

Abstract
Background & Aims:
Human genetic variation is thought to guide the outcome of HCV infection, but model systems within which to dissect these host genetic mechanisms are limited. Norway rat hepacivirus, closely related to HCV, causes chronic liver infection in rats but causes acute self-limiting hepatitis in typical strains of laboratory mice, which resolves in 2 weeks. The Collaborative Cross (CC) is a robust mouse genetics resource comprised of a panel of recombinant inbred strains, which model the complexity of the human genome and provide a system within which to understand diseases driven by complex allelic variation.
Approach & Results:
We infected a panel of CC strains with Norway rat hepacivirus and identified several that failed to clear the virus after 4 weeks. Strains displayed an array of virologic phenotypes ranging from delayed clearance (CC046) to chronicity (CC071, CC080) with viremia for at least 10 months. Body weight loss, hepatocyte infection frequency, viral evolution, T-cell recruitment to the liver, liver inflammation, and the capacity to develop liver fibrosis varied among infected CC strains.
Conclusions:
These models recapitulate many aspects of HCV infection in humans and demonstrate that host genetic variation affects a multitude of viruses and host phenotypes. These models can be used to better understand the molecular mechanisms that drive hepacivirus clearance and chronicity, the virus and host interactions that promote chronic disease manifestations like liver fibrosis, therapeutic and vaccine performance, and how these factors are affected by host genetic variation.

INTRODUCTION
HCV causes chronic infection of the liver in a majority of those infected. If untreated, chronic HCV infection can lead to the development of end-stage liver disease, including HCC and cirrhosis, and may require liver transplantation.1–4 Curative treatments, although costly, are available yet undiagnosed cases, access to treatment, reinfection, drug resistance development, and a lack of an effective vaccine have complicated efforts for the global elimination of HCV as a public health threat.2,5 In addition, it is not yet clear if the cure will prevent the subsequent development of HCC.1,2,4 While human genetic variation (eg, IFNL3 locus) is thought to guide both the kinetics of spontaneous clearance and chronic infection, animal model systems within which to dissect these host genetic mechanisms are not available.6 Thus, our understanding of how host genetic variation affects HCV pathogenesis, the development of chronicity, immunity, and vaccine efficacy is hampered by the lack of robust immune-competent small animal models.
HCV-related hepaciviruses have been discovered in multiple animal species, including horses, nonhuman primates, wild rodents, and bats.7 In 2014, Norway rat hepacivirus (NrHV) was discovered in rats in New York City to cause chronic liver infection.8 Many basic aspects of NrHV biology are shared with HCV, including hepatotropism, genome organization, dependence on the liver-specific micro-RNA, miR-122, for replication, and the use of scavenger receptor B-1 for entry.9–11 In common lab strains of mice, NrHV causes a subclinical self-limiting hepatotropic infection that is cleared in 1–2 weeks. NrHV mouse adaptation delays clearance until after 3–5 weeks, whereas transient depletion of CD4 T-cells before infection leads to persistent infection.12,13 Models of chronic hepacivirus infection in fully immune-competent mice without immune manipulation are not yet available.
The Collaborative Cross (CC) is a robust mouse genetics resource created by the systematic breeding of 8 founder strains encompassing classic inbred, disease model, and wild-derived models resulting in the generation of a panel of recombinant inbred strains, which together model the complexity of the human genome and provide a system within which to understand disease driven by complex allelic variation.14 The CC contains greater than 90% of the common genetic variation in mouse resources through the presence of uniformly distributed SNPs (>40 million) and indels (>4 million) across the various genomes.15,16 When applied to virology, the CC has been a powerful tool to identify better models of human disease, as shown with the Ebola virus,17 severe acute respiratory syndrome-associated COV (SARS-CoV),18 West Nile virus,19,20 SARS-CoV-2,21 and influenza A virus,22 and also to map the genetic determinants responsible for complex phenotypic traits.15
Here, we describe multiple CC mouse models of chronic hepacivirus pathogenesis. We show that host genetic variation affects hepacivirus chronicity, the host response to infection, hepatocyte infection frequency, liver inflammation, liver fibrosis, and viral evolution, thus recapitulating many aspects of HCV disease biology. This system can be used to better understand the host genes and molecular mechanisms that guide hepacivirus pathogenesis, liver fibrosis, and therapeutic and vaccine performance.
METHODS
Recombinant virus production
DNA from consensus clones NrHV-A, NrHV-B, and the mouse-adapted NrHV-BSLIS, carrying the T190S, V353L, F369I, and N550S mutations,13 were linearized using MIuI and purified using DNAclean and concentrator (Zymo). RNA was transcribed from 1 ug of MIuI-linearized DNA using T7 RiboMAX Express Large Scale RNA Production System (Promega). Template DNA was degraded using RQ1 DNAse for 30 min at 4˚C, and RNA was purified using an RNAeasy mini kit (Qiagen). Four-week-old NRG mice were injected intra-hepatically using 10 µg of RNA in a maximum volume of 50 µL of PBS + RNA, and 1 week after injection, a terminal bleed was performed, and serum was pooled, titrated by qRT-PCR and stored in -80˚C freezer. These passage zero virus stocks were amplified through i.v. infection of new 4-week-old NRG mice (N = 10/virus strain to be amplified) with 104 genomic equivalents, after which serum harboring the new passage #1 stocks was harvested, titrated, and stored as above. All experiments were performed using passage #1 of each strain mixed in equal amounts (1:1:1).
Mouse infection studies
All Collaborative Cross (CC) mice were obtained in 2015–2021 from the Systems Genetics Core Facility at the University of North Carolina at Chapel Hill.23 For the initial CC-strain screen, 9–12-week-old female CC004/TauUnc, CC017/Unc, CC021/Unc, CC024/GeniUnc, CC040/TauUnc, CC044/Unc, CC045/GeniUnc, CC046/Unc, CC071/TauUnc, and CC080/TauUnc were obtained N = 8/CC strain. Age and sex-matched WT C57BL/6J mice were purchased from Jackson Labs. Mice were anesthetized with a mixture of ketamine/xylazine and then infected through retroorbital injection of 100 µl containing 1 × 105 genome equivalents (G.E.) of recombinant NrHV diluted in DPBS (Gibco) (N = 5/strain). As a negative control, 3 mice/strain were mock infected with DPBS. Mice were weighed daily for the first week of infection and then weekly thereafter. To measure virus replication in live mice, whole blood was collected by means of submandibular bleed, was allowed to clot at room temperature in snap cap tubes, centrifuged at 9600 × g for 10 minutes, and then serum was transferred to a new tube and stored at -80˚C until analysis. A complete blood count was also performed using a Vetscan HM5c automated veterinary blood analyzer using whole blood collected into EDTA tubes. At the end of week 4, animals were euthanized by isoflurane overdose, and whole blood and serum were collected and treated as above. Liver tissue was collected from each animal and stored in RNAlater (Thermo) at -80˚C or in 10% formalin before processing for histology.
For the subsequent 2 follow-up studies, 7–14-week-old female CC046/Unc (CC046) (N = 17 NrHV-infected, 13 mock-infected), CC071/TauUnc (CC071) (N = 28 NrHV-infected, 19 mock-infected) and CC080/TauUnc (CC080) (N = 15 NrHV-infected, 14 mock-infected) were purchased from the UNC Systems Genetics Core in January through March 2019. Age and sex-matched WT C57BL/6J mice were purchased from Jackson Labs (N = 23 NrHV-infected, N = 17 mock-infected). Mice were infected and followed for the first 6 weeks as described above. Bleeds and weights were then performed every other week for 8 weeks followed then by monthly assessments. These studies were terminated after 37–38 weeks, and blood and tissue were collected, as noted above.
