

Abstract
We have previously found that both CaMKII-mediated phosphorylation and calmodulin (CaM) binding to the channels are required for maintaining basal activity of the Cav1.2 Ca2+ channels. In this study, we investigated the hypothetical CaMKII phosphorylation site on Cav1.2 that contributes to the channel regulation. We found that CaMKII phosphorylates the Thr1603 residue (Thr1604 in rabbit) within the preIQ region in the C-terminal tail of the guinea-pig Cav1.2 channel. Mutation of Thr1603 to Asp (T1603D) slowed the run-down of the channel in inside-out patch mode and abolished the time-dependency of the CaM’s effects to reverse run-down. We also found that CaMKII-mediated phosphorylation of the proximal C-terminal fragment (CT1) increased, while dephosphorylation of CT1 decreased its binding with CaM. These findings suggest that CaMKII regulates the CaM binding to the channel, and thereby maintains basal activity of the Cav1.2 Ca2+ channel.
Keywords: CaMKII, CaM, Ca2+ channel, Phosphorylation
Introduction
L-type Ca2+ channels support Ca2+ entry into cells triggering cardiac contraction, hormone secretion and gene expression [2, 14]. Activity of the L-type Ca2+ channels is regulated by intracellular Ca2+ through feedback mechanisms. Increasing intracellular concentrations of Ca2+ ([Ca2+]i) result in Ca2+-dependent facilitation (CDF) and inactivation (CDI) [14, 24, 31, 32]. Recent studies indicate that calmodulin (CaM) plays an important role as a Ca2+ sensor for both CDF and CDI [3, 12]. CaM is suggested to be associated with IQ-motif region and/or preIQ region (or CB domain) in the C-terminal tail of the α1 subunit and promotes Ca2+-dependent conformation changes of the channel during CDF and CDI.
Other lines of evidence support that the Ca2+/CaM-dependent protein kinase II (CaMKII) mediates CDF via phosphorylation of a certain site in the channel. CaMKII inhibitors suppress CDF, and application of exogenous CaMKII enhances activity of Cav1.2 Ca2+ channels [1, 4, 7, 9, 13, 21, 25, 26]. However, the roles of CaM and CaMKII and their interactions in the regulation of the Cav1.2 channels are still controversial and remain to be established.
In our recent studies, we have found that CaM applied with ATP can reactivate (reprime) the channels in a run-down state in inside-out mode, suggesting important roles of CaM not only in CDF and CDI, but also in basal activity of the channels [27]. We have also reported that CaM inhibitors abolish both CDF and CDI, while CaMKII inhibitors delay the development of CDF in Ca2+-overloaded myocytes. These findings support the hypothesis that CaM and CaMKII have distinct roles in CDF and CDI: CaM is a key molecule to bifurcate the Ca2+ signal to CDF and CDI, while CaMKII plays a modulatory role in CDF [18]. However, the phosphorylationa site responsible for the modulation of the CaM’s effect remains to be established.
There are many potential phosphorylation sites in the Cav1.2 channel based on the consensus sequence for CaMKII-mediated phosphorylation. Phosphorylation of the β2a subunit responsible for CaMKII-mediated facilitation of the channel has been found [7], and it is suggested that this phosphorylation modulates the interaction between the β subunit and the C-terminal tail of the α1 subunit, thereby manifesting CDF [7, 21, 29]. On the other hand, a site in I-II loop (rabbit Ser439) and two sites in CT1 (rabbit Ser1517 and 1575) have been shown to be functionally phosphorylated by CaMKII [5, 13]; the roles of these sites in the Ca2+-dependent regulations are not clear. In this study, we have focused on the potential phosphorylation sites within the CaM-binding regions in the C-terminal tail of the α1C subunit and explored its possible role in the regulation of channel activity. It has been found that Thr1603 in a CaM-binding region (preIQ region) can be phosphorylated by CaMKII, and this phosphorylation modulates the interaction between CaM and the channel.
Methods
Preparation of fragment peptides of the Cav1.2 channel
The cDNAs corresponding to three C-terminal fragments of guinea-pig Cav1.2 channel (CT1, a.a.1509-1789; CT2, a.a.1778-2003; CT3, a.a.1942-2169; GenBank AB016287) were expressed as glutathione-S-transferase (GST) fusion proteins in E. coli BL21 (Stratagene, La Jolla, CA). For purification of GST-CT2 and CT3, E. coli suspension in Tris buffer was freeze-thawed (ten cycles) and centrifuged (15,000×g, 10 min). The fusion proteins were retrieved by glutathione-Sepharose-4B beads (GE Healthcare, Uppsala, Sweden). After dissolving with Tris buffer containing 10 mM glutathione, GST-CT2 and CT3 were quantified by the Bradford method using bovine serum albumin (BSA) as standard.
