

Abstract
ORFanage is a system designed to assign open reading frames (ORFs) to known and novel gene transcripts while maximizing similarity to annotated proteins. The primary intended use of ORFanage is the identification of ORFs in the assembled results of RNA sequencing experiments, a capability that most transcriptome assembly methods do not have. Our experiments demonstrate how ORFanage can be used to find novel protein variants in RNA-seq datasets, and to improve the annotations of ORFs in tens of thousands of transcript models in the human annotation databases. Through its implementation of a highly accurate and efficient pseudo-alignment algorithm, ORFanage is substantially faster than other ORF annotation methods, enabling its application to very large datasets. When used to analyze transcriptome assemblies, ORFanage can aid in the separation of signal from transcriptional noise and the identification of likely functional transcript variants, ultimately advancing our understanding of biology and medicine.
Introduction
Approximately 20,000 protein-coding genes have been annotated for the human genome1–5. While a single isoform is often the source of the dominant protein6–8, many human gene loci express isoforms that encode different protein sequences, some of which may be tissue-specific9–12. The NCBI RefSeq database, for example, contains an average of 6.9 isoforms for each human protein coding gene, which encode an average of 4.4 distinct protein sequences. The RefSeq annotation of the model organism Arabidopsis thaliana has on average 1.8 isoforms with 1.5 unique protein variants respectively.
RNA sequencing technology has allowed an unprecedented look at the transcriptome in a wide variety of species, with multiple studies reporting large numbers of previously unknown transcripts for protein coding genes3, 13–16. Consistent with previous reports about alternative splicing events17, most of the novel transcripts reported in RNA-seq studies are observed in protein-coding regions18, 19. Alternative splicing events can alter the translated protein through exon skipping, frame-shifting, and other changes20. These events and their effects on translated proteins are an essential component of genome biology9.
Changes in protein sequences may also be characteristic of disease states10, 21–24 or of specific tissues9, 25, 26. For example, splicing-induced changes in protein sequences have been associated with cancer development and progression, from activation of proto-oncogenes27 to genome-wide splicing alteration in certain cancer types28, 29. One example of why it is important to annotate all protein isoforms in the human genome is the widespread usage of exome sequencing in clinical settings. Exome capture methods have been extensively used to interrogate genetic variants and their associations with diseases, such as finding the genetic cause of a rare form of pediatric epilepsy30, or identifying driver mutations in cancer31. The technology is heavily dependent on the correct annotation of coding regions, and any exons that are unannotated will simply be missed by exome studies.
However, many observed novel transcripts are likely to represent transcriptional noise32; e.g., the original CHESS database assembled ~29 million transcript variants from 10,000 RNA sequencing experiments, of which fewer than 2% were kept in the final annotation3, 4. The ability to accurately identify non-functional isoforms can be a valuable tool in differentiating signal from noise in RNA-seq data, which is currently complicated by artifacts from computational methods, such as alignment and assembly errors, as well as the amount of noise inherently present in the data32.
Although many methods have been implemented for searching and assembling transcripts from RNA-seq data33, 34, none of them identifies open reading frames (ORFs) based on similarity to the original protein at the locus. A number of methods including TransDecoder34 and GeneMarkS-T35, 36 have been developed for ab initio ORF annotation (Methods, Table 2), but these methods were designed to find ORFs without the use of reference annotation as a guide. Other previous approaches only identified the longest ORF, sometimes requiring it to have the same start or stop codon positions as a reference35, 37–39. None of these approaches consider the similarity of the resulting protein to previously-known translations of the transcript.
Table 2.
Comparison of the True Positive Rate (TPR) of ORF annotation methods based on concordance with the GENCODE and RefSeq datasets. TPR was computed as the percentage of all ORFs in each dataset which were reconstracted identically by the method.
| GENCODE | RefSeq | |||||
|---|---|---|---|---|---|---|
| Execution Time (minutes) | TPR | Execution Time (minutes) | TPR | |||
| Multi-threaded | Single-threaded | Multi-threaded | Single-threaded | |||
| ORFanage | 0.28 | 0.6 | 0.88 | 0.33 | 1.1 | 0.94 |
| TransDecoder | 115 | - | 0.65 | 175 | - | 0.82 |
| GeneMarkS-T | 100 | 100 | 0.58 | 85 | 85 | 0.71 |
GeneMarkS-T times do not include conversion from reported format to genomic GTF style.
In this study, we present ORFanage, a highly efficient and sensitive method to search for open reading frames in protein-coding transcripts, guided by reference annotation to maximize protein similarity within genes.
Results
Accuracy of Reference ORF reconstruction
ORFanage utilizes protein-coding gene annotation by identifying ORFs in query transcripts that have the maximal sequence identity with a user-provided set of reference ORFs. This approach presumes that proteins produced by different transcripts at the same locus should be as similar as possible8, 40. In our first set of experiments, we tested ORFanage’s ability to reconstruct the GENCODE and RefSeq protein-coding annotation given an annotation that includes one canonical ORF at each protein-coding gene locus. For these experiments, we used the MANE database to define the canonical ORFs, because MANE was created by the developers of GENCODE and RefSeq to be a “universal standard”6 of human protein-coding genes, and because both GENCODE and RefSeq contain every gene in MANE. These experiments illustrate how ORFanage can produce a set of ORFs at a locus that better agree with a chosen reference annotation, conserving the protein sequences and making annotation more internally consistent.
As shown in Figure 1a, many gene transcripts in both RefSeq and GENCODE are annotated with ORFs that differ from the canonical variant; e.g., 65% of ORFs in the RefSeq human annotation and 36% in GENCODE differ from the MANE ORF (Figure 1a). In principle, the presence of an ORF that differs from MANE does not imply an error; however, if another ORF can be found in the same transcript that has closer identity to MANE, then an error seems possible. Furthermore, 8% of RefSeq and 43% of GENCODE transcripts in protein-coding loci have no ORFs annotated at all. By re-annotating each of the reference datasets using ORFanage, we identified numerous cases where a different ORF was more similar to the canonical protein. One example, from the ZNF180 gene, is shown in Figure 1f.
Figure 1.