RNA isolation, qRT-PCR, and illumina deep sequencing
For reverse transcription—quantitative PCR (qRT-PCR) quantitation of viral RNA in serum, RNA was isolated from 5 µl of mouse serum using the RNA Clean & Concentrator-5 (Zymo) or High Pure Viral Nucleic Acid kit (Roche) kits. qRT-PCR was performed using 5 µl of eluted RNA, TaqMan Fast Virus 1-step Master Mix (Applied Biosystems), and primer/probes targeting NS3 primers/probe (IDT).10 The sequence of the NrHV NS3 primers and probes were: Primer 1: AAGCGCAGCACCAATTCC, Primer 2: TACATGGCTAAGCAATACGG, and Probe: /56-FAM/CTCACGTAC/ZEN/ATGACGTACGGCATG/3IABkFQ/. NrHV standard curve RNA (MEGAscript, Thermo) was produced by PCR amplification of NS3 downstream of a T7 promoter. Reactions were performed in LightCycler 480 Multiwell Plates (Roche) in a LightCycler 480 (Roche) using the following program: 50°C for 30 minutes, 95°C for 5 minutes, followed by 40 cycles of 95°C for 15 seconds, 56°C for 30 seconds, and 60°C for 45 seconds, then 40°C for 10 seconds. For qRT-PCR to quantitate NrHV RNA, interferon-stimulated gene expression, and mRNA sequencing in liver tissue, the liver tissue was homogenized in TRIzol reagent (Thermo), and RNA was isolated according to protocol. In all, 500 ng total RNA was utilized for each qRT-PCR reaction. interferon-stimulated gene MX dynamin-like GTPase 1 (Mx1, Thermo Fisher, Mm00487796_m1) expression was assessed in select samples. Relative expression of Mx1 to housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH, Thermo Fisher, Mm99999915_g1) was calculated by ∆∆CT method.24 Illumina total RNA-seq was performed by the UNC High Throughput Sequencing Facility (HTSF). Stranded mRNA libraries were prepared (Kapa mRNA) and read on the Illumina HiSeq. 4000 platform (single end 1x50 read length). For analysis of raw reads, sequenced reads were mapped to the mouse reference transcriptome (Ensemble; Mus musculus version 108) using Kallisto (version 0.46.0). Transcript quantification data were normalized using the TMM method in EdgeR (version 3.38.4), and differentially expressed genes (p.Adj.val < 0.05; logLC > 1.5) were identified using linear modeling with limma (version 3.52.2) using R (version 4.2.0) in R Studio (version 2022-04-19). Gene ontology analysis was carried out using the Ingenuity Pathway Analysis (Qiagen). Heat maps were generated using Broad Institute’s Morpheus web application (https://software.broadinstitute.org/morpheus/). Hierarchical clustering was performed by One Minus Pearson Correlation with Average Linkage clustering by columns and rows. Sequence data is available on the Gene Expression Omnibus (accession # GSE230468).
Liver pathology and digital quantitation of viral RNA in liver tissue sections
To assess liver pathology, we formalin fixed, paraffin-embedded liver tissues, generated sections (~5 µm), and stained them with hematoxylin/eosin (Richard-Allen Scientific) on an Autostainer XL from Leica Biosystems. To assess liver fibrosis, sections were stained using Masson’s Trichrome (Richard-Allen Scientific). Dual Chromogenic Immunohistochemistry was accomplished using CD4 (ab183685, Abcam) and CD8 (14-0808-82, Invitrogen) antibodies on the Ventana Discovery platform (Roche). The slides were dewaxed and hydrated. Heat-induced Ag retrieval was accomplished using Ventana’s CC1 (pH 8.5). After pretreatment, slides were incubated with primary antibodies as follows: CD8 at 1:100 and CD4 1:1000, using Discovery Casein Diluent (760-219, Roche). Ready-to-use secondary antibodies Discovery OmniMap anti-Rabbit HRP (760-4311, Roche) or Discovery OmniMap anti-Rat HRP (760-4457, Roche) were used, followed by DAB or Discovery Purple (760-229, Roche) development and Hematoxylin II staining. Stained slides were dehydrated and coverslipped with Cytoseal 60 (8310-4, Thermo Fisher Scientific). A positive control was included for each run. Tissue sections were evaluated by a Board Certified Veterinary pathologist.
NrHV viral RNA was visualized in liver tissue sections by ISH using RNAscope probes (Item # V-NrHV-PP) designed by Advanced Cell Diagnostics based on the whole genome sequence of NrHV (Genebank Accession # MF113386.1). ISH on liver tissue sections was performed on a Leica Rx autostainer (Leica Biosystems) with a hematoxylin counterstain. For quantitation, slides were scanned on a Versa slide scanner (Leica Biosystems) with a 40X power objective and a Point Gray camera (8-bit image at 0.137152 microns/pixel) and imported to Definiens Architect XD 2.7 for analysis with Tissue Studio version 4.4.2. Per tissue section per animal, the total tissue area was calculated and the total numbers of cells per section was calculated using the nuclear counterstain as a guide. Similar numbers of cells were assessed in each tissue section per timepoint. NrHV-positive cells were identified by ISH positivity. The spot size threshold was 1.5 µm². Spot areas were ranked as none/low (2 µm²), low/medium (6 µm²), or medium/high (10 µm²). Resultant data was used to determine the infection frequency per tissue section.
Deep sequencing NrHV genomes
Serum from NrHV-infected mice was diluted in PBS (25 µL in 225 µL PBS), added to a 2 mL Phasemaker tube (Thermo Fisher Scientific), and mixed with 750 µL TRIzol LS Reagent (Thermo Fisher Scientific), after which 200 μL chloroform was added, shaken for 15 seconds, incubated for 3 min at room temperature, and centrifugated at 12,000 g for 15 min at 4°C. The aqueous phase was removed and mixed with 450 μL ethanol and transferred to an RNA Clean & Concentrator-5 column (Zymo Research) for downstream RNA purification and concentration. Reverse transcription (RT) was performed with Maxima H Minus Reverse Transcriptase (Thermo Fisher Scientific). Samples were pre-incubated in the presence of RNase inhibitors (Promega) at 65°C for 2 min before the addition of the RT enzyme, followed by incubation at 50°C for 2 h using 0.1 μM primer TS-O-00319 (GCTTCCTGGAGCGGGCTAGATACTG). Amplification of the complete open reading frame was performed using Q5 Hot Start High-Fidelity DNA Polymerase (New England Biolabs), including high GC Enhancer, and the primer pair Reverse TS-O-00318 (CCAAGCCCCAATGCCGTCCGGCACCGCTGCCCTTTTCGG) and Forward TS-O-00361 (AGGTGAAGGGGGCATCGATG) (0.5 μM each). The PCR cycling parameters were 98°C for 30 s, followed by 37 cycles of 98°C for 10 s, 65°C for 10 s and 72°C for 8 min, with a final extension at 72°C for 10 min. Amplified DNA was purified using DNA Clean & Concentrator-25 columns (Zymo Research), and libraries for deep sequencing were prepared using the NEBNext Ultra II FS DNA Library Prep Kit for Illumina (New England Biolabs). The quality of DNA libraries was validated using a 2100 Bioanalyzer Instrument (Agilent). DNA libraries were quantitated using the Qubit dsDNA High-Sensitivity Assay Kit (Thermo Fisher Scientific), and libraries were pooled in equimolar concentrations before denaturation. Pooled libraries were loaded on a MiSeq v3 150 cycle flow-cell, and sequencing was performed on a MiSeq benchtop sequencer (Illumina). Analysis was performed as described.11,13 In short, pair-end reads were trimmed and filtered by Sickle and subsequently mapped by BWA onto the NrHV-B reference (GenBank: ON758386) using the MEM algorithm. Samtools were used to process the alignment files, and Lofreq was applied to call SNPs that were translated by SNPEffect. Multiple mutations in the same specific codons were subsequently resolved by LinkGE. Pairwise distance and dN/dS ratios were calculated from the vcf files by SNPGenie.
RESULTS
Host genetics is a determinant of acute hepacivirus clearance
Given the profound impact human genetic variation can have on HCV infection, we aimed to determine if host genetic variation would affect NrHV pathogenesis and infection kinetics in mice (Figure 1A). We infected 10 different CC mouse strains with a mixture of 2 sub-populations of mouse-derived NrHV inoculum, NrHV-A and -B along with the mouse-adapted NrHV-BSLIS at a ratio of 1:1:1 (A:B:BSLIS) (Supplemental Figure S1, http://links.lww.com/HEP/H952).13 We utilized this mixture to give the best possible foundation for persistency, not knowing which variant would be optimal in the CC. After infection, we monitored viremia for 4 weeks which is 1–2 weeks after WT C57BL/6J [wild-type (WT)] mice typically clear. All strains of mice became infected following NrHV inoculation (Supplemental Figure S2, http://links.lww.com/HEP/H952), but most CC strains and WT had cleared the infection by week 4. In contrast, CC040, CC046, CC071, and CC080 remained viremic at week 4 (Figure 1B), and both CC071 (p = 0.02) and CC080 (p = 0.002) had significantly elevated levels of viremia. Concurrently, CC071 (p = < 0.0001) and CC080 (p = 0.001) intrahepatic viral RNA was significantly elevated over WT (Figure 1C). Gene expression of Mx1, a typical interferon-stimulated gene, was elevated in the livers of CC strains that remained viremic, a sign that sustained virus replication continued to instigate the host (Figure 1D). All together, these data demonstrate that host genetic variation influences acute hepacivirus clearance.
