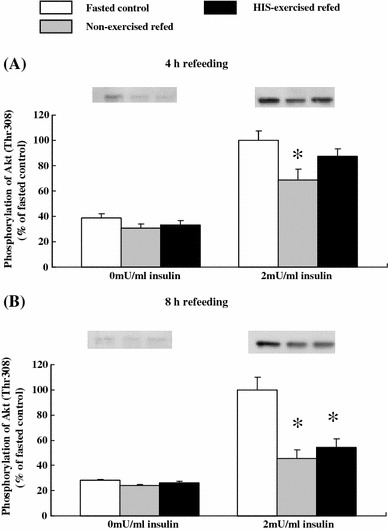
Basal and insulin-stimulated Thr308 phosphorylation of Akt in epitrochlearis muscles after 4 or 8 h of refeeding in time-matched fasted controls, non-exercised refed rats, and HIS-exercised refed rats. Open bars time-matched fasted controls, gray bars non-exercised refed rats, filled bars HIS-exercised refed rats. The mean values for insulin-stimulated phosphorylation of time-matched fasted controls were set at 100.0 arbitrary OD units. Representative Western blots of Thr308 phosphorylation of Akt are shown at the top of the figure. See the legend of Fig. 1 for a description of the animal treatment. Values are expressed as the mean ± SE; n = 6 muscles/group for basal phosphorylation and n = 6–8 muscles/group for insulin-stimulated phosphorylation. *Significant differences P < 0.05, non-exercised refed or HIS-exercised refed rats versus time-matched fasted controls
Abundance of Akt
The total amount of Akt protein in the epitrochlearis muscles of non-exercised fasted controls, non-exercised refed rats, and HIS-exercised refed rats was determined after 4 or 8 h of refeeding. No difference in the amount of Akt protein was observed among these three groups throughout the refeeding period. This result demonstrated that impaired Akt phosphorylation in glycogen supercompensated muscle was not due to changes in the abundance of Akt protein.
Abundance of SHIP2, PTEN, and SKIP
The total amount of SHIP2, PTEN, and SKIP proteins in epitrochlearis muscles of time-matched fasted controls, non-exercised refed rats, and HIS-exercised refed rats were determined after 4 or 8 h of refeeding. As shown in Fig. 5, there was no difference in SHIP2, PTEN, or SKIP among these three groups during this period.
Fig. 5.
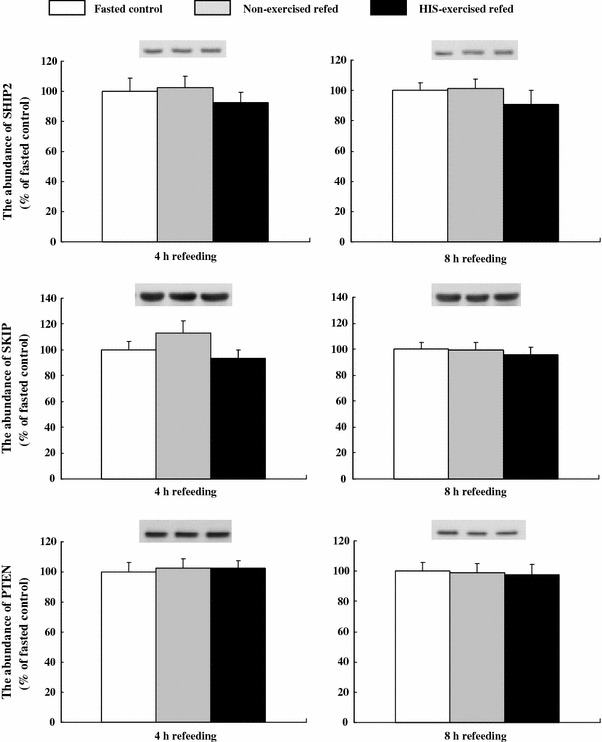
The amount of SHIP2, SKIP, and PTEN protein in epitrochlearis muscles after 4 or 8 h of refeeding in time-matched fasted controls and exercised refed rats. Open bars time-matched fasted controls, gray bars non-exercised refed rats, filled bars HIS-exercised refed rats. The mean values for time-matched fasted controls were set at 100.0 arbitrary OD units. Representative Western blots of SHIP2, SKIP, and PTEN are shown at the top of the figure. See the legend of Fig. 1 for a description of the animal treatment. Values are expressed as the mean ± SE; n = 6–8 muscles/group for SHIP2, and n = 8 muscles/group for SKIP and PTEN. SHIP2 SH2-domain containing protein, SKIP skeletal muscle and kidney enriched inositol phosphatase, PTEN phosphatase and tensin homolog deleted on chromosome 10
Abundance of GLUT4
As it has been well established that an abundance of GLUT4 protein is a determinant of glucose uptake in response to various stimuli in skeletal muscles [17], we determined the total amount of GLUT4 protein in the epitrochlearis muscles of non-exercised fasted controls, non-exercised refed rats, and HIS-exercised refed rats after 4, 8, and 16 h of refeeding. No difference in the abundance of GLUT4 protein was observed among these three groups at the three time points during refeeding. This result demonstrated that the impairment in insulin-stimulated 2DG uptake in glycogen supercompensated muscles was not due to changes in the abundance of GLUT4 protein.
