

Abstract
Indole is a heterocyclic compound formed by the fusion of a benzene ring and pyrrole ring, which has rich biological activity. Many indole-containing compounds have been sold on the market due to their excellent pharmacological activity. For example, vincristine and reserpine have been widely used in clinical practice. The diverse structures and biological activities of natural products provide abundant resources for the development of new drugs. Therefore, this review classifies natural products by structure, and summarizes the research progress of indole-containing natural product derivatives, their biological activities, structure–activity relationship and research mechanism which has been studied in the past 13 years, so as to provide a basis for the development of new drug development.
Indole is a heterocyclic compound formed by the fusion of a benzene ring and pyrrole ring, which has rich biological activity.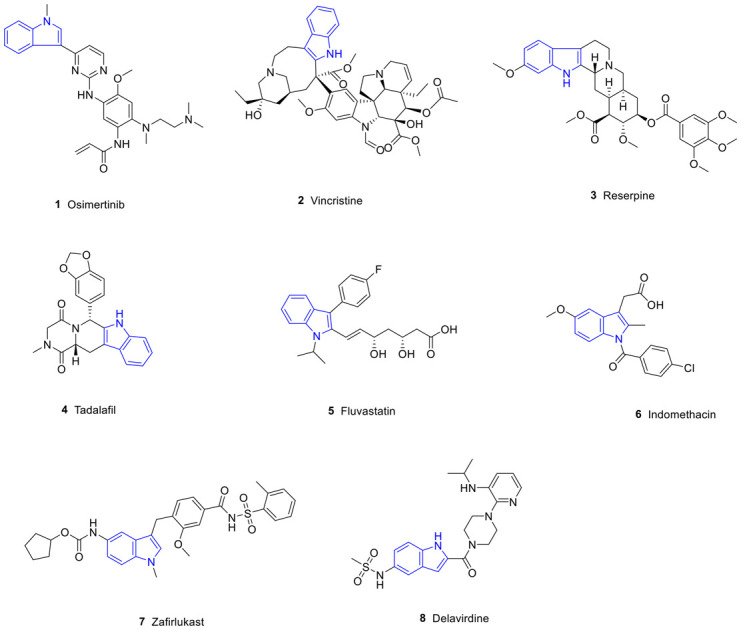
1. Introduction
Heterocyclic structures are present in many agents owing to their versatility and biodiversity.1 Among them, the indole ring is one of the most prevalent heterocycles occurring naturally and in synthetic bioactive compounds.2,3 Indole is a compound of pyrrole and benzene in parallel, also known as benzopyrrole, and its chemical formula is C8H7N. The lone pair of electrons of the nitrogen atom in the indole ring can form a conjugated system with the surrounding π electrons, which enhances the stability and reactivity of the molecule. Therefore, indole is widely used in organic synthesis and drug research.4 In addition, the N–H group in the indole ring can also form hydrogen bonds with biological macromolecules such as proteins and nucleic acids, thereby affecting biological activity.5 It is widely distributed in plants such as Isatidis radix,6Euodia rutaecarpa,7Rauvolfia serpentina,8 and Catharanthus roseus.9 Indole derivatives have a variety of pharmacological activities,10 including anti-tumor,11,12 anti-inflammatory,13–15 anti-oxidant,14–17 anti-Alzheimer's,18 and anti-diabetic19 activities; therefore, the indole ring plays an important role in the development of new agents. In addition, there are many agents containing indole structures, such as osimertinib, vincristine (VCR), reserpine, tadalafil, fluvastatin, indomethacin, zafirlukast and delavirdine which have been used in clinical research (Fig. 1, Table 1).
Fig. 1. FDA-approved drugs containing indole structures.

FDA-approved drugs containing indole structures.
| Compd. | Names | Activity | Targets or mechanisms |
|---|---|---|---|
| 1 | Osimertinib | Anti-tumor | Mutant-selective EGFR inhibitor |
| 2 | VCR | Anti-tumor | Tubulin inhibitor |
| 3 | Reserpine | Anti-hypertensive | Blocking the transmission of sympathetic impulses |
| 4 | Tadalafil | Anti-pulmonary hypertension | Phosphodiesterase inhibitor |
| 5 | Fluvastatin | Hypolipidemic | Inhibition of endogenous cholesterol synthesis |
| 6 | Indomethacin | Anti-inflammatory | Cyclooxygenase inhibitor |
| 7 | Zafirlukast | Anti-asthma | Leukotriene receptor antagonist |
| 8 | Delavirdine | Anti-HIV | Reverse transcriptase inhibitor |
Natural products have been used to treat various diseases for thousands of years. In 1578, a large number of cases of using plants to treat human diseases were recorded in the Compendium of Materia Medica. For example, the fruit of Eriobotrya japonica was used to relieve cough, Lonicera japonica was used to cure fever, and so on. Therefore, medicinal natural products are the basis of many early medicines. Natural products extracted and isolated from living organisms play an important role in the discovery and development of new agents and are also important sources of drug candidates.2 Natural products have great potential for the research and development of new drugs owing to their biodiversity and structural diversity. The use of natural products as lead compounds, combined with structure–bioactivity relationships and metabolic studies for further structural modification and synthesis of derivatives, has become a hot topic in the design and development of new drugs, which often possess higher safety, more efficacy, and cheaper preclinical evaluation.20 Many natural products are used in their natural form as lead compounds for the treatment of various diseases. Various natural products and their derivatives being used in clinical application include paclitaxel, VCR, morphine, indirubin, codeine, penicillin, etc.21 An increasing number of indole-natural product derivatives are being studied, based on the indole ring exhibiting significant biological activity in pharmaceuticals, which shows the importance of this field.22–24 Therefore, this paper summarises the research reports on indole-natural product derivatives that have been synthesized in the past 13 years. This review focuses on three aspects i.e. biological activity, structure–activity relationship, and mechanism of action of promising leads.
2. Methods
In this paper, the keywords “indole”, “terpene-indole”, “phenylpropanoid-indole”, “flavonoid-indole”, “alkaloid-indole”, “steroid-indole”, “polyphenol-indole”, “quinone-indole”, “structural modification”, “pharmacological properties”, etc. were extensively investigated in the databases such as Pubmed, Web of Science and SciFinder. The research progress of indole-containing natural product derivatives from 2011 to 2023 was summarized in three aspects: structural modification, pharmacological activities and structure–activity relationships.
3. Terpene-indole derivatives
Terpenoids and their derivatives are compounds derived from mevalonate and have isoprene units (C5 units) as the basic structural unit of the molecular backbone.25 Terpenoids are structurally complex and can be classified into eight groups according to the number of isoprene units: hemiterpenoids, monoterpenes, sesquiterpenes, diterpenes, sesterterpenoids, triterpenes, tetraterpenes, and polyterpenoids. Terpenoids are widely found in nature, with a wide variety of species and skeletons, and they are the most abundant compounds in natural products.26 More than 60 000 terpenoids have been discovered to date, and these compounds have a variety of biological activities, including anti-tumor,27 anti-oxidant,28 anti-bacterial,29 hypolipidaemic30 and other pharmacological activities.
Genipin is a natural product of iridoids, obtained by the hydrolysis of gardenia glycosides by β-glucosidase, with pharmacological activities such as anti-tumor activity.31 Fang et al. synthesized 34 genipin-indole derivatives and determined their cytotoxic activities against SW480, A-549, HL60, SMMC-7721 and MCF-7 cancer cells. Derivatives 9 (Fig. 2, Table 2, IC50: 0.9–3.64 μM) and 10 (Fig. 2, Table 2, IC50: 0.43–1.18 μM) showed strong anti-proliferative activity against SW480, A549, HL60, SMMC-7721 and MCF-7 cancer cells, with inhibition rates above 98%. Further experiments showed that compound 10 induced apoptosis and arrested the cell cycle.32
Fig. 2. Chemical structures of monoterpenoid-indole derivatives 9–10 and sesquiterpenoid-indole derivatives 11–16.

Biological activity of monoterpenoid-indole derivatives 9–10 and sesquiterpenoid-indole derivatives 11–16.
| Compd. | Activity | Ref. |
|---|---|---|
| 9 | HL-60 cells: IC50 = 0.9 μM | 21 |
| A549 cells: IC50 = 3.64 μM | ||
| SMMC-7721 cells: IC50 = 3.52 μM | ||
| MCF-7 cells: IC50 = 3.10 μM | ||
| SW480 cells: IC50 = 2.53 μM | ||
| 10 | HL-60 cells: IC50 = 0.43 μM | 21 |
| A549 cells: IC50 = 1.18 μM | ||
| SMMC-7721 cells: IC50 = 0.69 μM | ||
| MCF-7 cells: IC50 = 0.58 μM | ||
| SW480 cells: IC50 = 0.89 μM | ||
| 11 | RPMI-8226 cells: GI50 = 0.26 μM | 23 |
| CCRF-CEM cells: GI50 = 0.28 μM | ||
| 12 | RPMI-8226 cells: GI50 = 0.04 μM | 23 |
| CCRF-CEM cells: GI50 = 0.05 μM | ||
| 13 | RPMI-8226 cells: GI50 = 0.03 μM | 23 |
| CCRF-CEM cells: GI50 = 0.07 μM | ||
| M9-ENL1 AML cells: EC50 = 0.72 μM | ||
| Parthenolide | RPMI-8226 cells: GI50 = 8.20 μM | 23 |
| CCRF-CEM cells: GI50 = 9.16 μM | ||
| M9-ENL1 AML cells: EC50 = 15 μM | ||
| 14 | MCF-7 cells: IC50 = 5.25 μM | 14, 26 |
| A549 cells: IC50 = 6.17 μM | ||
| T. gondii-infected HeLa cells: IC50 = 85.3 μM | ||
| 15 | MCF-7 cells: IC50 = 14.48 μM | 26 |
| A549 cells: IC50 = 13.39 μM | ||
| 16 | MCF-7 cells: IC50 = 6.78 μM | 26 |
| A549 cells: IC50 = 12.92 μM |
Parthenolide is a natural product of sesquiterpene lactones, which is derived from the flower buds of Tanacetum balsamita and possesses anti-tumor activity.33 Bommagani et al. designed and synthesized a series of parthenolide-indole derivatives, hoping to obtain promising drugs against hematological tumors. Derivative 11 (Fig. 2, Table 2, GI50: 0.26–0.28 μM) showed considerable growth inhibitory activity against RPMI-8226 and CCRF-CEM cancer cell lines and was 32 fold more potent than parthenolide (GI50: 8.20–9.16 μM). Further modification showed that the introduction of a methoxy group as well as halogen groups such as fluorine and chlorine on the benzene ring was not conducive to anti-proliferative activity. Indole-3-carboxylate (compound 12) (Fig. 2, Table 2, GI50: 0.04–0.05 μM) displayed much lower growth inhibitory activity on RPMI-8226 and CCRF-CEM, suggesting that the connection to the indole C-3 position can improve the cells of the compound toxicity. Furthermore, the introduction of Michael receptors is beneficial for anti-proliferative activity. Compound 13 (Fig. 2, Table 2, GI50: 0.03–0.07 μM), obtained by replacing the carboxylic acid group with acrylic acid, exhibited stronger anti-proliferative activity. The results of the M9-ENL1 AML cell assay showed that compound 13 (EC50: 0.72 μM) exhibited the highest anti-proliferative activity against M9-ENL1 AML cancer cells and was 20 fold more potent than parthenolide (EC50: 15 μM).34
Artemisinin is a natural product of sesquiterpene lactones, which is derived from the stems and leaves of Artemisia carvifolia and possesses anti-tumor35 and anti-Toxoplasma gondii activities.36 Hu et al. designed and synthesized a series of artemisinin-indole derivatives and evaluated their cytotoxic activities against A549, MCF-7, HepG-2 and MDA-MB-231 cancer cells. Derivatives 14 (Fig. 2, Table 2, IC50: 5.25–6.17 μM), 15 (Fig. 2, Table 2, IC50: 13.39–14.48 μM) and 16 (Fig. 2, Table 2, IC50: 6.78–12.92 μM) showed high activity against MCF-7 and A549 cancer cell lines. Compound 14 with an electron-withdrawing group was generally more potent than compounds 15 and 16 against MCF-7 and A549 cells, indicating that the electron-withdrawing group on the benzene ring contributed more to the anti-proliferative activity. Further studies illustrated that compound 14 (reversal fold (RF): 117) could reverse the multidrug resistance of MCF-7/doxorubicin cells and induce G2/M cell cycle arrest in MCF-7 cells. Therefore, compound 14 can serve as a potential anticancer drug for reversing multidrug resistance and is worthy of further study.37 In addition, Deng's study showed that compound 14 exhibited promising anti-T. gondii activity with a selectivity of 1.58 and IC50 value of 85.3 in T. gondii-infected HeLa cells, being more potent than spiramycin with a selectivity of 0.72 and IC50 value of 262.2 in T. gondii-infected HeLa cells.23
Retinoic acid is a monocyclic diterpene natural product metabolised from vitamin A, which possesses anti-tumor activity.38 Gurkan-Alp et al. designed and synthesized five retinoic acid-indole derivatives and determined their anti-tumor activity in vitro. Derivative 17 (Fig. 3, Table 3, IC50: <0.01–1.83 μM) exhibited low-concentration anti-proliferative activity against Cama1, MCF-7, MDA-MB-453, SK-BR-3, MDA-MB-361 and MDA-MB-231 breast cancer cell lines as well as being comparable to camptothecin (IC50: <0.01–0.17 μM). In addition, compound 17 showed the strongest anti-proliferative activity against Cama1 and MDA-MB-453 (IC50 values below 0.01 μM), while the less toxic effect on normal epithelial breast cancer cell line MCF-12A was relatively small with an IC50 value of 3.92 μM. Interestingly, compound 17 displayed potent anti-proliferative activity against MDA-MB-231, a triple-negative breast cancer cell line, with an IC50 value of 1.83 μM. Further studies indicated that compound 17 induced apoptosis in MDA-MB-231 cells, which may be related to the inhibition of the RXRa receptors. Therefore, compound 17 can be used as a candidate agent for the treatment of triple-negative breast cancer, and is worthy of further study.39
Fig. 3. Chemical structures of diterpenoid-indole derivatives 17–22.

Biological activity of diterpenoid-indole derivatives 17–22.
| Compd. | Activity | Ref. |
|---|---|---|
| 17 | Cama1 cells: IC50 < 0.01 μM | 28 |
| MCF-7 cells: IC50 = 1.71 μM | ||
| MDA-MB-453 cells: IC50 < 0.01 μM | ||
| SK-BR-3 cells: IC50 = 1.29 μM | ||
| MDA-MB-361 cells: IC50 = 0.06 μM | ||
| MDA-MB-231 cells: IC50 = 1.83 μM | ||
| MCF-12A cells: IC50 = 3.92 μM | ||
| Camptothecin | Cama1 cells: IC50 = 0.07 μM | 28 |
| MCF-7 cells: IC50 < 0.01 μM | ||
| MDA-MB-453 cells: IC50 < 0.01 μM | ||
| SK-BR-3 cells: IC50 < 0.01 μM | ||
| MDA-MB-361 cells: IC50 = 0.17 μM | ||
| MDA-MB-231 cells: IC50 < 0.01 μM | ||
| 18 | MCF-7 cells: IC50 = 1.85 μM | 30 |
| HCT116 cells: IC50 = 1.22 μM | ||
| 19 | SMMC-7721 cells: IC50 < 1.39 μM | 32 |
| HepG2 cells: IC50 = 0.51 μM | ||
| Hep3B cells: IC50 < 0.73 μM | ||
| 20 | HCT116 cells: IC50 = 0.81 μM | 15 |
| Bel7402 cells: IC50 = 0.83 μM | ||
| MCF-7 cells: IC50 = 0.39 μM | ||
| 21 | HCT116 cells: IC50 = 0.68 μM | 15 |
| Bel7402 cells: IC50 = 1.73 μM | ||
| MCF-7 cells: IC50 = 0.83 μM | ||
| 22 | HCT116 cells: IC50 = 0.16 μM | 15 |
| Bel7402 cells: IC50 = 2.18 μM | ||
| MCF-7 cells: IC50 = 1.52 μM | ||
| Oridonin | HCT116 cells: IC50 = 6.84 μM | 15 |
| 5-Fu | HCT116 cells: IC50 = 24.8 μM | 15 |
Andrographolide is a bicyclic diterpene lactone natural product derived from Andrographis paniculata that possesses anti-tumor activity.40,41 Song et al. synthesized a series of novel indolo [3,2-b] andrographolide derivatives and determined their cytotoxic activities against three human cancer cell lines MCF7, HCT116 and DU145. Derivative 18 (Fig. 3, Table 3, IC50: 1.22–1.85 μM) showed considerable anti-proliferative activity against MCF-7 and HCT116 cancer cell lines. SAR analysis indicated that the anti-proliferative activity of the bisesterified compounds of the C-14 and C-19 hydroxyl groups was stronger than that of the monoesterified compounds, and the steric hindrance of the ester group also affected the anti-proliferative activity of the compounds. A smaller volume of acetyl groups exhibited stronger anti-proliferative activity than benzoyl groups. Further experiments showed that compound 18 induced HCT116 cell arrest in the S phase and induced apoptosis in a concentration-dependent manner.42
Dehydroabietic acid is a tricyclic diterpenoid oxygenated compound, which is widely present in all types of Pinaceae plants and exhibits antitumour activity.43 Chen et al. designed and synthesized a series of novel dehydroabietic acid-indole derivatives, and evaluated their anticancer activities against SMMC-7721, HepG2 and Hep3B cancer cells. Derivative 19 (Fig. 3, Table 3, IC50: 0.51–1.39 μM) showed strong cytotoxic activities against SMMC-7721, HepG2 and Hep3B cancer cell lines. Mechanistic studies indicated that compound 19 inhibited the MAPK signalling pathway, increased ROS generation, arrested the cell cycle in the G2/M phase, and damaged membrane integrity. Overall, compound 19 is a promising anticancer agent candidate.44
Oridonin is a tetracyclic diterpene natural product derived from the whole plant of Isodon rubescens with anti-tumor activity.45 Shen et al. designed and synthesized a series of oridonin derivatives, and found that when bearing an indole ring on the 14-OH position, the oridonin derivatives had the best anti-tumour activity. Derivatives 20 (Fig. 3, Table 3, IC50: 0.39–0.83 μM), 21 (Fig. 3, Table 3, IC50: 0.68–1.73 μM) and 22 (Fig. 3, Table 3, IC50: 0.16–2.18 μM) exhibited considerable activities against HCT116, Bel7402 and MCF-7 cancer cells. In particular, compound 22 showed extremely potent anti-proliferative activity against HCT116 cancer cells with an IC50 value of 0.16 μM, and was 43 times higher than oridonin (IC50: 6.84 μM) as well as 155 times the positive control drug 5-fluorouracil (5-Fu) (IC50: 24.8 μM). The selectivity of compound 22 for HCT116 cells (selection coefficient: 23.63) was stronger than that for normal L02 cells, and the selection effect was better than that of oridonin (selection coefficient: 1.02) and 5-Fu (selection coefficient: 0.65). SAR analysis showed that the properties and positions of the substituents on the indole ring were closely related to the anti-proliferative activity of the compounds, and the activity sequence was: 6-OCH3 > 6-F > 6-Br > 5-OCH3 > 6-Cl> 5-Cl > 5-H. Furthermore, compound 22 inhibited colony formation by HCT116 cells in a concentration-dependent manner. Further experiments revealed that compound 22 may induce G2/M phase arrest and apoptosis in HCT116 cells via the p53-MDM2 pathway. In vivo experiments showed that compared with the control group, the tumour volume and weight after compound 22 treatment were significantly decreased. Overall, compound 22 is a highly potential anti-tumor drug that deserves further study.24
Malabaricol is a tricyclic triterpenoid natural product derived from the gum exudates of the trunk of Ailanthus triphysa and exhibits anti-tumour activity.46 Bommakanti et al. synthesized a series of malabaricol derivatives and determined their cytotoxic activity against A549 cancer cells. Derivative 23 (Fig. 4, Table 4, IC50: 13.40 μM) showed promising activity against A549 cancer cells, being comparable to malabaricol (IC50: 10.91 μM).22
Fig. 4. Chemical structures of triterpenoid-indole derivatives 23–36.

