

Abstract
When a cardiac muscle is held in a stretched position, its [Ca2+] transient increases slowly over several minutes in a process known as stress-induced slow increase in intracellular Ca2+ concentration ([Ca2+]i) (SSC). Transient receptor potential canonical (TRPC) 3 forms a non-selective cation channel regulated by the angiotensin II type 1 receptor (AT1R). In this study, we investigated the role of TRPC3 in the SSC. Isolated mouse ventricular myocytes were electrically stimulated and subjected to sustained stretch. An AT1R blocker, a phospholipase C inhibitor, and a TRPC3 inhibitor suppressed the SSC. These inhibitors also abolished the observed SSC-like slow increase in [Ca2+]i induced by angiotensin II, instead of stretch. Furthermore, the SSC was not observed in TRPC3 knockout mice. Simulation and immunohistochemical studies suggest that sarcolemmal TRPC3 is responsible for the SSC. These results indicate that sarcolemmal TRPC3, regulated by AT1R, causes the SSC.
Electronic supplementary material
The online version of this article (doi:10.1007/s12576-016-0519-3) contains supplementary material, which is available to authorized users.
Keywords: Transient receptor potential canonical 3, Angiotensin II type 1 receptor, Ca2+ handling, Stretch, Cardiomyocyte, Mathematical model
Introduction
The mechanical environment affects cardiac muscle contractility via cellular Ca2+ handling in the heart, which is required to control cardiac output and meet physiological demands [1]. The myocardial stretch increases the force of contraction immediately, though there is no associated transient change in the intracellular Ca2+ concentration ([Ca2+]i) [2]. This is referred to as the Frank–Starling effect, and is caused by a rapid increase in myofilament Ca2+ sensitivity. Meanwhile, a long time-course stretch, over several minutes, causes a subsequent slow increase in the force of contraction. This is the result of stress-induced, slow increase in the [Ca2+]i (SSC) [3, 4]. The SSC, accompanied by the slow increase in contraction force, corresponds to the Anrep effect in the heart, which physiologically constitutes an autoregulation mechanism by which the heart adapts to an abrupt increase in afterload (e.g., an increase in aortic pressure), occurring after the Frank–Starling effect appears [5]. To date, various signaling pathways and mechanisms have been reported to explain the SSC. The stretch-induced autocrine and paracrine release of angiotensin II (Ang II) and endothelin-1 (ET-1) has been implicated in the SSC [6–8]. Ang II and ET-1 release causes an increase in intracellular Na+ concentration ([Na+]i) mediated by stretch-activated, non-selective cation channels, with or without involvement of the Na+/H+ exchanger-1. This is followed by an increase in [Ca2+]i via the Na+/Ca2+ exchanger (NCX) [9, 10]. Two subtypes of the transient receptor potential canonical (TRPC) channel, TRPC3 and TRPC6, have been proposed as stretch-activated, non-selective cation channel candidates involved in the SSC [10–13]. These receptor-operated channels are stimulated by the activation of the Ang II type 1 receptor [AT1R, a Gq protein-coupled receptor (GqPCR)] and subsequent diacylglycerol (DAG) synthesis through phospholipase C (PLC) [13–15]. Seo et al. reported that TRPC6 contributes to the SSC [16], but the involvement of TRPC3 in the SSC remains unclear. Studies investigating the effects of TRPC channels on the SSC are complicated by uncertainty about their subcellular localization, and results of immunochemical studies of TRPC channels are controversial. Some studies have indicated that TRPC channels are located in the membrane of the sarcoplasmic reticulum (SR) in cardiomyocytes [17, 18], and others have reported that they are located in the sarcolemma and transverse tubules of ventricular myocytes [19, 20].
Here, we have focused on clarifying the role of TRPC3 in the SSC. Using isolated ventricular myocytes from C57BL/6 J, 129/Sv wild-type (WT), and TRPC3 knockout (KO; Trpc3−/−) mice, we investigated whether TRPC3 activation, via AT1R-induced PLC, contributes to the SSC. Additionally, we investigated the location of TRPC3s and their location-dependent role on the SSC using an immunochemical method and mathematical cardiomyocyte model, with cation channels (representing TRPC3) on the sarcolemma or the SR.
Methods
Myocyte isolation
All experiments were conducted in accordance with the Guiding Principles for the Care and Use of Animals approved by the Council of the Physiological Society of Japan. Animal protocols were approved by the Animal Subjects Committee of Okayama University Graduate School of Medicine, Dentistry, and Pharmaceutical Sciences. Myocyte isolation was performed as described [21–23] with modifications. Mice (129/Sv WT, Trpc3 −/−, and Trpc6 −/−) were provided by the National Institute of Environmental Health Sciences (NC, USA) [24, 25]. TRPC3 and TRPC6 gene knockouts were confirmed by real-time PCR. Ventricular myocytes were isolated from 54 hearts of C57BL/6 J mice, 7 hearts of 129/Sv WT mice, 5 hearts of Trpc3 −/− mice, and 4 hearts of Trpc6 −/− mice (aged 8–15 weeks). After inhalation anesthesia with isoflurane (DS Pharma Animal Health, Osaka, Japan), adult mice were injected intraperitoneally with 100 IU of heparin sodium (AY Pharmaceuticals, Tokyo, Japan) and euthanized with isoflurane 30 min post injection. The heart was quickly excised and cannulated through the aorta using an 18G cannula. The heart was mounted on a Langendorff apparatus and perfused at 36–37 °C with oxygenated solution A (128 mM NaCl, 2.6 mM KCl, 1.18 mM MgSO4, 1.18 mM KH2PO4, 10 mM HEPES, 20 mM taurine, and 11 mM glucose; pH 7.4, adjusted with NaOH) containing 10 mM 2,3-butanedione monoxime (Sigma-Aldrich, St. Louis, MO, USA) at a rate of 4 ml min−1 for 1–2 min. Then, the flow rate was changed to 3 ml min−1 for 3–4 min. Subsequently, the perfusate was switched to 27 ml of solution A containing 12.5 μM CaCl2 and 2.5 mg of the enzyme Liberase TM Research Grade (Roche, Basel, Switzerland), to digest the myocardium for 3–5 min. The ventricles were then excised and cut into several pieces that were further digested in the enzyme solution for 15–25 min at 37 °C. The tissue was gently agitated, using a transfer pipette, in 10 ml of solution A containing 10% fetal bovine serum and 12.5 μM CaCl2. The cell suspension was filtered with a 100-μm nylon mesh filter (Becton, Dickinson and Company, Franklin Lakes, NJ, USA) and centrifuged in a 15-ml plastic conical tube at 15×g for 3 min. The cell pellet was re-suspended in solution A containing 600 μM CaCl2. The cells were then incubated at room temperature for 8 min, centrifuged at 15×g for 3 min, and re-suspended in solution A containing 1.2 mM CaCl2. The centrifugation process was repeated and the final pellet was re-suspended in a storage solution (solution A containing 1.8 mM CaCl2). The cells were kept in the storage solution at room temperature for at least 30 min before measurement.