Discussion
When rats underwent a bout of very short-duration (160 s) HIS or prolonged (3 h) low-intensity swimming (LIS) exercise just before the start of refeeding, glycogen supercompensation occurred at higher levels in the muscles of exercised refed rats than in those of non-exercised refed rats (Fig. 1). Furthermore, the peak glycogen level during refeeding after HIS exercise was about 85 μmol/g tissue (8 h after exercise), and this level was not different from that after LIS (Fig. 1), suggesting that sprint interval exercise supercompensates the muscle glycogen level in response to carbohydrate refeeding as well as prolonged endurance exercise does.
Muscle glycogen was depleted immediately after cessation of exercise, but was rapidly synthesized during 0–4 h of carbohydrate refeeding (Fig. 1). Glycogen synthesis rate seems to be higher after HIS than LIS during this initial 4 h of refeeding. We found that HIS increased insulin-independent glucose transport activity to much higher level than LIS immediately after cessation of exercise (Table 1), suggesting that glucose flux into muscle was larger in HIS than in LIS rats during initial phase of refeeding. This may explain the higher glycogen synthesis rate after HIS than LIS during 0–4 h of refeeding, since the rate of muscle glucose transport is one of rate-limiting for glycogen synthesis. On the other hand, the glycogen synthesis rate seems to be lower after HIS than LIS during 4–8 h of refeeding. This may be due to the lower glucose transport rate after HIS than LIS during this refeeding period. In fact, we found that insulin-dependent glucose transport activity was lower in muscles of HIS rats than in those of LIS rats at 4 h after cessation of exercise (Fig. 2). Consequently, it is likely that the total amount of glucose flooded into the muscles during 0–8 h of refeeding following HIS was not different from that following LIS, leading to the same extent of muscle glycogen accumulation during refeeding after each type of exercise.
Our present study also showed that just carbohydrate refeeding following 16 h of fasting increased the glycogen level to more than 50 μmol/g tissue, even in the muscles of non-exercised refed animals (Fig. 1). Since the glycogen level in the muscles of normal fed rats was around 24 μmol/g tissue (data not shown), these results show that muscle glycogen is supercompensated in response to carbohydrate refeeding after prolonged fasting without exercise. We previously showed that 24 h fasting induced a large increase in insulin-stimulated glucose transport in rat epitrochlearis muscle [7]. As a result of the fasting-induced increase in glucose transport, glucose floods into the muscles. This probably led to muscle glycogen supercompensation in the muscles of non-exercised refed animals.