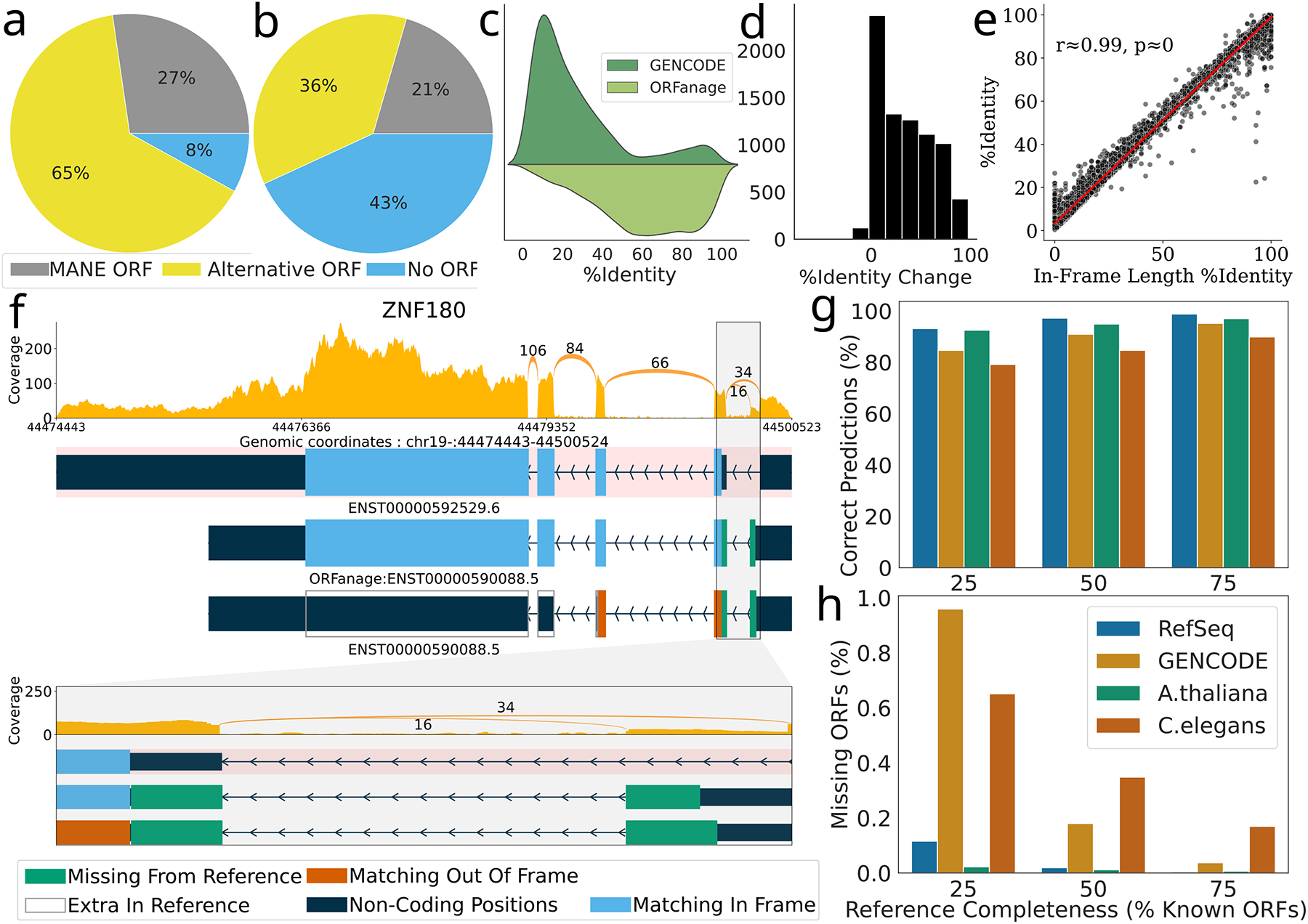
Overview of irregularities in reference database ORF annotation. a-b) Differences in ORFs at MANE loci as currently annotated for a) RefSeq and b) GENCODE annotations. Circular charts show, for each dataset, the proportions of transcripts annotated with the same ORF as MANE (grey), those with an alternative ORF not matching MANE (yellow), and transcripts in MANE loci that lack an annotated ORF (blue). c) Percent identity computed between the MANE protein and alternative ORFs as predicted by GENCODE (dark green) and ORFanage (light green). d) Histogram of the change in percent identity when replacing the GENCODE ORF with the ORFanage ORF. e) Pearson correlation coefficient (r) and p-value (two-sided, t-distribution) between percent identity computed via traditional alignment and In-Frame Length Percent Identity computed by ORFanage, illustrating the close similarity between the two metrics (10,000 random samples). f) A detailed look at alternative ORFs annotated by GENCODE and ORFanage for the ZNF180 gene. At top is the MANE isoform, shaded in pink, with its ORF shown in blue. Below it are two versions of an alternative isoform, with the ORFs annotated by ORFanage (middle) and GENCODE (bottom). Blue regions show where the protein sequence matches the MANE isoform, while green and orange show regions that are additional (green) or out of frame (orange) compared to MANE. At bottom is a zoomed-in view of the first intron and flanking ORF regions. g-h) Overview of the impact that completeness of reference annotation has on the accuracy of ORFanage. g) The percent of correctly inferred ORFs given different fractions of known reference ORFs for 4 organisms. h) Percentage of known ORFs that ORFanage failed to identify for different levels of reference completeness.
While we found that ORFs in a large majority of transcripts in the RefSeq human annotation were in agreement with those predicted by ORFanage (117,212 out of 135,694) there were some striking differences, as illustrated in Table 1 and Supplementary Table 2. For example, we identified 2,122 transcripts in which an ORF annotated by RefSeq could be replaced by the canonical version from MANE without alterations. Similarly, 786 of the ORFs in the GENCODE human annotation could be replaced by their canonical variants from MANE. Even though alternative translations may be present at those transcripts, because GENCODE and RefSeq both recognize MANE as a standard6, it seems appropriate to choose the MANE ORFs over the alternative variants in accordance with established curation guidelines41.
Table 1.
Summary of differences between ORFs found by ORFanage and the originally annotated ORFs for all transcripts in RefSeq and GENCODE protein-coding genes. Comparisons to the MANE annotation refer to the ORFs from the MANE gene set, which is fully contained within both RefSeq and GENCODE.
| Reference annotation | RefSeq | GENCODE |
|---|---|---|
| ORFanage finds the same ORF as reference | 117,212 | 63,966 |
| ORFanage finds a different ORF that matches MANE perfectly | 2,212 | 786 |
| No ORF annotated on reference transcript, ORFanage finds an ORF that matches MANE | 1,194 | 147 |
| No ORF annotated on reference transcript, ORFanage finds an ORF that is different from MANE | 9,240 | 35,393 |
| Other combinations | 5,836 | 27,994 |
| Total number of protein-coding transcripts | 135,694 | 128,286 |
In our analysis we purposefully refrained from filtering candidate ORFs, opting to report one best candidate ORF for every transcript where some sequence similarity was observed to the reference annotation. This allowed us to investigate all cases where analyzed annotations were inconsistent with the MANE reference at the cost of potential false discoveries. However, our software provides users with the ability to fine-tune the results through parameter settings such as the percent identity (PI) score, matching translation initiation site (TIS), and other customizable criteria. These options enable users to refine the identification of valid ORFs and limit the number of false positives.
As a result, we also found thousands of transcripts for which no ORF was listed, even though they were annotated under protein-coding genes and even though a candidate ORF was identified by ORFanage, such as examples illustrated in Supplementary Figures 3–4. In GENCODE, we found an ORF that at least partially overlapped the MANE ORF in 35,540 out of 55,328 of these transcripts, including 147 transcripts that contained a perfect match to the MANE ORF. Although the RefSeq database had fewer protein-coding transcripts with no ORF listed, we still found 10,434 transcripts for which our method predicted an ORF, including 1,194 with a perfect match to MANE (Table 1, Supplementary Table 2).