Transient [Ca2+]i measurement
[Ca2+]i was measured using IonOptix hardware and software (IonOptix Corporation, Milton, MA, USA) and an inverted microscope (IX-70; Olympus, Tokyo, Japan) with an oil immersion objective lens (UAPON 40 × O340; Olympus, Japan) as previously described [26]. To record [Ca2+]i, 2.5 μM Fura-4F acetoxymethyl ester (Fura-4F AM; Life Technologies, Carlsbad, CA, USA) was loaded onto the cells with 0.1% Pluronic F-127 (AnaSpec, Fremont, CA, USA) for 10 min. Cells were placed on a poly-HEMA (2-hydroxyethyl methacrylate; Sigma-Aldrich, USA)-coated coverslip to prevent firm cell attachment to the bottom of the chamber. Due to the short working distance of the objective lens, both cell ends were held by a pair of carbon fibers (CF) to stretch cells without lifting them off the coverslip. For the ratiometric recordings, excitation wavelengths of 340 and 380 nm (alternated at 250 Hz) were used, and the fluorescence emission was recorded at 480–520 nm with a photomultiplier tube. The background emission, obtained in the same area after each experiment, was subtracted from the raw emission signal. The ratio of the emission signals at the two wavelengths (340/380 nm) was taken to represent changes in [Ca2+]i.
Stretch protocol
Fura-4F-loaded myocytes were set on the cardiomyocyte stretch system mounted on the IX-70 inverted microscope as described above. A schematic representation of the stretch system is shown in Fig. 1a. Sarcomere length changes were recorded using IonOptix hardware and software (IonOptix Corporation, USA). After establishing firm CF attachment, the cell was perfused in normal Tyrode’s (NT) solution (140 mM NaCl, 5.4 mM KCl, 1.8 mM CaCl2, 1.0 mM MgCl2, 5.0 mM HEPES, and 11 mM glucose; pH 7.4, adjusted with NaOH) at room temperature and electrically stimulated at 1 Hz at slack length until steady-state was obtained [Fig. 1b (top panel)]. Stretch was applied by moving a computer-controlled piezoelectric translator (P-621.1CL; Physik Instrument, Karlsruhe/Palmbach, Germany) to achieve an 8% increase in sarcomere length [Fig. 1b (lower panel)]. In the stretch protocol, SSC was recorded 300 s after completion of the stretch, since our result (Fig. S1) and many other previous studies of stretch-induced increase in twitch force show that steady contractions were usually achieved within 300 s after stretch [10, 16, 27]. Thus, stretch was maintained for longer than 300 s and then released. The Fura-4F ratios at 10 s after the stretch were recorded, and at 300 s were recorded as the SSC.
Fig. 1.
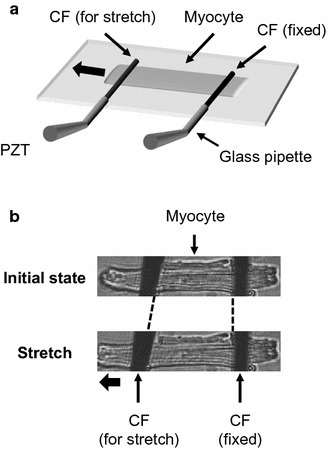
Overview of the stretch control system used to apply an axial stretch to the cell. a Both cell ends were held by a pair of carbon fibers (CF). The left CF was mounted on piezoelectric translators (PZT). b The myocyte was stretched from the initial state by moving the left CF with the PZT
Ang II protocol
After establishing firm CF attachment, the cell was perfused with NT solution and electrically stimulated at 1 Hz at slack length until steady-state was obtained. Then, the cell was perfused with NT solution containing 1 μM Ang II (A9525; Sigma-Aldrich, USA) without stretch [28]. The Fura-4F ratios observed 10 and 300 s after Ang II treatment correspond to the responses 10 s after the stretch and the SSC in the stretch protocol, respectively.
Mathematical modeling
We compared the cellular behaviors in SSC between cells with TRPC3 channels on the sarcolemma and those with TRPC3 channels on the SR, using a published mathematical cardiomyocyte model [26]. The model was modified by adding a cation current via the sarcolemma or a Ca2+ flux via the SR membrane. The net cellular current via TRPC3 channels on the sarcolemma (I trpc) is described as follows:
where I trpcNa, I trpcK, and I trpcCa are the Na+, K+, and Ca2+ current components of I trpc; G trpcNa, G trpcK, and G trpcCa are the Na+, K+, and Ca2+ conductance; V is the membrane potential; and E Na, E K, and E Ca are the Na+, K+, and Ca2+ reversal potentials. We used mimicking to simulate stretch-induced increase in Ca2+ release via TRPC3 in the SR by adding a Ca2+ leak factor in the SR Ca2+ release equation:
where J rel is the Ca2+ release flux from the SR; K rel is the maximum Ca2+ release flux; F rel is the fraction of ryanodine receptors related to the Ca2+ release; K leak is the intrinsic SR Ca2+ leak rate; K TRPC is the TRPC3 Ca2+ release rate; [Ca2+]SR is the SR Ca2+ concentration; and [Ca2+]iSL is the sub-sarcolemmal (SL) cytosolic Ca2+ concentration. Except for the above modification, all parameters were identical to those of the original published model. A full CellML version of the model is available as an online supplementary file and can be run using suitable modeling environments (such as OpenCOR; http://www.opencor.ws/).
Immunohistochemistry
We performed immunochemical studies as described [29] with modifications. Hearts were excised from 129/Sv WT and Trpc3 −/− mice (two of each genotype), to prepare paraffin sections. Mice were euthanized with isoflurane, and the hearts quickly excised and washed with phosphate-buffered saline (PBS). The specimens were fixed in 4% paraformaldehyde and embedded in paraffin and serial 5-μm-thick sections were cut from the paraffin-embedded tissue blocks. The paraffin sections were incubated with Blocking One (Nacalai Tesque, Kyoto, Japan) for 1 h at room temperature and incubated with the anti-TRPC3 antibody (1:100; ab51560; Abcam, Tokyo, Japan) overnight at 4 °C. Then, the sections were incubated with Fab Fragment Goat Anti-Rabbit IgG (H + L) (1:10; Jackson Immuno Research Laboratories, West Grove, PA, USA) for 1 h at room temperature. Subsequently, the sections were incubated with an Alexa Fluor 555-conjugated secondary antibody (1:1000; Life Technologies) for 1 h at room temperature. The sections were then incubated with the anti-sodium potassium ATPase (NK-ATPase) antibody (1:500; ab76020; Abcam) or the sarcoplasmic reticulum calcium ATPase (SERCA) 2 antibody (1:500; sc-8095; Santa Cruz Biotechnology, Dallas, TX, USA) overnight at 4 °C. Finally, the sections were incubated with an Alexa Fluor 488-conjugated secondary antibody (1:1000; Life Technologies) for 1 h at room temperature. At all staining steps, the sections were washed with PBS containing 0.1% Tween-20. After staining, the sections were mounted onto a cover glass with VECTASHIELD Mounting Medium (Vector Laboratories, Burlingame, CA, USA). Sections stained only with the secondary antibodies served as negative control. To confirm the specificity of the primary anti-TRPC3 antibody, it was pre-incubated with the antigenic-blocking peptide (ab65597; Abcam) overnight at 4 °C prior to use. Fluorescence images were obtained using a confocal microscope (FV 1000; Olympus, Japan). To estimate the co-localization ratio of TRPC3 and NK-ATPase or SERCA2, the Manders’ overlap coefficient was calculated using Fiji/ImageJ software with the Coloc2 plug-in.
Statistical analysis
Values are expressed as the mean ± standard error of the mean (SEM). All parameters were compared using Student’s paired t-test or one-way analysis of variance with Bonferroni’s post hoc as appropriate. A value of P < 0.05 was considered significant. Prism software (Graphpad Software, La Jolla, CA, USA) version 6.0 was used for the analysis.
Results
Changes in sarcomere length, before and after the stretch and Ang II application, are summarized in Table 1. The stretch significantly increased the sarcomere length by 8.85 ± 0.828%. There was no significant difference in control sarcomere length between the stretch and Ang II groups.