If rats were not provided with sufficient carbohydrate after exercise or fasting, muscle glycogen supercompensation would not occur and enhancement of insulin-stimulated glucose transport activity would be sustained as long as muscle glycogen level is kept low [2]. On the other hand, when muscle glycogen supercompensation is achieved with sufficient carbohydrate provision, insulin resistance of muscle glucose transport is developed [8–10]. These earlier findings are consistent with our present observation that insulin-stimulated glucose transport in the muscles of refed rats was reduced concomitantly with an increase in the muscle glycogen level (Figs. 1, 2). The glycogen supercompensation-associated insulin resistance prevents the excess accumulation of glycogen in muscle cells. However, in the present study, at the end of 4 h of refeeding, the insulin-stimulated glucose transport was higher in the muscles of both HIS- and LIS-exercised refed rats than in the muscles of non-exercised refed rats, in spite of the larger accumulation of glycogen in the muscles of exercised refed rats (Figs. 1, 2). Thus, both HIS and LIS exercise protect against the development of insulin resistance during the early phase of muscle glycogen supercompensation, resulting in the sustained increase of glucose flux into the exercised muscles. This is probably one aspect of the mechanism of enhanced muscle glycogen supercompensation after exercise.
In our present study, the decrease in insulin-stimulated Akt phosphorylation and glucose transport occurred concurrently during refeeding in the muscles of both non-exercised and HIS-exercised rats (Figs. 3, 4). Previous studies also demonstrated that decreased insulin-stimulated glucose transport in glycogen supercompensated muscles was accompanied by decreased insulin-stimulated Akt phosphorylation [8, 18]. As Akt is a key enzyme in insulin signaling that stimulates glucose transport in skeletal muscles [19–21], our and previous results strongly suggest that the decrease in insulin-stimulated glucose transport in glycogen supercompensated muscles is attributable to impaired Akt signaling. It might be also possible that exercise-induced protection against insulin resistance at the end of 4 h of refeeding was due to the prevention of decreased Akt signaling.
Following insulin stimulation, PI3 K-generated PI(3,4,5)triphosphate (PIP3) recruits Akt from the cytosol to the plasma membrane, leading to activation of Akt [22]. There is evidence that, in glycogen supercompensated muscles, activation of Akt by insulin stimulation is decreased in the absence of a reduction in PI3-kinase activity [8, 18]. This lack of an effect of glycogen supercompensation on PI3-kinase, the enzyme that catalyzes PIP3 production, raises the possibility that supercompensation promotes the degradation of PIP3. The phosphoinositide phosphatases SHIP2, SKIP, and PTEN have been shown to negatively regulate insulin signaling and glucose uptake in skeletal muscles by degrading PIP3 into PIP2 [23–26]. It may be possible that increased activity or expression of SHIP2, SKIP, or PTEN leads to decreased Akt phosphorylation in glycogen supercompensated muscles with a reduced insulin-stimulated glucose uptake. However, we found no increase in the abundance of SHIP2, SKIP, or PTEN protein in glycogen supercompensated muscles of either non-exercised rats or HIS-exercised refed rats in our study (Fig. 5). The activity rather than the expression of SHIP2, SKIP, or PTEN might be possibly altered in glycogen supercompensated muscles. Clearly, more research is needed to elucidate the mechanism responsible for the decrease in Akt phosphorylation in glycogen supercompensated muscles.
In summary, the peak glycogen level during refeeding following HIS was not different from that following LIS exercise, suggesting that sprint interval exercise supercompensates the muscle glycogen level in response to carbohydrate refeeding as well as prolonged endurance exercise does. Although it is generally accepted that a large accumulation of muscle glycogen results in insulin resistance of glucose transport, carbohydrate refeeding after HIS or LIS exercise did not reduce insulin-stimulated muscle glucose transport at the end of 4 h of refeeding, in spite of the large accumulation of glycogen. Thus, both types of exercise protected against the development of insulin resistance during the early phase of muscle glycogen supercompensation in response to carbohydrate refeeding. This is probably one aspect of the mechanism of enhanced muscle glycogen supercompensation after exercise.
Acknowledgments
We are grateful to Dr. Tadaomi Takenawa (Kobe University, Kobe, Japan) for providing the SKIP antibodies used in this study. This research was supported by the Uehara Memorial Foundation (Tokyo, Japan), the Nakatomi Foundation (Tosu, Japan), a Grant-in-Aid from Niigata University of Health and Welfare, and a Grant-in-Aid for Scientific Research (KAKENHI) (C) no. 16500426 from the Japan Society for the Promotion of Science. K. Koshinaka was supported by Postdoctoral Fellowships from the Japan Society for the Promotion of Science.