Since GST-fusion CT1 was insoluble [11], we applied the glutathione beads directly into the freeze-thawed lysate without centrifugation, and the washed beads were stocked in Tris buffer. GST-CT1 was quantified by densitometry on the gel of SDS-PAGE using BSA as standard.
Preparation of CaMKIIT286D and CaM
With the rat CaMKIIα cDNA in pGEX-6p-1 as template, the CaMKIIα mutant Thr286Asp (CaMKIIT286D), a constitutively active type in the absence of CaM and Ca2+, was created with QuickChange site-directed mutagenesis kit (Stratagene). CaM was cloned from HEK293 cells. CaMKII and CaM were expressed as GST-fusion proteins, affinity purified, and the GST region was cleaved by PreScissionTM protease (GE Healthcare). CaMKII and CaM were quantified by the Bradford method using BSA as standard.
Phosphorylation and dephosphorylation of GST-fusion proteins
The immobilized GST-fusion proteins were phosphorylated in an assay reaction (0.4 μM CaMKIIT286D and 1 mM Mg2+ATP or 1.85 MBq [γ-32P]-ATP) for 30 min at 30°C in Tris buffer. Phosphorylation was detected either by 32P-autoradiography or by immunoblot using the anti-phospho-threonine antibody Q7 and the anti-phospho-serine antibody Q5 (Qiagen, Hilden, Germany). Dephosphorylation was done with 5 unit/ml calf intestinal alkaline phosphatase, (CIP, New England Biolabs, Ipswich, MA) in Tris buffer for 30 min at 37°C.
CaM pull-down assays
The GST-CT1 immobilized to glutathione-Sepharose beads were incubated with CaM (0.2–3 μM) at 4°C overnight in Tris buffer containing protease inhibitor cocktail (Roche, Mannhein, Germany) at 1 mM [Ca2+]. After washing three times with the same buffer containing 0.05% Tween-20, bound CaM was eluted with 1 × SDS sample buffer and resolved by SDS-PAGE (12.5% gel) [20]. Protein bands of GST-CT1 and CaM, stained by Coomassie blue, were scanned and quantified using concentration-density calibration curves using BSA as standard.
Site-directed mutagenesis and expression of Ca2+ channels
CT1T1603A, α1CT1603A and α1CT1603D were created by the site-directed mutagenesis with wild-type CT1 and α1C as templates, respectively. Chinese hamster ovary (CHO)-K1 cells were cultured under 5% CO2 at 37°C at a density of 2 × 105 cells/dish (35 mm) in Ham’s F12 containing 10% fetal bovine serum, 50 U/ml penicillin, 50 μg/ml streptomycin and 10 mM Hepes. The wild type or mutant α1C together with β2a (total 0.3 μg) were cotransfected into CHO-K1 cells (α1C:β2a = 10:7) by using the Transfast transfection kit (Promega, Madison, WI). Cotransfection with 1 μg green fluorescent protein (GFP) expression vector was used for identification of the transfected cells.
Patch-clamp and data analysis
Patch-clamp technique was used to monitor Ca2+ channel activity [8, 10, 27]. The patch pipettes (2–4 MΩ) contained (in mM) 50 BaCl2, TEA Cl 70, EGTA 0.5, Bay K8644 0.003 and Hepes–CsOH buffer 10 (pH 7.4). Channel openings elicited by repetitive depolarizing pulses to 0 mV at 0.5 Hz were recorded first in the cell-attached and then in the inside-out mode in the basic internal solution consisting of (mM): K-aspartate 90, KCl 30, KH2PO4 10, MgCl2 0.5, EGTA 1, CaCl2 0.5 (free [Ca2+] 80 nM), and Hepes–KOH buffer 10 (pH 7.4). The NP o value (N: number of channels, P o: time-averaged open probability) was calculated as a measure of channel activity. During application of CaM, NP o values of 3-min span (90 traces) were averaged every 1 min, and the maximum average value was taken as the effect of CaM.
Student’s t test was used to estimate statistical significance, and P values less than 0.05 were considered as significant.
Results
Thr1603 of Cav1.2 channel is phosphorylated by CaMKII
We first examined whether the C-terminal tail of the guinea-pig Cav1.2 channel was phosphorylated by CaMKII. Three GST-fusion peptides derived from the C-terminal tail, CT1, CT2 and CT3 (Fig. 1a) were incubated with permanently active CaMKII (CaMKIIT286D), and phosphorylation was detected with antibodies against phospho-threonine and -serine. CT1 and CT2, but not CT3 and GST (control), were recognized by the antibodies (Fig. 1b). Both phospho-threonine and -serine residues were detected in CT2, whereas only the phospho-threonine residue was detected in CT1 (Fig. 1b). CT1 contained EF-hand region and preIQ-IQ region, the latter of which was known to interact with CaM and thus to be important for the Ca2+-mediated regulation of the channel [6, 11, 12, 14, 17, 19, 20, 22–24, 31, 32]. There are five serine sites (Ser1516, Ser1574, Ser1626, Ser1699, and Ser1720) and only one threonine site (Thr1603) in the CT1 based on the consensus sequence of Arg-X-X-Ser/Thr and Ser-X-Asp for CaMKII phosphorylation. Thus, it was likely that Thr1603 was phosphorylated by CaMKII. Interestingly, Thr1603 is located within preIQ region, a CaM-binding site.