Table 1.
Sarcomere length in stretch and angiotensin II (Ang II) protocol
| Protocol | Before stretch or Ang II (μm) | During stretch (μm) |
|---|---|---|
| Stretch protocol (n = 12) | 1.74 ± 0.0257 | 1.98 ± 0.0297* |
| Ang II protocol (n = 8) | 1.73 ± 0.0381 |
Values are mean ± SEM
* P < 0.05 vs before stretch
Stretch protocol
The SSC was assessed by Fura-4F fluorescence ratio (340/380 nm) in single cardiomyocytes of C57BL/6 J mice (Fig. 2a). Compared with the recording obtained before applying an axial stretch, the [Ca2+] transient remained unchanged immediately after the initial stretch. Sustained stretch for 300 s resulted in the SSC. Results of stretch-induced change in the peak of [Ca2+] transient were obtained for nine cells (Fig. 2b). The sustained stretch caused a 43 ± 15% increase in the [Ca2+] transient.
Fig. 2.
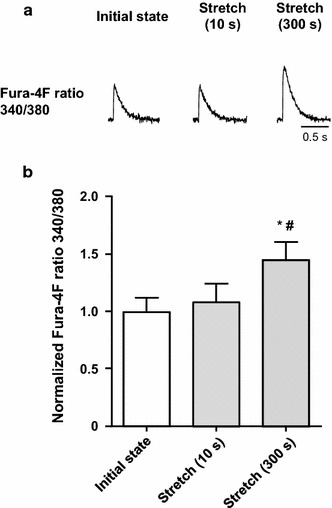
Changes in the [Ca2+] transient in single cardiomyocytes from C57BL/6 J mice in the stretch protocol. a A representative response to axial stretch in the [Ca2+] transient. Following the initial state, the [Ca2+] transient remained unchanged at 10 s after the stretch, and then showed a slow increase at 300 s after the stretch (a stress-induced slow increase in the [Ca2+]i: SSC). b Summarized data for the [Ca2+] transient response to the stretch (n = 9). # P < 0.05 vs initial state; *P < 0.05 vs stretch (10 s)
Ang II protocol
We measured the change in the [Ca2+] transient in the C57BL/6 J mouse cardiomyocytes during the Ang II protocol (Fig. 3). The application of Ang II for 10 s, which corresponds to the 10-s stretch in the stretch protocol, did not alter the [Ca2+] transient. However, the application of Ang II for 300 s, corresponding to the SSC in the stretch protocol, significantly increased the [Ca2+] transient (Fig. 3b). Therefore, the change in [Ca2+] transient induced by Ang II is consistent with the observed stretch-induced changes (Figs. 2, 3).
Fig. 3.
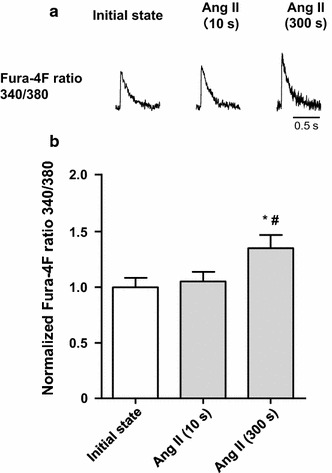
Change in [Ca2+] transient in cardiomyocytes from C57BL/6 J mice in the angiotensin II (Ang II) protocol. a Representative traces of Fura-4F fluorescence signals. After recording the initial state in normal Tyrode’s (NT) solution, the application of Ang II (1 μM) for 10 s left the [Ca2+] transient unchanged. However, application of Ang II for 300 s increased the [Ca2+] transient, similar to the SSC. b Summarized data for the change in [Ca2+] transient by Ang II application (n = 8). # P < 0.05 vs initial state; *P < 0.05 vs Ang II (10 s)
Involvement of TRPC3 in the SSC
To examine the role of TRPC3 in the [Ca2+] transient increase in C57BL/6 J mouse cardiomyocytes during the stretch protocol, we used BTP-2 (10 μM; Cayman Chemical, Ann Arbor, MI, USA), an inhibitor of TRPC channels [30], and Pyr3 (1 μM; Sigma-Aldrich), an inhibitor specific to TRPC3 [31]. Cells were incubated with BTP-2 or Pyr3 for at least 5 min before recording the initial state. Both BTP-2 and Pyr3 suppressed SSC (Fig. 4a). Additionally, we investigated whether TRPC3 activation during the sustained stretch was mediated by PLC-generated DAG (via the activation of AT1R). We inhibited AT1R and PLC by applying olmesartan (RNH 6270, 10 μM; ChemScene, Monmouth Junction, NJ, USA) [32] and U-73122 (10 μM; Cayman Chemical) [33], respectively. Both inhibitors abolished SSC (Fig. 4a). The specificity of U-73122 was confirmed by a negative control experiment using U-73343 (the inactive analogue of U-73122, 10 μM; Sigma-Aldrich), which did not affect the SSC (Fig. S2). Finally, we used BTP-2, Pyr3, and U-73122 to test the involvement of TRPC3 and PLC activation in the increase in [Ca2+] transient during the Ang II protocol. All three inhibitors suppressed the significant increase in [Ca2+] transient observed after the application of Ang II at 300 s to a similar extent to which they suppressed the response to 300 s stretch (SSC) (Fig. 4a, b). To confirm the effects of olmesartan, U-73122, BTP-2, and Pyr3 on the [Ca2+] transient at the initial state, Fura-4F ratios were compared before and after inhibitors application without stretch or applying Ang II. These inhibitors did not affect the [Ca2+] transient at the initial state (Fig. S3).
Fig. 4.
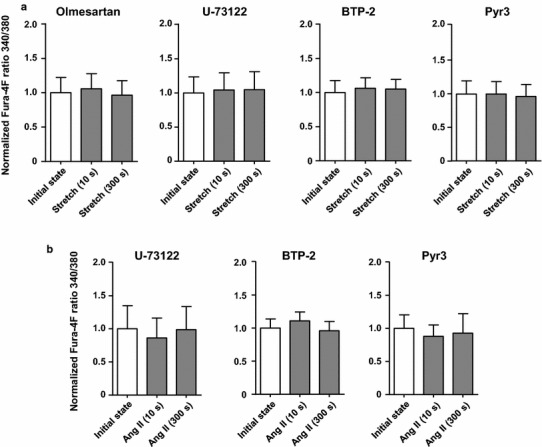
Effects of pharmacological inhibition of the AT1R/TRPC3 pathway on the [Ca2+] transient in cardiomyocytes from C57BL/6 J mice in the stretch and angiotensin II (Ang II) protocols. a The significant slow increase in [Ca2+] transient at 300 s after the stretch (SSC) was suppressed by the inhibitors olmesartan (AT1R blocker, 10 μM; n = 10), U-73122 (PLC inhibitor, 10 μM; n = 9), BTP-2 (TRPC channels inhibitor, 10 μM; n = 9), and Pyr3 (TRPC3 inhibitor, 1 μM; n = 8). These inhibitors did not affect [Ca2+] transient at initial state and 10 s after the stretch. b The SSC-like significant increase in [Ca2+] transient induced by the application of Ang II for 300 s was suppressed by U-73122 (10 μM; n = 6), BTP-2 (10 μM; n = 12), and Pyr3 (1 μM; n = 8). These inhibitors did not affect [Ca2+] transient at initial state and 10 s after Ang II application. AT1R angiotensin II type 1 receptor, PLC phospholipase C, TRPC3 transient receptor potential canonical subtype 3
To further examine the involvement of TRPC3 and TRPC6 in the SSC, we investigated the changes in [Ca2+] transient during the stretch protocol in cardiomyocytes isolated from 129/Sv WT, Trpc3 −/−, and Trpc6 −/− mice [Fig. 5a (top panel), 5b (n = 12)]. Although the amplitude of [Ca2+] transient did not change at 10 s after the stretch, it significantly increased 300 s after the stretch, similar to the response of [Ca2+]i in C57BL/6 J mice (Fig. 2). However, the response at 300 s after the stretch (SSC) was completely abolished in both the Trpc3 −/− and the Trpc6 −/− mice (Fig. 5a (middle and bottom panels), 5c).
