

Abstract
We investigated the effect of interleukin-1β (IL-1β) on activity of an inwardly rectifying K+ channel in cultured human proximal tubule cells (RPTECs), using the patch-clamp technique and Fura-2 Ca2+ imaging. IL-1β (15 pg/ml) acutely reduced K+ channel activity in cell-attached patches. This effect was blocked by the IL-1 receptor antagonist (20 ng/ml), an inhibitor of phospholipase C, neomycin (300 μM), and an inhibitor of protein kinase C (PKC), GF109203X (500 nM). The Fura-2 Ca2+ imaging revealed that IL-1β increased intracellular Ca2+ concentration even after removal of extracellular Ca2+, which was blocked by an inhibitor of inositol 1,4,5-trisphosphate receptors, 2-aminoethoxydiphenyl borate (2-APB, 1 μM). Moreover, IL-1β suppressed channel activity in the presence of 2-APB without extracellular Ca2+. These results suggest that IL-1β suppresses K+ channel activity in RPTECs through binding to its specific receptor and activation of the PKC pathway even though intracellular Ca2+ does not increase.
Keywords: Cytokine, Patch-clamp, Fura-2, Ca2+, PKC
Introduction
The kidney proximal tubule reabsorbs about 70 % of the filtered Na+ load [1, 2]. The basolateral K+ channels in proximal tubule cells provide a driving force for the transepithelial Na+ reabsorption by maintaining cell-negative potential and serving as the K+ recycling pathway for the basolateral Na+-K+-ATPase [1, 2]. We previously demonstrated that an inwardly rectifying K+ channel with an inward conductance of about 40 pS was predominantly observed in cultured human proximal tubule cells under the control condition, using the patch-clamp technique [3]. Since inwardly rectifying K+ channels in proximal tubule cells are reported to be mainly present in the basolateral membrane [4], this 40 pS K+ channel would also be basolateral in origin, even though the cultured cells we used were not well polarized [5].
In addition to their physiological importance, K+ channels in the proximal tubule seem to be involved in the pathogenesis of renal cell injury. Some investigators reported that increased K+ channel activity augmented hypoxic tissue damage in proximal tubules [6, 7], whereas others reported that reduction of K+ channel activity exacerbates ischemic/reperfusion injury [8] or endotoxemic renal failure [9].
It is widely accepted that proinflammatory cytokines are the key molecules of cell injury in various organs during inflammatory diseases [10]. In the kidney, acute renal failure is often caused by a severe bacterial infection [11, 12]. The bacteria-derived endotoxin stimulates production of proinflammatory cytokines, which in turn accelerate renal tubular cell injury [13, 14], as well as impairment of glomerular filtration [15]. Since the changes in K+ channel activity were involved in the renal cell injury as described above, it is possible that the adverse effect of proinflammatory cytokines on renal tubular cells would partly be mediated by modulation of K+ channel activity. However, there are only a few reports regarding the effects of proinflammatory cytokines on activity of renal K+ channels [16, 17]. In rat thick ascending limb, Wei et al. [16] reported that tumor necrosis factor acutely stimulated activity of the apical K+ channel. We demonstrated that interferon-γ possessed delayed suppressive and acute stimulatory effects on activity of the inwardly rectifying K+ channel in cultured human proximal tubule cells [17].
Interleukin-1β (IL-1β) is another well-known proinflammatory cytokine which plays important roles in promoting renal cell injury [13–15]. Part of the cytotoxic effect of IL-1β might be related to its effect on K+ channel activity. In addition, IL-1β reduces renal tubular transport of Na+ [18] and glucose [19], whose driving force is partly dependent on K+ channel activity. Thus, it is important to examine how IL-1β affects the activity of renal K+ channels.
In this study, we investigated the effect of IL-1β on activity of an inwardly rectifying K+ channel in cultured human proximal tubule cells, using the patch-clamp technique and Fura-2 Ca2+ imaging.
Materials and methods
Cell culture
Renal proximal tubule epithelial cells (RPTECs) of normal kidney origin (36-year-old man, lot 3F0307) were purchased from Lonza (Walkersville, MD, USA). The manufacturer guaranteed that >90 % of the cells were positive for γ-GTP, a marker protein specific to the proximal tubule [20]. These cells were provided as secondary cultures and maintained up to 6th passage in the REGM medium (Lonza) in a humidified atmosphere of 5 % CO2/95 % air at 37 °C. When the culture reached 70–80 % confluence, the RPTECs were dispersed with trypsin/EDTA and resuspended in REGM. Then, the cells were seeded on collagen-coated coverslips (Asahi Techno Glass, Tokyo, Japan) placed in a ϕ35-mm dish (Falcon, Franklin Lakes, NJ, USA) for the patch-clamp experiments or directly on a ϕ35-mm dish with a ϕ12-mm polymer bottom (BMS, Tokyo, Japan) for Fura-2 Ca2+ imaging at a density of 1 × 105 cells/dish. After a 3- to 7-h incubation, the coverslip was transferred to an open-bath heating chamber mounted on an inverted microscope for the patch-clamp. The polymer-bottom dish was similarly transferred to a heater platform mounted on an inverted fluorescent microscope for Ca2+ imaging. All experiments were performed at 33 °C.
Solutions
The control bath solution contained (in mM) 140 NaCl, 5 KCl, 1 CaCl2, 1 MgCl2, 5 glucose, and 10 HEPES. For the Ca2+-free bath solution, CaCl2 was omitted from the control bath solution. Patch pipettes were filled with a KCl solution, which contained (in mM) 145 KCl, 1 MgCl2, 1 EGTA, and 5 HEPES. In inside-out patches, the KCl solution containing 10−6 M Ca2+ was used as the bath solution. The solution with 10−6 M Ca2+ was prepared by adding an adequate amount of CaCl2 and 10 mM EGTA according to the computer program of Oiki and Okada [21], which was made by using the absolute values of stability constant of EGTA for the binding of Ca2+, Mg2+ and H+ [22]. These solutions were titrated to pH7.3 with NaOH or KOH.
Test substances
IL-1β and IL-1 receptor antagonist (IL-1ra) were purchased from PeproTech EC (London, UK) and ProSpec-Tany TechnoGene (Rehovot, Israel), respectively. Protein kinase C-α (PKC-α), an inhibitor of PKC, GF109203X, and an inhibitor of phospholipase C (PLC), neomycin, were purchased from Merck (Darmstadt, Germany). MgATP, phorbol-12-myristate-13-acetate (PMA), and 2-aminoethoxydiphenyl borate (2-APB) were from Sigma (St. Louis, MO, USA). GF109203X, PMA and 2-APB were dissolved in dimethyl sulfoxide (DMSO) as stock solutions, whereas the others were dissolved in water. These stock solutions were diluted with the bath solution before use and added to the bath by hand pipetting. The final concentration of DMSO in the bath was 0.023 % or less, which did not affect channel activity and intracellular Ca2+ concentration.