We also looked at transcripts where both ORFanage and the reference annotation differed from MANE (5,301 in RefSeq and 7,957 in GENCODE). For these transcripts we computed the percentage of in-frame positions shared between the annotated proteins and the MANE protein and observed that in 613 RefSeq and 7,005 GENCODE transcripts, ORFanage produced a protein that was closer to MANE (Figure 1c,d). In many cases the differences were minor, affecting only start coordinates or conserving different segments of the reference protein. In some cases, though, such as ZNF180 as shown in Figure 1f, ORFanage identified an ORF that conserved nearly all of the MANE protein sequence, while the protein encoded by the GENCODE ORF had no overlap with MANE. However, higher similarity of ORFs is not the only criterion for assessing ORF validity and other methods may be necessary to validate any novel sequences. Yet, in the absence of additional data, the similarity criterion can be successfully applied, as shown in our evaluation.
When ORFanage found an ORF that differed from the one chosen by RefSeq or GENCODE, the ORFanage sequence had an equal or higher proportion of codons that matched MANE (Figure 1c,d), a property that is guaranteed by the algorithm. We confirmed these results by performing global alignments of the proteins to the MANE variants using EMBOSS Stretcher42. The higher percent identity is a consequence of the metric that ORFanage maximizes, which we term In-frame Length Percent Identity (ILPI). Following ORF identification via the algorithm described in Figure 2, to compute ILPI, our method first computes the total number of positions in an ORF that are in the same frame as the reference, thus coding for the same codons, which determines the In-frame Length (IL). Then ILPI is computed as the fraction of IL of the total length of the reference coding sequence. As illustrated in Figure 1e, the correlation between ILPI and percent identity computed via the Smith-Waterman algorithm is very high.
Figure 2.
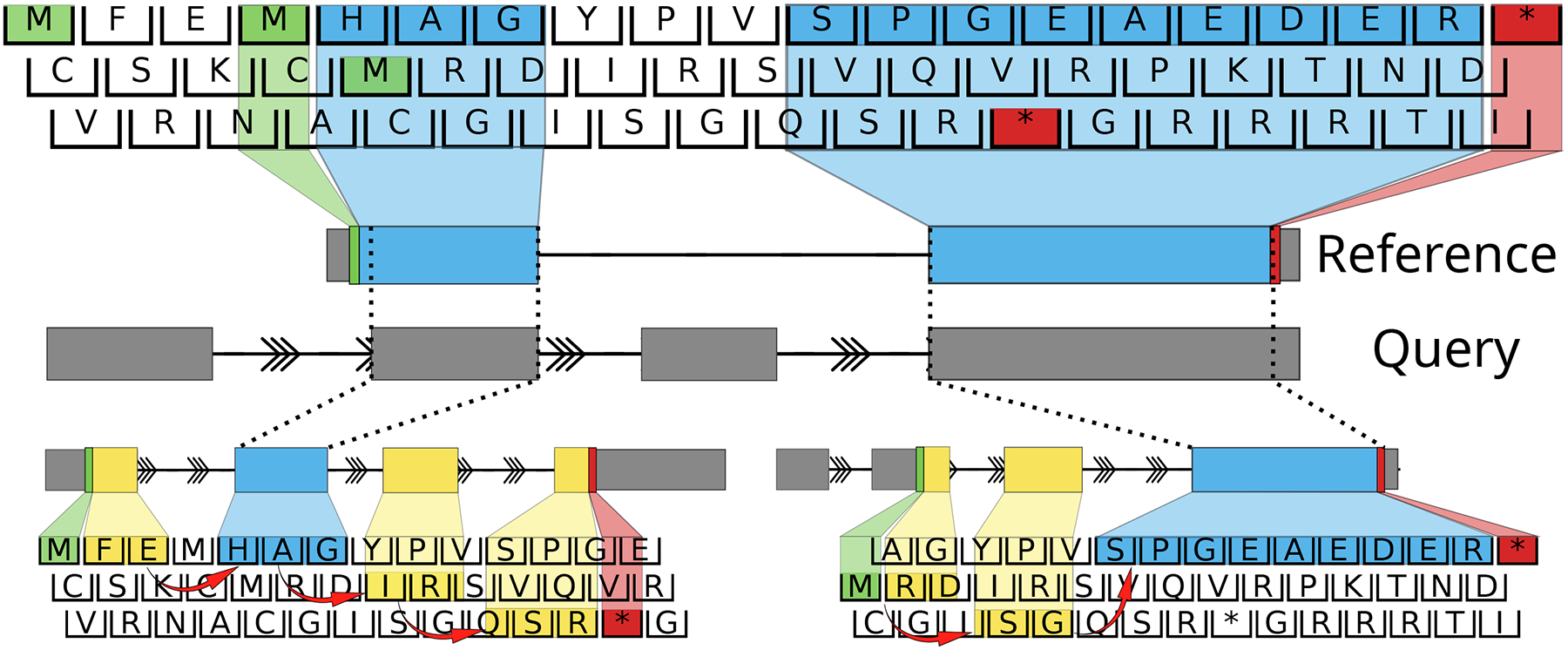
Diagram illustrating the algorithm implemented in ORFanage. ORFanage begins by computing overlaps between a reference ORF and query transcript. In the figure, dashed lines are used to connect matching intervals. For each overlap it extends coordinates towards the 3’ and 5’ ends based on suitable parameters. During extension, any changes to the exon structure may introduce shifting of the original frame (as indicated by red arrows). Once all intervals have been evaluated, ORFanage compares the results and reports the one with the highest score. In the figure, matching residues to the reference are highlighted in blue, and mismatching residues are highlighted in yellow. In this example, ORFanage selects the longer ORF on the lower right, which has 10 out of 14 matching residues, compared to the ORF on the lower left with only 3 out of 14 matching residues.
We then took a closer look at the 44,532 GENCODE transcripts where ORFanage found a different ORF. We found that ORFs identified by ORFanage often contained many novel positions (i.e., not matching MANE). More specifically, nearly 22% of positions in these novel ORFs are marked as potentially coding only by our method, and while many of these positions could be artifacts of partial transcript models included in the GENCODE annotation, some are likely to represent new functional variants of known proteins14, 23.
It is also worth noting here that when guided by protein-coding annotation such as MANE, ORFanage can reconstruct the ORFs present in GENCODE or RefSeq faster and more accurately than ab initio ORF finders like TransDecoder or GeneMarkS-T (see Table 2 and Methods).
The large number of missing annotations and overall observed improvements demonstrate the potential use of ORFanage at finding consistent ORFs in novel transcripts at protein-coding loci.
Impact of Reference Transcripts on Accuracy
In the next set of experiments, we set out to investigate how well our method can reconstruct a full set of protein sequences from subsets of reference data. We wanted to establish 1) how accuracy improves with an increase in the number of annotated ORFs at a locus and 2) the effects of choosing different subsets of known ORF variants on the accuracy of prediction. To answer the first question, we incrementally increased the number of ORFs provided to ORFanage as a reference. To address the second question, we repeated the experiment but randomly chose different sets of reference ORFs.