Fig. 1.
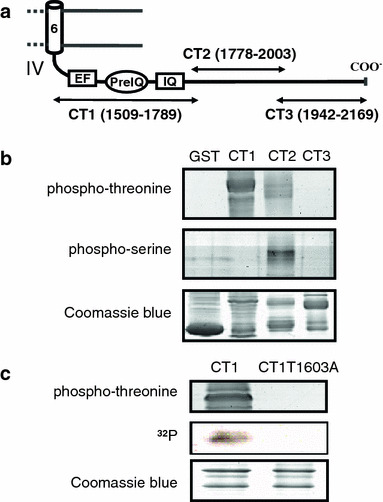
Thr1603 of Cav1.2 channel is phosphorylated by CaMKII. a Schematic diagram of the C-terminal tail of the guinea-pig cardiac Cav1.2 channel. EF-hand, preIQ and IQ-motif regions are shown. Fragment peptides, CT1, CT2 and CT3, are shown with a.a. numbers. b CaMKII-mediated phosphorylation of GST-fusion CT1, CT2 and CT3 and GST (negative control) detected with immunoblot blot analysis using anti-phospho-threonine (upper panel) and anti-phospho-serine (middle panel) antibodies. The bottom panel shows Coomassie staining of the gel. c Phosphorylation of CT1 and its mutant CT1T1603A detected by anti-phospho-threonine antibody (upper panel) and autoradiography (middle panel). Coomassie staining is in bottom panel. Each experiment was performed at least three times with equivalent results
To verify that Thr1603 was actually phosphorylated by CaMKII, we examined phosphorylation of a CT1 mutant, CT1T1603A. As expected, CT1T1603A was not phosphorylated with CaMKIIT286D (Fig. 1c), confirming that Ser residues were not phosphorylated. The result strongly suggested that Thr1603 was the unique site for CaMKII-mediated phosphorylation in the preIQ-IQ region. Therefore, we hereafter focused our investigation on Thr1603 in the C-terminal tail.
CaMKII phosphorylation is essential for basal activity of Cav1.2 channel
It has been shown that CaM produces activity of the Ca2+ channel in inside-out patches [27]. We examined interactions in the effects of CaM and CaMKII in inside-out patches, where CaM and CaMKII could be applied in a controlled manner. First, the effects of CaM on the Cav1.2 channels were re-examined in inside-out patch mode. As shown in Fig. 2, channel activity (evaluated by NP o) decreased rapidly in the inside-out mode due to run-down, and activity of the wild type channel decayed to nearly null (NP o = 0.8 ± 0.4%, n = 8, relative to that in the cell-attached mode; cf. Fig. 3A) at 5 min after patch excision. CaM (1 μM) + ATP (3 mM), when applied at 1 min after run-down, produced relative NP o of 177 ± 26% (n = 8, Fig. 2A). However, when applied at 5 min, CaM produced the relative NP o of only 17 ± 2% (n = 5, Fig. 2B), confirming the time-dependent attenuation of the CaM’s effect [27]. Then, we examined the effect of CaMKII. As shown in Figs. 2C, 1 μM CaMKIIT286D and 3 mM ATP, applied at 1 min after patch excision, produced channel activity of a low level (25 ± 12%, n = 9), and subsequent application of CaM + ATP produced the relative NP o of 149 ± 16% (n = 6, Fig. 2C). These results suggested that CaMKII-mediated phosphorylation of the channel could produce channel activity in inside-out patches and counteracted the time-dependent attenuation of the CaM’s effect.
Fig. 2.