Patch-clamp technique
Single channel currents were recorded with cell-attached and inside-out patches applied to the surface membrane of single RPTECs. Patch pipettes were fabricated from glass capillaries (GC150-7.5, Warner, Hamden, CT, USA), with the resistance ranging from 3 to 4 MΩ when filled with the KCl solution. The pipette holding potential (V p) was set at 0 mV for cell-attached patches and +50 mV for inside-out patches. Current signals were recorded with a patch-clamp amplifier Axopatch 700B (Molecular Devices, Sunnyvale, CA, USA) and stored on a DAT recorder (RD-120TE, TEAC, Tokyo, Japan). The recorded signals were then low-pass filtered (3611 Multifunction Filter, NF electronic instruments, Tokyo, Japan) at 500 Hz and digitized at a rate of 2.5 kHz through an interface (Digidata 1440A, Molecular Devices). The acquired data were analyzed with pCLAMP10 software (Molecular Devices). Current traces of downward deflections represented inward currents. Channel activity was determined by NP o, which was calculated from an amplitude histogram as
where N is the maximum number of open channels observed in the patch, P o is the open probability, n is the number of channels observed at the same time, and t n is the probability that n channels are simultaneously open. Since the control values of NP o varied among patches, we calculated normalized channel activity (NP o,e/NP o,c) to conveniently compare the channel activity in experimental conditions with controls. NP o,c and NP o,e are the channel activities under control and experimental conditions, respectively. Routinely, we determined NP o,c from a 20-s sampling period just before adding the substance when the steady state lasted for at least 60 s. NP o,e was determined from a 20-s sampling period extracted from the steady state for at least 20–30 s made by the experimental substance.
Fluorescent Ca2+ imaging
Intracellular concentration of Ca2+ ([Ca2+]i) was measured by using the InCyt Basic IM imaging system (Intracellular Imaging, Cincinnati, OH, USA), as reported previously by Kubokawa et al. [23]. RPTECs on the polymer-bottom dishes were loaded with Fura-2 by a 20-min incubation in REGM medium containing 5 μM Fura-2AM (Dojindo, Kumamoto, Japan) at 37 °C. After loading, dishes were thoroughly washed with the control bath solution to remove excess dye and placed on the heater platform mounted on a fluorescent microscope. The loaded Fura-2 was sequentially excited by two wavelengths at 340 and 380 nm. The fluorescent emissions at 510 nm in response to both excitations were captured as paired signals every 5 s by a monochrome CCD camera, with the objective set at 40×.
Standard calibration of single RPTECs was carried out, and [Ca2+]i was calculated by using the equation described by Grynkiewicz et al. [24] as
where R represents the ratio of F 340:F 380, with the former and latter being fluorescent intensities excited at 340 and 380 nm, respectively; R min and R max are the F 340:F 380 ratios corresponding to the minimum and maximal Ca2+ concentrations; S f2 represents the F 380 value measured when Ca2+ is not bound to Fura-2 and S b2 represents the F 380 value measured when Fura-2 is fully bound to Ca2+; and K d is the Ca2+ dissociation constant of the indicator. R max was obtained by adding the Ca2+ ionophore, ionomycin (5 μM, Wako, Osaka, Japan), to the control bath solution with 1 mM Ca2+, and R min was obtained by adding ionomycin to the Ca2+-free bath solution with 5 mM EGTA. The K d value of 230 nM for Fura-2 was adopted in this study.
Statistical analysis
Data are expressed as mean ± SE. Student’s t-test or ANOVA in conjunction with Bonferroni t-test was used for statistical comparisons. A P value less than 0.05 was considered to be significant.
Results
Effect of IL-1β on K+ channel activity
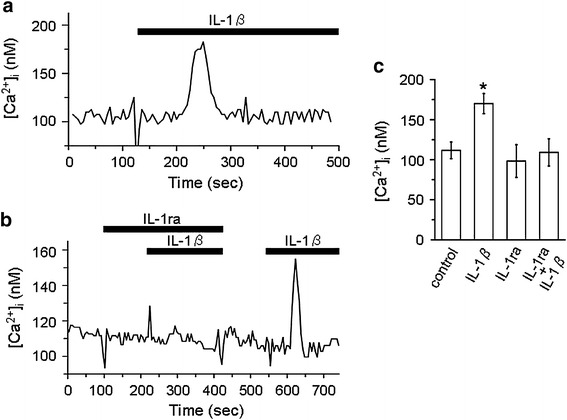
Figure 1a shows a representative current recording in response to IL-1β, which was obtained from a cell-attached patch. IL-1β added to the bath at 15 pg/ml suppressed activity of an inwardly rectifying K+ channel in a few minutes. To confirm that the acute suppressive effect of IL-1β was mediated by its specific receptor, we tested the IL-1ra. As shown in Fig. 1b, prior addition of IL-1ra to the bath at 20 ng/ml blocked the suppressive effect of IL-1β. Furthermore, the channel activity suppressed by IL-1β was restored by the subsequent addition of IL-1ra (Fig. 1c). Data were pooled and are summarized in Fig. 1d. IL-1β reduced channel activity to 33.5 ± 9.2 % of the control. IL-1ra almost completely blocked the suppressive effect of IL-1β, with the channel activity remaining at the control level. In addition, IL-1ra restored the channel activity suppressed by IL-1β to a level which was not significantly different from the control. These results suggested that the acute suppressive effect of IL-1β was receptor-mediated, but not a non-specific action.
Fig. 1.
