

Abstract
Pancreatic cancer (PC) presents challenges due to limited treatment options and patients seek complementary therapies alongside conventional treatments to improve well-being. This study uses computational drug discovery approaches to find potential phytochemicals from S. singueana for PC treatment. Among the 38 phytochemicals screened from S. singueana, specific inhibitors against PC were selected. Protein–protein interaction (PPI) network analysis highlighted key targets with high degrees, including PTEN (8) and PTK2 (7) genes, along with their respective proteins 5BZX and 3BZ3, which were employed for molecular docking studies. 1-methylchrysene and 3-methyl-1,8,9-anthracenetriol showed strong binding affinities of − 9.2 and − 8.1 Kcal/mol, respectively. Molecular dynamics simulations lasting 300 ns assessed structural stability and interaction energy of compound-target dockings: 1-methylchrysene-PTEN and 3-methyl-1,8,9-anthracenetriol-PTK2. In molecular dynamics simulations, the 3-methyl-1,8,9-anthracenetriol-PTK2 complex showed lower RMSD, RMSF, radius of gyration, solvent-accessible surface area, and more hydrogen bonds than the 1-methylchrysene-PTEN complex. The 3-methyl-1,8,9-anthracenetriol-PTK2 complex exhibited significantly stronger binding with a binding free energy (ΔG) of − 21.92 kcal/mol compared to the less favourable ΔG of − 10.65 kcal/mol for the 1-methylchrysene-PTEN complex. The consistent and stable binding interaction observed in the 3-methyl-1,8,9-anthracenetriol-PTK2 complex highlights its potential as a potent inhibitor of Focal Adhesion Kinase 1. Consequently, it emerges as a promising lead compound for the development of pancreatic cancer therapeutics. Conversely, the fluctuations observed in the 1-methylchrysene-PTEN complex indicate a less stable binding interaction. This indicates the potential of 3-methyl-1,8,9-anthracenetriol as a primary candidate for pancreatic cancer treatment. These findings improve our grasp of S. singueana’s multi-target effects and its promise in addressing pancreatic cancer. Nevertheless, additional in-vivo and in-vitro studies are required to validate their effectiveness and therapeutic potential.
Supplementary Information
The online version contains supplementary material available at 10.1007/s40203-023-00179-9.
Keywords: Molecular docking, MD simulation, Pancreatic cancer, PPI network, Senna singueana
Introduction
Over the past two decades, the number of new cases of pancreatic cancer has doubled, rising from 196,000 patients worldwide in 1990 to 441,000 in 2017 (Klein 2021). In 2020 according to the 2020 global cancer statistics total of 495,773 new cases of pancreatic cancer were reported (Sung et al. 2021). Pancreatic cancer rates are expected to rise soon because of changing lifestyles globally and in India (Gaidhani and Balasubramaniam 2021). Pancreatic ductal adenocarcinoma (PDAC) is the most common exocrine neoplasm and the most common cancer overall among pancreatic cancer histological subtypes. Slightly more common in men, and usually affects older people (Siegel et al. 2017). In the United States, PDAC has surpassed breast cancer to rank third among cancer-related causes of death. Unfortunately, PDAC is expected to surpass colorectal cancer before 2040 (Daina et al. 2017). Diabetes mellitus is also thought to play a role in the development of pancreatic cancer, though the exact mechanism by which this occurs is unknown (Larsson et al. 2005). Alcoholism, obesity, and various genetic syndromes are also known risk factors for pancreatic cancer, though they account for less than 10% of all cases (Adler 2021).
The treatment regime for pancreatic cancer typically involves a combination of different modalities depending on the stage and extent of the disease. The primary treatment options include surgery, chemotherapy, radiation therapy, targeted therapy, and immunotherapy. Among them, surgery is considered the primary curative treatment for pancreatic cancer, particularly for patients with localized disease that has not spread to distant organs. However, due to the limited routine screening methods for early pancreatic cancer detection, only 15–20% of patients are eligible for the corresponding surgery (Strobel et al. 2019). In preclinical research and clinical trials, promising therapies like immunotherapy and targeted therapy were also tested; however, due to the prevalence of therapeutic resistance, almost all these approaches show only marginally significant benefits over conventional chemotherapy when used to treat pancreatic cancer (Huang et al. 2022b). It indicates that there is an urgent need for highly effective pancreatic cancer treatment alternatives.
