

Abstract
Cancer is one of the major causes of mortality and morbidity worldwide. Substantial research efforts have been made to develop new chemical entities with improved anticancer efficacy. 2-Aminobenzothiazole is an important class of heterocycles containing one sulfur and two nitrogen atoms, which is associated with a broad spectrum of medical and pharmacological activities, including antitumor, antibacterial, antimalarial, anti-inflammatory, and antiviral activities. In recent years, an extraordinary collection of potent and low-toxicity 2-aminobenzothiazole compounds have been discovered as new anticancer agents. Herein, we provide a comprehensive review of this class of compounds based on their activities against tumor-related proteins, including tyrosine kinases (CSF1R, EGFR, VEGFR-2, FAK, and MET), serine/threonine kinases (Aurora, CDK, CK, RAF, and DYRK2), PI3K kinase, BCL-XL, HSP90, mutant p53 protein, DNA topoisomerase, HDAC, NSD1, LSD1, FTO, mPGES-1, SCD, hCA IX/XII, and CXCR. In addition, the anticancer potentials of 2-aminobenzothiazole-derived chelators and metal complexes are also described here. Moreover, the design strategies, mechanism of actions, structure-activity relationships (SAR) and more advanced stages of pre-clinical development of 2-aminobenzothiazoles as new anticancer agents are extensively reviewed in this article. Finally, the examples that 2-aminobenzothiazoles showcase an advantage over other heterocyclic systems are also highlighted.
Keywords: 2-Aminobenzothiazole, Anticancer, Inhibitors, Kinase, Epigenetic enzymes, Drug design, Chelator, Metal complexes
1. Introduction
Cancer, characterized by the uncontrolled and aggressive proliferation of abnormal cells, is one of the deadliest diseases in the world. In 2020, 19.3 million people around the world suffered from cancer, and approximately 10.0 million died from this disease.[1] Unfortunately, the morbidity and mortality of cancer will continuously increase, and there will be an estimated 29-37 million new cancer cases by 2040.[2] Among various cancer treatments, chemotherapy continues to represent the most effective treatment of most cancers, and to date almost 332 anticancer drugs have been approved by U.S. Food and Drug Administration (FDA). Unfortunately, resistance often develops to the majority of these drugs.[3-5] Besides, most chemotherapy drugs are nonspecific and very toxic, and demonstrate a narrow therapeutic index.[3] Thus, the discovery and development of new, potent, and selective anticancer agents with low toxicity still represent an urgent medical need.
Nitrogen/sulfur-containing heterocycles are biologically important scaffolds, and they are widely present in a number of natural products and commercially available drugs.[6-8] As a crucial family of such heterocycles, 2-aminobenzothiazole has attracted vast attention due to its broad application as a privileged structure in medicinal chemistry and drug discovery research. Structurally, the 2-aminobenzothiazole moiety is comprised of a benzothiazole ring and an amino group. Benzothiazole is a heterocycle containing a benzene ring fused to the 4,5-positions of thiazole ring, which exerts a wide range of biological activities.[9-11] The amino group of 2-aminobenzothiazole is an active and useful functionality, which could be tethered to many structural fragments or form various fused heterocycles.[12] In addition, the 2-aminobenzothiazole core (exocyclic nitrogen, cyclic sulfur, and cyclic nitrogen) could provide suitable coordination site(s) for metals. Furthermore, 2-aminobenzothiazole acts as a bioisostere for aniline, 2-aminothiazole, 2-aminobenzimidazole, and other nitrogen- or oxygen-containing heterocycles. At the structural level, the 2-aminobenzothiazole fragment can be involved in formation of hydrogen bonds (as a hydrogen bond acceptor and/or donor), chalcogen bonds, as well as π-π stacking/van der Waals contacts with the specific amino acid residues on target proteins, which contribute to inhibitory activity.
The 2-aminobenthiazole scaffold has been extensively explored to construct the structurally diverse analogues with excellent biological activity against various biological targets. Significantly, several therapeutic agents containing this fragment have been approved for clinical application. For instance, riluzole (Fig. 1) is an important 2-aminobenzothiazole-based drug used for the treatment of amyotrophic lateral sclerosis, a lethal neurodegenerative disease.[13, 14] Multiple studies have shown that it also manifests promising antitumor effects on a panel of human solid cancer cell lines.[15-17] Frentizole (Fig. 1) is a nontoxic antiviral and immune suppressive agent used clinically in rheumatoid arthritis and systemic lupus erythematosus.[18] Tioxidazole (Fig. 1) is an anthelmintic drug utilized for curing parasitic infections.[19] The rational study on 2-aminobenzothiazoles as anticancer agents involves the investigation of the SAR and mechanisms of action of these compounds. Such study can provide insights into how their chemical structures can be modified to improve the potency and selectivity. To date, no comprehensive overview of 2-aminobenzothiazole compounds as new potential anticancer agents has been reported. To fill this gap, this review focuses on summarizing the recent developments (2015-2022) of 2-aminobenzothiazole derivatives as new antineoplastic agents based on their protein targets, including tyrosine kinases (CSF1R, EGFR, VEGFR-2, FAK, and MET), serine/threonine kinases (Aurora, CDK, CK, RAF, and DYRK2), PI3K kinase, BCL-XL, HSP90, mutant p53 protein, HDAC, NSD1, LSD1, FTO, DNA topoisomerases, mPGES-1, SCD, hCA IX/XII, and CXCR receptor. In addition, the anticancer potentials of 2-aminobenzothiazole-derived chelators and 2-aminobenzothiazole-metal complexes are also presented here. Moreover, the design strategies, mechanism of actions and SAR studies of these anticancer agents along with their preclinical development are described. Furthermore, the advantages of 2-aminobenzothiazoles over other heterocyclic systems are also highlighted. We hope this work will shed light on rational drug design and lead optimization to afford more potent 2-aminobenzothiazole-containing anticancer agents.
Fig. 1.

Representative examples of 2-aminobenzothiazole scaffold in commercial drugs.
2. 2-Aminobenzothiazole derivatives as anticancer agents
2.1. Inhibition of tyrosine kinases
Protein tyrosine kinases (PTKs) catalyze transfer of the γ-phosphate of ATP to hydroxyl groups of tyrosine residues on target proteins.[20] They are mainly divided into receptor tyrosine kinases (RTKs) and non-receptor tyrosine kinases. RTKs play an important role in fundamental cellular processes including cell proliferation, migration, metabolism, and cell cycle.[20-22] All RTKs share a similar protein structure which includes a ligand-binding extracellular domain, a single transmembrane helix, and a catalytic intracellular kinase domain.[21, 23] Dysregulation, aberrant activation, and mutations in the RTKs are linked to human cancers as well as resistance to anticancer therapies.
2.1.1. Inhibition of CSF1R kinase
The colony-stimulating factor 1 receptor (CSF1R or c-FMS) belongs to the type of class III RTK family that also contains stem-cell factor receptor (c-KIT), FMS-like tyrosine kinase-3 (FLT3), and platelet-derived growth factor receptor (PDGFR) α/β.[24, 25] Activation of CSF1R occurs upon binding one of its ligands, macrophage colony stimulating factor (CSF1) or IL-34, followed by transphosphorylation of its intracellular domain, subsequently stimulating autophosphorylation for signal transduction. Tumor-associated macrophages (TAMs), as the critical regulatory immune cells, could promote tumor progression, resulting in poor prognosis of a variety of cancers. There are two phenotypes of TAMs, M1 and M2, which have antitumor and protumor functions, respectively. Inhibiting the polarization or survival of M2 macrophages through blocking CSF-1R signal transduction has emerged as a promising strategy for cancer immunotherapy.[24, 26]
Compound 1 (BLZ945, Fig. 2) developed by Novartis was a potent small-molecule CSF1R inhibitor (IC50 = 1 nM) with a >3200-fold selectivity for CSF1R over other related RTK kinases (e.g., cKIT IC50 = 3.2 μM and FLT3 IC50= 9.1 μM).[27] Compound 1 was able to reduce M2 macrophage polarization and effectively block tumor progression and improve survival. Currently, 1 is undergoing first-in-human phase I/II clinical trials as a single agent or in combination with a monoclonal antibody PDR001 for treatment of advanced solid tumors (NCT02829723). Compound 2 (Fig. 2), an active isomer of 1, was generated by CYP2C8 and CYP3A4 mediated-oxidation followed by aldo–keto cytosolic reductases catalyzed-reduction.[28] It also displayed potent inhibitory activity against CSF1R (IC50 = 5.5 nM) and excellent selectivity profile over the PDGFRβ kinase (IC50 = 13 μM).
Fig. 2.

Chemical structures of 2-aminobenzothiazole-based CSF1R inhibitors 1–3.
2-Aminobenzothiazole 3 (Fig. 2) was found to exhibit highly potent suppressive activity against CSF1R kinase (IC50 = 1.4 nM) and had an acceptable selectivity profile when tested against a panel of 468 kinases.[29] It also demonstrated suitable in vivo PK profiles across species and had good oral bioavailability. Treatment of PANC02 tumors with 3 decreased tumor macrophages and CSF1R protein levels to a similar extent as those treated with BLZ945. In an MC38 xenograft model, 3 reduced tumor growth by 62% at a subcutaneous dose of 200 mg/kg.[29]
The 4-methoxylated analogue 4 (Fig. 3) maintained single-digit nanomolar inhibitory activity toward CSF1R (IC50 = 4.0 nM) and manifested a remarkable enhancement in selectivity for related type III kinases (Kd = 30 μM), but it demonstrated unfavorable metabolic turnover profiles (e.g., rat Clint = 98 mL/min/kg).[29] The presence of F or Cl atom at the C7 position of 2-aminobenzothiazole scaffold in compound 4 was tolerated for potency but impaired the selectivity for CSF1R, and replacing the methoxyl group with a trifluoromethoxy motif resulted in a drop in CSF1R inhibition activity. Decreasing the cLogP by substitution of 2-amino-3-chloropyridine with 2-aminopyrimidine in the hinge region resulted in enhanced microsomal stability, as seen with compound 5 (IC50 = 21 nM, Kd = 6.7 nM, and rat Clint = 24 mL/min/kg). Reversing the stereochemistry of the amino and hydroxyl groups on the cyclohexyl ring delivered compound 6 with increased potency while maintaining excellent selectivity (IC50 = 7.0 nM and Kd = 4.1 nM), as illustrated in Fig. 3. Introducing a Cl atom at the C7 position of benzothiazole core led to further improvement in potency, with compound 7 having IC50 values of 2 nM and 60 nM against CSF1R kinase and MSNF60 cells, respectively. Encouragingly, both compounds 6 and 7 possessed suitable in vivo PK properties with reasonable half-life and excellent oral bioavailability in rats and mice, but only moderate oral bioavailability in dogs.[29] To further boost the potency of this class of inhibitors, the 2-aminopyrimidine fragment was then functionalized through N-arylation, instead of N-acylation (amide was hydrolytically unstable).[29] Among them, the N-methylpyrazole-containing compound 8 (IACS-9439) stood out as the most promising CSF1R inhibitor (IC50 = 1.7 nM and Kd = 1.0 nM) and elicited outstanding antiproliferative activity against MSNF60 cells with an IC50 value of 7 nM (Fig. 3). It not only showed improved potency in the pCSF1R cellular target engagement assay relative to BLZ945 but also displayed superior selectivity across RTK kinases (cKIT Kd = 17000 nM; FLT3 Kd = 9500 nM; PDGFRα Kd = 1900 nM; PDGFRβ Kd = 450 nM).[29] Notably, chlorination of 8 gave rise to analogue 9, which maintained the low nanomolar CSF1R suppressive activity (IC50 = 1.2 nM) but showed higher clearance in rat and human microsomes. Given its excellent potency and selectivity profile, 8 was further evaluated. It was found that 8 possessed desirable in vivo PK profiles and superior oral bioavailability across several species, especially rat and mouse (F = 81 and 100%, respectively). Moreover, 8 could significantly restrain the migration of macrophages and reprogram M2-like macrophages to the M1 phenotype. Concomitantly, subcutaneous administration of 8 remarkably attenuated the MC38 murine colon tumor growth and reversed the immunosuppressive tumor microenvironment with the increased M1/M2 ratio, highlighting 8 as a promising candidate for cancer immunotherapy. [29]
Fig. 3.

Medicinal chemistry strategy for the discovery of CSF1R inhibitor 8 (IACS-9439).
2.1.2. Suppression of EGFR kinase
EGFR (HER1/ErbB1), a transmembrane glycoprotein, belongs to the tyrosine kinase family that also consists of ErbB2 (HER2/neu), ErbB3 (HER3), and ErbB4 (HER4).[30] It acts a key mediator in activating multiple cellular signaling pathways involved in cell survival and proliferation, migration, angiogenesis, and cell death.[30] Upregulation or mutation of EGFR can aberrantly trigger EGFR-dependent pathways, resulting in various types of solid malignancies, such as non-small-cell lung cancer (NSCLC), prostate, breast, stomach, ovarian, and cervical cancer.[30, 31] Thus, targeting EGFR has been extensively pursued, with the development of a number of promising inhibitors utilized in clinical oncology.
Mokhtar and coworkers developed a new set of EGFR inhibitors using a structure-based design strategy.[32] Among them, 2-aminobenzothiazoles 10 and 11 exerted robust inhibitory activity against EGFR kinase with IC50 values of 94.7 and 54.0 nM, respectively (Fig. 4). However, these two compounds only displayed moderate suppressive activity towards HeLa, HCT-116, MCF-7, HepG2, and PC-3 cell lines. It was found that replacement of 2,5-dimethoxylphenyl group in compound 11 with 2-thienyl, 5-bromo-2-thienyl and 9-anthracenyl moieties dramatically compromised the antiproliferative potency. In a similar study, Allam et al. reported 2-aminobenzothiazole congener 12 as a potent EGFR inhibitor (IC50 = 96 nM) (Fig. 4).[33] Introducing a nitro or ethoxyl group at the C6 position of 2-aminobenzothiazole scaffold in compound 12 decreased the EGFR inhibition activity. In contrast to compounds 10 and 11, analogue 12 showed significantly improved antiproliferative activity against MCF-7 cells (IC50 = 2.49 ± 0.12 μM). Especially, compound 12 was active against PC9 and HCC827 cell lines harboring mutant EGFR (IC50 values of 1.05 ± 0.02 μM and 3.43 ± 0.066 μM, respectively), but exerted minimal cytotoxic activity towards the normal WI38 fibroblast cells (IC50 = 82.8 ± 4.14 μM). Flow cytometry analysis suggested 12 could induce apoptosis and G2/M phase arrest. Computational analysis of 12 revealed that the nitrogen atom within the pyridyl ring forms a hydrogen bond with residue Met793 and the benzothiazole ring is deeply inserted into the hydrophobic pocket of EGFR kinase. The amino group at C2 position of benzothiazole core was involved in hydrogen bonding interactions with the residue Asp855.[33]
Fig. 4.

Chemical structures of 2-aminobenzothiazole derivatives 10–18 as EGFR inhibitors.
In 2020, Sever et al. synthesized a new series of 2-aminobenzothiazole derivatives and investigated their antitumor potential.[34] In this series, compound 13 (Fig. 4) emerged as the most potent antiproliferative agent, exhibiting IC50 values of 6.43 ± 0.72, 9.62 ± 1.14 and 8.07 ± 1.36 μM against HCT116, A549, and A375 cell lines, respectively. It also exerted the least cytotoxicity against normal cell line PBMCs (IC50 > 300 μM), indicative of superior cellular selectivity. Meanwhile, compound 13 effectively blocked enzymatic activity of EGFR (IC50 = 2.80 μM). In this study, they found that replacement of the 2-aminobenzothiazole moiety with a 2-aminothiazole motif severely impaired the antiproliferative activity, supporting the crucial role of 2-aminobenzothiazole scaffold in improving the antiproliferative potency. In addition, the rank order of cytotoxicity for the substitution on the benzothiazole scaffold was found to be OEt > H > Me > NO2.
In the same year, Abdellatif et al. developed a new class of 2-aminobenzothiazole compounds as potential antitumor agents.[35] Compounds 14-18 (Fig. 4) manifested the most pronounced cell growth inhibition in PC3, MCF-7, A549, HCT-116, and MDA-MB-231 cell lines, having IC50 values ranging from 0.315 to 2.66 μM. These analogues also potently blocked EGFR activity with IC50 values in the range of 0.173-1.08 μM, which could shed light on its good cellular potency.
2.1.3. Blockade of VEGFR-2 kinase
Angiogenesis is the process of new blood vessel growth, which plays a crucial role in physiological conditions, such as embryonic development, pregnancy, and menstruation.[36] Nevertheless, deregulated angiogenesis is closely related with several pathologies including cancer, since it is pivotal to the rapid proliferation and migration of tumor cells.[37] VEGFR-2 (also known as FLK1), a type III transmembrane tyrosine kinase receptor, is the major signal transducer for angiogenesis.[36, 38] Hence, inhibition of angiogenesis via blockade of VEGFR-2 kinase activity has emerged as an effective approach for anticancer therapy.
Based upon a structure-based drug design strategy, Bhanushali et al. identified 2-aminobenzothiazole derivative 19 as a potent VEGFR-2 inhibitor with an IC50 value of 0.5 μM (Fig. 5).[39] Compound 19 also exhibited good inhibitory effect on chick chorioallantoic membrane and vasculogenic vessel formation. The SAR studies suggested that substituting the benzothiazole moiety with other aryl motifs (e.g., phenyl, pyridyl, thiazoyl or oxazoyl group) significantly compromises the anti-angiogenic activity and introducing a methyl group on the benzothiazole scaffold results in the reduced activity. The molecular docking study revealed that the benzothiazole ring of 19 penetrates the hydrophobic cavity formed by the DFG loop of the VEGFR-2 kinase, and the amide NH and benzothiazole nitrogen are engaged in the hydrogen bonding interactions with the side chain carbonyl of Glu883 and the backbone NH of Asp1044, respectively.[39]
Fig. 5.

Chemical structures of 2-aminobenzothiazole derivatives 19–23 as VEGFR-2 inhibitors.
Molecular hybridization as an important drug design strategy involves the rational design of new chemical entities by covalent fusion of two or more existing pharmacophores.[40-42] Thiazolidinedione (TZD) and cyanothiouracil (CT) fragments have been widely utilized in the design of novel anticancer drugs. Hence, El-Helby et al. incorporated the TZD or CT moiety with classic 2-aminobenzothiazole scaffold for the design of new VEGFR-2 inhibitors.[43] All synthesized hybrid molecules were investigated for their antiproliferative activities using HepG2, HCT-116 and MCF-7 cell lines. For the 2-aminobenzothiazole-TZD series, compound 20 (Fig. 5) demonstrated the strongest inhibitory activity against these three tumor cell lines with IC50 values of 9.99, 7.44 and 8.27 μM, respectively. The SAR results revealed that introduction of a substituent on the phenyl ring significantly enhances the cytotoxic activity, while shifting the substituent from C4 position to C2 position leads to a remarkable decline in activity. With regard to the 2-aminobenzothiazole-CT series, it was found that the methyl group on the phenyl ring was optimal for the antiproliferative potency and the corresponding compound 21 (Fig. 5) suppressed the proliferation of tumor cells with IC50 values ranging from 10.34 to 12.14 μM. It should be noted that compounds 20 and 21 potently inhibited VEGFR-2 kinase with IC50 values of 0.15 and 0.19 μM respectively, which were comparable to the clinical drug sorafenib.[43]
To further investigate the SAR of 2-aminobenzothiazole-TZD hybrids targeting angiogenesis, an essential hallmark of cancer, Upadhyay et al. installed diverse substituents on the benzothiazole scaffold and replaced the phenyl group with a pyridyl motif at the TZD terminal portion.[44] All hybrid compounds were screened to determine their effects on proliferation of human umbilical vein endothelial cells (HUVEC), and the results suggested that compounds with substituted phenyl group generally display more potent HUVEC inhibitory activity than those with the 2-pyridyl moiety. Further studies revealed that compound 22 (Fig. 5) with chlorophenyl and 6-methylbenzothiazole motifs shows the most potent inhibition of VEGFR-2 (IC50 = 0.6 μM), which could effectively reduce the cellular migration and capillary-like tube formation of HUVECs and block the formation of new capillaries on the growing chick chorioallantoic membranes.[44]
In an attempt to explore new VEGFR inhibitors as potential anticancer agents, twenty 2-aminobenzothiazole derivatives were designed and synthesized by Reddy et al.[45] Amongst them, compound 23 stood out as the most potent VEGFR-2 inhibitor (IC50 = 97 nM, Fig. 5). In agreement with its good enzymatic inhibition, 23 displayed excellent antiproliferative activity towards all tested cancer cell lines (HT-29, PC-3, A549, and U87MG) but marginal cytotoxicity against normal cells (HEK-293T). In a transgenic zebrafish model, 23 could dose-dependently inhibit the formation of intersegmental vessels.[45] Interestingly, the presence of electron-withdrawing groups (R1 and R2) at the C6 position of benzothiazole skeleton and phenyl ring was found to be beneficial for the antiproliferative potency.
2.1.4. Inhibition of FAK kinase
Focal adhesion kinase (FAK), also known as PTK2, is a non-receptor tyrosine kinase that primarily transduces signaling from cell adhesion to multiple biological cellular functions, including cell survival, proliferation, migration, and embryonic development.[46] FAK is a 125kD protein that is comprised of three domains (N-terminal FERM domain, central catalytic kinase domain, and C-terminal domain).[46, 47] FAK overexpression has been found in many solid tumors, in which FAK kinase is also implicated in promoting metastasis. Therefore, it is considered as a promising target for the development of novel anticancer drugs.
The Altintop group designed and synthesized a set of 2-aminobenzothiazoles containing 1,3,4-oxadiazole moiety and investigated their anticancer activity against C6 rat glioma and A549 human lung adenocarcinoma cell lines.[48] The most active compound 24 (Fig. 6) exhibited appreciable antiproliferative effects on these two cell lines, with IC50 values of 4.63 ± 0.85 and 39.33 ± 4.04 μM, respectively. The cytotoxicity data revealed that changing the 6-methoxyl group on the 2-aminobenzothiazole nucleus in compound 24 with a Cl atom substantially reduces the inhibitory potency. Compound 24 could also promote apoptosis, trigger mitochondrial membrane depolarization, and induce caspase-3 activation in both cell lines. Moreover, 24 effectively blocked the enzymatic activity of FAK with an IC50 value of 19.5 ± 2.12 μM. In silico molecular docking study indicated that 24 could form π-π stacking with Phe568 residue and present a salt bridge and π-cation with Lys457 residue in the active site of FAK.[48] Overall, these studies suggested that 24 represents a new class of ATP-competitive FAK inhibitors.
Fig. 6.