Biological activity of triterpenoid-indole derivatives 23–36.
| Compd. | Activity | Ref. |
|---|---|---|
| 23 | A549 cells: IC50 = 13.40 μM | 13 |
| 24 | Protein tyrosine phosphatase 1 B: IC50 = 4.3 μM | 36 |
| 25 | α-Glucosidase: IC50 = 4.02 μM | 39 |
| 26 | Hyaluronidase: IC50 = 9.97 μM | 40 |
| 27 | SMMC-7721 cells: IC50 = 0.56 μM | 42 |
| HepG2 cells: IC50 = 0.91 μM | ||
| 28 | SMMC-7721 cells: IC50 = 1.08 μM | 45 |
| HepG2 cells: IC50 = 1.26 μM | ||
| 29 | HepG2 cells: IC50 = 3.12 μM | 44 |
| SGC7901 cells: IC50 = 10.22 μM | ||
| Paclitaxel | HepG2 cells: IC50 = 7.87 μM | 44 |
| SGC7901 cells: IC50 = 14.35 μM | ||
| Ursolic acid | HepG2 cells: IC50 > 20 μM | 44 |
| SGC7901 cells: IC50 > 20 μM | ||
| 31 | Bel7402 cells: IC50 = 0.02 μM | 47 |
| 32 | H4 cells: IC50 = 2.03 μM | 47 |
| Bel7402 cells: IC50 = 0.01 μM | ||
| Celastrol | H4 cells: IC50 = 2.09 μM | 47 |
| Bel7402 cells: IC50 = 1.55 μM | ||
| 33 | α-Glucosidase: IC50 = 1.8 μM | 50 |
| 34 | α-Glucosidase: IC50 = 0.04 μM | 51 |
| Acarbose | α-Glucosidase: IC50 = 189.2 μM | 51 |
| 35 | HIV-1 cells: IC50 = 0.05 μM | 52 |
| Bevirimat | HIV-1 cells: IC50 = 0.065 μM | 52 |
| 36 | RAW 264.7 cells: IC50 = 22.6 μM | 54 |
| Lupeol | RAW 264.7 cells: IC50 = 37.3 μM | 54 |
Glycyrrhetinic acid is a natural product of pentacyclic triterpenes, which is derived from the root of Glycyrrhiza uralensis and possesses hypoglycaemic activity.47 De-la-Cruz-Martínez et al. designed and synthesized a series of glycyrrhetinic acid derivatives, expecting to obtain potential new hypoglycemic drugs. Derivative 24 (Fig. 4, Table 4, IC50: 4.3 μM) showed considerable inhibitory activity against PTP1B. The SAR illustrated that the introduction of trifluoromethyl, methoxy, and fluorine atoms enhanced the inhibitory activity, whereas the introduction of methyl and chlorine atoms reduced the activity. Enzyme kinetics experiments showed that compound 24 (Ki: 3.9 μM) is a non-competitive inhibitor with stronger binding ability to the enzyme than suramin (Ki: 7.1 μM).48
Oleanolic acid is a pentacyclic triterpenoid natural product derived from the fruit of Ligustrum lucidum and has pharmacological activities such as inhibition of glucosidase,49 anti-oxidant,50 and anti-hyaluronidase.50 Therefore, researchers modified the C2 and C3 positions of oleanolic acid to obtain compounds with better pharmacological activity. Derivative 25 (Fig. 4, Table 4, IC50:4.02 μM) showed considerable inhibitory activity against α-glucosidase. The SAR results showed that the introduction of an indole group at the C-3 position of oleanolic acid could enhance the biological activity of the compound, the carboxyl group at the C-28 position was crucial to the activity of the compound, and esterification of the carboxyl group greatly reduced its activity. The results of enzyme kinetics analysis revealed that compound 25 is a mixed type of α-glucosidase inhibitor, and the mechanism of action is to inhibit the formation of the α-glucosidase-p-NPG intermediate by binding to α-glucosidase and to the substrate p-NPG of α-glucosidase directly.51
Derivative 26 (Fig. 4, Table 4, IC50: 9.97 μM) exhibited high inhibitory activity against hyaluronidase. Compared to heterocycles such as quinoline and pyridine, the introduction of an indole ring can improve the biological activity of the compound. Further studies illustrated that compound 26 was a mixed non-competitive enzyme inhibitor, which reduced enzyme activity by binding to the aromatic amino acid side chain of the hyaluronidase protein. Moreover, compound 26 exhibited skin permeability under weakly acidic conditions (pH = 6.5). In vitro studies showed that, compared with oleanolic acid, compound 26 significantly reduced ROS levels in HaCAT cells and ameliorated H2O2-induced oxidative stress. Overall, compound 26 is a potential agent with anti-hyaluronidase activity, which has further research value.52
Ursolic acid is a pentacyclic triterpenoid natural product derived from the fruit of Arctostaphylos uva-ursi with anti-tumor activity.53 Therefore, researchers have modified the C2, C3 positions and carboxyl group of ursolic acid, hoping to obtain anti-tumor drugs with research prospects. Derivatives 27 (Fig. 4, Table 4, IC50: 0.56–0.91 μM)54 and 28 (Fig. 4, Table 4, IC50: 1.08–1.26 μM)55 showed strong inhibitory activity against SMMC-7721 and HepG2. Among them, compound 28 was comparable to doxorubicin (IC50: 0.62–0.77 μM) and compound 27 was less toxic to human normal hepatocytes L02 with an IC50 value of 10.58 μM. SAR analysis of compound 27 indicated that when the number of carbon atoms in the alkyl linker between the amide group and the amino group was three, the best tumour inhibitory effect was exhibited. The type of nitrogen-containing structure in the side chain is also closely related to the anti-proliferative activity of the compound. The introduction of methylpiperazine, –NH2, –N(Me)2, –N(Et)2, and piperidine increased the anti-proliferative activity of the compounds. Among them, the effect of N-methylpiperazine was the most obvious, whereas the anti-proliferative activity of compounds containing morpholine structures was not obvious. The SAR analysis of compound 28 indicated that the introduction of a substituent at the C-5 position of the indole ring could enhance anti-tumor activity, while the introduction of a substituent at the C-7 position greatly reduced anti-proliferative activity.
Further studies showed that compound 27 not only inhibited the relaxation of supercoiled DNA by inhibiting the activity of topoisomerase II but also induced the production of ROS and decreased membrane potential in SMMC-7721 cells in a dose-dependent manner to induce apoptosis. Compound 27 had a significant topoisomerase II inhibitory effect, strong anti-cancer activity, and selectivity, and can be used as an anti-cancer drug for further research.
Derivative 29 (Fig. 4, Table 4, IC50: 3.12–10.22 μM) exhibited promising activity against HepG2 and SGC7901 cancer cells, being more potent than ursolic acid (IC50: >20 μM) and paclitaxel (IC50: 7.87–14.35 μM). In addition, at a concentration of 10 μM, compound 29 (48.96% and 54.6% inhibition, respectively) inhibited the growth of HepG2 and SGC7901 cells compared to paclitaxel (52.75% and 50.82% inhibition, respectively), and was superior to ursolic acid (12.62% and 13.34% inhibition, respectively). SAR results showed that haloalkanes with branched chains were more active than straight-chain haloalkanes; meanwhile, the anti-tumor activity of haloalkanes increased with the growth of their carbon chains.56
Derivative 30 (Fig. 4) exhibited considerable anti-proliferative activity against U251 and C6 glioma cells (the proliferation rate was reduced to 17% and 21% at 10 μM, respectively). Mechanistic studies revealed that compound 30 could downregulate cAMP levels and induce G0/G1 phase arrest and apoptosis in U251 glioma cells. Therefore, compound 30 could be further studied as a potential anticancer drug.57
Celastrol is a pentacyclic triterpenoid natural product derived from the whole plant or peeled xylem of Tripterygium wilfordii, possessing anti-tumor activity.58 Tang et al. synthesized a series of C(6)-indole-substituted celastrol derivatives and explored their antitumor activity against Bel7402 cells. Derivatives 31 (Fig. 4, Table 4, IC50: 0.02 μM) and 32 (Fig. 4, Table 4, IC50: 0.01 μM) showed considerable anti-proliferative activity against human hepatocellular carcinoma Bel7402. Although most compounds substituted at the C-5 position of the indole ring are more cytotoxic than those substituted at the C-6 position, the C-6 substituted celastrol-indole derivative 31 is highly cytotoxic to human hepatocellular carcinoma Bel7402 which showed excellent anti-proliferative activity and was 77 fold more potent than celastrol (IC50: 1.55 μM). In addition, the N-substituted indole derivative 32 not only showed good anti-proliferative activity against Bel7402 cancer cells but also exhibited similar anti-proliferative activity to celastrol (IC50: 2.09 μM) against the human glioblastoma cell line H4 (IC50: 2.03 μM).59
Betulinic acid is a common pentacyclic triterpenoid derived from Betula platyphylla, which has hypoglycaemic60 and antiviral activities.61 Therefore, researchers have designed and synthesized a series of betulinic acid-indole derivatives, expecting to obtain new drugs with excellent biological activity. The inhibitory activity of derivative 33 (Fig. 4, Table 4, IC50: 1.8 μM) against α-glucosidase being 10 fold more potent active than betulinic acid (IC50: 18.4 μM) illustrated that the C-2 and C-3 positions of betulinic acid fused into the indole ring can significantly increase the activity.62 Further structural modifications revealed that compound 34 (Fig. 4, Table 4, IC50: 0.04 μM), with the replacement of the carboxyl group at C-28 with a carboxamide group, was 4730 fold more potent active than acarbose (IC50: 189.2 μM), probably because the carboxamide group provided more hydrogen bonds than the carboxyl group.63 In addition, derivative 35 (Fig. 4, Table 4, IC50: 0.05 μM) exhibited considerable activity against HIV-1, being more potent than bevirimat (IC50: 0.065 μM).64
Lupeol is a triterpenoid derived from the cuticle of lupin seeds that exhibits anti-inflammatory pharmacological activity.65 Bhandari et al. synthesized a series of lupeol indole derivatives, expecting to obtain new drugs with excellent anti-inflammatory activity. Lupeol-indole derivative 36 (Fig. 4, Table 4, IC50: 22.6 μM on RAW 264.7 cells) was more active in inhibiting NO production than lupeol (IC50: 37.3 μM on RAW 264.7 cells), and the SAR studies showed that substituted electron donating groups on the indole benzene ring promote the activity. Furthermore, at the concentration of 20 μg mL−1, the TNF-α and IL-1β inhibitory activities of compound 36 on RAW 264.7 cells were 42.3% and 22.9%, respectively, while the inhibitory activities of curcumin on TNF-α and IL-1β were 1.25% and 6.44%, respectively.66
Summary: there were many reports on the anti-tumor activity of indole-modified terpenoid natural products. Among them, oridonin-indole derivative 22 show 43 times higher anti-HCT116 cancer cell activity than oridonin, and the IC50 value was 0.16 μM. The activity of the substituent at the C6 position was significantly stronger than that of C5, and the electron-donating group improved the activity of the compound. Therefore, compound 22 had further research value.
4. Phenylpropanoid-indole derivatives
Phenylpropanoids are a class of compounds consisting of a benzene ring linked to three straight-chain carbon atoms (C6–C3 groups), including phenylpropylenes and their derivatives with different degrees of oxidation, coumarins, and lignans, which have pharmacological activities such as anti-tumor,67 anti-bacterial,68 hypolipidaemic69 and anti-oxidant.70
Podophyllotoxin is an aryl tetrahydronaphtholide lignan natural product, which is derived from the rhizome of Podophyllum versipelle and possesses anti-tumor activity.71 Therefore, researchers modified the C4 position of podophyllotoxin in order to obtain antitumor drugs with research value. Derivative 37 (Fig. 5, Table 5, IC50: 0.07–0.1 μM) showed excellent anti-tumor activity against HepG2, HeLa, A549 and MCF-7 cancer cell lines, and was 26–58 fold more potent than podophyllotoxin (IC50: 2.4–6.9 μM) as well as 24–86 fold more potent than colchicine (IC50: 9.7–14.3 μM), respectively. Further studies illustrated that compound 37 induced G2/M phase arrest and inhibited tubulin polymerisation. In vivo experiments showed that compound 29 significantly inhibited tumour growth. Therefore, compound 37 has further research value as a potential tubulin inhibitor.72
Fig. 5. Chemical structures of podophyllotoxin-indole derivatives 37–43.

Biological activity of podophyllotoxin-indole derivatives 37–42.
| Compd. | Activity | Ref. |
|---|---|---|
| 37 | HepG2 cells: IC50 = 0. 1 μM | 60 |
| HeLa cells: IC50 = 0.08 μM | ||
| A549 cells: IC50 = 0.08 μM | ||
| MCF-7 cells: IC50 = 0.07 μM | ||
| Podophyllotoxin | HepG2 cells: IC50 = 2.4 μM | 60 |
| HeLa cells: IC50 = 6.9 μM | ||
| A549 cells: IC50 = 2.6 μM | ||
| MCF-7 cells: IC50 = 2.4 μM | ||
| Colchicine | HepG2 cells: IC50 = 5.8 μM | 60 |
| HeLa cells: IC50 = 10.2 μM | ||
| A549 cells: IC50 = 9.7 μM | ||
| MCF-7 cells: IC50 = 14.3 μM | ||
| 38 | HepG2 cells: IC50 = 1.93 μM | 61 |
| HeLa cells: IC50 = 2.2 μM | ||
| Podophyllotoxin | HepG2 cells: IC50 = 6.21 μM | 61 |
| HeLa cells: IC50 = 9.32 μM | ||
| 39 | A549 cells: IC50 = 1.87 μM | 62 |
| 40 | A549 cells: IC50 = 2.24 μM | 62 |
| Podophyllotoxin | A549 cells: IC50 = 3.76 μM | 62 |
| VP-16 | A549 cells: IC50 = 2.27 μM | 62 |
| 41 | K562 cells: IC50 = 0.1 μM | 63 |
| 42 | K562 cells: IC50 = 0.034 μM | 64 |
| VP-16 | K562 cells: IC50 = 0.764 μM | 64 |
| VCR | K562 cells: IC50 = 0.178 μM | 64 |
The anti-proliferative activity of derivative 38 (Fig. 5, Table 5, IC50: 1.93–2.2 μM) was stronger than podophyllotoxin (IC50: 6.21–9.32 μM) against HepG2 and HeLa cells; it was also less toxic against LO2 cells (IC50: 75.66 μM) and PBMC cells (IC50: 88.97 μM). Further experiments showed that compound 38 induced cell arrest in the G2/M phase and apoptosis through the mediation of Bcl-2.73
Derivatives 39 (Fig. 5, Table 5, IC50: 1.87 μM) and 40 (Fig. 5, Table 5, IC50: 2.24 μM) showed strong anti-proliferative activity against A549 cancer cells, being more potent than podophyllotoxin (IC50: 3.76 μM) and etoposide (VP-16) (IC50: 2.27 μM). Further studies illustrated that compounds 39 and 40 induced cell G2/M phase arrest, inhibited topoisomerase II activity, and bound to DNA. Therefore, compounds 39 and 40 deserve further study as potential topoisomerase II inhibitors.74
Derivative 41 (Fig. 5, Table 5, IC50: 0.1 μM) showed promising anti-proliferative activity against K562 cancer cells, which was comparable to podophyllotoxin (IC50: 0.025 μM) and VCR (IC50: 0.178 μM). In addition, compound 41 (resistance factor value: 2.27) also displayed a lower resistance factor in K562/VCR cells, which was comparable to podophyllotoxin (resistance factor value: 2.00) but much lower than VP-16 (resistance factor value: 15.497) and VCR (resistance factor value: 26.612).75 Derivative 42 (Fig. 5, Table 5, IC50: 0.034 μM; the resistance factor values of compound 42, podophyllotoxin, VP-16, and VCR were 2.23, 2.00, 15.49, and 26.61, respectively) showed a lower resistance factor against K562/VCR, and exhibited promising anti-proliferative activity against K562 cancer cells, being more potent than VP-16 (IC50: 0.764 μM) and VCR (IC50: 0.178 μM), and comparable to podophyllotoxin (IC50: 0.025 μM).76 In addition, compound 41 induced autophagy in K562/VCR cells by inhibiting the PI3K/AKT/mTOR signalling pathway, while compound 42 induced autophagy in K562/VCR cells by increasing the levels of Beclin1 and LC3-II.
Derivative 43 (Fig. 5) (resistance factor value: 3.5) showed lower resistance against K562/ADR cancer cells than podophyllotoxin (resistance factor value: 14.166) and VP-16 (resistance factor value: 4.903). Preliminary experiments showed that compound 43 disrupted the tubulin structure, induced cell G2 arrest and apoptosis, and reversed the drug resistance of tumour cells by downregulating the expression levels of P-gp and MRP1 proteins.77
Derivative 44 (Fig. 6, Table 6, IC50: 5.87 μM) showed considerable anti-proliferative activity against Bel-7402 cancer cells, which was comparable to that of 5-Fu (IC50: 5.94 μM). Moreover, compound 44 (resistance factor value: 0.36) exhibited promising resistance to Bel-7402 cancer cells, lower than podophyllotoxin (resistance factor value: 0.84) and 5-Fu (resistance factor value: 16.83), which may be related to the ability of compound 44 to inhibit ERK1/2 phosphorylation levels.78
Fig. 6. Chemical structures of podophyllotoxin-indole derivatives 44–49.

