

Abstract
Our previous work has shown that interleukin-6 (IL-6) implements its neuroprotective effect by inhibiting the intracellular Ca2+ overload in neurons. Here, we examined whether regulation of L-type calcium channels (LCCs) activities is involved in the neuroprotective action of IL-6. In cultured cerebellar granule neurons (CGNs), patch-clamp recording showed that the whole-cell Ca2+ current and LCC current were significantly reduced by IL-6 pretreatment (120 ng/ml, for 24 h). Calcium imaging data indicated that IL-6 significantly suppressed high K+-induced intracellular Ca2+ overload and LCC Ca2+ influx. Moreover, expression of the LCC subunit, Cav1.2, was remarkably downregulated by IL-6 in cultured CGNs. These findings suggest that IL-6 exerts a neurotrophic effect by preventing Ca2+ overload, at least partly through inhibition of LCC activity in cultured CGNs.
Keywords: Interleukin-6, L-type calcium channels, Whole-cell recording, Calcium imaging, Cerebellar granule neurons
Introduction
Interleukin-6 (IL-6), a member of pleiotropic cytokine family, has complex effects on the central nervous system (CNS) [1]. Under normal physiological conditions, the IL-6 level in the CNS is low. In neural functional disorders, such as brain diseases and injuries, IL-6 expression increases greatly [2–7]. The increased IL-6 may reflect a harmful process as an injurious mediator. For example, IL-6 is a detrimental player in the CNS, contributing to pathogenesis of neurodegenerative diseases, e.g., Alzheimer’s and Parkinson’s disease [8, 9]. However, the IL-6 increase may also represent a compensative mechanism for neural repair. For instance, IL-6 regulates neuronal function and development in the innate response of the CNS to injury and diseases [10, 11], and exerts neurotrophic and neuroprotective effects on glutamate- and N-methyl-d-aspartic acid (NMDA)-induced neuronal damage [12–15]. Hereby, further exploration is needed to understand the roles of IL-6 in brain physiology and pathology.
It is well known that Ca2+ is not only an important signaling molecule in neurons, but also a mediator leading to neuronal injury and death when it accumulates in the cytosol of cells, termed Ca2+ overload. Neuronal Ca2+ overload mainly involves three mechanisms: Ca2+ influx through ligand-gated channels, Ca2+ influx through voltage-gated Ca2+ channels (VGCCs) activated by membrane depolarization, and Ca2+ release from intracellular store induced by an increase in cytosolic Ca2+ [16]. By means of confocal laser scanning microscope (CLSM), we previously found that IL-6 suppressed neuronal intracellular Ca2+ overload induced by glutamate or NMDA, and exerted a neuroprotective effect [13, 15]. However, the mechanism underlying the IL-6 suppression of intracellular Ca2+ overload is not clear. We hypothesized that IL-6 exerts its neuroprotective function by suppressing the expression of VGCCs in cerebellar granule neurons (CGNs).
VGCCs are expressed in neurons and have multiple types, such as L-, N-, P/Q-, R-, and T-type Ca2+ channels [17–21]. Among these various types of VGCCs, L-type calcium channels (LCCs) are widely distributed on the cell body of neurons in mammalian CNS, including CGNs [22–24]. Calcium influx through LCCs in response to membrane depolarization serves essential functions in the regulation of intracellular Ca2+ homeostasis and neuronal excitability [25, 26]. Excessive Ca2+ influx through LCCs results in intracellular Ca2+ overload, which has been implicated in the pathogenesis of neurodegenerative disorders resulting from brain ischemia [16, 27, 28]. Therefore, in the present study, we firstly focused on LCCs to clarify the mechanism of the neuroprotective effect of IL-6 on LCCs by means of whole-cell patch clamp methods and calcium imaging.
Materials and methods
Isolation and culture of rat CGNs
Primary cultures of CGNs were obtained from neonatal Sprague-Dawley rats (The Center of Experimental Animals, Nantong University, China) at 8 days of age using previously described procedures [29]. Briefly, the cerebellum was removed from rats and minced with sterile surgical blades. The minced cerebellum was chemically dissociated in the presence of trypsin (Amresco, USA) and DNase I (Worthington, USA), and resuspended in the following culture medium: basal Eagle’s medium (Sigma, USA), 10 % fetal bovine serum (Amresco, USA), 25 mM KCl, 0.1 g/l gentamicin, and 2.2 g/l NaHCO3, 2.385 g/l HEPES. The samples were plated onto poly-l-lysine-coated glass coverslips (0.32 × 106 cells/ml) for electrophysiological recording, or seeded at a density of 0.8 × 106 cells/ml in 96 wells for calcium imaging or at 2.0 × 106 cells/ml in 6 wells for Western blot, respectively. The cells were incubated at 37 °C with a humidified 5 % CO2/95 % air atmosphere in an incubator (ESPEC BNA-311, Japan). To inhibit glial proliferation, cytosine arabinoside (Sigma, USA, 10 μM) was added to the cultures 18–24 h after the cells were plated. Rat recombinant IL-6 (R&D Systems, USA) at a concentration of 120 ng/ml was added to the cultures of CGNs for at least 24 h incubation. All experiments described below were performed using the CGNs cultured for 8 days.
