

Abstract
Advancements in DNA sequencing technologies within the last decade have stimulated an unprecedented interest in the human microbiome, largely due the broad diversity of human diseases found to correlate with microbiome dysbiosis. As a direct consequence of these studies, a vast number of understudied and uncharacterized microbes have been identified as potential drivers of mucosal health and disease. The looming challenge in the field is to transition these observations into defined molecular mechanistic studies of symbiosis and dysbiosis. In order to meet this challenge, many of these newly identified microbes will need to be adapted for use in experimental models. Consequently, this review presents a comprehensive overview of the molecular microbiology tools and techniques that have played crucial roles in genetic studies of the bacteria found within the human oral microbiota. Here, we will use specific examples from the oral microbiome literature to illustrate the biology supporting these techniques, why they are needed in the field, and how such technologies have been implemented. It is hoped that this information can serve as a useful reference guide to help catalyze molecular microbiology studies of the many new understudied and uncharacterized species identified at different mucosal sites in the body.
Keywords: oral microbiome, genetic system, transformation, mutagenesis, reporter genes, molecular microbiology
This review presents a comprehensive overview of the molecular microbiology tools and techniques that have played crucial roles in genetic studies of the bacteria found within the human oral microbiota.
Introduction
A century before the terms ‘microbiome’ and ‘dysbiosis’ became commonplace in the scientific lexicon, pioneer oral microbiologists were already developing our current understanding of the connection between oral ecology and oral disease. For example, in Goadby's seminal 1903 publication, ‘Mycology of the mouth: a text-book of oral bacteria’, different aspects of oral bacterial metabolism were presented to describe the etiology of dental caries, which represented a substantial paradigm shift from the traditional view of this disease. According to Goadby's classification scheme, cariogenic bacteria are comprised of two classes: bacteria that are either involved in producing acid or promoting liquefaction (i.e. proteolysis) (Goadby 1903). With further advancements in our understanding of the species involved in oral health and disease, the field inevitably became increasingly focused upon the specific molecular mechanisms that could account for these activities. Prior to the development of recombinant DNA technologies, such studies often had to rely upon the generation of spontaneous or chemically-induced mutations in organisms of interest (Kuramitsu 2003). These approaches were inherently limited by the problems associated with correlating genotype to phenotype. Unlike today, genetic complementation or genome sequencing were not practical options at the time. Thus, the potential introduction of secondary mutations was an ever-present source of uncertainty. However, with the advent of the genetic engineering revolution of the 1970 s, a new era in oral microbiome molecular microbiology research was soon to follow. These technologies were essential for the development of the microbial genetic systems that presently form the foundations of nearly all mechanistic studies of oral microbiome commensalism and pathogenesis. With the recent explosion of microbiome OMICS studies, the list of important drivers of health and disease has grown exponentially for all mucosal sites. It seems inevitable that a substantial number of new genetic systems will be required by microbiome researchers before we can expect to fully understand the molecular mechanisms responsible for modulating mucosal homeostasis. Consequently, in the following review, we will describe the principal tools currently utilized for oral microbiome molecular microbiology studies to help guide future efforts to broaden the genetic tractability of the human microbiome. Due to the breadth of this topic, we will focus specifically upon the bacterial component of the oral microbiome, which constitutes the majority of oral biofilm biomass. However, it is certainly worth noting that the oral microbiome also contains a broad diversity of archaea, eukaryotes, and viruses, most of which are similarly understudied and/or uncharacterized (Bandara et al. 2019, Belmok et al. 2020, de Cena et al. 2021, Diaz 2021, Martinez et al. 2021, Szafranski et al. 2021).
General requirements for targeted mutagenesis of microbiome species
Most of the targeted chromosomal mutations previously engineered into microbiome species have incorporated three fundamental components: (i) a mechanism for transformation (exogenous DNA uptake), (ii) a selectable marker, and (iii) a strategy for homologous recombination. When creating a new genetic system for an organism of interest, the transformation protocol tends to be the most vexing and critical component. Without first knowing the optimal approach to introduce foreign DNA into an organism, it can be exceptionally challenging to interpret negative transformation results. Furthermore, the overall success or failure of a genetic system is most often dictated by the practicality and efficiency of the available transformation options. Once an efficacious transformation strategy has been established, the other major components of the genetic toolbox are typically quickly adapted for use. A wide variety of selectable markers like antibiotic resistance cassettes are already available, and these are commonly inserted onto bacterial chromosomes via two types of homologous recombination strategies. The first is synonymously referred to as insertion duplication mutagenesis, single crossover recombination, or a Campbell-type recombination. In the context of genetic engineering, all refer to the same mechanism whereby circular plasmid DNA containing a homologous fragment of the genome is integrated to the chromosome through recombination (Campbell 2007). Because this mechanism inserts the entire plasmid, these constructs result in a chromosomal duplication of the homologous DNA contained on the plasmid (Fig. 1A). Consequently, insertion duplication mutations are reversible and require constant selective pressure to prevent the inserted plasmid from looping out through spontaneous recombination between the duplicated fragments. Since these mutations only require a single recombination event, they can be quite efficient to generate. Also, as we will discuss later, the inherent instability of single crossover mutations can be exploited for the creation of markerless mutations. The second common type of homologous recombination-based mutagenesis is referred to as an allelic replacement, which is typically achieved via double crossover homologous recombination. If available for use, double crossover allelic replacement mutations are usually preferred because they are compatible with linear DNA constructs (Court et al. 2002). Hence, it is possible to assemble these mutagenesis constructs using cloning-independent strategies like overlap extension PCR (OE-PCR) and/or Gibson assembly, both of which can substantially reduce the time and effort required to produce them (Xie et al. 2011, Zhang et al. 2017). Most double crossover allelic replacement constructs are created by attaching homologous DNA fragments flanking the intended mutation site onto the 5′ and 3′ ends of a selectable marker (Court et al. 2002). Once the homologous fragments have both recombined with the chromosome, all of the intervening chromosomal DNA will be replaced by the selectable marker contained on the mutagenesis construct (Fig. 1B). Unlike single crossover mutations, double crossover allelic replacements are typically stable and do not require selective pressure to maintain the desired genotypes. Allelic replacement strategies can also be adapted for the creation of markerless mutations. Lastly, in the oral commensal species Streptococcus parasanguinis, double crossover allelic replacement mutations have been shown to occur at much higher frequencies than single-crossovers (Fenno et al. 1993). Therefore, when first attempting either single or double crossover mutagenesis in an organism of interest, it is advisable to try both approaches if one fails to yield the desired recombinant.
Figure 1.
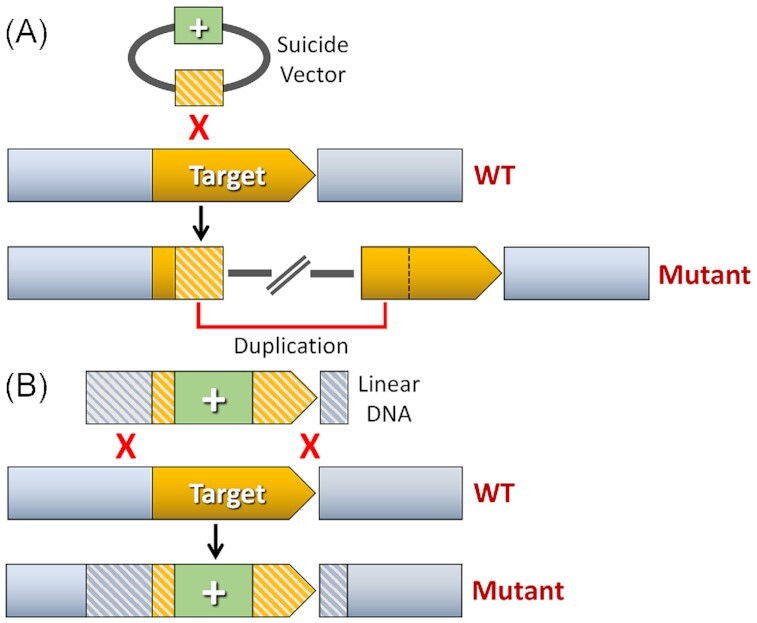
Targeted mutagenesis using homologous recombination. (A) Insertion duplication mutagenesis. An internal homologous fragment (illustrated in orange stripes) of the target gene is ligated to a suicide vector containing a positive selection marker (‘+’; illustrated in green). Transformation of this construct using selective media will isolate clones containing a plasmid insertion within the target gene, disrupting the function of its encoded protein. Insertion of the plasmid also creates a duplication of the homologous fragment on the chromosome. (B) Allelic replacement mutagenesis. Homologous fragments (illustrated in grey and orange stripes) flanking the intended mutation site of the target gene are ligated to the corresponding 5′ and 3′ ends of a positive selection marker (‘+’; illustrated in green). Following transformation of this construct, both homologous fragments recombine with the chromosome, which deletes all of the intervening chromosomal DNA located between the homologous segments and replaces it with the positive selection marker.
Bacterial transformation strategies
The abundance and diversity of oral microbiome transformation literature offers many template protocols that may be adaptable for use in novel and/or understudied microbiome species, with conjugation, electroporation, and natural transformation being the most frequently utilized approaches (Table 1). Conjugation was more commonly employed in the past when fewer transformation options were widely available. Conjugation-based approaches are also more complex to implement and can require considerable optimization. However, conjugation often succeeds in organisms that are recalcitrant to other transformation approaches. Electroporation is the most commonly employed transformation approach and is fairly simple to execute. As discussed later, DNA transformed via electroporation is also more likely to be degraded upon entry into recipient cells. When available, natural transformation is the preferred approach because it normally circumvents the major limitations encountered with conjugation and electroporation. Unfortunately, only a small minority of species have established natural transformation protocols, and there is no guarantee that a novel organism is even capable of natural transformation. Each of these transformation approaches has unique advantages and disadvantages to consider when attempting to create a new transformation protocol.
Table 1.
Transformation strategies in selected oral microbiome species.*
| Species | Conjugation | Electroporation | Natural Transformation |
|---|---|---|---|
| Aggregatibacter actinomycetemcomitans | (Goncharoff et al. 1993, Mintz and Fives-Taylor 2000) | (Sreenivasan et al. 1991) | (Wang et al. 2002) |
| Actinomyces oris | (Yeung and Kozelsky 1994) | ||
| Actinomyces viscosus | (Yeung and Kozelsky 1994) | ||
| Corynebacterium matruchotii | (Luong et al. 2018) | ||
| Capnocytophaga ochracea | (Hosohama-Saito et al. 2016) | ||
| Campylobacter rectus | (Wang et al. 2000) | ||
| Enterococcus faecalis | (Kristich et al. 2007) | (Dunny et al. 1991, Shepard and Gilmore 1995) | |
| Filofactor alocis | (Mishra et al. 2020) | ||
| Fusobacterium nucleatum | (Haake et al. 2000) | ||
| Lactobacillus casei | (Natori et al. 1990) | ||
| Porphyromonas gingivalis | (Dyer et al. 1992, Maley et al. 1992) | (Yoshimoto et al. 1993) | (Tribble et al. 2012) |
| Prevotella intermedia | (Naito et al. 2021) | ||
| Prevotella melaninogenica | (Kondo et al. 2018) | ||
| Parvimonas micra | (Higashi et al. 2022) | ||
| Streptococcus anginosus | (Salvadori et al. 2017) | ||
| Streptococcus cristatus | (Correia et al. 1995) | ||
| Streptococcus gordonii | (Gaustad et al. 1979) | ||
| Streptococcus infantis | (Ween et al. 2002) | ||
| Streptococcus mitis | (Salvadori et al. 2016) | ||
| Streptococcus mutans | (LeBlanc et al. 1978) | (Lee et al. 1989) | (Perry and Kuramitsu 1981) |
| Streptococcus salivarius | (LeBlanc et al. 1978) | (Fontaine et al. 2010) | |
| Streptococcus sobrinus | (Li et al. 2021) | ||
| Treponema denticola | (Li and Kuramitsu 1996) | ||
| Tannerella forsythia | (Honma et al. 2001) | (Honma et al. 2007, Sakakibara et al. 2007) | (Nishikawa and Tanaka 2013) |
| Veillonella atypica | (Liu et al. 2012) | ||
| Veillonella parvula | (Liu et al. 2011) | (Knapp et al. 2017) |
Due to space limitations, this table is not a comprehensive list of all representative studies using oral microbes.
Conjugation
Conjugation refers to the ability of certain bacteria to directly transfer transforming DNA between cells via a specialized pilus encoded by a type IV secretion system (Goessweiner-Mohr et al. 2014, Zechner et al. 2017, Costa et al. 2021). The best characterized conjugation system is encoded by the F plasmid of E. coli. The F plasmid is self-transmissible, meaning it encodes a type IV secretion system as well as the mobilization machinery required to translocate it through the conjugative pilus to a naïve recipient cell (Arutyunov and Frost 2013). If a bacterial cell concurrently hosts additional plasmids together with the F plasmid, these too can be transferred to a recipient cell provided the plasmids contain an origin of transfer compatible with the F plasmid-encoded type IV secretion system (Wong et al. 2012). The origin of transfer is a non-coding segment of DNA that serves as a nick site cleaved by the relaxase enzyme of a type IV secretion system (Wong et al. 2012, Guzman-Herrador and Llosa 2019). The 5′ end of this initial single-stranded DNA (ssDNA) cleavage is subsequently covalently attached to the relaxase enzyme via a phosphotyrosyl linkage (Wong et al. 2012, Guzman-Herrador and Llosa 2019). The plasmid DNA is then unwound and a single DNA strand is transferred to the recipient cell via the conjugative pilus. The ability of conjugative systems to transfer DNA in trans forms the basis for conjugation-based transformation strategies. A variety of conjugative E. coli donor strains have been constructed to facilitate mating with recipient organisms of interest. E. coli strains SM10 and S17-1 are two of the more commonly employed donor strains, as they both host chromosomally integrated versions of the self-transmissible RP4 plasmid, which encodes the requisite conjugation machinery (Strand et al. 2014). When using these strains for conjugation, donor plasmid DNA can be transferred to recipient cells provided that the DNA constructs are cloned onto mobilizable plasmid vectors containing the appropriate oriT origin of transfer (Strand et al. 2014, Ramsay and Firth 2017). Since conjugation reactions contain a mixture of both donor E. coli cells and recipient, it is typically necessary to incorporate a strategy to specifically isolate the transformants and eliminate the donor E. coli. This is often achievable based upon differential antibiotic resistance phenotypes, but in some cases nutritional selection or various growth conditions may be employed. The fact that the conjugation machinery only transfers a single strand of transforming DNA is one of the keys to its success in many organisms. Double-stranded DNA (dsDNA) is subject to efficient cleavage by bacterial restriction enzymes, which typically target DNA lacking host methylation patterns (Pingoud et al. 2005, Youell and Firman 2012, Rao et al. 2014), or in some cases, specifically target DNA containing foreign methylations (Tock and Dryden 2005, Loenen and Raleigh 2014). However, ssDNA and hemimethylated dsDNA are both poor substrates for most restriction enzymes (Tock and Dryden 2005). Therefore, ssDNA transferred via conjugation is usually resistant to restriction as is the hemimethylated dsDNA that is formed after the complementary DNA strand has been synthesized by the conjugation recipient.
Electroporation
As shown in Table 1, electroporation is the most commonly utilized transformation approach for oral microbiome genetic studies, and this is true for most other bacteria as well. Electroporation utilizes a brief, high-intensity electric field pulse to create pores within the cell membranes of target organisms. During the short window of time while these pores are present, DNA (and/or other molecules) can pass through the cell membrane primarily via diffusion (Tryfona and Bustard 2006). If the electroporation parameters are appropriate, these pores will collapse and reseal after the electric field has dissipated without triggering irreparable damage to the cells. Thus, it is normally necessary to optimize the electric pulse parameters to generate the ideal electric field for an individual organism. Most electroporation units allow the user to modulate the field strength, capacitance, and resistance settings for this purpose. To prepare cells for electroporation, cultures are typically washed several times in ice-cold non-ionic buffers that protect against osmotic shock, often glycerol or sucrose-based (Aukrust et al. 1995, McLaughlin and Ferretti 1995, Saulis 1999, Green and Sambrook 2020). These washes severely dilute the free salt concentration within the bacterial suspension, which is essential for maintaining the electric field during electroporation (Tryfona and Bustard 2006). When electroporating obligate anaerobes, which comprise a substantial fraction of the human microbiome (Eckburg et al. 2005, Mark Welch et al. 2016), these preparatory steps will require additional considerations to limit oxidative stress to the bacteria. In addition, most electroporation protocols require the bacteria to remain at 4 °C for the entirety of the procedure. This protects the cells from overheating while in the presence of an electric current and it also helps to stabilize the membrane pores that form in the electric field (Tryfona and Bustard 2006). After the electric pulse has been delivered, cells are first temporarily incubated in nonselective growth medium to both repair cellular damage and to express selection markers before subsequently plating on selective medium to identify the transformants. Typically, cells are grown to mid-logarithmic phase prior to prepping for electroporation, but in some cases, stationary-phase cells may perform better (Wang et al. 2020). It is worth noting that Gram-positive bacteria are frequently cultured in the presence of glycine to weaken the cell wall prior to electroporation (Shepard and Gilmore 1995, Buckley et al. 1999, Ruan et al. 2015, Welker et al. 2015). This step can increase the electroporation efficiency of Gram-positive bacteria by multiple orders of magnitude. It is also important to note that foreign DNA transformed via electroporation is likely sensitive to bacterial restriction enzymes, as electroporated DNA is typically double-stranded. The selectivity of the restriction barrier against foreign dsDNA can be extreme in many organisms, but often times certain strains of a given species will be more or less permissive than others (Welker et al. 2015, Wang et al. 2020). Therefore, it can be useful to test a panel of strains to identify those that may be amenable to electrotransformation.