We repeated the iterative selection of reference transcripts 10 times, providing 25%, 50% and 75% of the reference ORFs as a guide each time. We ran our analysis on the human genome annotation as well as Arabidopsis thaliana and Caenorhabditis elegans using the same protocol. For the human genome, we evaluated both RefSeq and GENCODE, because the two databases differ substantially in their ORF annotations. For each test run, we ensured that at least one transcript remained unannotated at each locus and that any non-coding transcripts were removed prior to the evaluation.
The diversity of transcripts annotated for A. thaliana and C. elegans is much lower compared to human reference annotations, with 1.8 and 1.4 transcripts per coding gene respectively, compared to 6 and 8 for RefSeq and GENCODE human annotations. Worth noting is that for C. elegans, only 4,440 suitable loci were identified based on the aforementioned criteria.
As expected, we observed an increase in accuracy as more reference annotation was provided. For the human genome, if we provided just a single reference ORF per locus (equivalent to 11% of all ORFs in RefSeq and 18% of all ORFs in GENCODE), ORFanage was able to correctly re-create 85% of the RefSeq ORFs and 81% of GENCODE ORFs. When we provided 75% of the reference ORFs, ORFanage correctly re-created close to 99% of RefSeq and 95% of GENCODE ORFs (Figure 1g–h).
Even when ORFs were not identical to the original sources, the predictions produced by ORFanage were highly similar, averaging 81% for the non-identical predictions in the RefSeq dataset and 77% in GENCODE respectively.
Because A. thaliana and C. elegans have fewer annotated reference ORFs per locus, our random permutations had smaller effects on the results. Nevertheless, in A. thaliana ORFanage was able to correctly identify 91–97% of reference ORFs. For C. elegans the values were lower, ranging from 77% when a single random reference ORF was provided to 90% when guided by more complete annotations.
Finding novel ORFs in assembled RNA-seq data
One of the main applications of ORFanage is to search for ORFs in datasets containing large numbers of transcripts that have not been assigned open reading frames. ORFanage can annotate transcriptome assemblies from RNA-seq experiments, which often contain many novel splice variants, even for well-annotated genomes3, 4, 43. In these cases, ORFanage can identify candidate ORFs for protein-coding transcripts based on conservation of known protein sequences at the locus using reference annotation as a guide.
We next applied ORFanage to search for novel ORFs in experimental data, using data from the GTEx project44, a high-quality collection of poly-A selected RNA-seq samples across multiple human tissue types. We focused our experiments on 1,448 samples from brain tissue because these represented the most diverse collection of samples in the dataset. We ran ORFanage on the complete, unfiltered set of assemblies containing 6,674,316 isoforms that were assembled originally for the CHESS human annotation database3, 4.
We computed ORFs for all transcripts using the MANE annotation as the guide. For every MANE gene, we first identified all assembled transcripts overlapping that gene using gffcompare45 (similarity codes “=“, “c”, “k”, “m”, “n”, “j”, “e”), yielding 4,256,346 transcripts. We then computed the total gene expression for each transcript using the sum of transcripts per million (TPM) values for that transcript across all samples.
In our search for novel ORFs, we took a conservative approach: if a transcript could accommodate an ORF from either RefSeq or GENCODE, we assigned that ORF to the transcript. Additionally, we removed ORFanage predictions for all transcripts marked as non-coding by either RefSeq or GENCODE. Because multiple distinct transcripts can contain the same novel ORF, we simplified our analysis by computing the total TPM aggregated across transcripts sharing the same ORF. In transcripts for which no ORF was assigned, we computed the total TPM as the sum of TPMs for that transcript across all samples. This selection left us with a total of 3,046,286 novel transcripts representing 1,006,547 ORF variants.
Next to focus on highly expressed cases, we considered 4,190 loci where more than 50% of expression came from novel transcripts and ORFs (Figure 3a). Many of the transcripts at these loci either had no valid ORF or else contained an ORF that was highly dissimilar from the canonical MANE protein. We therefore narrowed our focus to 462 loci where over 50% of expression was due to a single novel ORF. Of those, only 24 loci (Supplementary Table 1) were at least 70% identical to the MANE protein and had cumulative expression greater than 1000 TPM across all samples (Figure 3b–d, Supplementary Figure 1,3). For instance, in the PLGLB gene, an exon skipping event via a novel intron leads to the loss of the original start codon and a different, slightly longer N-terminal amino acid sequence. Interestingly, we observed very similar exon skipping events in two different paralogs of this gene, PLGLB1 and PLGLB2, shown in Figure 3c–d. In both cases, the alternative protein contains a different initial coding exon that replaces exon 1 of the MANE isoform, and in both cases, the majority of the expression comes from the alternative (non-MANE) isoform, suggesting that the MANE isoforms are not the dominant ones.
Figure 3.
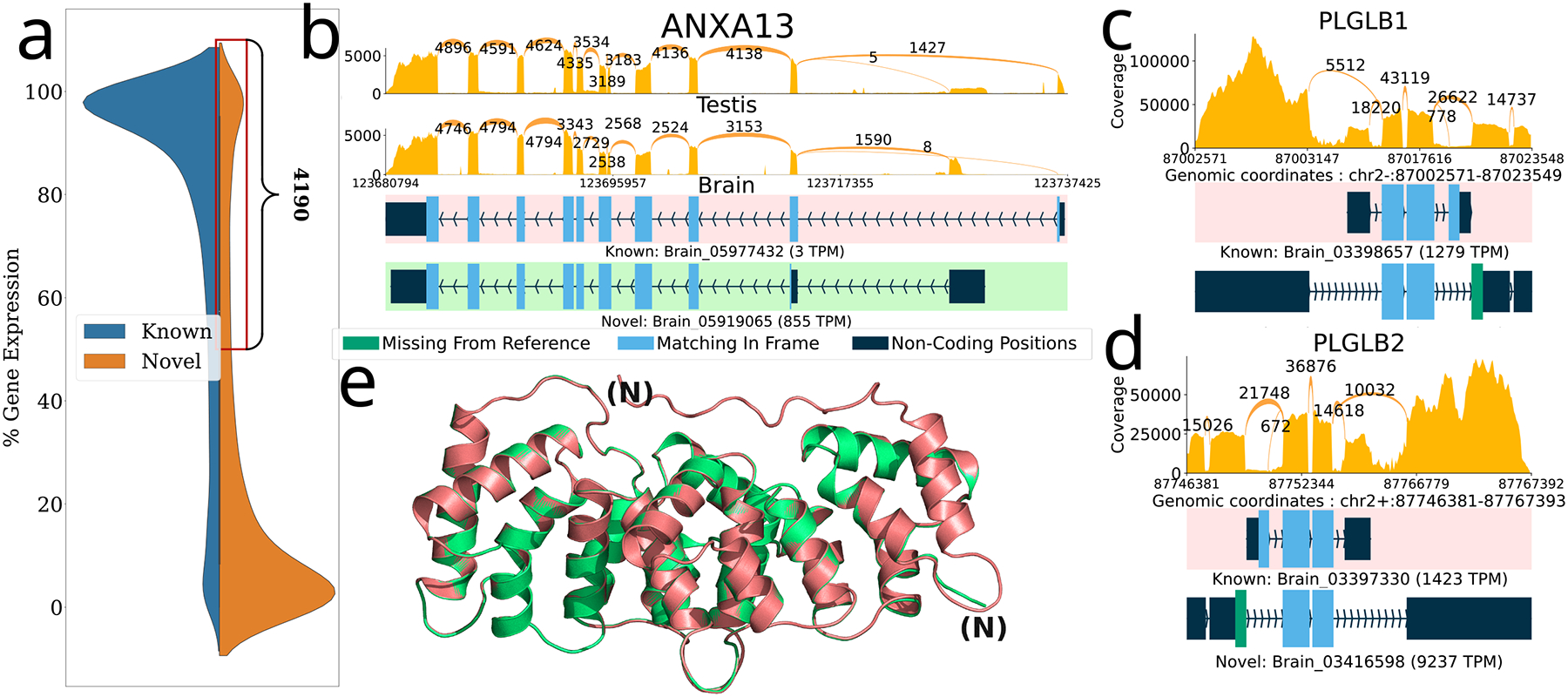
Novel ORFs in the GTEx dataset inferred using ORFanage. a) Overall distribution of loci by percent gene expression (y-axis) that comes from novel (orange) and known (blue) transcripts and a zoomed-in view of the region containing 4,190 loci where >=50% of the total expression comes from transcripts with novel ORFs or novel transcripts without an ORF. b,c,d) Sashimi plots illustrating selected examples of novel ORFs that were identified by ORFanage, each depicting a different type of variation. In each plot, coverage and splice junction values are cumulative across all samples60. The uppermost transcript, highlighted with a pink background, shows the MANE annotation. Expression levels measured in TPM are shown for each transcript. b) An alternative 5’ exon in ANXA13 that changes the start codon and shortens the ORF. e) The 3D alignment of the MANE protein (pink) to the novel ORF (green) computed by Alphafold2 and visualized via PyMOL70 is shown below with the N-termini labeled for each. c,d) Two plots show similar novel ORFs for two paralogous genes PLGLB1 and PLGB2, where skipping of the 1st reference coding exon is effectively offset by the introduction of an upstream novel exon with an alternative start codon.