FIGURE 1.
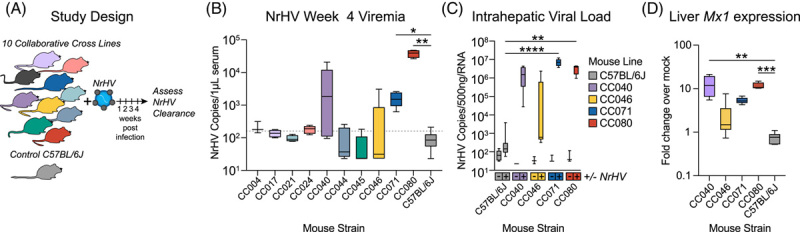
Host genetics is a determinant of acute hepacivirus clearance. (A) Study design. Ten Collaborative Cross (CC) strains and control C57BL/6J female mice 9–13 weeks in age were infected with 1 × 105 genome equivalents of recombinant NrHV or negative control PBS through retroorbital injection and bled weekly to monitor viremia. Mouse numbers per strain were: C57BL/6J N = 8, CC004 N = 4, CC017 N = 4, CC021 N = 4, CC024 N = 5, CC040 N = 5, CC044 N = 5, CC045 N = 5, CC046 N = 6. CC071 N = 5, CC080 N = 4. For all strains PBS mock infected N = 3. (B) Week 4 viremia as determined by qRT-PCR of viral RNA isolated from serum. The dotted line indicates the limit of quantitation. (C) Intrahepatic viral load 4 weeks after infection by qRT-PCR using 500 ng total liver RNA. Numbers of mice per group: C57BL/6J (8 NrHV, 4 Mock), CC040 (4 NrHV, 3 mock), CC046 (6 NrHV, 3 mock), CC071 (5 infected, 3 mock), CC080 (4 infected, 3 mock). (D) Mx1 intrahepatic gene expression 4 weeks by qRT-PCR using 500 ng total RNA. Data are expressed as fold change over mock infected by ∆∆CT method. For B and D, asterisks indicate statistical significance as determined by the Kruskal-Wallis test. For C, asterisks indicate statistical significance by 2-Way ANOVA with a Dunnett’s multiple comparison test. Abbreviations: CC, Collaborative Cross; NrHV, Norway rat hepacivirus.
Host genetics is a determinant of hepacivirus chronicity
We then further characterized a subset of CC strains with sustained NrHV replication at 4 weeks. CC046, CC071, and CC080 are genetically distinct with notably different genomic architectures, including at loci likely to be important for the antiviral response such as major histocompatibility complex (MHC), T-cell activation, and type I and III interferons (Supplemental Figure S3, http://links.lww.com/HEP/H952).25 We infected WT, CC046, CC071, and CC080 mouse strains with NrHV as done above and monitored them for 10 months (Figure 2A). Interestingly, body weight loss was only observed in CC071-infected mice and only during the first week (Supplemental Figure S4, http://links.lww.com/HEP/H952 and Supplemental Figure S5, http://links.lww.com/HEP/H952). As expected, WT cleared the virus within 3 weeks (Figure 2B). In contrast, CC046 had sustained replication during weeks 1–4 but ultimately all mice cleared the virus by week 7 (Figure 2B). Notably, the majority of CC071 mice became chronically infected with quantifiable viremia until the termination of the study at weeks 38–39 (Figure 2B). CC080 mice had a variety of virologic outcomes where some mice cleared the virus in the middle (4–7 wk) or late (8–22 wk) times, but only a minority became chronically infected (Figure 2B). During the first 2 weeks of infection, the levels of viremia in CC071 (week 1 p = 0.007, week 2 p = 0.001) and CC080 (week 1 p = 0.036, week 2 p = 0.02) were significantly higher than WT and by week 3, viremia in all 3 CC strains exceeded that in WT mice (CC046 p = 0.006, CC071 p = 0.007, CC080 p = 0.04) (Figure 2C). While all CC strains had measurable viremia in week 5, only CC071 (p = 0.01) remained significantly elevated over WT (Figure 2C).
FIGURE 2.
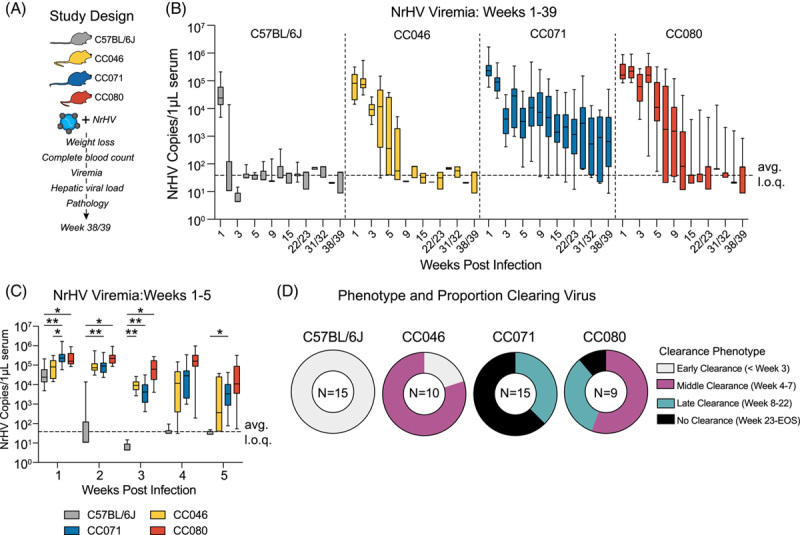
Host genetics is a determinant of hepacivirus chronicity. (A) Study Design. C57BL/6J, CC046, CC071, and CC080 mice were infected with 1 × 105 genome equivalents of recombinant NrHV or negative control PBS using retroorbital injection, and viremia was monitored weekly or monthly. (B) NrHV viremia for weeks 1–39 determined by qRT-PCR of RNA from serum. See Table S2 for the number of animals per strain per time. (C) NrHV Viremia for weeks 1–5. Levels of viral RNA in serum were measured by qRT-PCR. C57BL/6J Time (N): week 1 (19), week 2 (15), week 3 (15), week 4 (15), week 5 (15). CC046 Time (N): week 1 (13), week 2 (10), week 3 (10), week 4 (10), week 5 (10). CC071 Time (N): week 1 (22), week 2 (20), week 3 (18), week 4 (19), week 5 (19). CC080 Time (N): week 1 (13), week 2 (10), week 3 (10), week 4 (10), and week 5 (10). Asterisks indicate statistically significant differences by 2-Way ANOVA Tukey’s multiple comparisons test. (D) Proportion of mice per strain that clear virus early ( < 3 wk), middle (week 4–7), late (week 8–22), or do not clear (viremic week 23 to end of study). Only mice that survived to the end of the study were considered for this analysis. For B and C, the dotted line indicates the average limit of quantitation. All data for B–D, was compiled from 2 independent experiments. Abbreviations: CC, Collaborative Cross; NrHV, Norway rat hepacivirus.
To assess infectivity in serum, we passively transferred serum from 10 weeks after infection to naïve WT animals and then monitored viremia over time (Supplemental Figure S6, http://links.lww.com/HEP/H952). As expected, the transfer of serum from aviremic mice (WT and CC046) did not transfer the infection to recipient animals (Supplemental Figures S6B, S6C, http://links.lww.com/HEP/H952), but the transfer of serum from viremic CC071 (Supplemental Figure S6D, http://links.lww.com/HEP/H952) and CC080 (Supplemental Figure S6E, http://links.lww.com/HEP/H952) mice resulted in an infection in recipient animals. Thus, viral RNA associated with significant viremia is infectious. Interestingly, when examining viremia per animal, chronically infected animals experienced periods of relatively consistent levels of viremia followed by drastic increases or decreases, suggesting an interplay between the virus and the host response (Supplemental Figure S7, http://links.lww.com/HEP/H952). Altogether, by varying host genetics, a spectrum of virologic outcomes was observed following NrHV infection where all WT mice clear early (<3 wk), most CC046 mice clear between weeks 4–7, the majority of CC071 became chronically infected, and CC080 displayed phenotypes seen in both CC046 and CC071(Figure 2D).