Biological activity of podophyllotoxin-indole derivatives 44–49.
| Compd. | Activity | Ref. |
|---|---|---|
| 44 | Bel-7402 cells: IC50 = 5.87 μM | 66 |
| 45 | HeLa cells: IC50 = 1.96 μM | 67 |
| MCF-7 cells: IC50 = 3.13 μM | ||
| VP-16 | HeLa cells: IC50 = 2.56 μM | 67 |
| MCF-7 cells: IC50 = 8.61 μM | ||
| 46 | K562 cells: IC50 = 1.72 μM | 68 |
| HeLa cells: IC50 = 3.92 μM | ||
| VP-16 | K562 cells: IC50 = 6.26 μM | 68 |
| HeLa cells: IC50 = 5.74 μM | ||
| 47 | K562 cells: IC50 = 1.13 μM | 69 |
| HeLa cells: IC50 = 0.1 μM | ||
| VP-16 | K562 cells: IC50 = 2.02 μM | 69 |
| HeLa cells: IC50 = 1.98 μM | ||
| 48 | HeLa cells: IC50 = 0.79 μM | 70 |
| VP-16 | HeLa cells: IC50 = 21.44 μM | 70 |
| 49 | K562 cells: IC50 = 0.4 μM | 71 |
| HeLa cells: IC50 = 0.15 μM | ||
| VP-16 | K562 cells: IC50 = 0.9 μM | 71 |
| HeLa cells: IC50 = 2.11 μM |
Derivative 45 (Fig. 6, Table 6, IC50: 1.96–3.13 μM) exhibited broad-spectrum anti-tumor activity against HeLa and MCF-7 cancer cells, being more potent than VP-16 (IC50: 2.56–8.61 μM). Mechanistic experiments showed that compound 45 achieved anti-tumor activity by inhibiting topoisomerase II and inducing apoptosis.79
Derivative 46 (Fig. 6, Table 6, IC50: 1.72–3.92 μM) showed better anti-tumor activity against K562 and HeLa, stronger than VP-16 (IC50: 5.74–6.26 μM). Although its anti-tumor activity against K562 was not as good as that of the parent podophyllotoxin (IC50: 0.008 μM), it showed higher resistance to K562/A02 (resistance factor value: 0.73, resistance factor value of podophyllotoxin: 35), which could be further studied as a potential anti-tumor multidrug resistance drug.80
Derivative 47 (Fig. 6, Table 6, IC50: 0.1–1.13 μM) showed considerable activity against HeLa and K562 cancer cells, being more potent than VP-16 (IC50: 1.98–2.02 μM). The resistant factor of compound 47 (resistance factor value: 1.97) was lower than VP-16 (resistance factor value: 11.29) and the IC50 value of compound 47 (IC50: 2.23 μM) against K562/AO2 was lower than VP-16 (IC50: 22.81 μM), indicating that compound 47 possessed weak multidrug resistance.81 Derivative 48 (Fig. 6, Table 6, IC50: 0.79 μM) exhibited promising activity against HeLa cancer cells, being 27 fold more potent than VP-16 (IC50: 21.44 μM).82 Derivative 49 (Fig. 6, Table 6, IC50: 0.15–0.4 μM) showed good activity against HeLa and K562 cancer cells, being more potent than VP-16 (IC50: 0.9–2.11 μM).83 Further research illustrated that the connection site of podophyllotoxin and the indole ring had a certain influence on the degree of anti-proliferative activity.
Coumarin is a lactone compound with a basic skeleton of the parent nucleus of benzo-alpha-pyrone, mainly distributed in Apiaceae, Fabaceae, Asteraceae, and Ruta graveolens, with anti-tumor,84,85 anti-bacterial,86 anti-leishmaniasis,87 anti-oxidant,88 hypoglycaemic,89 anti-hyperlipidemia,90 anti-Alzheimer's disease91 and other pharmacological activities. Therefore, researchers modified the structure of coumarin, hoping to obtain new drugs with better biological activity. Derivative 50 (Fig. 7, Table 7, IC50: 7.4 μM) exhibited significant anti-cancer activity against MCF-7 cancer cells as well as was lower cytotoxicity to VERO cells with an IC50 value >100 μM. SAR studies suggested that the introduction of aldehyde or carboxyl groups into the indole nucleus could enhance the anti-proliferative activity of compounds. Among them, compounds with halogen substitution in the coumarin ring exhibited better anticancer activity than the unsubstituted or hydroxy-substituted compounds, which may be due to the enhanced lipophilicity of the compounds by the halogen atoms. The presence of bromine atoms increased the anti-proliferative activity more than substitution with fluorine or chlorine atoms.92
Fig. 7. Chemical structures of coumarin-indole derivatives 50–55.

Biological activity of coumarin-indole derivatives 50–55.
| Compd. | Activity | Ref. |
|---|---|---|
| 50 | MCF-7 cells: IC50 = 7.4 μM | 79 |
| 51 | MCF-7 cells: IC50 = 8.01 μM | 80 |
| 52 | MCF-7 cells: IC50 = 17.5 μM | 81 |
| 53 | HOP-92 cells: GI50 = 0.95 μM | 82 |
| 54 | MRSA: MIC = 0.25 μg mL−1 | 83 |
| E. faecalis: MIC = 0.25 μg mL−1 | ||
| S. aureus ATCC 25922: MIC = 0.25 μg mL−1 | ||
| S. aureus ATCC 29213: MIC = 0.25 μg mL−1 | ||
| E. coli: MIC = 0.25 μg mL−1 | ||
| A. baumannii: MIC = 0.25 μg mL−1 | ||
| Norfloxacin | MRSA: MIC = 4 μg mL−1 | 83 |
| E. faecalis: MIC = 4 μg mL−1 | ||
| S. aureus ATCC 25922: MIC = 4 μg mL−1 | ||
| S. aureus ATCC 29213: MIC = 2 μg mL−1 | ||
| E. coli: MIC = 2 μg mL−1 | ||
| A. baumannii: MIC = 4 μg mL−1 | ||
| 55 | M. tuberculosis H37Rv strain: MIC = 2.02 μM | 84 |
Derivative 51 (Fig. 7, Table 7, IC50: 8.01 μM) showed good anti-proliferative activity against MCF-7 cancer cells. The highest anti-proliferative activity was observed when the alkyl carbon chain length between the indole and thiazole rings was 3. The wound healing experiment showed that the migration ability of MCF-7 cells treated with compound 51 was significantly decreased, indicating that compound 51 can resist the metastasis of MCF-7 cancer cells.93 Compound 52 (Fig. 7, Table 7, IC50: 17.5 μM) showed considerable anti-proliferative activity to MCF-7 cancer cells, and the western blotting experiments showed that the expression level of CDK2 protein in cells was significantly decreased after compound 52 treatment.94 Derivative 53 (Fig. 7, Table 7) exhibited good antimitotic activity, with a GI50 value of 0.95 μM.95
Derivative 54 (Fig. 7, Table 7) showed a broad anti-bacterial spectrum against methicillin-resistant Staphylococcus aureus, Enterococcus faecalis, Staphylococcus aureus ATCC 25922, Staphylococcus aureus ATCC 29213, Escherichia coli and Acinetobacter baumannii with a minimum inhibitory concentration (MIC) value of 0.25 μg mL−1, and was potent more than norfloxacin (MIC: 2–4 μg mL−1). SAR analysis showed that the introduction of the indole ring could greatly enhance the antibacterial activity of the compound, and the presence of the NH group of the indole ring was crucial for the antibacterial activity of the compound. Further studies showed that compound 54 caused the leakage of proteins, nucleic acids, and other substances by damaging the cell membrane of bacteria, thereby affecting their metabolism. Furthermore, compound 54 promotes excessive accumulation of intracellular ROS and induces lipid peroxidation to inhibit bacterial growth.96
The absence of substituents on the indole benzene ring or introduction of electron-withdrawing substituents can enhance the activity of the compounds. Derivative 55 (Fig. 7, Table 7, MIC: 12.5 μg mL−1) showed good anti-tuberculosis activity to the Mycobacterium tuberculosis H37Rv strain (M. tuberculosis H37Rv strain);97 derivatives 56 (Fig. 8, Table 8, IC50: 95.5 μg mL−1) and 57 (Fig. 8, Table 8, IC50: 95 μg mL−1) exhibited good anti-Leishmania activity. Furthermore, compound 56 and 57 also showed good anti-oxidant activity, and the IC50 values for anti-oxidant activity of compounds 56 and 57 were 12.4 μg mL−1 and 13.49 μg mL−1, respectively. Their anti-Leishmania activity was approximately 5 fold more potent than that of sodium stibogluconate (IC50: 490 μg mL−1), and the anti-oxidant activity was comparable to that of sodium ascorbate (IC50: 12.8 μg mL−1).98
Fig. 8. Chemical structures of coumarin-indole derivatives 56–61.

Biological activity of coumarin-indole derivatives 56–60.
| Compd. | Activity | Ref. |
|---|---|---|
| 56 | Anti-Leishmania activity: IC50 = 95.5 μg mL−1 | 85 |
| DPPH: IC50 = 12.4 μM | ||
| 57 | Anti-Leishmania activity: IC50 = 95 μg mL−1 | 85 |
| DPPH: IC50 = 13.49 μM | ||
| Sodium stibogluconate | Anti-Leishmania activity: IC50 = 490 μg mL−1 | 85 |
| Sodium ascorbate | DPPH: IC50 = 12.8 μM | 85 |
| 58 | α-Glucosidase: IC50 = 85 μM | 86 |
| Acarbose | α-Glucosidase: IC50 = 750 μM | 86 |
| 59 | HMG-CoA reductase: inhibition rate = 56% | 87 |
| 60 | Acetylcholinesterase: IC50 = 0.16 μM | 88 |
| 61 | HepG2 cells: IC50 = 4.63 μM | 89 |
| SMMC-7721 cells: IC50 = 3.11 μM |
Derivative 58 (Fig. 8, Table 8, IC50: 85 μM) displayed good inhibitory activity against α-glucosidase and was more potent than acarbose (IC50: 750 μM). Enzyme kinetic experiments have shown that compound 58 inhibits α-glucosidase in a competitive manner.99
At a concentration of 10 μM, the inhibitory activity of derivative 59 (Fig. 8, Table 8, inhibition rate: 56%) against HMG-CoA reductase was comparable to that of atorvastatin (inhibition rate: 60%). In vivo experiments showed that compound 59 reduced the body weight of rats, reversed the levels of TC and TG in VLDL and LDL in hyperlipidaemic rats, and increased LCAT activity, LPL lipolytic activity, and receptor-mediated LDL catabolism to balance lipid metabolism.100 In addition, the inhibitory activity SAR of derivative 60 (Fig. 8, Table 8, IC50: 0.16 μM) against acetylcholinesterase illustrated that the benzyloxy group at the C-7 position of coumarin is critical for activity.101 Derivative 61 (Fig. 8, Table 8, IC50: 3.11–4.63 μM) showed significant anti-proliferative activity against HepG-2 and SMMC-7721 cells. Further experiments showed that compound 8f induced apoptosis in a concentration-dependent manner and down-regulated the expression of PKM2, PFKM, HK2 and tumor angiogenesis-related vascular endothelial growth factor.102
Cinnamic acid is an organic acid isolated from Cinnamomum verum bark that possesses anti-tumor,103 anti-oxidant,104 tyrosinase inhibitory105 anti-bacterial106 and other bioactivities. Pharmacochemistry researchers have modified the structure of cinnamic acid, hoping to obtain new drugs with research value such as anti-tumor, anti-oxidation and anti-bacterial activities. Derivative 62 (Fig. 9, Table 9, IC50: 8.1 nM) exhibited comparable inhibitory activity against histone deacetylase enzyme and was potent more than vorinostat which served as the positive control (IC50: 160.8 nM). The in vitro experiments showed that compound 62 (IC50: 0.04–1.09 μM) exhibited good anti-proliferative activity on MOLT-4, HEL, K562, HeLa, and PC-3 cancer cells, being potent more than vorinostat (IC50: 0.21–16.28 μM).107
Fig. 9. Chemical structures of cinnamic-indole derivatives 62–68 and arctigenin-indole derivatives 69–70.

Biological activity of cinnamic-indole derivatives 62–68 and arctigenin-indole derivatives 69–70.
| Compd. | Activity | Ref. |
|---|---|---|
| 62 | HeLa extract: IC50 = 8.1 nM | 94 |
| MOLT-4 cells: IC50 = 0.04 μM | ||
| HEL cells: IC50 = 0.15 μM | ||
| K562 cells: IC50 = 0.32 μM | ||
| HELA cells: IC50 = 0.31 μM | ||
| PC-3 cells: IC50 = 1.09 μM | ||
| Vorinostat | HeLa extract: IC50 = 160 nM | 94 |
| MOLT-4 cells: IC50 = 0.33 μM | ||
| HEL cells: IC50 = 0.21 μM | ||
| K562 cells: IC50 = 1.49 μM | ||
| HELA cells: IC50 = 1.72 μM | ||
| PC-3 cells: IC50 = 16.28 μM | ||
| 63 | ORAC value = 8.71 | 95 |
| 64 | ORAC value = 7.32 | 96 |
| Melatonin | ORAC value = 2.45 | 96 |
| 65 | DPPH: EC50 = 8.1 μM | 97 |
| Ascorbic acid | DPPH: EC50 = 23 μM | 97 |
| 66 | M. bovis strain: inhibition rate = 100% | 98 |
| 67 | M. tuberculosis strain: inhibition rate = 90.4% | 98 |
| Rifampin: inhibition rate = 99.5% | ||
| 68 | DPPH: IC50 = 50.98 μM | 99 |
| 70 | T. gondii-infected HeLa cells: IC50 = 187.2 μM | 103 |
| Arctigenin | T. gondii-infected HeLa cells: IC50 = 586.4 μM | 103 |
| Spiramycin | T. gondii-infected HeLa cells: IC50 = 262.2 μM | 103 |
Compared with the positive control, ferulic acid (ORAC value: 3.74), derivatives 63 (Fig. 9, Table 9, ORAC value: 8.71)108 and 64 (Fig. 9, Table 9, ORAC value: 7.32)109 showed higher stronger anti-oxidant activity and were 2.99 fold more potent than melatonin (ORAC value: 2.45), respectively. Derivative 65 (Fig. 9, Table 9) showed considerable DPPH radical scavenging activity with an EC50 value of 8.1 μM, which is more potent than ascorbic acid (EC50: 23 μM).110 Derivative 66 (Fig. 9, Table 9) exhibited good inhibitory against the M. bovis strain with 100 inhibition in 10 μg mL−1. At a concentration of 30 μg mL−1, derivative 67 (Fig. 9, Table 9) showed 90.4% inhibition against the M. tuberculosis strain, and rifampin was 99.5%.111 Derivative 68 (Fig. 9, Table 9, IC50: 50.98 μM) showed excellent DPPH radical scavenging activity, and was found to have a higher ability to neutralize free radical cation ABTS˙+ than Trolox with an IC50 value of 19.49 μM, while the IC50 value of Trolox was 29.62 μM.112
Arctigenin is a natural lignan product derived from the dried and ripe fruit of Arctium lappa and possesses anti-tumor113 and anti-T. gondii activities.114 Pharmacochemistry researchers have synthesized a series of arctigenin indole derivatives, expecting to obtain new drugs with research value such as anti-tumor and anti-Toxoplasma activities. In the H22 xenograft mouse model, derivative 69 (Fig. 9) showed significantly stronger inhibitory activity against tumour growth, and the tumour growth inhibitory rate was 45.04% at a dose of 40 mg kg−1, which was more potent than arctigenin, with a tumour growth inhibitory rate of 27.86%. Serological data exhibited that compared with arctigenin (IL-2: 27.49 pg mL−1; IL-6: 71.17 pg mL−1; TNF-α: 59.86 pg mL−1; IFN-γ: 40.07 pg mL−1), the levels of IL-2, IL-6, TNF-α and IFN-γ in the serum of tumor-bearing mice treated with compound 69 (IL-2: 39.87 pg mL−1; IL-6: 82.62 pg mL−1; TNF-α: 70.65 pg mL−1; IFN-γ: 45.17 pg mL−1) were significantly increased at a dose of 40 mg kg−1. In addition, at a dose of 40 mg kg−1, the values of ALT, AST, BUN, and Cre in mice were comparable to those in the blank group, indicating that the high dose of compound 69 had little toxicity to the main functions of the liver and kidney in mice. Further study showed that compound 69 induced apoptosis, which may be related to the upregulation of BAX and Caspase-3 expression and the downregulation of Bcl-2 and VEGF expression.115
Derivative 70 (Fig. 9, Table 9, IC50: 187.2 μM) exhibited promising anti-T. gondii activity and was potent more than arctigenin (IC50: 586.4 μM) as well as spiramycin (IC50: 262.2 μM). In addition, the selection index (SI) of compound 70 (SI: 3.2) was higher than that of arctigenin (SI: 0.98) and spiramycin (SI: 0.72), indicating that the introduction of the indole group enhanced the anti-T. gondii activity of arctigenin.116
Summary: indole-modified phenylpropanoid derivatives have been widely reported in podophyllotoxin, coumarin and cinnamic acid. The activity of podophyllotoxin-indole derivative 37 against HepG2, HeLa, A549 and MCF-7 tumor cells was more potent than podophyllotoxin as well as colchicine. When podophyllotoxin was attached to the C-4 position of the indole ring, toxicity was moderate. When it was attached to C-5 or C-6, the anti-proliferative activity of the compound was enhanced, whereas when it was attached to C-7 or C-2, the toxicity sharply decreased. Furthermore, the longer the alkyl chain attached to the indole, the less toxic the compound.
A variety of biological activities have been reported on indole-modified coumarin derivatives. The introduction of an aldehyde or carboxyl group in the indole nucleus can enhance the anti-proliferative activity of the compound, such as the IC50 of compound 50 was 7.4 μM, and the introduction of a benzyloxy group at the C7 position of the coumarin could increase the inhibitory activity of the compound against acetylcholinesterase, such as compound 60. The nature of the linker between natural products and cinnamic acid also affects the activity of the compound. When the linker was benzamide, the activity was the strongest, and the substituent on the benzamide was an electron-donating group, which enhanced the activity of the compound more than the electron-withdrawing group. For example, cinnamic acid-indole derivative 62 showed nanomolar activity against histone deacetylase enzyme with an IC50 value of 8.1 nM.
5. Flavonoid-indole derivatives
Flavonoids are a series of compounds consisting of two phenolic hydroxyl groups interconnected by a central triple carbon atom, that is, compounds composed of C6–C3–C6 units, which have anti-tumor,117 anti-inflammatory118 and anti-bacterial activities.119
Chalcone is a flavonoid widely found in Asteraceae, Fabaceae and other plants, and possesses anti-tumor120 and anti-bacterial activities.121 Pharmacochemistry researchers worked on synthesizing chalcone linked indole derivatives in the hope to obtain promising anti-tumor and anti-bacterial drugs. Derivative 71 (Fig. 10, Table 10, IC50: 0.84 μM) with an N1-methyl-substituted indole ring displayed considerable anti-proliferative activity against HepG2 cancer cells. In addition, the introduction of ester, alkoxy, and sulfonate groups also enhanced the anti-cancer activity. In particular, derivative 72 (Fig. 10, Table 10, IC50: 0.22 μM) showed high anti-proliferative activity against HepG2 cancer cells, and low anti-proliferative activity against human normal cell line LO2 (IC50: >10 μM). Mechanistic studies illustrated that compound 72 induced cancer cell arrest in the G2/M phase and apoptosis, and inhibited tubulin polymerisation. In vivo experiments showed that compound 72 showed significant anti-tumor activity and had little effect on the growth of animals.122
Fig. 10. Chemical structures of chalcone-indole derivatives 71–85.