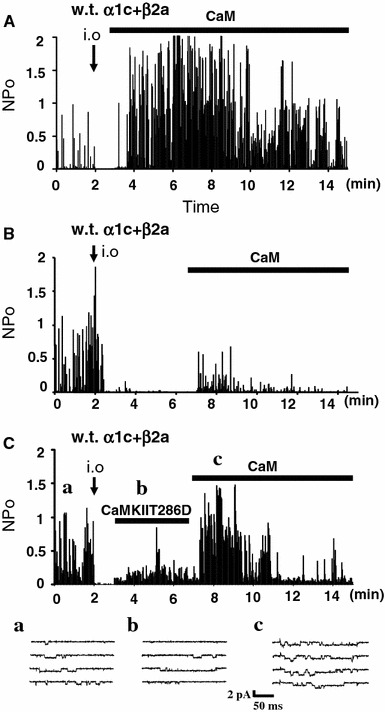
CaMKII facilitates CaM’s effect on channel activity. Ca2+ channel activity was recorded in CHO cells expressing wild type (w.t.) α1C and β2a in the cell-attached and inside-out mode. The open-state probability (NP o) of the channels for each repetitive depolarization was plotted against time. A Application of CaM (1 μM) + ATP (3 mM) 1 min after patch excision (shown by i.o. and an arrow) restored channel activity. B Channel activity was relatively low when CaM + ATP were applied 5 min after patch excision, indicating time-dependent attenuation of the CaM’s effect. C Application of CaMKIIT286D (1 μM) + ATP (3 mM) in the inside-out mode produced channel activity at a low level and retained the CaM’s effect. Examples of current traces recorded in the cell-attached mode (a) and during application of CaMKIIT286D + ATP (b) and CaM + ATP (c) in the inside-out mode are shown
Fig. 3.
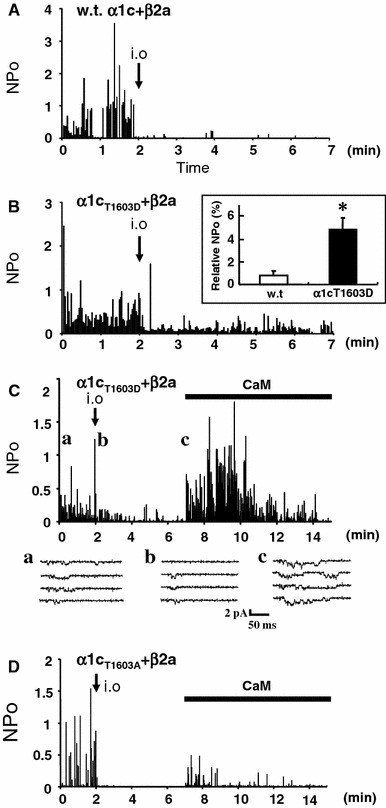
Mutant channel α1CT1603D is resistant to the slow run-down. Representative time courses of run-down for the wild-type (A) and α1CT1603D (B) channels coexpressed with β2a in CHO cells. Inset graph in b shows relative NPo values for the wild-type and the mutant channels in the inside-out mode. Effects of CaM on α1CT1603D (C) and α1CT1603A (D) channels in the inside-out patch mode. CaM (1 μM) + ATP (3 mM) produced marked channel activity in α1CT1603D, but little in α1CT1603A. Representative current traces recorded in the cell-attached mode (a) and in the inside-out mode before (b) and after (c) application of CaM + ATP are shown in (C)
α1CT1603D abolishes the time-dependency of the CaM’s effect on run-down
To assess the role of phosphorylation of Thr1603, the unique phosphorylation site for CaMKII in the preIQ-IQ region, we examined channel activity of two mutants, α1CT1603A and α1CT1603D, and the CaM’s effects on these mutants. The wild type channels showed marked run-down in the inside-out patch mode (Fig. 3A), and α1CT1603A also showed a similar time course of run-down (figure not shown). On the other hand, the α1CT1603D channel showed a low but certain activity of 4.9 ± 1% (n = 10) at 5–8 min after patch excision, and it was significantly higher compared with 0.8 ± 0.4% (n = 8) for the wild-type channels (Fig. 3B, inset). Thus, the result implied that the negative charge at Thr1603 seemed to promote channel activity to some extent, although the T1603D mutation itself did not produce full channel activity without CaM in the inside-out mode.
We then examined the effects of CaM on the mutant channels. Application of CaM (1 μM) at 5 min after patch excision induced α1CT1603D channel activity with relative NP o of 121 ± 23% (n = 6, Fig. 3C). However, CaM induced α1CT1603A channel activity of 19 ± 2% (n = 8, Fig. 3D), comparable to that for the wild type (Fig. 2B). These results suggested that the negative charge at a.a. 1603 was important for the facilitatory effect of CaM on the channel.