Medicinal plants are a great hope for the development of new drugs because they meet the need for treatment and are very beneficial to human health. Several medicinal herbs including Senna singueana (Delile) Lock, Kaempferia parviflora Wall. ex Baker, and Ziziphus nummularia (Burm.f.) Wight & Arn. have been reported for the treatment of cancer (Mesmar et al. 2021; Sun et al. 2021). Among these plants, Senna singueana is also reported for the treatment of pancreatic cancer by various African ethnic groups. S. singueana, commonly known as Wild Cassia or Pambisuma (Margi) and rumfu (Hausa) in northern Nigeria, is a traditional African medicinal plant. In Nigeria, it has been traditionally utilized in the treatment of cancer, as well as for managing conditions like hernias, constipation, snakebites, and urinary schistosomiasis (Adzu et al. 2003; Gidado et al. 2019). S. singueana is also known as Cassia singueana, and has been previously studied for its antidiabetic, antipyretic, antinociceptive, antioxidant, anticancer, antiplasmodial/antimalarial, hypolipidemic, and hepatoprotective properties. Anthraquinones, proanthocyanidins, other phenolics, fatty acids, amino acids, and triterpenoids have been also investigated from the various parts of this plant during phytochemical investigations (Adedoyin et al. 2019).
Computational approaches have gained prominence in herbal drug discovery due to their ability to rapidly screen and analyze large libraries of herbal compounds. Several studies have utilized these techniques to identify potential herbal drugs and explore their therapeutic properties. Protein–protein interactions (PPIs) are important in many biological processes and are appealing drug discovery targets, including herbal drug discovery (Ni et al. 2019). Molecular docking and molecular dynamics (MD) simulations are computational techniques used to study PPIs and aid in the discovery of potential herbal drugs (Salo-Ahen et al. 2021). Molecular docking algorithms evaluate the complementarity of the ligand and target protein, considering factors such as shape, electrostatics, and hydrophobic interactions. Molecular dynamic simulations can be employed to study the stability, flexibility, and conformational changes of protein-ligand complexes, as well as the binding energetics and thermodynamics (Ibrahim et al. 2022). It is worth noting that while these computational techniques provide valuable insights, experimental validation through in-vitro and in-vivo studies is essential to confirm the efficacy, safety, and pharmacological properties of herbal compounds before they can be considered for clinical use (Majeed et al. 2021).
Pancreatic cancer poses a significant challenge in terms of effective treatment options due to its high mortality rate. Thus, there is a pressing need to identify suitable therapeutic approaches to combat this disease. In traditional Nigerian medicine, S. singueana has been historically employed for the treatment of cancer by various ethnic groups. With this outset, the present study aims to investigate the therapeutic potential of S. singueana phytochemicals in attenuating pancreatic cancer through the exploration of protein-protein interactions, molecular docking, and molecular dynamics simulations. The study is also steered to analyze the interactions between the phytochemicals derived from S. singueana and target proteins involved in pancreatic cancer. To investigate the pharmacological characteristics and pharmacokinetics of the phytochemicals, the ADMET (absorption, distribution, metabolism, excretion, and toxicity) of the substances was examined. It seeks to assess the binding affinities, stability, and dynamics of these protein-ligand complexes using computational techniques. The ultimate goal is to unravel the mechanisms by which the phytochemicals from S. singueana could potentially modulate pancreatic cancer progression and provide insights into their therapeutic effects, aiding in the development of novel herbal-based interventions for pancreatic cancer treatment.
Materials and methods
Screening and selection of phytochemicals and potential targets
The selection of potential targets for pancreatic cancer and phytochemicals was based on an extensive review of published literature and GeneCard, focusing on the keyword “Pancreatic cancer.“ From this study, a set of 38 phytochemicals was identified as mentioned in the previous study investigated on gas chromatography-mass spectrometry (GC–MS) and selected these phytochemicals as potential inhibitors for pancreatic cancer targets. The PubChem compound database was utilized to retrieve the structures of the compounds in three-dimensional structure data file (SDF) format. Ligand optimization, energy minimization, and conversion of the retrieved two-dimensional (2D) ligands to 3D PDB format were performed using Open Babel. Specifically, the proteins encode by Phosphatase and Tensin homolog (PTEN) and Protein Tyrosine Kinase 2 ( PTK2) were chosen as targets to investigate the inhibitory potential. The corresponding three-dimensional (3D) structures of these compounds were obtained from the Protein Data Bank (PDB) database. Belzutifan, an FDA-approved drug, was used as a positive control in the study.
