

Abstract
The aim of this study was to investigate the effect of sirtinol, as an inhibitor of sirtuin NAD-dependent histone deacetylases, on myocardial ischemia reperfusion injury following early and late ischemia preconditioning (IPC). Rats underwent sustained ischemia and reperfusion (IR) alone or proceeded by early or late IPC. Sirtinol (S) was administered before IPC. Arrhythmias were evaluated based on the Lambeth model. Infarct size (IS) was measured using triphenyltetrazolium chloride staining. The transcription level of antioxidant-coding genes was assessed by real-time PCR. In early and late IPC groups, IS and the number of arrhythmia were significantly decreased (P < 0.05 and P < 0.01 vs IR, respectively). In S + early IPC, incidences of arrhythmia and IS were not different compared with the early IPC group. However, in S + late IPC the IS was different from the late IPC group (P < 0.05). In late IPC but not early IPC, transcription levels of catalase (P < 0.01) and Mn-SOD (P < 0.05) increased, although this upregulation was not significant in the S + late IPC group. Our results are consistent with the notion that different mechanisms are responsible for early and late IPC. In addition, sirtuin NAD-dependent histone deacetylases may be implicated in late IPC-induced cardioprotection.
Keywords: Myocardial ischemia reperfusion, Early ischemia preconditioning, Late ischemia preconditioning, Sirtuin1, Sirtuin2, Sirtinol
Introduction
Ischemia preconditioning (IPC) is an innate protective phenomenon in which the induction of short sublethal episodes of ischemia strengthens the heart against subsequent lethal ischemic attacks. IPC is followed by two cardioprotection phases: (1) the early phase or first window IPC that protects the heart against infarction for 1–3 h; and (2) the late or delayed phase, also called second window IPC, which indicates the induction of cardioprotection starting from 24 h to 3–4 days following IPC [1, 2]. The cellular and molecular mechanisms involved in different phases of IPC are not accurately known. However, it seems that the mechanisms responsible for the early and late phases of IPC are different; researchers think that protein post-translational modifications are responsible in the emergence of early phase IPC, and that changes in gene expression and protein synthesis contribute to the appearance of late IPC [3, 4]. Regarding the significance of the issue, in order to design a totally cardioprotective model, the identification of mechanisms responsible for IPC phases seems mandatory. Lysine deacetylation is one of the protein modifications in the heart that has recently attracted attention [5]. Sirtuins (SIRT1–SIRT7) are prominent among the important lysine deacetylases [6, 7]. SIRT1 is an important deacetylase of histones and non-histonic proteins in the heart [8] which affects various functions of cardiomyocytes by deacetylating proteins such as NF-Κb [9], endothelial nitric oxide synthase [10], and transcription factors [11]. Extensive studies have been conducted on the significance of SIRT1 in protecting the heart against damage due to ischemia reperfusion, hypertrophy and cardiac failure [12, 13]. The role of SIRT2 in programmed necrosis following myocardial ischemia reperfusion and in diabetic cardiomyopathy has also been reported previously [14, 15].
Nonetheless, few studies have been carried out on the significance of sirtuins in IPC most of which have focused on early IPC. Thus, this study investigated the effect of sirtinol as the inhibitor of sirtuin NAD-dependent histone deacetylases SIRT1 and 2 on infarct size (IS), reperfusion-induced arrhythmias and the antioxidant factors transcription levels in the heart following early and late IPC.
Animals, materials and methods
Experimental design
Male Wistar rats (250–300 g) were divided into seven experimental groups as shown in Fig. 1. The experimental protocols were approved by the Animal Ethics Care and Use Committee of Shahid Sadoughi University of Medical Sciences.
Fig. 1.