Fig. 5.
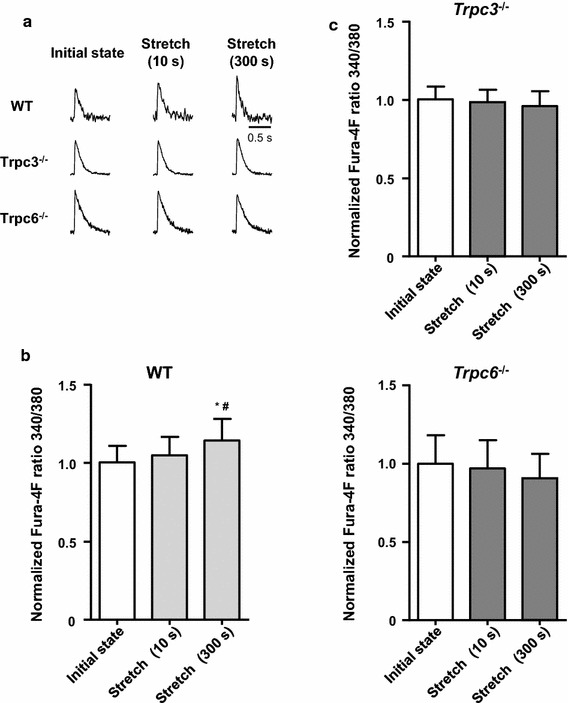
Effects of genetic deletion of TRPC3 and TRPC6 on the [Ca2+] transient in cardiomyocytes from 129/Sv mice during the stretch protocol. a Representative traces of the slow increase in [Ca2+] transient during the sustained stretch (SSC) on single cardiomyocytes isolated from 129/Sv wild-type (WT), Trpc3 −/−, and Trpc6 −/− mice. b Summarized data for the stretch-induced change in [Ca2+] transient in cardiomyocytes from WT mice (n = 12). c The stretch-induced slow increase in [Ca2+] transient at 300 s after the stretch (SSC), seen in the WT mice, was suppressed in the Trpc3 −/− and Trpc6 −/− mice (n = 11 and 8). Deletion of TRPC3 and TRPC6 did not affect [Ca2+] transient at initial state and 10 s after the stretch. # P < 0.05 vs initial state; * P < 0.05 vs stretch (10 s). TRPC transient receptor potential canonical, Trpc3 −/− transient receptor potential canonical subtype 3 knockout, Trpc6 −/− transient receptor potential canonical subtype 6 knockout
Localization of TRPC3 (mathematical modeling study)
To investigate the intracellular location of the TRPC3 channels, we performed a simulation study using our mathematical model of a single cardiomyocyte with stretch-activated cation channels located on either the sarcolemma or the SR. This model is used to simulate [Ca2+] transient during the stretch protocol with a stretch-induced cation influx that mimics TRPC3 channels on the sarcolemma (Fig. 6a). Additionally, this model can simulate [Ca2+] transient during the stretch protocol with stretch-induced Ca2+ leak from the SR, mimicking Ca2+ flux via SR membrane TRPC3 channels (Fig. 6b). For TRPC3 channels on the sarcolemma, the stretch was simulated by increasing the Na+, K+, and Ca2+ conductance of the TRPC3 channels. All conductance values (G trpcNa, G trpcK, and G trpcCa) were set to 0 nS before the stretch, and increased to 0.07 nS after the stretch. This scenario successfully reproduced a stretch-induced slow increase in [Ca2+] transient during the sustained stretch (SSC) without significant changes immediately after the stretch (Fig. 6a). To investigate which cation component of the TRPC3 current is responsible for the SSC, each of the three cation conductance values (G trpcNa, G trpcK, and G trpcCa) were changed independently. Increases in G trpcNa and G trpcCa contributed to the increase in [Ca2+] transient during the sustained stretch, but increases in G trpcK did not (Fig. 6c). Increases in G trpcNa decrease Ca2+ extrusion via NCX. This leads to an increase in SR Ca2+ uptake to accumulate intracellular Ca2+, causing SSC. Increases in G trpcCa increase cytosolic Ca2+ concentration. This also increases SR Ca2+ uptake, therefore SR Ca2+ load, causing SSC. Diastolic Ca2+ concentration is determined by the balance of these influx/efflux factors. Although change in diastolic Ca2+ concentration in our model shown in Fig. 6a is too small to recognize from the figure, it slightly increased after sustained stretch (0.0051 μM at initial state, and 0.0064 μM after sustained stretch). For TRPC3 channels on the SR, the stretch was simulated by increasing the Ca2+ leak rate factor of TRPC3 channels (K TRPC) from 0 to 0.007 s−1. This scenario failed to reproduce the change in [Ca2+] transient associated with the SSC (Fig. 6b). Further increase of the K TRPC also did not reproduce the SSC (data not shown). These results suggest that sarcolemmal TRPC3s are responsible for the SSC, by increasing cation (Na+ and Ca2+) influx.
Fig. 6.
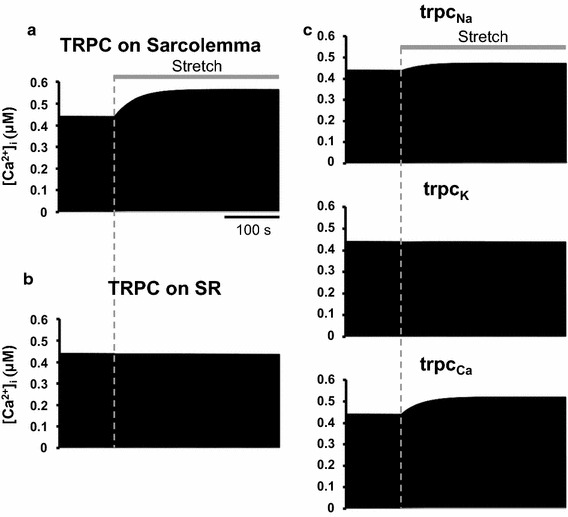
Simulated [Ca2+]i response to stretch with different TRPC3 locations. a Stretch-induced slow increase in [Ca2+]i was observed with the increase in conductance of all cations (Na+, K+, and Ca2+) when TRPC3 was located on the sarcolemma (no stretch: G trpcNa, G trpcK, G trpcCa = 0 nS; with stretch: G trpcNa, G trpcK, G trpcCa = 0.07 nS, respectively). b The [Ca2+]i was unchanged with increasing cation flux when TRPC3 was located on the SR (no stretch: K TRPC = 0 s−1; with stretch: K TRPC = 0.007 s−1). c Increasing only the Na+ or Ca2+ conductance (no stretch: G trpcNa, G trpcCa = 0 nS; with stretch: G trpcNa, G trpcCa = 0.07 nS, respectively) caused SSC (trpcNa and trpcCa, respectively), but increasing only the K+ conductance (no stretch: G t rpcK = 0 nS; with stretch: G trpcK = 0.07 nS) did not (trpcK)
Localization of TRPC3 (immunofluorescence study)
We investigated the intracellular localization of TRPC3 in cardiomyocytes using immunofluorescence. Heart sections were labeled with antibodies for TRPC3 and NK-ATPase, as a sarcolemma and transverse tubules marker, or SERCA2, as an SR marker. TRPC3 partially co-localized with NK-ATPase (Fig. 7b) and SERCA2 (Fig. 7c). The ratio of co-localization was estimated using a Manders’ overlap coefficient-based technique. The coefficient values obtained were 0.17 and 0.45 for TRPC3 and NK-ATPase, and TRPC3 and SERCA2, respectively (Table 2). Therefore, 17% of the TRPC3 signals in sections co-labeled with NK-ATPase were observed on the sarcolemma and transverse tubules, and 45% of the TRPC3 signals in sections co-stained with SERCA2 were observed on the SR. The validity of the staining was confirmed by using Trpc3 −/− mice cardiomyocytes (Fig. 7a), pre-incubation of the antigenic-blocking peptide, and incubation only with the secondary antibody as controls. None of the negative control treatments showed a TRPC3 immunofluorescence signal.