Protein–protein interaction (PPI)
To investigate more potential genes of PTEN family protein-protein interaction (PPI) was used. STRING (http://string-db.org/cgi/input.pl), an online PPI database that is popular in bioinformatics and network pharmacology and can provide all PPI network data for free, scored each interaction between input proteins (Szklarczyk et al. 2019). The higher the score, the more reliable the data. The targeted protein PTEN was entered in the protein retrieval box after clicking “Search” on the first page of the STRING database. Homo sapiens was the only species. Using Cityscape 3.8.0 software, the top ten interacting proteins with the highest scores were chosen for PPI network construction and a medium confidence score with a correlation degree of ≥ 0.007.
Gene Ontology and pathway analysis
Gene Ontology (GO) is a conceptual framework and set of concepts for describing the functions of gene products in all organisms. For GO enrichment analysis, the biological process (BP), cellular component (CC), and molecular function (MF) were carried out using R-package (R x64 4.3) with Bioconductor Cluster Profiler.
Molecular docking
To examine the binding interactions between the selected compounds and the target proteins, docking methods were employed. Molecular docking allowed for the prediction of the binding modes and affinities of the compounds with the target proteins. In this study, molecular docking was performed to generate a population of possible ligand conformations and orientations at the binding site with the help of AutoDock Vina (Trott and Olson 2009). The Protein Data Bank of the RCSB (Research Collaboratory for Structural Bioinformatics) (https://www.rcsb.org/) was used to retrieve the three-dimensional crystal structure of the PTEN gene protein PDB ID 5BZX with a resolution of 2.50 Å and PTK2 gene protein PDB ID 3BZ3 with a resolution 2.20 Å. 5BZX protein was containing four chains with a total of 314 amino acids and 3BZ3 protein contained one chain with a sequence length of 276. Proteins were prepared with AutoDock. From the crystal structure, the other three chains were removed from 5BZX. Only chain A was selected and from the chain, heteroatoms and water molecules were also removed. The UCSF Chimera integrated with Modeller was used to add missing residues in the structures and the best model was selected. This approach allows researchers to select the most suitable model with added missing residues, enhancing the completeness and accuracy of the protein structure (Webb and Sali 2016).
Molecular dynamics simulation
In the present study, molecular dynamics (MD) simulations were conducted using GROMACS software (Version 2023.1) on a Linux operating system (Van Der Spoel et al. 2005). The visualization of results was performed using Microsoft Excel software. To establish the protein topology, the charmm36 force field was employed, while the ligand topology and parameters were generated using the CHARMM all atoms force field, suitable for both CHARMM and GROMACS. Each system was solvated using the TIP 3-point solvent model, followed by neutralization using appropriate quantities of Na+ and Cl−. Energy minimization techniques were applied to reduce the overall potential energy of the protein and ligand. Specifically, the steepest descent minimization algorithm was used, terminating at more than 50,000 steps and a maximum force threshold of 10.0 kJ/mol. Subsequently, each system underwent a 1000-ps NVT equilibration and a 1000-ps NPT equilibration process. The NPT equilibration was performed under 1 bar pressure and 300 K temperature. The MD simulations were conducted for each equilibrated system, spanning 300 ns with a time step of 2 fs. Structural coordinates were saved every 20 ps during the simulation. Following the removal of constraints, a 300 ns molecular dynamics simulation was carried out. The particle mesh Ewald (PME) algorithm was employed to accurately compute long-range electrostatic interactions (Reddy et al. 2018). The obtained results were analyzed using various parameters, such as root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), hydrogen bonds (HB), and solvent accessible surface area (SASA). Each complex was performed in replicate to enhance the reliability, robustness, and accuracy of the findings. These analyses were performed using the built-in tools of Gromacs (Version 2023.1) and data visualization was accomplished by utilizing XMGRACE, VMD and Microsoft Excel tools.
Free energy calculations
To compute the binding free energy (ΔG) of ligand–protein complexes, we employed the gmx_MMPBSA package (version 1.6.2) utilizing the MM/PBSA (molecular mechanics/Poisson–Boltzmann surface area) method (Miller et al. 2012; Valdés-Tresanco et al. 2021). This method utilized Gromacs trajectory, topology, and index files of the target complexes. The total binding free energy (ΔG total) was determined by subtracting ΔG receptor from ΔG complex and then subtracting ΔG ligand from the result. Subsequently, the data was analyzed using the gmxMMPBSA ana module.
Drug-likeness and ADMET prediction
The SwissADME (http://www.swissadme.ch) server was used to check the drug-likeness properties as well as physicochemical properties, pharmacokinetics, drug-likeness, and medicinal chemistry friendliness, including proprietary methods such as the BOILED-Egg, iLOGP, and Bioavailability Radar of S. singueana natural compounds (Daina et al. 2017). The ADMETlab 2.0 web server was also utilized to assess the toxicity, and absorption characteristics of the phytochemicals derived from S. singueana (Xiong et al. 2021).