Phosphorylation of CT1 increases CaM binding to the channel
Based on the above results, we hypothesized that CaMKII-mediated phosphorylation at Thr1603 might affect the CaM-binding to a site near Thr1603, i.e., preIQ-IQ region. We therefore examined effects of CaMKII-mediated phosphorylation of the C-terminal peptides on its binding with CaM. CT1, but not CT2 or CT3 peptide bound to CaM (Fig. 4a), consistent with previous reports [20, 23, 31, 32]. Phosphorylation of CT1 with CaMKIIT286D increased the CaM binding to this peptide, while treatment of CT1 with an alkaline phosphatase CIP decreased the CaM binding at 1 mM [Ca2+] (Fig. 4b). A similar tendency was also observed at 80 nM [Ca2+] (data not shown), although amounts of bound CaM were relatively low. Treatment of CT1 first with CIP and then with CaMKIIT286D increased CaM binding (Fig. 4b), supporting that these effects are mediated by phosphorylation or dephosphorylation of the peptide. Figure 4c shows the concentration-dependent binding of CaM to CT1, indicating that the treatment with CaMKIIT286D increased, while the treatment with CIP decreased an apparent maximum binding (B max) of CaM (P < 0.01 and <0.05, respectively). The B max for the kinase-treated CT1 was approximately 2 mole/mole, suggesting that two (or more) CaM molecules might bind to CT1. A change in the apparent dissociation constant (0.02–0.2 μM) was implied, but it was not examined intensively.
Fig. 4.
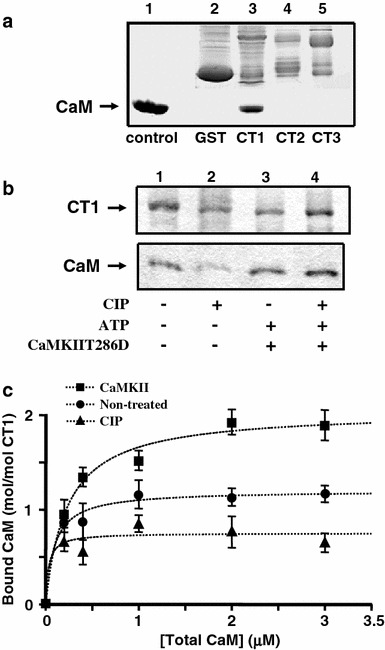
CaMKII-mediated phosphorylation enhances CaM binding to CT1. a Pull-down assay for CaM binding to GST-fusion CT1, CT2 and CT3 at 1 mM [Ca2+]. Lane 1 calibrating CaM (5 μg), lane 2 GST alone (negative control), lane 3 GST-CT1, lane 4 GST-CT2, lane 5 GST-CT3. b Pull-down assay for CaM binding to CT1 after various treatments at 1 mM [Ca2+]. Lane 1 non-treated, lane 2 calf intestinal alkaline phosphatase 1 (CIP), lane 3 CaMKIIT286D + ATP; or lane 4 first with CIP then with CaMKIIT286D + ATP. c Concentration-dependent binding of CaM to GST-CT1 treated with CaMKII (squares), none (control, circles) and CIP (triangles). Data were from three independent experiments
Discussion
This study shows that CaMKII phosphorylates the Thr1603 residue within the preIQ region, a CaM-binding region of the Cav1.2 channel, and this phosphorylation modifies the CaM’s effect on channel activity. This finding may provide a molecular mechanism by which CaMKII regulates the Cav1.2 channel.
Roles of CaM and CaMKII phosphorylation in the regulation of Ca2+ channel
CaM modulates, through direct binding to channel proteins, various types of ion channels, including Ca2+ channels [14, 15, 21, 31, 32]. In the L-type Cav1.2 channels, many reports have suggested that CaM is tethered to the channel and functions as a Ca2+ sensor for Ca2+-dependent facilitation (CDF) and inactivation (CDI). It has been shown that CaM binds to regions in the N-terminal tail, I-II linker, and the C-terminal tail (preIQ and IQ regions) in vitro [3, 6, 11, 12, 14, 17, 19, 20, 22, 24]. We have suggested, however, that CaM may not be tightly associated with the channel, but dynamically interacts with its site, based on the finding that CaM produces channel activity in a concentration-dependent manner in inside-out patches [27]. We think that precise mechanisms for CDF and CDI are still to be established. It is also unclear whether CaM binding to the channels is modulated by any signaling systems, such as phosphorylation.
CaMKII is also shown to modulate cardiac L-type Ca2+ channels, and this is suggested as an underlying mechanism of CDF [1, 4, 7, 9, 21, 25, 26, 28, 29]. A phosphorylation site for CaMKII responsible for CDF has been identified in the β2a subunit [7]. Furthermore, CaMKII is shown to associate with the β2a and the C-terminal tail of the α1 subunit of the channel [7, 9, 21]. However, possible involvement of phosphorylation of the C-terminal tail of the α1C subunit was to be examined. There are 17 potential phosphorylation sites in the C-terminal tail of the guinea-pig Cav1.2 channel, based on the consensus sequences Arg-X-X-Ser/Thr and Ser-X-Asp. Erxleben et al. [5] have reported that CaMKII phosphorylates Ser439 in I-II loop and Ser1517 in CT1 (proximal region of the C-terminal tail), thereby promoting mode 2 activity in the rabbit cardiac Cav1.2 channel. Lee et al. [13] have also reported that CaMKII phosphorylates Ser1512 and Ser1570 (Ser1575 in cardiac type) in the C-terminal tail of the rabbit smooth muscle Ca2+ channel and thereby promotes voltage-dependent facilitation. However, phosphorylation of Ser1512/1517 or Ser1570/1575 was not detected by another study [9]. In this study, we failed to detect phosphorylation of Ser1516 and Ser1574 (rabbit Ser1517 and 1575), but found that Thr1603 (rabbit Thr1604) was phosphorylated by CaMKII. Although the reason for this discrepancy is not clear, we speculate that a difference in the higher structure of CT1 might be involved, considering that CT1 is poorly soluble in water [11].