Natural transformation
Naturally transformable bacteria actively internalize exogenous environmental DNA through a process referred to as natural competence (Dubnau and Blokesch 2019). Many species express their natural competence systems only when exposed to specific growth conditions, and these conditions may be quite challenging to identify (Seitz and Blokesch 2013, Attaiech and Charpentier 2017). For example, chitin-dependent natural competence in Vibrio cholerae was first reported in 2005, decades after widespread genetic studies first began with this organism (Meibom et al. 2005). Chitin is an unobvious culture supplement in the context of human V. cholerae infections, but it does play a major ecological role for V. cholerae within the aquatic environment (Vezzulli et al. 2008). DNA uptake by natural competence systems is achieved via retractable type IV pili or via specialized natural competence-specific pseudopili (Bakkali 2013, Dubnau and Blokesch 2019). Both Gram-positive and Gram-negative bacteria are capable of developing natural competence, and both types of bacteria produce highly homologous DNA uptake apparatuses, despite the exceptionally diverse array of environmental stimuli and sensory systems that regulate their production (Dubnau and Blokesch 2019). Several features of natural competence make this the preferred approach for the transformation of foreign DNA. (i) Natural transformation is simple to execute and normally functions well with both circular and linear DNA, although perhaps not at identical efficiencies (Knapp et al. 2017). Once cells have entered the naturally competent state, one only needs to add transforming plasmids, gDNA, or PCR products to the culture, incubate, and then plate on a selective medium. (ii) Naturally competent bacteria typically couple the degradation of one strand of transforming DNA together with its energy-dependent import into the cell (Dubnau and Blokesch 2019). Consequently, naturally transformed DNA enters the cell single-stranded and is usually resistant to the activity of most restriction enzymes for the same reasons that were previously described for conjugation. Naturally transformed DNA may even be methylated during import, which adds an additional layer of protection against degradation (Johnston et al. 2013). (iii) Many naturally competent bacteria synchronize competence development with the activation of recombination machinery (Okinaga et al. 2010, Kidane et al. 2012, Khan et al. 2016). This point is often overlooked, but it can be one of the principal advantages of natural transformation for targeted mutagenesis. With both conjugation and electroporation, transformed DNA that manages to escape restriction may still fail to recombine with the chromosome if the cell's recombination machinery has not been appropriately primed for action. As the saying goes, ‘you can lead a horse to water, but you can't make it drink’. Simply introducing DNA into a cell does not ensure its subsequent recombination, even if that DNA is resistant to restriction. For naturally competent bacteria, this is rarely an issue. For example, in the naturally competent oral microbe Streptococcus mutans, the constitutively expressed recombinase gene recA contains a second promoter that is only recognized by the natural competence-specific alternative sigma factor ComX (Okinaga et al. 2010). Consequently, recA gene expression is stimulated in S. mutans (and many other naturally competent streptococci) concurrently with the induction of natural competence (Okinaga et al. 2010, Khan et al. 2016). While natural competence is generally considered to be a specialized ability of a small subset of organisms, there is reason to suspect that this ability is far more common than is currently appreciated, perhaps even typical (Kovacs et al. 2009, Attaiech and Charpentier 2017). We have recently demonstrated natural competence from low passage clinical isolates of both Veillonella parvula and Parvimonas micra, two distantly related oral microbiome species that were previously considered to be genetically intractable, but in fact, are highly amenable to genetic manipulation via natural transformation (Knapp et al. 2017, Higashi et al. 2022). In the case of V. parvula, orthologous natural competence loci are widespread within the entire Veillonellaceae family, despite the fact that no additional members of this family have been reported as naturally competent (Knapp et al. 2017). Furthermore, we currently have ongoing studies of several additional ‘genetically intractable’ oral microbiome species for which we have found compelling evidence of natural transformability (unpublished results). If natural competence does indeed prove to be common among prokaryotes, natural transformation could eventually supplant electroporation as the standard transformation technique used for microbiome genetic studies.
Marked mutagenesis
Antibiotic resistance cassettes are the most commonly employed selectable markers used for genetically manipulating the microbiome. Most of the commonly employed resistance cassettes were originally discovered on mobile genetic elements, especially transposons and plasmids (Roberts and Mullany 2011, Clewell et al. 2014, Santoro et al. 2014, Kohler et al. 2018). The majority of the resistance cassettes used for oral microbiome research confer resistance to kanamycin, erythromycin/clindamycin, spectinomycin/streptomycin, chloramphenicol/thiamphenicol, tetracycline, and gentamycin (Table 2). There is also a wide diversity of resistance cassettes available for use with these antibiotics, as most resistance genes only function in a subset of species (Table 2). Therefore, it may be necessary to examine several different resistance cassettes to determine which may be suitable for an organism of interest. In many or most cases, it should be possible to identify useful resistance cassettes for novel organisms by selecting resistance genes previously shown to function in phylogenetically related species (Table 2). Evidently, the species-specificity of some resistance cassettes is simply a consequence of the endogenous resistance cassette promoters, rather than as a functional failure of the encoded resistance proteins. For example, the catA1 gene encoding chloramphenicol resistance was previously widely employed as a reporter gene in numerous organisms (Kain and Ganguly 2001, Arnone et al. 2004). Promoterless versions of the gene were sometimes even included on shuttle vectors to facilitate transcriptional studies (Hudson and Curtiss 1990, Kili et al. 1999). By transcriptionally fusing a constitutive promoter derived from a novel organism, one could conceivably utilize the catA1 open reading frame (ORF) as a chloramphenicol resistance cassette specifically tailored for use in that same organism. This concept was employed to create a novel tetracycline resistance cassette for Veillonella species by fusing a tetM ORF with the constitutively expressed Veillonella atypica DNA gyrase promoter PgyrA (Liu et al. 2012). In the unlikely event that none of the resistance cassettes listed in Table 2 function in a novel organism of interest, replacing a resistance cassette promoter may address the issue. Alternatively, one could easily create synthetic versions of any of these cassettes by having them synthesized with the appropriate codon optimizations and desired promoters. However, if no wild strains of a pathogenic organism of interest have ever been demonstrated to exhibit resistance to a particular antibiotic, it is generally not advisable to introduce an antibiotic resistance cassette for that same antibiotic. This is in the best interests of global public health and responsible antibiotic stewardship.
Table 2.
Antibiotic resistance cassettes used in selected oral microbiome species.
| Antibiotic Resistance | Oral Microbiome Species | Reference |
|---|---|---|
| Gentamycin, Kanamycin | ||
| aacA-aphD | S. gordonii, S. mitis | (Behnke et al. 1981, Johnsborg et al. 2008) |
| Gentamycin | ||
| aacC1 | T. denticola | (Yang et al. 2008) |
| Spectinomycin/Streptomycin | ||
| aad9 | A. actinomycetemcomitans, C. rectus, E. faecalis, S. anginosus, S. cristatus, S. gordonii, S. mutans, S. salivarius, S. sobrinus | (LeBlanc et al. 1991, Sreenivasan et al. 1991, Correia et al. 1995, Higuchi et al. 1999, Wang et al. 2000, Upton et al. 2001, Shinozaki-Kuwahara et al. 2005, Li et al. 2021) |
| strA | A. viscosus, A. oris | (Yeung and Kozelsky 1994) |
| Kanamycin | ||
| aph3Ia | A. actinomycetemcomitans, C. matruchotii | (Brogan et al. 1996, Takayama et al. 2003) |
| aphA7 | S. gordonii | (Tenover et al. 1992) |
| aphAII | A. viscosus, A. oris, T. denticola | (Yeung and Kozelsky 1994, Li et al. 2015) |
| aphAIII | E. faecalis, P. micra, S. anginosus, S. cristatus, S. gordonii, S. mutans, S. salivarius, S. sobrinus | (Dunny et al. 1991, Buckley et al. 1995, Correia et al.1996, Gutierrez et al. 1996, Weaver et al. 2000, Petersen et al. 2006, Li et al. 2021, Higashi et al. 2022) |
| Chloramphenicol/Thiamphenicol | ||
| catA1 | A. actinomycetemcomitans, P. gingivalis, T. forsythia | (Goncharoff et al. 1993, Shi et al. 1999, Sakakibara et al. 2007) |
| catA7 | S. gordonii | (Macrina et al. 1980) |
| catA9 | S. infantis, S. mitis, S. sobrinus, T. denticola | (Ween et al. 2002, Slivienski-Gebhardt et al. 2004, Johnsborg et al. 2008, Li et al. 2021) |
| catP | F. nucleatum, L. casei, V. parvula | (Perez-Arellano et al. 2001, Kaplan et al. 2005, Bechon et al. 2020) |
| Erythromycin/Clindamycin | ||
| ermB | E. faecalis, L. casei, P. micra, S. anginosus, S. cristatus, S. gordonii, S. mitis, S. mutans, S. salivarius, T. denticola | (LeBlanc and Hassell 1976, LeBlanc et al. 1978, Natori et al. 1990, Fukushima et al. 1992, Correia et al. 1996, Bryan et al. 2000, Chen et al. 2000, Johnsborg et al. 2008, Goetting-Minesky and Fenno 2010, Higashi et al. 2022) |
| ermF | C. ochracea, F. alocis, F. nucleatum, P. gingivalis, P. intermedia, P, melaninogenica, T. forsythia | (Hoover et al. 1992, Haake et al. 2000, Honma et al. 2007, Hosohama-Saito et al. 2016, Kondo et al. 2018, Mishra et al. 2020, Naito et al. 2021) |
| Tetracycline | ||
| tetL | E. faecalis, S. gordonii | (Burdett 1980, Banai and LeBlanc 1983) |
| tetM | S. mitis, S. mutans, V. atypica, V. parvula | (Lindler and Macrina 1986, Hannan et al. 2010, Liu et al. 2012, Knapp et al. 2017) |
| tetQ | P. gingivalis, T. forsythia | (Maley et al. 1992, Honma et al. 2001) |
It is worth mentioning that marked mutations sometimes create unwanted artifacts complicating the interpretation of mutant phenotypes. The most common of these is a polar effect, which is an altered expression of genes downstream of an inserted element like a selectable marker (Cherepanov and Wackernagel 1995). Since resistance cassettes and other selectable markers typically contain constitutive promoters, transcriptional read-through from these genes can trigger the overexpression of downstream genes, unless transcription terminators have been incorporated into the cassettes. However, the inclusion of transcription terminators can also prevent transcriptional read-through that might normally occur within a locus, such as within operons. It is possible to circumvent both types of polar effects by only utilizing the ORF of a particular marker (i.e. no promoter or transcription terminator) (Bian et al. 2012). However, this approach will only function in a locus with sufficient basal expression to ensure that the marker ORF can confer continual selection. Antibiotic resistance cassettes may also trigger additional artifacts due to the enzymatic functions of the encoded resistance proteins. Most antibiotic resistance is conferred through enzymes that either modify components of the host cell or modify the antibiotic. For example, erythromycin resistance is typically conferred by specific methylases targeting the ribosome, whereas kanamycin and various other aminoglycoside antibiotics are directly inactivated via O-phosphorylation or other modifications (Davies and Wright 1997, Wright and Thompson 1999, Golkar et al. 2018). During normal growth, these activities rarely cause noticeable deleterious effects. However, we have encountered situations where some mutant phenotypes were only observable when using particular antibiotic resistance markers (unpublished results). One can usually mitigate all of these aforementioned issues by creating markerless mutations.
Markerless mutagenesis
As the name suggests, markerless mutations do not leave selectable marker genes on the chromosome. These types of mutations are similarly targeted via homologous recombination and can be employed for the creation of all types of desired mutations, including deletions, insertions, and point mutations. However, markerless mutations are far less prone to artifactually impacting a mutant phenotype because they do not encode foreign transcription regulatory elements or enzymes. Markerless mutations are also particularly useful when multiple mutations are to be engineered within a single strain, as the same mutagenesis approach can be repeatedly administered to generate a virtually limitless number of unique mutations (Xie et al. 2011). With marked mutagenesis, the number of mutations that can be engineered within a strain is inherently limited by the number of available selectable markers. The vast majority of markerless mutations are created using an initial positive selection step to insert a mutagenesis construct onto the chromosome followed by a subsequent recombination-based approach to remove the selectable marker, yielding the final markerless mutant strain (Fig. 2A-C). Markerless mutations made in this manner can be engineered via a counterselection-based approach or with a site-specific recombinase. Counterselection typically employs a combination of both positive and negative selection markers, which are often combined into a single counterselectable cassette (Reyrat et al. 1998, Xie et al. 2011). Unfortunately, there are very few negative selection markers available for use in wild-type bacteria, which is an inherent limitation to this approach. For species that lack the galK (galactokinase) or sacB (levansucrase) genes, either of these can be utilized as negative selection markers to confer sensitivity to media supplemented with galactose or sucrose, respectively (Table 3). Both markers similarly trigger the toxic accumulation of carbohydrates in species that lack the appropriate catabolic enzymes (Ueki et al. 1996, Reyrat et al. 1998). Thus, negative selection with these genes is most often useful for asaccharolytic organisms, as many saccharolytic species naturally encode galK and/or sacB as well as the corresponding downstream catabolic machinery (Merritt et al. 2007). Recently, a more universal negative selection approach was developed based upon acquired sensitivity to the amino acid analog p-chloro-phenylalanine (4-CP). This approach utilizes a mutant form of the endogenous pheS gene, encoding the highly conserved phenylalanyl-tRNA synthetase alpha subunit (Kast and Hennecke 1991). To utilize 4-CP negative selection, one only needs to engineer the equivalent of an E. coli PheS A294G mutation using an ectopic copy of the endogenous pheS gene from an organism of interest (Kristich et al. 2007, Xie et al. 2011). In some cases, it can be advantageous to also introduce a second PheS mutation corresponding to T251A, which further increases the sensitivity to 4-CP (Miyazaki 2015, Zhang et al. 2017). Both mutant forms of PheS have relaxed stringencies relative to the wild-type version, sensitizing the cells to aminoacylation with 4-CP and subsequent pleiotropic translational defects. Even though this approach is theoretically adaptable for use in most bacteria, it is important to note that individual mutant pheS genes may need to be created for each particular species of interest. For example, we have found that a mutant pheS gene from S. mutans loses its selectivity when used in other closely related oral streptococci, whereas negative selection cassettes derived from the endogenous pheS genes function well in those organisms (Xie et al. 2011, Cheng et al. 2018, Hall et al. 2019). It is also worth noting that fluorescent proteins can be employed as alternatives to negative selection markers for counterselection (Table 3). This approach circumvents the limited availability of negative selection markers for markerless mutagenesis and should be broadly applicable for aerotolerant organisms. While this approach only yields a small fraction of colonies having the desired mutant genotype, the loss of colony fluorescence provides a practical mechanism to identify the correct mutants among thousands of colonies (Wu and Ton-That 2010, Vickerman et al. 2015).
Figure 2.