Electrophysiological recording
Current through the Ca channel was isolated by blocking the Na channel with TTX and recorded using an Axopatch 200B patch-clamp amplifier (Axon, USA) at room temperature (20–22 °C). The bath solution was composed of TEA-Cl 144, BaCl2 10, MgCl2 2, CsCl 3, HEPES 10, glucose 10, 4-aminopyridine 2, and TTX 0.001 (all in mM), and adjusted to pH 7.4 with TEA-OH. Patch pipettes were pulled on a micropipette puller (pp830, Narishige, Japan) to a tip resistance of 3–5 MΩ when filled with internal solution. The pipette solution contained CsCl 140, HEPES 10, EGTA 10, TEA-Cl 5, and Na2-ATP 2 (all in mM), and was adjusted to pH 7.2 with CsOH. Current responses were low-pass filtered at 1 kHz and analyzed with pClamp10.2 (Axon, USA). Linear components of capacitive and leak currents were subtracted using the P/4 protocol. I Ca, carried by Ba2+, was elicited by a series of command potentials from −60 to +40 mV for 250 ms in 10-mV steps from a holding potential of −80 mV. The whole-cell current densities were defined as peak current amplitude divided by cell capacitance. Nifedipine (Sigma), a blocker for LCCs, was used to determine the proportion of LCC current in the whole-cell current. It was added to 2 ml of bath solution with a final concentration of 10 μM, and 2-min later, the non-L-type channel current was recorded [30]. To determine the voltage-dependent activation property of LCCs, values of currents obtained were normalized to conductance with the form g = I/(V m − V rev), and fitted to a single Boltzmann function of the form g/g max = 1 − {1 + exp[(V m − V 1/2)/K]}−1, where g is conductance, I is the amplitude of whole-cell LCC current, V m is the membrane voltage, V rev is the reversal potential, k is the slope factor, and g max is the maximal conductance.
Measurement of intracellular Ca2+ fluorescence intensity
Intracellular Ca2+ level was quantified by single cell fluo-3 fluorescence intensity as described previously [29] with a small modification. Briefly, cultured CGNs were rinsed twice with balanced salt solution (BSS), then incubated at 37 °C for 45 min in the presence of 5 μM fluo-3/acetoxymethyl ester (Fluo-3/AM, Calbiochem), washed twice again with BSS, and incubated for an additional 20 min prior to imaging. The BSS was composed of (in mM): 145 NaCl, 5.6 KCl, 5 HEPES, 3.6 NaHCO3, 5.6 glucose, and 2.3 CaCl2. Calcium imaging was recorded by CLSM (Leica TCS SPE, Germany). Successive images were collected at 5-s intervals. Fluo-3 fluorescence was excited at 488 nm, and emitted light was measured at 530 nm. Quantification of the fluorescence intensity was performed using TCS-SPE software from Leica. To depolarize neurons and activate VGCCs, neurons were stimulated with high K+-solution (150 mM KCl), whose composition was the same as that of BSS, but Na+ was replaced by K+. When the high-K+ solution was applied to stimulate neurons, 100 μl of solution containing 150 mM KCl was added to 100 μl of BSS, and therefore the high K+ concentration was about 75 mM. Because the concentration of other constituents than K+ in the high-K+ solution was the same as that in BSS, the addition of the high-K+ solution to BSS did not alter the concentration of other constituents such as HEPES, NaHCO3, glucose, and CaCl2. Nifedipine (10 μM) was applied to neurons 25 min before high K+ stimulation. In one-scanned visual field, 30 neurons were randomly selected to obtain their dynamic intracellular Ca2+ levels. Neuronal basal Ca2+ fluorescence intensity before high K+ stimulation was firstly recorded for about 90 s, and then these neurons were stimulated by high K+ and scanned for 6 min. Neuronal maximal fluo-3 fluorescence intensity after high K+ stimulation was statistically analyzed. The same experiment was repeated four times.
Western blot assay
For measurement of expression of the LCC subunit, pore-forming α1c (also known as Cav1.2), the cultured CGNS were lysed by boiling sample buffer (125 mM Tris-HCl, pH 6.8, containing 4 % SDS, 12 % β-mercaptoethanol, and 20 % glycerol). The cell extracts were boiled for 5 min and loaded onto gels in each electrophoresis. After SDS-PAGE, the separated proteins in the gel were electrotransferred onto a PVDF membrane (Millipore) in tris-glycine-methanol buffer. The membrane was blocked in blocking solution (5 % non-fat dry milk in TBS), and then incubated with primary antibody in blocking solution (rabbit anti-α1c, 1:200; Alomone) overnight at 4 °C. After washing with TBS/Tween-20, the membrane was incubated in secondary antibody (1:5,000 dilution) coupled to HRP, washed as above, and visualized by chemiluminescence using the ECL system.