Host genetics determines infection frequency and viral dynamics in the liver
To complement our virologic measures in peripheral blood, we next extended our studies to the liver. Labeling viral genomic RNA through in situ hybridization (ISH) revealed a variety of phenotypes. First, clearance phenotypes observed in the periphery were similarly observed in WT and CC046, and most CC071 and some CC080 remained viral RNA ISH-positive for the duration of the study (Figure 3A). Second, the amount of viral RNA per cell varied widely, especially at 2 dpi, where the amount of viral RNA per cell was greatest in CC071 (Figure 3A). Third, the average infection frequency varied greatly among the different strains. On 2 dpi, the infection frequency for CC046 was 22%, more than 10-fold greater than other strains (Figure 3B). By 7 dpi, the infection frequency in CC071 mice had increased to similar levels, still significantly greater than both WT and CC080 mice. By 28 dpi, the infection frequency in CC071 mice had increased even further (average frequency = 38.1%) but decreased to levels seen at earlier times by the end of the study (Figure 3B). In paired tissue samples, we quantitated viral RNA by means of qRT-PCR. As seen with ISH, CC046 had significantly elevated levels of NrHV RNA at 2 dpi (p = 0.03–0.04) compared with other CC strains (Figure 3C). Like the viremia and ISH data, CC071 had elevated levels of intrahepatic viral RNA over WT and CC046 at both 28 dpi and 272 dpi. Altogether, the intrahepatic virologic data were concordant with that observed in the periphery but also revealed new insights into the impact of host genetic variation on infection frequency and replication dynamics.
FIGURE 3.
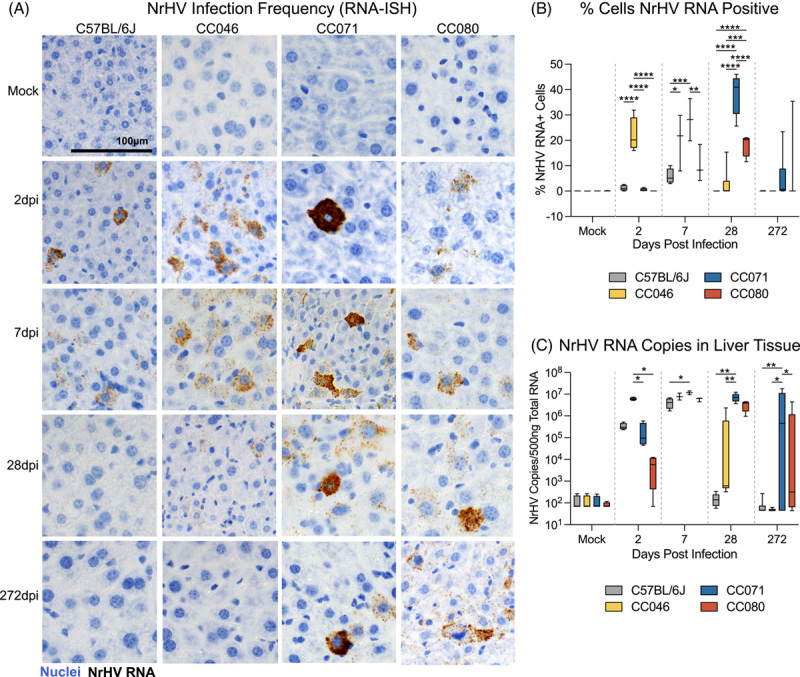
Host genetics determines infection frequency and viral dynamics in the liver. (A) NrHV viral RNA in situ hybridization in liver tissue sections from mock or NrHV-infected C57BL/6J, CC046, CC071, or CC080 mice 2, 7, 28, or 272 dpi. Nuclei are stained blue, and viral RNA is labeled brown. (B) Percent cells NrHV RNA positive using RNAscope in situ hybridization quantified using Definiens Architect. C57BL/6J Time (N): Mock (6), 2 dpi (4), 7 dpi (4), 28 dpi (5), and 272 dpi (7). CC046 Time (N): Mock (7), 2 dpi (4), 7 dpi (3), 28 dpi (6), and 272 dpi (5). CC071 Time (N): Mock (7), 2 dpi (4), 7 dpi (2), 28 dpi (5), and 272 dpi (15). CC080 Time (N): Mock (6), 2 dpi (4), 7 dpi (3), 28 dpi (4), and 272 dpi (8). (C) NrHV genome copy number in 500ng total liver RNA by qRT-PCR. C57BL/6J Time (N): Mock (4), 2dpi (4), 7 dpi (4), 28 dpi (5), and 272 dpi (7). CC046 Time (N): Mock (4), 2dpi (4), 7 dpi (3), 28 dpi (6), and 272 dpi (5). CC071 Time (N): Mock (4), 2 dpi (4), 7 dpi (2), 28 dpi (5), and 272 dpi (15). CC080 Time (N): Mock (4), 2 dpi (4), 7 dpi (3), 28 dpi (4), and 272 dpi (8). Asterisks in B and C indicate statistical significance by 2-Way ANOVA Tukey’s multiple comparisons test. Abbreviations: CC, Collaborative Cross; NrHV, Norway rat hepacivirus.
Immune dysregulation is associated with chronic hepacivirus infection
Given the disparate outcomes we had observed thus far, we next aimed to determine if host genetics differentially affected immune responses. First, we performed a completed blood count on longitudinal peripheral blood samples from the studies described above. The numbers of total lymphocytes were largely unaffected over time, comparing infected and strain-matched mock mice (Supplemental Figure S8A and B, http://links.lww.com/HEP/H952). In contrast, neutrophils were significantly and consistently elevated in NrHV-infected CC071 compared with strain-matched mock mice in weeks 1 and 2 (Supplemental Figures S8C and D, http://links.lww.com/HEP/H952). We hypothesize that innate immune cells like neutrophils may be responding in earnest as an attempt to compensate for inadequate innate or adaptive immune responses in CC071 mice. To better understand intrahepatic responses, we labeled liver tissue sections for CD4 and CD8 Ags since CD4 and CD8 T-cells were previously shown to be important for clearance in WT mice.12 On 2 dpi, clusters of CD4+ and CD8+ cells were observed in the parenchyma and on portal tracts in WT mice, who most rapidly clear the virus, whereas the labeling pattern remained unchanged from mock for all infected CC strains (Figure 4). By 7 dpi, periportal CD4 + and CD8 + cells were prominent in WT, CC046, and CC071 mice, yet the staining pattern for CC080 remained like that in the uninfected mice. By 28 dpi, clusters of CD4+ and CD8+ cells in periportal and parenchymal space were readily apparent in all infected CC strains but were diminished from the previous time point in WT mice. By 39 weeks after infection, staining in WT mice was unremarkable, but clusters of CD4+ and CD8+ cells around portal tracts remained in all chronically infected CC strains and even in CC046 that had cleared after week 7.
FIGURE 4.
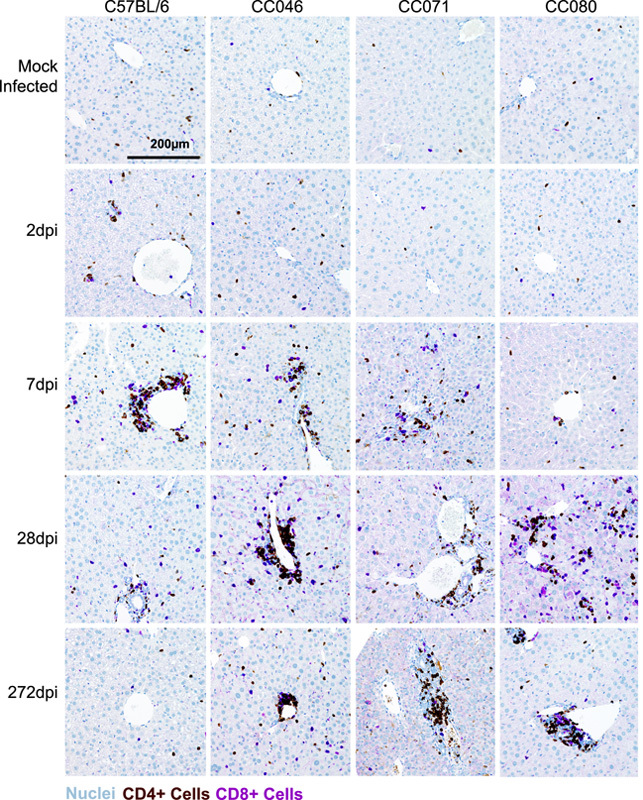
CD4 and CD8 cell recruitment is delayed in CC mouse strains. CD4 and CD8 cell labeling in liver tissue sections from mock and NrHV-infected animals at 2, 7, 28, and 272 dpi with CD4 cells (brown), CD8 cells (purple), and nuclei (blue). Representative images of mouse strains and time points are shown. Abbreviations: CC, Collaborative Cross; NrHV, Norway rat hepacivirus.