Biological activity of chalcone-indole derivatives 71–85.
| Compd. | Activity | Ref. |
|---|---|---|
| 71 | HepG2 cells: IC50 = 0.82 μM | 109 |
| 72 | HepG2 cells: IC50 = 0.22 μM | 109 |
| 73 | HeLa cells: IC50 = 2.4 μM | 110 |
| HT29 cells: IC50 = 1 μM | ||
| MCF-7 cells: IC50 = 0.59 μM | ||
| 74 | HeLa cells: IC50 = 0.33 μM | 110 |
| HT29 cells: IC50 = 0.39 μM | ||
| MCF-7 cells: IC50 = 0.46 μM | ||
| 75 | HeLa cells: IC50 = 0.37 μM | 110 |
| HT29 cells: IC50 = 0.16 μM | ||
| MCF-7 cells: IC50 = 0.17 μM | ||
| 76 | HeLa cells: IC50 = 0.84 μM | 110 |
| HT29 cells: IC50 = 0.13 μM | ||
| MCF-7 cells: IC50 = 0.24 μM | ||
| 77 | HeLa cells: IC50 = 2 μM | 110 |
| HT29 cells: IC50 = 7.3 μM | ||
| MCF-7 cells: IC50 = 1.1 μM | ||
| 78 | HeLa cells: IC50 = 2.1 μM | 110 |
| HT29 cells: IC50 = 0.44 μM | ||
| MCF-7 cells: IC50 = 1.8 μM | ||
| 79 | HepG2 cells: IC50 = 0.023 μM | 111 |
| A549 cells: IC50 = 0.029 μM | ||
| HeLa cells: IC50 = 0.059 μM | ||
| 80 | A549 cells: IC50 = 4.3 μg mL−1 | 112 |
| VP-16 | A549 cells: IC50 = 9.9 μg mL−1 | 112 |
| 81 | T. mentagrophytes: MIC = 8 μg mL−1 | 113 |
| M. fulvum: MIC = 4 μg mL−1 | ||
| A. benhamiae: MIC = 8 μg mL−1 | ||
| 82 | T. mentagrophytes: MIC = 8 μg mL−1 | 113 |
| M. fulvum: MIC = 1 μg mL−1 | ||
| A. benhamiae: MIC = 4 μg mL−1 | ||
| 83 | T. verucco: MIC = 2 μg mL−1 | 113 |
| T. mentagrophytes: MIC = 4 μg mL−1 | ||
| M. fulvum: MIC = 0.25 μg mL−1 | ||
| 84 | T. verucco: MIC = 32 μg mL−1 | 113 |
| T. mentagrophytes: MIC = 16 μg mL−1 | ||
| M. fulvum: MIC = 1 μg mL−1 | ||
| 85 | HCT-116: IC50: 3.2 nM | 114 |
Derivatives 73 (Fig. 10, Table 10, IC50: 0.59–2.4 μM), 74 (Fig. 10, Table 10, IC50: 0.33–0.46 μM), 75 (Fig. 10, Table 10, IC50: 0.16–0.37 μM) and 76 (Fig. 10, Table 10, IC50: 0.13–0.84 μM) exhibited promising anti-proliferative activity against HeLa, HT29 and MCF-7 cancer cell lines. The SAR showed that the position of the methoxy group introduced on the phenyl ring of the indole moiety had an important effect on anti-cancer activity. The anti-tumor activity of 5-methoxy-substituted compounds 73 and 74 and 6-methoxy-substituted compounds 75 and 76 was significantly stronger than compounds 77 (Fig. 10, Table 10, IC50: 1.1–7.3 μM) and 78 (Fig. 10, Table 10, IC50: 0.44–2.1 μM) without substituents on the benzene ring and the biological activity of 6-methoxy-substituted compounds was better than 5-methoxy-substituted compounds. Compound 74 exhibited stronger inhibitory activity against HeLa, HT29, and MCF-7 cancer cells than compound 73, indicating that the introduction of ethoxycarbonyl at the C-2 position of indole had a more pronounced effect on promoting the anticancer activity of the compound. As mentioned above, the steric effect of the N1 position of the indole ring had a significant influence on biological activity. The introduction of alkyl, benzyl, and other substituents at the N1 position of the indole ring would reduce or even eliminate anti-cancer activity. Mechanistic studies showed that compounds 75 and 76 bound to the colchicine-binding domain to inhibit tubulin aggregation. Further experiments showed that compounds 74, 75, and 76 arrested the cell cycle in the G2/M phase and promoted apoptosis by inhibiting the JNK/SAPK pathway.123
Derivative 79 (Fig. 10, Table 10, IC50: 0.023–0.059 μM) exhibited promising activity against HepG2, A549, and HeLa cancer cells.124 Derivative 80 (Fig. 10, Table 10, IC50: 4.3 μg mL−1) showed promising activity against A549 cancer cells as well as being more potent than VP-16 (IC50: 9.9 μg mL−1). Further studies have shown that compound 80 may induce cell death by inhibiting the aggregation of tubulin, reducing mitochondrial thiol content, and inducing mitochondrial apoptosis.125
The SAR study showed that 4-methoxy-substituted chalcone-indole derivatives 81 (Fig. 10, Table 10, MIC: 4–8 μg mL−1) and 82 (Fig. 10, Table 10, MIC: 1–8 μg mL−1) had better inhibitory effects on Trichophyton mentagrophytes, Microsporum fulvum, and Arthroderma benhamiae. However, the antibacterial activity of the trimethoxy-substituted compounds did not improve, and indole N-methylation and N-ethylation did not improve the biological activity of the compounds. Interestingly, the antifungal activity of compounds substituted with bromine at the 3-position was generally better than that of trimethoxy-substituted compounds. The type of alkoxy group substituted on the benzene ring also affected the antibacterial activity of Trichophyton verrucosum, T. mentagrophytes, and M. fulvum. The antibacterial activity of compound 83 (Fig. 10, Table 10, MIC: 0.25–4 μg mL−1) substituted by 4-ethoxy was 4–16 fold more potent than that of compound 84 (Fig. 10, Table 10, MIC: 1–32 μg mL−1) obtained by 4-methoxy base-substitution.126 Derivative 85 (Fig. 10, Table 10, IC50: 3.2 nM) showed significant anti-proliferative activity against HCT-116 cells. In vitro experiments showed that compound 85 induced G2/M phase arrest by regulating the expression of cyclin B1, produced ROS, and targeted tubulin in HCT-116 cells. In vivo experiments showed that compound 85 could significantly inhibit tumor growth, which was higher than that of paclitaxel.127
Chrysin is a natural flavonoid present in the seeds and stem bark of Oroxylum indicum and has pharmacological activities such as anti-inflammatory and analgesic activities.128 Researchers synthesized chrysin linked indole derivatives in the hope to obtain promising anti-inflammatory and analgesic drugs. Derivatives 86 (Fig. 11, Table 11, IC50: 0.001–7 μM), 87 (Fig. 11, Table 11, IC50: 0.007–60 μM) and 88 (Fig. 11, Table 11, IC50: 0.07 μM) showed promising inhibitory activity against COX-2, being more potent than indomethacin (IC50: 0.96 μM), and was comparable to bischlorothiophene (IC50: 0.02 μM) as well as celecoxib (IC50: 0.04 μM). Compared with COX-1, the inhibitory activities of compounds 86 and 87 against COX-2 were more obvious and exhibited certain selectivity. In particular, (rac) 86 (SI: 250) and (S) 86 (SI: 340) showed excellent COX-2 selectivity, with selectivity coefficients comparable to those of celecoxib (SI: 375). The SAR showed that the stereochemistry on the asymmetric carbon exhibited greater effect on the inhibition of COX-2 activity, and the compound with S configuration had more obvious anti-inflammatory activity, (R) 86 (IC50: 7 μM) showed far less inhibitory effect against COX-2 than (rac) 86 (IC50: 0.002 μM) and (S) 86 (IC50: 0.001 μM), and (R) 87 (IC50: 60 μM) exhibited lower anti-inflammatory activity than (rac) 87 (IC50: 0.01 μM) and (S) 87 (IC50: 0.007 μM) in the 87 series. The type of linker between indole and chrysin also affects the activity; compound (S) 87 with a linker of 2-propanol inhibited COX-2 10 times more than compound 88 (IC50: 0.07 μM) with an alkyl chain as a linker. Further studies showed that compound (S) 86 might exert its anti-inflammatory effect by inhibiting the cyclooxygenase and lipoxygenase pathways.129
Fig. 11. Chemical structures of chrysin-indole derivatives 86–89, xanthone-indole derivative 90, aurone-indole derivative 91 and morphine-indole derivative 92.

Biological activity of chrysin-indole derivatives 86–89, xanthone-indole derivative 90, aurone-indole derivative 91 and morphine-indole derivative 92.
| Compd. | Activity | Ref. |
|---|---|---|
| (rac) 86 | COX-2: IC50 = 0.002 μM | 116 |
| (S) 86 | COX-2: IC50 = 0.001 μM | 116 |
| (R) 86 | COX-2: IC50 = 7 μM | 116 |
| (rac) 87 | COX-2: IC50 = 0.01 μM | 116 |
| (S) 87 | COX-2: IC50 = 0.007 μM | 116 |
| (R) 87 | COX-2: IC50 = 60 μM | 116 |
| 88 | COX-2: IC50 = 0.07 μM | 116 |
| 89 | COX-2: IC50 = 0.7 μM | 117 |
| 90 | S. aureus: MIC = 16 μg mL−1 | 120 |
| B. subtilis: MIC = 16 μg mL−1 | ||
| E. coli: MIC = 15 μg mL−1 | ||
| K. pneumonia: MIC = 22 μg mL−1 | ||
| A. niger: MIC = 21 μg mL−1 | ||
| C. albicans: MIC = 20 μg mL−1 | ||
| F. oxysporum: MIC = 20 μg mL−1 | ||
| Anti-inflammatory activity: IC50 = 26 μg mL−1 | ||
| Gentamicin | S. aureus: MIC = 26 μg mL−1 | 120 |
| B. subtilis: MIC = 28 μg mL−1 | ||
| E. coli: MIC = 26 μg mL−1 | ||
| K. pneumonia: MIC = 26 μg mL−1 | ||
| Bavistin | A. niger: MIC = 27 μg mL−1 | 120 |
| C. albicans: MIC = 28 μg mL−1 | ||
| F. oxysporum: MIC = 27 μg mL−1 | ||
| 91 | NS5B protein polymerase: IC50 = 2.4 μM | 122 |
| Aurone | NS5B protein polymerase: IC50 = 5.4 μM | 122 |
| 92 | Mu opioid receptor: EC50 = 0.21 nM | 124 |
Derivative 89 (Fig. 11, Table 11, IC50: 0.7 μM) showed promising inhibitory activity on COX-2, which was 36 fold more potent than chrysin (IC50: 25.5 μM), and injection of 10 mg kg−1 of compound 89 into capsaicin-treated mice produced the same analgesic effect as 25 mg kg−1 of diclofenac. Hence, compound 89 has anti-inflammatory and analgesic potential, and merits further study.130
Xanthone is a benzochromone natural product, which mainly exists in microorganisms and has pharmacological activities such as antibacterial131 and anti-inflammatory.132 Chen et al. synthesized 23 xanthone derivatives and evaluated their antibacterial and anti-inflammatory activities. Derivative 90 (Fig. 11, Table 11, MIC: 15–22 μg mL−1) showed good antibacterial activity against Staphylococcus aureus (S. aureus), Bacillus subtilis (B. subtilis), E. coli and Klebsiella pneumonia (K. pneumonia), being more potent than gentamicin (MIC: 26–28 μg mL−1), and exhibited good antifungal activity against Aspergillus niger (A. niger), Candida albicans (C. albicans) and Fusarium oxysporum (F. oxysporum) (MIC: 20–21 μg mL−1), being more potent than bavistin (MIC: 27–28 μg mL−1). In addition, compound 90 (IC50: 26 μg mL−1) exhibited strong anti-inflammatory activity. Therefore, compound 90 has potential antibacterial and anti-inflammatory activities and deserves further investigation.133
Aurone is a natural product of flavonoids that contain a benzofuranone structure. It is a secondary product of plant metabolism, existing in Scrophulariaceae, Asteraceae and other plants, and has antiviral and other pharmacological activities.134 Meguellati et al. modified the structure of the B-ring moiety of aurone. They designed and synthesized 37 new aurone derivatives in order to obtain drugs with better inhibitory effect on NS5B protein polymerase. Derivative 91 (Fig. 11, Table 11, IC50: 2.4 μM) exhibited considerable inhibitory activity against NS5B protein polymerase, which was more potent than aurone (IC50: 5.4 μM). Analysis of the SAR results showed that the indole group containing an unsubstituted benzene ring was very important for the enhancement of the anti-viral activity of aurone-indole derivatives. In addition, the introduction of benzyl and alkyl groups at the N1 position of the indole can enhance activity.135
Morphine, derived from the capsules of Papaveraceae, is a powerful opioid receptor regulator.136 Obeng et al. synthesized 18 morphine indole derivatives, expecting to obtain new drugs with stronger binding ability to opioid receptors. Derivative 92 (Fig. 11) showed promising affinity for the mu opioid receptor, with an EC50 value of 0.21 nM. A warm water immersion assay showed that compound 92 was the opioid agonist with the highest antinociception.137
Summary: chalcone and chrysin-indole derivatives have good biological activities. Among them, chalcone-indole derivative 79 has strong anti-proliferative activity against HepG2, A549 and HeLa with IC50 values of 0.023 μM, 0.029 μM and 0.059 μM, respectively. The attachment site of chalcone to the indole ring was closely related to its anticancer activity. When chalcone was connected to the indole ring C-3 or C-5, the activity was the best; when it was connected to C-4 or C-6, the activity was moderate; and when it was connected to C-7, the activity decreased sharply. The steric effect of the N1 position of the indole ring is crucial for the anti-cancer activity; the larger the volume of the substituent on N1, the lower the anti-tumor activity. Chrysin-indole derivative (S) 87 has good anti-inflammatory activity with an IC50 of 0.007 μM. When the linker between indole and chrysin is isopropanol, the activity is stronger than that of alkyl.
6. Alkaloid-indole derivatives
Alkaloids are cyclic compounds containing nitrogen atoms in the negative oxidation state and are present in biological organisms. They are mainly classified into pyridine alkaloids, scopolamine alkaloids, isoquinoline alkaloids, indole alkaloids, and organic amine alkaloids, which have pharmacological activities such as anti-tumor activity.138
Sophoridine is a double-condensed piperidine alkaloid that exists in Sophora alopecuroides and has anti-tumor activity.139 Researchers have designed and synthesized a series of sophoridine derivatives, hoping to obtain new drugs with excellent anti-tumor activity. Most sophoridine-indole derivatives showed considerable cytotoxic activity against HepG2 cancer cells, and the anti-proliferative activity of sophoridine derivatives modified with an indole group was much higher than that of sophoridine, indicating that the introduction of an indole group enhanced the anti-proliferative activity of sophoridine derivatives. Among them, derivative 93 (Fig. 12, Table 12, IC50: 3.1 μM) showed considerable anti-proliferative activity against HepG2 cancer cells, which was potent more than the control drug camptothecin (IC50: 13.2 μM) and lead compound sophoridine (IC50: 4670 μM). The SAR analysis showed that the presence of an NH bond on indole N1 was not conducive to the anti-tumor activity, and substitution of benzyl and alkyl groups greatly enhanced the anti-tumor activity. When the ester group was substituted by an alcoholic hydroxyl group with better water solubility, the anti-tumor activity was stronger. Further experiments showed that compound 93 induced cell G1 arrest and inhibited topoisomerase I in the human hepatocellular carcinoma xenograft mouse model; treatment with compound 93 at 40 mg kg−1 resulted in a significant inhibition of tumour growth with a T/C value of 55.99% without loss of body weight, while the sophoridine group induced 93.38%. Therefore, compound 93 deserves further study as a potential anti-tumor drug agent.140
Fig. 12. Chemical structures of alkaloid-indole derivatives 93–98.

Biological activity of alkaloid-indole derivatives 93–98.
| Compd. | Activity | Ref. |
|---|---|---|
| 93 | HepG2 cells: IC50 = 3.1 μM | 127 |
| Camptothecin | HepG2 cells: IC50 = 13.2 μM | 127 |
| Sophoridine | HepG2 cells: IC50 = 4670 μM | 127 |
| 94 | HepG2 cells: IC50 = 0.93 μM | 128 |
| SMMC-7721 cells: IC50 = 1.32 μM | ||
| HeLa cells: IC50 = 1.44 μM | ||
| MCF-7 cells: IC50 = 0.94 μM | ||
| Camptothecin | HepG2 cells: IC50 = 1.36 μM | 128 |
| 95 | SMMC-7721 cells: IC50 = 3.95 μM | 130 |
| A549 cells: IC50 = 4.96 μM | ||
| Matrine | SMMC-7721 cells: IC50 = 6591 μM | 130 |
| A549 cells: IC50 = 5725 μM | ||
| Cisplatin | SMMC-7721 cells: IC50 = 6.08 μM | 130 |
| A549 cells: IC50 = 8.56 μM | ||
| 96 | HeLa cells: IC50 = 0.9 μM | 131 |
| MCF-7 cells: IC50 = 1.2 μM | ||
| SGC-7901 cells: IC50 = 1.1 μM | ||
| HepG2 cells: IC50 = 1.2 μM | ||
| 97 | DPPH: IC50 = 17.09 μg mL−1 | 133 |
| Berberine | DPPH: IC50 = 41.87 μg mL−1 | 133 |
| 98 | S. aureus: MIC = 3.125 μg mL−1 | 135 |
| MRSA: MIC = 3.125 μg mL−1 | ||
| B. cereus: MIC = 3.125 μg mL−1 | ||
| R. solanacearum: MIC = 1.5625 μg mL−1 | ||
| Fosfomycin sodium | S. aureus: MIC = 100 μg mL−1 | 135 |
| MRSA: MIC = 50 μg mL−1 | ||
| B. cereus: MIC = 25 μg mL−1 | ||
| R. solanacearum: MIC = 50 μg mL−1 |
Derivative 94 (Fig. 12, Table 12, IC50: 0.93–1.44 μM) exhibited promising anti-tumor activity against HepG2, SMMC-7721, HeLa and MCF-7 cancer cell lines. The anti-proliferative activity of HpeG2 cells was stronger than that of camptothecin (IC50: 1.36 μM). The results of the SAR analysis indicated that the α,β-unsaturated ketone structure in sophoridine derivatives was important for cytotoxic activity. When the C-5 or C-6 position of the indole ring was replaced by –Me, –Cl, and –Br, the cytotoxic activity decreased, and when the C-5 position of the indole ring was replaced by –OCH3, the cytotoxic activity was enhanced. The results of the enzymatic assay showed that compound 94 inhibited topoisomerase I activity. In addition, activation of caspase-3 and reduction of the Bcl-2/Bax ratio suggest that this compound may be a potential anticancer agent.141
Matrine is a double-condensed piperidine alkaloid derived from the dried root of Sophora flavescens and has anti-tumor activity.142 Researchers designed and synthesized a series of matrine derivatives and evaluated the anti-tumor activity of these compounds. Derivative 95 (Fig. 12, Table 12, IC50: 3.95–4.96 μM) showed considerable anti-proliferative activity against SMMC-7721 and A549 cancer cells, being more potent than matrine (IC50: 5725–6591 μM) and cisplatin (IC50: 6.08–8.56 μM). Further studies showed that compound 95 induced apoptosis of CNE2 and SMMC-7721 cells in a dose-dependent manner. Therefore, compound 95 is worthy of further exploration as a potential anti-tumor drug.143 Derivative 96 (Fig. 12, Table 12, IC50: 0.9–1.2 μM) showed strong anti-proliferative activity against HeLa, MCF-7, SGC-7901, HepG2 cancer cell lines and was 3 orders of magnitude stronger than matrine. Further studies demonstrated that compound 96 strongly induced mitochondrial stress, manifested as impaired energy metabolism, mitochondrial membrane potential depolarization, mitochondrial calcium overload and increased ROS production. SAR analysis showed that the properties of the substituents on the indole ring had a great influence on the antiproliferative activity of the compounds, and the activity sequence was: 6-Cl > 6-Br > 6-Me > 6-F > 6-OCH3.144
Berberine is a quaternary ammonium alkaloid derived from the rhizome of Coptis chinensis that has anti-oxidant activity.145 Mistry et al. combined a variety of heterocyclic nitrogen nuclei in the form of indole structural units with berberine to obtain new drugs with better antioxidant activity. They synthesized 10 different types of berberine-indole derivatives. The anti-oxidant activity of derivative 97 (Fig. 12, Table 12, IC50: 17.09 μg mL−1) was comparable to that of ascorbic acid (IC50: 15.59 μg mL−1), and was stronger than berberine (IC50: 41.87 μg mL−1). Therefore, compound 97 could be further studied as a novel anti-oxidant.146
Carboline is an alkaloid with a tricyclic structure, formed by the fusion of a pyridine ring with the pyrrole ring of an indole. Dai et al. designed and synthesized 32 carboline-indole derivatives with different substituents, expecting to obtain new drugs with good antibacterial activity. It was first discovered in the seeds of Peganum harmala and exhibited good antibacterial activity.147 Derivative 98 (Fig. 12, Table 12, MIC: 3.125–1.56 μmol L−1) was more potent than fosfomycin sodium (MIC: 25–100 μmol L−1) against S. aureus, MRSA, Bacillus cereus (B. cereus) and Ralstonia solanacearum (R. solanacearum). Further studies implied that compound 98 damages the cell membrane and increases permeabilization.148
Summary: sophoridine, matrine, and berberine indole derivatives all showed stronger biological activity than their parent compounds, among which are derivatives 93, 95, 96. The anti-tumor activity of these derivatives was stronger than that of the parent by three orders of magnitude. Therefore, compounds 93, 95, 96 were worthy of further study.
7. Steroid-indole derivatives
Steroids are a class of naturally widespread chemical components, a class of compounds containing cyclopentano-perhydrophenanthrene, including phytosterols, bile acids, C21 steroids, steroid saponins, etc., which have pharmacological activities such as anti-tumor,149 anti-Alzheimer's disease,150 and α-glucosidase activity inhibition.151
Lithocholic acid is a secondary bile acid, also known as cholelithic acid, which is produced by the bacterial metabolism of chenodeoxycholic acid in the intestine and has anti-tumor activity.152 Most researchers have modified the carboxyl group of lithocholic acid, and they have synthesized many lithocholic acid-indole derivatives, hoping to obtain antitumor drugs with research prospects. Derivative 99 (Fig. 13, Table 13, IC50: 0.8 μM) showed significant inhibitory activity against the EphA2 receptor, which was about 98 fold more potent than lithocholic acid (IC50: 79 μM). SAR analysis revealed that free carboxyl groups are critical for the ability of the compound to inhibit EphA2-ephrin-A1 binding. The introduction of a single aryl group to the compound did not improve its inhibitory activity. In contrast, the introduction of heterocycles, such as naphthalene rings, indole rings, and benzothiophenes, improved the inhibitory activity. Since the NH group on the indole ring can form weak hydrogen bonds with the EphA2 receptor, the compounds with the indole ring showed a strong ability to inhibit the binding of EphA2-ephrin-A1 among the tested compounds. In a pharmacokinetic study, compound 99 not only had good solubility and plasma stability, but also had high metabolic stability, which may be related to the fact that the acyloxy group at the C-3 position of compound 99 is not easily oxidised.153
Fig. 13. Chemical structures of steroid-indole derivatives 99–107.