Statistical analysis
Data were analyzed using pClamp 10.2 (Axon Instruments). One-way analysis or Student’s t test was used for comparisons, with p < 0.05 indicating statistical difference. All data were presented as mean ± standard deviation (M ± SD).
Results
Influence of IL-6 on whole-cell LCC current
Under the condition of Ba2+ instead of Ca2+ in the bath solution, which reduced the influence of Ca2+ current rundown [31], the whole-cell current through the Ca channel, evoked by depolarization from −60 to +40 mV at a holding potential of −80 mV, in neurons pretreated with IL-6 (120 ng/ml) was smaller than that in control neurons (Fig. 1a, b). Statistical analysis of current density displayed that the effect of IL-6 diminishing Ca-channel current was significant between −20 and +10 mV of depolarization (Fig. 1c).
Fig. 1.
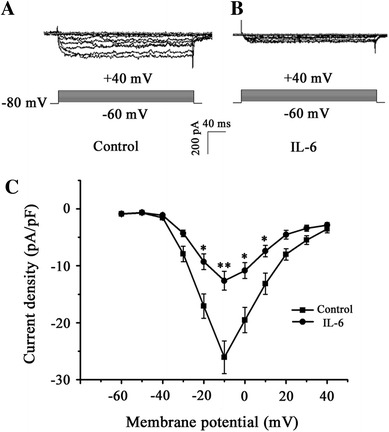
Effect of IL-6 on whole-cell Ca-channel current in cultured CGNs. The whole-cell inward currents through the Ca channel were evoked by depolarization from −60 to +40 mV at a holding potential of −80 mV. A typical whole-cell inward Ca-channel current in control neuron (a) and in IL-6-pretreated neuron (b) was exhibited. Statistical analysis of current density displayed that the effect of IL-6 diminishing the Ca-channel current was significant between −20 and +10 mV of depolarization (c). *p < 0.05, **p < 0.01, compared with relative membrane potential of control (n = 10)
The effect of IL-6 on LCC current was examined using the selective LCC antagonist, nifedipine. In control neurons, depolarization from a holding potential of −80 mV to a test potential of −10 mV evoked an inward Ca-channel current, and perfusion with nifedipine (10 μM) diminished the Ca-channel current (Fig. 2a). This demonstrated that opening of LCCs contributed to the inward current through the Ca channel. In IL-6-pretreated neurons, the depolarization from −80 to −10 mV also evoked an inward Ca-channel current, but the current was smaller than that in control neurons (Fig. 2a), demonstrating an inhibitory effect of IL-6 on Ca-channel current. The nifedipine perfusion also decreased the current through Ca-channel in IL-6-pretreated neurons (Fig. 2a). However, between IL-6-treated and control neurons, the nifedipine-insensitive Ca-channel current was not significantly different (Fig. 2b), indicating that IL-6 did not alter the non-L-type Ca-channel current. On the other hand, the nifedipine-sensitive Ca-channel current was remarkably suppressed by IL-6 exposure (Fig. 2c). This revealed that the suppressive effect of IL-6 on the Ca-channel current was a result of its inhibition of LCCs. Moreover, to examine whether the voltage-dependent activation property of I LCC was modified by IL-6 exposure, we calculated normalized conductance of LCCs using Boltzmann’s equation. The value of the reversal potential was close to 60 mV. The fitted values of V 1/2 were −25.05 ± 1.93 and −26.84 ± 1.64 mV, and the slope factors were −5.84 ± 1.81 and −4.75 ± 1.30 in control and IL-6-treated neurons, respectively. These data showed that neuronal voltage dependence on activation was not changed following incubation of the neurons with IL-6 (Fig. 2c3).
Fig. 2.