Results
Protein–protein interaction (PPI) network of targeted gene
The PPI network of targeted genes was constructed to find out more significant genes which were having highest degree of connectivity with the other genes, responsible for causing pancreatic cancer. The top ten genes with the highest degree were PTEN (8), PTK2 (7), GRB2 (6), PIK3R1 (6), PXN (6), BCAR1 (5), ITGB1 (6), SLC9A33R1 (1), TP53 (2), and NEDD4 (1) (Fig. 1a, b). PTK2 along with PTEN genes were chosen for this study because they had the highest degree scores.
Fig. 1.

a PPI network of PTEN gene with other proteins; b top 10 genes with their degree score
Gene Ontology enrichment analysis of targets
The enrichment analysis uncovered significant associations in various Gene Ontology (GO) terms related to biological processes (BP), cellular components (CC), and molecular functions (MF). In terms of BP, GO terms like negative regulation of synapse organization, negative regulation of cell junction assembly, central nervous system neuron axonogenesis, negative regulation of organ growth, and regulation of focal adhesion assembly displayed strong associations, with high enrichment scores ranging from 5.81 to 5.13. In the category of cellular components, GO terms such as the cytoplasmic side of the plasma membrane, dendritic spine, neuron spine, cytoplasmic side of the membrane, and apical plasma membrane exhibited varying degrees of enrichment, with scores ranging from 4.3 to 3.4. Additionally, terms like Schmidt–Lanterman incisure, compact myelin, postsynaptic cytosol, and region of cytosol showed enrichment, though with lower scores. Regarding molecular functions, GO terms including inositol phosphate phosphatase activity, platelet-derived growth factor receptor binding, phosphatidylinositol-3-phosphatase activity, and phosphatidylinositol monophosphate phosphatase activity displayed enrichment scores ranging from 2.89 to 2.8 (Fig. 2a).
Fig. 2.


a Graphical representation of gene ontology enrichment analysis of genes, b represents the enrichment analysis of genes in the biological process by network interaction, c represents the enrichment analysis of genes in cellular component by network interaction, d represents the enrichment analysis of genes in molecular function by network interaction
The results of the network analysis revealed significant associations between specific genes and various biological processes, cellular components, and molecular functions. In terms of biological processes (BP), the PTEN gene was found to be highly involved in the response to nutrient levels, endomembrane system organization, muscle system processes, and regulation of DNA-binding transcription factor activity. These associations were statistically significant, with P values of 0.049515827, 0.048998979, 0.048895592, and 0.047654516, respectively. On the other hand, the PTK2 gene was highly involved in the regulation of cell-cell adhesion, the establishment of organelle localization, and the regulation of lipid metabolic processes, with the highest P values being 0.00097033, 0.000960813, and 0.000960552, respectively (Fig. 2b). Regarding the Cellular component analysis, both PTEN and PTK2 genes showed significant involvement in several cellular locations. PTEN was highly associated with the apical part of the cell, apical plasma membrane, cytoplasmic side of the membrane, neuron spine, dendritic spine, and cytoplasmic side of the plasma membrane. The corresponding P values for these associations were 0.002200458, 0.001834562, 0.000620343, 0.000620343, 0.000620343, and 0.000620343, respectively (Fig. 2c). In terms of molecular function, PTEN was found to be highly involved in inositol phosphate phosphatase activity, platelet-derived growth factor receptor binding, and phosphatidylinositol-3-phosphatase activity. These associations had the highest P values of 0.010848351. Additionally, PTK2 showed significant involvement in SH2 domain binding, non-membrane spanning protein tyrosine kinase activity, and protein tyrosine kinase activity, with the highest P values being 0.010848351, 0.010848351, and 0.020023136, respectively (Fig. 2d). Overall, these results suggest that the PTEN and PTK2 genes play important roles in various biological processes, cellular components, and molecular functions. The associations identified through the network analysis provide valuable insights into the functional significance of these genes in the context of the studied system.
KEGG pathway enrichment analysis
In the KEGG pathway enrichment analysis, a total of 10 signaling pathways including small cell lung cancer, focal adhesion, human papillomavirus infection, PI3K-Akt signaling pathway, VEGF signaling pathway, and Central carbon metabolism in cancer were identified to be statistically significant with a P value less than 0.05. Among the genes studied, FAK (Focal Adhesion Kinase) emerged as the highest associated gene within the focal adhesion pathway. This finding suggests that FAK plays a crucial role in focal adhesion, which is consistent with its known involvement in cell adhesion and migration. In addition to FAK, another important gene identified in the focal adhesion pathway are PTEN (Phosphatase and Tensin Homolog), known for its tumor-suppressive functions and its involvement in regulating cell adhesion, migration, and invasion. The presence of PTEN in the focal adhesion pathway suggests its contribution to the modulation of adhesion-related processes. The enrichment analysis highlights the significance of both FAK and PTEN in the focal adhesion pathway, emphasizing their potential roles in cellular processes related to adhesion, migration, and potential cancer progression (Fig. 3a).