Biological activity of steroid-indole derivatives 99–107.
| Compd. | Activity | Ref. |
|---|---|---|
| 99 | EphA2 receptor: IC50 = 0.8 μM | 140 |
| Lithocholic acid | EphA2 receptor: IC50 = 79 μM | 140 |
| 100 | MCF-7 cells: IC50 = 8.8 μM | 141 |
| MCF-7/ADR cells: IC50 = 4.28 μM | ||
| Lithocholic acid | MCF-7 cells: IC50 = 150.03 μM | 141 |
| 101 | HepG2 cells: IC50 = 10.63 μM | 143 |
| SK-Hep1 cells: IC50 = 9.38 μM | ||
| MCF-7 cells: IC50 = 15.78 μM | ||
| MDA-MB-231 cells: IC50 = 10.66 μM | ||
| Ergosterol | HepG2 cells: IC50 = 16.8 μM | 143 |
| SK-Hep1 cells: IC50 = 17.25 μM | ||
| MCF-7 cells: IC50 = 23.31 μM | ||
| MDA-MB-231 cells: IC50 = 19.17 μM | ||
| 102 | HepG2 cells: IC50 = 7.97 μM | 144 |
| MCF-7 cells: IC50 = 9.05 μM | ||
| HeLa cells: IC50 = 5.69 μM | ||
| Ergosterol peroxide | HepG2 cells: IC50 = 23.35 μM | 144 |
| MCF-7 cells: IC50 = 18.00 μM | ||
| HeLa cells: IC50 = 19.54 μM | ||
| 104 | MDA-MB-453 cells: IC50 = 10.64 nM | 146 |
| MCF-7 cells: IC50 = 11.6 nM | ||
| MDA-MB-231 cells: IC50 = 27.00 nM | ||
| 105 | MDA-MB-453 cells: IC50 = 10.01 nM | 146 |
| MCF-7 cells: IC50 = 1.06 nM | ||
| MDA-MB-231 cells: IC50 = 4.81 nM | ||
| 106 | HeLa cells: IC50 < 5 μM | 148 |
| Cisplatin | HeLa cells: IC50 = 10 μM | 148 |
| 107 | α-Glucosidase: IC50 = 115.2 μM | 151 |
| Cyclomusalenone | α-Glucosidase: IC50 = 603.8 μM | 151 |
| Acarbose | α-Glucosidase: IC50 = 420.2 μM | 151 |
Derivative 100 (Fig. 13, Table 13, IC50: 8.8 μM) showed considerable anti-proliferative activity against MCF-7 cells, which was comparable to that of the positive control drug tamoxifen (IC50: 7.05 μM), and was more potent than the parent lithocholic acid (IC50: 150.3 μM). Compound 100 (IC50: 4.28 μM) was also able to reverse the multidrug resistance of MCF-7/ADR cells. In addition, the presence of the alkenyl group at the C-11 position can significantly improve anti-tumor activity.154
Ergosterol is a plant sterol compound widely distributed in fungi, with anti-tumor activity.155 Researchers have devoted themselves to the synthesis of indole derivatives linked to ergosterol and evaluated the anti-tumor activity of these derivatives. The SAR showed that derivative 101 (Fig. 13, Table 13, IC50: 9.38–15.78 μM) containing an indole structure exhibited promising anti-proliferative activity against HepG2, SK-Hep1, MCF-7 and MDA-MB-321 cancer cell lines, being more potent than parental ergosterol (IC50: 16.8–23.31 μM).156 Derivative 102 (Fig. 13, Table 13, IC50: 5.69–9.05 μM) displayed considerable anti-proliferative activity against HepG2, MCF-7 and HeLa cancer cell lines, being more potent than ergosterol (IC50: 18.00–23.35 μM). In addition, the presence of electron-withdrawing groups on the indole ring contributed more to the anti-tumor activity than the electron-withdrawing groups. Further experiments showed that compound 102 induced G1 phase arrest and inhibited colony growth in HepG2 cells. Therefore, compound 102 can be used as a lead compound for anti-tumor agents, which is worthy of further study.157
Arenobufagin is a cardiac glycoside steroid derived from the dry secretions of Bufo gargarizans and has anti-tumor activity.158 Researchers have designed and synthesized a series of arenobufagin-indole derivatives, hoping to obtain new drugs with good anti-tumor activity. Derivative 103 (Fig. 13, Table 13, Vmax: 0.682 μmol min−1 mg−1, S50: 10.36 μmol L−1, CL int: 65.89 mL min−1 mg−1) is a prodrug activated by the FAPα enzyme which is only expressed in tumour tissues. Further study revealed that compound 103 could be cleaved by hFAPα with the highest enzymatic hydrolysis efficiency among arenobufagin-indole derivatives. Furthermore, compound 103 (IC50: 10.64–27.00 nM) exhibited considerable activity comparable to that of the lead product arenobufagin (IC50: 8.88–22.45 nM) against MDA-MB-435, MCF-7, and MDA-MB-231 cancer cell lines and displayed significantly reduced cardiotoxicity. More importantly, the active form of compound 104 (Fig. 13, Table 13, IC50: 1.06–10.01 nM) showed strong activity against MDA-MB-435, MCF-7, and MDA-MB-231 cells. In the MDA-MB-231 xenografted mouse model, compound 103 exhibited a tumor regression effect at a dose of 12 μmol kg−1 with tumors weighing 0.43 ± 0.18 g, while the control group was 0.67 ± 0.14 g. Hence, compound 103 provides a new candidate drug for alleviating cardiotoxicity of clinical anti-cancer drugs.159
Dehydroepiandrosterone is a C21 steroid, an intermediate in testosterone biosynthesis, with anti-tumor activity.160 Cui et al. synthesized a series of dehydroepiandrosterone derivatives with different aromatic heterocyclic structures at the 17-position side chain of the steroid nucleus and evaluated their anti-tumor activity. Derivative 105 (Fig. 13, Table 13, IC50: <5 μM) showed good anti-proliferative activity against HeLa cells and was stronger than cisplatin (IC50: 10 μM).161
Diosgenin is a steroidal saponin, which is widely present in the tubers of Dioscoreaceae plants, such as Discorea nipponica and Dioscorea zingiberensis, possessing neuroprotective activity.162 Zhou et al. designed and synthesized 10 diosgenin derivatives linked to indole groups, expecting to obtain new drugs for multi-target anti-Alzheimer's disease. At a concentration of 10 μM, derivative 106 (Fig. 13, Table 13, cell viability: 52.9–54.4%) exhibited considerable neuroprotective activity against H2O2 and Aβ injury. The SAR results showed that the properties of the benzene ring substituents were closely related to the enhanced anti-oxidant activities of the compounds. The active sequence of substituents on the benzene ring was as follows: 5-OCH3 > 5-CH3 > 5-F > 5-Cl > 5-Br > 5-H. The introduction of a methoxy group has a stronger effect on activity enhancement than the introduction of a methyl group, which may be caused by the conjugation of the methoxy group and indole ring. Substitution of C-5 of the indole ring is beneficial for enhancing neuroprotective activity. In addition, carbamates are significantly more effective than esters as linkers between indole and diosgenin. In vivo studies showed that compound 106 improved spatial memory and learning impairment in mice injected with Aβ. Overall, compound 106 is a potential anti-Alzheimer's agent with neuroprotective activity that deserves further study.163
Cyclomusalenone is found in the peel of Musa balbisiana and has hypoglycaemic activity. Smirnova et al. designed and synthesized a series of cyclomusalenone derivatives, expecting to obtain hypoglycemic drugs with research value. Derivative 107 (Fig. 13, Table 13, IC50: 115.2 μM) was an effective inhibitor of α-glucosidase being 5.2-and-3.6 fold more active than cyclomusalenone and acarbose.164
Summary: compounds 99 and 100 showed strong anti-tumor activity with IC50 values of 0.8 μM and 8.8 μM, respectively, which were 98 and 17 times stronger than the parent. The carboxyl group on the linker between lithocholic acid and indole enhanced the activity of compound 100, while compounds 103 and 104 showed nanomolar anti-proliferative activity against MDA-MB-435, MCF-7, and MDA-MB-231, so compounds 99, 100, 103 and 104 were valuable compounds for research.
8. Polyphenol-indole derivatives
Polyphenols are a collective name for a group of chemicals in plants, named for having multiple phenolic groups, with pharmacological activities, such as anti-tumor,165 anti-Alzheimer's disease166 and anti-inflammatory.167
Curcumin is a polyphenolic compound extracted from the rhizome of Curcuma longa and has anti-tumor,168 anti-Alzheimer's disease169 and anti-inflammatory activities.170 Researchers have designed and synthesized many curcumin-indole derivatives, expecting to obtain anti-tumor, anti-Alzheimer's disease and anti-inflammatory drugs with good biological activity. Derivative 108 (Fig. 14, Table 14, IC50: 3.15–3.31 μM) showed strong anti-tumor activity against PC-3 and HeLa cancer cell lines, being more potent than curcumin (IC50: 12.22–25.41 μM). Further research revealed that compound 108 significantly inhibited tubulin polymerisation, reduced the mitochondrial inner membrane, induced cell G2/M cycle arrest and apoptosis. In addition, the presence of electron-donating groups on the benzene ring is important for anti-tumor activity.171
Fig. 14. Chemical structures of curcumin-indole derivatives 108–113.

Biological activity of curcumin-indole derivatives 108–113.
| Compd. | Activity | Ref. |
|---|---|---|
| 108 | PC-3 cells: IC50 = 3.15 μM | 158 |
| HeLa cells: IC50 = 3.31 μM | ||
| Curcumin | PC-3 cells: IC50 = 12.22 μM | 158 |
| HeLa cells: IC50 = 25.41 μM | ||
| 109 | Caco-2 cells: IC50 = 3.3 μM | 159 |
| CHO-K1 cells: IC50 = 7.9 μM | ||
| Curcumin | Caco-2 cells: IC50 = 28 μM | 159 |
| CHO-K1 cells: IC50 = 21.3 μM | ||
| 110 | Caco-2 cells: IC50 = 8 μM | 160 |
| Curcumin | Caco-2 cells: IC50 = 47.6 μM | 160 |
| 111 | Aβ: IC50 = 0.42 μM | 161 |
| Tau: IC50 = 0.44 μM | ||
| Curcumin | Aβ: IC50 = 5.4 μM | 161 |
| Tau: IC50 = 3 μM | ||
| 112 | Hep 3B: IC50 = 14 μM | 162 |
| 113 | IL-6: 1.06 μM | 163 |
Derivative 109 (Fig. 14, Table 14, IC50: 3.3–7.9 μM) showed promising broad-spectrum anti-tumor activity against Caco-2 and CHO-K1 cancer cell lines, being more potent than curcumin (IC50: 21.3–28 μM). Further experiments indicated that compound 109 might induce apoptosis through the accumulation of ROS in tumour cells.172
Replacing the phenyl moiety of curcumin with an indole group is important for enhancing the Caco-2 inhibitory activity of curcumin and the pharmacological activity of curcumin against Alzheimer's disease. Derivative 110 (Fig. 14, Table 14, IC50: 8 μM) showed promising anti-tumor activity against Caco-2 cancer cells, being 6 fold more potent than curcumin (IC50: 47.6 μM).173 Derivative 111 (Fig. 14, Table 14, IC50: 0.42–0.44 μM) exhibited considerable inhibitory activity against Aβ and tau protein aggregation, being 8–10 fold more potent than curcumin (IC50: 3.0–5.4 μM).174 Derivative 112 (Fig. 14, Table 14, IC50: 14 μM) showed strong anti-proliferative activity against Hep3B cancer cells, inhibiting cell migration in a time- and dose-dependent manner and decreasing MMP-9 activity.175 Derivative 113 (Fig. 14, Table 14, IC50: 1.05 μM) showed strong inhibitory activity against IL-6, which was superior to curcumin and indomethacin and had no obvious cytotoxic side effects. Further experiments showed that compound 113 significantly inhibited LPS-induced inflammatory response, reduced the number of neutrophils and lung wet-to-dry weight ratio. Therefore, compound 11 is a promising anti-inflammatory drug.176
Resveratrol, a polyphenolic natural product, was first isolated from the roots of Veratrum grandiflorum in 1940 and has anti-tumor pharmacological activity.177 Many resveratrol-indole derivatives have been synthesized by researchers, who expect to obtain anti-tumor drugs with good biological activity. The anti-proliferative activity of derivative 114 (Fig. 15, Table 15, IC50: 0.022–0.056 μM) against A549, HeLa, PC-3, Bel-7402, Lovo, A2780, and MCF-7 cancer cell lines illustrated that N-methylation significantly enhanced the anti-proliferative activity. A mechanistic study illustrated that compound 114 inhibited tubulin polymerisation, disrupted microtubule organisation, and induced G2/M cell cycle arrest and apoptosis in A549 cells. Hence, compound 114 might potentially be a new tubulin-targeting agent that merits further study.178
Fig. 15. Chemical structures of resveratrol-indole derivatives 114–121.

Biological activity of resveratrol-indole derivatives 114–121.
| Compd. | Activity | Ref. |
|---|---|---|
| 114 | A549 cells: IC50 = 0.022 μM | 165 |
| HeLa cells: IC50 = 0.056 μM | ||
| PC-3 cells: IC50 = 0.047 μM | ||
| Bel-7402 cells: IC50 = 0.039 μM | ||
| Lovo cells: IC50 = 0.049 μM | ||
| A2780 cells: IC50 = 0.037 μM | ||
| MCF-7 cells: IC50 = 0.051 μM | ||
| 115 | MV4-11 cells: IC50 = 6 nM | 166 |
| THP-1 cells: IC50 = 2 nM | ||
| A549 cells: IC50 = 2 nM | ||
| PC-3 cells: IC50 = 3.5 nM | ||
| 116 | HeLa cells: IC50 = 0.07 μM | 167 |
| CA-4 | HeLa cells: IC50 = 0.21 μM | 167 |
| 117 | HT-29 cells: IC50 = 0.03 μM | 168 |
| A549 cells: IC50 = 0.02 μM | ||
| CA-4 | HT-29 cells: IC50 = 0.13 μM | 168 |
| A549 cells: IC50 > 10 μM | ||
| 118 | MDA-MB-468 cells: IC50 = 37 nM | 169 |
| MDA-MB-436 cells: IC50 = 62 nM | ||
| MDA-MB-23 cells: IC50 = 39 nM | ||
| PC-3 cells: IC50 = 0.3 nM | ||
| RD cells: IC50 = 0.2 nM | ||
| HepG2 cells: IC50 = 0.1 nM | ||
| 119 | MDA-MB-468 cells: IC50 = 33 nM | 169 |
| MDA-MB-436 cells: IC50 = 75 nM | ||
| MDA-MB-23 cells: IC50 = 47 nM | ||
| PC-3 cells: IC50 = 19 nM | ||
| RD cells: IC50 = 16 nM | ||
| HepG2 cells: IC50 = 62 nM | ||
| Vinblastine | PC-3 cells: IC50 = 766 nM | 169 |
| RD cells: IC50 = 53 nM | ||
| HepG2 cells: IC50 = 81 nM | ||
| Paclitaxel | PC-3 cells: IC50 = 4900 nM | 169 |
| RD cells: IC50 > 10 000 nM | ||
| HepG2 cells: IC50 = 2600 nM | ||
| 120 | HepG2 cells: IC50 = 0.63 μM | 170 |
| HeLa cells: IC50 = 5.89 μM | ||
| MCF-7 cells: IC50 = 0.64 μM | ||
| A549 cells: IC50 = 0.08 μM | ||
| 121 | A549 cells: IC50 = 0.015 μM | 171 |
| HeLa cells: IC50 = 0.053 μM | ||
| MCF-7 cells: IC50 = 0.031 μM | ||
| HCT116 cells: IC50 = 0.033 μM | ||
| CA-4 | A549 cells: IC50 = 0.031 μM | 171 |
| HeLa cells: IC50 = 0.058 μM | ||
| MCF-7 cells: IC50 = 0.058 μM | ||
| HCT116 cells: IC50 = 0.398 μM |
The SAR revealed that the introduction of a heterocyclic ring at the C-6 position of indole was favourable for anti-proliferative activity, and derivative 115 (Fig. 15, Table 15, IC50: 2–6 nM) showed excellent anti-proliferative activity against PC-3, A-549, THP-1, and MV4-11 cancer cell lines. Further study revealed that compound 115 arrested the cell cycle at the G2/M phase and fully inhibited tubulin polymerisation at 50 nM. Overall, compound 109 may potentially be used as a lead agent for the development of novel tubulin inhibitors.179
Derivative 116 (Fig. 15, Table 15, IC50: 0.07 μM) was 3 fold more potent than combretastatin A-4 (CA-4) (IC50: 0.21 μM) against HeLa cancer cells, and further study revealed that compound 116 greatly disrupted microtubule maturation in a concentration-dependent manner, induced G2/M cell cycle arrest, increased the ROS level and induced apoptosis in HeLa cells, which was associated with dissipation of mitochondrial membrane potential. Furthermore, compound 116 inhibited the migration of cancer cells, and in a HeLa xenograft mouse model, treatment with compound 116 significantly decreased tumour volume and inhibited tumour growth by 65.76% at a dose of 30 mg kg−1 without any weight loss. Consequently, compound 116 can act as a novel tubulin inhibitor for cancer therapy.180
Derivative 117 (Fig. 15, Table 15, IC50: 0.02–0.03 μM) was more potent than CA-4 (IC50: 0.13 ≥ 10 μM) against HT-29 and A549 cancer lines, comparable to vinblastine (IC50: 0.02–0.03 μM). Further experiments showed that compound 117 induced intracellular ROS generation and arrested HeLa cells in the G2/M phase by inducing apoptosis, which was associated with the loss of mitochondrial potential. Moreover, the compound exhibited high metabolic stability, with 48.6% remaining in human liver microsomes after 30 min, and possessed promising aqueous solubility with a solubility of 64.5 μM.181
Derivative 118 (Fig. 15, Table 15, IC50: 37–62 nM) and derivative 119 (Fig. 15, Table 15, IC50: 33–75 nM) inhibited the growth of MDA-MB-468, MDA-MB-23, and MDA-MB-436 cancer cell lines in a dose- and time-dependent manner. In addition, compounds 118 (IC50: 0.1–0.3 nM) and 119 (IC50: 16–62 nM) were generally more potent than vinblastine (IC50: 53–766 nM) and paclitaxel (IC50: 4900 to >10 000 nM) against PC-3, RD, and HeLa cancer cell lines, especially compound 118 at the micromolar level. Further studies showed that compounds 118 and 119 inhibited the Hh signalling pathway and tubulin polymerisation.182
Derivative 120 (Fig. 15, Table 15, IC50: 0.08–5.89 μM) possessed excellent activity against HepG2, MCF-7, HeLa and A549 cancer cell lines, comparable to CA-4 (IC50: 0.13–5.52 μM). SAR studies illustrated that the acylhydrazone group is essential for anti-proliferative activity. Mechanistic studies demonstrated that compound 120 induced significant ROS generation, mitochondrial depolarisation, G2/M cell cycle arrest, and apoptosis in A549 cancer cells. Western blot studies illustrated that compound 120 upregulated the levels of Bax, cleaved caspase-3 and PARP, and downregulated the levels of Bcl-2 and Bcl-xl. In the A549 xenografted mouse model, compound 120 strongly suppressed tumour growth by 70% at a dose of 30 mg kg−1, and the weight showed no significant changes during the whole treatment period. Overall, compound 120 underwent further preclinical evaluation.183
Derivative 121 (Fig. 15, Table 15, IC50: 0.015–0.053 μM) was more potent than CA-4 (IC50: 0.031–0.058 μM) against the A549, HCT116, HeLa, and MCF-7 cancer cell lines. Further studies illustrated that compound 121 arrested the cell cycle in the G2/M phase, inhibited tubulin polymerisation, and bound to the colchicine-binding site of the tubulin polymer.184
Summary: curcumin and resveratrol-indole derivatives showed strong anti-tumor and anti-Alzheimer's disease activities. Among them, compound 108 had good antitumor activity against PC3, IC50: 3.15–3.31 μM; derivative 111 showed 8–10 times higher inhibitory activity against Aβ and tau protein aggregation than curcumin, and derivative 115 showed nanomolar-level antitumor activity against PC-3, A-549, THP-1, and MV4-11. Therefore, compounds 108, 111 and 115 are worthy of further study.
9. Quinone-indole derivatives
Quinones are relatively important active ingredients in natural products, which are natural products with an unsaturated cyclic diketone structure within the molecule or easily transformed into this quinone structure, including benzoquinone, naphthoquinone, and anthraquinone. Naphthoquinone is widely distributed in many kinds of higher plants, among which the high content includes Asteraceae, Lamiaceae, Ebenaceae, etc. Naphthoquinones are structurally divided into 1,4-naphthoquinone, 1,2-naphthoquinone, and 2,6-naphthoquinone.
1,4-Naphthoquinone is found in a variety of natural products and has various biological activities. It is considered a pharmacophore of bioactive compounds because of its various properties such as redox,185 nucleophilic acceptance186 and DNA intercalation.187 Many 1,4-naphthoquinone derivatives have been synthesised and exhibit pharmacological activities, such as anti-tumor188 and anti-viral activities.189 Córdova-Rivas et al. designed and synthesized six 1,4-naphthoquinone derivatives with different substituents and determined their antitumor activities. Derivative 122 (Fig. 16, Table 16, IC50: 0.0034–3.20 μM) exhibited promising activity against SiIHa, CaLo, and C33-A cancer cells. Further studies have shown that hydrophobic and aromatic amino acids have better inhibitory effects on cell proliferation.190
Fig. 16. Chemical structures of quinone-indole derivatives 122–126.