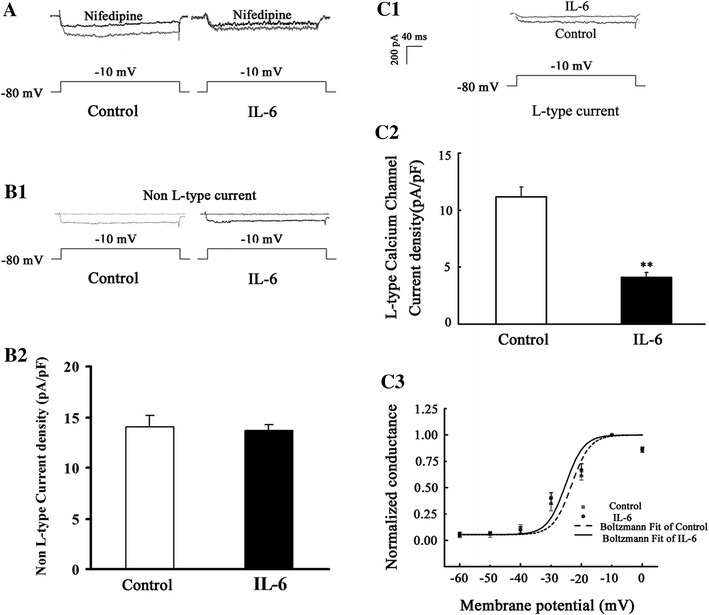
Influence of IL-6 on whole-cell LCC current in cultured CGNs. Depolarization voltage was set to −10 mV from a holding potential of −80 mV, and whole-cell inward current through the Ca channel was recorded in control and IL-6-exposed neurons. Perfusion of control or IL-6-exposed neurons with 10 μM of nifedipine, a blocker for LCCs, reduced the inward current through the Ca channel (a). The inward current after nifedipine action was non-L-type Ca-channel current (b1), and it was not significantly different between IL-6-pretreated and control neurons (b2). The inward current blocked by nifedipine was the LCC current (c1). The LCC current density was evidently lower in IL-6-exposed neurons than in controls (c2). **p < 0.01, compared with controls (n = 8). Voltage-dependent activation curves were obtained by the Boltzmann equation, g/g max = 1 − {1 + exp[(V m − V 1/2)/K]}−1. The fitted values of V 1/2 were −25.05 ± 1.93 and −26.84 ± 1.64 mV, and the k (slope factor) was −5.84 ± 1.81 and −4.75 ± 1.30 in control and IL-6-treated neurons, respectively. No significant differences in the data were found between IL-6-treated and control neurons (c3, n = 6)
Effect of IL-6 on high K+-evoked [Ca2+]i increase
To further demonstrate the effect of IL-6 on LCCs, we measured dynamic changes of intracellular Ca2+ fluorescence intensity in cultured CGNs by CLSM. In control neurons, depolarization stimulation by high K+ evoked an acute elevation of intracellular Ca2+ level (Fig. 3). In IL-6-pretreated neurons, high K+ stimulation evoked significantly less elevation of the intracellular Ca2+ level than in control neurons (Fig. 3), indicating that IL-6 suppressed high K+-induced intracellular Ca2+ overload. After exposure to nifedipine (10 μM), an LCC antagonist, for 25 min, high K+ stimulation resulted in a reduction of intracellular Ca2+ overload compared with control neurons lacking nifedipine exposure (Fig. 3). This suggests that the reduction of intracellular Ca2+ was attributable to a reduction of Ca2+ influx through LCCs. However, the inhibitory effect of nifedipine on high K+-induced intracellular Ca2+ overload did not have a notable difference in the presence and the absence of IL-6 (Fig. 3). This indicated that IL-6 did not significantly alter nifedipine-resistant Ca2+ overload components and therefore suggested that IL-6 exerted its suppressive effect on high K+-evoked intracellular Ca2+ overload by attenuating nifedipine-dependent LCC Ca2+ influx.
Fig. 3.
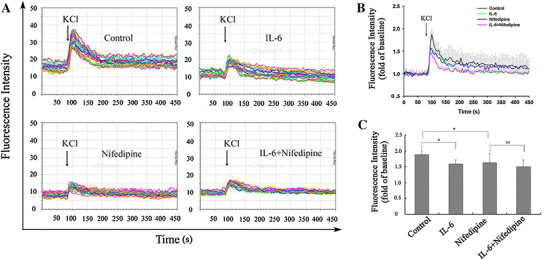
Role of LCCs in IL-6 suppressing high K+-trigged intracellular Ca2+ overload. LCC blocker nifedipine (10 μM) treated neurons for 25 min before high K+-stimulation. The neurons were incubated at 37 °C for 45 min in the presence of 5 μM of Fluo-3/AM, and then dynamic changes in intracellular Ca2+ levels were tested by CLSM during the whole 6-min high-K+ stimulation. In each treatment, 30 neurons were randomly selected to analyze dynamic intracellular Ca2+ levels (a). The compilation of data for the mean and SD of four separate experiments as in a is presented in b. The peak intracellular Ca2+ levels following high K+ stimulation were compared for statistical significance of the differences between the various treatments (c). The arrows denote the beginning time when KCl was applied. *p < 0.05 and NS means no significant difference
IL-6 downregulates protein expression of LCC subunit
Expression of the LCC subunit, pore-forming α1c (also known as Cav1.2), in cultured CGNs was measured in order to reveal the mechanism underlying IL-6 suppression of the LCC current and LCC Ca2+ influx. The LCC subunit protein expression was remarkably downregulated by IL-6 pretreatment (Fig. 4). This showed that via the downregulation, IL-6 carried out its inhibitory effect on LCC function.
Fig. 4.