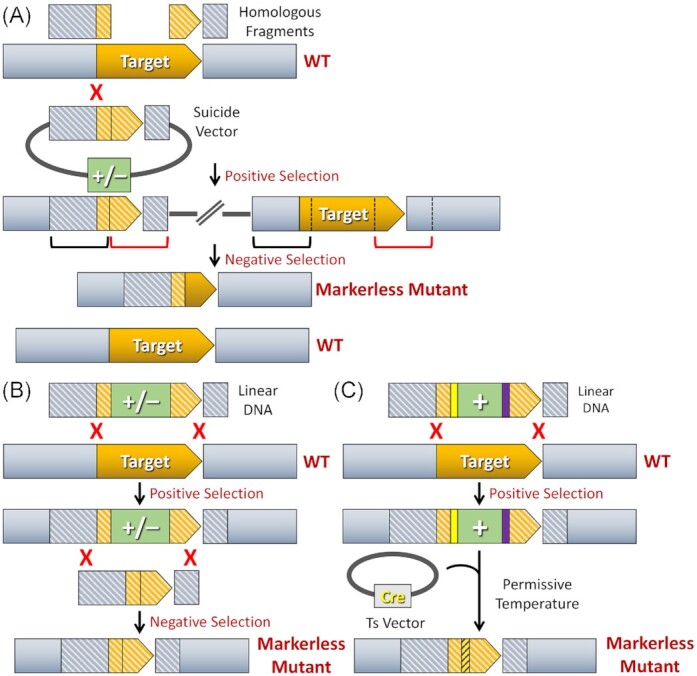
Markerless mutagenesis strategies. (A) Counterselection with insertion duplication mutagenesis. Two equally sized homologous fragments (illustrated in grey and orange stripes) flanking the intended mutation site are cloned adjacent to each other on a suicide vector containing both positive and negative selection markers (‘+/–’; illustrated in green). Following transformation of the construct, one of the two fragments will randomly insert to the chromosome via single crossover homologous recombination. The same final outcome is achieved irrespective of which of the two homologous fragments recombines. Therefore, only one option is illustrated. Since the suicide vector contains two homologous fragments, both of these segments will be duplicated on the chromosome after the plasmid has inserted (as indicated by the red and black brackets). Negative selection is used to isolate clones in which these homologous segments have randomly recombined to excise the inserted vector from the chromosome. In this example, a markerless mutant will be created following a recombination event between the homologous segments marked by red brackets. However, if recombination occurs between the homologous segments marked by black brackets, a wild-type genotype will result. Consequently, counterselection with insertion duplication mutagenesis yields a mixed population of clones consisting of 50% mutant and 50% wild-type genotypes. (B) Counterselection with allelic replacement mutagenesis. Two homologous fragments flanking the intended mutation site of the target gene are ligated to the corresponding 5′ and 3′ ends of a counterselection cassette (‘+/–’; illustrated in green). Following transformation of this construct, both homologous fragments recombine with the chromosome, which deletes all of the intervening chromosomal DNA between the homologous segments and replaces it with the counterselection cassette. The resulting strain is then transformed with an unmarked mutagenesis construct and subjected to negative selection to isolate the double crossover recombinants that have deleted the counterselection cassette. All of the resulting transformants should contain the desired markerless mutant genotype. (C) Recombinase-mediated markerless mutagenesis. A typical double crossover allelic replacement construct is assembled using a positive selection cassette (‘+’; illustrated in green) flanked by two Cre recombinase-dependent loxP sites (illustrated in yellow and purple). The allelic replacement mutant is next transformed with a temperature sensitive plasmid encoding the cre gene. Growth at the permissive temperature supports plasmid replication and ectopic production of the Cre recombinase. After a predetermined number of generations, the cells are shifted to the non-permissive temperature to trigger loss of the temperature sensitive cre expression plasmid. Plasmid-free clones are finally screened to identify those that have undergone Cre-mediated excision of the antibiotic cassette. Strains exhibiting the markerless mutant genotype will also retain a hybrid loxP site (illustrated in yellow and purple stripes) created via Cre-mediated recombination between the two original loxP sites.
Table 3.
Markerless mutagenesis in selected oral microbiome species.
| Species | Counterselection | Recombinase | Reference |
|---|---|---|---|
| A. actinomycetemcomitans | SacB (Levansucrase) | Cre-loxP | (Fujise et al. 2008, Juarez-Rodriguez et al. 2013) |
| A. oris | mCherry (Fluorescence) | (Wu and Ton-That 2010) | |
| C. matruchotii | SacB (Levansucrase) | (Luong et al. 2018) | |
| E. faecalis | PheS (Phe tRNA synth. α) | (Kristich et al. 2007) | |
| F. nucleatum | GalK (Galactokinase) | (Peluso et al. 2020) | |
| L. casei | Cre-loxP | (Xin et al. 2018) | |
| S. anginosus | Cre-loxP | (Bauer et al. 2018) | |
| S. gordonii | mTFP1 (Fluorescence), PheS (Phe tRNA synth. α) | (Vickerman et al. 2015, Hall et al. 2019) | |
| S. mutans | PheS (Phe tRNA synth. α) | Cre-loxP | (Banerjee and Biswas 2008, Xie et al. 2011) |
| V. atypica | PheS (Phe tRNA synth. α) | (Zhou et al. 2015) |
To create markerless mutations using counterselection, one can employ either insertion-duplication or double crossover allelic replacement mutagenesis (Fig. 2A and B). As previously mentioned, it is also possible to employ site-specific recombinases like Cre to remove selectable markers from the chromosome in lieu of negative selection (Banerjee and Biswas 2008). Cre-mediated excision of selectable markers requires them to be flanked by the appropriate loxP sequences (Fig. 2C). The two principal limitations to this approach are its requirement for ectopic cre expression as well as the loxP sites themselves, which are not completely removed from the chromosome following Cre-mediated excision (Van Duyne 2015). The presence of loxP scars on the chromosome can be problematic for the creation of precise gene truncations, point mutations, and fusion proteins, but they are generally not an issue for typical gene inactivation mutations. The most facile approach to create markerless mutations is to directly transform unmarked mutagenesis constructs. This approach is only practical for organisms with exceptionally high transformation efficiencies like many naturally competent streptococci because mutant identification usually requires PCR screening and/or sequencing (Junges et al. 2017). For example, using highly optimized transformation conditions for S. mutans, it is possible to achieve transformation rates of up to 60% of the population, which makes mutant identification a relatively painless process even without the aid of selectable markers (Morrison et al. 2015). It is also worth noting that markerless mutagenesis with CRISPR/Cas technology has been recently demonstrated in S. mutans (Gong et al. 2018). In the coming years, this approach may provide new mutagenesis options for many microbiome species, especially for those organisms that are difficult to manipulate using the traditional approaches (Ramachandran and Bikard 2019).
Reporter genes
Reporter genes are a fundamental component of any robust genetic system and numerous reporter systems have been adapted for use in oral microbiome research (Table 4). Depending upon the type of reporter gene fusion, it can be employed to measure RNA abundance, mRNA stability/translation, or protein abundance. Transcriptional reporter gene fusions are the most commonly employed and can be created using two approaches. The typical method is to fuse a copy of the promoter region of a gene of interest to an independently translated reporter gene ORF (Kreth et al. 2004, Hughes and Maloy 2007). The reporter fusion can either be integrated within the same locus as the target gene or inserted at an ectopic location on the chromosome. In either case, care should be taken to ensure that the construct does not create downstream polar effects due to unnatural transcriptional read-through or termination. In our experience, some particularly sensitive genes may also exhibit abnormal expression patterns if a promoter is duplicated (unpublished results). These issues are much less likely to occur if transcription fusions are made via the second approach: creation of an artificial operon with a target gene of interest. One can insert an independently translated reporter gene ORF upstream or downstream of a target gene such that it will be included within the same transcript, thus eliminating problems of downstream polar effects or promoter duplication (Zou et al. 2018, Qin et al. 2021). Regardless of the method employed to create a transcription fusion reporter strain, it should be noted that such constructs may be limited in their abilities to accurately measure down-regulation of gene expression. Each unique reporter enzyme has a characteristic turnover rate when expressed in an organism. If a reporter enzyme is particularly stable (i.e. slow turnover rate), it may continue to function long after its encoding gene has ceased to produce new proteins. In such instances, there can be significant discrepancies between target gene RNA abundance and the measured reporter signal. To determine whether this is an issue, one can simply compare the output of a reporter strain with qRT-PCR analysis of the target gene. For reporter studies examining changes in mRNA stability and/or translation, especially via regulatory elements encoded within 5′ untranslated regions (UTRs), one can compare the activity of a reporter construct containing a target gene 5′ UTR fusion vs. a transcription start site (+1 site)-reporter ORF fusion (Lenz et al. 2004). If different reporter activities are observed ± 5′ UTR, there is a strong likelihood that the target gene is regulated through a posttranscriptional mechanism. Normally, this type of reporter construct would also include the endogenous Shine-Dalgarno sequence found in the target gene because many common posttranscriptional regulatory elements modulate translation efficiencies by controlling access to ribosome binding sites located within 5′ UTRs (Merritt et al. 2014, Kreth et al. 2015, Meyer 2017, Evguenieva-Hackenberg 2021). Reporter genes can also be employed to interrogate protein abundance. The most direct approach is to create a chimeric fusion protein between a target protein of interest and a reporter (Hughes and Maloy 2007). For such constructs, we normally include a flexible poly-aspartate-serine (DSS) linker between the two proteins to limit potential steric interference that may affect the proper folding of the chimera (Marx et al. 2020). Ideally, such fusion protein reporter constructs should also be examined to determine whether the chimera retains the wild-type target protein function, as functional defects can sometimes feedback into target protein abundance (e.g. autoregulatory transcription factor).
Table 4.
Reporter proteins used in selected oral microbiome species.
| Species | Chromogenic | Fluorescent | Bioluminescent | Reference |
|---|---|---|---|---|
| A. actinomycetemcomitans | LacZ | GFP, dsRed | (Kolodrubetz et al. 1996, Lippmann et al. 1999, Maula et al. 2021) | |
| A. oris | mCherry | (Wu and Ton-That 2010) | ||
| E. faecalis | LacZ | GFP, mCherry, mTFP1 | Firefly, Bacterial | (Grissom-Arnold et al. 1997, Simon et al. 2001, Day et al. 2003, Hancock et al. 2003, Garcia-Cayuela et al. 2012, Vickerman et al. 2015) |
| L. casei | GusA | GFP, Evoglow | Bacterial | (Gosalbes et al. 1999, Oozeer et al. 2002, Perez-Arellano and Perez-Martinez 2003, Martinez-Fernandez et al. 2019) |
| P. micra | Renilla | (Higashi et al. 2022) | ||
| P. gingivalis | LacZ | Evoglow | Bacterial | (Lu and McBride 1998, Liu et al. 2000, Choi et al. 2011) |
| S. anginosus | mCherry, mTFP1 | (Vickerman et al. 2015) | ||
| S. gordonii | LacZ | Citrine, dsRed, GFP, mCherry, mTFP1 | Cypridina, Firefly, Luciola, Renilla | (Hansen et al. 2001, Loeliger et al. 2003, McCormick et al. 2011, Tao et al. 2011, Vickerman et al. 2015, Merritt et al. 2016, Shields et al. 2019) |
| S. mitis | mCherry, mTFP1 | Firefly | (Vickerman et al. 2015, Salvadori et al. 2016) | |
| S. mutans | LacZ, GusA | BFP2, Citrine, dsRed, GFP, mCherry, mRFP1, mTFP1 | Cypridina, Firefly, Luciola, Renilla | (Goodman and Gao 1999, Cvitkovitch et al. 2000, Yoshida and Kuramitsu 2002, Kreth et al. 2004, Tian et al. 2013, Li et al. 2015, Reck et al. 2015, Vickerman et al. 2015, Merritt et al. 2016, Shields et al. 2019) |
| T. denticola | LacZ | GFP | (Girons et al. 2000) | |
| V. atypica | Firefly | (Zhou et al. 2016) |
Reporter genes also play a critical role in measuring protein-protein interactions, especially via the two-hybrid assay. Two-hybrid assays require an ectopic host (typically Saccharomyces cerevisiae or E. coli) to simultaneously express two chimeric protein fusions between the test proteins of interest and separate halves of a reporter protein, which for S. cerevisiae is the Gal4 transcription factor, while adenylate cyclase is used in E. coli (Battesti and Bouveret 2012, Paiano et al. 2019). If a positive interaction occurs between the two proteins of interest, it will reconstitute the split reporter protein fragments fused to the two proteins, leading to a measurable reporter signal produced by the host cell. As a general rule, the strength of the two-hybrid reporter signal correlates with the strength of the interaction between the assayed proteins. A stronger interaction (i.e. more stable) will yield a greater response from the reporter and vice-versa. Two-hybrid studies have been employed in a variety of oral microbes using both S. cerevisiae (Baev et al. 2000, Seepersaud et al. 2010) and E. coli (Tian et al. 2013, Dou et al. 2021, Lara Vasquez et al. 2021) as host organisms. It is worth noting that the two-hybrid approach can also be adapted as a genetic screening tool to identify novel protein interactors and/or reveal protein interactomes (Parrish et al. 2006, Kondo et al. 2010). A more recent variation on the two-hybrid concept called bimolecular fluorescence complementation (BiFC) has been developed for microscopy-based studies of protein-protein interactions (Miller et al. 2015). BiFC is performed using a split fluorescent protein as the reporter. A major advantage of this approach is that it can be performed directly within an organism of interest, rather than in an ectopic host like the classic yeast and bacterial two-hybrid assays. Positive interactions between the test protein chimeras will reconstitute the fluorescent protein, leading to fluorescence that can be detected via microscopy. When combined with superresolution microscopy, a single BiFC experiment can be used to interrogate both protein-protein interactions as well as their subcellular contexts (Lu et al. 2019, Xie et al. 2020).
The majority of reporter genes commonly used for oral microbiome genetic studies can be grouped into three categories: chromogenic, fluorescent, and bioluminescent (Table 4). Chromogenic reporter genes are most often employed for blue/white screening on agar plates, which is particularly powerful when paired with library-scale genetic screens. The most commonly utilized chromogenic reporter is the beta-galactosidase-encoding gene lacZ. When lacZ-expressing bacteria are plated onto media supplemented with 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal), colonies will produce a blue precipitate, the intensity of which is proportional to LacZ abundance in the organism (Juers et al. 2012). Many saccharolytic bacteria naturally encode the lacZ gene, which can limit the utility of X-gal selection in these organisms. Consequently, the beta-glucuronidase-encoding gene gusA is frequently used as an alternative for blue/white screening (Chary et al. 2005). GusA will catalyze the formation of a similar blue precipitate on media supplemented with 5-bromo-4-chloro-3-indolyl-β-D-glucuronic acid (X-gluc) (Kreth et al. 2004, Chary et al. 2005). For both chromogenic substrates, the development of blue precipitate is critically dependent upon an oxidation reaction that occurs subsequent to the LacZ- or GusA-mediated cleavage of X-gal or X-gluc (Kiernan 2007). Consequently, reporter strains must be exposed to oxygen to allow the blue precipitate to develop. This requirement can complicate the use of blue/white screening with obligate anaerobes. For reporter assays performed in liquid cultures, fluorescent and bioluminescent reporters are typically preferred, even though LacZ and GusA activity can be measured in liquid as well. When visual assays are to be performed, such as microscopy-based imaging, fluorescent reporters are the optimal choice due to their bright signals that are easily captured via camera. A broad palette of different colored fluorescent proteins has been used for oral microbiome research and multiplexing with two or more fluorescent reporters is fairly common (Table 4), as discussed in greater detail in the Molecular Ecology review in this issue. Most of the popular fluorescent proteins like green fluorescent protein (GFP) and its derivatives have an obligate requirement for oxygen to complete the formation of the protein chromophores (Remington 2006, Craggs 2009). Excitation of these proteins also yields reactive oxygen species that can potentially impact the physiology of the reporter strain (Remington 2006). These issues are often highly problematic for the study of obligate anaerobes. Recently, a new class of flavin mononucleotide (FMN)-based fluorescent protein (FbFP) has been developed to circumvent the strict oxygen requirement of most other fluorescent proteins (Chia et al. 2019). These proteins, now referred to as evoglow® proteins, are commercially available and have been adapted for use in anaerobic imaging studies of a small number of oral microbiome species (Table 4). For quantitative reporter studies, luciferase-based reporters offer the greatest precision and dynamic range. Due to the superior signal-to-noise ratio of luciferases, we are able to accurately quantitate luciferase data in cultures containing as few as several hundred bacterial cells (Merritt et al. 2016, Higashi et al. 2022). Thus, luciferases are ideally suited for low-volume, high-throughput applications in addition to routine reporter studies (Merritt et al. 2005, Syed and Anderson 2021). Unlike fluorescent proteins, luciferases require specific substrates for light generation, which is the key to their exceptional signal-to-noise performance (Syed and Anderson 2021, Zambito et al. 2021). Luciferases are available with a range of different emission spectra and distinct substrate requirements, so there are a variety of approaches available for multiplexing (Merritt et al. 2016, Kreth et al. 2020, Zambito et al. 2021). The two most commonly used luciferase substrates, d-luciferin and coelenterazine, are both membrane-permeable, and as discussed further the Molecular Ecology review, these can both be repeatedly administered to cultures for temporal whole-cell reporter assays (Merritt et al. 2016, Kreth et al. 2020). For studies employing the bacterial luciferase operons (luxCDABEG) of Photorhabdus luminescens or bioluminescent Vibrio species, the luciferase substrates are produced in situ and therefore do not need to be added exogenously (Syed and Anderson 2021). It is worth noting that all known luciferases create bioluminescence through an oxidative mechanism (Widder 2010, Adams and Miller 2020). Thus, these reporters are similarly dependent upon oxygen to function. However, in stark contrast to most fluorescent protein reporters, we have had no difficulties to measure luciferase signals immediately after moving cultures from an anerobic chamber into ambient air for measurement (Higashi et al. 2022). Likewise, luciferase reporter assays were successfully performed using anaerobically grown cultures of Veillonella atypica (Zhou et al. 2016). Even though oxygen is essential for bioluminescence, the concentration required for luciferase catalysis is apparently quite low, which may also explain why luciferases are commonly produced by marine invertebrates of the deep ocean where the conditions are nearly anoxic (Rees et al. 1998).