Biological activity of quinone-indole derivatives 122–124.
| Compd. | Activity | Ref. |
|---|---|---|
| 122 | SiIHa cells: IC50 = 3.2 μM | 177 |
| CaLo cells: IC50 = 2.69 μM | ||
| C33-A cells: IC50 = 0.0034 μM | ||
| 123 | A578 cells: IC50 = 5 nM | 178 |
| Shikonin | A578 cells: IC50 = 0.46 μM | 178 |
| 124 | MCF-7 cells: IC50 = 11.33 μM | 181 |
| HepG2 cells: IC50 = 10.14 μM | ||
| MG-63 cells: IC50 = 13.85 μM | ||
| Naphthoquinone | MCF-7 cells: IC50 = 16.92 μM | 181 |
| HepG2 cells: IC50 = 15.89 μM | ||
| MG-63 cells: IC50 > 25 μM |
Shikonin is a natural product of 1,4-naphthoquinones, derived from the roots of Lithospermum species and has anti-tumor191 and anti-viral activities.192 Researchers designed and synthesized a series of shikonin derivatives, and evaluated the antitumor activity and antiviral activity of these derivatives. Derivative 123 (Fig. 16, Table 16, IC50: 5 nM) was 92 fold more potent than shikonin (IC50: 0.46 μM) against A578 cancer cells and induced G2/M cell cycle arrest.193 Furthermore, shikonin-indole derivative 123 (IC50: 0.011–0.052 μM) exhibited considerable activity against EV71, inhibited the expression levels of VP1 protein and phospho-p65 protein, and reduced the expression of mRNA in RD cells.194
1,2-Naphthoquinone, also known as β-naphthoquinone, is a nucleophilic receptor with anti-tumor and other pharmacological activities.195 Shukla et al. designed and synthesized 18 1,2-naphthoquinone derivatives, expecting to obtain drugs with high anti-tumor activity. Derivative 124 (Fig. 16, Table 16, IC50: 10.14–13.85 μM) exhibited comparable activity against MCF-7, HepG2 and MG-63 cancer cells, and was more potent than 1,2-naphthoquinone (IC50: >15.89 μM). SAR analysis showed that the electron-absorbing substituents on the benzene ring contributed more to the anti-tumor activity than the electron-donating substituents.196
Anthraquinone derivatives are widely found in plants and have various pharmacological activities, including anti-dyslipidaemic effects.197 Shattat et al. designed and synthesized seven different types of anthraquinone-indole derivatives, expecting to obtain hypolipidemic drugs with good biological activity. Because the whole molecule forms a conjugated system and C9 and C10 are at the highest oxygen level, natural anthraquinones are most commonly known as 9,10-anthraquinones. At a dose of 15 mg kg−1, derivatives 125 (Fig. 16) and 126 (Fig. 16) significantly reduced plasma total cholesterol levels (p < 0.0001) and plasma triglyceride levels (p < 0.0001) and significantly increased HDL cholesterol levels after 12 and 24 h compared to hyperlipidaemic controls. Therefore, compounds 125 and 126 deserve further investigation as potential antihyperlipidaemic drugs.198
Summary: when the substituent on the benzene ring was an electron-withdrawing group, the activity was better. For example, compound 124 showed good activity against MCF-7, HepG2 and MG-63 cancer cells with IC50 values of 10.14–13.85 μM. Shikonin-indole derivative 123 had strong anti-tumor activity with an IC50 value of 5 nM, which was 92 fold that of shikonin. Therefore, compound 123 had further research value.
10. Conclusion
In this review, indole scaffolds have shown strong biological activities in anti-tumor, anti-inflammatory, anti-oxidation, anti-bacterial, hypolipidemic, anti-diabetic, anti-viral, anti-Alzheimer's disease, and anti-parasitic studies, among which anti-tumor is the most popular research field. Observing indole as a basic anti-tumor derivative, it can be found that most of the compounds with good biological activity have certain similarities with the connection sites, methods, properties and volume of substituents of indole. For example, podophyllotoxin and chalcone have the best anti-tumor activity when they are connected to the C5 position of indole. In addition, some indole derivatives with better pharmacological activity than lead compounds are also introduced in this paper. For example, compounds 103, 104, 118, 119, and 123 possessed high anti-proliferative activity against the tested cancer cells with IC50 values at nanomolar levels; compounds 83 and 90 showed broad-spectrum antifungal and antibacterial activities, respectively; and compounds 22, 37, 59, 69, 72, 89, 93, 103 and 106 exhibited good in vivo efficacy; thus, these indole-modified natural product derivatives deserve further investigation. In summary, the indole structure will continue to be an important scaffold for the development of new highly bioactive drugs in future medicinal chemistry research, especially in the field of anti-tumor research. This review provides a comprehensive data resource of indole-natural product derivatives for researchers engaged in drug design and research, and helps medicinal chemistry researchers to design and develop new, efficient and low-toxic indole derivatives more rationally.
Conflicts of interest
The authors confirm that this research article has no conflict of interest.
Supplementary Material
Acknowledgments
This work was supported by the Higher Education Discipline Innovation Project (D18012), Key Projects of Jilin Province Science and Technology Development Plan (No. 20200404130YY), National Natural Science Foundation of China (No. 81960626, 82060628), Education Department Project of Jilin (No. JJKH20220559KJ), and Jilin Scientific and Technological Development Program (No. YDZJ202301ZYTS440). We would like to thank Editage (https://www.editage.cn) for English language editing.
References
- Kumari A. Singh R. K. Bioorg. Chem. 2020;96:103578. doi: 10.1016/j.bioorg.2020.103578. [DOI] [PubMed] [Google Scholar]
- Dadashpour S. Emami S. Eur. J. Med. Chem. 2018;150:9–29. doi: 10.1016/j.ejmech.2018.02.065. [DOI] [PubMed] [Google Scholar]
- Kumar S. Singh R. K. Patial B. Goyal S. Bhardwaj T. R. J. Enzyme Inhib. Med. Chem. 2016;31:173–186. doi: 10.3109/14756366.2015.1016513. [DOI] [PubMed] [Google Scholar]
- Xiong X. Zhang D. Li J. Sun Y. Zhou S. Yang M. Shao H. Li A. Chem. – Asian J. 2015;10:869–872. doi: 10.1002/asia.201403312. [DOI] [PubMed] [Google Scholar]
- Chaudhuri A. Haldar S. Sun H. Koeppe, 2nd R. E. Chattopadhyay A. Biochim. Biophys. Acta. 2014;1838:419–428. doi: 10.1016/j.bbamem.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F. Bi J. He L. Chen J. Zhang Q. Hou X. Xu H. Fitoterapia. 2021;153:104950. doi: 10.1016/j.fitote.2021.104950. [DOI] [PubMed] [Google Scholar]
- Wang M. Zhou B. Cong W. Zhang M. Li Z. Li Y. Liang S. Chen K. Yang D. Wu Z. Front. Pharmacol. 2021;12:797605. doi: 10.3389/fphar.2021.797605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey A. Roy D. Mohture V. M. Ghorai M. Rahman M. H. Anand U. Dewanjee S. Radha R. Kumar M. Prasanth D. A. Jha N. K. Jha S. K. Shekhawat M. S. Pandey D. K. Appl. Microbiol. Biotechnol. 2022;106:4867–4883. doi: 10.1007/s00253-022-12040-8. [DOI] [PubMed] [Google Scholar]
- Zhou M. L. Shao J. R. Tang Y. X. Biotechnol. Appl. Biochem. 2009;52:313–323. doi: 10.1042/BA20080239. [DOI] [PubMed] [Google Scholar]
- Kumari A. Singh R. K. Bioorg. Chem. 2019;89:103021. doi: 10.1016/j.bioorg.2019.103021. [DOI] [PubMed] [Google Scholar]
- Xu D. Xu Z. Curr. Top. Med. Chem. 2020;20:1938–1949. doi: 10.2174/1568026620666200622150325. [DOI] [PubMed] [Google Scholar]
- Dhiman A. Sharma R. Singh R. K. Acta Pharm. Sin. B. 2022;12:3006–3027. doi: 10.1016/j.apsb.2022.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob Í. T. T. Gomes F. O. S. de Miranda M. D. S. de Almeida S. M. V. da Cruz-Filho I. J. Peixoto C. A. da Silva T. G. Moreira D. R. M. de Melo C. M. L. de Oliveira J. F. de Lima M. C. A. Pharmacol. Rep. 2021;73:907–925. doi: 10.1007/s43440-021-00221-7. [DOI] [PubMed] [Google Scholar]
- Kumari A. Singh R. K. Med. Chem. 2023;19:163–173. doi: 10.2174/1573406418666220812152950. [DOI] [PubMed] [Google Scholar]
- Kumari A. Singh R. K. Chem. Biodiversity. 2022;19:e202200290. doi: 10.1002/cbdv.202200290. [DOI] [PubMed] [Google Scholar]
- Chen W. H. Li K. L. Lin X. P. Liao S. R. Yang B. Zhou X. F. Wang J. J. Liu Y. H. Wang J. F. Nat. Prod. Res. 2021;35:5266–5270. doi: 10.1080/14786419.2020.1749614. [DOI] [PubMed] [Google Scholar]
- Kumari A. Singh R. K. Comb. Chem. High Throughput Screening. 2023;26:2077–2084. doi: 10.2174/1386207326666230426093315. [DOI] [PubMed] [Google Scholar]
- Chang K. H. Lin C. H. Chen H. C. Huang H. Y. Chen S. L. Lin T. H. Ramesh C. Huang C. C. Fung H. C. Wu Y. R. Huang H. J. Lee-Chen G. J. Hsieh-Li H. M. Yao C. F. CNS Neurosci. Ther. 2017;23:45–56. doi: 10.1111/cns.12592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura Y. Morita I. Hinata Y. Kojima E. Ozasa H. Ikemoto H. Asano M. Wada T. Hayasaki-Kajiwara Y. Iwasaki T. Matsumura K. Bioorg. Med. Chem. Lett. 2022;68:128769. doi: 10.1016/j.bmcl.2022.128769. [DOI] [PubMed] [Google Scholar]
- Najmi A. Javed S. A. Al Bratty M. Alhazmi H. A. Molecules. 2022;27:349. doi: 10.3390/molecules27020349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chopra B. Dhingra A. K. Phytother. Res. 2021;35:4660–4702. doi: 10.1002/ptr.7099. [DOI] [PubMed] [Google Scholar]
- Bommakanti S. S. Kundeti L. S. R. Saddanapu V. Nagaiah K. ACS Omega. 2020;5:14069–14077. doi: 10.1021/acsomega.0c01525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H. Huang X. Jin C. Jin C. M. Quan Z. S. Bioorg. Chem. 2020;94:103467. doi: 10.1016/j.bioorg.2019.103467. [DOI] [PubMed] [Google Scholar]
- Shen Q. K. Deng H. Wang S. B. Tian Y. S. Quan Z. S. Eur. J. Med. Chem. 2019;173:15–31. doi: 10.1016/j.ejmech.2019.04.005. [DOI] [PubMed] [Google Scholar]
- Zhang T. L. Yu H. W. Ye L. D. ACS Synth. Biol. 2023;12:639–656. doi: 10.1021/acssynbio.2c00569. [DOI] [PubMed] [Google Scholar]
- Huang P. W. Wang L. R. Geng S. S. Ye C. Sun X. M. Huang H. Appl. Microbiol. Biotechnol. 2021;105:4919–4930. doi: 10.1007/s00253-021-11368-x. [DOI] [PubMed] [Google Scholar]
- El-Baba C. Baassiri A. Kiriako G. Dia B. Fadlallah S. Moodad S. Darwiche N. Apoptosis. 2021;26:491–511. doi: 10.1007/s10495-021-01684-y. [DOI] [PubMed] [Google Scholar]
- Grassmann J. Vitam. Horm. 2005;72:505–535. doi: 10.1016/S0083-6729(05)72015-X. [DOI] [PubMed] [Google Scholar]
- Yamaguchi T. Arch. Microbiol. 2022;204:520. doi: 10.1007/s00203-022-03142-y. [DOI] [PubMed] [Google Scholar]
- Syrov V. N. Abzalova M. K. Khushbaktova Z. A. Sultanov M. B. Pharm. Chem. J. 1985;19:270–272. doi: 10.1007/BF00833357. [DOI] [Google Scholar]
- Shanmugam M. K. Shen H. Tang F. R. Arfuso F. Rajesh M. Wang L. Kumar A. P. Bian J. Goh B. C. Bishayee A. Sethi G. Pharmacol. Res. 2018;133:195–200. doi: 10.1016/j.phrs.2018.05.007. [DOI] [PubMed] [Google Scholar]
- Fang J. Huang T. Xia M. Deng L. Hao X. Wang Y. Mu S. Org. Biomol. Chem. 2018;16:3026–3037. doi: 10.1039/C8OB00677F. [DOI] [PubMed] [Google Scholar]
- An T. Yin H. Lu Y. Liu F. Drug Des., Dev. Ther. 2022;16:1255–1272. doi: 10.2147/DDDT.S355059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bommagani S. Ponder J. Penthala N. R. Janganati V. Jordan C. T. Borrelli M. J. Crooks P. A. Eur. J. Med. Chem. 2017;136:393–405. doi: 10.1016/j.ejmech.2017.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slezakova S. Ruda-Kucerova J. Anticancer Res. 2017;37:5995–6003. doi: 10.21873/anticanres.12046. [DOI] [PubMed] [Google Scholar]
- D'Angelo J. G. Bordón C. Posner G. H. Yolken R. Jones-Brando L. J. Antimicrob. Chemother. 2009;63:146–150. doi: 10.1093/jac/dkn451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y. Li N. Zhang J. Wang Y. Chen L. Sun J. Bioorg. Med. Chem. Lett. 2019;29:1138–1142. doi: 10.1016/j.bmcl.2019.02.021. [DOI] [PubMed] [Google Scholar]
- Ni X. Hu G. Cai X. Crit. Rev. Food Sci. Nutr. 2019;59:S71–S80. doi: 10.1080/10408398.2018.1509201. [DOI] [PubMed] [Google Scholar]
- Gurkan-Alp A. S. Mumcuoglu M. Andac C. A. Dayanc E. Cetin-Atalay R. Buyukbingol E. Eur. J. Med. Chem. 2012;58:346–354. doi: 10.1016/j.ejmech.2012.10.013. [DOI] [PubMed] [Google Scholar]
- Farooqi A. A. Attar R. Sabitaliyevich U. Y. Alaaeddine N. de Sousa D. P. Xu B. Cho W. C. Cancers. 2020;12:2159. doi: 10.3390/cancers12082159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta S. Sharma A. K. Singh R. K. Mini-Rev. Med. Chem. 2021;21:1556–1577. doi: 10.2174/1389557521666210315162354. [DOI] [PubMed] [Google Scholar]
- Song Y. Xin Z. Wan Y. Li J. Ye B. Xue X. Eur. J. Med. Chem. 2015;90:695–706. doi: 10.1016/j.ejmech.2014.12.017. [DOI] [PubMed] [Google Scholar]
- Kim W. J. Kim W. Bae J. M. Gim J. Kim S. J. Plants. 2021;10:1047. doi: 10.3390/plants10061047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H. Qiao C. Miao T. T. Li A. L. Wang W. Y. Gu W. J. Enzyme Inhib. Med. Chem. 2019;34:1544–1561. doi: 10.1080/14756366.2019.1655407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X. Xu J. Zhou J. Shen Q. Genes Dis. 2021;8:448–462. doi: 10.1016/j.gendis.2020.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thongnest S. Boonsombat J. Prawat H. Mahidol C. Ruchirawat S. Phytochemistry. 2017;134:98–105. doi: 10.1016/j.phytochem.2016.11.007. [DOI] [PubMed] [Google Scholar]
- Zhang Y. Yang S. Zhang M. Wang Z. He X. Hou Y. Bai G. Nutrients. 2019;11:604. doi: 10.3390/nu11030604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De-la-Cruz-Martinez L. Duran-Becerra C. Gonzalez-Andrade M. Paez-Franco J. C. German-Acacio J. M. Espinosa-Chavez J. Torres-Valencia J. M. Perez-Villanueva J. Palacios-Espinosa J. F. Soria-Arteche O. Cortes-Benitez F. Molecules. 2021;26:4375. doi: 10.3390/molecules26144375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding H. Hu X. Xu X. Zhang G. Gong D. Int. J. Biol. Macromol. 2018;107:1844–1855. doi: 10.1016/j.ijbiomac.2017.10.040. [DOI] [PubMed] [Google Scholar]