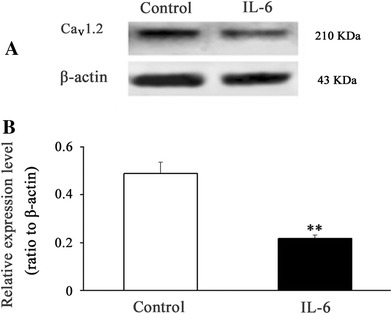
IL-6 downregulates LCC subunit expression in cultured CGNs. The CGNs from 8-day-old rats were incubated for 7 days and then exposed to IL-6 (120 ng/ml) for 24 h. The protein expression of the LCC subunit, pore-forming α1c (also known as Cav1.2), was significantly downregulated by IL-6 pretreatment (a). The data are from three separate experiments (b). **p < 0.01, compared with control
Discussion
In this study, IL-6 pretreatment of cultured CGNs significantly reduced the inward current through the Ca channel evoked by depolarization from −20 to +10 mV at a holding potential of −80 mV, suggesting that IL-6 inhibits VGCC opening. To examine the contribution of LCCs, a type of VGCCs, to the inward Ca-channel current, we used nifedipine to block LCCs and found that the inward Ca-channel current was diminished. This suggests that depolarizing stimulation causes opening of LCCs and consequent influx of Ca2+ current in cultured CGNs. The report that extracellular Ca2+ influx occurs not only directly through the glutamate-activated membrane channel, but also indirectly through activated VGCCs by membrane depolarization [32] supports our present results. Importantly, after neurons were pretreated with IL-6, the effect of the nifedipine-sensitive inward Ca-channel current was significantly suppressed. The result suggests that IL-6 inhibits LCC activity. Some other cytokines, such as interleukin-1β, tumor necrosis factor α, and ciliary neurotrophic factor, have been reported to modulate various types of VGCC currents in neurons [30, 33, 34]. Thus, our present data provide more evidence for IL-6 regulating the LCC current in cultured CGNs.
To further demonstrate the modulation of LCC activity by IL-6, we observed the influence of IL-6 on intracellular Ca2+ overload evoked by high K+-depolarization stimulation in cultured CGNs. The IL-6 pretreatment significantly reduced the high K+-evoked intracellular Ca2+ overload. The result is consistent with the data obtained from the patch-clamp experiments and demonstrates that IL-6 inhibits VGCC activity. In our previous work, we indicated that IL-6 suppresses glutamate- or NMDA-induced intracellular Ca2+ overload and neuronal apoptosis in cultured CGNs, and therefore suggest that IL-6 has a neuroprotective effect [13, 15, 29]. Here we add evidence for the IL-6 neuroprotection at the profile of its suppression of VGCCs. Further, we hypothesized that the inhibitory effect of IL-6 on VGCC-dependent Ca2+ influx is mediated by LCC-activity suppression. We observed that nifedipine attenuated intracellular Ca2+ overload triggered by high K+-depolarization stimulation, demonstrating that LCC opening is involved in the high K+-induced intracellular Ca2+ overload. The inhibitory effect of nifedipine on intracellular Ca2+ overload occurred similarly in IL-6-exposed and control neurons. It indicates that IL-6 does not significantly alter the nifedipine-insensitive Ca2+-influx component. Therefore, the suppression of intracellular Ca2+ overload by IL-6 is attributed to its suppression of the nifedipine-sensitive Ca2+-influx component. These findings are consistent with the conclusion from the whole-cell recording that IL-6 suppresses LCC activity. Thus, we suggest that IL-6 neuroprotection through suppression of intracellular Ca2+ overload is implemented, at least partly, by the inhibition of the LCC current.
Since the voltage-dependent property of I LCC was not modified by IL-6 pretreatment in the current study, the mechanism underlying the IL-6 inhibition of LCC activity needs to be explained. We found that expression of the LCC pore-forming subunit Cav1.2 was significantly downregulated by IL-6 exposure in cultured CGNs. The downregulation reached 60 %, and it was quite consistent with the reduction in I LCC peak current density in IL-6-treated neurons. On the basis of these findings, we suggest that the suppression of LCC function by IL-6 is related to a decrease in LCC protein expression.
As we previously reported [13, 15, 29], the present study represents a neuroprotective role of IL-6. However, since IL-6 is a pleiotropic cytokine, it exerts neurotrophic and neuroprotective effects, and yet can also function as a mediator of inflammation, demyelination, and astrogliosis, depending on the cellular context [35]. Therefore, the dosage of IL-6, degree of neuronal damage, type and environment of neurons, and existence of soluble IL-6 receptors can influence IL-6 effects [36, 37]. For example, Nelson et al. [38] showed that a lower dose of IL-6 (5 ng/ml) exposure enhances the mean amplitude of the Ca2+ signal in response to glutamate receptor agonists in cultured cerebellar Purkinje neurons, whereas a higher concentration of IL-6 (10 ng/ml) has no effect on the Ca2+ signal in response to the same agonists. On the other hand, Vereyken et al. [39] report that transient high-K+ stimulation (0.5 s) enhances the Ca2+ signal, but longer high-K+ stimulation (>1 s) attenuates the Ca2+ signal in IL-6-treated neurons. In addition, NMDA infusion into rat striatum results in a decrease in striatal cholinergic and GABAergic neurons, and co-infusion of IL-6 and NMDA reduces the loss of cholinergic neurons, but fails to prevent the loss of GABAergic neurons [37]. These differences of response to IL-6 among different IL-6 dosages, neuron-damaged degrees, and neuronal types explain the distinct and complex effects of IL-6, neuroprotective, neuroinjured, or non-effective. Further exploration is needed to clarify the mechanisms underlying the different effects of IL-6.