Regulated gene expression systems
There are a variety of situations in which genetic studies may benefit from or even require exogenous regulatory control of a gene of interest. One of the more common uses is for correlating target gene expression with the severity of a phenotypic output (Bertram et al. 2021). If controlled changes in target gene expression yield proportional changes in a measurable phenotype, one can establish a clear genetic linkage between the two. Additionally, regulated gene expression systems are crucial for the study of essential genes due to the limited options for knock-out mutagenesis. With a regulated gene expression system, it is possible to create a conditional lethal phenotype using transcriptional depletion, thus circumventing the problem of essentiality (Liu et al. 2017, Shields et al. 2020, Bertram et al. 2021). A transcriptional depletion approach is also far easier to engineer and study compared to the classic approach utilizing temperature-sensitive mutations.
Most regulated gene expression systems function via transcriptional control of inducible promoters, especially those promoters controlled by small molecule-sensing transcription repressors (Kim et al. 2020). The efficacy of such systems is determined by the utility of the inducer in an organism of interest as well as the expression characteristics of the regulated promoter. Ideally, an inducer should be non-toxic to the recipient organism, easy to acquire (i.e. commercially available), and efficient to administer (i.e. favorable uptake kinetics). For a regulated promoter, the key characteristic is its dynamic range of expression, which is simply a function of the difference in expression levels between the basal uninduced state of the promoter and its maximal expression when fully induced (Xie et al. 2013, Bertram et al. 2021). In most cases, it is the expression characteristics of the inducible promoter that determine the utility of the system. For example, exogenous control of a toxic gene product would likely require an expression system with the lowest basal expression, whereas a study aiming to determine a maximal phenotypic response would likely benefit from a system yielding the strongest induced target gene expression (Xie et al. 2013). Of the regulated gene expression systems employed in oral microbes, the majority utilize carbohydrates, peptides, or tetracycline as the inducer molecules (Table 5).
Table 5.
Regulated gene expression systems of selected oral microbiome species.
| Species | Type* | Inducer | Reference |
|---|---|---|---|
| A. actinomycetemcomitans | Trx | IPTG | (Bhattacharjee et al. 2007) |
| E. faecalis | Trx | Nisin | (Bryan et al. 2000) |
| Trx | Agmatine | (Linares et al. 2014) | |
| Trx | PQ Pheromone | (Weaver et al. 2017) | |
| L. casei | Trx | Temperature | (Binishofer et al. 2002) |
| Trx | Lactose | (Perez-Arellano and Perez-Martinez 2003) | |
| Trx | Nisin | (Martin et al. 2004) | |
| Trx | Chloride | (Chang and Yan 2014) | |
| Trx | Bile | (Martinez-Fernandez et al. 2019) | |
| P. micra | Post-Trx | Theophylline | (Higashi et al. 2022) |
| S. gordonii | Trx | Sucrose, Tetracycline | (Mallaley et al. 2006) |
| Trx | Xylose | (Xie et al. 2013) | |
| S. mutans | Trx | Sucrose | (Baev et al. 1999) |
| Trx | Tetracycline | (Wang and Kuramitsu 2005) | |
| Trx | Lactose | (Xie et al. 2010) | |
| Trx | Xylose | (Xie et al. 2013) | |
| Post-Trl | Xylose | (Liu et al. 2017) |
*Abbreviations: Trx (transcriptional), Post-Trx (posttranscriptional), Post-Trl (posttranslational)
It is important to note that transcriptional regulation is inherently leaky. Consequently, prokaryotic gene expression is normally regulated at multiple levels to ensure a precise control over protein abundance. Recently, some of these posttranscriptional regulatory mechanisms have been coopted as tools to improve the performance of regulated gene expression systems. One example is the riboswitch. A riboswitch is a small molecule-binding aptamer encoded within the 5′ UTR of an mRNA that is responsible for posttranscriptionally regulating gene expression by modulating downstream transcription termination and/or translation (Pavlova et al. 2019, Turnbough 2019). The key to riboswitch function is its RNA secondary structure, which changes in the presence or absence of the particular ligand bound by the riboswitch aptamer. Riboswitches can be useful additions to a genetic system because they tend to be modular, meaning they can be engineered onto many target mRNAs, and they are also normally quite small. Some riboswitches can be synthesized as part of a primer and directly incorporated onto a construct with PCR. We recently demonstrated the utility of the theophylline riboswitch as part of a new genetic system for the understudied oral microbe P. micra (Higashi et al. 2022). Theophylline is a small molecule from the xanthine family that is commonly found in nature and is nontoxic to most organisms (Wrist et al. 2020). By introducing several point mutations into this riboswitch, we were able to further improve its dynamic range of regulation by an order of magnitude in an improved version that we refer to as the Theo + riboswitch (Fig. 3) (Higashi et al. 2022). While we successfully employed this riboswitch as a standalone regulatory element, one could also combine a riboswitch with a transcription-based gene regulatory system to multiply the overall dynamic range of the system (Kato 2020). Antisense RNAs provide another strategy to incorporate posttranscriptional gene regulation. By expressing a sequence complementary to a gene of interest, it is possible to inhibit translation of a target mRNA without directly altering its transcription. This approach is potentially useful for the study of essential genes, especially when the antisense RNA is placed under the control of a transcription-based regulated gene expression system (Sato et al. 1998, Wang and Kuramitsu 2005). In addition to posttranscriptional regulatory elements, it is also possible to directly modulate protein abundance using a tunable proteolysis system. These systems are particularly useful when a wild-type gene expression pattern is required for a study and/or when studying an essential gene. While the previously described transcriptional depletion approach or an antisense RNA could be employed to control an essential gene, both approaches may function poorly for genes encoding proteins with high intrinsic stability and slow turnover rates (Liu et al. 2017). In such cases, target protein abundance may only exhibit nominal reductions after initiating its depletion. With a tunable proteolysis system, target gene expression is typically unaffected, whereas target protein abundance is specifically and rapidly depleted. In S. mutans, a tunable proteolysis system was developed using a hybrid N-terminal protein tag consisting of the small ubiquitin-like protein NEDD8 fused to an endogenous S. mutans degron (Liu et al. 2017). A degron is a short amino acid motif that targets a protein for proteolysis (Izert et al. 2021). Once this chimeric tag is added to a target protein of interest, one can ectopically express the highly specific NEDP1 endopeptidase to expose the buried degron, which immediately directs the target protein for rapid and potent Clp- and/or FtsH-dependent proteolysis (Fig. 4). In this system, production of the NEDP1 endopeptidase was placed under the transcriptional control of the xylose-inducible expression cassette Xyl-S1 (Xie et al. 2013, Liu et al. 2017). Thus, target protein abundance can be exogenously modulated by the addition of xylose without directly altering the expression of its encoding gene.
Figure 3.

Comparison of the original vs. Theo + theophylline riboswitches. (A) Unmodified theophylline riboswitch. The secondary structure of the ligand-free theophylline riboswitch was calculated using the mFold webserver (http://www.unafold.org/mfold/applications/rna-folding-form.php) (Zuker 2003). In the absence of free theophylline, the Shine-Dalgarno sequence (bold, red font) in the mRNA is sequestered within the secondary structure of the riboswitch, which prevents translation initiation at the downstream initiation codon (bold, green font). Upon binding theophylline, the riboswitch aptamer will adopt an alternate conformation (not pictured) that releases the Shine-Dalgarno sequence from sequestration, thus promoting translation initiation of the mRNA. (B) Theo + riboswitch. The secondary structure of the ligand-free Theo + riboswitch was calculated using the mFold webserver (http://www.unafold.org/mfold/applications/rna-folding-form.php) (Zuker 2003). The predicted secondary structure is nearly identical to the original theophylline riboswitch, except that it contains several point mutations (bold, blue font) that greatly improve its dynamic range of regulation.
Figure 4.
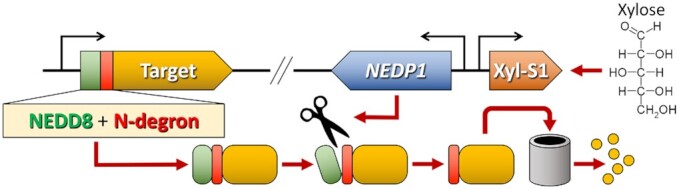
Tunable proteolysis in S. mutans. To engineer tunable proteolysis onto a target protein, the corresponding target gene (orange) is fused to a chimeric tag encoding the small ubiquitin-like protein NEDD8 (green) followed by an endogenous S. mutans degron (red). The protein chimera will remain stable until the NEDD8-specific endopeptidase NEDP1 (scissor icon) is produced. In this system, NEDP1 is ectopically expressed under the transcriptional control of the Xyl-S1 cassette (brown), which is induced by the sugar xylose. In the presence of xylose, repression of NEDP1 is relieved, NEDP1 is produced, and NEDD8 is subsequently cleaved from the target protein. This exposes the N-degron at the new N-terminus of the protein, which targets the protein for highly efficient Clp- and/or FtsH-mediated proteolysis.
Forward genetic screens
Most of the genetic tools described thus far are employed for reverse genetics (i.e. connecting phenotype to genotype). By mutating or expressing a gene of interest, one can reveal its function through the resulting phenotypes. However, in many instances, a phenotype of interest is already known, but the encoding gene(s) are not. For these situations, forward genetic screens can be immensely valuable tools for gene discovery. It is important to note that the success of a forward genetic screen is critically dependent upon the ability of a phenotype to unambiguously reveal candidate mutants of interest among thousands of bacterial colonies (Shuman and Silhavy 2003). If available, this is the ideal scenario to employ chromogenic reporter genes for a particular phenotype. The most common approach for forward genetic screening is the random insertion genetic library. These libraries are typically assembled using two strategies. The first was more commonly employed in the past and involves cloning a library of random chromosomal fragments into a suicide vector. These plasmids are then pooled and transformed into an organism of interest to collect a library of mutant strains containing random plasmid insertions created via single crossover homologous recombination (Colby et al. 1995, Polissi et al. 1998). As a general rule, one should target a library of mutants with at least a 3-fold coverage of the genome, meaning the sum total of the chromosomal fragments contained in the plasmid library is at least 3x that of the total length of the bacterial chromosome. These libraries are usually laborious to construct because they require classic cloning approaches to cut and ligate random genomic DNA fragments into plasmid vectors. In the past, plasmid libraries were also often limited by the biases created by the restriction enzymes used to construct the libraries. However, library creation was substantially improved with the introduction of the restriction enzyme CviKI (Tsang et al. 2005). This enzyme is blunt-end cutter with a flexible 4-base consensus consisting of RG/CY. The extreme variability of its consensus renders this enzyme as a nearly random cutter for most genomes, yielding blunt-ended fragments immediately available for cloning into a plasmid library. The principal limitation for the plasmid insertion mutant library is that many genes are difficult to inactivate via single crossover mutagenesis, especially small genes. Plasmid insertions also create significant polar effects that may need to be further examined to determine the actual genetic source of a mutant phenotype. The second common approach to create random insertion libraries is via transposon mutagenesis. A transposon is a mobile genetic element that inserts into a specific consensus sequence on a chromosome usually through a cut-and-paste mechanism catalyzed by a transposase enzyme (Plasterk 2013). The transposon itself consists of a sequence of DNA (often an antibiotic resistance cassette) flanked by inverted repeats recognized by its cognate transposase. As shown in Table 6, a wide variety of transposons has been employed for forward genetic screens of oral bacteria. The key considerations when implementing transposon mutagenesis are the efficiency of transposition and its overall genome coverage. Traditionally, transposition reactions were performed in vivo and were often difficult to employ, with relatively low transposition frequencies and highly biased distributions of transposon insertions around the chromosome. These issues explain why older transposon mutagenesis studies tended to employ a much greater diversity of transposons relative to today (Table 6). In contrast, recent transposon mutagenesis studies are likely to employ mutagenized versions of either the Tn5 or Himar/Mariner transposon systems. These optimized systems yield far higher transposition frequencies than would normally occur using the respective wild-type versions of the transposase enzymes (Lampe 2010, Li et al. 2020). Consequently, it has only recently become practical to perform in vitro transposon mutagenesis using the genomic DNA of an organism of interest, purified recombinant transposase enzyme, and transposon DNA (van Opijnen et al. 2014). Following in vitro mutagenesis, the genomic DNA is transformed into a wild-type organism to create a library of mutant strains. Importantly, both the Tn5 and Himar transposases exhibit very low insertion biases with their respective transposons, which means in most cases they can effectively sample an entire genome with near randomness (Lampe 2010, van Opijnen and Camilli 2013, Li et al. 2020). The utility of the in vitro transposition approach is largely dependent upon the efficiency of transformation protocol available for a particular organism. For example, using this approach with P. micra, we obtained > 6000 Mariner transposon mutants/µg of DNA (Higashi et al. 2022). We obtained comparable results using S. mutans as well (Zou et al. 2018). At these levels, a single in vitro transposition reaction using several µg of bacterial DNA would reliably yield highly dense transposon libraries suitable for most applications, including Tn-seq analysis (described below). With a less efficient transformation protocol, additional transposition reactions may need to be pooled to create comparably sized libraries. Alternatively, in vivo mutagenesis is also possible using the Himar/Mariner transposon system (Table 6).
Table 6.
Transposons employed in selected oral microbiome species.
| Species | Transposon | In vivo/vitro | Reference |
|---|---|---|---|
| Library Screens | |||
| A. actinomycetemcomitans | Tn5 | In vivo | (Kolodrubetz and Kraig 1994) |
| IS903 | In vivo | (Thomson et al. 1999) | |
| Mariner | In vivo | (Ding and Tan 2017) | |
| A. oris | Tn5 | In vitro | (Mashimo et al. 2013) |
| E. faecalis | Tn917 | In vivo | (Garsin et al. 2004) |
| Mariner | In vivo | (Kristich et al. 2008) | |
| F. nucleatum | Tn5 | In vitro | (Coppenhagen-Glazer et al. 2015) |
| L. casei | IS1223 | In vivo | (Licandro-Seraut et al. 2012) |
| P. micra | Mariner | In vitro | (Higashi et al. 2022) |
| P. gingivalis | Tn4351 | In vivo | (Hoover et al. 1992) |
| Tn4400 | In vivo | (Chen et al. 2000) | |
| S. cristatus | Tn916 | In vivo | (Correia et al. 1995) |
| S. gordonii | Tn4001 | In vivo | (Lunsford 1995) |
| Tn916 | In vivo | (Whittaker et al. 1996) | |
| Tn917 | In vivo | (Loo et al. 2003) | |
| S. mutans | MudE | In vivo | (Kuramitsu 1987) |
| Tn916 | In vivo | (Caufield and Shah 1995) | |
| Tn917 | In vivo | (Cvitkovitch et al. 2000) | |
| Iss1 | In vivo | (Zhang and Biswas 2009) | |
| Mariner | In vivo | (Nilsson et al. 2014) | |
| Tn4001 | In vivo | (Jalal et al. 2015) | |
| T. denticola | Mariner | In vivo | (Yang et al. 2008) |
| V. parvula | Mariner | In vivo | (Bechon et al. 2020) |
| Tn-seq | |||
| A. actinomycetemcomitans | Tn10 | In vivo | (Stacy et al. 2016) |
| Mariner | In vivo | (Narayanan et al. 2017) | |
| E. faecalis | Mariner | In vivo | (Dale et al. 2018) |
| P. gingivalis | Mariner | In vivo | (Klein et al. 2012) |
| S. mutans | Mariner | In vitro | (Shields et al. 2018) |
In addition to traditional forward genetic screening, certain transposons have been adapted for use in transposon sequencing (Tn-seq) studies. Tn-seq is an especially powerful forward genetic approach that combines high-density transposon mutagenesis with next-generation sequencing technology to provide a quantitative genome-level assessment of both positive and negative genetic interactions supporting a particular growth condition or phenotype (van Opijnen and Camilli 2013). By calculating the representation of transposon insertions for every non-essential gene in a transposon mutant library, one can determine the severity of transposon insertion biases created by a particular treatment or growth condition. In this way, it is possible to utilize Tn-seq for both gene discovery as well as for characterizing genetic networks. Most Tn-seq studies are now performed using a point mutant form of the Mariner transposon in which its inverted repeats each contain an engineered MmeI restriction site (van Opijnen and Camilli 2013). MmeI is a particularly useful restriction enzyme for Tn-seq because it cuts 20 bp downstream of each restriction site in the inverted repeats surrounding a transposon insertion. This provides a straightforward mechanism to identify and quantify the transposon insertion sites within an entire mutant library. By examining the ratio of total transposon insertions in each gene before and after a treatment, it is possible to assign a relative score for a gene's positive, negative, or neutral influence upon a given growth condition. This approach is ideally suited for studies of virulence in experimental models of pathogenesis, as both virulence factors and their genetic regulators can be assessed in a single experiment (Klein et al. 2015, Peek et al. 2020). Tn-seq has been recently employed to assess both cellular fitness and genetic networks in multiple Gram-positive and Gram-negative oral microbes (Table 6).