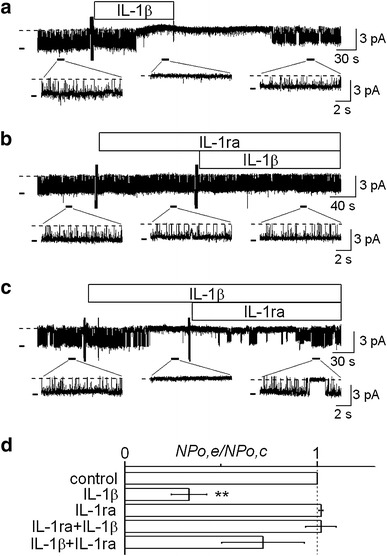
Effect of IL-1β on K+ channel activity and its abolishment by IL-1ra in cultured human proximal tubule cells. a–c Representative current traces showing acute suppression of channel activity by IL-1β (a), blockade of the suppressive effect by IL-1ra (b), and restoration of channel activity by IL-1ra in the presence of IL-1β (c). Dotted line represents the closed channel level. Short thick horizontal bar on the left of each trace represents the open channel level. The NP o values calculated from these current traces were 0.75–0.86 for controls, 0 for IL-1β alone, 0.82 for IL-1ra alone, 0.84 for IL-1ra + IL-1β, and 0.68 for IL-1β + IL-1ra. The doses of IL-1β and IL-1ra were 15 pg/ml and 20 ng/ml, respectively. Each trace was obtained from separate cell-attached patches at a V p of 0 mV. d Summary of the effects of IL-1β on channel activity and its blockade by IL-1ra. From the top, effects of IL-1β (n = 18), IL-1ra (n = 10), IL-1β in the presence of IL-1ra (n = 10), and IL-1ra added to the bath after IL-1β (n = 8). **P < 0.01, significantly different compared with the initial control level
Effect of a PKC inhibitor on the IL-1β-induced suppression of channel activity
It has been reported that the PKC-mediated phosphorylation processes suppressed activity of the native inwardly rectifying K+ channels in opossum kidney proximal tubule (OKP) cells [25], Ambystoma proximal tubule cells [26], and principal cells of rat cortical collecting duct (CCD) [27]. In addition, some investigators reported that IL-1β inhibited activity of a voltage-dependent Ca2+ channel in guinea-pig hippocampal neurons [28] or expression of epithelial sodium channel in human middle ear [29] through activation of PKC. Therefore, we examined whether the suppressive effect of IL-1β on K+ channel activity in RPTECs would have some relation to the PKC pathway. As shown in Fig. 2a, a PKC inhibitor, GF109203X, added to the bath at 500 nM blocked the suppressive effect of IL-1β on channel activity, while GF109203X itself did not affect channel activity. Figure 2b further shows that GF109203X reactivated the channel which was suppressed by IL-1β. Data on the effects of GF109203X are summarized in Fig. 2c. Channel activity in the presence of GF109203X alone was the same level as the control. IL-1β had no significant effect on channel activity when GF109203X had been added in advance. It was also evident that the IL-1β-induced significant reduction of channel activity (36.7 ± 8.1 % of the control) was reversed by the subsequent addition of GF109203X, with the channel activity returning to the level comparable to the control. Thus, it is highly likely that the suppressive effect of IL-1β was dependent on PKC activity.
Fig. 2.
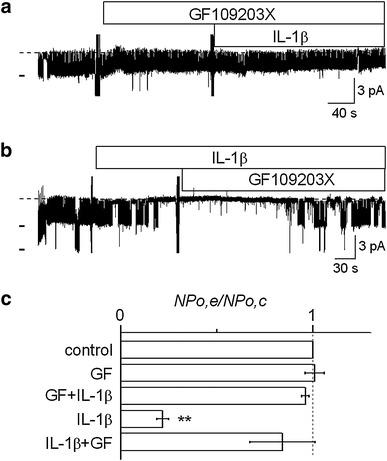
Involvement of PKC in the acute suppressive effect of IL-1β on channel activity. a Representative current trace showing that a PKC inhibitor, GF109203X, blocked the suppressive effect of IL-1β. b GF109203X restored the channel activity which was suppressed by IL-1β. The NP o values calculated from these current traces were 0.78–0.83 for controls, 0.85 for GF109203X alone, 0.83 for GF109203X + IL-1β, 0 for IL-1β alone, and 0.64 for IL-1β + GF109203X. The doses of IL-1β and GF109203X were 15 pg/ml and 500 nM, respectively. Each trace was obtained from separate cell-attached patches at a V p of 0 mV. c Summary of the effects of GF109203X on the IL-1β-induced suppression of channel activity. From the top, effects of GF109203X alone (n = 12), IL-1β in the presence of GF109203X (n = 12), IL-1β (n = 8), and GF109203X added after IL-1β (n = 8). **P < 0.01, significantly different compared with the initial control level
Direct effect of PKC on channel activity
In this experiment, we examined the effect of cytoplasmic PKC on channel activity in inside-out patches. Figure 3a is a representative current recording in response to PMA and PKC-α isoform. The bath solution was a high K+ (145 mM) solution, which contained Ca2+ of 10−6 M for the activation of PKC. This high K+ bath solution also contained 1 mM MgATP, since the activity of this K+ channel requires cytoplasmic ATP to maintain its activity in inside-out patches [3]. Although addition of PMA alone at 500 nM had no effect on channel activity, the subsequent addition of PKC-α at 1 U/ml suppressed it (Fig. 3a), which was abolished by GF109203X. Data were pooled and are summarized in Fig. 3b. There was no statistically significant difference between the control (MgATP alone) and PMA-treated groups, whereas PKC-α significantly reduced channel activity to 21.3 ± 6.0 % of the control in the presence of MgATP and PMA. The channel activity suppressed by PKC-α was restored by GF109203X to the control level. These results suggested that PKC directly suppressed the activity of the K+ channel in RPTECs.
Fig. 3.
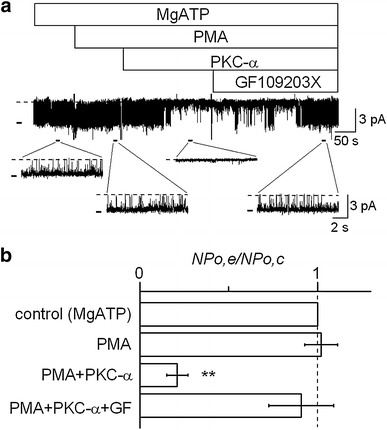
Direct suppressive effect of PKC on channel activity in inside-out patches. a Representative current trace showing that PKC-α suppressed channel activity in the presence of MgATP and PMA, with the subsequent addition of GF109203X restoring channel activity. The V p was held at +50 mV. The bath solution used was a KCl solution containing 10−6 M Ca2+. In this trace, the calculated NP o values were 0.83 for control, 0.81 for PMA alone, 0 for PMA + PKC-α, and 0.78 for PMA + PKC-α + GF109203X. The doses of MgATP, PMA, PKC-α, and GF109203X were 1 mM, 500 nM, 1 U/ml, and 500 nM, respectively. b Summary of the effects of PMA (n = 8), PKC-α in the presence of PMA (n = 8), and GF109203X added after PMA and PKC (n = 6). **P < 0.01, significantly different compared with the control level, which was maintained by MgATP alone
Effect of a PLC inhibitor on the IL-1β-induced suppression of channel activity
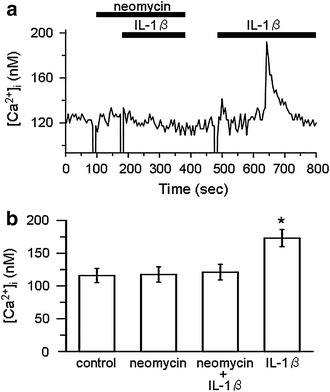
It is well known that PLC plays an important role in activating the PKC pathway [30]. PLC hydrolyzes phosphatidylinositol 4,5-bisphosphate (PIP2) into inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG), both of which contribute to activation of PKC [30, 31]. Therefore, we examined whether a PLC inhibitor would interfere with the suppressive effect of IL-1β on channel activity. As shown in Fig. 4a, a PLC inhibitor, neomycin, added to the bath at 300 μM blocked the suppressive effect of IL-1β, while the IL-1β-induced suppression of channel activity was observed in the same patch after washing out the neomycin. Data are summarized in Fig. 4b. Neomycin itself did not affect channel activity. IL-1β had no significant effect on channel activity in the presence of neomycin, although it reduced channel activity to 38.8 ± 11.5 % of the control after washing out the neomycin. These results suggest that the PKC-dependent suppressive effect of IL-1β is mediated by PLC.