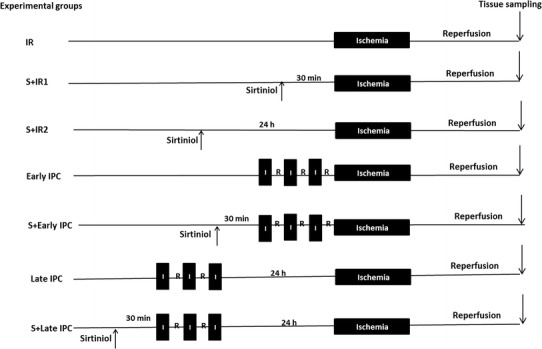
Schematic diagram depicting the experimental protocol. Rats underwent 30 min of ischemia followed by 120 min of reperfusion (IR). In ischemic preconditioning groups (IPC) three episodes of 5-min transient ischemia (I) and reperfusion (R) were induced before the onset of sustained ischemia. Sirtinol (S) was administered (IV) 30 min or 24 h before IR (S + IR1 and S + IR2 groups, respectively) or 30 min before IPC induction in IPC groups
Ischemia reperfusion model
In IR and early IPC groups, the animals were anesthetized (sodium thiopental 50 mg/kg i.p.) and plugged to ventilators after tracheotomies. Arterial blood pressure was measured by cannulation of the left carotid artery and an electrocardiogram was recorded by subcutaneous electrodes. For induction of sustained ischemia reperfusion, after opening the fifth intercostal spaces, 5–0 silk suture was passed under the left anterior descending (LAD) branch of the coronary artery. The artery was occluded for 30 min and reopened for 120 min to reperfuse the myocardium. In IPC groups, three episodes of transient ischemia reperfusion were induced. In delayed IPC groups, the animals were anesthetized with intraperitoneal injection of ketamine-xylazine and intubated intraorally. Twenty-four hours after thoracotomy and induction of IPC, the animals were anesthetized again and subjected to sustained ischemia reperfusion [16–18].
Infarct size measurement
In all groups, at the end of reperfusion Evans blue (0.5 %) was injected into the heart through the tail vein so the non-ischemic area of the heart stained blue. The area that remained unstained was defined as the area at risk (AAR). After irrigating with cold PBS, the heart was placed in a rat heart slicer matrix: 12 h after freezing, the heart was sliced into 2-mm slices from base to apex. The slices were incubated for 20–30 min in a solution containing 1 % tetrazolium chloride at 37 °C. The slices were scanned. IS was reported as a percentage of the ischemic area.
Evaluation of ischemia-reperfusion induced arrhythmias
Reperfusion-induced arrhythmias were defined and assessed according to the Lambeth convention. The episode and prevalence of single premature ventricular complex (PVC), bigeminy, salvos, ventricular tachycardia (VT), and ventricular fibrillation (VF) were identified and the number of each was counted.
Gene expression assessment
The total RNA was extracted using an RNeasy fibrous tissue mini kit (QIAGEN, USA), according to the protocol. After reading the absorption of mRNA in the NanoDrop spectrophotometer, a reverse transcription reaction was done based on instructions given in the cDNA kit (Fermentaz, USA). cDNA of experimental groups underwent amplification with master mix containing SYBR green and specific primers in a real-time PCR set (Rotor-Gene, Qiagen). Β-actin was used as the reference. Comparison of gene expressions was done by the 2−ΔΔCT method. Following are the nucleotide sequences of the PCR primers: catalase: F: 5′-CTGACGTCCACCCTGACT-3, R: 5′-GGCAGCTATGTGAGAGCC-3′; Mn-SOD: F: 5′-GGCCAAGGGAGATGTTACAA-3′, R: 5′-GCTTGATAGCCTCCAGCAAC-3′; β-actin: F: 5′-AACCCTAAGGCCAACCGTGAAAAGAT-3′, R: 5′ ACCGCTCGTTGCCAATAGTGATG-3′ [19].
Statistical analysis
The number of ventricular ectopic beats (VEBs), episodes of VT and blood pressure were analyzed by the Kruskal–Wallis test with Dunn’s post-test for multiple comparisons. Target gene transcription levels were analyzed by one-way ANOVA. Post hoc analysis was performed using the Tukey test. P < 0.05 was considered as statistically significant. Statistical analysis was performed using Prism software. Eight samples of each group were examined to measure the infarct area and five samples were studied to investigate the changes at the the mRNA level because no molecular examination was possible on the samples targeted for the measurement of infarct size. As ECGs were recorded for all samples, there were thirteen samples in each group for studying arrhythmias of the reperfusion episode.