Protein epitope tagging
The lack of commercially available antibodies for most microbiome species can present a serious challenge for protein studies in these organisms. While it is possible to have a custom antibody raised against a protein of interest, it can be quite time-consuming to do so, particularly when developing a highly specific antibody. For studies of genetic networks or protein interactomes, it may be especially impractical to develop individual antibodies for each unique protein of interest. In such situations, protein epitope tags offer a useful solution. Numerous highly specific antibodies are commercially available for a variety of protein epitopes. These epitopes are usually fairly small, with multiple tags being < 10 residues, and they can be engineered onto proteins of interest using the previously described mutagenesis approaches (Table 7) (Brizzard 2008). As with any protein fusion, there is always the possibility that the addition of an epitope disrupts the function of a protein of interest. Furthermore, an epitope tag may be only weakly detectable via western blot or perhaps even undetectable if the epitope is obscured by other portions of the protein. In these situations, one can always move the epitope to another location, usually the N- or C-terminus. We have also performed western blots using internal epitope tags without disrupting protein function (Marx et al. 2020). In our experience, internal epitope tags are usually well-tolerated if they are inserted within solvent-exposed flexible loops, which can be identified using a confirmed structural model or a predicted protein structure generated from reliable software like AlphaFold (Jumper et al. 2021). Besides the obvious utility of epitope tags for western blotting, these tags can also be valuable assets for protein-protein interaction studies using coimmunoprecipation (co-IP). Successful co-IP reactions are highly dependent upon the specificity of the antibodies employed, and commercial monoclonal antibodies to common epitopes like FLAG, HA, and c-Myc bind with exceptionally high avidity and specificity (Gerace and Moazed 2015, DeCaprio and Kohl 2019). In addition, there are a variety of commercial antibody affinity resins for these epitopes, which further simplifies co-IP studies. FLAG and HA antibody affinity resins have been recently employed for binary protein-protein interaction studies in oral bacteria (Dou et al. 2021, Mu et al. 2021). These same resins have also been employed to identify target protein interactomes (Mu et al. 2019, Qin et al. 2021). Interactome screening via co-IP is a powerful alternative to the previously described yeast and bacterial two-hybrid assays because all co-IP protein interactions are sampled in their native contexts, rather than in ectopic hosts. As recently demonstrated in S. mutans, the biochemical approach also offers the possibility of coimmunoprecipitating entire protein complexes, rather than just sampling binary protein interactions as with the two-hybrid approach (Mu et al. 2019, Qin et al. 2021). Furthermore, the availability of commercial anti-epitope affinity resins provides a straightforward strategy to incorporate tandem affinity purification during protein interactome studies. Tandem affinity purifications can tremendously increase both the specificity and sensitivity of protein interactome studies (DeCaprio and Kohl 2019). For this approach, a target protein of interest is tagged with two separate epitopes and then sequentially purified using the corresponding antibody affinity resins (Qin et al. 2021). Since two different antibodies are required for tandem affinity purifications, this approach is rarely employed using custom antibodies. Alternatively, tandem affinity purifications can also be performed using a variety of other affinity resins. For example, S. mutans protein fusions containing protein A IgG-binding domains and calmodulin-binding peptide have been successfully employed for protein interactome studies (Peng et al. 2016, Rainey et al. 2019).
Table 7.
Epitope tags used in selected oral microbiome species.
| Species | Epitope | Reference |
|---|---|---|
| A. actinomycetemcomitans | T7 | (Bhattacharjee et al. 2001) |
| E. faecalis | Strep | (Fujimoto and Ike 2001) |
| P. gingivalis | c-Myc | (Kadowaki et al. 2016) |
| S. gordonii | His | (Myscofski et al. 2000) |
| S. mutans | T7 | (Zhou et al. 2008) |
| FLAG | (Kim et al. 2013) | |
| HA | (Liu et al. 2017) | |
| His | (Murata et al. 2018) | |
| T. denticola | His | (Godovikova et al. 2010) |
Outlook
In the preceding review, we describe the key molecular microbiology tools and techniques that have facilitated our current mechanistic understanding of oral microbiome genetics. Even though the focus has been on oral microbiome species, it is important to note that these examples are applicable as a resource for all human microbiomes. The fact is, molecular microbiology studies of the human microbiota all currently share the same challenge: namely, a daunting array of uncharacterized species with largely unknown roles in mucosal homeostasis. As such, it may currently seem impractical or even unrealistic to breach the genetic barriers from the plethora of uncharacterized organisms, especially when many are assumed to be genetically intractable. The technologies and approaches described here offer many avenues to investigate these seemingly intractable organisms, which will hopefully provide the impetus to try. In the coming years, the field will also likely require researchers to advance beyond a singular focus upon individual species of interest in favor of mixed species molecular microbiology studies. It is already evident that numerous important microbiome phenotypes are the unique product of polymicrobial interactions (Murray et al. 2014, Flynn et al. 2016, Tay et al. 2016, Hajishengallis and Lamont 2021). If we expect to interrogate the molecular basis of such processes, it seems inevitable that in vitro model systems will require the simultaneous implementation of multiple genetic systems for the different organisms grown in coculture studies. The time required for this significant transition to occur in the field will likely depend heavily upon our progress in the quest to develop tractable genetic systems for many additional members of the microbiome.
Contributor Information
Justin Merritt, Department of Restorative Dentistry, School of Dentistry, Oregon Health and Science University, Portland, OR, United States; Department of Molecular Microbiology and Immunology, Oregon Health and Science University, Portland, OR 97239, United States.
Jens Kreth, Department of Restorative Dentistry, School of Dentistry, Oregon Health and Science University, Portland, OR, United States; Department of Molecular Microbiology and Immunology, Oregon Health and Science University, Portland, OR 97239, United States.
Funding
The authors would like to acknowledge the following National Institutes of Health Grants: DE022083 and DE028252 to JM and DE029612 and DE029492 to JK. We greatly appreciate the helpful comments and insights from Drs. Michiko Nakano, Hui Wu, and Peter Zuber during the preparation of this manuscript. We would also like to acknowledge the many references that could not be included due to editorial limitations.
Conflicts of interest statement
None declared.
References
- Adams ST Jr, Miller SC. Enzymatic promiscuity and the evolution of bioluminescence. FEBS J. 2020;287:1369–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnone MI, Dmochowski IJ, Gache C. Using reporter genes to study cis-regulatory elements. Methods Cell Biol. 2004;74:621–52. [DOI] [PubMed] [Google Scholar]
- Arutyunov D, Frost LS. F conjugation: back to the beginning. Plasmid. 2013;70:18–32. [DOI] [PubMed] [Google Scholar]
- Attaiech L, Charpentier X. Silently transformable: the many ways bacteria conceal their built-in capacity of genetic exchange. Curr Genet. 2017;63:451–5. [DOI] [PubMed] [Google Scholar]
- Aukrust TW, Brurberg MB, Nes IF. Transformation of Lactobacillus by electroporation. Methods Mol Biol. 1995;47:201–8. [DOI] [PubMed] [Google Scholar]
- Baev D, England R, Kuramitsu HK. Stress-induced membrane association of the Streptococcus mutans GTP-binding protein, an essential G protein, and investigation of its physiological role by utilizing an antisense RNA strategy. Infect Immun. 1999;67:4510–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baev D, Ohk SH, Kuramitsu HK. Protein interactions of SGP, an essential Streptococcus mutans gtpase, revealed by biochemical and yeast two-hybrid system analyses. FEMS Microbiol Lett. 2000;184:149–53. [DOI] [PubMed] [Google Scholar]
- Bakkali M. Could DNA uptake be a side effect of bacterial adhesion and twitching motility?. Arch Microbiol. 2013;195:279–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banai M, LeBlanc DJ. Genetic, molecular, and functional analysis of Streptococcus faecalis R plasmid pJH1. J Bacteriol. 1983;155:1094–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandara H, Panduwawala CP, Samaranayake LP. Biodiversity of the human oral mycobiome in health and disease. Oral Dis. 2019;25:363–71. [DOI] [PubMed] [Google Scholar]
- Banerjee A, Biswas I. Markerless multiple-gene-deletion system for Streptococcus mutans. Appl Environ Microbiol. 2008;74:2037–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battesti A, Bouveret E. The bacterial two-hybrid system based on adenylate cyclase reconstitution in Escherichia coli. Methods. 2012;58:325–34. [DOI] [PubMed] [Google Scholar]
- Bauer R, Mauerer S, Grempels Aet al. The competence system of Streptococcus anginosus and its use for genetic engineering. Mol Oral Microbiol. 2018;33:194–202. [DOI] [PubMed] [Google Scholar]
- Bechon N, Jimenez-Fernandez A, Witwinowski Jet al. Autotransporters drive biofilm formation and autoaggregation in the Diderm Firmicute Veillonella parvula. J Bacteriol. 2020;202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behnke D, Gilmore MS, Ferretti JJ. Plasmid pGB301, a new multiple resistance streptococcal cloning vehicle and its use in cloning of a gentamicin/kanamycin resistance determinant. Molecular and General Genetics MGG. 1981;182:414–21. [DOI] [PubMed] [Google Scholar]
- Belmok A, de Cena JA, Kyaw CMet al. The oral archaeome: a scoping review. J Dent Res. 2020;99:630–43. [DOI] [PubMed] [Google Scholar]
- Bertram R, Neumann B, Schuster CF. Status quo of tet regulation in bacteria. Microb Biotechnol. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharjee MK, Fine DH, Figurski DH. tfoX (sxy)-dependent transformation of Aggregatibacter (Actinobacillus) actinomycetemcomitans. Gene. 2007;399:53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharjee MK, Kachlany SC, Fine DHet al. Nonspecific adherence and fibril biogenesis by Actinobacillus actinomycetemcomitans: tadA protein is an atpase. J Bacteriol. 2001;183:5927–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian J, Fenno JC, Li C. Development of a modified gentamicin resistance cassette for genetic manipulation of the oral spirochete Treponema denticola. Appl Environ Microbiol. 2012;78:2059–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binishofer B, Moll I, Henrich Bet al. Inducible promoter-repressor system from the Lactobacillus casei phage phiFSW. Appl Environ Microbiol. 2002;68:4132–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brizzard B. Epitope tagging. BioTechniques. 2008;44:693–5. [DOI] [PubMed] [Google Scholar]
- Brogan JM, Lally ET, Demuth DR. Construction of pYGK, an actinobacillus actinomycetemcomitans-Escherichia coli shuttle vector. Gene. 1996;169:141–2. [DOI] [PubMed] [Google Scholar]
- Bryan EM, Bae T, Kleerebezem Met al. Improved vectors for nisin-controlled expression in gram-positive bacteria. Plasmid. 2000;44:183–90. [DOI] [PubMed] [Google Scholar]
- Buckley ND, Lee LN, LeBlanc DJ. Use of a novel mobilizable vector to inactivate the scrA gene of Streptococcus sobrinus by allelic replacement. J Bacteriol. 1995;177:5028–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley ND, Vadeboncoeur C, LeBlanc DJet al. An effective strategy, applicable to Streptococcus salivarius and related bacteria, to enhance or confer electroporation competence. Appl Environ Microbiol. 1999;65:3800–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdett V. Identification of tetracycline-resistant R-plasmids in Streptococcus agalactiae (group B). Antimicrob Agents Chemother. 1980;18:753–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell A. Phage integration and chromosome structure. A personal history. Annu Rev Genet. 2007;41:1–11. [DOI] [PubMed] [Google Scholar]
- Caufield PW, Shah G. Transformation of naturally competent Streptococcus mutans with replicative and non-replicative Tn916-containing plasmids: implications for a mechanism of transposition. Dev Biol Stand. 1995;85:19–25. [PubMed] [Google Scholar]
- Chang SM, Yan TR. Genetic engineering techniques for lactic acid bacteria: construction of a stable shuttle vector and expression vector for beta-glucuronidase. Biotechnol Lett. 2014;36:327–35. [DOI] [PubMed] [Google Scholar]
- Chary VK, Busuioc M, Renye JA Jret al. Vectors that facilitate the replacement of transcriptional lacZ fusions in Streptococcus mutans and Bacillus subtilis with fusions to gfp or gusA. FEMS Microbiol Lett. 2005;247:171–6. [DOI] [PubMed] [Google Scholar]
- Chen T, Dong H, Tang YPet al. Identification and cloning of genes from Porphyromonas gingivalis after mutagenesis with a modified Tn4400 transposon from Bacteroides fragilis. Infect Immun. 2000;68:420–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YY, Weaver CA, Burne RA. Dual functions of Streptococcus salivarius urease. J Bacteriol. 2000;182:4667–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X, Redanz S, Cullin Net al. Plasticity of the pyruvate node modulates hydrogen peroxide production and acid tolerance in multiple oral streptococci. Appl Environ Microbiol. 2018;84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherepanov PP, Wackernagel W. Gene disruption in Escherichia coli: tcR and KmR cassettes with the option of flp-catalyzed excision of the antibiotic-resistance determinant. Gene. 1995;158:9–14. [DOI] [PubMed] [Google Scholar]
- Chia HE, Marsh ENG, Biteen JS. Extending fluorescence microscopy into anaerobic environments. Curr Opin Chem Biol. 2019;51:98–104. [DOI] [PubMed] [Google Scholar]
- Choi CH, DeGuzman JV, Lamont RJet al. Genetic transformation of an obligate anaerobe, P. gingivalis for FMN-green fluorescent protein expression in studying host-microbe interaction. PLoS One. 2011;6:e18499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clewell DB, Weaver KE, Dunny GMet al. 2014; Extrachromosomal and mobile elements in enterococci: transmission, maintenance, and epidemiology. In: Enterococci: From Commensals to Leading Causes of Drug Resistant Infection,(Gilmore MS, Clewell DB, Ike Y, Shankar N, eds). Boston. [PubMed] [Google Scholar]
- Colby SM, Whiting GC, Tao L &et al. Insertional inactivation of the Streptococcus mutans dexA (dextranase) gene results in altered adherence and dextran catabolism. Microbiology. 1995;141:2929–36. [DOI] [PubMed] [Google Scholar]
- Coppenhagen-Glazer S, Sol A, Abed Jet al. Fap2 of fusobacterium nucleatum is a galactose-inhibitable adhesin involved in coaggregation, cell adhesion, and preterm birth. Infect Immun. 2015;83:1104–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correia FF, DiRienzo JM, Lamont RJet al. Insertional inactivation of binding determinants of Streptococcus crista CC5A using Tn916. Oral Microbiol Immunol. 1995;10:220–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correia FF, DiRienzo JM, McKay TLet al. scbA from Streptococcus crista CC5A: an atypical member of the lraI gene family. Infect Immun. 1996;64:2114–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correia FF, McKay TL, Farrow MFet al. Natural transformation of Streptococcus crista. FEMS Microbiol Lett. 1996;143:13–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa TRD, Harb L, Khara Pet al. Type IV secretion systems: advances in structure, function, and activation. Mol Microbiol. 2021;115:436–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Court DL, Sawitzke JA, Thomason LC. Genetic engineering using homologous recombination. Annu Rev Genet. 2002;36:361–88. [DOI] [PubMed] [Google Scholar]
- Craggs TD. Green fluorescent protein: structure, folding and chromophore maturation. Chem Soc Rev. 2009;38:2865–75. [DOI] [PubMed] [Google Scholar]
- Cvitkovitch DG, Gutierrez JA, Behari Jet al. Tn917-lac mutagenesis of Streptococcus mutans to identify environmentally regulated genes. FEMS Microbiol Lett. 2000;182:149–54. [DOI] [PubMed] [Google Scholar]