Chemical structure of 2-aminobenzothiazole derivative 24 as an FAK inhibitor.
2.1.5. Inhibition of MET kinase
MET, also termed as hepatocyte growth factor receptor (HGFR), encoded by MET proto-oncogene is a member of the receptor tyrosine kinase family.[49] Hepatocyte growth factor (HGF), also known as scatter factor (SF), is the high affinity and naturally occurring ligand of MET.[49] Activation of the MET/HGF signaling pathway results in multiple cellular responses including proliferation and survival, scattering, angiogenesis, motility, and invasion.[49, 50] The physiological function of the MET/HGF signaling pathway is restricted to embryonic development, wound healing, and tissue regeneration processes.[49, 50] Overexpression of MET and/or aberrant activation of this signaling pathway occurs in various solid tumors. In addition, this abnormal signaling is often associated with tumor aggressiveness, metastasis, and poor prognosis, and is involved in the development of therapeutic resistance, remarkably decreasing survival rates.[49, 51]
The Gong group designed and synthesized a new subset of 2-aminobenzothiazole derivatives bearing a 4-phenoxyquinoline moiety, with the goal of developing potent c-MET inhibitors. The cytotoxicity of these compounds against HT-29, MKN-450 and H460 cell lines was assessed.[52] All tested compounds demonstrated higher cytotoxic activity against MKN-450 and H460 cell lines as compared to HT-29 cell line. Among them, compound 25 was found to be the most potent molecule with IC50 values of 0.06 ± 0.01, 0.01 ± 0.003, and 0.18 ± 0.02 μM against MKN-45, H460 and HT-29 cells, respectively (Fig. 7). Also, 25 exhibited the strongest inhibitory activity against c-MET kinase (IC50 = 17.6 ± 1.17 nM) and manifested superior kinase selectivity. Notably, this compound displayed very weak potency against EGFR (IC50 > 20 μM) and moderate VEGFR-2 inhibition activity (IC50 = 3.36 μM), indicating superior kinase selectivity. In this study, the authors also found that the presence of 4,7-diCH3 (R1) group on the 2-aminobenzothiazole core was favorable for antiproliferative activity, and the potency trend for R1 substituent was 4,7-diCH3 > H > 5-Br > 4-OCF3 > 6-Cl > 4-F,6-Br. Additionally, it appeared that the R2 substituent had no significant influence on the antiproliferative potency.[52]
Fig. 7.

Chemical structures of 2-aminobenzothiazole derivatives 25–31 as MET inhibitors.
Using the promising anticancer agent 26 (SC745689)[53] as a template, Moosavi and coworkers developed a novel series of 2-aminobenzothiazoles containing 3,4-dihydropyrimidin-2(1H)-one motif as potential MET inhibitors using a scaffold hopping strategy (Fig. 7).[54] The SAR studies showed that substitution of benzothiazole scaffold with other aromatic rings (e.g., pyridine and benzene) significantly decreases the activity, indicating the importance of benzothiazole for antiproliferative activity. In addition, the introduction of 5,6-diMe or 6-EtO group (R1) on the 2-aminobenzothiazole nucleus was conducive to improving the cytotoxic activity. As for R2 group, the isopropyl was deleterious to the cytotoxicity. In this series, compounds 27-31 (Fig. 7) could block the proliferation of lung EBC-1 and gastric MKN-45 cancer cell lines harboring MET gene amplification, with IC50 values in the range of 14.6–18.8 μM and 15.0–18.7 μM, respectively. The homogenous time resolved fluorescence assay revealed that compounds 27, 29 and 30 suppress MET kinase at a concentration of 100 μM by 55.8%, 52.4% and 54.9%, respectively. Molecular docking studies confirmed the interactions of compound 27 with the active site of the MET kinase enzyme, such as two hydrogen bonding interactions with Asp1164 and Ile1084, and a π-π interaction with Tyr1230.[54] However, further improvement in MET inhibition activity for compound 27 is warranted.
In another study by Sanofi,[55] benzimidazole analogue 32 was claimed as a potent MET inhibitor with an IC50 value of 10 nM (Fig. 8). Nonetheless, this compound exhibited affinity toward tubulin (IC50 = 2 μM). Optimization of 32 through bioisosteric replacement afforded a highly selective and potent compound 33, which did not bind to tubulin (IC50 > 25 μM). In comparison to 32, benzothiazole analogue 33 was more lipophilic and demonstrated decreased aqueous solubility. To tune the physicochemical properties, 33 was further modified by substituting the carbamate unit with a urea moiety and introducing a variety of hydrophilic nitrogen-containing functionalities at the C2 position of the benzothiazole core. This decoration culminated in the discovery of compound 34 (SAR125844, Fig. 8), a development candidate with an IC50 value of 4 nM against MET tyrosine kinase. It also demonstrated very potent inhibitory activity against MET mutants (METL1195V IC50 = 64 nM; METM1250T IC50 = 6 nM; METY1230H IC50 = 204 nM) and robust antiproliferative activity against MKN45 cells (IC50 = 7 nM). Co-complex structural analysis indicated that 34 tightly interacts with MET Y1230H mutant residues Met1160, Lys1161 and Asp1222 through several hydrogen bonding interactions. Significantly, 34 exerted favorable druggability properties for intravenous administration and promoted sustained target engagement in a MET-amplified tumor model.[55] The evidence of anticancer efficacy fostered the evaluation of 34 in a phase II clinical study for NSCLC patients with MET amplification (NCT02435121).
Fig. 8.

Identification of 2-aminobenzothiazole derivative 34 (SAR125844) as a MET inhibitor. Crystal structure of 34 with the METY1230H (PDB code 5HO6).
2.2. Inhibition of serine/threonine kinases
Human serine/threonine kinases (STKs) are enzymes that modulate protein activity by phosphorylation of serine and threonine amino acid residues. STKs are involved in multiple signal transduction pathways controlling cell metabolism, cell division, and angiogenesis. Deregulated activation of these protein kinases has fueled tumor initiation and progression.[56] As such, the development of effective inhibitors against certain STKs has been regarded as an alternative approach for oncology therapeutics.
2.2.1. Inhibition of Aurora kinase
Aurora kinases (Aurora A, B and C), members of serine/threonine kinase family of enzymes, are critical regulators of cell cycle and mitosis.[57] They are highly overexpressed in various human cancer cell lines.[57] Deregulation of Aurora kinase activity causes mitotic abnormalities, genomic instability and ultimately tumorigenesis. Consequently, considerable efforts have been devoted to developing inhibitors of Aurora kinases.
Through bioisosteric replacement of 2-aminobenzoxazoles previously identified Aurora B kinase inhibitors, the Jeon group developed a new class of Aurora B inhibitors with a 2-aminobenzothiazole scaffold (Fig. 9).[58] These 2-aminobenzothiazoles exhibited significantly improved inhibitory activity and selectivity for Aurora B kinase as compared to the corresponding 2-aminobenzoxazole analogues. The SAR results are summarized as follows: 1. The para-substitution on the phenyl ring was beneficial for the Aurora B inhibition activity; 2. Replacing the urea group with amide or sulfonamide moiety compromised the inhibitory activity, while the thiourea motif was tolerated; 3. Substituting the morpholinyl fragment with dimethylamino or piperidinyl group decreased the potency. The representative compounds 35 and 36 can effectively inhibit the enzymatic activity of Aurora B with IC50 values of 0.09 and 0.12 μM, respectively. Molecular modeling study showed that the 2-aminobenzothiazole nucleus of 36 occupies the adenine-binding region of Aurora B kinase and interacts with Ala157 and Glu155 in the hinge backbone via two hydrogen bonds. Additional critical hydrogen bonding interaction was observed between the urea carbonyl oxygen and the catalytic Lys106. Moreover, the oxygen C6 alkoxyl portion that pointed toward solvent-exposed region formed a water-mediated hydrogen bond with Glu161.[58] A mechanistic study revealed that both compounds 35 and 36 could reduce the histone H3S10 phosphorylation levels and induce G2/M cell cycle arrest in HeLa cells.[58] Hence, 35 and 36 could be considered as promising lead compounds for further investigations.
Fig. 9.

Chemical structures of 2-aminobenzothiazole derivatives 35 and 36 as Aurora kinase B inhibitors.
2.2.2. Inhibition of CDK kinase
The cyclin-dependent protein kinases (CDKs) are serine/threonine protein kinases that belong to the CMGC family (CDKs, mitogen-activated protein kinases, glycogen synthase kinases, and CDK-like kinases).[59, 60] CDKs are involved not only in the cell cycle but also in the other critical cellular processes, such as gene transcription, insulin secretion, glycogen synthesis and neuronal functions.[61, 62] In view of the function of CDKs, a large number of CDK inhibitors with diverse structural features have been developed in the past few years, including 2-aminobenzothiazole class compounds.
In 2019, the Zhao group incorporated the aminopyridine motif into the 2-aminobenzothiazole scaffold for development of a new class of CDK2 kinase inhibitors as potential anticancer agents (Fig. 10).[63] Analogues 37 and 38 potently inhibited the CDK2 kinase, with IC50 values of 37.8 and 21.7 nM, respectively. Both compounds manifested submicromolar or single-digit micromolar activity against HeLa and HCT116 cell lines, but were not active or less effective in PC-3 and MDA-MB-231 cells. To further improve the anticancer potency, the authors introduced various substituents on the benzothiazole core and pyrimidine ring on the basis of the structure of compounds 37 and 38. The optimized compound 39 (Fig. 10) displayed significant enhancement in cellular potency relative to compounds 37 and 38, having IC50 values of 0.45, 0.70, 0.92, and 1.80 μM against these four tumor cell lines, respectively. Coherently, 39 elicited robust CDK2 inhibitory activity (IC50 = 15.4 nM), which was approximately 3-fold more potent than the positive control AZD-5438 (Fig.10). In HeLa cells, 39 could block the cell cycle progression at G2/M phase and induce apoptosis in a concentration-dependent manner. The SAR analysis suggested that the electron-withdrawing groups (e.g., methylsulfonyl and sulfamoyl groups) are preferred for the antiproliferative activity, whereas the electron-donating substituents are detrimental to the inhibitory potency. Besides, replacing the methyl group on the pyrimidine ring with a F atom dramatically compromised the cytotoxicity.[63]
Fig. 10.

Chemical structures of AZD-5438 and 2-aminobenzothiazole derivatives 37–41 as CDK2 kinase inhibitors.
In a similar study, Abdelazeem et al. devised another series of 2-aminobenzothiazole-derived CDK2 inhibitors, in which the 1,2,4-triazole moiety was fused with a benzothiazole core.[64] Compound 40 (Fig. 10) demonstrated low micromolar inhibitory activity against CDK2 (IC50 = 4.29 μM). 40 also exhibited potent cell-killing activity against A549, MCF-7 and Hep3B tumor cell lines with IC50 values of 3.55, 3.17 and 4.32 μM, respectively. In addition, 40 could efficiently trigger apoptosis through the activation of caspase-3/7 in A549 cells. It is essential to emphasize that replacement of 2-aminobenzothiazole fragment in 40 with other nitrogen-containing heterocycles led to a great loss of cytotoxic activity. [64]
Compound 41 (Fig. 10), a 2-aminobenzothiazole derivative, remarkably suppressed tumor cell proliferation and decreased tumor burden in vivo.[65] The mechanism studies revealed that 41 induces G2/M cell cycle arrest in leukemic cell lines (Nalm6, K562, REH, and Molt4) and breast cancer cell lines (MCF-7 and EAC), which is associated with the elevated levels of ROS and DNA double strand breaks. Moreover, 41 could cause the pronounced reduction in phosphorylation level of CDK1 and upregulate the expression of CyclinB1 protein (a G2/M phase specific marker) in Nalm6 cells. To probe the possibility of 41 targeting cyclin dependent kinases involved in cell cycle regulation, docking studies of 41 with CDK1 and CDK2 kinases were performed. The results indicated that 41 can bind to the catalytic site of CDK1 and CDK2 with binding energies of −10.36 and −9.02 kcal/mol, respectively.[65] Briefly, when binding to CDK1, the thiourea of 41 made two hydrogen bonds with residue Ile10, and 2-aminotetrahydrobenzothiazole of 41 formed another two hydrogen bonds with residue Leu83 and Glu81. When binding to CDK2, 41 formed three hydrogen bonds with residue Asp145 and Leu83. Notably, both binding patterns revealed the involvement of amino acid residue Leu83 in hydrogen bonding interactions which were crucial to the potency of CDK1 and CDK2 inhibitors. Thus, inactivation of CDK1 and CDK2 kinases could be the potential mechanism of compound 41 behind the observed G2/M cell cycle arrest, in addition to ROS generation and DNA breaks.[65]
2.2.3. Inhibition of CK kinase
Casein kinase 1 (CK1) is a highly conserved and ubiquitously expressed serine/threonine kinase in mammals, participating in the phosphorylation of a broad range of proteins. Six isoforms (α, γ1, γ2, γ3, δ, and ε) exist in humans and their various alternative splice variants have been characterized in the CK1 family that plays crucial regulatory roles in numerous biological processes including cell proliferation, cell apoptosis, DNA repair, inflammation, circadian rhythm as well as signaling pathways (e.g., Wnt/β-catenin signaling and hedgehog pathways).[66, 67] Since deregulation of CK1 expression and activity is linked to tumor progression, there is intensive interest to develop CK1-specific inhibitors as oncology tools.
Compound 42 (IWP-2, Fig. 11) exhibited potent inhibitory activity against wtCK1δ, wtCK1δKD (C-terminal truncated form of wtCK1δ) and wtCK1ε with IC50 values of 0.93, 0.32, and 4.03 μM, respectively.[68] Of note, compared to wild-type CK1δ, 42 demonstrated even stronger inhibitory effect on the gatekeeper mutant M82FCK1δ (IC50 = 0.04 μM). Analysis of the crystal structure of compound 42/CK1δ complex revealed that 2-aminobenzothiazole fragment fits snugly into the hydrophobic pocket I, with residue Leu85 forming the key hydrogen bonding interactions, and the thieno-pyrimidone moiety occupies the solvent-exposed hydrophobic region II (Fig. 11). Consistent with potent M82FCK1δ suppression, compound 42 strongly inhibited the proliferation of eight tumor cell lines within the single-digit micromolar range.[68]
Fig. 11.

Chemical structures of 2-aminobenzothiazole derivatives 42-45 as CK kinase inhibitors. Crystal structure of 42 with the CK1δ (PDB code 5OKT).
Compound 43 bearing a trifluoromethyl group (Fig. 11) exerted increased potency against wtCK1δ and wtCK1ε kinases (IC50 values of 0.09 and 0.56 μM, respectively) and showed high kinase selectivity.[68] Compound 44 (Fig. 11), an analogue of 43, strongly inhibited enzymatic activity of wtCK1δ (IC50 = 0.09 μM).[69] It also exhibited a significant repressive effect on cell viability of HCT-116, HT-29, SW480 and SW620 colon cell lines, having the greatest potency against SW620 cells derived from primary tumor and metastasis of the same patient (IC50 = 3.0 μM).
Casein kinase 2 (CK2) is highly expressed in cancer and has also been considered a potential therapeutic target.[70, 71] More recently, Wang et al. reported the design and synthesis of a series of 2-aminobenzothiazoles containing a chromone moiety as novel CK2 inhibitors.[72] Among these synthesized derivatives, compound 45 (Fig. 11) represented the most active CK2 inhibitor (IC50 = 0.08 μM), showing the strongest cell growth inhibition in HL-60 cancer cells (IC50 = 0.25 μM). Treatment of HL-60 cells with compound 45 resulted in the stabilization of CK2, implying that 45 is able to bind to CK2 in cells. Also, 45 was found to inhibit the proliferation of HL-60 cells by inducing apoptosis and G0/G1 cell cycle arrest in a dose- and time-dependent fashion. Moreover, 45 remarkably suppressed the expressions of p-α-catenin, p-AKT (S129), p-p21, Survivin and p-STAT3, and enhanced the cleavage of PARP level.[72] This work paved the way for further development of 2-aminobenzothiazole compounds as potent CK2 inhibitors.
2.2.4. Inhibition of RAF kinase
The RAF family of protein kinases (ARAF, BRAF, and CRAF) are critical effectors of the RAS/RAF/MEK/ERK pathway that regulates cell growth, proliferation, and survival.[73, 74] This pathway is activated by oncogenic mutations in many types of cancer. BRAF is one of the proteins frequently mutated to an active form during tumor development. Among various BRAF mutations, V600E (BRAFV600E) emerges as the most active mutant.[73, 75, 76] Its in vitro kinase activity is almost 10.7-fold greater than that of wild-type BRAF.[75] Similar to BRAF, CRAF (RAF-1) kinase is implicated in the abnormal proliferation of melanoma.[77] CRAF is also overexpressed in various solid cancers. Hence, discovery of BRAFV600E/CRAF inhibitors has received a great deal of attention. Intriguingly, a distinct feature for recently reported inhibitors is the common occurrence of a 2-aminobenzothiazole motif.
Compound 46 (TAK-632, Fig. 12) showed robust BRAFV600E inhibition activity (IC50 = 2.4 nM), good BRAF selectivity, and desirable PK profiles.[78] The cocrystal structure of 46 with BRAF kinase revealed favorable H-bonding and sulfur-carbonyl interactions. It should be noted that, in contrast to urea group, the methylene of acetamide could provide more flexibility to fit into the back pocket of BRAF. Significantly, 46 exhibited dose-dependent antitumor efficacy in human melanoma A375 (BRAFV600E) xenograft model with no signs of body weight loss.[78] Moreover, 46 could efficiently delay the HMVII (NRASQ61K/BRAFG469V) tumor growth and restore body weight loss associated with HMVII tumor-induced cachexia. Collectively, these results implied that 46 is a selective pan-RAF kinase inhibitor with excellent anticancer efficacy and could serve as a clinic candidate for the treatment of human cancer harboring either BRAFV600E or NRAS mutation.[78]
Fig. 12.

Chemical structures of 2-aminobenzothiazole derivatives 46-48 as RAF inhibitors. The crystal structure of 46 (TAK632) with BRAF (PDB code 4KSP).
Based upon the structure of the approved RAF inhibitor sorafenib, El-Damasy et al. developed a new series of 2-aminobenzothiazole congeners as dual BRAFV600E and CRAF inhibitors through isosteric replacement of the central phenyl ring with a benzothiazole scaffold.[79] Among them, compound 47 was found to be the most potent inhibitor with IC50 values of 0.095 μM and 0.015 μM against BRAFV600 and CRAF, respectively (Fig. 12). The cellular-based assay indicated that another interesting compound 48 (BRAFV600E IC50 = 0.131 μM and CRAF IC50 = 0.111 μM) manifests the best antiproliferative activity, surpassing sorafenib over 57 human cancer cell lines. The SAR studies indicated that the presence of Cl atom at the para-position of phenyl ring is favorable for RAF inhibition activity and compounds with di-substitutions are more cytotoxic than those with mono-substitution (Fig. 12). Further studies suggested that compounds 47 and 48 demonstrate minimized hERG and CYP450 inhibition, indicative of their desirable safety profiles.[79] Accordingly, these two compounds could be considered as promising leads for further studies.
2.2.5. Inhibition of DYRK2 kinase
Dual-specificity tyrosine-phosphorylation-regulated kinases (DYRKs) belong to the CMGC family of protein kinases and are pleiotropic regulators of cellular functions such as cell survival, cell differentiation and gene transcription.[80, 81] In humans, the DYRKs are comprised of five members: DYRK1A, DYRK1B, DYRK2, DYRK3, and DYRK4.[80, 81] Among them, DYRK2 is the most extensively studied as a potential target for cancer treatment. However, most small-molecule DYRK2 inhibitors lack selectivity over DYRK family members and exhibit poor drug-like properties.
To develop more potent and selective DYRK2 inhibitors, the Yang group performed a high-throughput screening (HTS) and identified the anti-DYRK2 hit 49 (IC50 = 263 nM and Kd = 4.21 μM),[81] as shown in Fig.13. Chemical optimization of 49 led to discovery of 2-aminobenzothiazole derivative 50 (YK-2-69) that manifested approximately 29-fold and 46-fold improvement in DYRK2 inhibitory potency and binding affinity (IC50 = 9 nM and Kd = 92 nM, respectively). Of particular note, changing the 2-aminobenzothiazole scaffold in compound 50 with pyrazolo[1,5-a]pyrimidine moiety abrogated the DYRK2 inhibition activity. Compound 50 also displayed exquisite selectivity over the DYRK subfamily and a panel of 370 kinases. Impressively, 50 demonstrated favorable safety properties with the maximum tolerated dose (MTD) of > 10,000 mg/kg and desirable pharmacokinetic profiles (e.g., F = 56%). In addition, 50 exerted more potent suppressive effect on tumor growth than first-line drugs enzalutamide and palbociclib in DU145 and PC-3 xenograft mouse models.[81] The mechanism studies suggested that 50 exhibits superior anti-prostate cancer efficacy via synergistic regulation on multiple pathways (e.g., DYRK2-4E-BP1 and DYRK2-RRS1-p21/27-CDK4/6) to promote apoptosis and block proliferation.[81] Collectively, the highly potent and selective DYRK2 inhibitor 50 with favorable druggability might be used as a valuable chemical probe for further biological studies and also considered as a potential candidate for treatment of prostate cancer.
Fig. 13.