Fig. 7.
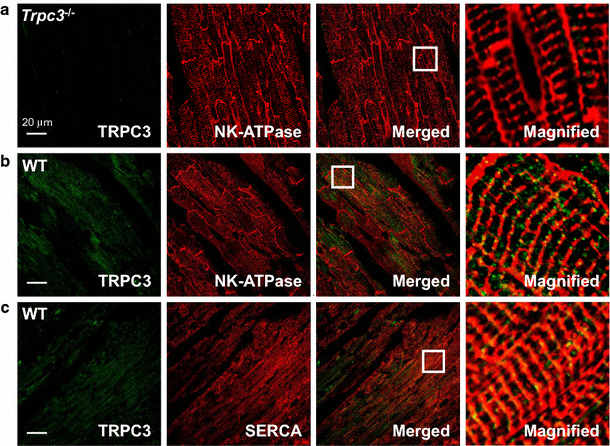
Localization of TRPC3 in mouse hearts. a Heart sections of Trpc3 −/− mice were stained with anti-TRPC3 antibodies (green) and anti-NK-ATPase antibodies (red) to confirm the staining specificity of anti-TRPC3 antibodies. b, c Double immunofluorescence images showing TRPC3 (green) and NK-ATPase (red) (b) or SERCA2 (red) (c) expression in ventricular cardiomyocytes of 129/Sv wild-type (WT) mice. The merged and magnified images show the small amount of co-localization of TRPC3 and NK-ATPase (b), compared to SERCA2 (c). The white inset is magnified in the right panel. Trpc3 −/− transient receptor potential canonical subtype 3 knockout, NK-ATPase sodium potassium ATPase, SERCA sarcoplasmic reticulum calcium ATPase
Table 2.
Manders’ overlap coefficient for TRPC3 fluorescence
| NK-ATPase | SERCA2 | |
|---|---|---|
| TRPC3 | 0.171 ± 0.012 | 0.450 ± 0.015 |
Values are mean ± SEM; values were calculated from nine cell regions of interest, from three slides, for each of two hearts
Discussion
The SSC has been studied for decades, yet the underlying mechanisms have remained unclear. It has been reported that the stretch-induced release of Ang II activates AT1R. This induces the activation of extracellular signal-regulated kinase 1/2 via reactive oxygen species (ROS) to mediate the increase in [Na+]i [34, 35]. The increase in Na+ influx leads to an increase in [Ca2+]i via NCX [36, 37]. Recently, several TRPC channel subtypes have been proposed to be involved with various types of stretch responses [10, 11, 16]. Here, we have demonstrated that stretch-induced cation (Na+ and Ca2+) influx, via TRPC3, is involved in the SSC.
This study confirms previous findings that TRPC6 is involved in the SSC [16], and presents the novel finding that TRPC3 is an equivalent contributor to this process. The results obtained in this study using pharmacological interventions (Pyr3) in C57BL/6 J mice and genetic deletions in 129/Sv mice, suggest that TRPC3 and TRPC6 cooperate in the stretch response. There is growing evidence to suggest that TRPC3 and TRPC6 can form functional heterotetramers [38–41]. If cation influx through either TRPC3 or TRPC6 monomeric channels alone is insufficient to potentiate contraction, influx through TRPC3/6 heteromeric channels may provide another cation source. TRPC3 and TRPC6 may also function sequentially, as observed in endothelial cells in which TRPC6 activation is followed by TRPC5 activation [41]. In such cases, loss of either component would result in loss of the final output.
TRPC3 and TRPC6 function as receptor-operated channels modulated by GqPCRs, such as AT1R [14]. The sustained stretch has been reported to release Ang II, which activates AT1R in cardiomyocytes [42, 43]. The activation of AT1R may induce cation influx via TRPC3 and TRPC6 [14, 44]. However, other studies report that TRPC3 and TRPC6 are primarily activated by the stretch [45]. Our results demonstrate that application of Ang II, instead of stretch, reproduced the slow increase in the [Ca2+] transient (Fig. 3), and that TRPC3 and PLC inhibition significantly suppressed the increase in [Ca2+]i induced by both the sustained stretch and Ang II (Fig. 4). These experimental results indicate that the activation of TRPC3/6 to sustained stretch is mediated via the AT1R–Gq–PLC–DAG signaling pathway. Additionally, it has been reported that AT1R activation stimulates NADPH oxidase (NOX), the main source of ROS in the myocardium, to cause SSC [35]. Furthermore, NOX-derived ROS has been reported to regulate TRPC6 in vascular endothelial cells, glomerular mesangial cells, and podocytes [46–48]. Therefore, it remains possible that TRPC3 is regulated by two different AT1R pathways in SSC: (1) the AT1R–Gq–PLC–DAG–TRPC3 signaling pathway; or (2) the AT1R–NOX–ROS–TRPC3 signaling pathway. While our results support the regulation of TRPC3 by the AT1R–Gq–PLC–DAG–TRPC3 signaling pathway, they do not exclude ROS-dependent activation of TRPC3 in SSC. The relationship between these pathways needs to be studied further. Another possible regulatory mechanism for Ca2+ handling involved in SSC is inositol trisphosphate (IP3), which is also affected by the AT1R signaling pathway via ET-1 [42, 49]. In atrial myocytes, IP3 receptors activation enhances systolic and diastolic Ca2+ levels and increases spontaneous Ca2+ release [50]. However, IP3 receptors expression level in ventricular myocytes is much smaller (1/6) than in atrial myocytes [51], and ryanodine receptors are the dominant mechanism of Ca2+ release in ventricular myocytes [52]. Additionally, our mathematical modeling study demonstrates that IP3-dependent Ca2+ release, which is the Ca2+ release from the SR, may not affect SSC. Therefore, the contribution of IP3-dependent Ca2+ release for SSC might be negligible in ventricular myocytes.
Our mathematical modeling demonstrated that wet-experimentally observed SSC could be reproduced only by the extracellular cation influx (Na+ and Ca2+) (Fig. 6). This predicts that TRPC3 is located on sarcolemma and/or transverse tubules. Although our simulation with TRPC3 on the SR did not reproduce SSC, it does not mean that TRPC3 is not located on the SR. Indeed, several studies reported that TRPC3 is localized on the sarcolemma and transverse tubules in rodent cardiomyocytes [17, 19, 20], while other studies have reported that it is also located on the SR membrane in rabbit or mouse cardiomyocytes [17, 18]. Further studies reported that some types of stimuli induce the translocation of TRPCs from the sub-sarcolemmal space to the sarcolemma [53, 54]. Therefore, TRPCs have been observed in both sarcolemma and non-sarcolemmal areas, including the SR. These findings are consistent with our immunohistochemical results, in which the TRPC3 fluorescent signal was found on the sarcolemma and transverse tubules (Fig. 7b), and on non-sarcolemmal areas including the SR (Fig. 7c).