Another striking example of a novel ORF among these 24 loci occurs in the ANXA13 gene (Figure 3b,e), which is a member of the family of annexin genes responsible for the production of calcium-dependent membrane-binding protein variants46. Proteins in this family contain two major domains, one at the C-terminus for the Ca2+ binding effect, and the other at the N-terminus responsible for the membrane interactions. While the core domain at the C-terminus is highly conserved across the gene family, the N-terminus is variable47, allowing for tissue-specific regulation48, 49 and localization50.The two known forms of the gene differ only in the length of the last helical structure, where the incorporation of additional peptides allows for an extension of the first helix.
In our results, most of the expression of ANXA13 came from a novel variant of the gene characterized by a mutually exclusively alternative splicing event which results in the switching of the start-codon-harboring exon for another one downstream, as shown in Figure 3b. The novel variant has an alternative methionine, followed by a glycine, which serves as its start codon, preserving much of the protein sequence with a new N-terminus. We also observed that this isoform was dominant in brain tissue, while the MANE isoform was dominant in testis (and other tissues).
We investigate how the change would impact the protein’s structure by folding it with AlphaFold2 via ColabFold51, 52. We observed an increase in the pLDDT score from 94 to 97, suggesting an even more stable structure for the new isoform, due to the removal of an unstructured segment at the N-terminus of the MANE isoform (Figure 3e). The alternative protein identified here matches a variant that was previously annotated as the third isoform of AXNA13 in Pan troglodytes53 and Papio anubis54.
Discussion
Our understanding of the transcriptional complexity of eukaryotic genomes has expanded dramatically over the years, yet the full extent and functional implications of alternative splicing are not yet entirely understood. A comprehensive evaluation of the proteome generated by alternative splicing is critical not only for identifying anomalies in disease states but also for identifying novel protein variants with distinct functions.
Our experiments demonstrate the effectiveness of ORFanage for identifying open reading frames in a set of transcripts by using reference annotation as a guide. ORFanage can recover most of the original annotation of the human genome using any of several widely used annotation databases, and it can also identify inconsistencies in those databases. More specifically, we showed that it can identify likely novel translations of transcripts with no previously assigned ORFs and find cases where an annotated ORF can be adjusted to match a canonical protein sequence. While increased similarity of ORFs to the reference is not a proof of correctness, our experiments demonstrate multiple examples where annotations can be improved via our method.
However, despite demonstrating the accuracy of our approach within the scope of this study, some important challenges remain. First, as previously discussed, ORFanage is designed to identify ORFs in a set of transcripts by using reference annotation as a guide. Therefore, it is incapable of finding translations at loci with no prior protein-coding annotations in the reference. While few protein-coding genes likely remain to be discovered in well-studied model organisms, signs of translation are being routinely reported at non-reference loci, and our method would not be suitable for protein discovery at such loci. This raises another important consideration, namely that non-model organisms may have fewer proteins annotated. While our experiments do demonstrate high accuracy of ORF reconstruction even in the presence of limited reference data, the low quality or absence of reference protein annotation in non-model organisms can present additional challenges. While not explicitly tested here, future research could combine our protocol with programs like Liftoff55 to facilitate comprehensive annotations of genomes of various ancestries that include not only transcripts but coding regions as well.
ORFanage can be used in conjunction with RNA-seq alignment and assembly to identify ORFs in novel transcripts, and to guarantee that those ORFs match the reference annotation as closely as possible. Whether using long-read alignments directly or assembled transcripts, this approach can uncover valuable insights into isoforms within protein-coding regions, leading to a better understanding of their effects on biological systems. And because RNA-seq datasets often produce large numbers of novel transcripts, the efficiency and scalability of ORFanage make it suitable for datasets of any size. We have recently applied our method to annotate ORFs in novel transcripts for the revised CHESS 34 catalog, and to help identify novel structurally stable isoforms that were then confirmed using AlphaFold256.
ORFanage can also be a valuable aid to isolating true signal from noisy transcriptome data. Assuming that proteins produced from alternative transcripts need to remain similar for genes to function correctly56, the ORF structures in the observed isoforms should be similar as well. Our approach can identify transcripts that cannot accommodate a similar ORF to the reference, serving as a noise filtering step in RNA-seq analysis.
Methods
ORFanage is based on the direct comparison of intervals that make up the exonic structures of query and reference transcripts. This optimization technique does not require sequence alignment or pre-computed genome indices, greatly reducing the computational burden of running the tool and making the analysis far more efficient than an alignment-based approach. We have tested ORFanage on datasets comprising tens of millions of transcripts assembled from thousands of RNA-seq experiments3, 4 and found that it runs robustly on these data.
Creating Bundles of Transcripts.
ORFanage operates on “bundles” of data, defined as the union of a set of overlapping reference ORFs with a set of query transcripts that overlap 1 or more of the reference ORFs. To reduce the impact of annotation errors such as readthrough transcription, ORFanage only loads coding sequence (CDS) coordinates for each reference transcript, discarding non-coding exonic coordinates.