Discovery of 2-aminobenzothiazole derivative 50 (YK-2-69) as an exquisitely potent and selective DYRK2 inhibitor.
2.3. Inhibition of PI3K kinases
Phosphoinositide 3-kinases (PI3Ks) are a family of lipid kinases that can catalyze the phosphorylation of the inositol ring of phosphoinositide and are secondary messengers which help to transduce signals.[82] PI3Ks are key regulators of the PI3K/AKT/mTOR (mammalian target of rapamycin) signaling pathway that is essential to control cell survival, growth, motility, and metabolism.[83] Deregulated activation of PI3Ks has been implicated in tumor angiogenesis, tumorigenesis, and conferring resistance to antineoplastic drugs.[84] PI3Ks can be divided into three classes (class I, II, and III) according to their structural differences and substrate specificity. Of these, the most extensively investigated members in cancer development are the class I PI3Ks (class IA: PI3Kα, PI3Kβ, and PI3Kδ; class IB: PI3Kγ) that are directly activated by cell surface receptors.[84, 85] In the past several years, a number of PI3K inhibitors with different structure types have been developed, including the privileged scaffold 2-aminobenzothiazole.
PIK-93 (Fig. 14) has been reported to exhibit potent inhibitory activity against PI4Ks and PI3Ks, especially for class Ib member PI3Kγ (IC50 = 16 nM and Ki = 7 nM).[86] Taking PIK-93 as a starting point, Collier et al. presented a structure-guided development of potent and isoform-selective 2-aminobenzothiazole-based PI3Kγ inhibitors.[87] The initial lead optimization efforts were focused on C6 position of benzothiazole scaffold derived from ring fusion of the phenylthiazole core. The representative compound 51 with 4,5-dimethoxypyridine showed the highest binding affinity toward PI3Kγ (Ki = 1 nM). This remarkable enhancement in potency was rationalized by several favorable hydrogen bonding interactions with the ATP binding site of PI3Kγ (Fig. 14). Subsequent substitution of the acetamide moiety with a chain-extended urea fragment resulted in compound 52 (Fig. 14) which demonstrated considerably improved selectivity for PI3Kγ over PI3Kα/δ while maintaining good PI3Kγ potency (Ki = 2 nM). Intriguingly, the PI3Kγ selectivity order for N-alkyl substituent was propyl > ethyl > methyl > H and extending the urea chain length gave rise to the improved PI3Kγ selectivity. Also, 52 suppressed MCP-1 induced chemotaxis of THP-1 cells and the proliferation of MCF-7 cells with IC50 values of 0.083 and 7.4 μM, respectively.[88] Notably, in the structure of 52-PI3Kγ complex, hydrogen bond between 5-methoxy substituent and residue Lys833 was not observed. In addition, the propyl group was found to occupy a newly formed binding cleft adjacent to the ATP binding site.[87]
Fig. 14.

Chemical structures of 2-aminobenzothiazole derivatives 51–54 as PI3K inhibitors. X-ray crystal structure of 51 and 52 bound to PI3Kγ (PDB code 4PS8 and 4PS3, respectively).
In a similar work, Gao et al. designed and synthesized a novel series of 2-aminobenzothiazole analogues as potential PI3Kβ inhibitors.[89] The typical compounds 53 exerted potent activity against PI3Kβ (IC50 = 0.02 μM), with excellent selectivity over other class I PI3K subunits and mTOR (Fig. 14). In agreement with its PI3Kβ inhibitory potency, 53 strongly repressed the growth of prostate PC-3 and DU145 cancer cells, with IC50 values of 0.35 and 0.62 μM, respectively. Additionally, it showed low cytotoxicity against the normal fibroblast MRC-5 cells (IC50 = 33.11 μM), indicating good cellular selectivity.
Very recently, the Yar group reported a set of new 2-aminobenzothiazole derivatives as new anticancer agents targeting PI3Kα kinase.[90] Compound 54 was characterized as the most potent PI3Kα inhibitor (IC50 = 1.03 nM, Fig. 14). 54 also exerted significant growth-inhibitory activity against MCF-7 cells. Moreover, it effectively suppressed the migration of MCF-7 cells and induced cell cycle arrest. Furthermore, 54 dose-dependently triggered apoptosis by decreasing the levels of antiapoptotic proteins BCL-XL and MCL-1.
2.4. Inhibition of topoisomerase
DNA topoisomerases are nuclear enzymes which make transient DNA strand breaks, allowing the cell to manipulate the topology of its DNA. They are essential for DNA replication, transcription, chromosomal segregation, and DNA recombination.[91] Topoisomerases are classified as type I and II in terms of their reaction mechanism, amino acid sequence, and structure. The type I enzymes cleave only one strand of duplex DNA, whereas the type II enzymes cut both strands.[92] The essential role of these enzymes in cell processes and their elevated level in solid tumors make topoisomerase inhibition as an important mechanism for cancer therapy.[93, 94] Current topoisomerase-targeted agents are limited by some side effects (e.g., dose-limiting toxicity, drug resistance, and cardiotoxicity), despite their efficiency in the clinic. To improve the potency and decrease adverse effects, new topoisomerase inhibitors have been developed in the past few years, including 2-aminobenzothiazole-based inhibitors of topoisomerases.
By incorporating the β-naphthol fragment and 2-aminobenzothiazole moiety, Nagaraju and colleagues designed and synthesized a library of 2-aminobenzothiazole derivatives as potential topoisomerase I inhibitors (Fig. 15).[95] Among them, compounds 55, 56 and 57 could efficiently bind to DNA and showed comparable topoisomerase I inhibition activity to camptothecin. Also, these compounds displayed strong cell growth inhibition towards HeLa cells with IC50 values of 5.20, 5.54 and 4.63 μM, respectively. The cytotoxicity data suggested that replacement of the methoxyl group at C6 position of 2-aminobenzothiazole scaffold with a Cl atom abolishes the activity, however, the methyl group is tolerated. In addition, installation of the electron-withdrawing group (R) on the phenyl ring significantly enhanced the antiproliferative activity.[95]
Fig. 15.

Chemical structures of 2-aminobenzothiazole derivatives 55–63 as topoisomerase inhibitors.
Sović et al. developed a set of new 2-aminobenzothiazole derivatives as potential topoisomerase inhibitors (Fig. 15).[96] Among them, compounds 58 and 59 demonstrated the highest cellular potency (IC50 values ranging from 0.15 to 1.0 μM), and compounds 60 and 61 exhibited the best DNA intercalation activity. Further topoisomerase assay suggested that compound 60, which exerts the highest topoisomerase I inhibitory activity, is more potent than campthotecin. Especially, both compounds 58 and 60 manifested notable topoisomerase II inhibitory properties, which were comparable to etoposide.
To explore new topoisomerase IIα inhibitors as promising anticancer agents, a total of thirty 2-aminobenzothiazoles bearing β-carboline moiety were rationally designed and synthesized by Tokala et al.[97] All derivatives showed cytotoxic potential against a panel of human tumor cell lines, wherein compounds 62 and 63 (Fig. 15) manifested the strongest inhibitory potency towards A549 cells (IC50 = 1.81 and 1.46 μM, respectively) and limited cytotoxic activity against human normal lung epithelial cells BEAS-2B (IC50 > 58 μM). In addition, these two compounds demonstrated potent intercalative topoisomerase-II inhibitory effect by blocking the conversion of catenated kDNA into decatenated DNA. The SAR studies revealed that introducing the methyl group (R1) on the benzothiazole ring decreases the cytotoxicity, and the substituent (R2) in the order of improved cytotoxicity is NO2 > Cl > H > Me. Additionally, changing the phenyl group with 2-thiophenyl moiety reduced the cytotoxic potency.
2.5. Inhibition of BCL-XL
Apoptosis, or programmed cell death, is a highly conserved process that is critical for cellular homeostasis and development. The proteins of B cell lymphoma protein 2 (BCL-2) family are the fundamental and key regulators of the intrinsic apoptosis pathway, which consists of anti-apoptotic (pro-survival) proteins (e.g., BCL-2, BCL-XL, MCL-1, BFL-1 and BCL-w), pro-apoptotic BH3-only proteins (e.g., BID, BIM, PUMA, NOXA, BAD, and HRK), and pro-apoptotic effectors (e.g., BAK and BAX).[98, 99] BCL-XL is frequently overexpressed in solid tumors and hematological malignancies, and its overexpression or activation is linked to poor prognosis and drug resistance.[98-100]
Tao et al at AbbVie took advantage of NMR fragment screening and structure-based design strategies, delivering compound 64 (A-1155463, Fig. 16) which demonstrated picomolar binding affinity to BCL-XL (Ki < 0.01 nM) and > 1000-fold selectivity over other BCL-2 family members (e.g., BCL-2 Ki = 80 nM).[101] The cocrystal structure revealed that the 2-aminobenzothiazole moiety of 64 interacts with Leu108 and Ser106 in the hydrophobic P2 pocket via essential hydrogen bonds. In addition, 2-fluoro atom in 64 was proximal to the side chains of Val141 and Phe97 within the hydrophobic P4 pocket and made favorable van der Waals contacts. Administration of 64 to tumor bearing SCID-Beige mice resulted in modest tumor growth inhibition, possibly due to its poor solubility and oral absorption. It should be noted that 64 represented a first reported selective BCL-XL inhibitor showing in vivo efficacy.[101, 102]
Fig. 16.

Structure-guided development of potent BCL-XL inhibitor 67 (A-1331852). X-ray crystal structure of 64 bound to BCL-XL (PDB code 4QVX).
To develop 2-aminobenzothiazole-based selective BCL-XL inhibitors with suitable oral exposure profiles, researchers at AbbVie selected pyridine-containing compound 65 (Ki = 164 nM) as a starting template for further SAR exploration.[103] Considering that nitrogen-containing heterocycles with fine-tuning physicochemical properties and decreasing the number of rotatable bonds could impart better drug-like properties such as oral absorption, they next introduced rigid 5-methylpyrazole linker to the pyridine ring of 65 instead of a conformationally flexible propyloxy unit. The resulting compound 66 showed substantial improvement in target affinity (Ki = 0.027 nM), low micromolar activity in MOLT-4 cells in the presence of 10% human serum (EC50 = 1610 nM), and a suitable level of oral absorption (F = 17%).[103] Further optimization of 66 through increasing the pharmacophore sp3-fraction and occupying lipophilic volume of the P4 pocket culminated in the identification of compound 67 (A-1331852, Ki < 0.01 nM, Fig. 16) which exhibited over 250-fold increase in antiproliferative activity against MOLT-4 cells (EC50 = 6.3 nM) and maintained high selectivity for BCL-2-dependent RS4;11 cells (EC50 > 5000 nM).[103] Compound 67 demonstrated modest bioavailability (F = 11-13%). Further in vivo studies demonstrated that 67 has the potential to augment the efficacy of docetaxel and irinotecan in xenograft solid tumor models and reduce neutropenia caused by inhibition of BCL-XL.[102, 103]
Very recently, the AbbVie group reported a novel series of BCL-XL inhibitors based upon structure-based design and molecular hybridization strategies (Fig. 17).[104] The promising compound 68 was featured by integrating the pharmacophores of selective BCL-XL inhibitor 64 and ABT-737, while another benchmark molecule 69 (A-1293102) contained the same structural elements of 64 and ABT-263 (navitoclax, Fig. 17). Both compounds displayed picomolar Ki value against BCL-XL (Ki values of 0.14 and 0.43 nM, respectively) and high selectivity over BCL-2 and MCL-1. Interestingly, 69 exhibited strong cellular potency against BCL-XL dependent MOLT-4 cells (EC50 = 80 nM), whereas 68 did not show any cell-killing activity in the same line (EC50 > 5000 nM). This can be explained by faster on- and off-rate binding kinetics and shorter residence time of 68 as compared to 69 (Kon 2.18 × 107 M−1S−1 vs 5.88 × 105 M−1S−1; Koff 1.09 × 10−2 S−1 vs 2.34 × 10−3 S-1; t1/2 1 min vs 49 min). Further investigation into the binding mode revealed that the benzothiazole moiety of 69 binds to the hydrophobic P2 pocket through two key hydrogen bonds as described previously, likely explaining the observed selectivity. The carbonyl oxygen and nitrogen atom of the central thiazole moiety interacted with respective residue Asn136 and Arg139 through hydrogen bonds.[104] Another two hydrogen bond interactions were observed between one of the sulfonyl oxygen atoms with residues Asn136 and Gly138. Taken together, the highly potent and selective BCL-XL inhibitor 69 could represent an invaluable tool compound for further biological studies.
Fig. 17.

Chemical structures of 2-aminobenzothiazole derivatives 68 and 69 as BCL-XL inhibitors. X-ray crystal structure of 69 bound to BCL-XL (PDB code 7LH7).
Proteolysis Targeting Chimeras (PROTACs) are an emerging modality with the potential to modulate protein targets which are challenging to target using traditional small molecules. PROTACs are bifunctional molecules wherein one ligand for a protein of interest (POI) is linked to another ligand for an E3 ligase such as von Hippel–Lindau (VHL) or cereblon (CRBN), leading to target poly-ubiquitination and proteasomal degradation of POI.[105-108] PROTACs have many advantages over small molecule inhibitors, such as the potential of overcoming drug resistance and low-dose toxicity as a result of catalytic properties.
To develop BCL-XL degrader, researchers at GlaxoSmithKline chose a well-established BCL-XL inhibitor 64 (A1155463, Fig. 16) which well occupied the bind site as a BCL-XL-targeting ligand.[109] The co-crystal structure of BCL-XL with 64 revealed a solvent-exposed vector through the propargylic amine, providing a potential tethering site for the design of degrader (Fig. 18). For this reason, degrader 70 was developed by conjugation of 64 to the VHL E3 ligase binder. 70 selectively degraded BCL-XL in THP-1 cells (DC50 = 4.8 nM, Dmax = 76%), which was equipotent to clinical candidate DT2216.[109]
Fig. 18.

Chemical structure of BCL-XL degrader 70. X-ray crystal structure of 64 (A1155463) bound to BCL-XL (PDB code 4QVX).
2.6. Inhibition of HSP90
Heat shock protein 90 (HSP90) is a molecular chaperone that plays an important role in cellular proteostasis and is responsible for the folding and maturation of client proteins including protein kinases (AKT, CDK4/6, RAF1, HER2, and SRC), transcription factors (p53, and Hif1) and telomerase, all of which are closely associated with ten hallmarks of cancer.[110, 111] This supports HSP90 as an attractive target for the discovery of novel anticancer agents. The HSP90 polypeptide is a homodimer, and each monomer contains an ATP-hydrolyzing N-terminal domain, middle domain, and C-terminal domain.[112] So far at least 18 HSP90 N-terminal inhibitors have entered clinical trials, yet none of them have been successful, due to the strong activation of the heat shock response (HSR) that upregulates the HSP90 protein level and ultimately results in cytoprotective effects. Therefore, recent focus of HSP90 research centered on the design of inhibitors has shifted towards C-terminal domain and protein–protein interactions (e.g., HSP90-CDC37).
The Blagg group reported a series of HSP90 C-terminal inhibitors derived from a 2-aminobenzothiazole-based scaffold.[113] Several compounds were found to exert potent antiproliferative activity against both MCF-7 and SKBr3 breast cancer cell lines. Among them, the most active compound 71 (Fig. 19) displayed an EC50 value of 0.86 and 7.03 μM against SKBr3 and MCF-7 cells, respectively. Besides, 71 could remarkably downregulate the expression of Hsp90 client proteins (e.g., AKT, CDK4, HER2 and RAF-1), a hallmark of HSP90 C-terminal inhibition, and did not cause HSR effect. The SAR trends of this class compounds can be summarized as follows: 1. Replacement of the ethyl oxalyl group with 2-oxo-2-phenylacetamide or acetamide moiety totally abrogated the cytotoxic activity; 2. Changing the indole ring with pyrimidine or phenyl ring reduced the inhibitory potency; 3. The substituent at the C5 position of indole ring in the order of improved antiproliferative potency was OMe > Me > Et; 4. Shifting the methoxyl group from C5 position to C6 position led to the reduced activity.
Fig. 19.

Chemicals structures of 2-aminobenzothiazole derivatives 71–73 as HSP90 C-terminal inhibitors and analogue 74 as a HSP90-CDC37 inhibitor.
Using a ligand-based design approach and structure-based pharmacophore model, the Tomašič group devised and synthesized a new library of 2-aminobenzothiazole derivatives as HSP90 C-terminal inhibitors.[114] Initial SAR elaboration of phenyl group revealed that the activity order for C4 substituent is I > Br > Cl > F > H > OMe and replacement of phenyl group with benzyl moiety significantly diminishes the antiproliferative activity. Subsequent SAR exploration of benzothiazole core indicated that 4-aminopiperidine fragment is optimal for activity. The representative compounds 72 and 73 (Fig. 19) exhibited the highest cellular potency with EC50 values of 2.8 ± 0.1 and 3.9 ± 0.1 μM, respectively. Mechanistically, compound 73 demonstrated dose-dependent downregulation of HSP90 client proteins (AKT, c-RAF, and Erα) without induction of HSR, which was consistent with the Blagg’s study[113].
Jin et al. rationally designed and synthesized a set of 2-aminobenzothiazole-18β-glycyrrhetinic acid hybrids targeting the HSP90-CDC37 interaction.[115] Most compounds showed low micromolar disruption activity against HSP90-CDC37, with compound 74 having the highest potency (IC50 = 0.14 ± 0.03 μM, Fig. 19). Structure and activity survey suggested that the F atom at C6 position of benzothiazole ring is optimal for the potency and altering the F atom from C6 position to C4 position substantially decreases the activity. Consistent with the strong HSP90-CDC37 inhibition, hybrid molecule 74 potently evaded the growth of A549 tumor cells (IC50 = 4.04 ± 0.66 μM). Also, 74 was capable of reversing multidrug-resistance in NCI-H460/DOX lung cells (resistant factor = 1.36). Moreover, 74 induced dose-dependent degradation of HSP90 client proteins, implying that the anticancer activity of 74 is, at least in part, ascribed to HSP90 inhibition. However, it should be noted that both HSP70 and HSP20 were upregulated upon treatment of tumor cells with 74. Hence, how to minimize the HSR toxicity during the optimization might be a concern for this class of compounds.
2.7. Reactivation of mutant p53
p53 is a tumor suppressor protein that acts crucial roles in cell cycle arrest, DNA damage response, apoptosis, and senescence.[116, 117] In human cancers, p53 is the most commonly mutated protein with more than half of cancers carrying a mutation in p53 (e.g., p53Y220C and p53R175H). Mutant p53 abolishes the tumor-suppressive function and manifests a dominant-negative effect on remaining wild-type p53, thereby blocking its anticancer property.[118, 119] Additionally, mutant p53 may interact with diverse transcriptional factors or co-factors to induce tumor-promoting responses such as inflammation and metabolic reprogramming. Thus, reactivation of mutant p53 to restore its wild-type function represents a promising strategy for the development of novel anticancer therapeutics.
In 2018, the Fersht group discovered a novel class of small-molecule Y220C stabilizers employing a structure-guided design strategy (Fig. 20).[120] A biophysical screen of a halogen-enriched fragment library identified a 2-iodophenol-containing compound 75 that bound to the Y220C pocket (Kd = 820 μM). The computational modeling studies revealed that the iodine atom at C3 position of phenyl ring forms a halogen bond with Leu145, and the hydroxyl group at C2 position of phenyl ring creates two hydrogen bonds with a water molecule bridging residues Val147 and Asp228. The carboxylate at C1 pointed toward solvent exposed region (subsite 1). Hydrophobic contacts were also observed between the iodine atom at C5 position and the hydrophobic channel towards subsite 2. Optimization of hit 75 targeting the subsite 3 (central pocket) and subsite 2 resulted in compound 76 with approximately 58-fold improved activity (Kd = 14 μM). The SAR studies suggested that substituting the carboxylic group with amide, hydroxamic acid, dihydroindazolone or dimethylamino motif dramatically reduces the potency. In addition, the flexible thiother linker was optimal. Furthermore, introduction of pyrrole functionality on the phenyl ring resulted in a significant increase in activity.
Fig. 20.