- Dale JL, Beckman KB, Willett JLEet al. Comprehensive functional analysis of the Enterococcus faecalis core genome using an ordered, sequence-Defined collection of insertional mutations in strain OG1RF. Msystems. 2018;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies J, Wright GD. Bacterial resistance to aminoglycoside antibiotics. Trends Microbiol. 1997;5:234–40. [DOI] [PubMed] [Google Scholar]
- Day AM, Cove JH, Phillips-Jones MK. Cytolysin gene expression in Enterococcus faecalis is regulated in response to aerobiosis conditions. Mol Genet Genomics. 2003;269:31–39. [DOI] [PubMed] [Google Scholar]
- de Cena JA, Zhang J, Deng Det al. Low-Abundant microorganisms: the Human microbiome's dark matter, a scoping review. Front Cell Infect Microbiol. 2021;11:689197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCaprio J, Kohl TO. Tandem immunoaffinity purification using anti-FLAG and anti-HA antibodies. Cold Spring Harb Protoc. 2019;2019:pdb.prot098657. [DOI] [PubMed] [Google Scholar]
- Diaz PI. Subgingival fungi, Archaea, and viruses under the omics loupe. Periodontol 2000. 2021;85:82–89. [DOI] [PubMed] [Google Scholar]
- Ding Q, Tan KS. Himar1 Transposon for efficient random mutagenesis in aggregatibacter actinomycetemcomitans. Front Microbiol. 2017;8:1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou Y, Rutanhira H, Schormann Net al. PG1659 functions as anti-sigma factor to extracytoplasmic function sigma factor RpoE in Porphyromonas gingivalis W83. Mol Oral Microbiol. 2021;36:80–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubnau D, Blokesch M. Mechanisms of DNA uptake by naturally competent bacteria. Annu Rev Genet. 2019;53:217–37. [DOI] [PubMed] [Google Scholar]
- Dunny GM, Lee LN, LeBlanc DJ. Improved electroporation and cloning vector system for gram-positive bacteria. Appl Environ Microbiol. 1991;57:1194–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer DW, Bilalis G, Michel JHet al. Conjugal transfer of plasmid and transposon DNA from Escherichia coli into Porphyromonas gingivalis. Biochem Biophys Res Commun. 1992;186:1012–9. [DOI] [PubMed] [Google Scholar]
- Eckburg PB, Bik EM, Bernstein CNet al. Diversity of the human intestinal microbial flora. Science. 2005;308:1635–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evguenieva-Hackenberg E. Riboregulation in bacteria: from general principles to novel mechanisms of the trp attenuator and its sRNA and peptide products. Wiley Interdiscip Rev RNA. 2021:e1696. [DOI] [PubMed] [Google Scholar]
- Fenno JC, Shaikh A, Fives-Taylor P. Characterization of allelic replacement in Streptococcus parasanguis: transformation and homologous recombination in a ‘nontransformable’ streptococcus. Gene. 1993;130:81–90. [DOI] [PubMed] [Google Scholar]
- Flynn KJ, Baxter NT, Schloss PD. Metabolic and community synergy of oral bacteria in colorectal cancer. Msphere. 2016;1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontaine L, Boutry C, de Frahan MHet al. A novel pheromone quorum-sensing system controls the development of natural competence in Streptococcus thermophilus and Streptococcus salivarius. J Bacteriol. 2010;192:1444–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto S, Ike Y. pAM401-based shuttle vectors that enable overexpression of promoterless genes and one-step purification of tag fusion proteins directly from Enterococcus faecalis. Appl Environ Microbiol. 2001;67:1262–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujise O, Wang Y, Chen Wet al. Adherence of Aggregatibacter actinomycetemcomitans via serotype-specific polysaccharide antigens in lipopolysaccharides. Oral Microbiol Immunol. 2008;23:226–33. [DOI] [PubMed] [Google Scholar]
- Fukushima K, Ikeda T, Kuramitsu HK. Expression of Streptococcus mutans gtf genes in Streptococcus milleri. Infect Immun. 1992;60:2815–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Cayuela T, de Cadinanos LP, Mohedano MLet al. Fluorescent protein vectors for promoter analysis in lactic acid bacteria and Escherichia coli. Appl Microbiol Biotechnol. 2012;96:171–81. [DOI] [PubMed] [Google Scholar]
- Garsin DA, Urbach J, Huguet-Tapia JCet al. Construction of an Enterococcus faecalis Tn917-mediated-gene-disruption library offers insight into Tn917 insertion patterns. J Bacteriol. 2004;186:7280–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaustad P, Eriksen J, Henriksen SD. Genetic transformation in Streptococcus sanguis. Spontaneous and induced competence of selected strains. Acta Pathol Microbiol Scand B. 1979;87B:117–22. [PubMed] [Google Scholar]
- Gerace E, Moazed D. Affinity pull-Down of proteins using anti-FLAG M2 agarose beads. Methods Enzymol. 2015;559:99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girons IS, Chi B, Kuramitsu H. Development of shuttle vectors for spirochetes. J Mol Microbiol Biotechnol. 2000;2:443–5. [PubMed] [Google Scholar]
- Goadby K. The mycology of the mouth: a text-book of oral bacteria. Longmans, Green, and Co: London: 1903. [Google Scholar]
- Godovikova V, Wang HT, Goetting-Minesky MPet al. Treponema denticola PrcB is required for expression and activity of the PrcA-PrtP (dentilisin) complex. J Bacteriol. 2010;192:3337–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goessweiner-Mohr N, Arends K, Keller Wet al. Conjugation in gram-Positive bacteria. Microbiology Spectrum. 2014;2:PLAS–0004-2013. [DOI] [PubMed] [Google Scholar]
- Goetting-Minesky MP, Fenno JC. A simplified erythromycin resistance cassette for Treponema denticola mutagenesis. J Microbiol Methods. 2010;83:66–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golkar T, Zielinski M, Berghuis AM. Look and outlook on enzyme-Mediated macrolide resistance. Front Microbiol. 2018;9:1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncharoff P, Yip JK, Wang Het al. Conjugal transfer of broad-host-range incompatibility group P and Q plasmids from Escherichia coli to actinobacillus actinomycetemcomitans. Infect Immun. 1993;61:3544–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong T, Tang B, Zhou Xet al. Genome editing in Streptococcus mutans through self-targeting CRISPR arrays. Mol Oral Microbiol. 2018;33:440–9. [DOI] [PubMed] [Google Scholar]
- Goodman SD, Gao Q. Firefly luciferase as a reporter to study gene expression in Streptococcus mutans. Plasmid. 1999;42:154–7. [DOI] [PubMed] [Google Scholar]
- Gosalbes MJ, Monedero V, Perez-Martinez G. Elements involved in catabolite repression and substrate induction of the lactose operon in Lactobacillus casei. J Bacteriol. 1999;181:3928–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green MR, Sambrook J. Transformation of Escherichia coli by electroporation. Cold Spring Harb Protoc. 2020;2020:pdb.prot101220. [DOI] [PubMed] [Google Scholar]
- Grissom-Arnold J, Alborn WE Jr, Nicas TIet al. Induction of VanA vancomycin resistance genes in Enterococcus faecalis: use of a promoter fusion to evaluate glycopeptide and nonglycopeptide induction signals. Microb Drug Resist. 1997;3:53–64. [DOI] [PubMed] [Google Scholar]
- Gutierrez JA, Crowley PJ, Brown DPet al. Insertional mutagenesis and recovery of interrupted genes of Streptococcus mutans by using transposon Tn917: preliminary characterization of mutants displaying acid sensitivity and nutritional requirements. J Bacteriol. 1996;178:4166–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman-Herrador DL, Llosa M. The secret life of conjugative relaxases. Plasmid. 2019;104:102415. [DOI] [PubMed] [Google Scholar]
- Haake SK, Yoder SC, Attarian Get al. Native plasmids of fusobacterium nucleatum: characterization and use in development of genetic systems. J Bacteriol. 2000;182:1176–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajishengallis G, Lamont RJ. Polymicrobial communities in periodontal disease: their quasi-organismal nature and dialogue with the host. Periodontol 2000. 2021;86:210–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall JW, Lima BP, Herbomel GGet al. An intramembrane sensory circuit monitors sortase A-mediated processing of streptococcal adhesins. Sci Signal. 2019;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock LE, Shepard BD, Gilmore MS. Molecular analysis of the Enterococcus faecalis serotype 2 polysaccharide determinant. J Bacteriol. 2003;185:4393–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannan S, Ready D, Jasni ASet al. Transfer of antibiotic resistance by transformation with eDNA within oral biofilms. FEMS Immunol Med Microbiol. 2010;59:345–9. [DOI] [PubMed] [Google Scholar]
- Hansen MC, Palmer RJ, Udsen Cet al. Assessment of GFP fluorescence in cells of Streptococcus gordonii under conditions of low pH and low oxygen concentration. Microbiology. 2001;147:1383–91. [DOI] [PubMed] [Google Scholar]
- Higashi DL, McGuire S, Abdelrahman YMet al. Development of the first tractable genetic system for parvimonas micra, a ubiquitous pathobiont in Human dysbiotic disease. Microbiology Spectrum. 2022;10:e0046522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi M, Yamamoto Y, Poole LBet al. Functions of two types of NADH oxidases in energy metabolism and oxidative stress of Streptococcus mutans. J Bacteriol. 1999;181:5940–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honma K, Inagaki S, Okuda Ket al. Role of a Tannerella forsythia exopolysaccharide synthesis operon in biofilm development. Microb Pathog. 2007;42:156–66. [DOI] [PubMed] [Google Scholar]
- Honma K, Kuramitsu HK, Genco RJet al. Development of a gene inactivation system for Bacteroides forsythus: construction and characterization of a BspA mutant. Infect Immun. 2001;69:4686–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover CI, Abarbarchuk E, Ng CYet al. Transposition of Tn4351 in Porphyromonas gingivalis. Plasmid. 1992;27:246–50. [DOI] [PubMed] [Google Scholar]
- Hosohama-Saito K, Kokubu E, Okamoto-Shibayama Ket al. Involvement of luxS in biofilm formation by capnocytophaga ochracea. PLoS One. 2016;11:e0147114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson MC, Curtiss R 3rd. Regulation of expression of Streptococcus mutans genes important to virulence. Infect Immun. 1990;58:464–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes KT, Maloy SR. Use of operon and gene fusions to study gene regulation in Salmonella. Methods Enzymol. 2007;421:140–58. [DOI] [PubMed] [Google Scholar]
- Izert MA, Klimecka MM, Gorna MW. Applications of bacterial degrons and degraders - Toward targeted protein degradation in bacteria. Front in Mol Biosci. 2021;8:669762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalal N, Tian XL, Dong Get al. Identification and characterization of SMU.244 encoding a putative undecaprenyl pyrophosphate phosphatase protein required for cell wall biosynthesis and bacitracin resistance in Streptococcus mutans. Microbiology. 2015;161:1857–70. [DOI] [PubMed] [Google Scholar]
- Johnsborg O, Eldholm V, Bjornstad MLet al. A predatory mechanism dramatically increases the efficiency of lateral gene transfer in Streptococcus pneumoniae and related commensal species. Mol Microbiol. 2008;69:245–53. [DOI] [PubMed] [Google Scholar]
- Johnston C, Martin B, Granadel Cet al. Programmed protection of foreign DNA from restriction allows pathogenicity island exchange during pneumococcal transformation. PLoS Pathog. 2013;9:e1003178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juarez-Rodriguez MD, Torres-Escobar A, Demuth DR. Construction of new cloning, lacZ reporter and scarless-markerless suicide vectors for genetic studies in Aggregatibacter actinomycetemcomitans. Plasmid. 2013;69:211–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juers DH, Matthews BW, Huber RE. LacZ beta-galactosidase: structure and function of an enzyme of historical and molecular biological importance. Protein Sci. 2012;21:1792–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper J, Evans R, Pritzel Aet al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junges R, Khan R, Tovpeko Yet al. Markerless genome editing in competent streptococci. Methods Mol Biol. 2017;1537:233–47. [DOI] [PubMed] [Google Scholar]
- Kadowaki T, Yukitake H, Naito Met al. A two-component system regulates gene expression of the type IX secretion component proteins via an ECF sigma factor. Sci Rep. 2016;6:23288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kain SR, Ganguly S. Overview of genetic reporter systems. Curr Protoc Mol Biol. 2001, Chapter 9: Unit9 6. [DOI] [PubMed] [Google Scholar]
- Kaplan CW, Lux R, Huynh Tet al. Fusobacterium nucleatum apoptosis-inducing outer membrane protein. J Dent Res. 2005;84:700–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kast P, Hennecke H. Amino acid substrate specificity of Escherichia coli phenylalanyl-tRNA synthetase altered by distinct mutations. J Mol Biol. 1991;222:99–124. [DOI] [PubMed] [Google Scholar]
- Kato Y. Extremely low leakage expression systems using dual transcriptional-Translational control for toxic protein production. Int J Mol Sci. 2020,705, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan R, Rukke HV, Hovik Het al. Comprehensive transcriptome profiles of Streptococcus mutans UA159 map core streptococcal competence genes. Msystems. 2016;1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidane D, Ayora S, Sweasy JBet al. The cell pole: the site of cross talk between the DNA uptake and genetic recombination machinery. Crit Rev Biochem Mol Biol. 2012;47:531–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiernan JA. Indigogenic substrates for detection and localization of enzymes. Biotech Histochem. 2007;82:73–103. [DOI] [PubMed] [Google Scholar]
- Kili AO, Herzberg MC, Meyer MWet al. Streptococcal reporter gene-fusion vector for identification of in vivo expressed genes. Plasmid. 1999;42:67–72. [DOI] [PubMed] [Google Scholar]
- Kim JN, Stanhope MJ, Burne RA. Core-gene-encoded peptide regulating virulence-associated traits in Streptococcus mutans. J Bacteriol. 2013;195:2912–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim NM, Sinnott RW, Sandoval NR. Transcription factor-based biosensors and inducible systems in non-model bacteria: current progress and future directions. Curr Opin Biotechnol. 2020;64:39–46. [DOI] [PubMed] [Google Scholar]
- Klein BA, Duncan MJ, Hu LT. Defining essential genes and identifying virulence factors of Porphyromonas gingivalis by massively parallel sequencing of transposon libraries (Tn-seq). Methods Mol Biol. 2015;1279:25–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein BA, Tenorio EL, Lazinski DWet al. Identification of essential genes of the periodontal pathogen Porphyromonas gingivalis. BMC Genomics. 2012;13:578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapp S, Brodal C, Peterson Jet al. Natural competence is common among clinical isolates of Veillonella parvula and is useful for genetic manipulation of this key member of the oral microbiome. Front Cell Infect Microbiol. 2017;7:139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler V, Vaishampayan A, Grohmann E. Broad-host-range Inc18 plasmids: occurrence, spread and transfer mechanisms. Plasmid. 2018;99:11–21. [DOI] [PubMed] [Google Scholar]
- Kolodrubetz D, Kraig E. Transposon Tn5 mutagenesis of actinobacillus actinomycetemcomitans via conjugation. Oral Microbiol Immunol. 1994;9:290–6. [DOI] [PubMed] [Google Scholar]
- Kolodrubetz D, Spitznagel J Jr, Wang Bet al. cis elements and trans factors are both important in strain-specific regulation of the leukotoxin gene in actinobacillus actinomycetemcomitans. Infect Immun. 1996;64:3451–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo Y, Ohara N, Sato Ket al. Tetratricopeptide repeat protein-associated proteins contribute to the virulence of Porphyromonas gingivalis. Infect Immun. 2010;78:2846–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo Y, Sato K, Nagano Ket al. Involvement of PorK, a component of the type IX secretion system, in Prevotella melaninogenica pathogenicity. Microbiol Immunol. 2018;62:554–66. [DOI] [PubMed] [Google Scholar]
- Kovacs AT, Smits WK, Mironczuk AMet al. Ubiquitous late competence genes in Bacillus species indicate the presence of functional DNA uptake machineries. Environ Microbiol. 2009;11:1911–22. [DOI] [PubMed] [Google Scholar]
- Kreth J, Abdelrahman YM, Merritt J. Multiplex imaging of polymicrobial communities-Murine models to study oral microbiome interactions. Methods Mol Biol. 2020;2081:107–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreth J, Liu N, Chen Zet al. RNA regulators of host immunity and pathogen adaptive responses in the oral cavity. Microbes Infect. 2015;17:493–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreth J, Merritt J, Bordador Cet al. Transcriptional analysis of mutacin I (mutA) gene expression in planktonic and biofilm cells of Streptococcus mutans using fluorescent protein and glucuronidase reporters. Oral Microbiol Immunol. 2004;19:252–6. [DOI] [PubMed] [Google Scholar]