In general, in the presence of IL-6 receptor, IL-6 acts on target cells and promotes dimerization of gp130, a signal-transducing subunit coupled with IL-6 receptor. CGNs have been reported to express IL-6 receptor and gp130 signal protein [40, 41]. In our previous work, anti-gp130 antibody blocked the inhibitory effect of IL-6 on glutamate-induced intracellular Ca2+ overload, indicating that the IL-6 receptor is involved in the neuroprotective effect of IL-6 [29]. On the basis of these findings, we suggest that the suppressed LCC activity caused by IL-6 is mediated by the IL-6 receptor.
In conclusion, we revealed that IL-6 inhibits the activity of LCCs in cultured CGNs and this inhibition is associated with downregulation of LCC protein expression. These results imply that a neuroprotective role of IL-6 in the CNS is implemented, at least partially, by suppression of the neuronal LCC current and therefore a reduction in intracellular Ca2+ overload.
Acknowledgments
This work was supported by grants 30870819 and 30870929 from the National Natural Science Foundation of China, BK2010278 and BK2011386 from the Natural Science Foundation of Jiangsu Province of China, 08KJD310014 from the Educational Department of Jiangsu Province of China, and A Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Contributor Information
Yu-Ping Peng, Phone: +86-513-85051714, FAX: +86-513-85051506, Email: yppeng@ntu.edu.cn.
Yi-Hua Qiu, Phone: +86-513-85051723, FAX: +86-513-85051876, Email: yhqiu@ntu.edu.cn.
References
- 1.Gadient RA, Otten UH. Interleukin-6 (IL-6)—a molecule with both beneficial and destructive potentials. Prog Neurobiol. 1997;52:379–390. doi: 10.1016/S0301-0082(97)00021-X. [DOI] [PubMed] [Google Scholar]
- 2.Ali C, Nicole O, Docagne F, Lesne S, MacKenzie ET, Nouvelot A. Ischemia-induced interleukin-6 as a potential endogenous neuroprotective cytokine against NMDA receptor-mediated excitotoxicity in the brain. J Cereb Blood Flow Metab. 2000;20:956–966. doi: 10.1097/00004647-200006000-00008. [DOI] [PubMed] [Google Scholar]
- 3.Baranzini SE, Elfstrom C, Chang SY, Butunoi C, Murray R, Higuchi R. Transcriptional analysis of multiple sclerosis brain lesions reveals a complex pattern of cytokine expression. J Immunol. 2000;165:6576–6582. doi: 10.4049/jimmunol.165.11.6576. [DOI] [PubMed] [Google Scholar]
- 4.Conroy SM, Nguyen V, Quina LA, Prieto AL, Gruol DL. Interleukin-6 produces neuronal loss in developing cerebellar granule neuron cultures. J Neuroimmunol. 2004;155:43–54. doi: 10.1016/j.jneuroim.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 5.Damm J, Luheshi GN, Gerstberger R, Roth J, Rummel C. Spatiotemporal nuclear factor interleukin-6 expression in the rat brain during lipopolysaccharide-induced fever is linked to sustained hypothalamic inflammatory target gene induction. J Comp Neurol. 2011;519:480–505. doi: 10.1002/cne.22529. [DOI] [PubMed] [Google Scholar]
- 6.Gruol DL, Nelson TE. Purkinje neuron physiology is altered by the inflammatory factor interleukin-6. Cerebellum. 2005;4:198–205. doi: 10.1080/14734220500199987. [DOI] [PubMed] [Google Scholar]
- 7.Liimatainen S, Fallah M, Kharazmi E, Peltola M, Peltola J. Interleukin-6 levels are increased in temporal lobe epilepsy but not in extra-temporal lobe epilepsy. J Neurol. 2009;256:796–802. doi: 10.1007/s00415-009-5021-x. [DOI] [PubMed] [Google Scholar]
- 8.Nagatsu T, Mogi M, Ichinose H, Togari A. Changes in cytokines and neurotrophins in Parkinson’s disease. J Neural Transm Suppl. 2000;60:277–290. doi: 10.1007/978-3-7091-6301-6_19. [DOI] [PubMed] [Google Scholar]
- 9.Qiu Z, Gruol DL. Interleukin-6, β-amyloid peptide and NMDA interactions in rat cortical neurons. J Neuroimmunol. 2003;139:51–57. doi: 10.1016/S0165-5728(03)00158-9. [DOI] [PubMed] [Google Scholar]