Fig. 4.
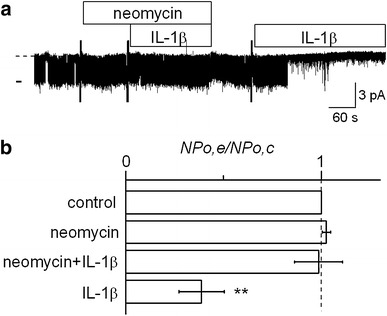
Effect of a PLC inhibitor on the IL-1β-induced suppression of channel activity. a Representative current trace showing that IL-1β did not suppress channel activity in the presence of neomycin, whereas IL-1β alone suppressed the channel after washing out neomycin. In this trace, the calculated NP o values were 0.78 for control, 0.82 for neomycin alone, 0.83 for neomycin + IL-1β, and 0.05 for IL-1β alone. The doses of neomycin and IL-1β were 300 μM and 15 pg/ml, respectively. b Summary of the effects of neomycin (n = 11), IL-1β in the presence of neomycin (n = 11), and IL-1β alone after washing-out (n = 7). **P < 0.01, significantly different compared with the initial control level
Effect of IL-1β on [Ca2+]i
One of the PLC-hydrolyzed PIP2 derivatives, IP3, induces Ca2+ release from the intracellular store by binding to IP3 receptors [30, 31]. The resultant increase in [Ca2+]i, as well as DAG, is necessary for activation of some PKC isoforms [32]. In the following set of experiments, we examined the effect of IL-1β on [Ca2+]i by using Fura-2 Ca2+ imaging. Figure 5a is a representative trace of Fura-2 Ca2+ imaging in response to IL-1β, showing that IL-1β added to the bath at 15 pg/ml caused a transient rise in [Ca2+]i. This effect of IL-1β was blocked by the prior addition of IL-1ra to the bath at 20 ng/ml (Fig. 5b). These data are summarized in Fig. 5c. The peak value of [Ca2+]i in response IL-1β (170 ± 12 nM) was significantly higher than the [Ca2+]i under control conditions (112 ± 10 nM). However, the effect of IL-1β was not observed in the presence of IL-1ra, with the [Ca2+]i remaining at the control level. IL-1ra alone had no significant effect on [Ca2+]i. In addition to IL-1ra, neomycin (300 μM) also blocked the effect of IL-1β on [Ca2+]i (Fig. 6a), which was consistent with the patch-clamp experiments. The [Ca2+]i values in response to neomycin alone and IL-1β in the presence neomycin were not significantly different from the control value (116 ± 11 nM, Fig. 6b), although IL-1β caused a significant rise in [Ca2+]i after washing out the neomycin (173 ± 13 nM, Fig. 6b). These results suggest that the effect of IL-1β on [Ca2+]i is also mediated by its specific receptor and PLC, as is the effect on channel activity.
Fig. 5.
Effect of IL-1β on intracellular Ca2+ concentration ([Ca2+]i) in cultured human proximal tubule cells. a Representative trace of Fura-2 Ca2+ imaging which shows that IL-1β caused a transient rise in [Ca2+]i. b The IL-1β-induced rise in [Ca2+]i did not occur in the presence of IL-1ra, although the effect of IL-1β was still observed after washing out IL-1ra. The doses of IL-1β and IL-1ra were 15 pg/ml and 20 ng/ml, respectively. Each trace was recorded with separate cells. c Summarized data showing [Ca2+]i under the control condition (n = 24) and [Ca2+]i in response to IL-1β (n = 24), IL-1ra (n = 10), and IL-1β in the presence of IL-1ra (n = 10). *P < 0.05, significantly different compared with the control
Fig. 6.
Effect of a PLC inhibitor, neomycin, on the IL-1β-induced rise in [Ca2+]i. a Representative trace of Fura-2 Ca2+ imaging which shows that neomycin blocked the IL-1β-induced rise in [Ca2+]i. IL-1β was still capable of increasing [Ca2+]i after washing out neomycin. The doses of neomycin and IL-1β were 300 μM and 15 pg/ml, respectively. b Summarized data showing [Ca2+]i under the control condition (n = 9) and [Ca2+]i in response to neomycin (n = 9), IL-1β in the presence of neomycin (n = 9), and IL-1β alone (n = 9). *P < 0.05, significantly different compared with the control
Involvement of intracellular Ca2+ store in the IL-1β-induced rise in [Ca2+]i
We further examined whether the IL-1β-induced rise in [Ca2+]i would be accounted for by Ca2+ release from the intracellular store. As shown in Fig. 7a, IL-1β caused transient rises in [Ca2+]i in both the absence and presence of extracellular Ca2+. The peak values of [Ca2+]i in response to IL-1β in the absence and presence of extracellular Ca2+ were 159 ± 5 and 152 ± 6 nM, respectively, both of which were significantly higher than the control value of 121 ± 11 nM (Fig. 7b). Removal of extracellular Ca2+ slightly reduced [Ca2+]i to 111 ± 6 nM, but this change was insignificant (Fig. 7b). Figure 7c is a representative trace showing that the effect of IL-1β on [Ca2+]i in the Ca2+-free bath was blocked by 2-APB (1 μM), while the effect was evident after washing out the 2-APB. Data on the effects of 2-APB in the Ca2+-free bath are summarized in Fig. 7d. Addition of 2-APB to the bath caused a slight but insignificant decrease in [Ca2+]i (98 ± 12 nM), compared to the control (113 ± 8 nM). The [Ca2+]i value in response to IL-1β in the presence of 2-APB (99 ± 9 nM) was also insignificant compared to the control, although IL-1β caused a significant rise in [Ca2+]i after removal of 2-APB (161 ± 9 nM). It has been reported that 2-APB inhibited IP3 receptors, as well as Ca2+ influx through the Ca2+ permeable channels in the plasma membrane [33]. Since our results showed that IL-1β increased [Ca2+]i in the absence of extracellular Ca2+, which was blocked by 2-APB, it seems highly likely that the Ca2+ release from the intracellular store would be involved in the IL-1β-induced rise in [Ca2+]i.