Results
Effect of sirtinol on blood pressure
As shown in Fig. 2, the administration of sirtinol had no effect on animals’ baseline mean arterial pressure.
Fig. 2.
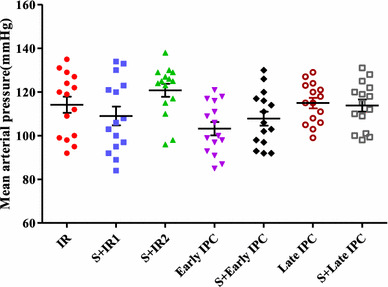
Mean arterial pressure (MAP) (mmHg) in experimental groups. Blood pressure was measured before LAD occlusion through a carotid artery cannula connected to the transducer. In ischemic preconditioning groups (IPC) three episodes of transient ischemia and reperfusion were induced before the onset of sustained ischemia reperfusion (IR). Sirtinol (S) was administered before IR or IPC induction
The effect of sirtinol on infarct size following early and late IPC
As displayed in Fig. 3, IS was 25.8 ± 5.5 % in the early IPC group and 29.7 ± 6.8 % in late IPC group which were statistically significant compared with the IR group at P < 0.001 and P < 0.05, respectively. In the S + IR1 and S + IR2 groups IS did not change significantly. In the next stage, the effect of sirtinol on changes in infarct size due to early and late IPC was evaluated. In the S + early the IS was not significantly different from the early IPC group. However, in the S + late IPC group the IS reached 39.8 ± 2.8 %, which was significantly different from the late IPC group (P < 0.05).
Fig. 3.
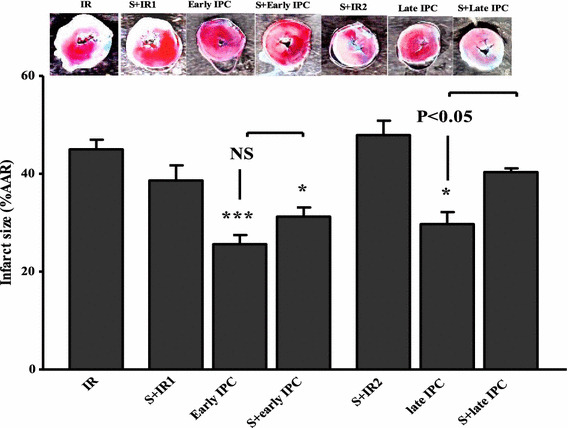
Infarct size evaluation in experimental groups. In ischemic preconditioning groups (IPC) three episodes of transient ischemia and reperfusion were induced before the onset of sustained ischemia reperfusion (IR). Sirtinol (S) was administered before IR or IPC induction. NS not significant; *P < 0.05, ***P < 0.001 vs IR. Data are expressed as mean ± SEM
The effect of sirtinol on reperfusion-induced arrhythmias following early and late IPC
In the IR model used in this study, a large number of arrhythmias appeared in ECGs with the onset of reperfusion. As shown in Fig. 4, the number of VEBs was about 171 ± 51 in the early IPC group and 228 ± 77 in the late IPC group, which were significantly different from the IR group (347 ± 65) at P < 0.001 and P < 0.05, respectively. Further, the number of VEBs in the S + early IPC group was 212 ± 62 which was not significantly different from the early IPC group. This number was 335 ± 64 beats in the S + late IPC group, which was significantly different from the late IPC group (P < 0.01). The number of VT episodes in the early IPC and S + early IPC groups was decreased significantly (P < 0.001 vs IR) and there was no significant difference between these two groups. Moreover, the number of episodes in the late IPC group was not significantly different from the IR group.
Fig. 4.