In our immunohistochemical study, the ratio of co-localization estimated by Manders’ coefficient for TRPC3 and NK-ATPase or SERCA2 are 17 and 45%, respectively. However, this does not necessarily mean that 17% of TRPC3 is located on sarcolemma and 45% is located on the SR. Overlapped confocal fluorescent signals does not always assure the co-localization of the target substances because the spatial resolution of a confocal microscope is limited (250 nm) [55]. Furthermore, even though our modeling study demonstrated that the sarcolemmal fraction of TRPC3 is responsible for causing SSC, it does not tell us whether the observed fraction (17%) is sufficient for causing SSC with an 8% stretch. To the best of our knowledge, there are no previous studies concerning dose-dependency between stretch and TRPC3 activity in cardiac muscle. Therefore, the parameters for the stretched conditions applied in the simulation were nominally set to cause SSC in our model. The quantitative relationship between stretch and TRPC3 activity may not be so simple because of other stretch-induced factors. For instance, the stretch-induced translocation of TRPC3 from the non-sarcolemmal area to the sarcolemma may affect its activity [56]. Despite the limitations mentioned, our combination of mathematical modeling and immunohistochemical studies reasonably suggests that a fraction of TRPC3 is located on the sarcolemma and/or transverse tubules, and that a stretch-induced cation (Na+ and Ca2+) influx via this TRPC3 fraction is required for the SSC.
Conclusion
Taken together, our results support the following model of the mechanisms involved in the SSC (Fig. 8). Stretch activates AT1R [57] and the downstream AT1R–Gq–PLC–DAG signaling cascade activates sarcolemmal TRPC3/6. This increases intracellular Ca2+ handling either by direct Ca2+ influx or by indirect effects of Na+ influx on NCX activity causing the accumulation of total intracellular Ca2+. Our mathematical modeling and immunohistochemical studies indicate that localization of TRPC3 on the sarcolemma causes the SSC in cardiomyocytes. In conclusion, this study shows that sarcolemmal TRPC3, regulated by AT1R, is involved in the SSC in cardiomyocytes.
Fig. 8.
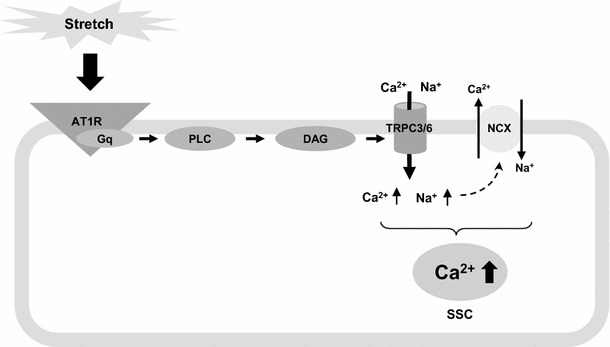
Proposed mechanism of the SSC. Stretch triggers AT1R activation of PLC via Gq to produce DAG. Increased DAG production activates TRPC3/6. The activation of sarcolemmal TRPC3/6 increases the intracellular cation (Na+ and Ca2+) influx. It contributes to intracellular Ca2+ accumulation via indirect effects of TRPC3/6-mediated Na+ influx on NCX activity, or via direct TRPC3/6-mediated Ca2+ influx. AT1R angiotensin II type 1 receptor, PLC phospholipase C, DAG diacylglycerol, NCX Na+/Ca2+ exchanger, TRPC3/6 transient receptor potential canonical subtype 3 and transient receptor potential canonical subtype 6
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
The authors would like to thank Ms. Keiko Kaihara (Department of Cardiovascular Physiology, Graduate School of Medicine, Dentistry and Pharmaceutical Sciences, Okayama University, Japan) for her skilled technical support, and Dr. Anastasia Khokhlova (Ural Federal University, Institute of Immunology and Physiology, Russia) for her support in improving the method of myocyte isolation. The authors also thank Ms. Yumiko Morishita (Central Research Laboratory, Okayama University Medical School, Japan) for technical assistance with the histological preparations. This study was funded by the Japan Society for the Promotion of Science (JSPS KAKENHI 23300167, 26282121 and 16K12878 to G.I.), by the Intramural Research Program of the NIH (Project Z01-ES-101864 to L.B.) and by the Life Science Foundation of Japan (as a travel grant to Y.Y.).
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
Animal protocols were approved by the Animal Subjects Committee of Okayama University Graduate School of Medicine, Dentistry, and Pharmaceutical Sciences. All experiments were conducted in accordance with the Guiding Principles for the Care and Use of Animals approved by the Council of the Physiological Society of Japan.
References
- 1.Parmley WW, Chuck L. Length-dependent changes in myocardial contractile state. Am J Physiol. 1973;224:1195–1199. doi: 10.1152/ajplegacy.1973.224.5.1195. [DOI] [PubMed] [Google Scholar]
- 2.Kobirumaki-Shimozawa F, Inoue T, Shintani SA, Oyama K, Terui T, Minamisawa S, Ishiwata S, Fukuda N. Cardiac thin filament regulation and the Frank–Starling mechanism. J Physiol Sci. 2014 doi: 10.1007/s12576-014-0314-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Allen DG, Kurihara S. The effects of muscle length on intracellular calcium transients in mammalian cardiac muscle. J Physiol. 1982 doi: 10.1113/jphysiol.1982.sp014221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hongo K, White E, Le Guennec JY, Orchard CH. Changes in [Ca2+]i, [Na+]i and Ca2+ current in isolated rat ventricular myocytes following an increase in cell length. J Physiol. 1996 doi: 10.1113/jphysiol.1996.sp021243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cingolani HE, Perez NG, Cingolani OH, Ennis IL. The Anrep effect: 100 years later. Am J Physiol Heart Circ Physiol. 2013 doi: 10.1152/ajpheart.00508.2012. [DOI] [PubMed] [Google Scholar]
- 6.Calaghan SC, White E. Contribution of angiotensin II, endothelin 1 and the endothelium to the slow inotropic response to stretch in ferret papillary muscle. Pflügers Arch. 2001 doi: 10.1007/s004240000458. [DOI] [PubMed] [Google Scholar]
- 7.Hunyady L, Turu G. The role of the AT1 angiotensin receptor in cardiac hypertrophy: angiotensin II receptor or stretch sensor? Trends Endocrinol Metab. 2004 doi: 10.1016/j.tem.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 8.Cingolani HE, Pérez NG, Aiello EA, de Hurtado MC. Intracellular signaling following myocardial stretch: an autocrine/paracrine loop. Regul Pept. 2005 doi: 10.1016/j.regpep.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 9.Kentish JC. A role for the sarcolemmal Na+/H+ exchanger in the slow force response to myocardial stretch. Circ Res. 1999 doi: 10.1161/01.RES.85.8.658. [DOI] [PubMed] [Google Scholar]