- Kristich CJ, Chandler JR, Dunny GM. Development of a host-genotype-independent counterselectable marker and a high-frequency conjugative delivery system and their use in genetic analysis of Enterococcus faecalis. Plasmid. 2007;57:131–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristich CJ, Nguyen VT, Le Tet al. Development and use of an efficient system for random mariner transposon mutagenesis to identify novel genetic determinants of biofilm formation in the core Enterococcus faecalis genome. Appl Environ Microbiol. 2008;74:3377–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuramitsu HK. Molecular genetic analysis of the virulence of oral bacterial pathogens: an historical perspective. Crit Rev Oral Biol Med. 2003;14:331–44. [DOI] [PubMed] [Google Scholar]
- Kuramitsu HK. Utilization of a mini-mu transposon to construct defined mutants in Streptococcus mutans. Mol Microbiol. 1987;1:229–32. [DOI] [PubMed] [Google Scholar]
- Lampe DJ. Bacterial genetic methods to explore the biology of mariner transposons. Genetica. 2010;138:499–508. [DOI] [PubMed] [Google Scholar]
- Lara Vasquez P, Mishra S, Kuppuswamy SKet al. Protein interactomes of Streptococcus mutans YidC1 and YidC2 membrane Protein insertases suggest SRP pathway-Independent- and -Dependent functions, respectively. Msphere. 2021;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBlanc DJ, Hassell FP. Transformation of Streptococcus sanguis Challis by plasmid deoxyribonucleic acid from Streptococcus faecalis. J Bacteriol. 1976;128:347–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBlanc DJ, Hawley RJ, Lee LNet al. “Conjugal” transfer of plasmid DNA among oral streptococci. Proc Natl Acad Sci. 1978;75:3484–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBlanc DJ, Lee LN, Inamine JM. Cloning and nucleotide base sequence analysis of a spectinomycin adenyltransferase AAD(9) determinant from Enterococcus faecalis. Antimicrob Agents Chemother. 1991;35:1804–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SF, Progulske-Fox A, Erdos GWet al. Construction and characterization of isogenic mutants of Streptococcus mutans deficient in major surface protein antigen P1 (I/II). Infect Immun. 1989;57:3306–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz DH, Mok KC, Lilley BNet al. The small RNA chaperone hfq and multiple small rnas control quorum sensing in Vibrio harveyi and Vibrio cholerae. Cell. 2004;118:69–82. [DOI] [PubMed] [Google Scholar]
- Li H, Kuramitsu HK. Development of a gene transfer system in Treponema denticola by electroporation. Oral Microbiol Immunol. 1996;11:161–5. [DOI] [PubMed] [Google Scholar]
- Li JW, Wyllie RM, Jensen PA. A novel competence pathway in the oral pathogen Streptococcus sobrinus. J Dent Res. 2021;100:542–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Jin K, Bai Yet al. Tn5 Transposase applied in genomics research. Int J Mol Sci. 2020,8329, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Liu Z, Zhang Yet al. Live-cell and super-resolution imaging reveal that the distribution of wall-associated protein A is correlated with the cell chain integrity of Streptococcus mutans. Mol Oral Microbiol. 2015;30:376–83. [DOI] [PubMed] [Google Scholar]
- Li Y, Ruby J, Wu H. Kanamycin resistance cassette for genetic manipulation of treponema denticola. Appl Environ Microbiol. 2015;81:4329–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licandro-Seraut H, Brinster S, van de Guchte Met al. Development of an efficient in vivo system (Pjunc-TpaseIS1223) for random transposon mutagenesis of Lactobacillus casei. Appl Environ Microbiol. 2012;78:5417–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linares DM, Perez M, Ladero Vet al. An agmatine-inducible system for the expression of recombinant proteins in Enterococcus faecalis. Microb Cell Fact. 2014;13:169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindler LE, Macrina FL. Characterization of genetic transformation in Streptococcus mutans by using a novel high-efficiency plasmid marker rescue system. J Bacteriol. 1986;166:658–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippmann JE, Froeliger EH, Fives-Taylor PM. Use of the Actinobacillus actinomycetemcomitans leukotoxin promoter to drive expression of the green fluorescent protein in an oral pathogen. Oral Microbiol Immunol. 1999;14:321–5. [DOI] [PubMed] [Google Scholar]
- Liu J, Merritt J, Qi F. Genetic transformation of Veillonella parvula. FEMS Microbiol Lett. 2011;322:138–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Xie Z, Merritt Jet al. Establishment of a tractable genetic transformation system in Veillonella spp. Appl Environ Microbiol. 2012;78:3488–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Chaudhry MT, Xie Zet al. Identification of new degrons in Streptococcus mutans reveals a novel strategy for engineering targeted, controllable proteolysis. Front Microbiol. 2017;8:2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Abaibou H, Fletcher HM. Development of a noninvasive reporter system for gene expression in Porphyromonas gingivalis. Plasmid. 2000;44:250–61. [DOI] [PubMed] [Google Scholar]
- Loeliger B, Caldelari I, Bizzini Aet al. Antibiotic-dependent correlation between drug-induced killing and loss of luminescence in Streptococcus gordonii expressing luciferase. Microb Drug Resist. 2003;9:123–31. [DOI] [PubMed] [Google Scholar]
- Loenen WA, Raleigh EA. The other face of restriction: modification-dependent enzymes. Nucleic Acids Res. 2014;42:56–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo CY, Mitrakul K, Voss IBet al. Involvement of the adc operon and manganese homeostasis in Streptococcus gordonii biofilm formation. J Bacteriol. 2003;185:2887–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B, McBride BC. Expression of the tpr protease gene of Porphyromonas gingivalis is regulated by peptide nutrients. Infect Immun. 1998;66:5147–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu K, Vu CQ, Matsuda Tet al. Fluorescent protein-Based indicators for functional super-Resolution imaging of biomolecular activities in living cells. Int J Mol Sci. 2019,5784, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunsford RD. A Tn4001 delivery system for Streptococcus gordonii (Challis). Plasmid. 1995;33:153–7. [DOI] [PubMed] [Google Scholar]
- Luong TT, Tirgar R, Reardon-Robinson MEet al. Structural basis of a thiol-Disulfide oxidoreductase in the hedgehog-Forming actinobacterium Corynebacterium matruchotii. J Bacteriol. 2018;200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macrina FL, Jones KR, Wood PH. Chimeric streptococcal plasmids and their use as molecular cloning vehicles in Streptococcus sanguis (Challis). J Bacteriol. 1980;143:1425–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maley J, Shoemaker NB, Roberts IS. The introduction of colonic-Bacteroides shuttle plasmids into Porphyromonas gingivalis: identification of a putative P. gingivalis insertion-sequence element. FEMS Microbiol Lett. 1992;93:75–81. [DOI] [PubMed] [Google Scholar]
- Mallaley PP, Halperin SA, Morris Aet al. Expression of a pertussis toxin S1 fragment by inducible promoters in oral Streptococcus and the induction of immune responses during oral colonization in mice. Can J Microbiol. 2006;52:436–44. [DOI] [PubMed] [Google Scholar]
- Mark Welch JL, Rossetti BJ, Rieken CWet al. Biogeography of a human oral microbiome at the micron scale. Proc Natl Acad Sci. 2016;113:E791–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin MC, Fernandez M, Martin-Alonso JMet al. Nisin-controlled expression of Norwalk virus VP60 protein in Lactobacillus casei. FEMS Microbiol Lett. 2004;237:385–91. [DOI] [PubMed] [Google Scholar]
- Martinez A, Kuraji R, Kapila YL. The human oral virome: shedding light on the dark matter. Periodontol 2000. 2021;87:282–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Fernandez JA, Bravo D, Peiroten Aet al. Bile-induced promoters for gene expression in Lactobacillus strains. Appl Microbiol Biotechnol. 2019;103:3819–27. [DOI] [PubMed] [Google Scholar]
- Marx P, Sang Y, Qin Het al. Environmental stress perception activates structural remodeling of extant Streptococcus mutans biofilms. Npj Biofilms and Microbiomes. 2020;6:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashimo C, Kamitani H, Nambu Tet al. Identification of the genes involved in the biofilm-like structures on actinomyces oris K20, a clinical isolate from an apical lesion. J Endod. 2013;39:44–48. [DOI] [PubMed] [Google Scholar]
- Maula T, Vahvelainen N, Tossavainen Het al. Decreased temperature increases the expression of a disordered bacterial late embryogenesis abundant (LEA) protein that enhances natural transformation. Virulence. 2021;12:1239–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick NE, Halperin SA, Song Fet al. Regulation of D-alanylation of lipoteichoic acid in Streptococcus gordonii. Microbiology. 2011;157:2248–56. [DOI] [PubMed] [Google Scholar]
- McLaughlin RE, Ferretti JJ. Electrotransformation of streptococci. Methods Mol Biol. 1995;47:185–93. [DOI] [PubMed] [Google Scholar]
- Meibom KL, Blokesch M, Dolganov NAet al. Chitin induces natural competence in Vibrio cholerae. Science. 2005;310:1824–7. [DOI] [PubMed] [Google Scholar]
- Merritt J, Chen Z, Liu Net al. Posttranscriptional regulation of oral bacterial adaptive responses. Current Oral Health Reports. 2014;1:50–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merritt J, Kreth J, Qi Fet al. Non-disruptive, real-time analyses of the metabolic status and viability of Streptococcus mutans cells in response to antimicrobial treatments. J Microbiol Methods. 2005;61:161–70. [DOI] [PubMed] [Google Scholar]
- Merritt J, Senpuku H, Kreth J. Let there be bioluminescence: development of a biophotonic imaging platform for in situ analyses of oral biofilms in animal models. Environ Microbiol. 2016;18:174–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merritt J, Tsang P, Zheng Let al. Construction of a counterselection-based in-frame deletion system for genetic studies of Streptococcus mutans. Oral Microbiol Immunol. 2007;22:95–102. [DOI] [PubMed] [Google Scholar]
- Meyer MM. The role of mRNA structure in bacterial translational regulation. WIREs RNA. 2017;8. [DOI] [PubMed] [Google Scholar]
- Miller KE, Kim Y, Huh WKet al. Bimolecular fluorescence complementation (BiFC) analysis: advances and recent applications for genome-Wide interaction studies. J Mol Biol. 2015;427:2039–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mintz KP, Fives-Taylor PM. impA, a gene coding for an inner membrane protein, influences colonial morphology of Actinobacillus actinomycetemcomitans. Infect Immun. 2000;68:6580–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra A, Aja E, Fletcher HM. Role of superoxide reductase FA796 in oxidative stress resistance in filifactor alocis. Sci Rep. 2020;10:9178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki K. Molecular engineering of a PheS counterselection marker for improved operating efficiency in Escherichia coli. BioTechniques. 2015;58:86–88. [DOI] [PubMed] [Google Scholar]
- Morrison DA, Khan R, Junges Ret al. Genome editing by natural genetic transformation in Streptococcus mutans. J Microbiol Methods. 2015;119:134–41. [DOI] [PubMed] [Google Scholar]
- Mu R, Anderson D, Merritt Jet al. Post-translational modification of Streptococcus sanguinis SpxB influences protein solubility and H2 O2 production. Mol Oral Microbiol. 2021;36:267–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu R, Shinde P, Zou Zet al. Examining the protein interactome and subcellular localization of rnase J2 complexes in Streptococcus mutans. Front Microbiol. 2019;10:2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata T, Ishikawa M, Shibuya Ket al. Method for functional analysis of a gene of interest in Streptococcus mutans: gene disruption followed by purification of a polyhistidine-tagged gene product. J Microbiol Methods. 2018;155:49–54. [DOI] [PubMed] [Google Scholar]
- Murray JL, Connell JL, Stacy Aet al. Mechanisms of synergy in polymicrobial infections. J Microbiol. 2014;52:188–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myscofski DM, Dutton EK, Bolken TCet al. Expression and purification of histidine-tagged proteins from the gram-positive Streptococcus gordonii SPEX system. Protein Expression Purif. 2000;20:112–23. [DOI] [PubMed] [Google Scholar]
- Naito M, Belvin BR, Shoji Met al. Insertional inactivation of Prevotella intermedia OxyR results in reduced survival with oxidative stress and in the presence of host cells. Microorganisms. 2021,551, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan AM, Ramsey MM, Stacy Aet al. Defining genetic fitness determinants and creating genomic resources for an oral pathogen. Appl Environ Microbiol. 2017;83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natori Y, Kano Y, Imamoto F. Genetic transformation of Lactobacillus casei by electroporation. Biochimie. 1990;72:265–9. [DOI] [PubMed] [Google Scholar]
- Nilsson M, Christiansen N, Hoiby Net al. A mariner transposon vector adapted for mutagenesis in oral streptococci. Microbiologyopen. 2014;3:333–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa K, Tanaka Y. A simple mutagenesis using natural competence in Tannerella forsythia. J Microbiol Methods. 2013;94:378–80. [DOI] [PubMed] [Google Scholar]
- Okinaga T, Xie Z, Niu Get al. Examination of the hdrRM regulon yields insight into the competence system of Streptococcus mutans. Mol Oral Microbiol. 2010;25:165–77. [DOI] [PubMed] [Google Scholar]
- Oozeer R, Goupil-Feuillerat N, Alpert CAet al. Lactobacillus casei is able to survive and initiate protein synthesis during its transit in the digestive tract of human flora-associated mice. Appl Environ Microbiol. 2002;68:3570–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paiano A, Margiotta A, De Luca Met al. Yeast two-Hybrid assay to identify interacting proteins. Curr Proto Protein Sci. 2019;95:e70. [DOI] [PubMed] [Google Scholar]
- Parrish JR, Gulyas KD, Finley RL Jr. Yeast two-hybrid contributions to interactome mapping. Curr Opin Biotechnol. 2006;17:387–93. [DOI] [PubMed] [Google Scholar]
- Pavlova N, Kaloudas D, Penchovsky R. Riboswitch distribution, structure, and function in bacteria. Gene. 2019;708:38–48. [DOI] [PubMed] [Google Scholar]
- Peek CT, Ibberson CB, Cassat JE. Identification of virulence determinants during host-Pathogen interaction using tn-Seq technology. Methods Mol Biol. 2020;2069:155–75. [DOI] [PubMed] [Google Scholar]
- Peluso EA, Scheible M, Ton-That Het al. Genetic manipulation and virulence assessment of fusobacterium nucleatum. Curr Proto Microbiol. 2020;57:e104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng X, Zhang Y, Bai Get al. Cyclic di-AMP mediates biofilm formation. Mol Microbiol. 2016;99:945–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Arellano I, Perez-Martinez G. Optimization of the green fluorescent protein (GFP) expression from a lactose-inducible promoter in Lactobacillus casei. FEMS Microbiol Lett. 2003;222:123–7. [DOI] [PubMed] [Google Scholar]
- Perez-Arellano I, Zuniga M, Perez-Martinez G. Construction of compatible wide-host-range shuttle vectors for lactic acid bacteria and Escherichia coli. Plasmid. 2001;46:106–16. [DOI] [PubMed] [Google Scholar]
- Perry D, Kuramitsu HK. Genetic transformation of Streptococcus mutans. Infect Immun. 1981;32:1295–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen FC, Ahmed NA, Naemi Aet al. LuxS-mediated signalling in Streptococcus anginosus and its role in biofilm formation. Antonie Van Leeuwenhoek. 2006;90:109–21. [DOI] [PubMed] [Google Scholar]
- Pingoud A, Fuxreiter M, Pingoud Vet al. Type II restriction endonucleases: structure and mechanism. Cell Mol Life Sci. 2005;62:685–707. [DOI] [PubMed] [Google Scholar]
- Plasterk RHA. Brenner's Encyclopedia of Genetics. Elsevier; 2013. [Google Scholar]
- Polissi A, Pontiggia A, Feger Get al. Large-scale identification of virulence genes from Streptococcus pneumoniae. Infect Immun. 1998;66:5620–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin H, Zou Z, Anderson Det al. The transcription regulator BrsR serves as a network hub of natural competence protein-protein interactions in Streptococcus mutans. Proc Natl Acad Sci. 2021;118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainey K, Wilson L, Barnes Set al. Quantitative proteomics uncovers the interaction between a virulence factor and mutanobactin synthetases in Streptococcus mutans. Msphere. 2019;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran G, Bikard D. Editing the microbiome the CRISPR way. Philosop Transact Royal Soc B: Biolog Sci. 2019;374:20180103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsay JP, Firth N. Diverse mobilization strategies facilitate transfer of non-conjugative mobile genetic elements. Curr Opin Microbiol. 2017;38:1–9. [DOI] [PubMed] [Google Scholar]