Dysregulation of the transcriptome in strains susceptible to prolonged infection
To determine if early defects in the host response were associated with chronic infection, we analyzed the transcriptional responses (Figure 5) in the liver of WT and CC mouse strains on 7 dpi when significant virologic and immunologic differences were observed. Importantly, using principal component analysis (PCA), we confirmed that the global responses among mock and infected groups differed and related samples clustered together (Figure 5A). The numbers of significantly regulated genes varied with host genetics, with CC071 regulating more genes than all other strains (Figure 5B). Pathway analysis of significantly regulated genes (<1.5 Log2 fold change over mock, p < 0.05) revealed the differential regulation of canonical pathways associated with innate and adaptive immunity among WT and CC strains (Figure 5C). When focusing on select canonical pathways, including pathogen-induced cytokine storm signaling, LXR/IL-1 mediated inhibition of RXR function, Th1 signaling, and LXR/RXR activation (Figure 5D), the gene expression patterns for CC071 were different compared with WT and other CC strains. Compared with other strains, CC071 animals lacked induction of innate immune (eg, Tlr1, 6, 7, and 13), chemokine (eg, Ccl6, Cxcl1, Cxcl10), adaptive immune (eg, Cd247, Cd3), and Il-1/inflammasome (eg, Il1b, Il1rn, IL18rap, NLRP3, etc.) related genes. Coupled with the CD4/CD8 + cell labeling data above, collectively these data show that CC strains with prolonged and chronic infection are dysregulated in key innate and adaptive immune pathways compared with WT mice, which most rapidly clear the virus.
FIGURE 5.
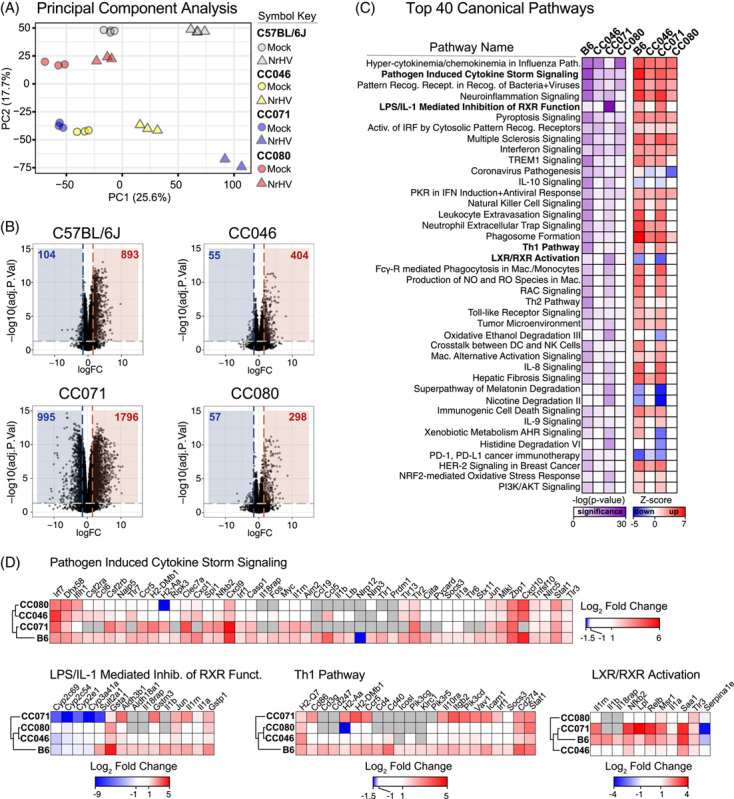
Dysregulation of the transcriptome in strains susceptible to chronic infection. Total RNA from mock and NrHV-infected liver tissue was deep sequenced by Illumina HiSeq. 4000. For all groups, N = 3 except for CC071 NrHV-infected N = 2. (A) Principal component analysis of expression data for mock and infected groups. (B) Top 40 Regulated Canonical Pathways by Ingenuity IPA. The significance (-log(p-value) and predicted regulation (Z-score) for each pathway are indicated by the scale bar. (C) Expression of significantly regulated genes ( > 1.5 Log2 fold change p < 0.05) over mock in select Canonical pathways. Heat maps were generated in Morpheus. Abbreviations: CC, Collaborative Cross; NrHV, Norway rat hepacivirus.
Chronic infection drives continual viral evolution
To determine if viral evolutionary patterns varied with host genetics, we deep sequenced the complete viral open reading frame in longitudinal serum samples from select mice that either cleared the virus or developed a chronic infection (Supplemental Figure S9, http://links.lww.com/HEP/H952). Virus populations from chronically infected animals (CC071 #41, 43, 46; CC080 #58, 63) evolved over time, as evidenced by the continued branching and increased branch length in the dendrogram (Supplemental Figure S10A, http://links.lww.com/HEP/H952). In contrast, virus populations from CC046 that cleared the virus the earliest among CC strains evolved the least (Supplemental Figure S10A, http://links.lww.com/HEP/H952). Proteins/regions of the genome under positive selection accumulate non-synonymous changes at increased frequency compared with synonymous changes. We therefore compared the non-synonymous and synonymous changes (dN/dS) across the viral open reading frame. Regardless of the host genetic background and unlike other portions of the genome, the dN/dS ratio for structural proteins E1 and E2, as well as p7 was increased at all times, suggestive of positive selection (Supplemental Figure S10B, http://links.lww.com/HEP/H952).
We next analyzed the specific mutational patterns over time per mouse (Figure 6). Virus circulating in CC071 (Figure 6A) mice generally had more amino acid consensus changes compared with CC080 (Figure 6B) or CC0046 (Figure 6C) mice at similar times after infection. This was significantly different at late times of infection (Figure 6D). In all 3 CC mouse strains, the mouse-adapted NrHV-BSLIS variant was rapidly selected for, likely due to its mutations T190S, V353L, F369I, and N550S, which adapt the virus to WT B6 mice.12,13 Several mutations residing in an MHC-I epitope conserved between mice and rats (T184A, S191F, V196A) were observed in viral genomes from all CC strains.26 In addition, either A698V or C715S appeared to be selected in p7 but rarely together. Disruption of the E2 glycosylation sites, N476 or N550, was previously linked to mouse adaptation.12,13 Here, N550S reverted in 2 of 3 chronically infected CC071 mice concomitantly with the disruption of another predicted glycosylation site at residues 504–506. One CC071 mouse also selected N437D, thus suggesting a link between disruption of any of the 4 predicted E2 glycosylation sites and mouse adaptation of NrHV.13 Further, unique changes were observed in CC071 (R984H in NS3) and CC080 (P1446S/L), the latter of which was linked to persistence. Thus, viral evolution, clearance, and chronicity were linked to host genetics.
FIGURE 6.
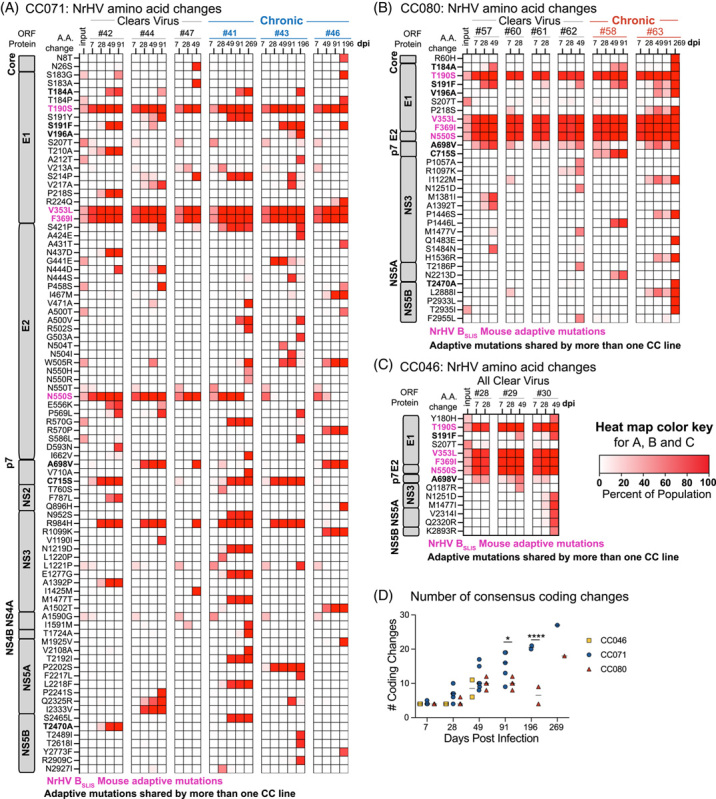
Chronic infection drives continual viral evolution. Consensus amino acid changes observed in the NrHV ORF and frequency in virus populations for (A) CC071 animals that clear (#42, 44, 47) and become chronically infected (#41, 43, 46), (B) CC080 animals that clear (#57, 60, 61, 62) and become chronically infected (#58 and 63) and (C) CC046 animals that clear (#28, 29, 30). The heat map color intensity indicates mutation frequencies (%). Amino acid changes were included in the heat map if the position was changed at a frequency > 50% in at least 1 sample per mouse line. Mouse adaptive mutations present in the inoculum (T190S, V353L, F369I, and N550S) are highlighted in hot pink. Changes shared by more than 1 CC strain are in bold. (D) The number of amino acid consensus changes over time. Only changes occurring at a frequency of > 50% were included in the analysis. Each symbol represents the number of coding changes in virus populations from a single mouse. Asterisks indicate statistical significance by 2-way ANOVA with Sidaks’s multiple comparison test. ****p = < 0.0001, *p = 0.0107. Abbreviations: CC, Collaborative Cross; NrHV, Norway rat hepacivirus; ORF, open reading frame.