- 10.Ward ML, Williams IA, Chu Y, Cooper PJ, Ju YK, Allen DG. Stretch-activated channels in the heart: contributions to length-dependence and to cardiomyopathy. Prog Biophys Mol Biol. 2008 doi: 10.1016/j.pbiomolbio.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 11.Inoue R, Jian Z, Kawarabayashi Y. Mechanosensitive TRP channels in cardiovascular pathophysiology. Pharmacol Ther. 2009 doi: 10.1016/j.pharmthera.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 12.Shibasaki K. Physiological significance of TRPV2 as a mechanosensor, thermosensor and lipid sensor. J Physiol Sci. 2016 doi: 10.1007/s12576-016-0434-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bandyopadhyay BC, Swaim WD, Liu X, Redman RS, Patterson RL, Ambudkar IS. Apical localization of a functional TRPC3/TRPC6-Ca2+-signaling complex in polarized epithelial cells. Role in apical Ca2+ influx. J Biol Chem. 2005 doi: 10.1074/jbc.M410013200. [DOI] [PubMed] [Google Scholar]
- 14.Onohara N, Nishida M, Inoue R, Kobayashi H, Sumimoto H, Sato Y, Mori Y, Nagao T, Kurose H. TRPC3 and TRPC6 are essential for angiotensin II-induced cardiac hypertrophy. EMBO J. 2006 doi: 10.1038/sj.emboj.7601417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spassova MA, Hewavitharana T, Xu W, Soboloff J, Gill DL. A common mechanism underlies stretch activation and receptor activation of TRPC6 channels. Proc Natl Acad Sci USA. 2006 doi: 10.1073/pnas.0606894103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seo K, Rainer PP, Lee DI, Hao S, Bedja D, Birnbaumer L, Cingolani OH, Kass DA. Hyperactive adverse mechanical stress responses in dystrophic heart are coupled to transient receptor potential canonical 6 and blocked by cGMP-protein kinase G modulation. Circ Res. 2014 doi: 10.1161/CIRCRESAHA.114.302614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goel M, Zuo CD, Sinkins WG, Schilling WP. TRPC3 channels colocalize with Na+/Ca2+ exchanger and Na+ pump in axial component of transverse-axial tubular system of rat ventricle. Am J Physiol Heart Circ Physiol. 2006 doi: 10.1152/ajpheart.00785.2006. [DOI] [PubMed] [Google Scholar]
- 18.Hu Q, Thomas MG, Sachse FB. Localization and role of transient receptor potential cation channels in rabbit ventricular myocytes. Biophys J. 2014 doi: 10.1016/j.bpj.2013.11.4153. [DOI] [Google Scholar]
- 19.Fauconnier J, Lanner JT, Sultan A, Zhang SJ, Katz A, Bruton JD, Westerblad H. Insulin potentiates TRPC3-mediated cation currents in normal but not in insulin-resistant mouse cardiomyocytes. Cardiovasc Res. 2007 doi: 10.1016/j.cardiores.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 20.Kojima A, Kitagawa H, Omatsu-Kanbe M, Matsuura H, Nosaka S. Ca2+ paradox injury mediated through TRPC channels in mouse ventricular myocytes. Br J Pharmacol. 2010 doi: 10.1111/j.1476-5381.2010.00986.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O’Connell TD, Rodrigo MC, Simpson PC. Isolation and culture of adult mouse cardiac myocytes. Methods Mol Biol. 2007 doi: 10.1385/1-59745-214-9:271. [DOI] [PubMed] [Google Scholar]
- 22.Shioya T. A simple technique for isolating healthy heart cells from mouse models. J Physiol Sci. 2007 doi: 10.2170/physiolsci.RP010107. [DOI] [PubMed] [Google Scholar]
- 23.Iribe G, Kaneko T, Yamaguchi Y, Naruse K. Load dependency in force-length relations in isolated single cardiomyocytes. Prog Biophys Mol Biol. 2014 doi: 10.1016/j.pbiomolbio.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 24.Dietrich A, Mederos y Schnitzler M, Gollasch M, Gross V, Storch U, Dubrovska G, Obst M, Yildirim E, Salanova B, Kalwa H, Essin K, Pinkenburg O, Luft FC, Gudermann T, Birnbaumer L. Increased vascular smooth muscle contractility in TRPC6–/– mice. Mol Cell Biol. 2005 doi: 10.1128/MCB.25.16.6980-6989.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hartmann J, Dragicevic E, Adelsberger H, Henning HA, Sumser M, Abramowitz J, Blum R, Dietrich A, Freichel M, Flockerzi V, Birnbaumer L, Konnerth A. TRPC3 channels are required for synaptic transmission and motor coordination. Neuron. 2008 doi: 10.1016/j.neuron.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iribe G, Kohl P. Axial stretch enhances sarcoplasmic reticulum Ca2+ leak and cellular Ca2+ reuptake in guinea pig ventricular myocytes: experiments and models. Prog Biophys Mol Biol. 2008 doi: 10.1016/j.pbiomolbio.2008.02.012. [DOI] [PubMed] [Google Scholar]
- 27.von Lewinski D, Kockskämper J, Zhu D, Post H, Elgner A, Pieske B. Reduced stretch-induced force response in failing human myocardium caused by impaired Na+-contraction coupling. Circ Heart Fail. 2009 doi: 10.1161/CIRCHEARTFAILURE.108.794065. [DOI] [PubMed] [Google Scholar]
- 28.Watanabe A, Endoh M. Relationship between the increase in Ca2+ transient and contractile force induced by angiotensin II in aequorin-loaded rabbit ventricular myocardium. Cardiovasc Res. 1998 doi: 10.1016/S0008-6363(97)00287-3. [DOI] [PubMed] [Google Scholar]
- 29.Piao H, Takahashi K, Yamaguchi Y, Wang C, Liu K, Naruse K. Transient receptor potential melastatin-4 is involved in hypoxia-reoxygenation injury in the cardiomyocytes. PLoS One. 2015 doi: 10.1371/journal.pone.0121703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bon RS, Beech DJ. In pursuit of small molecule chemistry for calcium-permeable non-selective TRPC channels—mirage or pot of gold? Br J Pharmacol. 2013 doi: 10.1111/bph.12274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kiyonaka S, Kato K, Nishida M, Mio K, Numaga T, Sawaguchi Y, Yoshida T, Wakamori M, Mori E, Numata T, Ishii M, Takemoto H, Ojida A, Watanabe K, Uemura A, Kurose H, Morii T, Kobayashi T, Sato Y, Sato C, Hamachi I, Mori Y. Selective and direct inhibition of TRPC3 channels underlies biological activities of a pyrazole compound. Proc Natl Acad Sci USA. 2009 doi: 10.1073/pnas.0808793106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iribe G, Kaihara K, Ito H, Naruse K. Effect of azelnidipine and amlodipine on single cell mechanics in mouse cardiomyocytes. Eur J Pharmacol. 2013 doi: 10.1016/j.ejphar.2013.05.030. [DOI] [PubMed] [Google Scholar]
- 33.Antigny F, Jousset H, König S, Frieden M. Thapsigargin activates Ca2+ entry both by store-dependent, STIM1/Orai1-mediated, and store-independent, TRPC3/PLC/PKC-mediated pathways in human endothelial cells. Cell Calcium. 2011 doi: 10.1016/j.ceca.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 34.Caldiz CI, Garciarena CD, Dulce RA, Novaretto LP, Yeves AM, Ennis IL, Cingolani HE, Chiappe de Cingolani G, Pérez NG. Mitochondrial reactive oxygen species activate the slow force response to stretch in feline myocardium. J Physiol. 2007 doi: 10.1113/jphysiol.2007.141689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cingolani HE, Ennis IL, Aiello EA, Pérez NG. Role of autocrine/paracrine mechanisms in response to myocardial strain. Pflügers Arch. 2011 doi: 10.1007/s00424-011-0930-9. [DOI] [PubMed] [Google Scholar]