- 10.Braida D, Sacerdote P, Panerai AE, Bianchi M, Aloisi AM, Iosue S. Cognitive function in young and adult IL (interleukin)-6 deficient mice. Behav Brain Res. 2004;153:423–429. doi: 10.1016/j.bbr.2003.12.018. [DOI] [PubMed] [Google Scholar]
- 11.Sallmann S, Juttler E, Prinz S, Petersen N, Knopf U, Weiser T, Schwaninger M. Induction of interleukin-6 by depolarization of neurons. J Neurosci. 2000;20:8637–8642. doi: 10.1523/JNEUROSCI.20-23-08637.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carlson NG, Wieqqel WA, Chen J, Bacchi A, Roqers SW, Gahring LC. Inflammatory cytokines IL-1 alpha, IL-1 beta, IL-6, and TNF-alpha impart neuroprotection to an excitotoxin through distinct pathways. J Immunol. 1999;163:3963–3968. [PubMed] [Google Scholar]
- 13.Liu Z, Qiu YH, Li B, Ma SH, Peng YP. Neuroprotection of interleukin-6 against NMDA-induced apoptosis and its signal-transduction mechanisms. Neurotox Res. 2011;19:484–495. doi: 10.1007/s12640-010-9215-x. [DOI] [PubMed] [Google Scholar]
- 14.Pizzi M, Sarnico I, Boroni F, Benarese M, Dreano M, Garotta G. Prevention of neuron and oligodendrocyte degeneration by interleukin-6 (IL-6) and IL-6 receptor/IL-6 fusion protein in organotypic hippocampal slices. Mol Cell Neurosci. 2004;25:301–311. doi: 10.1016/j.mcn.2003.10.022. [DOI] [PubMed] [Google Scholar]
- 15.Wang XQ, Peng YP, Lu JH, Cao BB, Qiu YH. Neuroprotection of interleukin-6 against NMDA attack and its signal transduction by JAK and MAPK. Neurosci Lett. 2009;450:122–126. doi: 10.1016/j.neulet.2008.11.051. [DOI] [PubMed] [Google Scholar]
- 16.Lee JM, Zipfel GJ, Choi DW. The changing landscape of ischemic brain injury mechanisms. Nature. 1999;399:A7–A14. doi: 10.1038/399a007. [DOI] [PubMed] [Google Scholar]
- 17.Bauer EP, Schafe GE, Ledoux JE. NMDA receptors and L-type voltage-gated calcium channels contribute to long-term potentiation and different components of fear memory formation in the lateral amygdale. J Neurosci. 2002;22:5239–5249. doi: 10.1523/JNEUROSCI.22-12-05239.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Catterall WA, Few AP. Calcium channel regulation and presynaptic plasticity. Neuron. 2008;59:882–901. doi: 10.1016/j.neuron.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 19.Jarvis SE, Zamponi GW. Trafficking and regulation of neuronal voltage-gated calcium channels. Curr Opin Cell Biol. 2007;19:474–482. doi: 10.1016/j.ceb.2007.04.020. [DOI] [PubMed] [Google Scholar]
- 20.Minor DL, Jr, Findeisen F. Progress in the structural understanding of voltage-gated calcium channel (CaV) function and modulation. Channels (Austin) 2010;4:459–474. doi: 10.4161/chan.4.6.12867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ishibashi H, Rhee JS, Akaike N. Effect of nifedipine on high-voltage activated Ca2+ channels in rat CNS neurons. Neuroreport. 1997;8:853–857. doi: 10.1097/00001756-199703030-00009. [DOI] [PubMed] [Google Scholar]
- 22.Davare MA, Hell JW. Increased phosphorylation of the neuronal L-type Ca2+ channel CaV1.2 during aging. Proc Natl Acad Sci USA. 2003;100:16018–16023. doi: 10.1073/pnas.2236970100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Forti L, Pietrobon D. Functional diversity of L-type calcium channels in rat cerebellar neurons. Neuron. 1993;10:437–450. doi: 10.1016/0896-6273(93)90332-L. [DOI] [PubMed] [Google Scholar]
- 24.Hirota K, Lambert DG. A comparative study of L-type voltage sensitive Ca2+ channels in rat brain regions and cultured neuronal cells. Neurosci Lett. 1997;223:169–172. doi: 10.1016/S0304-3940(97)13434-6. [DOI] [PubMed] [Google Scholar]
- 25.Dolmetsch RE, Pajvani U, Fife K, Spotts JM, Greenberg ME. Signaling to the nucleus by an L-type calcium channel–calmodulin complex through the MAP kinase pathway. Science. 2001;294:333–339. doi: 10.1126/science.1063395. [DOI] [PubMed] [Google Scholar]
- 26.Fisher TE, Bourque CW. The function of Ca channel subtypes in exocytotic secretion: new perspectives from synaptic and non-synaptic release. Prog Biophys Mol Biol. 2001;77:269–303. doi: 10.1016/S0079-6107(01)00017-7. [DOI] [PubMed] [Google Scholar]