Discovery of 2-aminobenzothiazole 78 (MB725) as a mutant p53 reactivator.
To boost the hydrophobic contacts and reduce unfavorable entropy through impeding the C-S bond rotation, they devised and synthesized compound 77 (Kd = 4 μM). This new chemical probe stabilized p53-Y220C in vitro and exhibited 3.5-fold increase in affinity relative to 76. However, its potent binding affinity did not translate into cellular activity as compound 77 showed relatively low cytotoxicity against all cell lines tested, possibly because of its low cell permeability.[120] Further amidation of 77 resulted in compound 78 (MB725, Fig. 20), which induced selective viability reduction in several tumor cell lines with a p53-Y220C mutation while being well tolerated in control cell lines, and upregulated transcription of specific p53 target genes responsible for apoptosis and cell cycle arrest in a p53-Y220C dependent fashion.[120] These data implied that 2-aminobenzothiazole derivative 78 could restore the p53-Y220C transcriptional activity, paving the way for development of potent and non-toxic Y220C-mutant p53 reactivators in antineoplastic therapy.
Given the limitations of thiosemicarbazones (toxicity and poor solubility), the Carpizo group sought to exploit an alternative ZMC scaffold as potential clinical lead candidates.[121] Taking ZMC1 as a lead, they replaced the thiosemicarbazone moiety with benzothiazolyl hydrazone, resulting in compound 79 (KZn = 119 nM), as shown in Fig. 21. It effectively killed ovarian carcinoma cells TOV-112D (R175H mutant) with an EC50 value of 0.055 μM but was ineffective for p53 wild type and p53 null cell lines, suggesting excellent allele cellular selectivity. Unfortunately, 79 also suffered from low aqueous solubility. Optimization of 79 through adding a solubilizing polyether or alkylamine fragment led to the respective compound 80 and 81, which displayed the improved zinc binding affinity over 79 (KZn = 22 nM for compound 80 and KZn = 55 nM for compound 81). Nevertheless, alkylamine 81 strongly inhibited the potassium channel hERG that was correlated with serious cardiovascular risks. Further elaboration of 81 by removal of the lipophilic base (dimethylamine) resulted in compounds 82 and 83 devoid of hERG liabilities (Fig. 21), maintaining potent zinc-binding affinity (KZn = 22 and 79 nM, respectively). Unexpectedly, these two compounds were unable to initiate mutant p53 refolding.[121] Further in vivo study revealed that 79 is well tolerated up to 30 mg/kg without significant weight loss, whereas the MTD of ZMC1 is only 5 mg/kg. 79 also significantly suppressed tumor growth by 72% at 5.73 mg/kg. Hence, this work validated the general design principle that replacing the thiocarbamoyl group with a benzothiazole moiety could attenuate toxicity and retain anticancer efficacy, and meanwhile provide a framework for the development of more potent zinc-binding and p53 refolding agents.[121]
Fig. 21.

Development of 2-aminobenzothiazole derivatives as mutant p53 reactivators.
2.8. Inhibition of HDAC
Histone deacetylases (HDACs), a class of epigenetic enzymes, regulate the expression and activity of numerous proteins involved in cancer initiation and progression. By removal of acetyl groups from histones, HDACs create a non-permissive chromatin conformation which prevents the transcription of genes that encode proteins involved in tumorigenesis.[122, 123] There are 18 human HDACs divided into four classes: class I (HDAC1, 2, 3 and 8), class II (IIa: HDAC4, 5, 7, 9; IIb: HDAC6, 10) and class IV (HDAC11) that are Zn2+-dependent, while class III (Sirtuin1-7) is NAD+-dependent.[124, 125] The overexpression and aberrant recruitment of HDACs (especially class I and II) are positively correlated with tumorigenesis and tumor development. Inhibition of HDACs has been demonstrated to trigger apoptosis, cell differentiation, and cell cycle arrest.
Vorinostat (SAHA) is first approved HDAC inhibitor for the treatment of several types of lymphoma. Riluzole is a drug used to treat amyotrophic lateral sclerosis, the antitumor potency of which has been well documented.[16, 17] Taking advantage of the molecular hybridization strategy, Xu et al. devised and discovered 2-aminobenzothiazole-SAHA conjugate 84 as a potential anticancer agent targeting HDAC (Fig. 22).[126] In comparison with SAHA, this hybrid molecule displayed 2-fold and 7-fold more potent total HDAC and HDAC6 isoform inhibitory activity with IC50 values of 0.12 ± 0.01 μM and 0.012 ± 0.002 μM, respectively. In addition, 84 demonstrated 27.5-fold and 275-fold selectivity for HDAC6 over HDAC2 and HDAC8, respectively. Significantly, in an MDA-MB-231 xenograft model, 84 demonstrated better in vivo efficacy than SAHA (tumor growth inhibition (TGI): 59% vs 33%) at a dose of 30 mg/kg once daily without noticeable toxicity.
Fig. 22.

Chemical structures of 2-aminobenzothiazole derivatives 84–88 as HDAC inhibitors.
In another study, a series of 2-aminotetrahydrobenzothiazole-SAHA hybrids were designed and synthesized.[127] Of these, compound 85 (Fig. 22) was proved to be the most potent inhibitor with IC50 values of 1.4, 12.1, 5.6, and 4.6 nM against class I HDAC1, HDAC2, HDAC3 and class IIb HDAC6, respectively. Meanwhile, 85 was capable of increasing the expression of acetylated histone H3 and acetylated α-tubulin in a dose-dependent fashion. Docking simulations on 85 revealed that the hydroxamate moiety forms favorable bidentate interactions with the Zn2+ ion of HDAC6 and it interacts with Asp460 and His574 via two hydrogen bonds. In line with its strong anti-HDACs activity, 85 demonstrated potent antiproliferative activity against all 6 tested cancer cell lines with IC50 values in the single digit micromolar range, which was more active or comparable relative to SAHA. Notably, minimal cytotoxicity was observed in normal cell lines treated with 85. In addition, 85 demonstrated good in vivo efficacy in an A549 zebrafish xenograft model.[127]
In search of non-hydroxamate HDAC inhibitors, the Ramaa group used the same fragment assembly strategy and identified benzothiazole-thiazolidinedione hybrid 86 (Fig. 22) as a selective HDAC8 inhibitor (HDAC8 IC50 = 9.3 μM; HDAC1/3/4/5/7 IC50 > 50 μM; HDAC2 IC50 = 41 μM).[128] In a follow-up study, compound 87 was characterized as the most potent HDAC4 inhibitor with good isoform selectivity (HDAC4 IC50 = 0.75 μM; HDAC1 IC50 = 7.4 μM; HDAC3 IC50 = 3.1 μM; HDAC6 IC50 = 15 μM; HDAC7 IC50 = 13 μM; HDAC8 IC50 = 12 μM).[129] However, in contrast to compound 87, the ethoxylated analogue 88 (Fig. 22) displayed much better antiproliferative activity and broader coverage in panel of tumor cell lines, albeit with less potent HDACs inhibition (e.g., HDAC4 IC50 = 4.9 μM and HDAC7/8 IC50 > 50 μM). Interestingly, 88 could induce the accumulation of acetylated H3 but not acetylated α-tubulin, supporting its HDAC4 selectivity.[129]
2.9. Inhibition of NSD1
The nuclear receptor-binding SET domain (NSD) family, a class of histone methyltransferases (HMTs), is categorized into three types, NSD1, NSD2 (MMSET/WHSC1) and NSD3 (WHSC1L1).[130] These structurally similar enzymes mono- and di-methylate histone H3 lysine 36 (H3K36), which contribute to regulate the chromatin integrity and gene expression.[131] Aberrant expression or mutation of NSDs is associated with the occurrence of some human malignancies.[130] The role of NSD1 in cancer is complicated and increased expression of NSD1 drives a particular hematological cancer, whereas loss-of function mutation in NSD1 characterizes a wide variety of solid human cancers.[132, 133] The well-recognized oncogenic role of NSD1 is linked to its translocation with the NUP98 gene, which predominantly occurs in pediatric patients with acute leukemia.[132, 133] All NSD HMTs have highly conserved catalytic SET domain, which features a unique autoinhibitory loop that blocks the access of histone lysine substrates to the active site.[134]
Recently, we developed and characterized first-in-class irreversible small-molecule inhibitors of NSD1 targeting the catalytic SET domain (Fig. 23).[135] By conducting fragment-based screening of almost 1600 fragment-like molecules, we identified 2-aminobenzothiazole 89 (BT1) that bound to the SET domain of NSD1. An analogue of 89, 2-amino-4-hydroxy-6-bromobenzothiazole 90 (BT2) suppressed NSD1 activity with an IC50 value of 66 μM. Mapping the binding of 90 to the NSD1 SET domain by 1H-15N HSQC spectrum showed strong chemical shift perturbations for residues in the autoinhibitory loop including Cys2062. Thus, we developed an irreversible ligand 91 (BT3). Analysis of the crystal structure of NSD1 bound to 91 confirmed the covalent binding interaction of 91 to Cys2062. 91 fitted well into the binding pocket of the SET domain and formed well-defined interactions including four key hydrogen bonds and two favorable chalcogen bonds (Fig. 23).
Fig. 23.

Discovery of 2-aminobenzothiazole 93 (BT5) as a first-in-class NSD1 inhibitor. X-ray crystal structure of 91 (BT3) bound to NSD1 SET domain (PDB code 6KQQ).
To explore the irreversible inhibitors, the small electrophile aziridine was next introduced at the C6 position of 2-aminobenzothiazole core.[135] The resulting compound 92 (BT4) covalently bound to NSD1 as validated by MS and NMR. Further optimization of 92 gave rise to compound 93 (BT5) with significantly improved NSD1 inhibitory activity (IC50 = 5.8 μM). Of note, compounds containing acrylamide moiety weakly interacted with NSD1 and replacement of 2-methylaziridine by 2-ethylaziridine decreased the binding affinity. In addition, replacement of hydroxyl group with methoxyl group abrogated the activity. Encouragingly, 93 was highly selective against epigenetic enzymes and protein kinases.[135] Moreover, 93 effectively blocked the proliferation of NUP98-NSD1 cells (GI50 = 0.87 μM). Further mechanism of action studies revealed that 93 could downregulate the expression levels of leukemia-relevant target genes (HOXA9 and MEIS1), decrease global H3K36me2 levels and impair the colony formation of acute myeloid leukemia (AML) cells with NUP98-NSD1 translocation but not with MLL-ENL translocation or in normal CD34+ progenitor cells. Altogether, our efforts led to the discovery of 93, a valuable chemical probe, that can irreversibly bind to all three NSD SET domains but showed distinct preference toward NSD1. Further optimization of 93 to develop next generation of potent and selective NSD inhibitors with drug-like properties is underway.
2.10. Inhibition of LSD1
Lysine-specific demethylase 1 (LSD1), also known as KDM1A, AOF2, BHC110 or KIAA0601, is a flavin adenine dinucleotide (FAD)-dependent amino oxidase that catalyzes the demethylation of H3K4 and H3K9 via amine oxidation.[136, 137] LSD1 is aberrantly expressed in diverse cancers, where it promotes proliferation, impedes differentiation, and enhances cell motility.[137, 138] In addition, the high level of LSD1 is correlated with poor prognosis.[137, 138] Its pivotal role in cancer therapy compelled researchers to develop a new class of LSD1 inhibitors.
Alnabulsi et al. reported the discovery of novel reversible LSD1 inhibitors by employing a computational fragment-based drug design approach.[139] Two sets of chemically diverse fragments from the Maybridge Ro3 2000 diversity fragment library were docked into two active LSD1 sites: a hydrophobic F pocket adjacent to the FAD binding pocket, and a hydrophilic negatively charged S pocket. On the basis of docking scores and total binding energy values, a set of 2-aminobenzothiazole derivatives were synthesized and their enzymatic inhibitory activities were evaluated. Compound 94 exerted an IC50 value of 43.8 μM against LSD1 enzyme (Fig. 24). Optimization of 94 by replacing the methylene unit with a rigid phenyl group led to compound 95, achieving almost 2.4-fold gain in potency (IC50 = 18.4 μM). The authors have suggested that further development of potent LSD1 inhibitors with potential anticancer activity is currently undergoing, using 95 as a lead compound.
Fig. 24.

Identification of 2-aminobenzothiazole derivative 95 as an LSD1 inhibitor.
2.11. Inhibition of FTO
N6-methyladenosine (m6A) is the most abundant epitranscriptomic modification of mRNA. This dynamic modification is regulated by three classes of proteins, known as “writers” (e.g., METTL3, METTL14 and METTL16), “readers” (YTHDF1–3 and YTHDC1–2), and “erasers” (FTO and ALKBH5).[140] FTO, the fat mass obesity-associated protein, is an Fe(II)- and 2-oxoglutarate-dependent dioxygenase that predominantly catalyzes m6A demethylation.[141] Dysregulation of FTO is linked to tumorigenesis and poor prognosis. In multiple cancer types, FTO is upregulated and plays a crucial tumor-promoting role.[142, 143] Thus, intense interest has been focused on the development of small-molecule inhibitors targeting FTO for cancer therapy.
In 2021, the Rana group reported the design and biochemical characterization of a new class of FTO inhibitors with improved physicochemical properties over existing inhibitor meclofenamic acid (MA).[144] Compound 96 (FTO-04, Fig. 25) was found to selectively block the enzymatic activity of FTO (IC50 = 3.39 μM), which was almost 3.7-fold more potent than MA. It should be noted that replacement of 2-aminobenzothiazole scaffold in compound 96 with other aromatic rings such as 2-methoxynaphthalene, 2-methylquinoline or (2-methoxyphenyl)methanol dramatically reduced the FTO inhibition activity. Significantly, 96 inhibited neurosphere formation in multiple patient-derived glioblastoma stem cell (GSC) lines but had no effect on the growth of healthy neural stem cell-derived neurospheres. Consistent with its potent FTO inhibition, 107 could markedly augment m6A and m6Am levels in GSCs. Overall, these data supported 2-aminobenzothiazole 96 as a potential anti-FTO lead for further medicinal chemistry investigation.
Fig. 25.

Chemical structures of MA and 2-aminobenzothiazole-based FTO inhibitor 96 (FTO-04).
2.12. Inhibition of mPGES-1
Microsomal prostaglandin E synthase-1 (mPGES-1) is the terminal enzyme responsible for the production of the pro-tumorigenic prostaglandin E2 (PGE2).[145, 146] mPGES-1 is overexpressed in various human cancers, and it is considered a validated and promising target in cancer treatment.
In 2020, the Bifulco group developed a new class of mPGES-1 inhibitors featuring a 2-aminobenzothiazole scaffold based upon a combinatorial virtual screening campaign approach.[145] Compounds 97-99 exerted the most potent inhibitory activity against mPGES-1 in a cell-free system with IC50 values of 1.7 ± 0.2, 1.4 ± 0.2, and 0.7 ± 0.1 μM, respectively (Fig. 26). These compounds could concentration-dependently reduce the cytokine-induced PGE2 production in A549 cells where mPGES-1 was overexpressed. They also showed good cell growth inhibition and induced G0/G1 cell cycle arrest in a time- and concentration-dependent fashion in A549 cells. Thus, these compounds could be reckoned as anti-mPGES-1 leads for a further medicinal chemistry campaign.
Fig. 26.

Chemical structures of 2-aminobenzothiazole derivatives 97–99 as mPGES-1 inhibitors.
2.13. Inhibition of SCD
An increase in the expression of stearoyl-CoA desaturase (SCD), an enzyme that converts saturated fatty acids to monounsaturated fatty acids, has been reported in a wide range of cancer cells, and this increase is positively correlated with tumor growth rates.[147, 148] Inhibition of SCD represents a potential approach for cancer treatment.[149] However, clinical development of SCD inhibitors has been impeded by the on-target toxicity. Particularly, SCD inactivation in the skin results in the atrophy of sebocytes.[150] Thus, identification of new chemotypes as SCD inhibitors with minimized skin toxicity has gained considerable attention.
Scientists from UT Southwestern Medical Center identified the hit 100 (Fig. 27), an irreversible SCD inhibitor, through an unbiased HTS campaign using 12 distinct NSCLC cell lines.[150, 151] Compound 100 displayed IC50 values < 1 μM against a subset of NSCLC cell lines (e.g., H2122 cells IC50 = 0.058 μM) but > 5 μM against other NSCLC lines (e.g., H2009 cells IC50 = 22 μM). Single-digit nanomolar activity against H2122 cells was achieved when the 4-amino group was substituted with a phenone group (compound 101, IC50 = 8 nM). Despite excellent in vitro potency, 101 suffered from poor PK profiles (e.g., Cmax = 0.3 μM and AUC = 0.6 μM.h). Further modification resulted in compound 102 (SW206638, Fig. 27) with low logP and significant improvement in plasma pharmacokinetic properties (e.g., Cmax = 1.5 μM and AUC = 3.0 μM.h). One noteworthy finding was that the antiproliferative activity of compound 102 was substantially reduced when the benzothiazole core was replaced with benzimidazole, benzoxazole, thiazole, or phenyl-thiazole. Compound 102 potently blocked the enzymatic activity of SCD with an IC50 value of 54 nM. Consistently, a significant reduction in H2122-derived tumor growth was observed following a 25 mg/kg intraperitoneal dose of 102 twice a day (BID). In addition, 102 displayed higher antitumor efficacy and lower skin toxicity than the canonical SCD inhibitor Xenon-45. The enantiomer (R)-102 exerted robust antiproliferative activity against H2122 cells (IC50 = 21 nM, Fig. 27).[151] Mechanistically, (R)-102 functioned as a prodrug that could be transformed into active analogue (R)-103 (H2122 cells IC50 = 8 nM) mediated by CYP4F11 which was expressed in sensitive NSCLC cell lines but not expressed in insensitive NSCLC cell lines. The SCD inhibitor (R)-104 showed a similar selectivity pattern, and the same metabolite (R)-103 was also monitored in H2122 cells. In the modified tumor xenograft model, (R)-103 significantly arrested tumor growth at the dose of 10 mg/kg via subcutaneous BID administration, and no obvious toxicity was observed. Collectively, this tumor-activated prodrug strategy offered the promise to exploit potent SCD inhibitors and avoid toxicity correlating with systemic SCD suppression.[151]
Fig. 27.

Chemical structures of 2-aminobenzothiazole derivatives 100–104 as SCD inhibitors.
2.14. Inhibition of hCA IX/XII
Human carbonic anhydrases (hCAs) are ubiquitous zinc-metalloenzymes which catalyze the reversible interconversion between carbon dioxide and bicarbonate.[152, 153] hCAs are categorized into 15 isoforms on the basis of their tissue distribution and kinetic properties, such as cytosolic (hCA I, II, III, VII, and XIII), mitochondrial (hCA VA and VB), membrane-bound (hCA IV, IX, XII, XIV, and XV), secreted (hCA VI) isoenzymes.[153] Amongst them, hCA IX and hCA XII are overexpressed in many tumors but sparsely present in normal tissues. In addition, hCA IX and hCA XII are related with hypoxic solid tumors inducing tumor growth and metastasis. Pharmacological inhibition of hCA IX/XII has become a promising approach for cancer treatment.[154] As a consequence, many small-molecule hCA IX/XII inhibitors belonging to diverse classes have been investigated for their antineoplastic potentials, including 2-aminobenzothiazole derivatives.
Compound 105 (Fig. 28) exerted good hCA IX/XII suppressive activity (hCA IX Ki = 0.079 μM and hCA XII Ki = 0.76 μM) as well as excellent selectivity over the cytosolic hCA I and II (Ki > 50 μM).[155] Compound 106 (Fig. 28) bearing a sulfonamide moiety effectively blocked the enzymatic activity of hCA IX and XII, with Ki values of 10 nM and 46.5 nM, respectively.[156] 2-Aminobenzothiazole derivative 107 (Fig. 28) was reported as a highly potent hCA IX/XII inhibitor (Ki = 4.1 and 6.7 nM, respectively).[157] However, both compounds 106 and 107 also showed pronounced inhibitory potency towards physiologically dominant hCA I and II subtypes. Compound 108 that featured a 4-methoxyphenyl substituted piperazine-dithiocarbamate tail displayed strong and selective inhibition activity against hCA IX isoform (Fig. 28), with a Ki value of 30.9 nM.[158] This compound was found to be active against MCF-7 cells. Very recently, the Supuran group developed a set of new 2-aminobenzothiazole-based inhibitors of hCA IX/XII.[159] The representative compound 109 manifested excellent potency and selectivity towards cancer-related isozymes (hCA IX Ki = 48.9 nM and hCA XII Ki = 57.5 nM), as shown in Fig.28. Consistent with the hCA IX/XII suppression, 109 elicited good antiproliferative activity against breast cancer T-47D and MCF-7 cells, having IC50 values of 6.73 and 9.16 μM, respectively.[159] These data revealed the potential of hCA IX/XII inhibition for designing 2-aminobenzothiazole-derived anticancer agents.
Fig. 28.

Chemical structures of 2-aminobenzothiazole derivatives 105-109 as hCA IX/XII inhibitors.
2.15. Inhibition of CXCR
CXCR1 and CXCR2, known as interleukin-8 receptor A (IL-8RA) and interleukin-8 receptor B (IL-8RB) respectively, are members of the cell surface G protein-coupled receptors (GPCR) family.[160] Both of them mediate the initiation and development of various cancers. Accumulating evidence shows that CXCR1/2 inhibition profoundly suppresses the progression of cancers and augments the therapeutic effect.[160-162]
Compound 110 (Fig. 29) that possessed electron-withdrawing substituents on both benzothiazole and phenyl rings manifested pronounced CXCR2 inhibition activity (IC50 = 0.3 μM).[163] In addition, 110 exerted potent antiproliferative activity against ovarian OVCAR-8 and CXCR2-U2OS cell lines with IC50 values of 4.9 and 2.9 μM, respectively. Compound 111 (Fig. 29), an analogue of 110, presented strong inhibitory activity against the solid and hematological tumor cell lines with IC50 values ranging from 2 to 5 μM, especially renal cell carcinoma (RCC) and head and neck squamous cell carcinoma (HNSCC) where CXCR1/2 were highly expressed.[164] This compound showed low cytotoxicity against normal primary kidney cells 15S and Mel202 (IC50 > 10 μM). Notably, it was found to be equally efficient on parental and resistant RCC and HNSCC cells. Moreover, 111 demonstrated reasonable PK properties (e.g., t1/2 = 191 min and Tmax = 30 min). Further in vivo studies indicated that 111 remarkably reduced the growth of experimental RCC or HNSCC tumors with no weight loss observed. [164] Altogether, these results suggested that 111 may represent a therapeutic tool for further RCC and HNSCC investigation.
Fig. 29.