Once both the reference and query datasets are loaded into memory and sorted internally, bundling is done in linear time by iterating over transcripts and collecting groups of all overlapping transcripts. This technique is insensitive to any information on gene boundaries, and readthrough transcription, commonly present in RNA-seq assemblies, may lead to several genes being combined into a single locus. In some cases, genes may genuinely overlap, and in such instances ORFanage might compare the ORFs of unrelated genes, possibly leading to incorrect inferences. To combat this problem, ORFanage gives users the option to group transcripts by gene IDs.
Interval Comparison.
For each query transcript in a bundle, ORFanage performs a comparison to each reference CDS. For each pair being compared, an intersection is computed to identify all intervals that belong to both the query and the reference. The process is performed for all reference transcripts and duplicate intervals are removed.
After a set of candidate overlaps is identified, ORFanage continues to search for the optimal start and end coordinates for each interval, discarding any incomplete ORFs in the process. We define a valid open reading frame as an uninterrupted sequence of 3-base codons that begins with a start codon (usually ATG in humans), ends with a stop codon (TAA, TAG, and TGA in humans), and does not contain any other stop codons other than the final one. While only one valid stop codon can be found by extending any given ORF, multiple start (ATG) codons may be present in a single ORF. In ORFanage, an optimal start codon is the one that maximizes the number of bases which are in the same frame as the reference ORF while minimizing the number of coordinates that do not match or that match out-of-frame (Figure 2).
After all intervals have been examined, if multiple distinct ORFs are plausible, ORFanage performs a heuristic selection of the optimal ORF based on a series of configurable steps. Internally, for every unique ORF, the software computes three scores which are applied successively to each set of candidate ORFs to find the best result:
the Inframe Length (IL), defined as the number of positions that are shared with the reference in the same coding frame,
ILPI, defined as the fraction of IL with respect to the length of the reference ORF, and
the length of the ORF.
When maximizing ILPI, ORFanage will prioritize ORFs that have as little novel sequence present as possible, where “novel” is defined as sequence that is not present in the reference ORF. When maximizing IL instead, ORFanage might select longer ORFs with more novel sequence if that choice increases the number of matches with the reference. Alternatively, users may specify via optional parameters that conserving the position of the start codon takes priority over conservation of the remaining protein sequence, forcing the algorithm to select ORFs whose start codon matches the reference protein whenever possible. Worth noting here is that 568 out of 19,058 ORFs in the MANE database use a start codon that is not the longest ORF.
Additional parameters.
As shown in our analysis, the ILPI metric is an effective function to assess which ORF to pick for a given isoform and corresponds closely to percent identity. It is not identical to the familiar percent identity measure, which would be more expensive to compute. For applications that might require it, ORFanage includes support for computing a Smith-Waterman alignment between the reference ORF and the ORFs identified by ORFanage, as part of the final validation of the open reading frames. ORFanage also includes an option to measure evolutionary conservation of any ORF by computing PhyloCSF scores. This option is implemented via an integrated PhyloCSF++ module57, 58. Finally, ORFanage contains a multi-threading option, under which it can process each bundle in parallel, further speeding its runtime. In our tests, ORFanage was able to process 4,256,346 collected from 1,448 brain samples of the GTEx dataset, using the MANE annotation as a reference, in 7 minutes using 24 cores of an Intel Xeon 6248R 3GHz processor, with all other parameters set to defaults. A random individual sample from the same dataset (SRR598396) was processed in 8 seconds.
Datasets
Studies of the human genome account for a large proportion of transcriptomic data being generated today, and several annotation databases are available for these studies. For our evaluation of ORFanage on the human genome, we used both the RefSeq (release 110) and GENCODE (release 41) annotations1, 2.
To investigate the utility of ORFanage on other organisms, we focused on the well-studied A. thaliana and C. elegans genomes, both of which have highly curated annotations of the transcriptome and proteome. Since for each of these two genomes only a single reference annotation was available, we chose to investigate how well ORFanage could reconstruct the ORFs using a bootstrapping technique, which allowed us to evaluate the concordance of annotated ORFs with the ones inferred by ORFanage.
For our evaluations on GTEx data, we used 1,448 poly-A selected RNA-seq samples representing 13 brain regions (age ≥ 20) from GTEx release 744. Samples were aligned with HISAT259, assembled with StringTie 233, and merged with gffcompare. Coverage and splice junction summaries were extracted using the TieBrush suite60.
Data preparation.
While ORFanage can handle several types of exceptions to the normal rules governing ORFs, such as alternative (non-ATG) start codons, selenoproteins, and otherwise overlapping genes, for our evaluations we removed these exceptions in order to measure accuracy on genes that conform to standard rules.
We began by choosing a set of genes to be used as a reference for human annotation. The MANE database6 was created by the developers of RefSeq and GENCODE as a resource of human genes where both databases agree precisely on the complete exon-intron structure as well as on the coding sequence of every gene in the database. MANE contains one canonical transcript for nearly every protein coding gene, plus a small number (62 in release 1.0) of medically-relevant transcripts that differ from the canonical ones. In our reference set, we included all genes in MANE except for 1) genes with non-ATG start codons, 2) selenoproteins, and 3) polycistronic genes. For our evaluations of both RefSeq and GENCODE, we retained only transcripts corresponding to the remaining MANE genes.
In some cases, manual curation might have altered RefSeq or GENCODE to create unusual ORFs. For instance, some partial transcripts have been manually curated to show usage of an alternative start codon, despite other ORFs at the locus containing a canonical start codon. Because we do not know whether such exceptions are intentional, we decided to avoid penalizing RefSeq or GENCODE and filter out such cases, as follows. First, we used gffread45 to identify and remove all transcripts that did not contain valid start and stop codons. Second, we searched for all pairs of overlapping ORFs that were labeled with different gene IDs and removed all such occurrences. In addition, for the RefSeq dataset we also removed 846 genes that had transcripts with known exceptions as annotated by NCBI. In the end, our filtering resulted in the removal of 1,423 genes out of 20,442 genes from RefSeq (release 110) and 1,869 genes out of 20,427 from GENCODE (version 41).
For the A. thaliana and C. elegans annotation datasets61, 62, we used the primary model organism annotation as the reference, after filtering out genes with non-ATG start codons, selenoproteins, and polycistronic genes.
Comparison of ORF-Finding Methods
To evaluate various ORF annotation methods against ORFanage, we generated two transcript sets from the GENCODE and RefSeq datasets using the process outlined in the “Data Preparation” section. For each set, we created two files: one with original ORFs preserved, and another with only transcript models, devoid of any CDS records. The first file served as our control, while the second was used as input for all ORF annotation methods. Detailed results of this comparison are provided in Table 2.
Worth noting here is that both TransDecoder and GeneMarkS-T ORF finding methods used in our comparison are designed for finding ORFs de novo, without a need for guide annotation, and as such serve a different niche of applications than ORFanage; e.g., annotation of species for which no previous annotation is available.
TransDecoder
TransDecoder, part of the Trinity package63 , can also find open reading frames in a set of transcripts. Although originally intended as an ab initio method for finding ORFs in de-novo transcriptome assemblies, the results can be improved by using homology searching against a protein database of choice. Since its original release, the software has been adapted for use with transcript models that are assembled by programs such as StringTie64 or Cufflinks65.