- 36.von Lewinski D, Stumme B, Maier LS, Luers C, Bers DM, Pieske B. Stretch-dependent slow force response in isolated rabbit myocardium is Na+ dependent. Cardiovasc Res. 2003 doi: 10.1016/S0008-6363(02)00830-1. [DOI] [PubMed] [Google Scholar]
- 37.Villa-Abrille MC, Caldiz CI, Ennis IL, Nolly MB, Casarini MJ, Chiappe de Cingolani GE, Cingolani HE, Pérez NG. The Anrep effect requires transactivation of the epidermal growth factor receptor. J Physiol. 2010 doi: 10.1113/jphysiol.2009.186619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dietrich A, Kalwa H, Rost BR, Gudermann T. The diacylgylcerol-sensitive TRPC3/6/7 subfamily of cation channels: functional characterization and physiological relevance. Pflügers Arch. 2005 doi: 10.1007/s00424-005-1460-0. [DOI] [PubMed] [Google Scholar]
- 39.Hofmann T, Schaefer M, Schultz G, Gudermann T. Subunit composition of mammalian transient receptor potential channels in living cells. Proc Natl Acad Sci USA. 2002 doi: 10.1073/pnas.102596199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hirschler-Laszkiewicz I, Tong Q, Conrad K, Zhang W, Flint WW, Barber AJ, Barber DL, Cheung JY, Miller BA. TRPC3 activation by erythropoietin is modulated by TRPC6. J Biol Chem. 2009 doi: 10.1074/jbc.M804734200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chaudhuri P, Colles SM, Bhat M, Van Wagoner DR, Birnbaumer L, Graham LM. Elucidation of a TRPC6-TRPC5 channel cascade that restricts endothelial cell movement. Mol Biol Cell. 2008 doi: 10.1091/mbc.E07-08-0765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alvarez BV, Pérez NG, Ennis IL, Camilión de Hurtado MC, Cingolani HE. Mechanisms underlying the increase in force and Ca(2+) transient that follow stretch of cardiac muscle : a possible explanation of the Anrep effect. Circ Res. 1999 doi: 10.1161/01.RES.85.8.716. [DOI] [PubMed] [Google Scholar]
- 43.Pérez NG, de Hurtado MC, Cingolani HE. Reverse mode of the Na+–Ca2+ exchange after myocardial stretch: underlying mechanism of the slow force response. Circ Res. 2001 doi: 10.1161/01.RES.88.4.376. [DOI] [PubMed] [Google Scholar]
- 44.Nishioka K, Nishida M, Ariyoshi M, Jian Z, Saiki S, Hirano M, Nakaya M, Sato Y, Kita S, Iwamoto T, Hirano K, Inoue R, Kurose H. Cilostazol suppresses angiotensin II-induced vasoconstriction via protein kinase A-mediated phosphorylation of the transient receptor potential canonical 6 channel. Arterioscler Thromb Vasc Biol. 2011 doi: 10.1161/ATVBAHA.110.221010. [DOI] [PubMed] [Google Scholar]
- 45.Quick K, Zhao J, Eijkelkamp N, Linley JE, Rugiero F, Cox JJ, Raouf R, Gringhuis M, Sexton JE, Abramowitz J, Taylor R, Forge A, Ashmore J, Kirkwood N, Kros CJ, Richardson GP, Freichel M, Flockerzi V, Birnbaumer L, Wood JN. TRPC3 and TRPC6 are essential for normal mechanotransduction in subsets of sensory neurons and cochlear hair cells. Open Biol. 2012 doi: 10.1098/rsob.120068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ding Y, Winters A, Ding M, Graham S, Akopova I, Muallem S, Wang Y, Hong JH, Gryczynski Z, Yang SH, Birnbaumer L, Ma R. Reactive oxygen species-mediated TRPC6 protein activation in vascular myocytes, a mechanism for vasoconstrictor-regulated vascular tone. J Biol Chem. 2011 doi: 10.1074/jbc.M111.248344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Anderson M, Roshanravan H, Khine J, Dryer SE. Angiotensin II activation of TRPC6 channels in rat podocytes requires generation of reactive oxygen species. J Cell Physiol. 2014 doi: 10.1002/jcp.24461. [DOI] [PubMed] [Google Scholar]
- 48.Graham S, Gorin Y, Abboud HE, Ding M, Lee DY, Shi H, Ding Y, Ma R. Abundance of TRPC6 protein in glomerular mesangial cells is decreased by ROS and PKC in diabetes. Am J Physiol Cell Physiol. 2011 doi: 10.1152/ajpcell.00014.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Signore S, Sorrentino A, Ferreira-Martins J, Kannappan R, Shafaie M, Del Ben F, Isobe K, Arranto C, Wybieralska E, Webster A, Sanada F, Ogórek B, Zheng H, Liu X, del Monte F, D’Alessandro DA, Wunimenghe O, Michler RE, Hosoda T, Goichberg P, Leri A, Kajstura J, Anversa P, Rota M. Inositol 1, 4, 5-trisphosphate receptors and human left ventricular myocytes. Circulation. 2013 doi: 10.1161/CIRCULATIONAHA.113.002764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li X, Zima AV, Sheikh F, Blatter LA, Chen J. Endothelin-1-induced arrhythmogenic Ca2+ signaling is abolished in atrial myocytes of inositol-1,4,5-trisphosphate(IP3)-receptor type 2-deficient mice. Circ Res. 2005 doi: 10.1161/01.RES.0000172556.05576.4c. [DOI] [PubMed] [Google Scholar]
- 51.Lipp P, Laine M, Tovey SC, Burrell KM, Berridge MJ, Li W, Bootman MD. Functional InsP3 receptors that may modulate excitation-contraction coupling in the heart. Curr Biol. 2000 doi: 10.1016/S0960-9822(00)00624-2. [DOI] [PubMed] [Google Scholar]
- 52.Kockskämper J, Zima AV, Roderick HL, Pieske B, Blatter LA, Bootman MD. Emerging roles of inositol 1,4,5-trisphosphate signaling in cardiac myocytes. J Mol Cell Cardiol. 2008 doi: 10.1016/j.yjmcc.2008.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rosenbaum MA, Chaudhuri P, Graham LM. Hypercholesterolemia inhibits re-endothelialization of arterial injuries by TRPC channel activation. J Vasc Surg. 2015 doi: 10.1016/j.jvs.2014.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Riazanski V, Gabdoulkhakova AG, Boynton LS, Eguchi RR, Deriy LV, Hogarth DK, Loaëc N, Oumata N, Galons H, Brown ME, Shevchenko P, Gallan AJ, Yoo SG, Naren AP, Villereal ML, Beacham DW, Bindokas VP, Birnbaumer L, Meijer L, Nelson DJ. TRPC6 channel translocation into phagosomal membrane augments phagosomal function. Proc Natl Acad Sci USA. 2015 doi: 10.1073/pnas.1518966112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schermelleh L, Heintzmann R, Leonhardt H. A guide to super-resolution fluorescence microscopy. J Cell Biol. 2010 doi: 10.1083/jcb.201002018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jemal I, Soriano S, Conte AL, Morenilla C, Gomis A. G protein-coupled receptor signalling potentiates the osmo-mechanical activation of TRPC5 channels. Pflügers Arch. 2014 doi: 10.1007/s00424-013-1392-z. [DOI] [PubMed] [Google Scholar]
- 57.Zou Y, Akazawa H, Qin Y, Sano M, Takano H, Minamino T, Makita N, Iwanaga K, Zhu W, Kudoh S, Toko H, Tamura K, Kihara M, Nagai T, Fukamizu A, Umemura S, Iiri T, Fujita T, Komuro I. Mechanical stress activates angiotensin II type 1 receptor without the involvement of angiotensin II. Nat Cell Biol. 2004 doi: 10.1038/ncb1137. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.