Chemical structures of 2-aminobenzothiazole derivatives 110 and 111 as CXCR inhibitors.
2.16. Chelators
Transition metals such as iron, copper, and zinc are essential for critical biological processes, including gene expression, oxygen transport, and respiration.[165] Tumor cells require these nutrients for their rapid proliferation, particularly iron. As such, trapping the iron in tumors provides new perspectives for antineoplastic treatments.[166, 167] In the past few years, a library of chelators as promising anticancer agents including 2-aminobenzothiazole analogues have been disclosed.
Triapine, a tridentate N-pyridyl thiosemicarbazone (Fig. 30), is a potent ribonucleotide reductase inhibitor currently undergoing phase I and II clinical trials. In accordance with the binding mode of triapine (N–N–S terdentate coordination), the Zhu group utilized a cyclization strategy and identified compound 112 with robust cellular potency.[168] Of note, substitution of the 2-aminobenzothiazole moiety in compound 112 with a 2-aminobenzimidazole motif significantly compromised the antiproliferative activity. 112 showed good mouse liver microsomal stability and acceptable pharmacokinetic properties (e.g., Cmax = 673 μg/L and F = 25%).
Fig. 30.

Lead optimization development of chelator 112 based on triapine.
In an MV4;11 xenograft model, treatment of mice with 112 at an oral dose of 30 mg/kg (BID) significantly inhibited tumor growth, with no impact on body weight. Further studies revealed that, unlike triapine, 112 preferentially suppresses the proliferation of hematopoietic tumor cells by inducing copper-dependent mitochondria ROS production and mitochondrial dysfunction. It was also able to enhance thioredoxin-interacting protein expression, attenuate the thioredoxin activity, and activate the JNK and p38 MAPK pathway, which potentiated its antitumor activity.[168]
2-aminobenzothiazole derivative 113 (Fig. 31), an NNS donor chelator, was substantially more effective for MDR tumor cell lines as compared to the corresponding parental cells lines, having IC50 values ranging from 0.014 to 0.28 μM.[169] The ONS donor chelator 114 demonstrated 2-fold more potent cytotoxicity against MES-SA_mCh/doxorubicin cells than the corresponding sensitive cell line (MES-SA_mCh).[169] Another interesting chelator 115 (Fig. 31), in which three substituents were installed on the benzothiazole scaffold, exerted strong suppressive activity against MDR Colo320 colonic adenocarcinoma cells (IC50 = 0.188 μM).[170] Similarly, 115 was more effective for MDR Colo320 cells than that for doxorubicin-sensitive Colo205 colon tumor cells. These results suggested that development of 2-aminobenzothiazole-derived chelators could be a potential strategy to conquer MDR.
Fig. 31.

Exploration of 2-aminobenzothiazole-derived chelators for overcoming multidrug resistance.
2.17. Ligands of metal-based anticancer agents
Platinum-based drugs, such as cisplatin, carboplatin, and oxaliplatin are used in the treatment of approximately 50% of cancers.[171, 172] Following the success of platinum-based drugs and considering their adverse effects and drug resistance, new metal-based anticancer agents have been developed through integration of the heterocyclic motif and metal.[173] Of these heterocycles, functionalized 2-aminobenzothiazole as a ligand has captured intensive attention.
Compounds 116-118 (Fig. 32) manifested potent antiproliferative activity against a panel of tumor cancer cell lines with IC50 values ranging from 3.77 to 7.43 μM, which were equipotent to the standard anticancer drug cisplatin.[174] The mechanistic study suggested that the inhibition of cell viability is attributed to the cleavage or fragmentation of DNA of these cancer cells resulting from active species (hydroxyl radicals). Rao and coworkers reported several metal complexes as potential anticancer agents.[175] Among them, compound 119 (Cu complexes) was found to intercalate stronger and promote DNA cleavage more efficiently than other metal complexes (Fig. 32). Consistently, 119 exerted the strongest in vitro cytotoxic activity against HeLa and MCF-7 cell lines. Interestingly, all metal complexes showed higher cytotoxicity than the parent ligand in both cell lines.
Fig. 32.

Chemical structures of metal-based complexes 116–123.
The Mostafa group developed several new 2-aminobenzothiazole-based metal complexes.[176] Among them, compounds 120 and 121 (Fig. 32) were demonstrated to be more active than cisplatin, with IC50 values of 1.26 and 1.78 μM against MDA-MB231 and OVCAR-8 cell lines, respectively. These two compounds were capable of binding to CT-DNA through intercalation and inhibition of DNA synthesis. In a follow-up study, a subset of new complexes of V(IV), Ru(III), Pd(II), Pt(II) and Ag(I) derived from 2-hydrazinobenzothiazole were designed and synthesized.[177] Compound 122 (Fig. 32) emerged as the strongest metal complex, achieving IC50 values of 5.15, 9.9, 13.1 and 17.7 μg/mL against EAC, HepG2, MCF-7 and HCT-116 cell lines, respectively. More recently, Nádia et al. reported that compound 123 (Fig. 32) exerted higher antiproliferative activity than 5-FU against HCT-116 cells, with an IC50 value of 16.6 μM.[178] It induced the cell cycle arrest at S phase and effectively suppressed cell migration. Overall, these results indicated that development of aminobenzothiazole-metal complexes is an important strategy to explore new chemical entities as anticancer agents.
2.18. Miscellaneous
The Liu group at Peking University carried out a substructure virtual searching and identified a new class of 2-aminobenzothiazole compounds as promising antineoplastic agents.[179] In this study, 2-aminobenzothiazole 124 (Fig. 33), deemed as the most potent compound, showed potent and specifically suppressive activity against HPV18-positive cervical cancer cell lines (HeLa and C4-1) with IC50 values of 0.38 and 0.44 μM, respectively. Notably, the amino group on the benzothiazole scaffold was indispensable for antiproliferative potency. The following mechanism study suggested that 124 could repress transcription, especially those related to the oncoprotein E7 cellular pathway (e.g., E7/Rb/E2F-1/DNMT1), which are crucial to tumorigenesis. Further proteomics investigation indicated that E7 might be degraded through E3 ubiquitin ligases by activating the NEDD8. Importantly, in the HeLa xenograft model, 124 manifested robust inhibition of tumor growth without noticeable adverse effects.[179]
Fig. 33.