References
- 1.Wallberg-Henriksson H, Constable SH, Young DA, Holloszy JO. Glucose transport into rat skeletal muscle: interaction between exercise and insulin. J Appl Physiol. 1988;65:909–913. doi: 10.1152/jappl.1988.65.2.909. [DOI] [PubMed] [Google Scholar]
- 2.Cartee GD, Douen AG, Ramlal T, Klip A, Holloszy JO. Stimulation of glucose transport in skeletal muscle by hypoxia. J Appl Physiol. 1991;70:1593–1600. doi: 10.1152/jappl.1991.70.4.1593. [DOI] [PubMed] [Google Scholar]
- 3.Gulve EA, Cartee GD, Zierath JR, Corpus VM, Holloszy JO. Reversal of enhanced muscle glucose transport after exercise: roles of insulin and glucose. Am J Physiol. 1990;259:E685–E691. doi: 10.1152/ajpendo.1990.259.5.E685. [DOI] [PubMed] [Google Scholar]
- 4.Richter EA, Garetto LP, Goodman MN, Ruderman NB. Muscle glucose metabolism following exercise in the rat. Increased sensitivity to insulin. J Clin Invest. 1982;69:785–793. doi: 10.1172/JCI110517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bergstrom J, Hultman E. Muscle glycogen synthesis after exercise: an enhancing factor localized to the muscle cells in man. Nature. 1966;210:309–310. doi: 10.1038/210309a0. [DOI] [PubMed] [Google Scholar]
- 6.Koshinaka K, Sano A, Howlett KF, Yamazaki T, Sasaki M, Sakamoto K, Kawanaka K. Effect of high intensity intermittent swimming on post-exercise insulin sensitivity in rat epitrochlearis muscle. Metabolism. 2008;57:749–756. doi: 10.1016/j.metabol.2008.01.014. [DOI] [PubMed] [Google Scholar]
- 7.Koshinaka K, Kawasaki E, Hokari F, Kawanaka K. Effect of acute high intensity intermittent swimming on post-exercise insulin responsiveness in epitrochlearis of fed rats. Metabolism. 2009;58:246–253. doi: 10.1016/j.metabol.2008.09.021. [DOI] [PubMed] [Google Scholar]
- 8.Derave W, Hansen BF, Lund S, Kristiansen S, Richter EA. Muscle glycogen content affects insulin-stimulated glucose transport and protein kinase B activity. Am J Physiol. 2000;279:E947–E955. doi: 10.1152/ajpendo.2000.279.5.E947. [DOI] [PubMed] [Google Scholar]
- 9.Host HH, Hansen PA, Nolte LA, Chen MM, Holloszy JO. Glycogen supercompensation masks the effect of a training-induced increase in GLUT-4 on muscle glucose transport. J Appl Physiol. 1998;85:133–138. doi: 10.1152/jappl.1998.85.1.133. [DOI] [PubMed] [Google Scholar]
- 10.Kawanaka K, Han D-H, Nolte LA, Hansen PA, Nakatani A, Holloszy JO. Decreased insulin-stimulated GLUT4 translocation in glycogen supercompensated muscles of exercised rats. Am J Physiol. 1999;276:E907–E912. doi: 10.1152/ajpendo.1999.276.5.E907. [DOI] [PubMed] [Google Scholar]
- 11.Kawanaka K, Tabata I, Higuchi M. Effects of high-intensity intermittent swimming on glucose transport in rat epitrochlearis muscle. J Appl Physiol. 1998;84:1852–1857. doi: 10.1152/jappl.1998.84.6.1852. [DOI] [PubMed] [Google Scholar]
- 12.Ueyama A, Sato T, Yoshida H, Magata K, Koga N. Nonradioisotope assay of glucose uptake activity in rat skeletal muscle using enzymatic measurement of 2-deoxyglucose 6-phosphate in vitro and in vivo. Biol Signals Recept. 2000;9:267–274. doi: 10.1159/000014649. [DOI] [PubMed] [Google Scholar]
- 13.Passonneau JV, Lowry OH. Enzymatic analysis: a practical guide. Totowa: Humana; 1993. [Google Scholar]
- 14.Hansen PA, Gulve EA, Holloszy JO. Suitability of 2-deoxyglucose for in vitro measurement of glucose transport activity in skeletal muscle. J Appl Physiol. 1994;76:979–985. doi: 10.1152/jappl.1994.76.2.979. [DOI] [PubMed] [Google Scholar]