- Rao DN, Dryden DT, Bheemanaik S. Type III restriction-modification enzymes: a historical perspective. Nucleic Acids Res. 2014;42:45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reck M, Tomasch J, Wagner-Dobler I. The alternative Sigma factor SigX controls bacteriocin synthesis and competence, the two quorum sensing regulated traits in Streptococcus mutans. PLos Genet. 2015;11:e1005353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees JF, de Wergifosse B, Noiset Oet al. The origins of marine bioluminescence: turning oxygen defence mechanisms into deep-sea communication tools. J Exp Biol. 1998;201:1211–21. [DOI] [PubMed] [Google Scholar]
- Remington SJ. Fluorescent proteins: maturation, photochemistry and photophysics. Curr Opin Struct Biol. 2006;16:714–21. [DOI] [PubMed] [Google Scholar]
- Reyrat JM, Pelicic V, Gicquel Bet al. Counterselectable markers: untapped tools for bacterial genetics and pathogenesis. Infect Immun. 1998;66:4011–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AP, Mullany P. Tn916-like genetic elements: a diverse group of modular mobile elements conferring antibiotic resistance. FEMS Microbiol Rev. 2011;35:856–71. [DOI] [PubMed] [Google Scholar]
- Ruan Y, Zhu L, Li Q. Improving the electro-transformation efficiency of Corynebacterium glutamicum by weakening its cell wall and increasing the cytoplasmic membrane fluidity. Biotechnol Lett. 2015;37:2445–52. [DOI] [PubMed] [Google Scholar]
- Sakakibara J, Nagano K, Murakami Yet al. Loss of adherence ability to human gingival epithelial cells in S-layer protein-deficient mutants of Tannerella forsythensis. Microbiology. 2007;153:866–76. [DOI] [PubMed] [Google Scholar]
- Salvadori G, Junges R, Khan Ret al. Natural transformation of oral streptococci by use of synthetic pheromones. Methods Mol Biol. 2017;1537:219–32. [DOI] [PubMed] [Google Scholar]
- Salvadori G, Junges R, Morrison DAet al. Overcoming the barrier of low efficiency during genetic transformation of Streptococcus mitis. Front Microbiol. 2016;07:1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro F, Vianna ME, Roberts AP. Variation on a theme; an overview of the Tn916/Tn1545 family of mobile genetic elements in the oral and nasopharyngeal streptococci. Front Microbiol. 2014;5:535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Wu J, Kuramitsu H. The sgp gene modulates stress responses of Streptococcus mutans: utilization of an antisense RNA strategy to investigate essential gene functions. FEMS Microbiol Lett. 1998;159:241–5. [DOI] [PubMed] [Google Scholar]
- Saulis G. Cell electroporation: estimation of the number of pores and their sizes. Biomed Sci Instrum. 1999;35:291–6. [PubMed] [Google Scholar]
- Seepersaud R, Bensing BA, Yen YTet al. Asp3 mediates multiple protein-protein interactions within the accessory Sec system of Streptococcus gordonii. Mol Microbiol. 2010;78:490–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seitz P, Blokesch M. Cues and regulatory pathways involved in natural competence and transformation in pathogenic and environmental gram-negative bacteria. FEMS Microbiol Rev. 2013;37:336–63. [DOI] [PubMed] [Google Scholar]
- Shepard BD, Gilmore MS. Electroporation and efficient transformation of Enterococcus faecalis grown in high concentrations of glycine. Methods Mol Biol. 1995;47:217–26. [DOI] [PubMed] [Google Scholar]
- Shi Y, Ratnayake DB, Okamoto Ket al. Genetic analyses of proteolysis, hemoglobin binding, and hemagglutination of Porphyromonas gingivalis. Construction of mutants with a combination of rgpA, rgpB, kgp, and hagA. J Biol Chem. 1999;274:17955–60. [DOI] [PubMed] [Google Scholar]
- Shields RC, Kaspar JR, Lee Ket al. Fluorescence tools adapted for real-Time monitoring of the behaviors of Streptococcus species. Appl Environ Microbiol. 2019;85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields RC, O'Brien G, Maricic Net al. Genome-Wide screens reveal new gene products that influence genetic competence in Streptococcus mutans. J Bacteriol. 2018;200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields RC, Walker AR, Maricic Net al. Repurposing the Streptococcus mutans CRISPR-Cas9 system to understand essential gene function. PLoS Pathog. 2020;16:e1008344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinozaki-Kuwahara N, Shiroza T, Hayakawa Met al. Expression of the gtfI gene from Streptococcus sobrinus in Streptococcus anginosus using integration-mediated transformation system. Biochimica Et Biophysica Acta (BBA) - General Subjects. 2005;1722:189–99. [DOI] [PubMed] [Google Scholar]
- Shuman HA, Silhavy TJ. The art and design of genetic screens: escherichia coli. Nat Rev Genet. 2003;4:419–31. [DOI] [PubMed] [Google Scholar]
- Simon L, Fremaux C, Cenatiempo Yet al. Luminescent method for the detection of antibacterial activities. Appl Microbiol Biotechnol. 2001;57:757–63. [DOI] [PubMed] [Google Scholar]
- Slivienski-Gebhardt LL, Izard J, Samsonoff WAet al. Development of a novel chloramphenicol resistance expression plasmid used for genetic complementation of a fliG deletion mutant in Treponema denticola. Infect Immun. 2004;72:5493–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreenivasan PK, LeBlanc DJ, Lee LNet al. Transformation of actinobacillus actinomycetemcomitans by electroporation, utilizing constructed shuttle plasmids. Infect Immun. 1991;59:4621–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacy A, Fleming D, Lamont RJet al. A commensal bacterium promotes virulence of an opportunistic pathogen via cross-Respiration. Mbio. 2016;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strand TA, Lale R, Degnes KFet al. A new and improved host-independent plasmid system for RK2-based conjugal transfer. PLoS One. 2014;9:e90372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed AJ, Anderson JC. Applications of bioluminescence in biotechnology and beyond. Chem Soc Rev. 2021;50:5668–705. [DOI] [PubMed] [Google Scholar]
- Szafranski SP, Slots J, Stiesch M. The human oral phageome. Periodontol 2000. 2021;86:79–96. [DOI] [PubMed] [Google Scholar]
- Takayama K, Hayes B, Vestling MMet al. Transposon-5 mutagenesis transforms Corynebacterium matruchotii to synthesize novel hybrid fatty acids that functionally replace corynomycolic acid. Biochem J. 2003;373:465–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao L, Pavlova SI, Ji Xet al. A novel plasmid for delivering genes into mammalian cells with noninvasive food and commensal lactic acid bacteria. Plasmid. 2011;65:8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay WH, Chong KK, Kline KA. Polymicrobial-Host interactions during infection. J Mol Biol. 2016;428:3355–71. [DOI] [PubMed] [Google Scholar]
- Tenover FC, Fennell CL, Lee Let al. Characterization of two plasmids from Campylobacter jejuni isolates that carry the aphA-7 kanamycin resistance determinant. Antimicrob Agents Chemother. 1992;36:712–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson VJ, Bhattacharjee MK, Fine DHet al. Direct selection of IS903 transposon insertions by use of a broad-host-range vector: isolation of catalase-deficient mutants of actinobacillus actinomycetemcomitans. J Bacteriol. 1999;181:7298–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian XL, Dong G, Liu Tet al. MecA protein acts as a negative regulator of genetic competence in Streptococcus mutans. J Bacteriol. 2013;195:5196–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tock MR, Dryden DT. The biology of restriction and anti-restriction. Curr Opin Microbiol. 2005;8:466–72. [DOI] [PubMed] [Google Scholar]
- Tribble GD, Rigney TW, Dao DHet al. Natural competence is a major mechanism for horizontal DNA transfer in the oral pathogen Porphyromonas gingivalis. Mbio. 2012;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tryfona T, Bustard MT. Enhancement of biomolecule transport by electroporation: a review of theory and practical application to transformation of Corynebacterium glutamicum. Biotechnol Bioeng. 2006;93:413–23. [DOI] [PubMed] [Google Scholar]
- Tsang P, Merritt J, Nguyen Tet al. Identification of genes associated with mutacin I production in Streptococcus mutans using random insertional mutagenesis. Microbiology. 2005;151:3947–55. [DOI] [PubMed] [Google Scholar]
- Turnbough CL Jr. Regulation of bacterial gene expression by transcription attenuation. Microbiol Mol Biol Rev. 2019;83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueki T, Inouye S, Inouye M. Positive-negative KG cassettes for construction of multi-gene deletions using a single drug marker. Gene. 1996;183:153–7. [DOI] [PubMed] [Google Scholar]
- Upton M, Tagg JR, Wescombe Pet al. Intra- and interspecies signaling between Streptococcus salivarius and Streptococcus pyogenes mediated by SalA and SalA1 lantibiotic peptides. J Bacteriol. 2001;183:3931–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Duyne GD. Cre recombinase. Microbiology Spectrum. 2015;3:MDNA3–0014-2014. [DOI] [PubMed] [Google Scholar]
- van Opijnen T, Camilli A. Transposon insertion sequencing: a new tool for systems-level analysis of microorganisms. Nat Rev Microbiol. 2013;11:435–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Opijnen T, Lazinski DW, Camilli A. Genome-Wide fitness and genetic interactions determined by tn-seq, a high-Throughput massively parallel sequencing method for microorganisms. Curr Proto Mol Biol. 2014;106: 7 16 11-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzulli L, Guzman CA, Colwell RRet al. Dual role colonization factors connecting Vibrio cholerae's lifestyles in human and aquatic environments open new perspectives for combating infectious diseases. Curr Opin Biotechnol. 2008;19:254–9. [DOI] [PubMed] [Google Scholar]
- Vickerman MM, Mansfield JM, Zhu Met al. Codon-optimized fluorescent mTFP and mCherry for microscopic visualization and genetic counterselection of streptococci and enterococci. J Microbiol Methods. 2015;116:15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Kraig E, Kolodrubetz D. Use of defined mutants to assess the role of the Campylobacter rectus S-layer in bacterium-epithelial cell interactions. Infect Immun. 2000;68:1465–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Kuramitsu HK. Inducible antisense RNA expression in the characterization of gene functions in Streptococcus mutans. Infect Immun. 2005;73:3568–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Cui Y, Qu X. Optimization of electrotransformation (ETF) conditions in lactic acid bacteria (LAB). J Microbiol Methods. 2020;174:105944. [DOI] [PubMed] [Google Scholar]
- Wang Y, Goodman SD, Redfield RJet al. Natural transformation and DNA uptake signal sequences in actinobacillus actinomycetemcomitans. J Bacteriol. 2002;184:3442–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver CA, Chen YM, Burne RA. Inactivation of the ptsI gene encoding enzyme I of the sugar phosphotransferase system of Streptococcus salivarius: effects on growth and urease expression. Microbiology. 2000;146:1179–85. [DOI] [PubMed] [Google Scholar]
- Weaver KE, Chen Y, Miiller EMet al. Examination of Enterococcus faecalis toxin-Antitoxin system toxin fst function utilizing a pheromone-Inducible expression vector with tight repression and broad dynamic range. J Bacteriol. 2017;199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ween O, Teigen S, Gaustad Pet al. Competence without a competence pheromone in a natural isolate of Streptococcus infantis. J Bacteriol. 2002;184:3426–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welker DL, Hughes JE, Steele JLet al. High efficiency electrotransformation of Lactobacillus casei. FEMS Microbiol Lett. 2015;362:1–6. [DOI] [PubMed] [Google Scholar]
- Whittaker CJ, Clemans DL, Kolenbrander PE. Insertional inactivation of an intrageneric coaggregation-relevant adhesin locus from Streptococcus gordonii DL1 (Challis). Infect Immun. 1996;64:4137–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widder EA. Bioluminescence in the ocean: origins of biological, chemical, and ecological diversity. Science. 2010;328:704–8. [DOI] [PubMed] [Google Scholar]
- Wong JJ, Lu J, Glover JN. Relaxosome function and conjugation regulation in F-like plasmids - a structural biology perspective. Mol Microbiol. 2012;85:602–17. [DOI] [PubMed] [Google Scholar]
- Wright GD, Thompson PR. Aminoglycoside phosphotransferases: proteins, structure, and mechanism. Front Biosci. 1999;4:D9–21. [DOI] [PubMed] [Google Scholar]
- Wrist A, Sun W, Summers RM. The theophylline aptamer: 25 years as an important tool in cellular engineering research. ACS Synthetic Biology. 2020;9:682–97. [DOI] [PubMed] [Google Scholar]
- Wu C, Ton-That H. Allelic exchange in Actinomyces oris with mCherry fluorescence counterselection. Appl Environ Microbiol. 2010;76:5987–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Okinaga T, Niu Get al. Identification of a novel bacteriocin regulatory system in Streptococcus mutans. Mol Microbiol. 2010;78:1431–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Okinaga T, Qi Fet al. Cloning-independent and counterselectable markerless mutagenesis system in Streptococcus mutans. Appl Environ Microbiol. 2011;77:8025–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Qi F, Merritt J. Development of a tunable wide-range gene induction system useful for the study of streptococcal toxin-antitoxin systems. Appl Environ Microbiol. 2013;79:6375–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Zou Z, Raz Aet al. Regulatory control of the Streptococcus mutans HdrRM LytTR Regulatory System functions via a membrane sequestration mechanism. Mol Microbiol. 2020;114:681–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin Y, Guo T, Mu Yet al. A single-Plasmid genome editing system for metabolic engineering of Lactobacillus casei. Front Microbiol. 2018;9:3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Stewart PE, Shi Xet al. Development of a transposon mutagenesis system in the oral spirochete Treponema denticola. Appl Environ Microbiol. 2008;74:6461–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung MK, Kozelsky CS. Transformation of Actinomyces spp. by a gram-negative broad-host-range plasmid. J Bacteriol. 1994;176:4173–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida A, Kuramitsu HK. Streptococcus mutans biofilm formation: utilization of a gtfB promoter-green fluorescent protein (PgtfB::gfp) construct to monitor development. Microbiology. 2002;148:3385–94. [DOI] [PubMed] [Google Scholar]
- Yoshimoto H, Takahashi Y, Hamada Net al. Genetic transformation of Porphyromonas gingivalis by electroporation. Oral Microbiol Immunol. 1993;8:208–12. [DOI] [PubMed] [Google Scholar]
- Youell J, Firman K. Mechanistic insight into type I restriction endonucleases. Front Biosci. 2012;17:2122–39. [DOI] [PubMed] [Google Scholar]
- Zambito G, Chawda C, Mezzanotte L. Emerging tools for bioluminescence imaging. Curr Opin Chem Biol. 2021;63:86–94. [DOI] [PubMed] [Google Scholar]
- Zechner EL, Moncalian G, de la Cruz F. Relaxases and plasmid transfer in gram-Negative bacteria. Curr Top Microbiol Immunol. 2017;413:93–113. [DOI] [PubMed] [Google Scholar]
- Zhang J, Biswas I. 3'-Phosphoadenosine-5'-phosphate phosphatase activity is required for superoxide stress tolerance in Streptococcus mutans. J Bacteriol. 2009;191:4330–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Zou Z, Kreth Jet al. Recombineering in Streptococcus mutans using direct repeat-Mediated cloning-Independent markerless mutagenesis (DR-CIMM). Front Cell Infect Microbiol. 2017;7:202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Fives-Taylor P, Wu H. The utility of affinity-tags for detection of a streptococcal protein from a variety of streptococcal species. J Microbiol Methods. 2008;72:249–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P, Li X, Qi F. Establishment of a counter-selectable markerless mutagenesis system in Veillonella atypica. J Microbiol Methods. 2015;112:70–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P, Li X, Qi F. Identification and characterization of a haem biosynthesis locus in Veillonella. Microbiology. 2016;162:1735–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Z, Qin H, Brenner AEet al. LytTR Regulatory Systems: a potential new class of prokaryotic sensory system. PLos Genet. 2018;14:e1007709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–15. [DOI] [PMC free article] [PubMed] [Google Scholar]