The pathologic hallmarks of chronic liver infection vary with time and host genetics
To determine the pathological consequences of acute and chronic NrHV infection, we evaluated hematoxylin and eosin-stained liver tissue sections. The hepatic architecture did not vary among mock-infected WT, CC046, CC071, and CC080 mice (Figure 7). For WT mice, the kinetics of liver pathology largely mirrored that of virus replication. On 2 dpi, randomly distributed foci of inflammatory cells associated with single cell necrosis of hepatocytes were noted but by 7 dpi, inflammation was notably increased with moderate lymphocytic inflammation predominantly focused on portal triads along with focal, random small clusters of inflammatory cells in the parenchyma all of which was largely resolved by 28 dpi (Figure 7 and Supplemental Figure S11 http://links.lww.com/HEP/H952). In infected CC046, a mild lymphocytic periportal inflammation was noted at 7 dpi, which became moderate by 28 dpi and persisted although more mildly to the end of the study, 8 months after mice had cleared the virus (Figure 7 and Supplemental Figure S12 http://links.lww.com/HEP/H952). Although liver tissue sections from CC071 mice were unremarkable on 2 dpi, by 7 dpi, moderate periportal inflammation was observed in infected CC071 mice with increased lymphocytes in circulation (Figure 7 and Supplemental Figure S13, http://links.lww.com/HEP/H952). By 28 dpi, periportal and intraparenchymal inflammation was noted, and hepatic nuclear inclusions (Supplemental Figure S13 Inset, http://links.lww.com/HEP/H952), known to be associated with liver diseases such as HCC and NAFLD, were observed.27,28 In addition, karyomegaly (ie, enlargement of nuclei), known to be associated with abnormal liver function, was observed in CC071-infected mice at 28 dpi (Supplemental Figure S13, http://links.lww.com/HEP/H952).29 Unlike WT and CC046, which clear infection early, by the end of the study, moderate periportal inflammation remained in infected CC071 mice. Like CC071, at 2 dpi, liver sections from CC080 infected mice were unremarkable (Figure 7 and Supplemental Figure S14, http://links.lww.com/HEP/H952). Over time, periportal inflammation and foci of intraparenchymal inflammation increased in frequency and severity, which by the end of the study was like that in CC071. Lastly, we performed Masson’s Trichrome stain on liver tissue sections from the end of the study (ie, 272 d after infection) to determine if the chronic infection and inflammation were associated with liver fibrosis (Figure 8). Remarkably, early bridging fibrosis was observed in chronically infected CC071 mice but not in chronically infected CC080 mice. While we have not mapped the causal genetic loci, these data suggest that genetic variation can guide disparate pathogenic outcomes.
FIGURE 7.
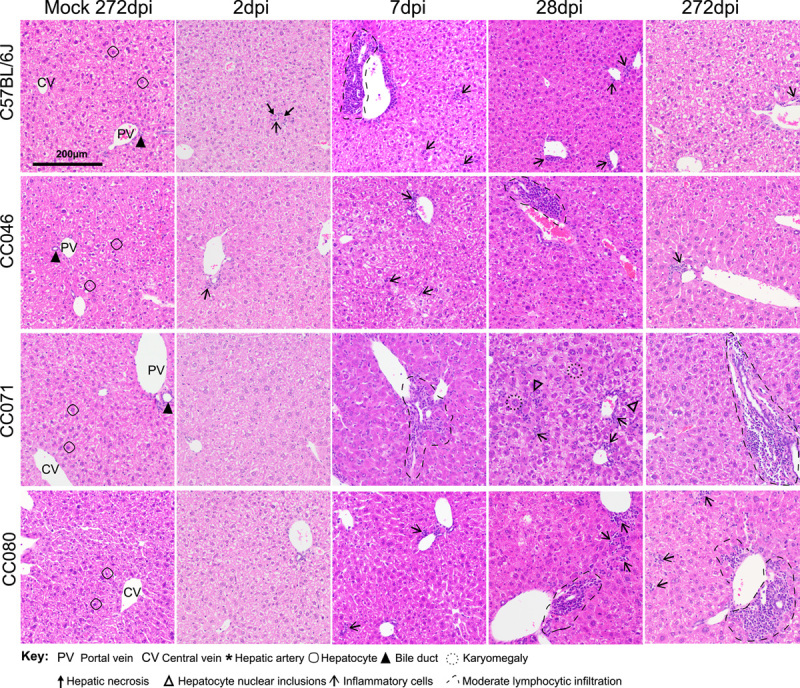
The pathologic hallmarks of chronic liver infection vary with time and host genetics. Liver tissue sections from mock or NrHV-infected WT, CC046, CC071, and CC080 mice at 2, 7, 28, and 272 dpi were stained with hematoxylin and eosin. Hallmark features of normal liver architecture, including portal vein, bile duct, hepatic artery, central vein, and hepatocytes, are noted in each mock panel. Abbreviations: CC, Collaborative Cross; WT, wild-type; NrHV, Norway rat hepacivirus.
FIGURE 8.
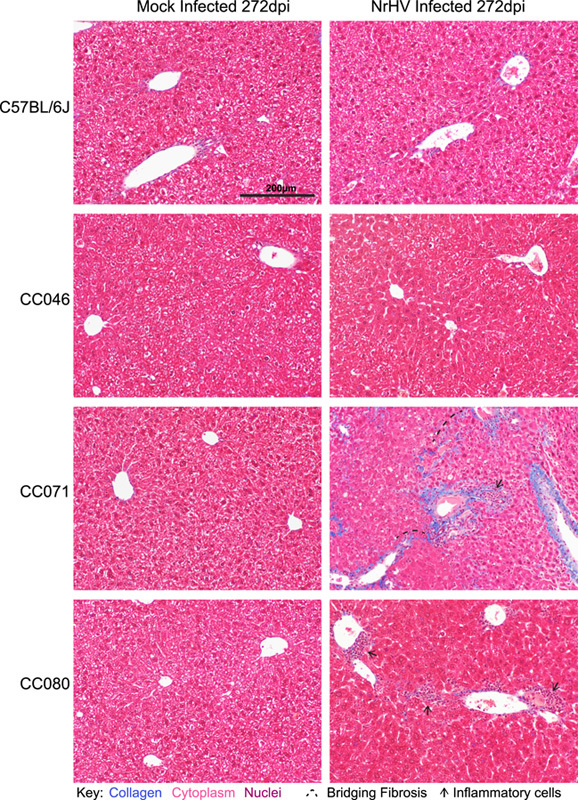
Liver fibrosis is associated with chronic liver infection. Liver tissue sections from mock or NrHV-infected WT, CC046, CC071, and CC080 mice at 272 dpi were stained with Masson’s Trichrome, which stains collagen blue. Inflammatory cells and early bridging fibrosis are noted. Abbreviations: CC, Collaborative Cross; NrHV, Norway rat hepacivirus; WT, wild-type.
DISCUSSION
Here we describe new CC mouse models of chronic hepacivirus infection that recapitulate multiple aspects of human HCV infection, including high titer viremia and replication in the liver, persistent infection with virus evolution over time, chronic liver inflammation, and liver fibrosis (Supplemental Figure S15, http://links.lww.com/HEP/H952).30,31 Aside from providing new models for the mechanistic study of acute and chronic infection, pathogenesis, immunity, and vaccination, these models should provide insights into how host genetics affects acute and chronic infection at an unprecedented resolution. Susceptibility alleles for multiple pathogens have been successfully identified using the CC mouse strains,15,18,21,32 and mapping these for NrHV will be an important future direction. Although we here demonstrate that host genetic variation can have profound effects on the virologic and pathologic outcomes, with the current suite of data, it is not possible to disentangle the genetics mediating persistent infection and that which shape the immune response and pathogenesis.