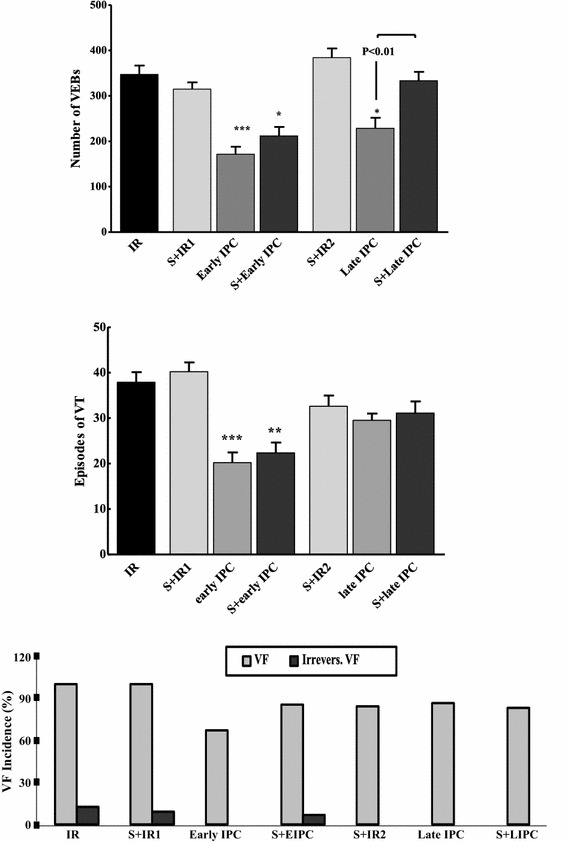
Evaluation of ischemia reperfusion-induced arrhythmias according to the Lambeth convention. Number of ventricular ectopic beats (VEBs), episodes of ventricular tachycardia (VT) and ventricular fibrillation (VF) in experimental groups. In ischemic preconditioning groups (IPC) three episodes of transient ischemia and reperfusion were induced before the onset of sustained ischemia reperfusion (IR). Sirtinol (S) was administered before IR or IPC induction. *P < 0.05, **P < 0.01, ***P < 0.001 vs IR. Data are presented as mean ± SEM
One of the lethal ventricular arrhythmias is fibrillation, which is often reversible; yet, in some cases it is irreversible and causes the death of the animal. In this study, the percentages of reversible and irreversible VF was not significantly different among the experimental groups.
The effect of sirtinol on transcriptional levels of catalase and Mn-SOD following early and late IPC
In the early IPC group, the transcription level of catalase and Mn-SOD was not significantly different from the IR group. However, in late IPC the mRNA levels relating to catalase and Mn-SOD increased by 67.2 ± 4 and 52.3 ± 3.5 %, respectively, which was significant compared to the IR group (P < 0.001). Interestingly, the increase in the level of catalase mRNA in the S + late IPC group was not significant compared to the IR group (Fig. 5).
Fig. 5.
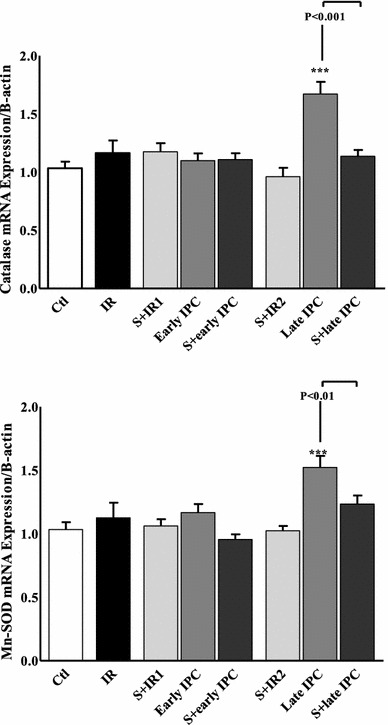
Catalase and Mn-SOD transcription level in experimental groups. Rats underwent 30 min of ischemia followed by 120 min of reperfusion (IR). In ischemic preconditioning groups (IPC) three episodes of 5-min transient ischemia (I) and reperfusion (R) were induced before the onset of sustained ischemia. Sirtinol (S) was administered before IR or IPC induction. ***P < 0.001 vs control (Ctl). Data are presented as mean ± SEM
Discussion
The results of the first part of our study showed that both early and late IPC are able to protect the heart against the ischemia reperfusion damage and reduce the infarct size and the incidence of ventricular arrhythmias. For late IPC, these cardioprotective effects were associated with increased expression of antioxidant factors. This is fully consistent with previous studies conducted on the IPC model [20, 21]. The mechanisms involved in this innate protection are not fully understood yet, though research shows that during short episodes of ischemia, the production of molecules such as nitric oxide, adenosine, bradykinin, and opioids increase in the myocardium and activate the G-protein coupled receptor leading to activation of protein kinase C (PKC) and ultimately, activate the cardioprotective paths in the heart [4, 22, 23].