Since all transcripts in our analysis had confident strand assignment, we made sure to use the “-S” option to ensure the software did not consider ORFs on the opposite strand to the one annotated. Secondly, we built a protein database using the MANE dataset for blastp search against the candidate ORFs predicted by TransDecoder. These protein alignments were used to select the best candidate ORF during the second stage of the TransDecoder execution.
GeneMarkS-T
GeneMarkS-T36 is another ab initio ORF finding method included in several prominent pipelines for annotating ORFs in transcriptome assemblies. Contrary to the TransDecoder, the method relies less on the longest ORFs to initiate search and more on other features, refining it’s choice the 5’ AUG as the translation initiation site (TIS).
We applied GeneMarkS-T to both RefSeq and GENCODE datasets, similarly ensuring the strand information is kept true to the reference via the “--strand direct” option.
Execution Time
Some methods include multiple separate steps and commands that need to be executed to annotate ORFs. When measuring runtime we recorded the total time it took to run all commands specified by each method. However, since GeneMarkS-T reports CDS coordinates relative to the transcript in which they were found, we developed our own custom script to convert transcriptomic coordinates to genomic ones. While we did not add the conversion time to the total runtime of the method, depending on the implementation, this step could considerably increase the runtime.
Both ORFanage and the costly blastp alignment step in TransDecoder can make proper use of multithreading, yet GeneMarkS-T can not. Nonetheless, primarily because of how slow TransDecoder was without multithreading enabled, we allowed both ORFanage and TransDecoder to use 30 threads concurrently. For ORFanage we provide both single and multi-threaded performance measurements (Table 2).
Supplementary Material
Acknowledgements
This work was supported in part by the U.S. National Institutes of Health under grants R01 HG006677 (S.L.S.), R01 MH123567 (S.L.S.), and R35 GM130151 (S.L.S.); by the U.S. National Science Foundation under grant DBI-1759518 (M.P.).
We would also like to thank Christopher Pockrandt for helpful discussions on phyloCSF++ implementation and usage.
Competing Interests
The authors have no conflicts of interest to declare.
Code availability
The core method is implemented in C++ and based on the GFFutils45 and KSW266, 67 libraries. The code and test data are available for download at: https://github.com/alevar/ORFanage/releases/tag/1.0 (https://doi.org/10.5281/zenodo.8102912)68. Jupyter notebooks used to generate all results described in the manuscript are provided separately at https://github.com/alevar/ORFanage_tests (https://doi.org/10.5281/zenodo.8102918)69. All additional software methods used in this study and their versions and appropriate references are listed in the methods section.
Data availability
Source data for Figs. 1 and 2 are provided with this manuscript where possible. All code required to reproduce the data generated within the study from public sources is provided at https://github.com/alevar/ORFanage_tests. No new sequencing data were created for this study. Sequencing data used in this study is available through the GTEx project (phs000424.v7.p2). GTEx data was first analyzed as part of the CHESS project and the details can be found in the corresponding resources and publications (http://ccb.jhu.edu/chess/). The datasets analyzed in this study are 1. GENCODE annotation build version 41 (https://www.gencodegenes.org/human/release_41.html); 2. RefSeq annotation build 110 (https://www.ncbi.nlm.nih.gov/genome/annotation_euk/Homo_sapiens/110/); 3. MANE joint annotation build version 1.0 (https://ftp.ncbi.nlm.nih.gov/refseq/MANE/MANE_human/); 4. A. thaliana annotation https://ftp.ncbi.nlm.nih.gov/genomes/refseq/plant/Arabidopsis_thaliana/all_assembly_versions/GCF_000001735.3_TAIR10/) and 5. C. elegans genome annotion (https://ftp.ncbi.nlm.nih.gov/genomes/refseq/invertebrate/Caenorhabditis_elegans/all_assembly_versions/GCF_000002985.6_WBcel235/).
References
- 1.O’Leary NA et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res 44, 733 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frankish A et al. GENCODE: reference annotation for the human and mouse genomes in 2023. Nucleic Acids Res 51, D942–D949 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pertea M et al. CHESS: a new human gene catalog curated from thousands of large-scale RNA sequencing experiments reveals extensive transcriptional noise. Genome Biol 19, 1–14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Varabyou A et al. CHESS 3: an improved, comprehensive catalog of human genes and transcripts based on large-scale expression data, phylogenetic analysis, and protein structure. bioRxiv, 2022.12. 21.521274 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Salzberg SL Open questions: How many genes do we have? BMC biology 16, 1–3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morales J et al. A joint NCBI and EMBL-EBI transcript set for clinical genomics and research. Nature 604, 310–315 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rodriguez JM et al. APPRIS: annotation of principal and alternative splice isoforms. Nucleic Acids Res 41, D110–D117 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tress ML, Abascal F & Valencia A Alternative splicing may not be the key to proteome complexity. Trends Biochem. Sci 42, 98–110 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang ET et al. Alternative isoform regulation in human tissue transcriptomes. Nature 456, 470–476 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Djebali S et al. Landscape of transcription in human cells. Nature 489, 101–108 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reyes A & Huber W Alternative start and termination sites of transcription drive most transcript isoform differences across human tissues. Nucleic Acids Res 46, 582–592 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sinitcyn P et al. Global detection of human variants and isoforms by deep proteome sequencing. Nat. Biotechnol, 1–11 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Glinos DA et al. Transcriptome variation in human tissues revealed by long-read sequencing. Nature 608, 353–359 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park E, Pan Z, Zhang Z, Lin L & Xing Y The expanding landscape of alternative splicing variation in human populations. The American Journal of Human Genetics 102, 11–26 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang S et al. New insights into Arabidopsis transcriptome complexity revealed by direct sequencing of native RNAs. Nucleic Acids Res 48, 7700–7711 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roach NP et al. The full-length transcriptome of C. elegans using direct RNA sequencing. Genome Res 30, 299–312 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao S Alternative splicing, RNA-seq and drug discovery. Drug Discov. Today 24, 1258–1267 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Kiyose H et al. Comprehensive analysis of full-length transcripts reveals novel splicing abnormalities and oncogenic transcripts in liver cancer. PLoS Genetics 18, e1010342 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leung SK et al. Full-length transcript sequencing of human and mouse cerebral cortex identifies widespread isoform diversity and alternative splicing. Cell reports 37, 110022 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matlin AJ, Clark F & Smith CW Understanding alternative splicing: towards a cellular code. Nature reviews Molecular cell biology 6, 386–398 (2005). [DOI] [PubMed] [Google Scholar]
- 21.Tazi J, Bakkour N & Stamm S Alternative splicing and disease. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease 1792, 14–26 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garcia-Blanco MA, Baraniak AP & Lasda EL Alternative splicing in disease and therapy. Nat. Biotechnol 22, 535–546 (2004). [DOI] [PubMed] [Google Scholar]
- 23.Cummings BB et al. Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Science translational medicine 9, eaal5209 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Merkin J, Russell C, Chen P & Burge CB Evolutionary dynamics of gene and isoform regulation in Mammalian tissues. Science 338, 1593–1599 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boulet A et al. The mammalian phosphate carrier SLC25A3 is a mitochondrial copper transporter required for cytochrome c oxidase biogenesis. J. Biol. Chem 293, 1887–1896 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim HK, Pham MHC, Ko KS, Rhee BD & Han J Alternative splicing isoforms in health and disease. Pflügers Archiv-European Journal of Physiology 470, 995–1016 (2018). [DOI] [PubMed] [Google Scholar]