Fig. 7.
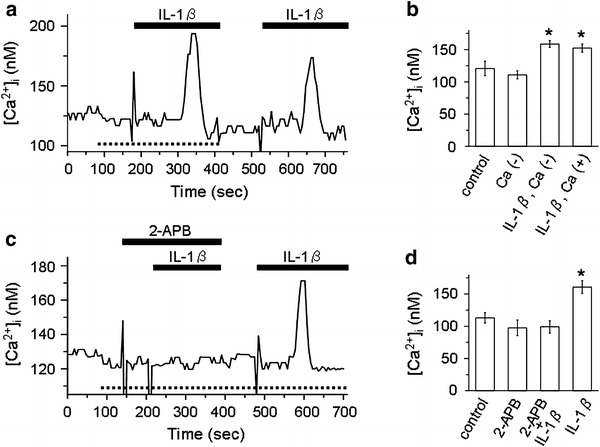
Involvement of intracellular Ca2+ store in the IL-1β-induced rise in [Ca2+]i. a Representative trace of Fura-2 Ca2+ imaging which shows that IL-1β increased [Ca2+]i in both the Ca2+-free bath solution (dotted line at the bottom of trace) and the control bath solution containing 1 mM Ca2+. b Summarized data showing [Ca2+]i under the control condition (n = 10) and [Ca2+]i in response to removal of extracellular Ca2+ (n = 10) and IL-1β with (n = 10) or without (n = 10) extracellular Ca2+. *P < 0.05, significantly different compared with the control. c Representative trace of Fura-2 Ca2+ imaging which shows that 2-APB blocked the IL-1β-induced rise in [Ca2+]i in the absence of extracellular Ca2+. Dotted line at the bottom of trace represents the period during which the Ca2+-free bath solution was used. The doses of 2-APB and IL-1β were 1 μM and 15 pg/ml, respectively. d Summarized data showing [Ca2+]i under the control condition (n = 8) and [Ca2+]i in response to 2-APB (n = 8), IL-1β in the presence of 2-APB (n = 8), and IL-1β alone (n = 8). *P < 0.05, significantly different compared with the control
Effect of blocking IP3 receptors on the IL-1β-induced suppression of channel activity in the absence of extracellular Ca2+
Finally, we examined the effect of 2-APB on the IL-1β-induced suppression of channel activity in the absence of extracellular Ca2+, using the cell-attached configuration of the patch-clamp technique. As shown in Fig. 8a, IL-1β suppressed channel activity even in the presence of 2-APB in the Ca2+-free bath solution. When GF109203X was concomitantly present, IL-1β did not suppress the channel (Fig. 8b), suggesting that the effect of IL-1β in the presence of 2-APB would actually be mediated by PKC. Data are summarized in Fig. 8c. There was no appreciable change in channel activity in response to 2-APB alone. IL-1β reduced channel activity even in the presence of 2-APB to 36.2 ± 9.8 % of the control, whereas this effect was abolished by the additional use of GF109203X, with the channel activity being around the control level.
Fig. 8.
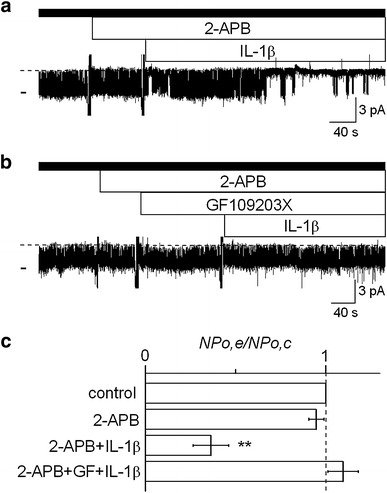
Effect of 2-APB on the IL-1β-induced suppression of channel activity in the Ca2+-free bath solution. a Representative current trace showing that the IL-1β suppressed channel activity in the presence of 2-APB with the Ca2+-free bath solution. b The effect of IL-1β in the presence of 2-APB was blocked by GF109203X. Filled bar above the trace indicates the use of Ca2+-free bath solution. The NP o values calculated from these current traces were 0.77–0.82 for controls, 0.76–0.82 for 2-APB alone, 0.19 for 2-APB + IL-1β, 0.76 for 2-APB + GF109203X, and 0.81 for 2-APB + GF109203X + IL-1β. The doses of 2-APB, IL-1β, and GF109203X were 1 μM, 15 pg/ml, and 300 μM, respectively. Each trace was obtained from separate cell-attached patches at a V p of 0 mV. c Summary of the effects of 2-APB (n = 17), IL-1β in the presence of 2-APB (n = 7), and IL-1β in the presence of 2-APB and GF109203X (n = 10). ** P < 0.01, significantly different compared with the initial control level
Discussion
In this study, we demonstrated that IL-1β acutely suppressed K+ channel activity in a PKC-dependent manner in RPTECs. As we reported previously [3], this K+ channel was most frequently observed under the control conditions, and exhibited inward rectification (Gi/Go = 40 pS/7 pS). Because we used the cultured cells in a configuration with poor polarity [5], it was not certain whether this K+ channel would represent the apical or basolateral one in native cells. However, it has been reported that inwardly rectifying K+ channels in proximal tubule cells are mainly distributed in the basolateral membrane [4]. Furthermore, the activity of the RPTECs’ K+ channel was pH-sensitive [3], stimulated by protein kinase A [3], and suppressed by PKC, consistent with the properties of basolateral K+ channels in animal proximal tubule cells [2, 26]. Thus, it seems likely that the inwardly rectifying K+ channel observed in this study in RPTECs would correspond to the basolateral one.
IL-1β is a representative member of the IL-1 superfamily which comprises at least 11 molecules [34, 35]. It plays important roles in promoting inflammatory processes and cell injury in various organs by inducing many effector proteins [36, 37]. In the kidney, the cytotoxicity of IL-β might involve K+ channel activity. Some investigators reported that changes in activity of the K+ channel resulted in renal cell injury during ischemia [6–8] or endotoxemia [9]. Thus, it is possible that the cytotoxic effects of IL-1β on kidney functions might partly be accounted for by its modulatory action on renal K+ channels. In addition to promoting cell injury, IL-1β impaired renal transport of Na+ [18] or glucose [19] during severe inflammation. These findings suggest that renal transporters of Na+ or glucose would be the targets of this proinflammatory cytokine. Since the transport of Na+ and glucose in the kidney is partly dependent on the activity of renal tubular K+ channels [1, 2], it is possible that the impaired transport would be due to inhibition of renal K+ channels by IL-1β. However, so far there has been no information available regarding the effects of IL-1β on renal tubular K+ channels.