- 27.Liu Y, Hou XY, Zhang GY, Xu TL. L-type voltage-gated calcium channel attends regulation of tyrosine phosphorylation of NMDA receptor subunit 2A induced by transient brain ischemia. Brain Res. 2003;972:142–148. doi: 10.1016/S0006-8993(03)02519-8. [DOI] [PubMed] [Google Scholar]
- 28.Stevens TR, Kureger SR, Fitzsimonds RM, Picciotto MR. Neuroprotection by nicotine in mouse primary cortical cultures involves activation of calcineurin and L-type calcium channel inactivation. J Neurosci. 2003;23:10093–10099. doi: 10.1523/JNEUROSCI.23-31-10093.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peng YP, Qiu YH, Lu JH, Wang JJ. Interleukin-6 protects cultured cerebellar granule neurons against glutamate-induced neurotoxicity. Neurosci Lett. 2005;374:192–196. doi: 10.1016/j.neulet.2004.10.069. [DOI] [PubMed] [Google Scholar]
- 30.Zhou C, Tai C, Ye HH, Wang SQ, Chai Z. Interleukin-1β downregulates the L-type Ca2+ channel activity by depressing the expression of channel protein in cortical neurons. J Cell Physiol. 2006;206:799–806. doi: 10.1002/jcp.20518. [DOI] [PubMed] [Google Scholar]
- 31.Hagiwara S, Ohmori H. Studies of calcium channels in rat clonal pituitary cells with patch electrode voltage clamp. J Physiol. 1982;331:231–252. doi: 10.1113/jphysiol.1982.sp014371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Choi DW. Ionic dependence of glutamate neurotoxicity. J Neurosci. 1987;7:369–379. doi: 10.1523/JNEUROSCI.07-02-00369.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Holm NR, Christophersen P, Hounsgaard J, Gammeltoft S, Olesen SP. CNTF inhibits high voltage activated Ca2+ currents in fetal mouse cortical neurons. J Neurochem. 2002;82:495–503. doi: 10.1046/j.1471-4159.2002.00963.x. [DOI] [PubMed] [Google Scholar]
- 34.Motagally MA, Lukewich MK, Chisholm SP, Neshat S, Lomax AE. Tumor necrosis factor α activates nuclear factor kB signalling to reduce N-type voltage-gated Ca2+ current in postganglionic sympathetic neurons. J Physiol. 2009;587:2623–2634. doi: 10.1113/jphysiol.2009.172312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Van Wagoner NJ, Benveniste EN. Interleukin-6 expression and regulation in astrocytes. J Neuroimmunol. 1999;100:124–139. doi: 10.1016/S0165-5728(99)00187-3. [DOI] [PubMed] [Google Scholar]
- 36.Thier M, März P, Otten U, Weis J, Rose-John S. Interleukin-6 (IL-6) and its soluble receptor support survival of sensory neurons. J Neurosci Res. 1999;55:411–422. doi: 10.1002/(SICI)1097-4547(19990215)55:4<411::AID-JNR2>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 37.Toulmond S, Vige X, Fage D, Benavides J. Local infusion of interleukin-6 attenuates the neurotoxic effects of NMDA on rat striatal cholinergic neurons. Neurosci Lett. 1992;144:49–52. doi: 10.1016/0304-3940(92)90713-H. [DOI] [PubMed] [Google Scholar]
- 38.Nelson TE, Ur CL, Gruol DL. Chronic interleukin-6 exposure alters electrophysiological properties and calcium signaling in developing cerebellar Purkinje neurons in culture. J Neurophysiol. 2002;88:475–486. doi: 10.1152/jn.2002.88.1.475. [DOI] [PubMed] [Google Scholar]
- 39.Vereyken EJF, Bajova H, Chow S, Graan NE, Gruol DL. Chronic interleukin-6 alters the level of synaptic proteins in hippocampus in culture and in vivo. Eur J Neurosci. 2007;25:3605–3616. doi: 10.1111/j.1460-9568.2007.05615.x. [DOI] [PubMed] [Google Scholar]
- 40.Schobitz B, De Kloet ER, Sutanto W, Holsboer F. Cellular localization of interleukin-6 mRNA and interleukin-6 receptor mRNA in rat brain. Eur J Neurosci. 1993;5:1426–1435. doi: 10.1111/j.1460-9568.1993.tb00210.x. [DOI] [PubMed] [Google Scholar]
- 41.Ha BK, King JS. Localization of gp130 in the developing and adult mouse cerebellum. J Chem Neuroanat. 2000;19:129–141. doi: 10.1016/S0891-0618(00)00056-9. [DOI] [PubMed] [Google Scholar]