Fig. 3.

a KEGG enrichment analysis of genes in a network of multiple pathways, b key genes and their associations in the focal adhesion pathway
Molecular docking and interactions analysis
In this study, a total of 38 compounds from the plant S. singueana and the drug Belzutifan as positive control were selected to discover multi-targeting inhibitors that could effectively target key protein targets and utilize them as potential therapeutic agents. The phytochemicals derived from different parts of the plant, along with their corresponding CIDs, canonical smiles, and 2-D structures retrieved from PubChem, as depicted in supplementary file. The valuable insights were gained regarding the binding affinity and potential interactions between the S. singueana phytochemicals and the targeted 5BZX proteins, shedding light on their potential therapeutic applications and efficacy. It has been found that out of 38 compounds from S. singueana, only two compounds showed significant binding affinity (Table 1). The strongest binding affinity was achieved with a value of − 9.2 Kcal/mol for 1-methylchrysene and − 8.8 Kcal/mol for 9-benzylidenexanthene when interacting with 5BZX. 1-methylchrysene and 9-benzylidenexanthene showed higher binding affinity as compared to the standard drug Belzutifan (− 8.0 Kcal/mol). Belzutifan (CID: 117947097) and protein complex were stabilized with two conventional hydrogen bonds at ARG A:173, one halogen (fluorine) at PHE A:279, one Pi–Pi T-shaped at TYR A:180, one alkyl bond at LEU A:318 and two Pi-alkyl at ARG:173, TYR:177. The compound 1-methylchrysene (CID: 18779) and protein complex were stabilized by one Pi-donor hydrogen bond at ARG A:173, one Pi-sigma bond at ARG A:173, four Pi–Pi T-shaped bond at TYR:177, TYR:180, PHE: 279, one alkyl bond at ARG A:173 and two Pi-alkyl at ARG:173, TYR:176, and 9-benzylidenexanthene (CID: 623062) and complex of protein were stabilized by five Pi–Pi stacked bond at TYR:176, TYR:177, TYR:180 and one Pi-alkyl bond at ARG A:173.
Table 1.
Interaction indices of potential S. singueana compounds with 5BZX
| S. n. | Ligand name | Binding energy (kcal/mol) | Bond type | Bond length | Amino acid | Hydrogen bonds | Total bonds |
|---|---|---|---|---|---|---|---|
| 1 | Belzutifan | − 8.0 | Conventional hydrogen bond | 2.09, 2.14 | ARG:173 | 2 | 7 |
| Halogen (fluorine) | 3.45 | PHE:279 | |||||
| Pi–Pi T-shaped | 4.95 | TYR:180 | |||||
| Alkyl bond | 4.98 | LEU:318 | |||||
| Pi-alkyl | 4.84, 5.36 | ARG:173, TYR177 | |||||
| 2 | 1-methylchrysene | − 9.2 | Pi-donor hydrogen bond | 2.41 | ARG:173 | 1 | 9 |
| Pi-sigma | 3.96 | ARG:173 | |||||
| Pi–Pi T-shaped | 5.34, 5.24, 4.91, 5.19 | TYR:177, TYR:180, PHE: 279 | |||||
| Alkyl bond | 4.08 | ARG: 173 | |||||
| Pi-alkyl | 4.43, 4.77 | ARG:173, TYR:176 | |||||
| 3 | 9-benzylidenexanthene | − 8.8 | Pi–Pi stacked | 3.68, 4.05, 4.56, 4.91, 5.37 | TYR:176, TYR:177, TYR:180 | 0 | 6 |
| Pi-alkyl | 5.12 | ARG:173 |