The first event for the wide variety of biological actions of IL-1β is its binding to type 1 IL-1 receptors (IL-1R1) on the plasma membranes of target cells [38]. Upon ligand binding, IL-1R1 associates with the IL-1-receptor accessory protein (IL-1RAcP) to form a complex which then initiates various signal transduction pathways [38]. The binding of IL-1β to IL-1R1 is competitively blocked by a naturally occurring receptor antagonist, IL-1ra, which also belongs to the IL-1 superfamily [35, 38]. Although IL-1ra binds to I IL-1R1, it does not trigger signal transduction [37, 38]. From our data using IL-1ra, it is strongly suggested that the cultured human proximal cells expressed IL-1R1 and that the IL-1β acted on the channel through its binding to the specific receptor.
The downstream effectors activated by the IL-1R1/IL-1AcP complex include IL-1 receptor associating kinase (IRAK), phosphatidylinositol-3-kinase (PI3K), and PLC [38]. IRAK is generally thought to be involved in activation of several transcription factors, such as NFκB and AP-1 whose actions need a longer time to be expressed [38]. Thus, IRAK’s contribution to the acute effect of IL-1β on channel activity would be little, if any. In addition, we previously confirmed that PI3K inhibitors reduced the basal activity of the K+ channel in cultured human proximal tubule cells [17]. This indicated that PI3K had a positive effect on K+ channel activity. Thus, PI3K seemed not to mediate the acute suppressive effect of IL-1β. PLC activates PKC by providing DAG and IP3 which induces Ca2+ release from the intracellular store [30, 31]. The PKC-mediated protein phosphorylation is known to rapidly suppress activities of renal tubular K+ channels, such as the native 35 pS apical K+ channel in principal cells of CCD [27], the ROMK channel (Kir1.1) [39], Kir7.1 [40], the basolateral 25 pS K+ channel in Ambystoma proximal tubule cells [26], and the 90 pS K+ channel in cultured OKP cells [25]. In this study, we demonstrated that the suppressive effect of IL-1β on channel activity was blocked by inhibitors of PLC and PKC in cell-attached patches. Also, in inside-out patches, the K+ channel activity was directly suppressed by PKC. Moreover, the inside-out patch experiments showed that addition of GF109203X restored the PKC-mediated suppression of channel activity. The restoration of channel activity by the inhibition of PKC may result from dephosphorylation, dominated by membrane-bound phosphatases. In this regard, it was reported that protein phosphatase-2B (PP-2B) was anchored to the cytoplasmic membrane through binding to an A kinase anchoring protein (AKAP) complex, AKAP79/150 [41]. Furthermore, PP-2B was reported to dephosphorylate the Ca2+/calmodulin-dependent protein kinase II-mediated and probably the PKC-mediated phosphorylation processes to rescue the K+ channel activity suppressed by these kinases [23]. Therefore, it is possible that the membrane-bound PP-2B would be involved in the restoration of channel activity suppressed by PKC in inside-out patches in our study. Taken together, these results strongly suggested that the PLC/PKC-mediated processes were crucial for the effect of IL-1β on channel activity. Although there is no doubt that PKC would be involved in the suppressive effect of IL-1β on channel activity, it seemed that the PKC activity would be relatively low or rather silent under the control conditions in cultured human proximal tubule cells. This notion was supported by the observations that the PKC inhibitor, as well as the PLC inhibitor, had no apparent effect on the basal activity of the K+ channel. Similar ineffectiveness of PKC inhibitors was reported with the 35 pS K+ channel of rat CCD [42].
Fura-2 Ca2+ imaging clearly showed that IL-1β transiently increased [Ca2+]i in the receptor-specific and PLC-dependent manners. The peak values of [Ca2+]i in response to IL-1β were not significantly different between the Ca2+-free and Ca2+-containing bath solutions. In addition, 2-APB blocked the effect of IL-1β on [Ca2+]i in the absence of extracellular Ca2+. It has been reported that 2-APB inhibited both Ca2+ influx and the IP3 receptor-mediated Ca2+ release from the intracellular store, but not the ryanodine receptor-mediated Ca2+ release from the intracellular store [33]. Taken together, it was strongly suggested that the IL-1β-induced rise in [Ca2+]i would mostly be attributable to the IP3-mediated Ca2+ release. The involvement of IP3-mediated Ca2+ release in IL-1β signaling is consistent with the previous reports of other investigators [43, 44]. However, it seemed that the IL-1β-induced rise in [Ca2+]i was not necessarily required for the activation of PKC and the subsequent suppression of K+ channel activity in cultured human proximal cells. In fact, the suppressive effect of IL-1β on channel activity was evident in the presence of 2-APB in the Ca2+-free bath solution, where increase in [Ca2+]i did not occur. This effect of IL-1β on channel activity in the presence of 2-APB was unambiguously PKC-dependent, since a PKC inhibitor, GF109203X, abolished it. PKC is classified into three types that include the conventional PKC, novel PKC and atypical PKC, each of which further consists of multiple isoforms [32]. The conventional PKC requires Ca2+ for its activation as well as DAG, whereas others are independent of Ca2+ [32]. According to the report by Karim et al. [45] using Western blot analysis, PKC-α, δ, ε and ζ isoforms were present in the rat proximal tubule. Among these isoforms, the last three do not require Ca2+ for their activations [32]. Thus, it is possible that Ca2+-independent PKC isoforms would play roles in the IL-1β-induced suppression of channel activity in the presence of 2-APB without extracellular Ca2+. Nevertheless, the possibility that the Ca2+-dependent PKC isoform(s) might contribute to the IL-1β-induced suppression of channel activity under physiological conditions could not be excluded. Although the magnitude of the IL-1β-induced rise in [Ca2+]i was relatively small, a similar increase in [Ca2+]i was reported to activate conventional PKC isoforms in human cultured breast epithelial cells [46].
In summary, IL-1β acutely suppresses activity of an inwardly rectifying K+ channel in cultured human proximal tubule cells through its binding to the specific receptor and the subsequent activation of the PLC/PKC pathway. Since IL-1β suppressed channel activity despite the lack of increase in [Ca2+]i, Ca2+-independent PKC isoforms would be mainly involved, even though the participation of Ca2+-dependent PKC isoforms might not be excluded. The IL-1β-induced suppression of K+ channel activity would account for the impaired renal functions, such as diminished reabsorption of Na+ [18] and glucose [19] during inflammation. The causal relationship between the IL-1β-induced channel suppression and renal cell injury needs to be further clarified.