In addition to the phosphorylation of Thr1604, we found that CaMKII phosphorylated at least one Ser and one Thr site in CT2, while the distal C-terminal region (CT3) was not phosphorylated. Since there were eight potential sites in CT2 and homology in this region among species was not so high, we did not explore identifying these sites in this study.
In our previous study, CaMKII inhibitors (KN-62 and AIP) delayed the development of facilitation and inhibition produced by elevating bulk [Ca2+]i, but did not depress either of them [18]. We therefore hypothesized that CaMKII might play a modulatory role in CDF and CDI. Our present finding that CaMKII-mediated phosphorylation enhances the effect of CaM on channel activity may provide a possible molecular mechanism for this hypothesis.
A molecular mechanism for CaMKII regulation of the Cav1.2 channel
The phosphorylation of Thr1603 enhanced the CaM’s effect on Cav1.2 channel activity. Several possibilities are raised to account for this finding. The phosphorylation (1) enhances an affinity of the channel for CaM, (2) increases the number of CaM molecules bound to the channel, and (3) facilitates the putative machinery to link CaM-binding region to the channel-gating region. It is suggested that one molecule of CaM tethers to the Cav1.2 channel and regulates the channel function [16]. However, since there are at least four CaM-binding sites in the channel [3, 30], the possibility that multiple molecules of CaM bind to the channel may not be excluded. In this study, we found that the CT1 peptide increased the number of bound CaM molecules to 2 mol/mol after its phosphorylation. Therefore, it is suggested that phosphorylation of Thr1603 in the preIQ region increases the number of CaM molecules bound to preIQ-IQ region and/or enhances the interaction of CaM with preIQ-IQ, and thereby facilitates opening of the Cav1.2 channel.
It should be noted, however, that there are several CaMKII phosphorylation sites beside Thr1603, such as rabbit Ser439 in I-II loop and Ser1517 and 1575 in the C-terminal tail as mentioned above [5, 13]. It is possible CaMKII phosphorylates multiple sites of the channel depending on the conformation of the channel and finely regulates channel activity.
A model for basal activity of the Cav1.2 channel
We have previously found that basal activity of the channel is supported by CaM and ATP, and their washout leads to run-down of the channel. In addition, the run-down consists of the initial and late phases [8], and CaM + ATP effectively reverse the initial phase, but not the late phase [27]. In this study, we have found that CaMKII counteracts this attenuation of the CaM’s effect. Since CaM binding to CT1 is increased by CaMKII treatment and mutation of Thr1603 to aspartate mimicked the CaMKII effect, it is suggested that CaMKII-mediated phosphorylation of Thr1603 facilitates or supports the CaM’s effect on channel activity.
Based on the previous and present findings, a hypothesis for the mechanism underlying basal activity of the Cav1.2 channel may be proposed. CaMKII-mediated phosphorylation of certain site(s) (including Thr1603) facilitates the interaction of the channel with CaM and thereby produces basal activity of the channel. It is possible that binding of CaM to the phosphorylated channel prevents or decelerates dephosphorylation of the site(s). Clearly, further studies are needed to completely understand the role of CaMKII in the regulation of Cav1.2 channels.
Acknowledgments
We thank Ms. T. Imaichi for technical assistance and Ms. E. Iwasaki for secretarial assistance. This work was supported by research grants from the JSPS to L.-Y.H. and M.K., and from the NNSF of China to L.-Y.H. and the Kodama memorial Fund.