- 27.Frampton GM et al. Activation of MET via Diverse Exon 14 Splicing Alterations Occurs in Multiple Tumor Types and Confers Clinical Sensitivity to MET InhibitorsMET Exon 14 Alterations Confer Response to Targeted Therapy. Cancer discovery 5, 850–859 (2015). [DOI] [PubMed] [Google Scholar]
- 28.Kahles A et al. Comprehensive analysis of alternative splicing across tumors from 8,705 patients. Cancer cell 34, 211–224. e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brooks AN et al. A pan-cancer analysis of transcriptome changes associated with somatic mutations in U2AF1 reveals commonly altered splicing events. PloS one 9, e87361 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Allen AS et al. De novo mutations in epileptic encephalopathies. Nature 501, 217–221 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med 368, 2059–2074 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Varabyou A, Salzberg SL & Pertea M Effects of transcriptional noise on estimates of gene and transcript expression in RNA sequencing experiments. Genome Res 31, 301–308 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kovaka S et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol 20, 1–13 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haas BJ et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nature protocols 8, 1494–1512 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vitting-Seerup K & Sandelin A The Landscape of Isoform Switches in Human CancersIsoform Switches in Cancer. Molecular Cancer Research 15, 1206–1220 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Tang S, Lomsadze A & Borodovsky M Identification of protein coding regions in RNA transcripts. Nucleic Acids Res 43, e78 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vitting-Seerup K, Porse BT, Sandelin A & Waage J spliceR: an R package for classification of alternative splicing and prediction of coding potential from RNA-seq data. BMC Bioinformatics 15, 1–7 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kang Y et al. CPC2: a fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res 45, W12–W16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Singh U & Wurtele ES orfipy: a fast and flexible tool for extracting ORFs. Bioinformatics 37, 3019–3020 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tress ML, Abascal F & Valencia A Most alternative isoforms are not functionally important. Trends Biochem. Sci 42, 408–410 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cunningham F et al. Ensembl 2022. Nucleic Acids Res 50, D988–D995 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rice P, Longden I & Bleasby A EMBOSS: the European molecular biology open software suite. Trends in genetics 16, 276–277 (2000). [DOI] [PubMed] [Google Scholar]
- 43.Steijger T et al. Assessment of transcript reconstruction methods for RNA-seq. Nature methods 10, 1177–1184 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lonsdale J et al. The genotype-tissue expression (GTEx) project. Nat. Genet. 45, 580–585 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pertea G & Pertea M GFF utilities: GffRead and GffCompare. F1000Research 9, 304 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moss SE & Morgan RO The annexins. Genome Biol 5, 1–8 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gerke V & Moss SE Annexins: from structure to function. Physiol. Rev 82, 331–371 (2002). [DOI] [PubMed] [Google Scholar]
- 48.McCulloch KM et al. An alternative N-terminal fold of the intestine-specific annexin A13a induces dimerization and regulates membrane-binding. J. Biol. Chem 294, 3454–3463 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lillebostad PA et al. Structure of the ALS mutation target annexin A11 reveals a stabilising N-terminal segment. Biomolecules 10, 660 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fernández-Lizarbe S et al. Structural and lipid-binding characterization of human annexin A13a reveals strong differences with its long A13b isoform. Biol. Chem 398, 359–371 (2017). [DOI] [PubMed] [Google Scholar]
- 51.Mirdita M et al. ColabFold: making protein folding accessible to all. Nature methods 19, 679–682 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Varadi M et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 50, D439–D444 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Finstermeier K et al. A mitogenomic phylogeny of living primates. PloS one 8, e69504 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wall JD, Robinson JA & Cox LA High-resolution estimates of crossover and noncrossover recombination from a captive baboon colony. Genome biology and evolution 14, evac040 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shumate A & Salzberg SL Liftoff: accurate mapping of gene annotations. Bioinformatics 37, 1639–1643 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sommer MJ et al. Structure-guided isoform identification for the human transcriptome. Elife 11, e82556 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pockrandt C, Steinegger M & Salzberg SL PhyloCSF++: a fast and user-friendly implementation of PhyloCSF with annotation tools. Bioinformatics 38, 1440–1442 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lin MF, Jungreis I & Kellis M PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics 27, 275 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim D, Paggi JM, Park C, Bennett C & Salzberg SL Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 37, 907–915 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Varabyou A, Pertea G, Pockrandt C & Pertea M TieBrush: an efficient method for aggregating and summarizing mapped reads across large datasets. Bioinformatics 37, 3650–3651 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Swarbreck D et al. The Arabidopsis Information Resource (TAIR): gene structure and function annotation. Nucleic Acids Res 36, D1009–D1014 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.C. elegans Sequencing Consortium*. Genome sequence of the nematode C. elegans: a platform for investigating biology. Science 282, 2012–2018 (1998). [DOI] [PubMed] [Google Scholar]
- 63.Haas BJ et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nature protocols 8, 1494–1512 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kovaka S et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol 20, 1–13 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Trapnell C et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature protocols 7, 562–578 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li H Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Suzuki H & Kasahara M Introducing difference recurrence relations for faster semi-global alignment of long sequences. BMC Bioinformatics 19, 45 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Varabyou A ORFanage: reference guided ORF annotation (1.0.2). (2023). [Google Scholar]
- 69.Varabyou A ORFanage evaluation notebooks. (2023). [Google Scholar]
- 70.DeLano WL Pymol: An open-source molecular graphics tool. CCP4 Newsl.Protein Crystallogr 40, 82–92 (2002). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Source data for Figs. 1 and 2 are provided with this manuscript where possible. All code required to reproduce the data generated within the study from public sources is provided at https://github.com/alevar/ORFanage_tests. No new sequencing data were created for this study. Sequencing data used in this study is available through the GTEx project (phs000424.v7.p2). GTEx data was first analyzed as part of the CHESS project and the details can be found in the corresponding resources and publications (http://ccb.jhu.edu/chess/). The datasets analyzed in this study are 1. GENCODE annotation build version 41 (https://www.gencodegenes.org/human/release_41.html); 2. RefSeq annotation build 110 (https://www.ncbi.nlm.nih.gov/genome/annotation_euk/Homo_sapiens/110/); 3. MANE joint annotation build version 1.0 (https://ftp.ncbi.nlm.nih.gov/refseq/MANE/MANE_human/); 4. A. thaliana annotation https://ftp.ncbi.nlm.nih.gov/genomes/refseq/plant/Arabidopsis_thaliana/all_assembly_versions/GCF_000001735.3_TAIR10/) and 5. C. elegans genome annotion (https://ftp.ncbi.nlm.nih.gov/genomes/refseq/invertebrate/Caenorhabditis_elegans/all_assembly_versions/GCF_000002985.6_WBcel235/).