The delay IPC phase would start from 12 to 24 h after the ischemia episodes and would last for 3–4 days. Late IPC is a phenomenon with a polygenic nature induced by numerous signals [24]. The findings of our study revealed that induction of late IPC but not early IPC increased the rate of transcription of genes in the ischemic tissue of the myocardium that code antioxidant factors such as catalase and Mn-SOD. This confirms that during late IPC, real changes in gene expression can be responsible for the cardioprotective effects in late IPC. The cardioprotective effects of late IPC observed in this study confirm the previous studies [25, 26].
In our study, administration of sirtinol before IPC could weaken the anti-ischemic effects of late IPC and prevent the late IPC-induced survival factors’ upregulation, though it had no effect on the anti-ischemic effect of early IPC. Many in vivo and ex vivo studies have shown that SIRT1 can affect the transcription rate and function of many intracellular proteins through deacetylation proteins in the cardiovascular system, and low to moderate expression of SIRT1 can strengthen the heart against hypertrophy, apoptosis, and impairment of cardiac function due to increasing age [11, 27, 28]. The use of resveratrol as a SIRT1 activator can induce anti-hypertrophic [29, 30] and anti-ischemic effects [31, 32]. In other words, resveratrol mimics the IPC effects in the heart. Our previous study also showed that administration of this agent combined with 1,25-dihydroxyvitamin D can decrease infarct size and reinforce survival genes expression in the myocardium [32].
There has not been much research conducted on the importance of SIRT1 in IPC. Sergiy et al. showed that early IPC could protect the heart of normal mice against ischemia reperfusion damage, yet IPC could not induce its protective properties in SIRT1+/− mice. In their study, splitomicin was used as a SIRT1 inhibitor and it was shown that the rate of deacetylation and, subsequently, the protective effects observed in SIRT1+++ are weakened by this inhibitor. They found that activation of endothelial nitric oxide synthase, deacetylation and inhibition of NF-kB, and autophagy modulation can be among the functioning mechanisms of SIRT1 in acute IPC. In another study by this group conducted on mice with isolated hearts, it was shown that the deacetylated lysine level was higher in the IPC group though, of course, the protein level of SIRT1 did not change. The use of splitomicin as a SIRT1 inhibitor could reverse IPC-mediated cardioprotection [33, 34].