Chemical structures of 2-aminobenzothiazole derivatives 124–141.
To seek novel chemical entities with anticancer activity, Mistry et al. attached a 2-aminobenzothiazole moiety to alkaloid berberine using the molecular hybridization strategy.[180] Compound 125 (Fig. 33) presented the highest antiproliferative potency against HeLa, CaSki, and SK-OV-3 cell lines, with IC50 values of 5.474, 5.311, and 32.61 μg/mL, respectively. Dadmal et al. developed a number of triazole and isoxazole-tagged benzothiazole hybrids.[181] Among them, 126 and 127 (Fig. 33) stood out as the most promising molecules, inhibiting the growth of tumor cells with IC50 values in the range of 1.57-2.415 μM. These two compounds could promote apoptosis through caspase-dependent apoptotic process via the mitochondrial pathway. An interesting family of cytotoxic compounds was obtained by tethering a 2-aminobenzothiazole core and another bioactive heterocycle via copper catalyzed azide-alkyne cycloadditions (CuAAC). Compound 128 showed the strongest inhibitory activity against SW1882 colorectal cell line. Further studies indicated that 128 is able to intercalate with Ct-DNA through the minor groove (Fig. 33).[182]
The Hranjec group disclosed that compound 129 (Fig. 33) exhibited strong inhibitory activity against HeLa cells with an IC50 value of 1.16 μM, which was approximately 7.6-fold more potent than standard drug 5-FU.[183] In a follow-up study, they found that its analogue 130 (Fig. 33) potently inhibited the proliferation of MCF-7 cells (IC50 = 40 nM).[184] However, further modification of 130 led to derivatives 131 and 132 with a great loss of activity, indicating that the presence of amino protonated moiety on the benzo[b]thiophene skeleton is unfavorable for antiproliferative potency.[184]
Videnovic and colleagues developed new 2-aminobenzothiazole chemotypes and assessed their antiproliferative activities.[185] Compounds 133 and 134 (Fig. 33) exerted outstanding inhibitory potency against various tumor cells, especially NT2/D1 cells (IC50 of 0.2 μM and 0.1 μM, respectively). The amide analogue 135 had weaker antiproliferative activity than compounds 133 and 134 against NT2/D1 cells. Interestingly, compound 134 was noncytotoxic to MCF-7 cells (IC50 > 100 μM), but compound 135 showed moderate suppressive effect on MCF-7 cells. The possible mechanism of action of these compounds was then investigated. In MCF-7 cells, 2-aminobenzothiazole analogues 133 and 135 were found to exhibit antiproliferative activity by arresting cell cycle in G2/M phase and triggering apoptosis. In NT2/D1 cells, induction of apoptosis and migration together with invasiveness could contribute to the cytotoxic potential of compounds 133 and 134.[185] However, the exact target(s) of 2-aminobenzothiazoles 133 and 134 on NT2/D1 cells remains uncertain.
A new series of 2-aminobenzothiazole derivatives bearing a 2-anilinopyridyl functionality were designed and evaluated for their antineoplastic potentials.[186] Among them, 136 and 137 (Fig. 33) were found to be the most active compounds with IC50 values of 1.03 and 1.69 μM against MCF-7 cells, respectively. In a similar work, Saipriya et al. reported that compound 138 (Fig. 33) displayed excellent activity with an IC50 value of 2.517 μg/mL against HeLa cells, which was 6.8-fold more active than cisplatin (IC50 = 17.2 μg/mL).[187] Very recently, JawalePatil et al. integrated a 2-aminobenzothiazole pharmacophore and naphthalimide moiety, delivering compound 139 that possessed low micromolar inhibitory potency against B16F10 cells.[188] Compound 140 was found to repress the viability of a panel of cancer cell lines with IC50 values in the range of 2.23-3.75 μM (Fig. 33).[189] Especially, 140 displayed comparable activity to doxorubicin against MDA-MB-231 cells. The mechanistic study suggested that 140 induces apoptosis in MDA-MB-231 cells by downregulation of Bcl-2 and upregulation of Bax protein levels. [189]
Based on the structure of the Twist1 inhibitor harmine, the Zhai group devised and developed a novel class of 2-aminobenzothiazole-containing compounds employing the scaffold hopping strategy.[190] Among them, compound 141 (Fig. 33) possessed the most pronounced antiproliferative effect on A549 and H2228 cells with corresponding IC50 value of 2.03 μM and 9.80 μM, which was superior to the lead harmine. In a consistent manner, 141 demonstrated effective reduction of Twist1 level in a dose-dependent pattern. In this study, substituting the piperidine group in 141 with piperazine, pyrrole, azetidine, or alkylamines resulted in a significant decrease in antiproliferative activity. The reduction in activity was also observed when the linker length of 141 was shortened.
3. Conclusion and perspective
2-Aminobenzothiazoles have been used as privileged scaffolds in drug design and discovery. It is evident that 2-aminobenzothiazole derivatives demonstrate immense potential as anticancer agents. Intriguingly, a large number of 2-aminobenzothiazole analogues were identified through screening, bioisosteric replacement, scaffold hopping, structure/fragment-based drug design and molecular hybridization strategies. In this study, we systematically summarize the recent developments of 2-aminobenzothiazoles as the privileged structures in the design and discovery of anticancer agents targeting tumor-related proteins (Table 1). In particular, the anticancer potentials of 2-aminobenzothiazole-derived chelators and 2-aminobenzothiazole-metal complexes are also detailed. In addition, the examples in which the 2-aminobenzothiazoles exert better performance in enzymatic inhibition and/or cellular activity than other heterocycles are highlighted. In this review, we also summarize some key SARs of these 2-aminobenzothiazole derivatives. For instance, the important feature of NSD1 inhibitors possesses the 2-aminobenzothiazole scaffold. Compounds bearing acrylamide moiety weakly interacted with NSD1, and substitution of 2-methylaziridine with 2-ethylaziridine compromised the binding affinity. In addition, changing the hydroxyl group with a methoxyl group substantially abolished the binding to NSD1. Importantly, the 2-aminobenzothiazole motif formed two hydrogen bonds with residue Thr1994 and SAM, respectively, and the sulfur atom within benzothiazole ring made contacts with residues Asp2065 and Gly2066 through two key chalcogen bonds. These favorable interactions underscore the vital role of the 2-aminobenzothiazole scaffold in drug design.
Table 1.
Representative 2-aminobenzothiazole derivatives and their molecular targets.
| Compd. | Chemical structure | Target | Bioactivity | Reference |
|---|---|---|---|---|
| 1 (BLZ945) |
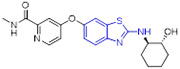
|
CSF1R | IC50 = 1.0 nM | [27] |
| 8 (IACS-9439) |
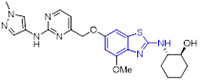
|
CSF1R | IC50 = 1.7 nM Kd = 1.0 nM | [29] |
| 12 |
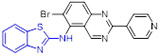
|
EGFR | IC50 = 96 nM | [33] |
| 23 |
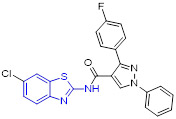
|
VEGFR-2 | IC50 = 97 nM | [45] |
| 24 |
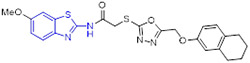
|
FAK | IC50 = 19.5 μM | [48] |
| 34 (SAR125844) |
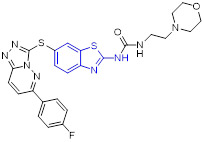
|
MET | IC50 = 4 nM | [55] |
| 35 |
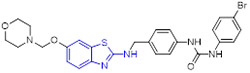
|
Aurora B | IC50 = 0.09 μM | [58] |
| 39 |
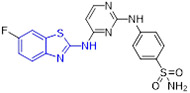
|
CDK2 | IC50 = 15.4 nM | [63] |
| 44 |
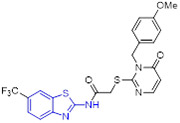
|
CK1δ | IC50 = 0.09 μM | [69] |
| 45 |
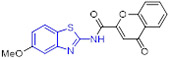
|
CK2 | IC50 = 0.08 μM | [72] |
| 46 (TAK632) |
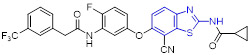
|
BRAFV600E | IC50 = 2.4 nM | [78] |
| 47 |
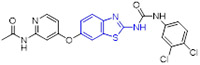
|
BRAFV600E | IC50 = 0.095 μM | [79] |
| 50 (YK-2-69) |
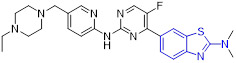
|
DYRK2 | IC50 = 9 nM Kd = 92 nM | [81] |
| 52 |
|
PI3Kγ | IC50 = 2 nM | [87] |
| 54 |
|
PI3Kα | IC50 = 1.03 nM | [90] |
| 57 |
|
Topoisomerase I | - | [95] |
| 63 |
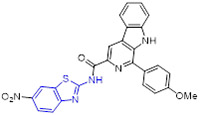
|
Topoisomerase IIα | - | [97] |
| 67 (A-1331852) |
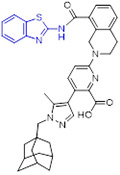
|
BCL-XL | Ki < 0.01 nM | [103] |
| 69 (A-1293102) |
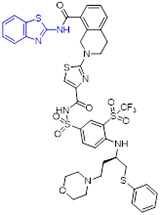
|
BCL-XL | Ki = 0.43 nM | [104] |
| 73 |
|
HSP90 | - | [114] |
| 74 |
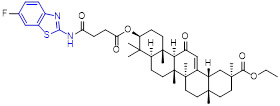
|
HSP90-CDC37 | IC50 = 0.14 μM | [115] |
| 78 (MB725) |
|
Mutant p53 | - | [120] |
| 79 |
|
Mutant p53 | Kzn = 119 nM | [121] |
| 84 |
|
HDAC6 | IC50 = 12 nM | [126] |
| 93 (BT5) |
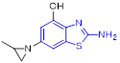
|
NSD1 | IC50 = 5.8 μM | [135] |
| 95 |
|
LSD1 | IC50 = 18.4 μM | [139] |
| 96 (FTO-04) |
|
FTO | IC50 = 3.39 μM | [144] |
| 99 |
|
mPGES-1 | IC50 = 0.7 μM | [145] |
| 102 |
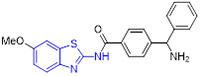
|
SCD | IC50 = 54 nM | [150] |
| 109 |
|
hCA IX/XII | Ki = 48.9/57.5 nM | [159] |
| 110 |
|
CXCR2 | IC50 = 0.3 μM | [163] |
| 111 |
|
CXCR1/2 | - | [164] |
However, the antiproliferative activity observed for some 2-aminobenzothiazole derivatives does not entirely correspond to their inhibition against a particular target, warranting more detailed studies to progress with their development. On the other hand, many 2-aminobenzothiazole derivatives have been reported to demonstrate excellent inhibitory activity against the specific target(s) (e.g., hCA IX/XII and EGFR). Nevertheless, there is no PK or in vivo data available for these inhibitors on the anticancer efficacy as of yet. Hybridization of 2-aminobenzothiazole motif with other pharmacophores could be an important approach to deliver new anticancer agents with higher potency. Besides, conversion of 2-aminobenzothiazole-based inhibitors into degraders and further development of 2-aminobenzothiazole-derived chelators would be new directions in the future to combat cancer drug resistance and achieve anticancer efficacy. We anticipate that, with the help of effective design strategies and in-depth SAR studies, safe and efficacious 2-aminobenzothiazole-based antitumor molecules with selective target engagement can be developed. This chemotype offers tremendous opportunities to explore new chemical space in future anticancer drug discovery programs.
Acknowledgements
This work was funded by the National Institute of Health (NIH) R01 grants (1R01 CA244254, 1R01 CA201204 and 5R01 CA160467) to J.G., and NIH R01 grants (1R01 CA226759 and 1R01 CA207272) to T. C. We thank Devon Hucek for proofreading this manuscript.
Abbreviations
- BCL-2
B cell lymphoma protein 2
- BID
twice a day
- CDKs
cyclin-dependent protein kinases
- CK1
casein kinase 1
- CK2
casein kinase 2
- CSF1R
colony-stimulating factor 1 receptor
- CT
cyanothiouracil
- DYRKs
dual-specificity tyrosine-phosphorylation-regulated kinases
- FAK
focal adhesion kinase
- FLT3
FMS-like tyrosine kinase-3
- FTO
fat mass obesity-associated protein
- 5-FU
5-fluorouracil
- HGF
hepatocyte growth factor
- HDACs
histone deacetylases
- hCAs
human carbonic anhydrases
- HSR
heat shock response
- HSP90
heat shock protein 90
- HTS
high-throughput screening
- HNSCC
head and neck squamous cell carcinoma
- LSD1
lysine-specific demethylase 1
- MDR
multidrug resistance
- mPGES-1
microsomal prostaglandin E synthase-1
- MTD
maximum tolerated dose
- NSCLC
non-small-cell lung cancer
- NSD
nuclear receptor-binding SET domain
- PDGFR
platelet-derived growth factor receptor
- ROS
reactive oxygen species
- PI3Ks
phosphoinositide 3-kinases
- PTKs
protein tyrosine kinases
- RCC
renal cell carcinoma
- SAR
structure-activity relationships
- SCD
stearoyl-CoA desaturase
- STKs
serine/threonine kinases
- TAMs
tumor-associated macrophages
- TZD
thiazolidinedione
- ZMCs
zinc metallochaperones
Footnotes
Declaration of competing interest
G.H. declares that he has no competing interests with this study. T.C. and J.G. received prior research support from Kura Oncology Inc. for unrelated project; served as consultants in Kura Oncology and have equity ownership in the company.
Reference
- [1].Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F, Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries, CA Cancer J. Clin 71(3) (2021) 209–249. 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- [2].WHO report on cancer: setting priorities, investing wisely and providing care for all. Geneva: World Health Organization; 2020. [Google Scholar]
- [3].Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG, Cancer drug resistance: an evolving paradigm, Nat. Rev. Cancer 13(10) (2013) 714–726. 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- [4].Vasan N, Baselga J, Hyman DM, A view on drug resistance in cancer, Nature 575(7782) (2019) 299–309. 10.1038/s41586-019-1730-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Huang G, Dong J, Zhang Q, Meng Q, Zhao H, Zhu B, Li S, Discovery and synthesis of sulfur-containing 6-substituted 5, 8-dimethoxy-1, 4-naphthoquinone oxime derivatives as new and potential anti-MDR cancer agents, Eur. J. Med. Chem 165 (2019) 160–171. 10.1016/j.ejmech.2019.01.005. [DOI] [PubMed] [Google Scholar]
- [6].Kerru N, Gummidi L, Maddila S, Gangu KK, Jonnalagadda SB, A review on recent advances in nitrogen-containing molecules and their biological applications, Molecules 25(8) (2020) 1909. 10.3390/molecules25081909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Akhtar J, Khan AA, Ali Z, Haider R, Yar MS, Structure-activity relationship (SAR) study and design strategies of nitrogen-containing heterocyclic moieties for their anticancer activities, Eur. J. Med. Chem 125 (2017) 143–189. 10.1016/j.ejmech.2016.09.023. [DOI] [PubMed] [Google Scholar]
- [8].Pathania S, Narang RK, Rawal RK, Role of sulphur-heterocycles in medicinal chemistry: An update, Eur. J. Med. Chem 180 (2019) 486–508. 10.1016/j.ejmech.2019.07.043. [DOI] [PubMed] [Google Scholar]
- [9].Keri RS, Patil MR, Patil SA, Budagumpi S, A comprehensive review in current developments of benzothiazole-based molecules in medicinal chemistry, Eur. J. Med. Chem 89 (2015) 207–251. 10.1016/j.ejmech.2014.10.059. [DOI] [PubMed] [Google Scholar]
- [10].Ammazzalorso A, Carradori S, Amoroso R, Fernßndez IF, 2-substituted benzothiazoles as antiproliferative agents: Novel insights on structure-activity relationships, Eur. J. Med. Chem 207 (2020) 112762. 10.1016/j.ejmech.2020.112762. [DOI] [PubMed] [Google Scholar]
- [11].Law CSW, Yeong KY, Current trends of benzothiazoles in drug discovery: a patent review (2015–2020), Expert Opin. Ther. Pat 32(3) (2022) 299–315. 10.1080/13543776.2022.2026327. [DOI] [PubMed] [Google Scholar]
- [12].Dadmal TL, Katre SD, Mandewale MC, Kumbhare RM, Contemporary progress in the synthesis and reactions of 2-aminobenzothiazole: a review, New J. Chem 42(2) (2018) 776–797. 10.1039/C7NJ03776G. [DOI] [Google Scholar]
- [13].Bensimon G, Lacomblez L, Meininger V, Group ARS, A controlled trial of riluzole in amyotrophic lateral sclerosis, N. Engl. J. Med 330(9) (1994) 585–591. 10.1056/NEJM199403033300901. [DOI] [PubMed] [Google Scholar]
- [14].Cheah B, Vucic S, Krishnan A, Kiernan M, Riluzole, neuroprotection and amyotrophic lateral sclerosis, Curr. Med. Chem 17(18) (2010) 1942–1959. 10.2174/092986710791163939. [DOI] [PubMed] [Google Scholar]
- [15].Blyufer A, Lhamo S, Tam C, Tariq I, Thavornwatanayong T, Mahajan SS, Riluzole: A neuroprotective drug with potential as a novel anti-cancer agent, Int. J. Oncol 59(5) (2021) 1–11. 10.3892/ijo.2021.5275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lemieszek MK, Stepulak A, Sawa-Wejksza K, Czerwonka A, Ikonomidou C, Rzeski W, Riluzole inhibits proliferation, migration and cell cycle progression and induces apoptosis in tumor cells of various origins, Anticancer Agents Med. Chem 18(4) (2018) 565–572. 10.2174/1871520618666180228152713. [DOI] [PubMed] [Google Scholar]
- [17].Speyer CL, Nassar MA, Hachem AH, Bukhsh MA, Jafry WS, Khansa RM, Gorski DH, Riluzole mediates anti-tumor properties in breast cancer cells independent of metabotropic glutamate receptor-1, Breast Cancer Res. Treat 157(2) (2016) 217–228. 10.1007/s10549-016-3816-x. [DOI] [PubMed] [Google Scholar]
- [18].Hatfield SM, Hartley LW, Schmidtke JR, The immunomodulatory action of frentizole, a novel immunosuppressive agent, Immunopharmacology 5(2) (1982) 169–179. 10.1016/0162-3109(82)90047-9. [DOI] [PubMed] [Google Scholar]
- [19].Minvielle M, Basualdo J, Ciarmela M, Niedfeld G, Anthelmintic efficacy of tinidazole against the progression of Toxocara canis larvae to the brain in mice, Parasitol. Res 85(10) (1999) 830–832. 10.1007/s004360050640. [DOI] [PubMed] [Google Scholar]
- [20].Hubbard SR, Till JH, Protein tyrosine kinase structure and function, Annu. Rev. Biochem 69(1) (2000) 373–398. 10.1146/annurev.biochem.69.1.373. [DOI] [PubMed] [Google Scholar]
- [21].Schlessinger J, Cell signaling by receptor tyrosine kinases, Cell 103(2) (2000) 211–225. 10.1016/j.cell.2010.06.011. [DOI] [PubMed] [Google Scholar]
- [22].Gschwind A, Fischer OM, Ullrich A, The discovery of receptor tyrosine kinases: targets for cancer therapy, Nat. Rev. Cancer 4(5) (2004) 361–370. 10.1038/nrc1360. [DOI] [PubMed] [Google Scholar]
- [23].Du Z, Lovly CM, Mechanisms of receptor tyrosine kinase activation in cancer, Mol. Cancer 17(1) (2018) 1–13. 10.1186/s12943-018-0782-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Peyraud F, Cousin S, Italiano A, CSF-1R inhibitor development: current clinical status, Curr. Oncol. Rep 19(11) (2017) 1–10. 10.1007/s11912-017-0634-1. [DOI] [PubMed] [Google Scholar]
- [25].Stanley ER, Chitu V, CSF-1 receptor signaling in myeloid cells, Cold Spring Harb. Perspect. Biol 6(6) (2014) a021857. 10.1101/cshperspect.a021857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Denny WA, Flanagan JU, Small-molecule CSF1R kinase inhibitors; review of patents 2015-present, Expert Opin. Ther. Pat 31(2) (2021) 107–117. 10.1080/13543776.2021.1839414. [DOI] [PubMed] [Google Scholar]
- [27].Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, Olson OC, Quick ML, Huse JT, Teijeiro V, Setty M, Leslie CS, Oei Y, Pedraza A, Zhang J, Brennan CW, Sutton JC, Holland EC, Daniel D, Joyce JA, CSF-1R inhibition alters macrophage polarization and blocks glioma progression, Nat. Med 19(10) (2013) 1264–1272. 10.1038/nm.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Krauser JA, Jin Y, Walles M, Pfaar U, Sutton J, Wiesmann M, Graf D, Pflimlin-Fritschy V, Wolf T, Camenisch G, Swart P, Phenotypic and metabolic investigation of a CSF-1R kinase receptor inhibitor (BLZ945) and its pharmacologically active metabolite, Xenobiotica 45(2) (2015) 107–123. 10.3109/00498254.2014.945988. [DOI] [PubMed] [Google Scholar]
- [29].Czako B, Marszalek JR, Burke JP, Mandal P, Leonard PG, Cross JB, Mseeh F, Jiang Y, Chang EQ, Suzuki E, Kovacs JJ, Feng N, Gera S, Harris AL, Liu Z, Mullinax RA, Pang J, Parker CA, Spencer ND, Yu SS, Wu Q, Tremblay MR, Mikule K, Wilcoxen K, Heffernan TP, Draetta GF, Jones P, Discovery of IACS-9439, a potent, exquisitely selective, and orally bioavailable inhibitor of CSF1R, J. Med. Chem 63(17) (2020) 9888–9911. 10.1021/acs.jmedchem.0c00936. [DOI] [PubMed] [Google Scholar]
- [30].Wheeler DL, Dunn EF, Harari PM, Understanding resistance to EGFR inhibitors—impact on future treatment strategies, Nat. Rev. Clin. Oncol 7(9) (2010) 493–507. 10.1038/nrclinonc.2010.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Gazdar A, Activating and resistance mutations of EGFR in non-small-cell lung cancer: role in clinical response to EGFR tyrosine kinase inhibitors, Oncogene 28(1) (2009) S24–S31. 10.1038/onc.2009.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Mokhtar AM, El-Messery SM, Ghaly MA, Hassan GS, Targeting EGFR tyrosine kinase: Synthesis, in vitro antitumor evaluation, and molecular modeling studies of benzothiazole-based derivatives, Bioorg. Chem 104 (2020) 104259. 10.1016/j.bioorg.2020.104259. [DOI] [PubMed] [Google Scholar]
- [33].Allam HA, Aly EE, Farouk AK, El Kerdawy AM, Rashwan E, Abbass SE, Design and Synthesis of some new 2, 4, 6-trisubstituted quinazoline EGFR inhibitors as targeted anticancer agents, Bioorg. Chem 98 (2020) 103726. 10.1016/j.bioorg.2020.103726. [DOI] [PubMed] [Google Scholar]
- [34].Sever B, Altıntop MD, Özdemir A, Akalın Çiftçi G, Ellakwa DE, Tateishi H, Radwan MO, Ibrahim MA, Otsuka M, Fujita M, Ciftci HI, Ali TFS, In vitro and in silico evaluation of anticancer activity of new indole-based 1, 3, 4-oxadiazoles as EGFR and COX-2 inhibitors, Molecules 25(21) (2020) 5190. 10.3390/molecules25215190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Abdellatif KR, Belal A, El-Saadi MT, Amin NH, Said EG, Hemeda LR, Design, synthesis, molecular docking and antiproliferative activity of some novel benzothiazole derivatives targeting EGFR/HER2 and TS, Bioorg. Chem 101 (2020) 103976. 10.1016/j.bioorg.2020.103976. [DOI] [PubMed] [Google Scholar]
- [36].Shibuya M, Vascular endothelial growth factor (VEGF) and its receptor (VEGFR) signaling in angiogenesis: a crucial target for anti-and pro-angiogenic therapies, Genes cancer 2(12) (2011) 1097–1105. 10.1177/1947601911423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hanahan D, Weinberg RA, Hallmarks of cancer: the next generation, cell 144(5) (2011) 646–674. 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- [38].Ivy SP, Wick JY, Kaufman BM, An overview of small-molecule inhibitors of VEGFR signaling, Nat. Rev. Clin. Oncol 6(10) (2009) 569–579. 10.1038/nrclinonc.2009.130. [DOI] [PubMed] [Google Scholar]
- [39].Bhanushali U, Rajendran S, Sarma K, Kulkarni P, Chatti K, Chatterjee S, Ramaa C, 5-Benzylidene-2, 4-thiazolidenedione derivatives: design, synthesis and evaluation as inhibitors of angiogenesis targeting VEGR-2, Bioorg. Chem 67 (2016) 139–147. 10.1016/j.bioorg.2016.06.006. [DOI] [PubMed] [Google Scholar]