Hepacivirus and hepatocyte interactions guide the trajectory of infection, likely involving but not limited to the interplay of viral innate immune antagonists, antiviral innate immune sensors, innate (eg, KCs) and adaptive immune cells (eg, T-cells), cytokines and chemokines, hepatocyte turnover, and host genetic variation (eg, IFNL3 SNPs).33–36 Here, we describe 3 genetically distinct mouse strains with a multitude of different hepacivirus infection phenotypes and disease trajectories. Interestingly, CC071 mice also have increased susceptibility to classical flaviviruses such as Zika virus (ZIKV),37 Powassan virus, and West Nile virus38 but not to the bunyavirus Rift Valley fever virus,39 indicating that this strain is particularly sensitive to Flaviviridae infection. JAK/STAT signaling, a key component of the interferon response, appears to function normally in CC071 as interferon treatment of MEFs from CC071 or WT results in similar levels of STAT1 phosphorylation, suggesting that defects in JAK/STAT signaling are not driving increased flavivirus susceptibility.37 Neither CC046 nor CC071 appears to have a broad immune deficiency as they are not more susceptible to Salmonella Typhimurium infection than WT mice (ie, 129, 129S2/ SvPasCrl).40
There is a dearth of small animal models of chronic hepacivirus infection within which to study the virus and host factors that contribute to pathogenesis, mechanisms of viral evolution, and evasion of adaptive immunity. Sophisticated transgenic mice, human liver chimeric mice, and even mice reconstituted with both human liver and immune cells have been employed to study HCV infection, but species incompatibility complicates the modeling of immunity and chronic disease.41 Here, we provide an orthogonal approach with NrHV.9,11,12 Although CD4 T-cell depletion before NrHV infection as well as persistent CD8 T-cell depletion in WT mice can facilitate chronic infection,12 using the CC resource, we provide a complementary approach demonstrating that chronic infection and fibrosis in mice is possible without immune manipulation and that host genetic variation affects viral evolution. Viral evolution was greatest in CC071 mice, with the majority of change occurring in E1 and E2, presumably the main targets of the neutralizing antibody response.11 The MHC and T-cell–related genetic loci of WT, CC046, CC071, and CC080 are different, suggesting the potential for differential adaptive immune responses. It is likely that acute resolving and chronic infection are complex processes involving infected cell intrinsic factors and innate and adaptive immunity. Genetic differences in all these elements could in principle determine the differential outcomes observed between strains. Unfortunately, MHC haplotypes in CC strains remain largely unknown unlike common WT strains of mice.
In summary, we provide new small animal models of chronic hepacivirus infection, evidence that host genetic variation can profoundly affect the outcome of infection and a model genetic system within which to map the genetic loci guiding virus clearance and chronicity. These models have the power to provide basic insights into hepacivirus biology, hepatocyte biology, innate and adaptive immunity, and mammalian genetics, as well as new immunocompetent systems within which to study hepacivirus therapeutic efficacy and vaccine performance. Thus, these and future studies should be of broad interest to the field of infectious disease biology, hepatology, and immunology.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the technical support from the UNC High Throughput Sequencing Facility. This facility is supported by the University Cancer Research Fund, Comprehensive Cancer Center Core Support grant (P30-CA016086), and UNC Center for Mental Health and Susceptibility grant (P30-ES010126). They also thank Louise B. Christensen (Department of Clinical Microbiology, University of Copenhagen) for laboratory assistance and the Department of Clinical Microbiology, Hvidovre Hospital, for access to Illumina miSeq equipment. Collaborative Cross (CC)23 mice were obtained from 2017 to 2021 from the Systems Genetics Core Facility at the University of North Carolina at Chapel Hill. They also thank Dr. Amit Kapoor from the University of Ohio and Dr. Brad Rosenberg from Mt. Sinai Medical School for a helpful and insightful discussion of this work.
Footnotes
Abbreviations: CC, Collaborative Cross; ISH, in situ hybridization; MHC, major histocompatibility complex; NrHV, Norway rat hepacivirus; ORF, open reading frame; WT, wild-type.
Supplemental Digital Content is available for this article. Direct URL citations are provided in the HTML and PDF versions of this article on the journal’s website, http://www.hepjournal.com.
Contributor Information
Ariane J. Brown, Email: ajbrown7@email.unc.edu.
John J. Won, Email: jjwon1203@gmail.com.
Raphael Wolfisberg, Email: raphael.w@hotmail.com.
Ulrik Fahnøe, Email: ulrik@sund.ku.dk.
Nicholas Catanzaro, Email: nick_catanzaro@med.unc.edu.
Ande West, Email: westande@email.unc.edu.
Fernando R. Moreira, Email: fernando.wisc@gmail.com.
Mariana Nogueira Batista, Email: mnogueirab@mail.rockefeller.edu.
Martin T. Ferris, Email: mtferris@email.unc.edu.
Colton L. Linnertz, Email: colton_linnertz@med.unc.edu.
Sarah R. Leist, Email: leist@email.unc.edu.
Cameron Nguyen, Email: came@unc.edu.
Gabriela De la Cruz, Email: zurcaled@email.unc.edu.
Bentley R. Midkiff, Email: midkiff1@email.unc.edu.
Yongjuan Xia, Email: yxia@email.unc.edu.
Mia D. Evangelista, Email: mdevange@email.unc.edu.
Stephanie A. Montgomery, Email: stephanie@dallastissue.com.
Eva Billerbeck, Email: eva.billerbeck@einsteinmed.edu.
Jens Bukh, Email: jbukh@sund.ku.dk.
Troels K.H. Scheel, Email: tscheel@sund.ku.dk.
Charles M. Rice, Email: ricec@rockefeller.edu.
Timothy P. Sheahan, Email: sheahan@email.unc.edu.
AUTHOR CONTRIBUTIONS
Timothy P. Sheahan, Martin T. Ferris, Charles M. Rice, Jens Bukh, and Troels K.H. Scheel designed the studies. Ariane J. Brown, John J. Won, Raphael Wolfisberg, Ulrik Fahnøe, Nicholas Catanzaro, Ande West, Fernando R. Moreira, Martin T. Ferris, Colton L. Linnertz, Sarah R. Leist, Cameron Nguyen, Gabriela De la Cruz, Bentley R. Midkiff, Yongjuan Xia, Stephanie A. Montgomery, and Timothy P. Sheahan performed the studies. Eva Billerbeck, Mariana Nogueira Batista, and Charles M. Rice provided recombinant NrHV for these studies. Timothy P. Sheahan, Raphael Wolfisberg, Ulrik Fahnøe, Eva Billerbeck, Stephanie A. Montgomery, Troels K.H. Scheel, Jens Bukh, and Charles M. Rice wrote and edited the manuscript. All authors read and approved this manuscript.
FUNDING INFORMATION
This work was funded by R01 grant from the National Institute of Allergy and Infectious Disease (NIAID, AI131688 to Charles M. Rice), a U19 grant from NIAID (U19AI100625 to Ralph Baric and Mark Heise), the Independent Research Fund Denmark (1030-00426 to Troels K.H. Scheel), Advanced Grant 4004-00598 to Jens Bukh), the Danish Cancer Society (R204-A12639 to Jens Bukh and Troels K.H. Scheel), the Novo Nordisk Foundation (Distinguished Investigator Grant NNF19OC0054518 and Tandem NNF19OC0055462 to Jens Bukh), and the European Research Council (Starting Grant 802899 to Troels K. H. Scheel). Raphael Wolfisberg was supported by an Early Postdoc Mobility Fellowship (P2BEP3_178527) and a Postdoc Mobility Fellowship (P400PB-183952) from the Swiss National Science Foundation.
CONFLICTS OF INTEREST
Timothy P. Sheahan received grants from GlaxoSmithKline, Regeneron, and ViiV Healthcare. The remaining authors have no conflicts to report.