Our finding regarding the ineffectiveness of sirtinol on early IPC is partly consistent with Adam et al.’s study. They showed that the use of SIRT1 inhibitor III does not weaken the IPC cardioprotective effect in young rats [35]. Our study surveyed the inhibition effect of sirtinol on both phases of IPC to provide a more accurate description with regard to exact mechanisms of IPC. One interesting finding of our study was that although the use of sirtinol before IPC could not attenuate the effects of acute IPC, it could weaken the IPC cardioprotective effects which manifest themselves 24 h later. Seeing that the mechanisms responsible for late IPC often act on the level of gene and protein, it can be suggested that the required time is provided for SIRT1 or SIRT2 performance during late IPC and the enzymes can increase the rate of expression of downstream targets such as catalase, thioredoxin, and Mn-SOD through deacetylation of FOXO transcription factors, and these survival factors, in turn, protect the heart against oxidative damage [11, 12]. In the present study the late IPC induction increases the mRNA level of catalase and Mn-SOD in the ischemic myocardial tissue and the use of sirtinol prevents this increase. This strengthens our group’s hypothesis that the enzyme SIRT1 is activated by the induction of IPC, gaining sufficient opportunity for deacetylating various factors which leads to an increase in anti-apoptotic factors in the heart and protecting the heart against ischemia reperfusion damage. One of the activating factors of SIRT1 is NAD [8]. NAD availability increases during preconditioning, so it is possible that in our study the increased NAD level during preconditioning has activated the enzyme SIRT1 and, consequently, its downstream protective paths. Hence, if this path is also activated, the role of SIRT1 in late IPC is more obvious. Regarding the inhibitory effect of sirtinol on SIRT2, it is possible that SIRT2 was involved in the late IPC-induced cardioprotection too. Therefore, to obtain better evidence, it is suggested to use specific inhibitors in future studies. More research is needed to determine the molecular and cellular mechanisms involved in early and late IPC.
Conclusion
Our study shows that a dose of sirtinol, which is ineffective on early IPC, can attenuate the cardioprotective effects of late IPC. Part of this effect can be attributed to inhibition of late IPC-induced survival factors upregulation.
Acknowledgments
This study was supported by Shahid Sadoughi University of Medical Sciences, Yazd, Iran. The authors thank the members of Department of Medical Physiology for their support and assistance.
Compliance with ethical standards
Conflict of interest
The authors declare that there are no conflicts of interest.
References
- 1.Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.CIR.74.5.1124. [DOI] [PubMed] [Google Scholar]
- 2.Tomai F, Crea F, Chiariello L, Gioffrè PA. Ischemic preconditioning in humans models, mediators, and clinical relevance. Circulation. 1999;100:559–563. doi: 10.1161/01.CIR.100.5.559. [DOI] [PubMed] [Google Scholar]
- 3.Yang X, Cohen MV, Downey JM. Mechanism of cardioprotection by early ischemic preconditioning. Cardiovasc Drugs Ther. 2010;24:225–234. doi: 10.1007/s10557-010-6236-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Downey JM, Davis AM, Cohen MV. Signaling pathways in ischemic preconditioning. Heart Fail Rev. 2007;12:181–188. doi: 10.1007/s10741-007-9025-2. [DOI] [PubMed] [Google Scholar]
- 5.Lu Z, Scott I, Webster BR, Sack MN. The emerging characterization of lysine residue deacetylation on the modulation of mitochondrial function and cardiovascular biology. Circ Res. 2009;105:830–841. doi: 10.1161/CIRCRESAHA.109.204974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lavu S, Boss O, Elliott PJ, Lambert PD. Sirtuins—novel therapeutic targets to treat age-associated diseases. Nat Rev Drug Discov. 2008;7:841–853. doi: 10.1038/nrd2665. [DOI] [PubMed] [Google Scholar]