- [40].Viegas-Junior C, Danuello A, da Silva Bolzani V, Barreiro EJ, Fraga CAM, Molecular hybridization: a useful tool in the design of new drug prototypes, Curr. Med. Chem 14(17) (2007) 1829–1852. 10.2174/092986707781058805. [DOI] [PubMed] [Google Scholar]
- [41].Ivasiv V, Albertini C, Gonçalves AE, Rossi M, Bolognesi ML, Molecular hybridization as a tool for designing multitarget drug candidates for complex diseases, Curr. Top. Med. Chem 19(19) (2019) 1694–1711. 10.2174/1568026619666190619115735. [DOI] [PubMed] [Google Scholar]
- [42].Huang G, Solano CM, Melendez J, Yu-Alfonzo S, Boonhok R, Min H, Miao J, Chakrabarti D, Yuan Y, Discovery of fast-acting dual-stage antimalarial agents by profiling pyridylvinylquinoline chemical space via copper catalyzed azide-alkyne cycloadditions, Eur. J Med. Chem 209 (2021) 112889. 10.1016/j.ejmech.2020.112889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].El-Helby AGA, Sakr H, Eissa IH, Al-Karmalawy AA, El-Adl K, Benzoxazole/benzothiazole-derived VEGFR-2 inhibitors: design, synthesis, molecular docking, and anticancer evaluations, Arch. Pharm 352(12) (2019) 1900178. 10.1002/ardp.201900178. [DOI] [PubMed] [Google Scholar]
- [44].Upadhyay N, Tilekar K, Safuan S, Kumar AP, Schweipert M, Meyer-Almes F-J, Ramaa C, Development and investigation of thiazolidinedione and pyrazoline compounds as antiangiogenic weapons targeting VEGFR-2, Future Med. Chem 13(22) (2021) 1963–1986. 10.4155/fmc-2021-0139. [DOI] [PubMed] [Google Scholar]
- [45].Reddy VG, Reddy TS, Jadala C, Reddy MS, Sultana F, Akunuri R, Bhargava SK, Wlodkowic D, Srihari P, Kamal A, Pyrazolo-benzothiazole hybrids: Synthesis, anticancer properties and evaluation of antiangiogenic activity using in vitro VEGFR-2 kinase and in vivo transgenic zebrafish model, Eur. J. Med. Chem 182 (2019) 111609. 10.1016/j.ejmech.2019.111609. [DOI] [PubMed] [Google Scholar]
- [46].Dawson JC, Serrels A, Stupack DG, Schlaepfer DD, Frame MC, Targeting FAK in anticancer combination therapies, Nat. Rev. Cancer 21(5) (2021) 313–324. 10.1038/s41568-021-00340-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Wu X, Wang J, Liang Q, Tong R, Huang J, Yang X, Xu Y, Wang W, Sun M, Shi J, Recent progress on FAK inhibitors with dual targeting capabilities for cancer treatment, Biomed. Pharmacother 151 (2022) 113116. 10.1016/j.biopha.2022.113116. [DOI] [PubMed] [Google Scholar]
- [48].Altıntop MD, Sever B, Çiftçi GA, Turan-Zitouni G, Kaplancıklı ZA, Özdemir A, Design, synthesis, in vitro and in silico evaluation of a new series of oxadiazole-based anticancer agents as potential Akt and FAK inhibitors, Eur. J. Med. Chem 155 (2018) 905–924. 10.1016/j.ejmech.2018.06.049. [DOI] [PubMed] [Google Scholar]
- [49].Cui JJ, Targeting receptor tyrosine kinase MET in cancer: small molecule inhibitors and clinical progress, J. Med. Chem 57(11) (2014) 4427–4453. 10.1021/jm401427c. [DOI] [PubMed] [Google Scholar]
- [50].Huang X, Li E, Shen H, Wang X, Tang T, Zhang X, Xu J, Tang Z, Guo C, Bai X, Liang T, Targeting the HGF/MET axis in cancer therapy: challenges in resistance and opportunities for improvement, Front. Cell Dev. Biol 8 (2020) 152. 10.3389/fcell.2020.00152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Parikh PK, Ghate MD, Recent advances in the discovery of small molecule c-Met Kinase inhibitors, Eur. J. Med. Chem 143 (2018) 1103–1138. 10.1016/j.ejmech.2017.08.044. [DOI] [PubMed] [Google Scholar]
- [52].Lei H, Hu G, Wang Y, Han P, Liu Z, Zhao Y, Gong P, Design, synthesis, and biological evaluation of 4-phenoxyquinoline derivatives containing benzo [d] thiazole-2-yl urea as c-Met kinase inhibitors, Arch. Pharm 349(8) (2016) 651–661. 10.1002/ardp.201600003. [DOI] [PubMed] [Google Scholar]
- [53].Nandekar PP, Tumbi KM, Bansal N, Rathod VP, Labhsetwar LB, Soumya N, Singh S, Sangamwar AT, Chem-bioinformatics and in vitro approaches for candidate optimization: a case study of NSC745689 as a promising antitumor agent, Med. Chem. Res 22(8) (2013) 3728–3742. 10.1007/s00044-012-0364-8. [DOI] [Google Scholar]
- [54].Moosavi F, Ebadi A, Mohabbati M, Damghani T, Mortazavi M, Miri R, Firuzi O, Antiproliferative effect, alteration of cancer cell cycle progression and potential MET kinase inhibition induced by 3, 4-dihydropyrimidin-2 (1H)-one C5 amide derivatives, Eur. J. Pharmacol 894 (2021) 173850. 10.1016/j.ejphar.2021.173850. [DOI] [PubMed] [Google Scholar]
- [55].Ugolini A, Kenigsberg M, Rak A, Vallée F, Houtmann J, Lowinski M, Capdevila C.c., Khider J, Albert E, Martinet N, Nemecek C, Grapinet S, Bacqué E, Roesner M, Delaisi C, Calvet L, Bonche F, Semiond D, Egile C, Goulaouic H, Schio L, Discovery and pharmacokinetic and pharmacological properties of the potent and selective MET kinase inhibitor 1-{6-[6-(4-fluorophenyl)-[1, 2, 4] triazolo [4, 3-b] pyridazin-3-ylsulfanyl] benzothiazol-2-yl}-3-(2-morpholin-4-ylethyl) urea (SAR125844), J. Med. Chem 59 (2016) 7066–7074. 10.1021/acs.jmedchem.6b00280. [DOI] [PubMed] [Google Scholar]
- [56].Capra M, Nuciforo PG, Confalonieri S, Quarto M, Bianchi M, Nebuloni M, Boldorini R, Pallotti F, Viale G, Gishizky ML, Frequent alterations in the expression of serine/threonine kinases in human cancers, Cancer Res. 66(16) (2006) 8147–8154. 10.1158/0008-5472.CAN-05-3489. [DOI] [PubMed] [Google Scholar]
- [57].Keen N, Taylor S, Aurora-kinase inhibitors as anticancer agents, Nat. Rev. Cancer 4(12) (2004) 927–936. 10.1038/nrc1502. [DOI] [PubMed] [Google Scholar]
- [58].Lee E, An Y, Kwon J, Kim KI, Jeon R, Optimization and biological evaluation of 2-aminobenzothiazole derivatives as Aurora B kinase inhibitors, Bioorg. Med. Chem 25(14) (2017) 3614–3622. 10.1016/j.bmc.2017.04.004. [DOI] [PubMed] [Google Scholar]
- [59].Malumbres M, Cyclin-dependent kinases, Genome Biol. 15(6) (2014) 1–10. 10.1186/gb4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Roskoski R Jr, Cyclin-dependent protein kinase inhibitors including palbociclib as anticancer drugs, Pharmacol. Res 107 (2016) 249–275. 10.1016/j.phrs.2016.03.012. [DOI] [PubMed] [Google Scholar]
- [61].Zhang M, Zhang L, Hei R, Li X, Cai H, Wu X, Zheng Q, Cai C, CDK inhibitors in cancer therapy, an overview of recent development, Am. J. Cancer Res 11(5) (2021) 1913. [PMC free article] [PubMed] [Google Scholar]
- [62].Lapenna S, Giordano A, Cell cycle kinases as therapeutic targets for cancer, Nat. Rev. Drug Discov 8(7) (2009) 547–566. 10.1038/nrd2907. [DOI] [PubMed] [Google Scholar]
- [63].Diao P, Lin W, Jian X, Li Y, You W, Zhao P, Discovery of novel pyrimidine-based benzothiazole derivatives as potent cyclin-dependent kinase 2 inhibitors with anticancer activity, Eur. J. Med. Chem 179 (2019) 196–207. 10.1016/j.ejmech.2019.06.055. [DOI] [PubMed] [Google Scholar]
- [64].Abdelazeem AH, Alqahtani AM, Omar HA, Bukhari SNA, Gouda AM, Synthesis, biological evaluation and kinase profiling of novel S-benzo [4, 5] thiazolo [2, 3-c][1, 2, 4] triazole derivatives as cytotoxic agents with apoptosis-inducing activity, J. Mol. Struct 1219 (2020) 128567. 10.1016/j.molstruc.2020.128567. [DOI] [Google Scholar]
- [65].Hegde M, Vartak SV, Kavitha CV, Ananda H, Prasanna DS, Gopalakrishnan V, Choudhary B, Rangappa KS, Raghavan SC, A benzothiazole derivative (5g) induces DNA damage and potent G2/M arrest in cancer cells, Sci. Rep 7(1) (2017) 1–14. 10.1038/s41598-017-02489-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Knippschild U, Krüger M, Richter J, Xu P, García-Reyes B, Peifer C, Halekotte J, Bakulev V, Bischof J, The CK1 family: contribution to cellular stress response and its role in carcinogenesis, Front. Oncol 4 (2014) 96. 10.3389/fonc.2014.00096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Cheong JK, Virshup DM, Casein kinase 1: Complexity in the family, Int. J. Biochem. Cell Biol 43(4) (2011) 465–469. 10.1016/j.biocel.2010.12.004. [DOI] [PubMed] [Google Scholar]
- [68].Garcia-Reyes B, Witt L, Jansen B.r., Karasu E, Gehring T, Leban J, Henne-Bruns D, Pichlo C, Brunstein E, Baumann U, Wesseler F, Rathmer B, Schade D, Peifer C, Knippschild U, Discovery of inhibitor of wnt production 2 (IWP-2) and related compounds as selective ATP-competitive inhibitors of casein kinase 1 (CK1) δ/ε, J. Med. Chem 61(9) (2018) 4087–4102. 10.1021/acs.jmedchem.8b00095. [DOI] [PubMed] [Google Scholar]
- [69].Liu C, Witt L, Ianes C, Bischof J, Bammert M-T, Baier J, Kirschner S, Henne-Bruns D, Xu P, Kornmann M, Peifer C, Knippschild U, Newly developed CK1-specific inhibitors show specifically stronger effects on CK1 mutants and colon cancer cell lines, Int. J. Mol. Sci 20(24) (2019) 6184. 10.3390/ijms20246184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Cozza G, Pinna LA, Casein kinases as potential therapeutic targets, Expert Opin. Ther. Targets 20(3) (2016) 319–340. 10.1517/14728222.2016.1091883. [DOI] [PubMed] [Google Scholar]
- [71].Qiao Y, Chen T, Yang H, Chen Y, Lin H, Qu W, Feng F, Liu W, Guo Q, Liu Z, Sun H, Small molecule modulators targeting protein kinase CK1 and CK2, Eur. J. Med. Chem 181 (2019) 111581. 10.1016/j.ejmech.2019.111581. [DOI] [PubMed] [Google Scholar]
- [72].Wang Q, Hu X, Shi W, Long H, Wang H, Design, synthesis and biological evaluation of chromone derivatives as novel protein kinase CK2 inhibitors, Bioorg. Med. Chem. Lett (2022) 128799. 10.1016/j.bmcl.2022.128799. [DOI] [PubMed] [Google Scholar]
- [73].Agianian B, Gavathiotis E, Current insights of BRAF inhibitors in cancer: miniperspective, J. Med. Chem 61(14) (2018) 5775–5793. 10.1021/acs.jmedchem.7b01306. [DOI] [PubMed] [Google Scholar]
- [74].Roskoski R Jr, RAF protein-serine/threonine kinases: structure and regulation, Biochem. Biophys. Res. Commun 399(3) (2010) 313–317. 10.1016/j.bbrc.2010.07.092. [DOI] [PubMed] [Google Scholar]
- [75].Madhunapantula SV, Robertson GP, Is B-Raf a good therapeutic target for melanoma and other malignancies?, Cancer Res. 68(1) (2008) 5–8. 10.1158/0008-5472.CAN-07-2038. [DOI] [PubMed] [Google Scholar]
- [76].Khan PS, Rajesh P, Rajendra P, Chaskar MG, Rohidas A, Jaiprakash S, Recent Advances in B-RAF Inhibitors as Anticancer Agents, Bioorg. Chem (2022) 105597. 10.1016/j.bioorg.2022.105597. [DOI] [PubMed] [Google Scholar]
- [77].Jilaveanu LB, Zito CR, Aziz SA, Conrad PJ, Schmitz JC, Sznol M, Camp RL, Rimm DL, Kluger HM, C-Raf Is Associated with Disease Progression and Cell Proliferation in a Subset of Melanomas C-Raf in Melanoma, Clin. Cancer Res 15(18) (2009) 5704–5713. 10.1158/1078-0432.CCR-09-0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Okaniwa M, Hirose M, Arita T, Yabuki M, Nakamura A, Takagi T, Kawamoto T, Uchiyama N, Sumita A, Tsutsumi S, Tottori T, Inui Y, Sang B-C, Yano J, Aertgeerts K, Yoshida S, Ishikawa T, Discovery of a selective kinase inhibitor (TAK-632) targeting pan-RAF inhibition: design, synthesis, and biological evaluation of C-7-substituted 1, 3-benzothiazole derivatives, J. Med. Chem 56(16) (2013) 6478–6494. 10.1021/jm400778d. [DOI] [PubMed] [Google Scholar]
- [79].El-Damasy AK, Lee J-H, Seo SH, Cho N-C, Pae AN, Keum G, Design and synthesis of new potent anticancer benzothiazole amides and ureas featuring pyridylamide moiety and possessing dual B-RafV600E and C-Raf kinase inhibitory activities, Eur. J. Med. Chem 115 (2016) 201–216. 10.1016/j.ejmech.2016.02.039. [DOI] [PubMed] [Google Scholar]
- [80].Aranda S, Laguna A, Luna S.d.l., DYRK family of protein kinases: evolutionary relationships, biochemical properties, and functional roles, FASEB J. 25(2) (2011) 449–462. 10.1096/fj.10-165837. [DOI] [PubMed] [Google Scholar]
- [81].Yuan K, Li Z, Kuang W, Wang X, Ji M, Chen W, Ding J, Li J, Min W, Sun C, Ye X, Lu M, Wang L, Ge H, Jiang Y, Hao H, Xiao Y, Yang P, Targeting dual-specificity tyrosine phosphorylation-regulated kinase 2 with a highly selective inhibitor for the treatment of prostate cancer, Nat. Commun 13(1) (2022) 1–15. 10.1038/s41467-022-30581-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Liu P, Cheng H, Roberts TM, Zhao JJ, Targeting the phosphoinositide 3-kinase pathway in cancer, Nat. Rev. Drug Discov 8(8) (2009) 627–644. 10.1038/nrd2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Bauer TM, Patel MR, Infante JR, Targeting PI3 kinase in cancer, Pharmacol. Ther 146 (2015) 53–60. 10.1016/j.pharmthera.2014.09.006. [DOI] [PubMed] [Google Scholar]
- [84].Akinleye A, Avvaru P, Furqan M, Song Y, Liu D, Phosphatidylinositol 3-kinase (PI3K) inhibitors as cancer therapeutics, J. Hematol. Oncol 6(1) (2013) 1–17. 10.1186/1756-8722-6-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Sabbah DA, Hajjo R, Bardaweel SK, Zhong HA, Phosphatidylinositol 3-kinase (PI3K) inhibitors: A recent update on inhibitor design and clinical trials (2016–2020), Expert Opin. Ther. Pat 31(10) (2021) 877–892. 10.1080/13543776.2021.1924150. [DOI] [PubMed] [Google Scholar]
- [86].Knight ZA, Gonzalez B, Feldman ME, Zunder ER, Goldenberg DD, Williams O, Loewith R, Stokoe D, Balla A, Toth B, Balla T, Weiss WA, Williams RL, Shokat KM, A pharmacological map of the PI3-K family defines a role for p110α in insulin signaling, Cell 125(4) (2006) 733–747. 10.1016/j.cell.2006.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Collier PN, Martinez-Botella G, Cornebise M, Cottrell KM, Doran JD, Griffith JP, Mahajan S, Maltais F, Moody CS, Huck EP, Wang T, Aronov AM, Structural basis for isoform selectivity in a class of benzothiazole inhibitors of phosphoinositide 3-kinase γ, J. Med. Chem 58(1) (2015) 517–521. 10.1021/jm500362j. [DOI] [PubMed] [Google Scholar]
- [88].Collier PN, Messersmith D, Le Tiran A, Bandarage UK, Boucher C, Come J, Cottrell KM, Damagnez V, Doran JD, Griffith JP, Khare-Pandit S, Krueger EB, Ledeboer MW, Ledford B, Liao Y, Mahajan S, Moody CS, Roday S, Wang T, Xu J, Aronov AM, Discovery of highly isoform selective thiazolopiperidine inhibitors of phosphoinositide 3-kinase γ, J. Med. Chem 58(14) (2015) 5684–5688. 10.1021/acs.jmedchem.5b00498. [DOI] [PubMed] [Google Scholar]
- [89].Cao S, Cao R, Liu X, Luo X, Zhong W, Design, synthesis and biological evaluation of novel benzothiazole derivatives as selective PI3Kβ inhibitors, Molecules 21(7) (2016) 876. 10.3390/molecules21070876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Haider K, Ahmad K, Najmi AK, Das S, Joseph A, Shahar Yar M, Design, synthesis, biological evaluation, and in silico studies of 2-aminobenzothiazole derivatives as potent PI3Kα inhibitors, Arch. Pharm (2022) e2200146. 10.1002/ardp.202200146. [DOI] [PubMed] [Google Scholar]
- [91].Khadka DB, Cho W-J, Topoisomerase inhibitors as anticancer agents: a patent update, Expert Opin. Ther. Pat 23(8) (2013) 1033–1056. 10.1517/13543776.2013.790958. [DOI] [PubMed] [Google Scholar]
- [92].Nitiss JL, DNA topoisomerase II and its growing repertoire of biological functions, Nat. Rev. Cancer 9(5) (2009) 327–337. 10.1038/nrc2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Nitiss JL, Targeting DNA topoisomerase II in cancer chemotherapy, Nat. Rev. Cancer 9(5) (2009) 338–350. 10.1038/nrc2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Pommier Y, Topoisomerase I inhibitors: camptothecins and beyond, Nat. Rev. Cancer 6(10) (2006) 789–802. 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- [95].Nagaraju B, Kovvuri J, Kumar CG, Routhu SR, Shareef MA, Kadagathur M, Adiyala PR, Alavala S, Nagesh N, Kamal A, Synthesis and biological evaluation of pyrazole linked benzothiazole-β-naphthol derivatives as topoisomerase I inhibitors with DNA binding ability, Bioorg. Med. Chem 27(5) (2019) 708–720. 10.1016/j.bmc.2019.01.011. [DOI] [PubMed] [Google Scholar]
- [96].Sović I, Jambon S, Pavelić SK, Markova-Car E, Ilić N, Depauw S, David-Cordonnier M-H, Karminski-Zamola G, Synthesis, antitumor activity and DNA binding features of benzothiazolyl and benzimidazolyl substituted isoindolines, Bioorg. Med. Chem 26(8) (2018) 1950–1960. 10.1016/j.bmc.2018.02.045 [DOI] [PubMed] [Google Scholar]
- [97].Tokala R, Mahajan S, Kiranmai G, Sigalapalli DK, Sana S, John SE, Nagesh N, Shankaraiah N, Development of β-carboline-benzothiazole hybrids via carboxamide formation as cytotoxic agents: DNA intercalative topoisomerase IIα inhibition and apoptosis induction, Bioorg. Chem 106 (2021) 104481. 10.1016/j.bioorg.2020.104481. [DOI] [PubMed] [Google Scholar]
- [98].Yap JL, Chen L, Lanning ME, Fletcher S, Expanding the Cancer Arsenal with Targeted Therapies: Disarmament of the antiapoptotic Bcl-2 proteins by small molecules: miniperspective, J. Med. Chem 60(3) (2017) 821–838. 10.1021/acs.jmedchem.5b01888. [DOI] [PubMed] [Google Scholar]
- [99].Zhang Z, Bai L, Hou L, Deng H, Luan S, Liu D, Huang M, Zhao L, Trends in targeting Bcl-2 anti-apoptotic proteins for cancer treatment, Eur. J. Med. Chem (2022) 114184. 10.1016/j.ejmech.2022.114184. [DOI] [PubMed] [Google Scholar]
- [100].Khan S, Zhang X, Lv D, Zhang Q, He Y, Zhang P, Liu X, Thummuri D, Yuan Y, Wiegand JS, Pei J, Zhang W, Sharma A, McCurdy CR, Kuruvilla VM, Baran N, Ferrando AA, Kim Y.-m., Rogojina A, Houghton PJ, Huang G, Hromas R, Konopleva M, Zheng G, Zhou D, A selective BCL-XL PROTAC degrader achieves safe and potent antitumor activity, Nat. Med 25(12) (2019) 1938–1947. 10.1038/s41591-019-0668-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Tao Z, Hasvold L, Wang L, Wang X, Petros AM, Park CH, Boghaert ER, Catron ND, Chen J, Colman PM, Czabotar PE, Deshayes K, Fairbrother WJ, Flygare JA, Hymowitz SG, Jin S, Judge RA, Nimmer P, Purkey HE, Oleksijew A, Phillips DC, Sleebs BE, Smith BJ, Smith ML, Tahir SK, Watson KG, Xiao Y, Xue J, Zhang H, Zobel K, Rosenberg SH, Tse C, Leverson JD, Elmore SW, Souers AJ, Discovery of a potent and selective BCL-XL inhibitor with in vivo activity, ACS Med. Chem. Lett 5(10) (2014) 1088–1093. 10.1021/ml5001867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Leverson JD, Phillips DC, Mitten MJ, Boghaert ER, Diaz D, Tahir SK, Belmont LD, Nimmer P, Xiao Y, Ma XM, Lowes KN, Kovar P, Chen J, Jin S, Smith M, Xue J, Zhang H, Oleksijew A, Magoc TJ, Vaidya KS, Albert DH, Tarrant JM, Li N, Wang L, Tao Z-F, Wendt MD, Sampath D, Rosenberg SH, Tse C, Huang DCS, Fairbrother WJ, Elmore SW, Souers AJ, Exploiting selective BCL-2 family inhibitors to dissect cell survival dependencies and define improved strategies for cancer therapy, Sci. Transl. Med 7(279) (2015) 279ra40. 10.1126/scitranslmed.aaa4642. [DOI] [PubMed] [Google Scholar]
- [103].Wang L, Doherty GA, Judd AS, Tao Z-F, Hansen TM, Frey RR, Song X, Bruncko M, Kunzer AR, Wang X, Wendt MD, Flygare JA, Catron ND, Judge RA, Park CH, Shekhar S, Phillips DC, Nimmer P, Smith ML, Tahir SK, Xiao Y, Xue J, Zhang H, Le PN, Mitten MJ, Boghaert ER, Gao W, Kovar P, Choo EF, Diaz D, Fairbrother WJ, Elmore SW, Sampath D, Leverson JD, Souers AJ, Discovery of A-1331852, a first-in-class, potent, and orally-bioavailable BCL-XL inhibitor, ACS Med. Chem. Lett 11(10) (2020) 1829–1836. 10.1021/acsmedchemlett.9b00568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Tao Z, Wang X, Chen J, Ingram JP, Jin S, Judge RA, Kovar PJ, Park C, Sun C, Wakefield BD, Zhou L, Zhang H, Elmore SW, Phillips DC, Judd AS, Leverson JD, Souers AJ, Structure-based design of A-1293102, a potent and selective BCL-XL inhibitor, ACS Med. Chem. Lett 12(6) (2021) 1011–1016. 10.1021/acsmedchemlett.1c00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Khan S, He Y, Zhang X, Yuan Y, Pu S, Kong Q, Zheng G, Zhou D, Proteolysis targeting chimeras (PROTACs) as emerging anticancer therapeutics, Oncogene 39(26) (2020) 4909–4924. 10.1038/s41388-020-1336-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Dale B, Cheng M, Park K-S, Kaniskan HÜ, Xiong Y, Jin J, Advancing targeted protein degradation for cancer therapy, Nat. Rev. Cancer 21(10) (2021) 638–654. 10.1038/s41568-021-00365-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Békés M, Langley DR, Crews CM, PROTAC targeted protein degraders: the past is prologue, Nat. Rev. Drug Discov 21(3) (2022) 181–200. 10.1038/s41573-021-00371-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Guenette RG, Yang SW, Min J, Pei B, Potts PR, Target and tissue selectivity of PROTAC degraders, Chem. Soc. Rev 51 (2022) 5740–5756. 10.1039/D2CS00200K. [DOI] [PubMed] [Google Scholar]
- [109].Chung C.-w., Dai H, Fernandez E, Tinworth CP, Churcher I, Cryan J, Denyer J, Harling JD, Konopacka A, Queisser MA, Tame CJ, Watt G, Jiang F, Qian D, Benowitz AB, Structural insights into PROTAC-mediated degradation of Bcl-xL, ACS Chem. Biol 15(9) (2020) 2316–2323. 10.1021/acschembio.0c00266. [DOI] [PubMed] [Google Scholar]
- [110].Kamal A, Thao L, Sensintaffar J, Zhang L, Boehm MF, Fritz LC, Burrows FJ, A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors, Nature 425(6956) (2003) 407–410. 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- [111].Neckers L, Hsp90 inhibitors as novel cancer chemotherapeutic agents, Trends Mol. Med 8(4) (2002) S55–S61. 10.1016/S1471-4914(02)02316-X. [DOI] [PubMed] [Google Scholar]
- [112].Gupta SD, Bommaka MK, Banerjee A, Inhibiting protein-protein interactions of Hsp90 as a novel approach for targeting cancer, Eur. J. Med. Chem 178 (2019) 48–63. 10.1016/j.ejmech.2019.05.073. [DOI] [PubMed] [Google Scholar]
- [113].Pugh KW, Zhang Z, Wang J, Xu X, Munthali V, Zuo A, Blagg BS, From bacteria to cancer: a benzothiazole-based DNA gyrase B inhibitor redesigned for Hsp90 C-terminal inhibition, ACS Med. Chem. Lett 11(8) (2020) 1535–1538. 10.1021/acsmedchemlett.0c00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Dernovšek J, Zajec Ž, Durcik M, Mašič LP, Gobec M, Zidar N, Tomašič T, Structure-activity relationships of benzothiazole-based Hsp90 C-terminal-domain inhibitors, Pharmaceutics 13(8) (2021) 1283. 10.3390/pharmaceutics13081283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Jin L, Huang R, Huang X, Zhang B, Ji M, Wang H, Discovery of 18β-glycyrrhetinic acid conjugated aminobenzothiazole derivatives as Hsp90-Cdc37 interaction disruptors that inhibit cell migration and reverse drug resistance, Bioorg. Med. Chem 26(8) (2018) 1759–1775. 10.1016/j.bmc.2018.02.021. [DOI] [PubMed] [Google Scholar]