Acknowledgments
This work was supported in part by a grant-in-aid for scientific research from the Japan Society for the Promotion of Science (to M.K; 23590264).
Conflict of interest
The authors declare that they have no conflict of interest.
References
- 1.Giebisch G. A long affair with renal tubules. Annu Rev Physiol. 2011;73:1–28. doi: 10.1146/annurev-physiol-012110-142241. [DOI] [PubMed] [Google Scholar]
- 2.Hebert SC, Desir G, Giebisch G, Wang W. Molecular diversity and regulation of renal potassium channels. Physiol Rev. 2005;85:319–371. doi: 10.1152/physrev.00051.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nakamura K, Hirano J, Kubokawa M. An ATP-regulated and pH-sensitive inwardly rectifying K+ channel in cultured human proximal tubule cells. Jpn J Physiol. 2001;51:523–530. doi: 10.2170/jjphysiol.51.523. [DOI] [PubMed] [Google Scholar]
- 4.Hamilton KL, Devor DC. Basolateral membrane K+ channels in renal epithelial cells. Am J Physiol Renal Physiol. 2012;302:F1069–F1081. doi: 10.1152/ajprenal.00646.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nakamura K, Habano W, Kojo T, Komagiri Y, Kubota T, Kubokawa M. Involvement of endogenous nitric oxide in the regulation of K+ channel activity in cultured human proximal tubule cells. J Physiol Sci. 2006;56:407–413. doi: 10.2170/physiolsci.RP003106. [DOI] [PubMed] [Google Scholar]
- 6.Reeves WB, Shah SV. Activation of potassium channels contributes to hypoxic injury in proximal tubules. J Clin Invest. 1994;94:2289–2294. doi: 10.1172/JCI117592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Engbersen R, Moons MM, Wouterse AC, Dijkman HB, Kramers C, Smits P, Russel FG. Sulphonylurea drugs reduce hypoxic damage in the isolated perfused rat kidney. Br J Pharmacol. 2000;130:1678–1684. doi: 10.1038/sj.bjp.0703469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rahgozar M, Willgoss DA, Gobé GC, Endre ZH. ATP-dependent K+ channels in renal ischemia reperfusion injury. Ren Fail. 2003;25:885–896. doi: 10.1081/JDI-120026024. [DOI] [PubMed] [Google Scholar]
- 9.Zager RA, Johnson AC, Lund S, Hanson SY, Abrass CK. Levosimendan protects against experimental endotoxemic acute renal failure. Am J Physiol Renal Physiol. 2006;290:F1453–F1462. doi: 10.1152/ajprenal.00485.2005. [DOI] [PubMed] [Google Scholar]
- 10.Feghali CA, Wright TM. Cytokines in acute and chronic inflammation. Front Biosci. 1997;2:12–26. doi: 10.2741/a171. [DOI] [PubMed] [Google Scholar]
- 11.Lopes JA, Jorge S, Resina C, Santos C, Pereira Á, Neves J, Antunes F, Prata MM. Acute renal failure in patients with sepsis. Crit Care. 2007;11:411. doi: 10.1186/cc5735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schrier RW, Wang W. Acute renal failure and sepsis. N Engl J Med. 2004;351:159–169. doi: 10.1056/NEJMra032401. [DOI] [PubMed] [Google Scholar]
- 13.Vesey DA, Cheung CW, Cuttle L, Endre ZA, Gobé G, Johnson DW. Interleukin-1β induces human proximal tubule cell injury, α-smooth muscle actin expression and fibronectin production. Kidney Int. 2002;62:31–40. doi: 10.1046/j.1523-1755.2002.00401.x. [DOI] [PubMed] [Google Scholar]
- 14.Vesey DA, Cheung C, Endre Z, Gobe G, Johnson D. Role of protein kinase C and oxidative stress in interleukin-1β-induced human proximal tubule cell injury and fibrogenesis. Nephrol. 2005;10:73–80. doi: 10.1111/j.1440-1797.2005.00363.x. [DOI] [PubMed] [Google Scholar]
- 15.Timoshanko JR, Kitching AR, Iwakura Y, Holdsworth SR, Tipping PG. Leukocyte-derived interleukin-1β interacts with renal interleukin-1 receptor I to promote renal tumor necrosis factor and glomerular injury in murine crescentic glomerulonephritis. Am J Pathol. 2004;164:1967–1977. doi: 10.1016/S0002-9440(10)63757-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wei Y, Babilonia E, Pedraza PL, Ferreri NR, Wang WH. Acute application of TNF stimulates apical 70-pS K+ channels in the thick ascending limb of rat kidney. Am J Physiol Renal Physiol. 2003;285:F491–F497. doi: 10.1152/ajprenal.00104.2003. [DOI] [PubMed] [Google Scholar]
- 17.Nakamura K, Komagiri Y, Kojo T, Kubokawa M. Delayed and acute effects of interferon-γ on activity of an inwardly rectifying K+ channel in cultured human proximal tubule cells. Am J Physiol. 2009;296:F46–F53. doi: 10.1152/ajprenal.00127.2008. [DOI] [PubMed] [Google Scholar]
- 18.Schmidt C, Höcherl K, Schweda F, Kurtz A, Bucher M. Regulation of renal sodium transporters during severe inflammation. J Am Soc Nephrol. 2007;18:1072–1083. doi: 10.1681/ASN.2006050454. [DOI] [PubMed] [Google Scholar]
- 19.Schmidt C, Höcherl K, Bucher M. Regulation of renal glucose transporters during severe inflammation. Am J Physiol Renal Physiol. 2007;292:F804–F811. doi: 10.1152/ajprenal.00258.2006. [DOI] [PubMed] [Google Scholar]
- 20.Hanigan MH, Frierson HF., Jr Immunohistochemical detection of gamma-glutamyl transpeptidase in normal human tissue. J Histochem Cytochem. 1996;44:1101–1108. doi: 10.1177/44.10.8813074. [DOI] [PubMed] [Google Scholar]
- 21.Oiki S, Okada Y. Ca-EGTA buffer in physiological solutions. Seitai-no-Kagaku. 1987;38:79–83. [Google Scholar]
- 22.Martell AE, Smith R (1977) Critical stability constants. In: Other organic ligands, vol 3. Plenum Press, New York
- 23.Kubokawa M, Kojo T, Komagiri Y, Nakamura K. Role of calcineurin-mediated dephosphorylation in modulation of an inwardly rectifying K+ channel in human proximal tubule cells. J Membr Biol. 2009;231:79–92. doi: 10.1007/s00232-009-9207-z. [DOI] [PubMed] [Google Scholar]