- 7.Kelly GS. A review of the sirtuin system, its clinical implications, and the potential role of dietary activators like resveratrol: part 2. Altern Med Rev J Clin Therap. 2010;15:313–328. [PubMed] [Google Scholar]
- 8.Tanno M, Sakamoto J, Miura T, Shimamoto K, Horio Y. Nucleocytoplasmic shuttling of the NAD+-dependent histone deacetylase SIRT1. J Biol Chem. 2007;282:6823–6832. doi: 10.1074/jbc.M609554200. [DOI] [PubMed] [Google Scholar]
- 9.Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA. Modulation of NF-κB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004;23:2369–2380. doi: 10.1038/sj.emboj.7600244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mattagajasingh I, Kim C-S, Naqvi A, Yamamori T, Hoffman TA, Jung S-B, et al. SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc Natl Acad Sci. 2007;104:14855–14860. doi: 10.1073/pnas.0704329104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 12.Alcendor RR, Gao S, Zhai P, Zablocki D, Holle E, Yu X, et al. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ Res. 2007;100:1512–1521. doi: 10.1161/01.RES.0000267723.65696.4a. [DOI] [PubMed] [Google Scholar]
- 13.Planavila A, Iglesias R, Giralt M, Villarroya F. Sirt1 acts in association with PPARα to protect the heart from hypertrophy, metabolic dysregulation and inflammation. Cardiovasc Res. 2011;90:276–284. doi: 10.1093/cvr/cvq376. [DOI] [PubMed] [Google Scholar]
- 14.Narayan N, Lee IH, Borenstein R, Sun J, Wong R, Tong G, Fergusson MM, Liu J, II, Rovira H-L Cheng, Wang G, Gucek M, Lombard D, Alt FW, Sack MN, Murphy E, Cao L, Finkel T. The NAD-dependent deacetylase SIRT2 is required for programmed necrosis. Nature. 2012;492:199–206. doi: 10.1038/nature11700. [DOI] [PubMed] [Google Scholar]
- 15.Yuan Q, Zhan L, Zhou Q-Y, Zhang L-L, Chen X-M, Hu X-M, Yuan X-C. SIRT2 regulates microtubule stabilization in diabetic cardiomyopathy. Eur J Pharmacol. 2015;764:554–561. doi: 10.1016/j.ejphar.2015.07.045. [DOI] [PubMed] [Google Scholar]
- 16.Liu Y, Downey JM. Ischemic preconditioning protects against infarction in rat heart. Am J Physiol. 1992;263:H1107–H1112. doi: 10.1152/ajpheart.1992.263.4.H1107. [DOI] [PubMed] [Google Scholar]
- 17.Klishadi MS, Zarei F, Hejazian SH, Moradi A, Hemati M, Safari F. Losartan protects the heart against ischemia reperfusion injury: sirtuin3 involvement. J Pharm Pharm Sci. 2015;18(1):112–123. doi: 10.18433/J3XG7T. [DOI] [PubMed] [Google Scholar]
- 18.Safari F, Anvari Z, Moshtaghioun S, Javan M, Bayat G, Forosh SS, Hekmatimoghaddam S. Differential expression of cardiac uncoupling proteins 2 and 3 in response to myocardial ischemia-reperfusion in rats. Life Sci. 2014;98(2):68–74. doi: 10.1016/j.lfs.2013.12.230. [DOI] [PubMed] [Google Scholar]
- 19.Colombrita C, Calabrese V, Stella AMG, Mattei F, Alkon DL, Scapagnini G. Regional rat brain distribution of heme oxygenase-1 and manganese superoxide dismutase mRNA: relevance of redox homeostasis in the aging processes. Exp Biol Med. 2003;228:517–524. doi: 10.1177/15353702-0322805-16. [DOI] [PubMed] [Google Scholar]
- 20.Juan-Zhang H-JB, Li X-X, Liu X-B, Sun J-P, Na-Li Y-Z, Ji X-P. ERK-MAPK signaling opposes rho-kinase to reduce cardiomyocyte apoptosis in heart ischemic preconditioning. Mol Med. 2010;16:307–315. doi: 10.2119/molmed.2009.00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang J, Liu X-B, Cheng C, Xu D-L, Lu Q-H, Ji X-P. Rho-kinase inhibition is involved in the activation of PI3-kinase/Akt during ischemic-preconditioning-induced cardiomyocyte apoptosis. Int J Clin Exp Med. 2014;7:4107. [PMC free article] [PubMed] [Google Scholar]