REFERENCES
- 1.Li DK, Chung RT. Impact of hepatitis C virus eradication on hepatocellular carcinogenesis. Cancer. 2015;121:2874–2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martinello M, Hajarizadeh B, Grebely J, Dore GJ, Matthews GV. Management of acute HCV infection in the era of direct-acting antiviral therapy. Nat Rev Gastroenterol Hepatol. 2018;15:412–424. [DOI] [PubMed] [Google Scholar]
- 3.Villanueva A, Hernandez-Gea V, Llovet JM. Medical therapies for hepatocellular carcinoma: a critical view of the evidence. Nat Rev Gastroenterol Hepatol. 2013;10:34–42. [DOI] [PubMed] [Google Scholar]
- 4.Zoulim F, Liang TJ, Gerbes AL, Aghemo A, Deuffic-Burban S, Dusheiko G, et al. Hepatitis C virus treatment in the real world: optimising treatment and access to therapies. Gut. 2015;64:1824–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walker CM, Grakoui A. Hepatitis C virus: why do we need a vaccine to prevent a curable persistent infection? Curr Opin Immunol. 2015;35:137–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gauthiez E, Habfast-Robertson I, Rueger S, Kutalik Z, Aubert V, Berg T, et al. A systematic review and meta-analysis of HCV clearance. Liver Int. 2017;37:1431–1445. [DOI] [PubMed] [Google Scholar]
- 7.Scheel TK, Simmonds P, Kapoor A. Surveying the global virome: identification and characterization of HCV-related animal hepaciviruses. Antiviral Res. 2015;115:83–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Firth C, Bhat M, Firth MA, Williams SH, Frye MJ, Simmonds P, et al. Detection of zoonotic pathogens and characterization of novel viruses carried by commensal Rattus norvegicus in New York City. MBio. 2014;5:e01933–01914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Trivedi S, Murthy S, Sharma H, Hartlage AS, Kumar A, Gadi SV, et al. Viral persistence, liver disease, and host response in a hepatitis C-like virus rat model. Hepatology. 2018;68:435–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wolfisberg R, Holmbeck K, Nielsen L, Kapoor A, Rice CM, Bukh J, et al. Replicons of a rodent Hepatitis C Model virus permit selection of highly permissive cells. J Virol. 2019;93:e00733–19. doi: 10.1128/jvi.00733-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wolfisberg R, Thorselius CE, Salinas E, Elrod E, Trivedi S, Nielsen L, et al. Neutralization and receptor use of infectious culture-derived rat hepacivirus as a model for HCV. Hepatology. 2022;76:1506–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Billerbeck E, Wolfisberg R, Fahnoe U, Xiao JW, Quirk C, Luna JM, et al. Mouse models of acute and chronic hepacivirus infection. Science. 2017;357:204–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wolfisberg R, Holmbeck K, Billerbeck E, Thorselius CE, Batista MN, Fahnoe U, et al. Molecular determinants of mouse adaptation of rat hepacivirus. J Virol. 2023;97:e0181222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Threadgill DW, Miller DR, Churchill GA, de Villena FP. The collaborative cross: a recombinant inbred mouse population for the systems genetic era. ILAR J. 2011;52:24–31. [DOI] [PubMed] [Google Scholar]
- 15.Noll KE, Ferris MT, Heise MT. The Collaborative Cross: a systems genetics resource for studying host-pathogen interactions. Cell Host Microbe. 2019;25:484–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Srivastava A, Morgan AP, Najarian ML, Sarsani VK, Sigmon JS, Shorter JR, et al. Genomes of the mouse Collaborative Cross. Genetics. 2017;206:537–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rasmussen AL, Okumura A, Ferris MT, Green R, Feldmann F, Kelly SM, et al. Host genetic diversity enables Ebola hemorrhagic fever pathogenesis and resistance. Science. 2014;346:987–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gralinski LE, Menachery VD, Morgan AP, Totura AL, Beall A, Kocher J, et al. Allelic Variation in the Toll-like receptor adaptor protein Ticam2 contributes to SARS-coronavirus pathogenesis in mice. G3 (Bethesda). 2017;7:1653–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Graham JB, Swarts JL, Wilkins C, Thomas S, Green R, Sekine A, et al. A mouse model of chronic West Nile Virus Disease. PLoS Pathog. 2016;12:e1005996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Graham JB, Thomas S, Swarts J, McMillan AA, Ferris MT, Suthar MS, et al. Genetic diversity in the collaborative cross model recapitulates human West Nile virus disease outcomes. MBio. 2015;6:e00493–00415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schafer A, Leist SR, Gralinski LE, Martinez DR, Winkler ES, Okuda K, et al. A multitrait locus regulates sarbecovirus pathogenesis. mBio. 2022;13:e0145422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ferris MT, Aylor DL, Bottomly D, Whitmore AC, Aicher LD, Bell TA, et al. Modeling host genetic regulation of influenza pathogenesis in the collaborative cross. PLoS Pathog. 2013;9:e1003196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Welsh CE, Miller DR, Manly KF, Wang J, McMillan L, Morahan G, et al. Status and access to the Collaborative Cross population. Mamm Genome. 2012;23:706–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. [DOI] [PubMed] [Google Scholar]
- 25.Leist SR, Baric RS. Giving the genes a shuffle: using natural variation to understand host genetic contributions to viral infections. Trends Genet. 2018;34:777–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hartlage AS, Walker CM, Kapoor A. Priming of antiviral CD8 T Cells without Effector Function by a Persistently Replicating Hepatitis C-Like Virus. J Virol. 2020;94:e00035–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schwertheim S, Kalsch J, Jastrow H, Schaefer CM, Theurer S, Ting S, et al. Characterization of two types of intranuclear hepatocellular inclusions in NAFLD. Sci Rep. 2020;10:16533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schwertheim S, Westerwick D, Jastrow H, Theurer S, Schaefer CM, Kalsch J, et al. Intranuclear inclusions in hepatocellular carcinoma contain autophagy-associated proteins and correlate with prolonged survival. J Pathol Clin Res. 2019;5:164–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thongthip S, Bellani M, Gregg SQ, Sridhar S, Conti BA, Chen Y, et al. Fan1 deficiency results in DNA interstrand cross-link repair defects, enhanced tissue karyomegaly, and organ dysfunction. Genes Dev. 2016;30:645–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pawlotsky JM, Feld JJ, Zeuzem S, Hoofnagle JH. From non-A, non-B hepatitis to hepatitis C virus cure. J Hepatol. 2015;62(1 Suppl):S87–S99. [DOI] [PubMed] [Google Scholar]
- 31.Burke KP, Cox AL. Hepatitis C virus evasion of adaptive immune responses: a model for viral persistence. Immunol Res. 2010;47:216–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gralinski LE, Ferris MT, Aylor DL, Whitmore AC, Green R, Frieman MB, et al. Genome wide identification of SARS-CoV susceptibility loci using the Collaborative Cross. PLoS Genet. 2015;11:e1005504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Prokunina-Olsson L, Muchmore B, Tang W, Pfeiffer RM, Park H, Dickensheets H, et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nature genetics. 2013;45:164–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thomas DL, Thio CL, Martin MP, Qi Y, Ge D, O’Huigin C, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature. 2009;461:798–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Urban TJ, Thompson AJ, Bradrick SS, Fellay J, Schuppan D, Cronin KD, et al. IL28B genotype is associated with differential expression of intrahepatic interferon-stimulated genes in patients with chronic hepatitis C. Hepatology. 2010;52:1888–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heim MH, Thimme R. Innate and adaptive immune responses in HCV infections. J Hepatol. 2014;61(1 Suppl):S14–S25. [DOI] [PubMed] [Google Scholar]
- 37.Manet C, Simon-Loriere E, Jouvion G, Hardy D, Prot M, Conquet L, et al. Genetic diversity of Collaborative Cross mice controls viral replication, clinical severity, and brain pathology induced by Zika Virus infection, independently of Oas1b. J Virol. 2020;94:e01034–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jasperse BA, Mattocks MD, Noll KM, Ferris MT, Heise MT, Lazear HM. Neuroinvasive flavivirus pathogenesis is restricted by host genetic factors in Collaborative Cross mice, independently of Oas1b. bioRxiv. 2022;97:e0071523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cartwright HN, Barbeau DJ, Doyle JD, Klein E, Heise MT, Ferris MT, et al. Genetic diversity of collaborative cross mice enables identification of novel rift valley fever virus encephalitis model. PLoS Pathog. 2022;18:e1010649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang J, Malo D, Mott R, Panthier JJ, Montagutelli X, Jaubert J. Identification of new loci involved in the host susceptibility to Salmonella Typhimurium in collaborative cross mice. BMC Genomics. 2018;19:303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bukh J. Animal models for the study of hepatitis C virus infection and related liver disease. Gastroenterology. 2012;142:1279–1287 e1273. [DOI] [PubMed] [Google Scholar]