- 24.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;25:3440–3450. [PubMed] [Google Scholar]
- 25.Mori Y, Kawasaki A, Takamaki A, Kitano I, Yoshida R, Kubokawa M, Kubota T. Ca2+-dependent inhibition of inwardly rectifying K+ channel in opossum kidney cells. Jpn J Physiol. 2001;51:371–380. doi: 10.2170/jjphysiol.51.371. [DOI] [PubMed] [Google Scholar]
- 26.Mauerer UR, Boulpaep EL, Segal AS. Regulation of an inwardly rectifying ATP-sensitive K+ channel in the basolateral membrane of renal proximal tubule. J Gen Physiol. 1998;111:161–180. doi: 10.1085/jgp.111.1.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang WH, Giebisch G. Dual modulation of renal ATP-sensitive K+ channel by protein kinases A and C. Proc Natl Acad Sci USA. 1991;88:9722–9725. doi: 10.1073/pnas.88.21.9722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Plata-Salaman CR, Ffrench-Mullen JM. Interleukin-1 β inhibits Ca2+ channel currents in hippocampal neurons through protein kinase C. Eur J Pharmacol. 1994;266:1–10. doi: 10.1016/0922-4106(94)90202-X. [DOI] [PubMed] [Google Scholar]
- 29.Choi JY, Choi YS, Kim SJ, Son EJ, Choi HS, Yoon JH. Interleukin-1β suppresses epithelial sodium channel β-subunit expression and ENaC-dependent fluid absorption in human middle ear epithelial cells. Eur J Pharmacol. 2007;567:19–25. doi: 10.1016/j.ejphar.2007.04.026. [DOI] [PubMed] [Google Scholar]
- 30.Rebecchi MJ, Pentyala SN. Structure, function, and control of phosphoinositide-specific phospholipase C. Physiol Rev. 2000;80:1291–1335. doi: 10.1152/physrev.2000.80.4.1291. [DOI] [PubMed] [Google Scholar]
- 31.Berridge MJ. Inositol trisphosphate and diacylglycerol as second messengers. Biochem J. 1984;220:345–360. doi: 10.1042/bj2200345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lipp P, Reither G. Protein kinase C: the “Masters” of calcium and lipid. Cold Spring Harb Perspect Biol. 2011;3:a004556. doi: 10.1101/cshperspect.a004556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peppiatt CM, Collins TJ, Mackenzie L, Conway SJ, Holmes AB, Bootman MD, Berridge MJ, Seo JT, Roderick HL. 2-Aminoethoxydiphenyl borate (2-APB) antagonises inositol 1,4,5-trisphosphate-induced calcium release, inhibits calcium pumps and has a use-dependent and slowly reversible action on store-operated calcium entry channels. Cell Calcium. 2003;34:97–108. doi: 10.1016/S0143-4160(03)00026-5. [DOI] [PubMed] [Google Scholar]
- 34.Sims JE, Smith DE. The IL-1 family: regulators of immunity. Nat Rev Immunol. 2010;10:89–102. doi: 10.1038/nri2691. [DOI] [PubMed] [Google Scholar]
- 35.Barksby HE, Lea SR, Preshaw PM, Taylor JJ. The expanding family of interleukin-1 cytokines and their role in destructive inflammatory disorders. Clin Exp Immunol. 2007;149:217–225. doi: 10.1111/j.1365-2249.2007.03441.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dinarello CA. Interleukin-1β. Crit Care Med. 2005;33:S460–S462. doi: 10.1097/01.CCM.0000185500.11080.91. [DOI] [PubMed] [Google Scholar]
- 37.Allan SM, Tyrrell PJ, Rothwell NJ. Interleukin-1 and neuronal injury. Nat Rev Immunol. 2005;5:629–640. doi: 10.1038/nri1664. [DOI] [PubMed] [Google Scholar]
- 38.Daun JM, Fenton MJ. Interleukin-1/Toll receptor family members: receptor structure and signal transduction pathways. J Interferon Cytokine Res. 2000;20:843–855. doi: 10.1089/10799900050163217. [DOI] [PubMed] [Google Scholar]
- 39.Zeng WZ, Li XJ, Hilgemann DW, Huang CL. Protein kinase C inhibits ROMK1 channel activity via a phosphatidylinositol 4,5-bisphosphate-dependent mechanism. J Biol Chem. 2003;278:16852–16856. doi: 10.1074/jbc.M300619200. [DOI] [PubMed] [Google Scholar]
- 40.Zhang W, Zitron E, Bloehs R, Müller-Krebs S, Scholz E, Zeier M, Katus H, Karle C, Schwenger V. Dual regulation of renal Kir7.1 potassium channels by protein kinase A and protein kinase C. BBRC. 2008;377:981–986. doi: 10.1016/j.bbrc.2008.10.110. [DOI] [PubMed] [Google Scholar]
- 41.Wong W, Scott JD. AKAP signalling complexes: focal points in space and time. Nature Rev Mol Cell Biol. 2004;5:959–970. doi: 10.1038/nrm1527. [DOI] [PubMed] [Google Scholar]
- 42.Sterling H, Lin DH, Chen YJ, Wei Y, Wang ZJ, Lai J, Wang WH. PKC expression is regulated by dietary K intake and mediates internalization of SK channels in the CCD. Am J Physiol Renal Physiol. 2004;286:F1072–F1078. doi: 10.1152/ajprenal.00425.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Beskina O, Miller A, Mazzocco-Spezzia A, Pulina MV, Golovina VA. Mechanisms of interleukin-1β-induced Ca2+ signals in mouse cortical astrocytes: roles of store- and receptor-operated Ca2+ entry. Am J Physiol Cell Physiol. 2007;293:C1103–C1111. doi: 10.1152/ajpcell.00249.2007. [DOI] [PubMed] [Google Scholar]
- 44.Zhu G, Okada M, Yoshida S, Mori F, Hirose S, Wakabayashi K, Kaneko S. Involvement of Ca2+-induced Ca2+ releasing system in interleukin-1βa-associated adenosine release. Eur J Pharmacol. 2006;532:246–252. doi: 10.1016/j.ejphar.2005.12.085. [DOI] [PubMed] [Google Scholar]
- 45.Karim Z, Defontaine N, Paillard M, Poggioli J. Protein kinase C isoforms in rat kidney proximal tubule: acute effect of angiotensin II. Am J Physiol Cell Physiol. 1995;269:C134–C140. doi: 10.1152/ajpcell.1995.269.1.C134. [DOI] [PubMed] [Google Scholar]
- 46.Greco S, Muscella A, Elia MG, Salvatore P, Storelli C, Marsigliante S. Activation of angiotensin II type I receptor promotes protein kinase C translocation and cell proliferation in human cultured breast epithelial cells. J Endocrinol. 2002;174:205–214. doi: 10.1677/joe.0.1740205. [DOI] [PubMed] [Google Scholar]