- Nema N. K. Maity N. Sarkar B. Mukherjee P. K. Arch. Dermatol. Res. 2011;303:247–252. doi: 10.1007/s00403-010-1103-y. [DOI] [PubMed] [Google Scholar]
- Wu P. He H. Ma H. Tu B. Li J. Guo S. Chen S. Cao N. Zheng W. Tang X. Li D. Xu X. Zheng X. Sheng Z. David Hong W. Zhang K. Bioorg. Chem. 2021;107:104580. doi: 10.1016/j.bioorg.2020.104580. [DOI] [PubMed] [Google Scholar]
- He H. Li H. Akanji T. Niu S. Luo Z. Li D. Seeram N. P. Wu P. Ma H. J. Enzyme Inhib. Med. Chem. 2021;36:1665–1678. doi: 10.1080/14756366.2021.1956487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khwaza V. Oyedeji O. O. Aderibigbe B. A. Int. J. Mol. Sci. 2020;21:5920. doi: 10.3390/ijms21165920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A. L. Hao Y. Wang W. Y. Liu Q. S. Sun Y. Gu W. Int. J. Mol. Sci. 2020;21:2876. doi: 10.3390/ijms21082876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W. Hao Y. Zhang G. Wang S. F. Miao T. T. Zhang K. P. Bioorg. Med. Chem. Lett. 2015;25:554–557. doi: 10.1016/j.bmcl.2014.12.021. [DOI] [PubMed] [Google Scholar]
- Pan H. S. Li L. Cui H. B. Yang L. N. Meng Y. Q. Acta Pharm. Sin. 2017;52:1890–1894. [Google Scholar]
- Fan H. Geng L. Yang F. Dong X. He D. Zhang Y. Eur. J. Med. Chem. 2019;176:61–67. doi: 10.1016/j.ejmech.2019.04.059. [DOI] [PubMed] [Google Scholar]
- Liu X. Zhao P. Wang X. Wang L. Zhu Y. Song Y. Gao W. J. Exp. Clin. Cancer Res. 2019;38:184. doi: 10.1186/s13046-019-1173-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang K. Huang J. Pan J. Zhang X. Lu W. RSC Adv. 2015;5:19620–19623. doi: 10.1039/C4RA15414B. [DOI] [Google Scholar]
- Lou H. Li H. Zhang S. Lu H. Chen Q. Molecules. 2021;26:5583. doi: 10.3390/molecules26185583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Q. X. Chen H. F. Luo X. R. Zhang Y. X. Yao X. Zheng X. Curr. Med. Sci. 2018;38:387–397. doi: 10.1007/s11596-018-1891-4. [DOI] [PubMed] [Google Scholar]
- Khusnutdinova E. F. Smirnova I. E. Kazakova O. B. Petrova A. V. Ha N. T. T. Viet D. Q. Med. Chem. Res. 2017;26:2737–2742. doi: 10.1007/s00044-017-1972-0. [DOI] [Google Scholar]
- Khusnutdinova E. F. Petrova A. V. Thu H. N. T. Tu A. L. T. Thanh T. N. Thi C. B. Babkov D. A. Kazakova O. B. Bioorg. Chem. 2019;88:102957. doi: 10.1016/j.bioorg.2019.102957. [DOI] [PubMed] [Google Scholar]
- Zhao Y. Chen C. H. Morris-Natschke S. L. Lee K. H. Eur. J. Med. Chem. 2021;215:113287. doi: 10.1016/j.ejmech.2021.113287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K. Zhang X. Xie L. Deng M. Chen H. Song J. Long J. Li X. Luo J. Pharmacol. Res. 2021;164:105373. doi: 10.1016/j.phrs.2020.105373. [DOI] [PubMed] [Google Scholar]
- Bhandari P. Patel N. K. Bhutani K. K. Bioorg. Med. Chem. Lett. 2014;24:3596–3599. doi: 10.1016/j.bmcl.2014.05.032. [DOI] [PubMed] [Google Scholar]
- Andrade M. A. Braga M. A. Cesar P. H. S. Trento M. V. C. Espósito M. A. Silva L. F. Marcussi S. Curr. Cancer Drug Targets. 2018;18:957–966. doi: 10.2174/1568009618666180102105843. [DOI] [PubMed] [Google Scholar]
- Auezova L. Najjar A. Kfoury M. Fourmentin S. Greige-Gerges H. J. Appl. Microbiol. 2020;128:710–720. doi: 10.1111/jam.14516. [DOI] [PubMed] [Google Scholar]
- Yang R. Chu X. Sun L. Kang Z. Ji M. Yu Y. Liu Y. He Z. Gao N. Phytother. Res. 2018;32:715–722. doi: 10.1002/ptr.6023. [DOI] [PubMed] [Google Scholar]
- Li F. Yan T. T. Fu Y. Y. Zhang N. L. Wang L. Zhang Y. B. Du J. Liu J. F. Chem. Biodiversity. 2021;18:e2001012. doi: 10.1002/cbdv.202001012. [DOI] [PubMed] [Google Scholar]
- Guo Q. Jiang E. Curr. Top. Med. Chem. 2021;21:1712–1724. doi: 10.2174/1568026621666210113163327. [DOI] [PubMed] [Google Scholar]
- Zhao W. He L. Xiang T. L. Tang Y. J. Eur. J. Med. Chem. 2019;170:73–86. doi: 10.1016/j.ejmech.2019.03.006. [DOI] [PubMed] [Google Scholar]
- Han H. W. Lin H. Y. He D. L. Ren Y. Sun W. X. Liang L. Du M. H. Li D. C. Chu Y. C. Yang M. K. Wang X. M. Yang Y. H. Chem. Biodiversity. 2018;15:e1800289. doi: 10.1002/cbdv.201800289. [DOI] [PubMed] [Google Scholar]
- Sathish M. Kavitha B. Nayak V. L. Tangella Y. Ajitha A. Nekkanti S. Alarifi A. Shankaraiah N. Nagesh N. Kamal A. Eur. J. Med. Chem. 2018;144:557–571. doi: 10.1016/j.ejmech.2017.12.055. [DOI] [PubMed] [Google Scholar]
- Zhang L. Zeng X. Ren X. Tao N. Yang C. Xu Y. Chen Y. Wang J. Med. Chem. Res. 2018;28:81–94. doi: 10.1007/s00044-018-2266-x. [DOI] [Google Scholar]
- Wang J. Long L. Chen Y. Xu Y. Zhang L. Bioorg. Med. Chem. Lett. 2018;28:1817–1824. doi: 10.1016/j.bmcl.2018.04.019. [DOI] [PubMed] [Google Scholar]
- Zhang L. Chen F. Wang J. Chen Y. Zhang Z. Lin Y. Zhu X. RSC Adv. 2015;5:97816–97823. doi: 10.1039/C5RA21217K. [DOI] [Google Scholar]
- Zhang L. Liu L. Zheng C. Wang Y. Nie X. Shi D. Chen Y. Wei G. Wang J. Eur. J. Med. Chem. 2017;131:81–91. doi: 10.1016/j.ejmech.2017.03.011. [DOI] [PubMed] [Google Scholar]
- Cao B. Chen H. Gao Y. Niu C. Zhang Y. Li L. Int. J. Mol. Med. 2015;35:771–776. doi: 10.3892/ijmm.2015.2068. [DOI] [PubMed] [Google Scholar]
- Ai T. Shi S.-Y. Chen L.-T. Li L. Cao B. Gao Y. Chen H. Zhou J. Chin. Chem. Lett. 2013;24:37–40. doi: 10.1016/j.cclet.2012.11.016. [DOI] [Google Scholar]
- Liu J. Cao B. Gao Y. Bai M. Mei X. Chen H. Jiang Y. G. Huang D. J. J. Asian Nat. Prod. Res. 2013;15:985–992. doi: 10.1080/10286020.2013.802688. [DOI] [PubMed] [Google Scholar]
- Zhang Z. H. Zhang L. M. Luo G. Zhang S. Chen H. Zhou J. J. Asian Nat. Prod. Res. 2014;16:527–534. doi: 10.1080/10286020.2014.913578. [DOI] [PubMed] [Google Scholar]
- Guo Y. E. Chen H. Zuo S. Liu D. L. Lu Y. L. Lv J. J. Wen S. P. Zhang T. C. J. Asian Nat. Prod. Res. 2011;13:417–424. doi: 10.1080/10286020.2011.568941. [DOI] [PubMed] [Google Scholar]
- Bhattarai N. Kumbhar A. A. Pokharel Y. R. Yadav P. N. Mini-Rev. Med. Chem. 2021;21:2996–3029. doi: 10.2174/1389557521666210405160323. [DOI] [PubMed] [Google Scholar]
- Bhatia R., Singh A., Kumar B. and Rawal R. K., Key Heterocyclic Cores for Smart Anticancer Drug–Design Part II, 2022, vol. 2, p. 35 [Google Scholar]
- Feng D. Zhang A. Yang Y. Yang P. Arch. Pharm. 2020;353:e1900380. doi: 10.1002/ardp.201900380. [DOI] [PubMed] [Google Scholar]
- Carneiro L. S. A. Almeida-Souza F. Lopes Y. S. C. Novas R. C. V. Santos K. B. A. Ligiero C. B. P. Calabrese K. D. S. Buarque C. D. Bioorg. Chem. 2021;114:105141. doi: 10.1016/j.bioorg.2021.105141. [DOI] [PubMed] [Google Scholar]
- Vianna D. R. Bubols G. Meirelles G. Silva B. V. Da Rocha A. Lanznaster M. Monserrat J. M. Garcia S. C. Von Poser G. Eifler-Lima V. L. Int. J. Mol. Sci. 2012;13:7260–7270. doi: 10.3390/ijms13067260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. Yao Y. Li L. J. Pharm. Pharmacol. 2017;69:1253–1264. doi: 10.1111/jphp.12774. [DOI] [PubMed] [Google Scholar]
- Sashidhara K. V. Kumar A. Kumar M. Sonkar R. Bhatia G. Khanna A. K. Bioorg. Med. Chem. Lett. 2010;20:4248–4251. doi: 10.1016/j.bmcl.2010.05.023. [DOI] [PubMed] [Google Scholar]
- Ali M. Y. Jannat S. Jung H. A. Choi R. J. Roy A. Choi J. S. Asian Pac. J. Trop. Med. 2016;9:103–111. doi: 10.1016/j.apjtm.2016.01.014. [DOI] [PubMed] [Google Scholar]
- Kamath P. R. Sunil D. Ajees A. A. Pai K. S. Das S. Bioorg. Chem. 2015;63:101–109. doi: 10.1016/j.bioorg.2015.10.001. [DOI] [PubMed] [Google Scholar]
- Kamath P. R. Sunil D. Joseph M. M. Abdul Salam A. A. Sreelekha T. T. Eur. J. Med. Chem. 2017;136:442–451. doi: 10.1016/j.ejmech.2017.05.032. [DOI] [PubMed] [Google Scholar]
- Pathoor R. Bahulayan D. New J. Chem. 2018;42:6810–6816. doi: 10.1039/C8NJ00032H. [DOI] [Google Scholar]
- Galayev O. Garazd Y. Garazd M. Lesyk R. Eur. J. Med. Chem. 2015;105:171–181. doi: 10.1016/j.ejmech.2015.10.021. [DOI] [PubMed] [Google Scholar]
- Yang X. C. Zhang P. L. Kumar K. V. Li S. Geng R. X. Zhou C. H. Eur. J. Med. Chem. 2022;232:114192. doi: 10.1016/j.ejmech.2022.114192. [DOI] [PubMed] [Google Scholar]
- Rathod A. S. Godipurge S. S. Biradar J. S. Int. J. Pharm. Pharm. Sci. 2017;9:233–240. doi: 10.22159/ijpps.2017v9i12.21970. [DOI] [Google Scholar]
- Sangshetti J. N. Kalam Khan F. A. Kulkarni A. A. Patil R. H. Pachpinde A. M. Lohar K. S. Shinde D. B. Bioorg. Med. Chem. Lett. 2016;26:829–835. doi: 10.1016/j.bmcl.2015.12.085. [DOI] [PubMed] [Google Scholar]
- Mollazadeh M. Mohammadi-Khanaposhtani M. Valizadeh Y. Zonouzi A. Faramarzi M. A. Kiani M. Biglar M. Larijani B. Hamedifar H. Mahdavi M. Hajimiri M. H. Med. Chem. 2021;17:264–272. doi: 10.2174/1573406416666200826101205. [DOI] [PubMed] [Google Scholar]
- Sashidhara K. V. Rao K. B. Sonkar R. Modukuri R. K. Chhonker Y. S. Kushwaha P. Chandasana H. Khanna A. K. Bhatta R. S. Bhatia G. Suthar M. K. Saxena J. K. Kumar V. Siddiqi M. I. MedChemComm. 2016;7:1858–1869. doi: 10.1039/C6MD00283H. [DOI] [Google Scholar]
- Ghanei-Nasab S. Khoobi M. Hadizadeh F. Marjani A. Moradi A. Nadri H. Emami S. Foroumadi A. Shafiee A. Eur. J. Med. Chem. 2016;121:40–46. doi: 10.1016/j.ejmech.2016.05.014. [DOI] [PubMed] [Google Scholar]
- Zou Y. Lu N. Yang X. Xie Z. Lei X. Liu X. Li Y. Huang S. Tang G. Wang Z. RSC Med. Chem. 2023;14:1172–1185. doi: 10.1039/D3MD00128H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X. Lin D. Jiang R. Li H. Wan J. Li H. Oncol. Rep. 2016;36:271–278. doi: 10.3892/or.2016.4804. [DOI] [PubMed] [Google Scholar]
- Zduńska K. Dana A. Kolodziejczak A. Rotsztejn H. Skin Pharmacol. Physiol. 2018;31:332–336. doi: 10.1159/000491755. [DOI] [PubMed] [Google Scholar]
- Yu Q. Fan L. Food Chem. 2021;352:129369. doi: 10.1016/j.foodchem.2021.129369. [DOI] [PubMed] [Google Scholar]
- Zolfaghari B. Yazdiniapour Z. Sadeghi M. Akbari M. Troiano R. Lanzotti V. Phytochem. Anal. 2021;32:84–90. doi: 10.1002/pca.2924. [DOI] [PubMed] [Google Scholar]
- Zang J. Shi B. Liang X. Gao Q. Xu W. Zhang Y. Bioorg. Med. Chem. 2017;25:2666–2675. doi: 10.1016/j.bmc.2016.12.001. [DOI] [PubMed] [Google Scholar]
- Benchekroun M. Ismaili L. Pudlo M. Luzet V. Gharbi T. Refouvelet B. Marco-Contelles J. Future Med. Chem. 2015;7:15–21. doi: 10.4155/fmc.14.148. [DOI] [PubMed] [Google Scholar]
- Pachón-Angona I. Martin H. Chhor S. Oset-Gasque M. J. Refouvelet B. Marco-Contelles J. Ismaili L. Future Med. Chem. 2019;11:3097–3108. doi: 10.4155/fmc-2019-0191. [DOI] [PubMed] [Google Scholar]
- Takao K. Toda K. Saito T. Sugita Y. Chem. Pharm. Bull. 2017;65:1020–1027. doi: 10.1248/cpb.c17-00416. [DOI] [PubMed] [Google Scholar]
- Desai N. C. Somani H. C. Mehta H. K. Jadeja D. J. Khasiya A. G. Khedkar V. M. SAR QSAR Environ. Res. 2022;33:89–109. doi: 10.1080/1062936X.2022.2032333. [DOI] [PubMed] [Google Scholar]
- Elkamhawy A. Oh N. K. Gouda N. A. Abdellattif M. H. Alshammari S. O. Abourehab M. A. S. Alshammari Q. A. Belal A. Kim M. Al-Karmalawy A. A. Lee K. Metabolites. 2023;13:141. doi: 10.3390/metabo13020141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao S. Lv C. Tao Y. Miao Y. Zhu Y. Zhang W. Sun D. Yun X. Xia Y. Wei Z. Dai Y. Cancer Lett. 2020;491:162–179. doi: 10.1016/j.canlet.2020.08.033. [DOI] [PubMed] [Google Scholar]
- Lu Y. N. Zhao X. D. Xu X. Piao J. Aosai F. Li Y. B. Shen L. X. Shi S. Y. Xu G. H. Ma J. Piao H. N. Jin X. Piao L. X. Int. Immunopharmacol. 2020;84:106539. doi: 10.1016/j.intimp.2020.106539. [DOI] [PubMed] [Google Scholar]
- Chen Q. Yang L. Han M. Cai E. Zhao Y. Biomed. Pharmacother. 2016;84:1792–1801. doi: 10.1016/j.biopha.2016.10.093. [DOI] [PubMed] [Google Scholar]
- Zhang H. B. Shen Q. K. Wang H. Jin C. Jin C. M. Quan Z. S. Eur. J. Med. Chem. 2018;158:414–427. doi: 10.1016/j.ejmech.2018.08.087. [DOI] [PubMed] [Google Scholar]
- Ponte L. G. S. Pavan I. C. B. Mancini M. C. S. da Silva L. G. S. Morelli A. P. Severino M. B. Bezerra R. M. N. Simabuco F. M. Molecules. 2021;26:2029. doi: 10.3390/molecules26072029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maleki S. J. Crespo J. F. Cabanillas B. Food Chem. 2019;299:125124. doi: 10.1016/j.foodchem.2019.125124. [DOI] [PubMed] [Google Scholar]
- Farhadi F. Khameneh B. Iranshahi M. Iranshahy M. Phytother. Res. 2019;33:13–40. doi: 10.1002/ptr.6208. [DOI] [PubMed] [Google Scholar]
- Xiao J. Gao M. Diao Q. Gao F. Curr. Top. Med. Chem. 2021;21:348–362. doi: 10.2174/1568026620666201022143236. [DOI] [PubMed] [Google Scholar]
- Xu M. Wu P. Shen F. Ji J. Rakesh K. P. Bioorg. Chem. 2019;91:103133. doi: 10.1016/j.bioorg.2019.103133. [DOI] [PubMed] [Google Scholar]
- Wang G. Li C. He L. Lei K. Wang F. Pu Y. Yang Z. Cao D. Ma L. Chen J. Sang Y. Liang X. Xiang M. Peng A. Wei Y. Chen L. Bioorg. Med. Chem. 2014;22:2060–2079. doi: 10.1016/j.bmc.2014.02.028. [DOI] [PubMed] [Google Scholar]
- Preti D. Romagnoli R. Rondanin R. Cacciari B. Hamel E. Balzarini J. Liekens S. Schols D. Estevez-Sarmiento F. Quintana J. Estevez F. J. Enzyme Inhib. Med. Chem. 2018;33:1225–1238. doi: 10.1080/14756366.2018.1493473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S. An B. Li J. Hu J. Huang L. Li X. Chan A. S. C. Org. Biomol. Chem. 2017;15:7404–7410. doi: 10.1039/C7OB01655G. [DOI] [PubMed] [Google Scholar]
- Mirzaei H. Shokrzadeh M. Modanloo M. Ziar A. Riazi G. H. Emami S. Bioorg. Chem. 2017;75:86–98. doi: 10.1016/j.bioorg.2017.09.005. [DOI] [PubMed] [Google Scholar]
- Mirzaei H. Abastabar M. Emami S. Comput. Biol. Chem. 2020;84:107189. doi: 10.1016/j.compbiolchem.2019.107189. [DOI] [PubMed] [Google Scholar]
- Du B. Liu X. Luan X. Zhang W. Zhuang C. Bioorg. Chem. 2023;135:106531. doi: 10.1016/j.bioorg.2023.106531. [DOI] [PubMed] [Google Scholar]
- Rauf A. Khan R. Raza M. Khan H. Pervez S. De Feo V. Maione F. Mascolo N. Fitoterapia. 2015;103:129–135. doi: 10.1016/j.fitote.2015.03.019. [DOI] [PubMed] [Google Scholar]
- Singh P. Kaur J. Singh G. Bhatti R. J. Med. Chem. 2015;58:5989–6001. doi: 10.1021/acs.jmedchem.5b00952. [DOI] [PubMed] [Google Scholar]