- 22.Ardehali H. Signaling mechanisms in ischemic preconditioning interaction of PKCε and MitoKATP in the inner membrane of mitochondria. Circ Res. 2006;99:798–800. doi: 10.1161/01.RES.0000247029.31997.a4. [DOI] [PubMed] [Google Scholar]
- 23.Lu XM, Zhang GX, Yu YQ, Kimura S, Nishiyama A, Matsuyoshi H, Shimizu J, Takaki M. The opposite roles of nNOS in cardiac ischemia-reperfusion-induced injury and in ischemia preconditioning-induced cardioprotection in mice. J Physiol Sci. 2009;59:253–262. doi: 10.1007/s12576-009-0030-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bolli R. The late phase of preconditioning. Circ Res. 2000;87:972–983. doi: 10.1161/01.RES.87.11.972. [DOI] [PubMed] [Google Scholar]
- 25.Zhai X, Zhou X, Ashraf M. Late ischemic preconditioning is mediated in myocytes by enhanced endogenous antioxidant activity stimulated by oxygen-derived free radicals. Ann N Y Acad Sci. 1996;793:156–166. doi: 10.1111/j.1749-6632.1996.tb33512.x. [DOI] [PubMed] [Google Scholar]
- 26.Hoshida S, Kuzuya T, Fuji H, Yamashita N, Oe H, Hori M, et al. Sublethal ischemia alters myocardial antioxidant activity in canine heart. Am J Physiol Heart Circ Physiol. 1993;26:H33–H39. doi: 10.1152/ajpheart.1993.264.1.H33. [DOI] [PubMed] [Google Scholar]
- 27.Luo J, Nikolaev AY, S-i Imai, Chen D, Su F, Shiloh A, et al. Negative control of p53 by Sir2α promotes cell survival under stress. Cell. 2001;107:137–148. doi: 10.1016/S0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- 28.Tanno M, Kuno A, Yano T, Miura T, Hisahara S, Ishikawa S, et al. Induction of manganese superoxide dismutase by nuclear translocation and activation of SIRT1 promotes cell survival in chronic heart failure. J Biol Chem. 2010;285:8375–8382. doi: 10.1074/jbc.M109.090266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dolinsky VW, Soltys C-LM, Rogan KJ, Chan AY, Nagendran J, Wang S, et al. Resveratrol prevents pathological but not physiological cardiac hypertrophy. J Mol Med. 2015;93:413–425. doi: 10.1007/s00109-014-1220-8. [DOI] [PubMed] [Google Scholar]
- 30.Dorri Mashhadi F, Zavvar Reza J, Jamhiri M, Hafizi Z, Zare Mehrjardi F, Safari F (2016) The effect of resveratrol on angiotensin II levels and the rate of transcription of its receptors in the rat cardiac hypertrophy model. J Physiol Sci. doi:10.1007/s12576-016-0465-0 [DOI] [PMC free article] [PubMed]
- 31.Liao Z, Liu D, Tang L, Yin D, Yin S, Lai S, et al. Long-term oral resveratrol intake provides nutritional preconditioning against myocardial ischemia/reperfusion injury: involvement of VDAC1 downregulation. Mol Nutr Food Res. 2015;59:454–464. doi: 10.1002/mnfr.201400730. [DOI] [PubMed] [Google Scholar]
- 32.Safari F, Zarei F, Shekarforoush S, Fekri A, Sharifi Klishadi M, Hekmatimoghaddam H. Combined 1,25-dihydroxyvitamin D and resveratrol: a novel therapeutic approach to ameliorate ischemia reperfusion-induced myocardial injury. Int J Vitam Nutr Res. 2015;85:174–184. doi: 10.1024/0300-9831/a000236. [DOI] [PubMed] [Google Scholar]
- 33.Nadtochiy SM, Yao H, McBurney MW, Gu W, Guarente L, Rahman I, et al. SIRT1-mediated acute cardioprotection. Am J Physiol Heart Circ Physiol. 2011;301:H1506–H1512. doi: 10.1152/ajpheart.00587.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nadtochiy SM, Redman E, Rahman I, Brookes PS. Lysine deacetylation in ischaemic preconditioning: the role of SIRT1. Cardiovasc Res. 2011;89:643–649. doi: 10.1093/cvr/cvq287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adam T, Sharp S, Opie LH, Lecour S. Loss of cardioprotection with ischemic preconditioning in aging hearts: role of sirtuin 1? J Cardiovasc Pharmacol Therap. 2013;18:46–53. doi: 10.1177/1074248412458723. [DOI] [PubMed] [Google Scholar]