Among the 38 compounds derived from S. singueana, only three compounds exhibited noteworthy binding affinity with 3BZ3. The highest binding affinity was obtained as − 8.9 Kcal/mol for 1-methylchrysene and − 8.6 Kcal/mol for 6-dehydroestradiol, − 8.1 for 3-methyl-1,8,9-anthracenetriol and for the positive control the binding affinity was obtained at − 7.6. It is clear from (Table 2) that 1-methylchrysene 6-dehydroestradiol and 3-methyl-1,8,9-anthracenetriol showed higher binding affinity against the known and active drug used Belzutifan (CID: 117947097) and protein complex were stabilized with one conventional hydrogen bond at ASP:151, six halogen (fluorine) at GLU:93, ARG:137, ARG:137, ASN:138, GLY:150, ASP:151, one Pi-sigma at LEU:154, one alkyl at ARG:137 and two Pi-alkyl bond at LEU:140, and LEU:154. The compound 1-methylchrysene (CID: 18779) and protein complex were stabilized by five Pi-sigma bonds at ILE:15, and LEU:140 and two Pi-alkyl bond at ALA:39, and LEU:140, 6-dehydroestradiol (CID: 245468) and complex of protein was stabilized by one Pi-sigma at ILE A:15 and nine alkyl bond at ILE:15, ALA:39, CYS:89, LEU:140, LEU:154, ILE:15, and VAL:23, and the compound 3-methyl-1,8,9-anthracenetriol (CID: 10288) and protein complex were stabilized with three conventional hydrogen bonds ILE:15, GLU:93, and CYS:89, two Pi-sigma at ILE:15, LEU:140 and two alkyl VAL:23, LEU:154 and six Pi-alkyl ILE:15, VAL:23, LEU:140, LEU:154 ALA:39. On the basis of molecular docking results, it was observed that the compound 1-methylchrysene (CID: 18779) with 5BZX and the compound 3-methyl-1,8,9-anthracenetriol (CID: 10288) with 3BZ3 were showing the best binding affinity with hydrogen interaction against positive control drug Belzutifan (Fig. 4a, b).
Table 2.
Interaction insights of potential S. singueana compounds with 3BZ3
| S. n. | Ligand name | Binding energy (kcal/mol) | Bond type | Bond length | Amino acid | Hydrogen bonds | Total bonds |
|---|---|---|---|---|---|---|---|
| 1 | Belzutifan | − 7.6 | Conventional hydrogen bond | 2.53 | ASP:151 | 1 | 11 |
| Halogen (fluorine) | 3.35, 3.09, 3.06, 3.60, 3.42, 3.60 | GLU:93, ARG:137, ARG:137, ASN:138, GLY:150, ASP:151 | |||||
| Pi-sigma bond | 3.60 | LEU:154 | |||||
| Alkyl bond | 4.94 | ARG:137 | |||||
| Pi-alkyl bond | 4.52, 5.19 | LEU:140, LEU:154 | |||||
| 2 | 1-methylchrysene | − 8.9 | Pi-sigma bond | 3.84, 3.80, 3.38, 3.36, 3.683 | ILE:15, LEU:140 | 0 | 7 |
| Pi-alkyl bond | 5.47, 4.38 | ALA:39, LEU:140 | |||||
| 3 | 6-dehydroestradiol | − 8.6 | Pi-sigma bond | 3.77 | ILE:15 | 0 | 10 |
| Alkyl bond | 4.47, 5.19, 3.62, 5.48, 4.18, 4.36, 5.47, 4.69, 4.31 | ILE:15, ALA:39, CYS:89, LEU:140, LEU:154, ILE:15, VAL:23 | |||||
| 4 | 3-methyl-1,8,9-anthracenetriol | − 8.1 | Conventional hydrogen bond | 2.58, 2.15, 3.08 | ILE:15, GLU:93, CYS:89 | 3 | 13 |
| Pi-sigma bond | 3.77, 3.73 | ILE:15, LEU:140 | |||||
| Alkyl bond | 4.73, 3.67 | VAL:23, LEU:154 | |||||
| Pi-alkyl bond | 4.82, 4.92, 4.41, 4.95, 4.79, 4.57 | ILE:15, VAL:23, LEU:140, LEU:154, ALA:39 |
Fig. 4.