References
- 1.Anderson ME, Braun AP, Schulman H, Premack BA. Multifunctional Ca/calmodulin-dependent protein kinase mediates Ca-induced enhancement of the L-type Ca current in rabbit ventricular myocytes. Circ Res. 1994;75:854–861. doi: 10.1161/01.res.75.5.854. [DOI] [PubMed] [Google Scholar]
- 2.Catterall WA. Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol. 2000;16:521–555. doi: 10.1146/annurev.cellbio.16.1.521. [DOI] [PubMed] [Google Scholar]
- 3.Dick IE, Tadross MR, Liang HY, Tay LH, Yang WJ, Yue DT. A modular switch for spatial Ca2+ selectivity in the calmodulin regulation of CaV channels. Nature. 2008;451:830–834. doi: 10.1038/nature06529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dzhura I, Wu Y, Colbran RJ, Balser JR, Anderson ME. Calmodulin kinase determines calcium-dependent facilitation of L-type calcium channels. Nat Cell Biol. 2000;2:173–177. doi: 10.1038/35004052. [DOI] [PubMed] [Google Scholar]
- 5.Erxleben C, Liao Y, Gentile S, Chin D, Gomez-Alegria C, Mori Y, Birnbaumer L, Armstrong DL. Cyclosporin and Timothy syndrome increase mode 2 gating of CaV1.2 calcium channels through aberrant phosphorylation of S6 helices. Proc Natl Acad Sci USA. 2006;103:3932–3937. doi: 10.1073/pnas.0511322103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fallon JL, Halling DB, Hamilton SL, Quiocho FA. Structure of calmodulin bound to the hydrophobic IQ domain of the cardiac Cav1.2 calcium channel. Structure. 2005;13:1881–1886. doi: 10.1016/j.str.2005.09.021. [DOI] [PubMed] [Google Scholar]
- 7.Grueter C, Abiria S, Dzhura I, Wu Y, Ham A, Mohler P, Anderson ME, Colbran R. L-type Ca2+ channel facilitation mediated by phosphorylation of the β subunit by CaMKII. Mol Cell. 2006;23:641–650. doi: 10.1016/j.molcel.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 8.Hao LY, Kameyma A, Kameyama M. A cytoplasmic factor, calpastatin and ATP together reverse run-down of Ca2+ channel activity in guinea-pig heart. J Physiol. 1999;514:687–699. doi: 10.1111/j.1469-7793.1999.687ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hudmon A, Schulman H, Kim J, Maltez JM, Tsien RW, Pitt GS. CaMKII tethers to L-type Ca2+ channels, establishing a local and dedicated integrator of Ca2+ signals for facilitation. J Cell Biol. 2005;171:537–547. doi: 10.1083/jcb.200505155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kameyama M, Kameyama A, Takano E, Maki M. Run-down of the cardiac L-type Ca2+ channel: partial restoration of channel activity in cell-free patches by calpastatin. Pflugers Arch. 1998;435:344–349. doi: 10.1007/s004240050521. [DOI] [PubMed] [Google Scholar]
- 11.Kim J, Ghosh S, Nunziato DA, Pitt GS. Identification of the components controlling inactivation of voltage-gated Ca2+ channels. Neuron. 2004;41:745–754. doi: 10.1016/S0896-6273(04)00081-9. [DOI] [PubMed] [Google Scholar]
- 12.Lee A, Zhou H, Scheuer T, Catterall WA. Molecular determinants of Ca2+/calmodulin-dependent regulation of CaV2.1 channels. Proc Natl Acad Sci USA. 2003;100:16059–16064. doi: 10.1073/pnas.2237000100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee TS, Karl R, Moosmang S, Lenhardt P, Klugbauer N, Hofmann F, Kleppisch T, Welling A. Calmodulin kinase II is involved in voltage-dependent facilitation of the L-type Cav1.2 calcium channel: identification of the phosphorylation sites. J Biol Chem. 2006;281:25560–25567. doi: 10.1074/jbc.M508661200. [DOI] [PubMed] [Google Scholar]
- 14.Liang H, DeMaria CD, Erickson MG, Mori MX, Alseikhan BA, Yue DT. Unified mechanisms of Ca2+ regulation across the Ca2+ channel family. Neuron. 2003;39:951–960. doi: 10.1016/S0896-6273(03)00560-9. [DOI] [PubMed] [Google Scholar]
- 15.Maier LS, Bers DM. Calcium, calmodulin, and calcium-calmodulin kinase II: heartbeat to heartbeat and beyond. J Mol Cell Cardiol. 2002;34:919–939. doi: 10.1006/jmcc.2002.2038. [DOI] [PubMed] [Google Scholar]
- 16.Mori MX, Erickson MG, Yue DT. Functional stoichiometry and locale enrichment of calmodulin interacting with Ca2+ channels. Sciences. 2004;304:432–435. doi: 10.1126/science.1093490. [DOI] [PubMed] [Google Scholar]