- [116].Bykov VJ, Eriksson SE, Bianchi J, Wiman KG, Targeting mutant p53 for efficient cancer therapy, Nat. Rev. Cancer 18(2) (2018) 89–102. 10.1038/nrc.2017.109. [DOI] [PubMed] [Google Scholar]
- [117].Gomes AS, Ramos H, Inga A, Sousa E, Saraiva L, Structural and drug targeting insights on mutant p53, Cancers 13(13) (2021) 3344. 10.3390/cancers13133344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Hu J, Cao J, Topatana W, Juengpanich S, Li S, Zhang B, Shen J, Cai L, Cai X, Chen M, Targeting mutant p53 for cancer therapy: Direct and indirect strategies, J. Hematol. Oncol 14(1) (2021) 1–19. 10.1186/s13045-021-01169-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Zhang S, Carlsen L, Hernandez Borrero L, Seyhan AA, Tian X, El-Deiry WS, Advanced strategies for therapeutic targeting of wild-type and mutant p53 in cancer, Biomolecules 12(4) (2022) 548. 10.3390/biom12040548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Baud MG, Bauer MR, Verduci L, Dingler FA, Patel KJ, Roy DH, Joerger AC, Fersht AR, Aminobenzothiazole derivatives stabilize the thermolabile p53 cancer mutant Y220C and show anticancer activity in p53-Y220C cell lines, Eur. J. Med. Chem 152 (2018) 101–114. 10.1016/j.ejmech.2018.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Gilleran JA, Yu X, Blayney AJ, Bencivenga AF, Na B, Augeri DJ, Blanden AR, Kimball SD, Loh SN, Roberge JY, Carpizo DR, Benzothiazolyl and benzoxazolyl hydrazones function as zinc metallochaperones to reactivate mutant p53, J. Med. Chem 64(4) (2021) 2024–2045. 10.1021/acs.jmedchem.0c01360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Glozak M, Seto E, Histone deacetylases and cancer, Oncogene 26(37) (2007) 5420–5432. 10.1038/sj.onc.1210610. [DOI] [PubMed] [Google Scholar]
- [123].Bondarev AD, Attwood MM, Jonsson J, Chubarev VN, Tarasov VV, Schiöth HB, Recent developments of HDAC inhibitors: Emerging indications and novel molecules, Br. J. Clin. Pharmacol 87(12) (2021) 4577–4597. 10.1111/bcp.14889. [DOI] [PubMed] [Google Scholar]
- [124].Zagni C, Floresta G, Monciino G, Rescifina A, The search for potent, small-molecule HDACIs in cancer treatment: a decade after vorinostat, Med. Res. Rev 37(6) (2017) 1373–1428. 10.1002/med.21437. [DOI] [PubMed] [Google Scholar]
- [125].Ho TC, Chan AH, Ganesan A, Thirty years of HDAC inhibitors: 2020 insight and hindsight, J. Med. Chem 63(21) (2020) 12460–12484. 10.1021/acs.jmedchem.0c00830. [DOI] [PubMed] [Google Scholar]
- [126].Xu Q, Liu C, Zang J, Gao S, Chou CJ, Zhang Y, Discovery of a novel hybrid of vorinostat and riluzole as a potent antitumor agent, Front. Cell Dev. Biol 8 (2020) 454. 10.3389/fcell.2020.00454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Sun S, Zhao W, Li Y, Chi Z, Fang X, Wang Q, Han Z, Luan Y, Design, synthesis and antitumor activity evaluation of novel HDAC inhibitors with tetrahydrobenzothiazole as the skeleton, Bioorg. Chem 108 (2021) 104652. 10.1016/j.bioorg.2021.104652. [DOI] [PubMed] [Google Scholar]
- [128].Upadhyay N, Tilekar K, Jänsch N, Schweipert M, Hess JD, Macias LH, Mrowka P, Aguilera RJ, Choe J.-y., Meyer-Almes F-J, Ramaa CS, Discovery of novel N-substituted thiazolidinediones (TZDs) as HDAC8 inhibitors: in-silico studies, synthesis, and biological evaluation, Bioorg. Chem 100 (2020) 103934. 10.1016/j.bioorg.2020.103934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Tilekar K, Hess JD, Upadhyay N, Schweipert M, Flath F, Gutierrez DA, Loiodice F, Lavecchia A, Meyer-Almes FJ, Aguilera RJ, Ramaa CS, HDAC4 inhibitors with cyclic linker and non-hydroxamate zinc binding group: design, synthesis, HDAC screening and in vitro cytotoxicity evaluation, ChemistrySelect 6(26) (2021) 6748–6763. 10.1002/slct.202102061. [DOI] [Google Scholar]
- [130].Morishita M, di Luccio E, Cancers and the NSD family of histone lysine methyltransferases, Biochim. Biophys. Acta Rev. Cancer 1816(2) (2011) 158–163. 10.1016/j.bbcan.2011.05.004. [DOI] [PubMed] [Google Scholar]
- [131].Topchu I, Pangeni RP, Bychkov I, Miller SA, Izumchenko E, Yu J, Golemis E, Karanicolas J, Boumber Y, The role of NSD1, NSD2, and NSD3 histone methyltransferases in solid tumors, Cell. Mol. Life Sci 79(6) (2022) 1–19. 10.1007/s00018-022-04321-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Yang C, Wang K, Liang Q, Tian T-T, Zhong Z, Role of NSD1 as potential therapeutic target in tumor, Pharmacol. Res 173 (2021) 105888. 10.1016/j.phrs.2021.105888. [DOI] [PubMed] [Google Scholar]
- [133].Tauchmann S, Schwaller J, Nsd1: A lysine methyltransferase between developmental disorders and cancer, Life 11(9) (2021) 877. 10.3390/life11090877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Qiao Q, Li Y, Chen Z, Wang M, Reinberg D, Xu R-M, The structure of NSD1 reveals an autoregulatory mechanism underlying histone H3K36 methylation, J. Biol. Chem 286(10) (2011) 8361–8368. 10.1074/jbc.M110.204115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Huang H, Howard CA, Zari S, Cho HJ, Shukla S, Li H, Ndoj J, González-Alonso P, Nikolaidis C, Abbott J, Rogawski DS, Potopnyk MA, Kempinska K, Miao H, Purohit T, Henderson A, Mapp A, Sulis ML, Ferrando A, Grembecka J, Cierpicki T, Covalent inhibition of NSD1 histone methyltransferase, Nat. Chem. Biol 16(12) (2020) 1403–1410. 10.1038/s41589-020-0626-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Fang Y, Liao G, Yu B, LSD1/KDM1A inhibitors in clinical trials: advances and prospects, J. Hematol. Oncol 12(1) (2019) 1–14. 10.1186/s13045-019-0811-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Yang G-J, Lei P-M, Wong S-Y, Ma D-L, Leung C-H, Pharmacological inhibition of LSD1 for cancer treatment, Molecules 23(12) (2018) 3194. 10.3390/molecules23123194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Dong J, Pervaiz W, Tayyab B, Li D, Kang L, Zhang H, Gong H, Ma X, Li J, Agboyibor C, Bi Y, Liu H, A comprehensive comparative study on LSD1 in different cancers and tumor specific LSD1 inhibitors, Eur. J. Med. Chem (2022) 114564. 10.1016/j.ejmech.2022.114564. [DOI] [PubMed] [Google Scholar]
- [139].Alnabulsi S, Al-Hurani EA, El-Elimat T, Amino-carboxamide benzothiazoles as potential LSD1 hit inhibitors. Part I: Computational fragment-based drug design, J. Mol. Graph 93 (2019) 107440. 10.1016/j.jmgm.2019.107440. [DOI] [PubMed] [Google Scholar]
- [140].Deng L-J, Deng W-Q, Fan S-R, Chen M-F, Qi M, Lyu W-Y, Qi Q, Tiwari AK, Chen J-X, Zhang D-M, Chen Z-S, m6A modification: recent advances, anticancer targeted drug discovery and beyond, Mol. Cancer 21(1) (2022) 1–21. 10.1186/s12943-022-01510-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Huang Y, Su R, Sheng Y, Dong L, Dong Z, Xu H, Ni T, Zhang ZS, Zhang T, Li C, Han L, Zhu Z, Lian F, Wei J, Deng Q, Wang Y, Wunderlich M, Gao Z, Pan G, Zhong D, Zhou H, Zhang N, Gan J, Jiang H, Mulloy JC, Qian Z, Chen J, Yang C-G, Small-molecule targeting of oncogenic FTO demethylase in acute myeloid leukemia, Cancer cell 35(4) (2019) 677–691. e10. 10.1016/j.ccell.2019.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Zuidhof HR, Calkhoven CF, Oncogenic and tumor-suppressive functions of the RNA demethylase FTO, Cancer Res. 82 (2022) 2201–2212. 10.1158/0008-5472.CAN-21-3710. [DOI] [PubMed] [Google Scholar]
- [143].Li Y, Su R, Deng X, Chen Y, Chen J, FTO in cancer: functions, molecular mechanisms, and therapeutic implications, Trends Cancer 8(7) (2022) 598–614. 10.1016/j.trecan.2022.02.010. [DOI] [PubMed] [Google Scholar]
- [144].Huff S, Tiwari SK, Gonzalez GM, Wang Y, Rana TM, m6A-RNA demethylase FTO inhibitors impair self-renewal in glioblastoma stem cells, ACS Chem. Biol 16(2) (2021) 324–333. 10.1021/acschembio.0c00841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Chini MG, Giordano A, Potenza M, Terracciano S, Fischer K, Vaccaro MC, Colarusso E, Bruno I, Riccio R, Koeberle A, Werz O, Bifulco G, Targeting mPGES-1 by a combinatorial approach: Identification of the aminobenzothiazole scaffold to suppress PGE2 levels, ACS Med. Chem. Lett 11(5) (2020) 783–789. 10.1021/acsmedchemlett.9b00618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Chang H-H, Meuillet EJ, Identification and development of mPGES-1 inhibitors: where we are at?, Future Med. Chem 3(15) (2011) 1909–1934. 10.4155/fmc.11.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Oatman N, Dasgupta N, Arora P, Choi K, Gawali MV, Gupta N, Parameswaran S, Salomone J, Reisz JA, Lawler S, Furnari F, Brennan C, Wu J, Sallans L, Gudelsky G, Desai P, Gebelein B, Weirauch MT, D’Alessandro A, Komurov K, Dasgupta B, Mechanisms of stearoyl CoA desaturase inhibitor sensitivity and acquired resistance in cancer, Sci. Adv 7(7) (2021) eabd7459. 10.1126/sciadv.abd7459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Kubota CS, Espenshade PJ, Targeting Stearoyl-CoA Desaturase in Solid Tumors, Cancer Res. 82(9) (2022) 1682–1688. 10.1158/0008-5472.CAN-21-4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Tracz-Gaszewska Z, Dobrzyn P, Stearoyl-CoA desaturase 1 as a therapeutic target for the treatment of cancer, Cancers 11(7) (2019) 948. 10.3390/cancers11070948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Theodoropoulos PC, Gonzales SS, Winterton SE, Rodriguez-Navas C, McKnight JS, Morlock LK, Hanson JM, Cross B, Owen AE, Duan Y, Moreno JR, Lemoff A, Mirzaei H, Posner BA, Williams NS, Ready JM, Nijhawan D, Discovery of tumor-specific irreversible inhibitors of stearoyl CoA desaturase, Nat. Chem. Biol 12(4) (2016) 218–225. 10.1038/nchembio.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Williams NS, Gonzales S, Naidoo J, Rivera-Cancel G, Voruganti S, Mallipeddi P, Theodoropoulos PC, Geboers S, Chen H, Ortiz F, Posner B, Nijhawan D, Ready JM, Tumor-activated benzothiazole inhibitors of stearoyl-CoA desaturase, J. Med. Chem 63(17) (2020) 9773–9786. 10.1021/acs.jmedchem.0c00899. [DOI] [PubMed] [Google Scholar]
- [152].Supuran CT, Carbonic anhydrases: novel therapeutic applications for inhibitors and activators, Nat. Rev. Drug Discov 7(2) (2008) 168–181. 10.1038/nrd2467 [DOI] [PubMed] [Google Scholar]
- [153].Alterio V, Di Fiore A, D’Ambrosio K, Supuran CT, De Simone G, Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms?, Chem. Rev 112(8) (2012) 4421–4468. 10.1021/cr200176r [DOI] [PubMed] [Google Scholar]
- [154].Supuran CT, Carbonic anhydrase inhibitors as emerging agents for the treatment and imaging of hypoxic tumors, Expert Opin. Investig. Drugs 27(12) (2018) 963–970. 10.1080/13543784.2018.1548608 [DOI] [PubMed] [Google Scholar]
- [155].Petrou A, Geronikaki A, Terzi E, Guler OO, Tuccinardi T, Supuran CT, Inhibition of carbonic anhydrase isoforms I, II, IX and XII with secondary sulfonamides incorporating benzothiazole scaffolds, J. Enzyme Inhib. Med. Chem 31(6) (2016) 1306–1311. 10.3109/14756366.2015.1128427 [DOI] [PubMed] [Google Scholar]
- [156].Abdoli M, Angeli A, Bozdag M, Carta F, Kakanejadifard A, Saeidian H, Supuran CT, Synthesis and carbonic anhydrase I, II, VII, and IX inhibition studies with a series of benzo [d] thiazole-5-and 6-sulfonamides, J. Enzyme Inhib. Med. Chem 32(1) (2017) 1071–1078. 10.1080/14756366.2017.1356295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Ibrahim DA, Lasheen DS, Zaky MY, Ibrahim AW, Vullo D, Ceruso M, Supuran CT, Abou El Ella DA, Design and synthesis of benzothiazole-6-sulfonamides acting as highly potent inhibitors of carbonic anhydrase isoforms I, II, IX and XII, Bioorg. Med. Chem 23(15) (2015) 4989–4999. 10.1016/j.bmc.2015.05.019 [DOI] [PubMed] [Google Scholar]
- [158].Manzoor S, Angeli A, Zara S, Carradori S, Rahman MA, Raza MK, Supuran CT, Hoda N, Development of benzene and benzothiazole-sulfonamide analogues as selective inhibitors of the tumor-associated carbonic anhydrase IX, Eur. J. Med. Chem 243 (2022) 114793. 10.1016/j.ejmech.2022.114793 [DOI] [PubMed] [Google Scholar]
- [159].Al-Warhi T, Elbadawi MM, Bonardi A, Nocentini A, Al-Karmalawy AA, Aljaeed N, Alotaibi OJ, Abdel-Aziz HA, Supuran CT, Eldehna WM, Design and synthesis of benzothiazole-based SLC-0111 analogues as new inhibitors for the cancer-associated carbonic anhydrase isoforms IX and XII, J. Enzyme Inhib. Med. Chem 37(1) (2022) 2635–2643. 10.1080/14756366.2022.2124409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Cheng Y, Mo F, Li Q, Han X, Shi H, Chen S, Wei Y, Wei X, Targeting CXCR2 inhibits the progression of lung cancer and promotes therapeutic effect of cisplatin, Mol. Cancer 20(1) (2021) 1–21. 10.1186/s12943-021-01355-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Singh JK, Farnie G, Bundred NJ, Simões BM, Shergill A, Landberg G, Howell SJ, Clarke RB, Targeting CXCR1/2 Significantly Reduces Breast Cancer Stem Cell Activity and Increases the Efficacy of Inhibiting HER2 via HER2-Dependent and-Independent MechanismsIL-8 Regulates Human Breast Cancer Stem Cell Activity, Clin. Cancer Res 19(3) (2013) 643–656. 10.1158/1078-0432.CCR-12-1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Grépin R, Guyot M, Giuliano S, Boncompagni M, Ambrosetti D, Chamorey E, Scoazec J-Y, Negrier S, Simonnet H, The CXCL7/CXCR1/2 axis is a key driver in the growth of clear cell renal cell carcinoma Cancer Res. 74(3) (2014) 873–883. 10.1158/0008-5472.CAN-13-1267. [DOI] [PubMed] [Google Scholar]
- [163].Mehanna WE, Lu T, Debnath B, Lasheen DS, Serya RA, Abouzid KA, Neamati N, Synthesis, ADMET properties, and biological evaluation of benzothiazole compounds targeting chemokine receptor 2 (CXCR2), ChemMedChem 12(13) (2017) 1045–1054. 10.1002/cmdc.201700229. [DOI] [PubMed] [Google Scholar]
- [164].Dufies M, Grytsai O, Ronco C, Camara O, Ambrosetti D, Hagege A, Parola J, Mateo L, Ayrault M, Giuliano S, Grépin R, Lagarde N, Montes M, Auberger P, Demange L, Benhida R, Pagès G, New CXCR1/CXCR2 inhibitors represent an effective treatment for kidney or head and neck cancers sensitive or refractory to reference treatments, Theranostics 9(18) (2019) 5332. 10.7150/thno.34681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Utterback RD, Tomat E, Developing ligands to target transition metals in cancer, Encycl. Inorg. Bioinorg. Chem (2011) 1–19. 10.1002/9781119951438.eibc2694. [DOI] [Google Scholar]
- [166].Richardson DR, Molecular mechanisms of iron uptake by cells and the use of iron chelators for the treatment of cancer, Curr. Med. Chem 12(23) (2005) 2711–2729. 10.2174/092986705774462996. [DOI] [PubMed] [Google Scholar]
- [167].Merlot AM, Kalinowski DS, Richardson DR, Novel chelators for cancer treatment: where are we now?, Antioxid. Redox Signal 18(8) (2013) 973–1006. 10.1089/ars.2012.4540. [DOI] [PubMed] [Google Scholar]
- [168].Chen G, Niu C, Yi J, Sun L, Cao H, Fang Y, Jin T, Li Y, Lou C, Kang J, Wei W, Zhu J, Novel triapine derivative induces copper-dependent cell death in hematopoietic cancers, J. Med. Chem 62(6) (2019) 3107–3121. 10.1021/acs.jmedchem.8b01996. [DOI] [PubMed] [Google Scholar]
- [169].Pape VF, Tóth S, Füredi A, Szebényi K, Lovrics A, Szabó P, Wiese M, Szakács G, Design, synthesis and biological evaluation of thiosemicarbazones, hydrazinobenzothiazoles and arylhydrazones as anticancer agents with a potential to overcome multidrug resistance, Eur. J. Med. Chem 117 (2016) 335–354. 10.1016/j.ejmech.2016.03.078. [DOI] [PubMed] [Google Scholar]
- [170].Besleaga I, Stepanenko I, Petrasheuskaya TV, Darvasiova D, Breza M, Hammerstad M, Marć MA, Prado-Roller A, Spengler G, Popović-Bijelić A, Triapine analogues and their copper (II) complexes: synthesis, characterization, solution speciation, redox activity, cytotoxicity, and mR2 RNR inhibition, Inorg. Chem 60(15) (2021) 11297–11319. 10.1021/acs.inorgchem.1c01275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Ghosh S, Cisplatin: The first metal based anticancer drug, Bioorg. Chem 88 (2019) 102925. 10.1016/j.bioorg.2019.102925. [DOI] [PubMed] [Google Scholar]
- [172].Dong J, Zhang Q, Wang Z, Huang G, Li S, Recent advances in the development of indazole-based anticancer agents, ChemMedChem 13(15) (2018) 1490–1507. 10.1002/cmdc.201800253. [DOI] [PubMed] [Google Scholar]
- [173].Simpson PV, Desai NM, Casari I, Massi M, Falasca M, Metal-based antitumor compounds: beyond cisplatin, Future Med. Chem 11(2) (2019) 119–135. 10.4155/fmc-2018-0248. [DOI] [PubMed] [Google Scholar]
- [174].Dar AM, Khan M, Mir S, Gatoo M, DNA binding, cleavage activity, molecular docking, cytotoxicity and genotoxicity studies of newly synthesized copper based metal complexes, Pharm. Anal. Acta 7(464) (2016) 1636–1616. 10.4172/2153-2435.1000464. [DOI] [Google Scholar]
- [175].Rao NN, Gopichand K, Nagaraju R, Ganai AM, Rao PV, Design, synthesis, spectral characterization, DNA binding, photo cleavage and antibacterial studies of transition metal complexes of benzothiazole Schiff base, Chem. Data Collect 27 (2020) 100368. 10.1016/j.cdc.2020.100368. [DOI] [Google Scholar]
- [176].Shabana AA, Butler IS, Gilson DF, Jean-Claude BJ, Mouhri ZS, Mostafa MM, Mostafa SI, Synthesis, characterization, anticancer activity and DNA interaction studies of new 2-aminobenzothiazole complexes; crystal structure and DFT calculations of [Ag (Habt) 2] ClO4, Inorganica Chim. Acta 423 (2014) 242–255. 10.1016/j.ica.2014.09.018. [DOI] [Google Scholar]
- [177].Elsayed SA, Saad EA, Mostafa SI, Development of new potential anticancer metal complexes derived from 2-hydrazinobenzothiazole, Mini-Rev. Med. Chem 19(11) (2019) 913–922. 10.2174/1389557518666181017143548. [DOI] [PubMed] [Google Scholar]
- [178].Ribeiro N, Farinha PF, Pinho JO, Luiz H, Mészáros JP, Galvão AM, Costa Pessoa J, Enyedy ÉA, Reis CP, Correia I, Gaspar MM, Metal Coordination and Biological Screening of a Schiff Base Derived from 8-Hydroxyquinoline and Benzothiazole, Pharmaceutics 14(12) (2022) 2583. 10.3390/pharmaceutics14122583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Jiao P, Wang Y, Mao B, Wang B, Zhong Y, Jin H, Zhang L, Zhang L, Liu Z, Discovery of 2-(2-aminobenzo [d] thiazol-6-yl) benzo [d] oxazol-5-amine derivatives that regulated HPV relevant cellular pathway and prevented cervical cancer from abnormal proliferation, Eur. J. Med. Chem 204 (2020) 112556. 10.1016/j.ejmech.2020.112556. [DOI] [PubMed] [Google Scholar]
- [180].Mistry B, Patel RV, Keum Y-S, Kim DH, Evaluation of the biological potencies of newly synthesized berberine derivatives bearing benzothiazole moieties with substituted functionalities, J. Saudi Chem. Soc 21(2) (2017) 210–219. 10.1016/j.jscs.2015.11.002. [DOI] [Google Scholar]
- [181].Dadmal TL, Appalanaidu K, Kumbhare RM, Mondal T, Ramaiah MJ, Bhadra MP, Synthesis and biological evaluation of triazole and isoxazole-tagged benzothiazole/benzoxazole derivatives as potent cytotoxic agents, New J. Chem 42(19) (2018) 15546–15551. 10.1039/C8NJ01249K. [DOI] [Google Scholar]
- [182].Aouad MR, Almehmadi MA, Rezki N, Al-blewi FF, Messali M, Ali I, Design, click synthesis, anticancer screening and docking studies of novel benzothiazole-1, 2, 3-triazoles appended with some bioactive benzofused heterocycles, J. Mol. Struct 1188 (2019) 153–164. 10.1016/j.molstruc.2019.04.005. [DOI] [Google Scholar]
- [183].Cindrić M, Jambon S, Harej A, Depauw S, David-Cordonnier M-H, Pavelić SK, Karminski-Zamola G, Hranjec M, Novel amidino substituted benzimidazole and benzothiazole benzo [b] thieno-2-carboxamides exert strong antiproliferative and DNA binding properties, Eur. J. Med. Chem 136 (2017) 468–479. 10.1016/j.ejmech.2017.05.014. [DOI] [PubMed] [Google Scholar]
- [184].Cindrić M, Perić M, Kralj M, Martin-Kleiner I, David-Cordonnier M-H, Paljetak HČ, Matijašić M, Verbanac D, Karminski-Zamola G, Hranjec M, Antibacterial and antiproliferative activity of novel 2-benzimidazolyl-and 2-benzothiazolyl-substituted benzo [b] thieno-2-carboxamides, Mol. Divers 22(3) (2018) 637–646. 10.1007/s11030-018-9822-7. [DOI] [PubMed] [Google Scholar]
- [185].Videnović M, Mojsin M, Stevanović M, Opsenica I, Srdić-Rajić T, Šolaja B, Benzothiazole carbamates and amides as antiproliferative species, Eur. J. Med. Chem 157 (2018) 1096–1114. 10.1016/j.ejmech.2018.08.067. [DOI] [PubMed] [Google Scholar]
- [186].Sultana F, Saifi MA, Syed R, Mani GS, Shaik SP, Osas E.G.s., Godugu C, Shahjahan S, Kamal A, Synthesis of 2-anilinopyridyl linked benzothiazole hydrazones as apoptosis inducing cytotoxic agents, New J. Chem 43(18) (2019) 7150–7161. 10.1039/C8NJ06517A. [DOI] [Google Scholar]
- [187].Saipriya D, Prakash A, Kini SG, Bhatt GV, Pai KSR, Biswas S, Shameer KM, Design, synthesis, antioxidant and anticancer activity of novel Schiff’s bases of 2-amino benzothiazole, Indian J. Pharm. Educ. Res 52(4) (2018) 333–342. 10.5530/ijper.52.4s.114. [DOI] [Google Scholar]
- [188].JawalePatil PD, Bhamidipati K, Damale MG, Sangshetti JN, Puvvada N, Bhosale RS, Ingle RD, Pawar RP, Bhosale SV, Bhosale SV, Synthesis of naphthalimide derivatives bearing benzothiazole and thiazole moieties: In vitro anticancer and in silico ADMET study, J. Mol. Struct 1263 (2022) 133173. 10.1016/j.molstruc.2022.133173. [DOI] [Google Scholar]
- [189].Liu DC, Gao MJ, Huo Q, Ma T, Wang Y, Wu CZ, Design, synthesis, and apoptosis-promoting effect evaluation of novel pyrazole with benzo [d] thiazole derivatives containing aminoguanidine units, J. Enzyme Inhib. Med. Chem 34(1) (2019) 829–837. 10.1080/14756366.2019.1591391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Zhao T, Yang Y, Yang J, Cui Y, Cao Z, Zuo D, Zhai X, Harmine-inspired design and synthesis of benzo [d] imidazo [2, 1-b] thiazole derivatives bearing 1, 3, 4-oxadiazole moiety as potential tumor suppressors, Bioorg. Med. Chem 46 (2021) 116367. 10.1016/j.bmc.2021.116367. [DOI] [PubMed] [Google Scholar]