a Interaction diagram of 1-methylchrysene with 5BZX, b interaction diagram of 3-methyl-1,8,9-anthracenetriol with 3BZ3
Structural stability and interaction energy analysis
The molecular dynamics simulations were conducted for the top two compound-target dockings: 3-methyl-1,8,9-anthracenetriol (PubChem ID: 10288) with Focal Adhesion Kinase 1 (PTK2; PDB ID: 3BZ3), and 1-methylchrysene (PubChem ID: 18779) with Human Phosphatase and Tensin homolog (PTEN; PDB ID: 5BZX). Following 300 ns of MD simulations, the structural stability and interaction energy of the 1-methylchrysene-PTEN and 3-methyl-1,8,9-anthracenetriol-PTK2 complexes were investigated at different MD endpoints (Fig. 5a–f). The backbone root mean square deviation (RMSD) average value was calculated for the 3-methyl-1,8,9-anthracenetriol-PTK2 complex of 0.3 nm (ranging from 0.1 to 0.5 nm) with low standard deviation (STD) of 0.05 nm, indicating stability throughout the simulation. In comparison, the backbone RMSD average value of 4.3 nm (ranging from 0.1 to 9.0 nm) with high standard deviation of 1.41 nm for 1-methylchrysene-PTEN exhibited larger fluctuations (ranging from 0.1 to 7.8 nm), indicating less stable binding between 1-methylchrysene and PTEN. The 3-methyl-1,8,9-anthracenetriol-PTK2 complex achieved a stable state within the first nanosecond and maintained stability throughout the entire MD simulation. However, structural stability could not be sustained by 1-methylchrysene-PTEN, with the most significant root mean square deviation (RMSD) occurring during time intervals spanning from 8 to 25 ns and from 177 to 196 ns (Fig. 5a). Additionally, the root mean square fluctuation (RMSF) curve was utilized to examine the variability and fluctuations in amino acid residues within the proteins. The RMSF values for residue numbers 2500–3000 in PTK2, when bound with 3-methyl-1,8,9-anthracenetriol, exhibited lesser flexibility compared to the PTEN bound with 1-methylchrysene. Furthermore, the RMSF average value for 3-methyl-1,8,9-anthracenetriol were calculated 0.1 nm (ranging from 0.1 to 1.1 nm) with SD of 0.13 nm, whereas 1-methylchrysene-PTEN has an average RMSF of 0.2 nm (ranging from 0.1 to 1.7 nm) with SD of 0.18 nm, indicating that its atoms experience slightly higher fluctuations. The minimum RMSF for both compounds are 0.1 nm, signifying regions of stability within their structures along with ligand residues (Fig. 5b).
Fig. 5.

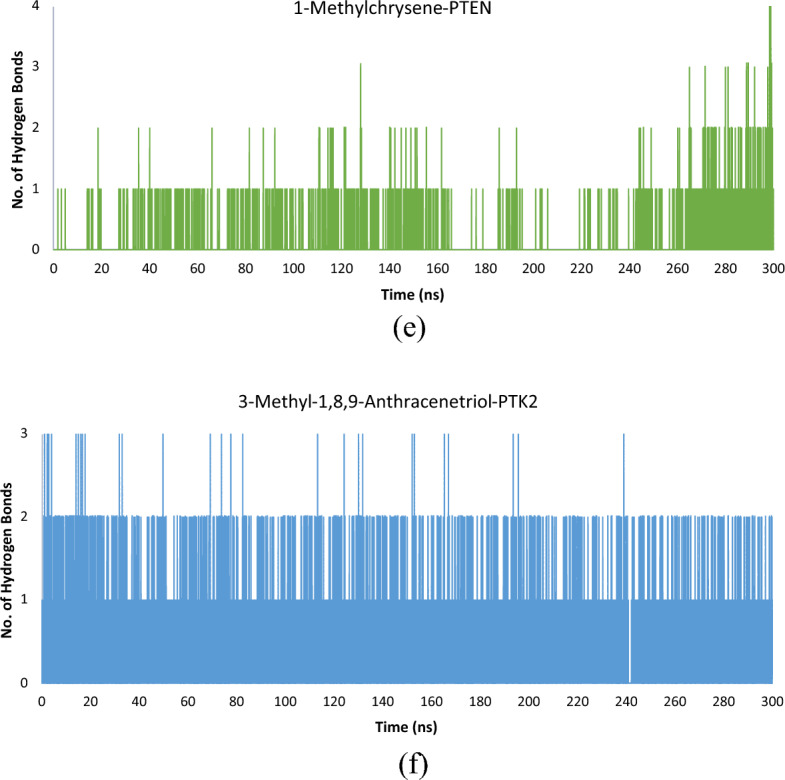
a–f Molecular dynamics simulations endpoints are shown as a root mean square deviation (RMSD), b root mean square fluctuation (RMSF), c radius of gyration (total and around axes), d solvent accessible surface, and e hydrogen bond number of complex 1-methylchrysene with Human Phosphatase and Tensin homolog (PTEN), f hydrogen bond number of complex 3-methyl-1,8,9-anthracenetriol with Focal Adhesion Kinase 1 (PTK2)
The radius of gyration (total and around axes) curve provides insight into the compactness or tightness of the protein’s overall structure. Both the 1-methylchrysene-PTEN and 3-methyl-1,8,9-anthracenetriol-PTK2 complexes exhibited stable rotation radii. Specifically, the 3-methyl-1,8,9-anthracenetriol-PTK2 complex exhibited a more compact folding having average radius of gyration (Rg) 2.0 nm (ranging from 1.99 to 2.08 nm) with SD of 0.02 nm compared to the 1-methylchrysene-PTEN complex having average Rg value of 2.3 nm (ranging from 2.18 to 2.32 nm) with SD value 0.04 nm (Fig. 5c). Additionally, solvent accessible surface area (SASA) refers to the area of a molecule’s surface that is accessible to the surrounding solvent molecules. For the 3-methyl-1,8,9-anthracenetriol-PTK2 complex, the average SASA values were observed 154.8 nm\S2\N (ranging from 143.39 to 170.62 nm\S2\N) with SD of 4.88 nm\S2\N, although average SASA values of 1-methylchrysene-PTEN complex exhibited 181.72 nm\S2\N (ranging from 161.43 to 183.76 nm\S2\N) with SD of 5.72 nm\S2\N. A lower SASA value of 3-methyl-1,8,9-anthracenetriol-PTK2 complex indicates that the ligand and protein are tightly bound together, resulting in a reduced exposure of their surface area to the surrounding solvent molecules. Moreover, the 3-methyl-1,8,9-anthracenetriol-PTK2 complex tends to have a more compact and buried binding interface, leading to a decrease in the SASA compared to the unbound or loosely bound state (Fig. 5d).