- 17.Mouton J, Feltz A, Maulet Y. Interactions of calmodulin with two peptides derived from the C-terminal cytoplasmic domain of the CaV1.2 Ca2+ channel provide evidence for a molecular switch involved in Ca2+-induced inactivation. J Biol Chem. 2001;276:22359–22367. doi: 10.1074/jbc.M100755200. [DOI] [PubMed] [Google Scholar]
- 18.Nie H, Hao LY, Xu JJ, Minobe E, Kameyama A, Kameyama M. Distinct roles of CaM and Ca2+/CaM-dependent protein kinase II in Ca2+-dependent facilitation and inactivation of cardiac L-type Ca2+channels. J Physiol Sci. 2007;57:167–173. doi: 10.2170/physiolsci.RP000507. [DOI] [PubMed] [Google Scholar]
- 19.Pate P, Mochca-Morales J, Wu Y, Zhang JZ, Rodney GG, Serysheva II, Williams BY, Anderson ME, Hamilton HL. Determinants for calmodulin binding on voltage-dependent Ca2+ channels. J Biol Chem. 2000;275:39786–39792. doi: 10.1074/jbc.M007158200. [DOI] [PubMed] [Google Scholar]
- 20.Peterson BZ, DeMaria CD, Adelman JP, Yue DT. Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of L-type calcium channels. Neuron. 1999;22:549–558. doi: 10.1016/S0896-6273(00)80709-6. [DOI] [PubMed] [Google Scholar]
- 21.Pitt GS. Calmodulin and CaMKII as molecular switches for cardiac ion channels. Cardiovasc Res. 2007;73:641–647. doi: 10.1016/j.cardiores.2006.10.019. [DOI] [PubMed] [Google Scholar]
- 22.Pitt GS, Zühlke RD, Hudmon A, Schulman H, Reuter H, Tsien RW. Molecular basis of calmodulin tethering and Ca2+-dependent inactivation of L-type Ca2+ channels. J Biol Chem. 2001;276:30794–30802. doi: 10.1074/jbc.M104959200. [DOI] [PubMed] [Google Scholar]
- 23.Qin N, Olcese R, Bransby M, Lin T, Birnbaumer L. Ca2+-induced inhibition of the cardiac Ca2+ channel depends on calmodulin. Proc Natl Acad Sci USA. 1999;96:2435–2438. doi: 10.1073/pnas.96.5.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Van Petegem F, Chatelain FC, Minor DL., Jr Insights into voltage-gated calcium channel regulation from the structure of the CaV1.2 IQ domain-Ca2+/calmodulin complex. Nat Struct Mol Biol. 2005;12:1108–1115. doi: 10.1038/nsmb1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu Y, Colbran RJ, Anderson ME. Calmodulin kinase is a molecular switch for cardiac excitation-contraction coupling. Proc Natl Acad Sci U S A. 2001;98:2877–2881. doi: 10.1073/pnas.051449198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu Y, Kimbrough JT, Colbran RJ, Anderson ME. Calmodulin kinase is functionally targeted to the action potential plateau for regulation of L-type Ca2+ current in rabbit cardiomyocytes. J Physiol. 2004;554:145–155. doi: 10.1113/jphysiol.2003.053314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu JJ, Hao LY, Kameyama A, Kameyama M. Calmodulin reverses rundown of L-type Ca2+ channels in guinea pig ventricular myocytes. Am J Physiol Cell Physiol. 2004;287:1717–1724. doi: 10.1152/ajpcell.00105.2004. [DOI] [PubMed] [Google Scholar]
- 28.Yuan W, Bers DM. Ca-dependent facilitation of cardiac Ca current is due to Ca-calmodulin dependent protein kinase. Am J Physiol. 1994;267:982–993. doi: 10.1152/ajpheart.1994.267.3.H982. [DOI] [PubMed] [Google Scholar]
- 29.Zhang R, Dzhura I, Grueter CE, Thiel W, Colbran RJ, Anderson ME. A dynamic α-β inter-subunit agonist signaling complex is a novel feedback mechanism for regulating L-type Ca2+ channel opening. FASEB J. 2005;19:1573–1575. doi: 10.1096/fj.04-3283fje. [DOI] [PubMed] [Google Scholar]
- 30.Zhou H, Yu K, McCoy KL, Lee A. Molecular mechanism for divergent regulation of Cav1.2 Ca2+ channels by calmodulin and Ca2+-binding protein-1. J Biol Chem. 2005;280:29612–29619. doi: 10.1074/jbc.M504167200. [DOI] [PubMed] [Google Scholar]
- 31.Zühlke RD, Pitt GS, Deisseroth K, Tsien RW, Reuter H. Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature. 1999;399:159–162. doi: 10.1038/20200. [DOI] [PubMed] [Google Scholar]
- 32.Zühlke RD, Pitt GS, Tsien RW, Reuter H. Ca2+-sensitive inactivation and facilitation of L-type Ca2+ channels both depend on specific amino acid residues in a consensus calmodulin-binding motif in the α1C subunit. J Biol Chem. 2000;275:21121–21129. doi: 10.1074/jbc.M002986200. [DOI] [PubMed] [Google Scholar]