- 15.Passonneau JV, Lauderdale VR. A comparison of three methods of glycogen measurements in tissue. Anal Biochem. 1974;60:405–412. doi: 10.1016/0003-2697(74)90248-6. [DOI] [PubMed] [Google Scholar]
- 16.Margolis B, Bellot F, Honegger AM, Ullrich A, Schlessinger J, Zilberstein A. Tyrosine kinase activity is essential for the association of phospholipase C-γ with the epidermal growth factor receptor. Mol Cell Biol. 1990;10:435–441. doi: 10.1128/mcb.10.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Holloszy JO, Hansen PA. Reviews of physiology, biochemistry and pharmacology. In: Blaustein MP, Grunicke H, Habermann E, Pette D, Schultz G, Schweiger M, editors. Regulation of glucose transport into skeletal muscle. Berlin: Spinger; 1996. [DOI] [PubMed] [Google Scholar]
- 18.Kawanaka K, Nolte LA, Han D-H, Hansen PA, Holloszy JO. Mechanism underlying impaired GLUT4 translocation in glycogen supercompensated muscles of exercised rats. Am J Physiol. 2000;279:E1311–E1318. doi: 10.1152/ajpendo.2000.279.6.E1311. [DOI] [PubMed] [Google Scholar]
- 19.Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu Q, Crenshaw EB, III, et al. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta) Science. 2001;292:1728–1731. doi: 10.1126/science.292.5522.1728. [DOI] [PubMed] [Google Scholar]
- 20.Garofalo RS, Orena SJ, Rafidi K, Torchia AJ, Stock JL, Hildebrandt AL, et al. Severe diabetes, age-dependent loss of adipose tissue, and mild growth deficiency in mice lacking Akt2/PKB beta. J Clin Invest. 2003;112:197–208. doi: 10.1172/JCI16885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McCurdy CE, Cartee GD. Akt2 is essential for the full effect of calorie restriction on insulin-stimulated glucose uptake in skeletal muscle. Diabetes. 2005;54:1349–1356. doi: 10.2337/diabetes.54.5.1349. [DOI] [PubMed] [Google Scholar]
- 22.Scheid MP, Marignani PA, Woodgett JR. Multiple phosphoinositide 3-kinase-dependent steps in activation of protein kinase B. Mol Cell Biol. 2002;22:6247–6260. doi: 10.1128/MCB.22.17.6247-6260.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clement S, Krause U, Desmedt F, Tanti JF, Behrends J, Pesesse X, Sasaki T, Penninger J, Doherty M, Malaisse W, Dumont JE, Le Marchand-Brustel Y, Erneux C, Hue L, Schurmans S. The lipid phosphatase SHIP2 controls insulin sensitivity. Nature. 2001;409:92–97. doi: 10.1038/35051094. [DOI] [PubMed] [Google Scholar]
- 24.Ijuin T, Yu YE, Mizutani K, Pao A, Tateya S, Tamori Y, Bradley A, Takenawa T. Increased insulin action in SKIP heterozygous knockout mice. Mol Cell Biol. 2008;28:5184–5195. doi: 10.1128/MCB.01990-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kagawa S, Soeda Y, Ishihara H, Oya T, Sasahara M, Yaguchi S, Oshita R, Wada T, Tsuneki H, Sasaoka T. Impact of transgenic overexpression of SH2-containing inositol 5’-phosphatase 2 on glucose metabolism and insulin signaling in mice. Endocrinology. 2008;149:642–650. doi: 10.1210/en.2007-0820. [DOI] [PubMed] [Google Scholar]
- 26.Wijesekara N, Konrad D, Eweida M, Jefferies C, Liadis N, Giacca A, Crackower M, Suzuki A, Mak TW, Kahn CR, Klip A, Woo M. Muscle-specific Pten deletion protects against insulin resistance and diabetes. Mol Cell Biol. 2005;25:1135–1145. doi: 10.1128/MCB.25.3.1135-1145.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]