- Singh P. Shaveta S. Sharma S. Bhatti R. Bioorg. Med. Chem. Lett. 2014;24:77–82. doi: 10.1016/j.bmcl.2013.11.080. [DOI] [PubMed] [Google Scholar]
- Ji Y. B. Chen W. J. Shan T. Z. Sun B. Y. Yan P. C. Jiang W. Chem. Biodiversity. 2020;17:e1900640. doi: 10.1002/cbdv.201900640. [DOI] [PubMed] [Google Scholar]
- Nhan N. T. Nguyen P. H. Tran M. H. Nguyen P. D. Tran D. T. To D. C. J. Asian Nat. Prod. Res. 2021;23:414–422. doi: 10.1080/10286020.2020.1767079. [DOI] [PubMed] [Google Scholar]
- Chen X. Leng J. Rakesh K. P. Darshini N. Shubhavathi T. Vivek H. K. Mallesha N. Qin H. L. MedChemComm. 2017;8:1706–1719. doi: 10.1039/C7MD00209B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haudecoeur R. Ahmed-Belkacem A. Yi W. Fortuné A. Brillet R. Belle C. Nicolle E. Pallier C. Pawlotsky J. M. Boumendjel A. J. Med. Chem. 2011;54:5395–5402. doi: 10.1021/jm200242p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meguellati A. Ahmed-Belkacem A. Yi W. Haudecoeur R. Crouillere M. Brillet R. Pawlotsky J. M. Boumendjel A. Peuchmaur M. Eur. J. Med. Chem. 2014;80:579–592. doi: 10.1016/j.ejmech.2014.04.005. [DOI] [PubMed] [Google Scholar]
- Xie N. Gomes F. P. Deora V. Gregory K. Vithanage T. Nassar Z. D. Cabot P. J. Sturgess D. Shaw P. N. Parat M. O. Brain, Behav., Immun. 2017;61:244–258. doi: 10.1016/j.bbi.2016.12.002. [DOI] [PubMed] [Google Scholar]
- Obeng S. Wang H. Jali A. Stevens D. L. Akbarali H. I. Dewey W. L. Selley D. E. Zhang Y. ACS Chem. Neurosci. 2019;10:1075–1090. doi: 10.1021/acschemneuro.8b00349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C. Yang S. Wang K. Bao X. Liu Y. Zhou S. Liu H. Qiu Y. Wang T. Yu H. Biomed. Pharmacother. 2019;120:109543. doi: 10.1016/j.biopha.2019.109543. [DOI] [PubMed] [Google Scholar]
- Ur Rashid H. Rasool S. Ali Y. Khan K. Martines M. A. U. Bioorg. Chem. 2020;99:103863. doi: 10.1016/j.bioorg.2020.103863. [DOI] [PubMed] [Google Scholar]
- Xu Y. Wu L. Rashid H. U. Jing D. Liang X. Wang H. Liu X. Jiang J. Wang L. Xie P. Eur. J. Med. Chem. 2018;156:479–492. doi: 10.1016/j.ejmech.2018.07.028. [DOI] [PubMed] [Google Scholar]
- Li Z. Luo M. Cai B. Haroon Ur R. Huang M. Jiang J. Wang L. Wu L. Eur. J. Med. Chem. 2018;157:665–682. doi: 10.1016/j.ejmech.2018.08.021. [DOI] [PubMed] [Google Scholar]
- Hu G. Cao C. Deng Z. Li J. Zhou X. Huang Z. Cen C. Oncol. Lett. 2021;21:66. doi: 10.3892/ol.2020.12327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z. Luo M. Cai B. Wu L. Huang M. Haroon Ur R. Jiang J. Wang L. Bioorg. Med. Chem. Lett. 2018;28:677–683. doi: 10.1016/j.bmcl.2018.01.017. [DOI] [PubMed] [Google Scholar]
- Li L. Li J. Ma L. Shang H. Zou Z. Bioorg. Chem. 2023;134:106341. doi: 10.1016/j.bioorg.2023.106341. [DOI] [PubMed] [Google Scholar]
- Ortiz L. M. Lombardi P. Tillhon M. Scovassi A. I. Molecules. 2014;19:12349–12367. doi: 10.3390/molecules190812349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistry B. Keum Y.-S. Kim D. H. Res. Chem. Intermed. 2015;42:3241–3256. doi: 10.1007/s11164-015-2208-x. [DOI] [Google Scholar]
- Liu H. W. Ji Q. T. Ren G. G. Wang F. Su F. Wang P. Y. Zhou X. Wu Z. B. Li Z. Yang S. J. Agric. Food Chem. 2020;68:12558–12568. doi: 10.1021/acs.jafc.0c02528. [DOI] [PubMed] [Google Scholar]
- Dai J. Dan W. Li N. Wang J. Eur. J. Med. Chem. 2018;157:333–338. doi: 10.1016/j.ejmech.2018.08.001. [DOI] [PubMed] [Google Scholar]
- Zolottsev A. V. Latysheva A. S. Pokrovsky V. S. Khan I. I. Misharin A. Y. Eur. J. Med. Chem. 2021;210:113089. doi: 10.1016/j.ejmech.2020.113089. [DOI] [PubMed] [Google Scholar]
- Yang G. X. Huang Y. Zheng L. L. Zhang L. Su L. Wu Y. H. Li J. Zhou L. C. Huang J. Tang Y. Wang R. Ma L. Eur. J. Med. Chem. 2020;187:111913. doi: 10.1016/j.ejmech.2019.111913. [DOI] [PubMed] [Google Scholar]
- Zhang H. Xu J. Wang M. Xia X. Dai R. Zhao Y. Steroids. 2020;161:108690. doi: 10.1016/j.steroids.2020.108690. [DOI] [PubMed] [Google Scholar]
- Kovács P. Csonka T. Kovács T. Sári Z. Ujlaki G. Sipos A. Karányi Z. Szeőcs D. Hegedűs C. Uray K. Jankó L. Kiss M. Kiss B. Laoui D. Virág L. Méhes G. Bai P. Mikó E. Cancers. 2019;11:1522. doi: 10.3390/cancers11091255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Incerti M. Russo S. Callegari D. Pala D. Giorgio C. Zanotti I. Barocelli E. Vicini P. Vacondio F. Rivara S. Castelli R. Tognolini M. Lodola A. J. Med. Chem. 2017;60:787–796. doi: 10.1021/acs.jmedchem.6b01642. [DOI] [PubMed] [Google Scholar]
- He X. L. Xing Y. Gu X. Z. Xiao J. X. Wang Y. Y. Yi Z. Qiu W. W. Steroids. 2017;125:54–60. doi: 10.1016/j.steroids.2017.06.009. [DOI] [PubMed] [Google Scholar]
- Tan W. Pan M. Liu H. Tian H. Ye Q. Liu H. OncoTargets Ther. 2017;10:3467–3474. doi: 10.2147/OTT.S139009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L. Bu M. Cao T. Li H. Guo M. Zhou Y. Zhang N. Zeng C. Wang Y. Heterocycles. 2017;94:691–701. doi: 10.3987/COM-17-13658. [DOI] [Google Scholar]
- Li H. Wang H. Wang J. Lin Y. Ma Y. Bu M. Steroids. 2020;153:108471. doi: 10.1016/j.steroids.2019.108471. [DOI] [PubMed] [Google Scholar]
- Chen L. Mai W. Chen M. Hu J. Zhuo Z. Lei X. Deng L. Liu J. Yao N. Huang M. Peng Y. Ye W. Zhang D. Pharmacol. Res. 2017;123:130–142. doi: 10.1016/j.phrs.2017.07.009. [DOI] [PubMed] [Google Scholar]
- Deng L. J. Wang L. H. Peng C. K. Li Y. B. Huang M. H. Chen M. F. Lei X. P. Qi M. Cen Y. Ye W. C. Zhang D. M. Chen W. M. J. Med. Chem. 2017;60:5320–5333. doi: 10.1021/acs.jmedchem.6b01755. [DOI] [PubMed] [Google Scholar]
- Pietri E. Massa I. Bravaccini S. Ravaioli S. Tumedei M. M. Petracci E. Donati C. Schirone A. Piacentini F. Gianni L. Nicolini M. Campadelli E. Gennari A. Saba A. Campi B. Valmorri L. Andreis D. Fabbri F. Amadori D. Rocca A. Oncologist. 2019;24:e197–e205. doi: 10.1634/theoncologist.2018-0243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J. Liu L. Zhao D. Gan C. Huang X. Xiao Q. Qi B. Yang L. Huang Y. Steroids. 2015;95:32–38. doi: 10.1016/j.steroids.2015.01.002. [DOI] [PubMed] [Google Scholar]
- Mahmoudi N. Kiasalari Z. Rahmani T. Sanaierad A. Afshin-Majd S. Naderi G. Baluchnejadmojarad T. Roghani M. Neuropsychobiology. 2021;80:25–35. doi: 10.1159/000507398. [DOI] [PubMed] [Google Scholar]
- Zhou L. C. Liang Y. F. Huang Y. Yang G. X. Zheng L. L. Sun J. M. Li Y. Zhu F. L. Qian H. W. Wang R. Ma L. Eur. J. Med. Chem. 2021;219:113426. doi: 10.1016/j.ejmech.2021.113426. [DOI] [PubMed] [Google Scholar]
- Smirnova I. E. Kazakova O. B. Viet D. Q. Thuc N. T. Linh P. T. Huong D. T. T. Med. Chem. Res. 2014;24:2177–2182. doi: 10.1007/s00044-014-1292-6. [DOI] [Google Scholar]
- Vervandier-Fasseur D. Latruffe N. Molecules. 2019;24:4506. doi: 10.3390/molecules24244506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan J. Zhang X. Ma C. Sun L. Feng Y. He Z. Zhang H. J. Food Sci. 2022;87:1244–1256. doi: 10.1111/1750-3841.16028. [DOI] [PubMed] [Google Scholar]
- Luque G. C. Moya M. Picchio M. L. Bagnarello V. Valerio I. Bolaños J. Vethencourt M. Gamboa S. H. Tomé L. C. Minari R. J. Mecerreyes D. Polymers. 2023;15:1076. doi: 10.3390/polym15051076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano A. Tommonaro G. Nutrients. 2019;11:2376. doi: 10.3390/nu11102376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang M. Taghibiglou C. J. Alzheimer's Dis. 2017;58:1003–1016. doi: 10.3233/JAD-170188. [DOI] [PubMed] [Google Scholar]
- Nayakula M. Jeengar M. K. Naidu V. G. M. Chella N. Eur. J. Drug Metab. Pharmacokinet. 2023;48:189–199. doi: 10.1007/s13318-023-00819-7. [DOI] [PubMed] [Google Scholar]
- Sri Ramya P. V. Angapelly S. Guntuku L. Singh Digwal C. Nagendra Babu B. Naidu V. G. M. Kamal A. Eur. J. Med. Chem. 2017;127:100–114. doi: 10.1016/j.ejmech.2016.12.043. [DOI] [PubMed] [Google Scholar]
- Theppawong A. Van de Walle T. Grootaert C. Bultinck M. Desmet T. Van Camp J. D'Hooghe M. ChemistryOpen. 2018;7:381–392. doi: 10.1002/open.201800029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Walle T. Theppawong A. Grootaert C. De Jonghe S. Persoons L. Daelemans D. Van Hecke K. Van Camp J. D'hooghe M. Monatsh. Chem. 2019;150:2045–2051. doi: 10.1007/s00706-019-02516-1. [DOI] [Google Scholar]
- Okuda M. Hijikuro I. Fujita Y. Teruya T. Kawakami H. Takahashi T. Sugimoto H. Bioorg. Med. Chem. Lett. 2016;26:5024–5028. doi: 10.1016/j.bmcl.2016.08.092. [DOI] [PubMed] [Google Scholar]
- Rajagopalan R. Subramanian S. Pajaniradje S. Tumdam R. Hoda M. Dasgupta A. J. Cancer Res. Ther. 2023;19:265–272. doi: 10.4103/jcrt.jcrt_1256_21. [DOI] [PubMed] [Google Scholar]
- Zheng Z. Li X. Chen P. Zou Y. Shi X. Li X. Young Kim E. Liao J. Yang J. Chattipakorn N. Wu G. Tang Q. Cho W.-J. Liang G. Bioorg. Chem. 2023;136:106557. doi: 10.1016/j.bioorg.2023.106557. [DOI] [PubMed] [Google Scholar]
- Rauf A. Imran M. Butt M. S. Nadeem M. Peters D. G. Mubarak M. S. Crit. Rev. Food Sci. Nutr. 2018;58:1428–1447. doi: 10.1080/10408398.2016.1263597. [DOI] [PubMed] [Google Scholar]
- Yan J. Hu J. An B. Huang L. Li X. Eur. J. Med. Chem. 2017;125:663–675. doi: 10.1016/j.ejmech.2016.09.056. [DOI] [PubMed] [Google Scholar]
- La Regina G. Bai R. Coluccia A. Naccarato V. Famiglini V. Nalli M. Masci D. Verrico A. Rovella P. Mazzoccoli C. Da Pozzo E. Cavallini C. Martini C. Vultaggio S. Dondio G. Varasi M. Mercurio C. Hamel E. Lavia P. Silvestri R. Eur. J. Med. Chem. 2018;152:283–297. doi: 10.1016/j.ejmech.2018.04.042. [DOI] [PubMed] [Google Scholar]
- Hua S. Chen F. Wang X. Gou S. Eur. J. Med. Chem. 2020;189:112041. doi: 10.1016/j.ejmech.2020.112041. [DOI] [PubMed] [Google Scholar]
- La Regina G. Bai R. Rensen W. M. Di Cesare E. Coluccia A. Piscitelli F. Famiglini V. Reggio A. Nalli M. Pelliccia S. Da Pozzo E. Costa B. Granata I. Porta A. Maresca B. Soriani A. Iannitto M. L. Santoni A. Li J. Miranda Cona M. Chen F. Ni Y. Brancale A. Dondio G. Vultaggio S. Varasi M. Mercurio C. Martini C. Hamel E. Lavia P. Novellino E. Silvestri R. J. Med. Chem. 2013;56:123–149. doi: 10.1021/jm3013097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Regina G. Bai R. Coluccia A. Famiglini V. Pelliccia S. Passacantilli S. Mazzoccoli C. Ruggieri V. Verrico A. Miele A. Monti L. Nalli M. Alfonsi R. Di Marcotullio L. Gulino A. Ricci B. Soriani A. Santoni A. Caraglia M. Porto S. Da Pozzo E. Martini C. Brancale A. Marinelli L. Novellino E. Vultaggio S. Varasi M. Mercurio C. Bigogno C. Dondio G. Hamel E. Lavia P. Silvestri R. J. Med. Chem. 2015;58:5789–5807. doi: 10.1021/acs.jmedchem.5b00310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Y. T. Man R. J. Tang D. J. Yao Y. F. Tao X. X. Yu C. Liang X. Y. Makawana J. A. Zou M. J. Wang Z. C. Zhu H. L. Sci. Rep. 2016;6:25387. doi: 10.1038/srep25387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamal A. Kumar G. B. Polepalli S. Shaik A. B. Reddy V. S. Reddy M. K. Reddy Ch R. Mahesh R. Kapure J. S. Jain N. ChemMedChem. 2014;9:2565–2579. doi: 10.1002/cmdc.201402256. [DOI] [PubMed] [Google Scholar]
- Shen C. C. Afraj S. N. Hung C. C. Barve B. D. Kuo L. Y. Lin Z. H. Ho H. O. Kuo Y. H. Bioorg. Med. Chem. Lett. 2021;41:127976. doi: 10.1016/j.bmcl.2021.127976. [DOI] [PubMed] [Google Scholar]
- Ibis C. Tuyun A. F. Bahar H. Ayla S. S. Stasevych M. V. Musyanovych R. Y. Komarovska-Porokhnyavets O. Novikov V. Med. Chem. Res. 2014;23:2140–2149. doi: 10.1007/s00044-013-0806-y. [DOI] [PubMed] [Google Scholar]
- Farias M. S. Pich C. T. Kviecinski M. R. Bucker N. C. Felipe K. B. Da Silva F. O. Günther T. M. Correia J. F. Ríos D. Benites J. Valderrama J. A. Calderon P. B. Pedrosa R. C. Mol. Med. Rep. 2014;10:405–410. doi: 10.3892/mmr.2014.2160. [DOI] [PubMed] [Google Scholar]
- Palacios J. Benites J. Owen G. I. Morales P. Chiong M. Nwokocha C. R. Paredes A. Cifuentes F. J. Cardiovasc. Pharmacol. 2021;77:245–252. doi: 10.1097/FJC.0000000000000940. [DOI] [PubMed] [Google Scholar]
- Tandon V. K. Singh R. V. Yadav D. B. Bioorg. Med. Chem. Lett. 2004;14:2901–2904. doi: 10.1016/j.bmcl.2004.03.047. [DOI] [PubMed] [Google Scholar]
- Cordova-Rivas S. Araujo-Huitrado J. G. Rivera-Avalos E. Escalante-Garcia I. L. Duron-Torres S. M. Lopez-Hernandez Y. Hernandez-Lopez H. Lopez L. de Loera D. Lopez J. A. Molecules. 2020;25:2058. doi: 10.3390/molecules25092058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulos J. C. Rahama M. Hegazy M. F. Efferth T. Cancer Lett. 2019;459:248–267. doi: 10.1016/j.canlet.2019.04.033. [DOI] [PubMed] [Google Scholar]
- Zhang Y. Han H. Qiu H. Lin H. Yu L. Zhu W. Qi J. Yang R. Pang Y. Wang X. Lu G. Yang Y. Biomed. Pharmacother. 2017;93:636–645. doi: 10.1016/j.biopha.2017.06.076. [DOI] [PubMed] [Google Scholar]
- Wang X. M. Lin H. Y. Kong W. Y. Guo J. Shi J. Huang S. C. Qi J. L. Yang R. W. Gu H. W. Yang Y. H. Chem. Biol. Drug Des. 2014;83:334–343. doi: 10.1111/cbdd.12247. [DOI] [PubMed] [Google Scholar]
- Zhang Y. Han H. Sun L. Qiu H. Lin H. Yu L. Zhu W. Qi J. Yang R. Pang Y. Wang X. Lu G. Yang Y. Braz. J. Med. Biol. Res. 2017;50:e6586–e6594. doi: 10.1590/1414-431x20176586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa R. Tateishi H. Radwan M. O. Chinen T. Ciftci H. Iwamaru K. Baba N. Tominaga Y. Koga R. Toma T. Inoue J. I. Umezawa K. Fujita M. Otsuka M. Chem. Pharm. Bull. 2022;70:477–482. doi: 10.1248/cpb.c21-01087. [DOI] [PubMed] [Google Scholar]
- Shukla S. Srivastava R. S. Shrivastava S. K. Sodhi A. Kumar P. Med. Chem. Res. 2012;22:1604–1617. doi: 10.1007/s00044-012-0150-7. [DOI] [Google Scholar]
- Mishra S. K. Tiwari S. Shrivastava A. Srivastava S. Boudh G. K. Chourasia S. K. Chaturvedi U. Mir S. S. Saxena A. K. Bhatia G. Lakshmi V. J. Nat. Med. 2014;68:363–371. doi: 10.1007/s11418-013-0810-z. [DOI] [PubMed] [Google Scholar]
- Shattat G. Al-Qirim T. Sheikha G. A. Al-Hiari Y. Sweidan K. Al-Qirim R. Hikmat S. Hamadneh L. Al-Kouz S. J. Enzyme Inhib. Med. Chem. 2013;28:863–869. doi: 10.3109/14756366.2012.692085. [DOI] [PubMed] [Google Scholar]