Hydrogen bonds play a crucial role in molecular dynamics (MD) simulations by contributing to molecular stability, protein folding, and function, influencing solvent interactions, facilitating conformational changes, and binding interactions. For this study, when investigating the interaction between 1-methylchrysene and the PTEN protein, a singular hydrogen bond was consistently observed, manifesting 971 occurrences. Furthermore, the occurrence of two, three, and four hydrogen bonds was notably less frequent, with 103, 12, and 3 instances, respectively. In total, 1089 instances were observed in which hydrogen bonds formed, propensity for less intricate bonding configurations and potentially weaker interactions within the protein–ligand association (Fig. 5e). Conversely, a total of 15,000 frames were recorded over a reaction time of 300 nanoseconds to analyse the hydrogen bonds formed during the MD simulations of both complexes. In the interaction between 3-methyl-1,8,9-anthracenetriol and the protein PTK2, a prominent occurrence of a single robust hydrogen bond formation was observed at 8585 instances out of 15,000 frames. This prevalence underscores the predominant presence of a strong hydrogen bond within the ligand and target protein. Moreover, the formation of two and three hydrogen bonds was less frequent, occurring 729 and 30 times, respectively, signifying sporadic additional interactions between the ligand and the target protein. Furthermore, a total of 9344 frames were observed where hydrogen bonds were formed, indicating a consistent and robust interaction between 3-methyl-1,8,9-anthracenetriol and PTK2 (Fig. 5f). The 3-methyl-1,8,9-anthracenetriol-PTK2 complex demonstrates a higher frequency of both stable and intermittent hydrogen bond formations compared to the 1-methylchrysene-PTEN complex. These results suggest that the 3-methyl-1,8,9-anthracenetriol-PTK2 complex is more favourable for drug discovery due to its stable interactions and higher occurrence of hydrogen bonds.
Binding free energies
The free energy calculations for 1-methylchrysene-PTEN and 3-methyl-1,8,9-anthracenetriol-PTK2 provided insights into their stabilities and binding affinities. For 1-methylchrysene-PTEN, a net binding free energy (ΔG) of − 10.65 kcal/mol was observed, indicating a slight preference for 1-methylchrysene binding to the PTEN receptor, but the interaction was relatively weak, possibly due to the influence of solvation (Table 3). When the energy contributions to the total binding free energy of 1-methylchrysene-PTEN were analyzed, it was evident that Van der Waals interactions (VDWAALS) contributed − 15.37 kcal/mol, while electrostatic interactions (EEL) contributed − 8.21 kcal/mol. Additionally, electrostatic solvation energy (EGB) was 15.01 kcal/mol, Surface area-based polar solvation energy (ESURF) was − 2.07 kcal/mol, Gas phase component of solvation energy (GGAS) was − 23.59 kcal/mol, and solvation energy in the solvent (GSOLV) was 12.94 kcal/mol. Collectively, these factors resulted in a net binding free energy of − 10.65 kcal/mol for 1-methylchrysene-PTEN (Fig. 6a). On the other hand, the binding free energy (ΔG) of the 3-methyl-1,8,9-anthracenetriol-PTK2 complex was − 21.92 kcal/mol, signifying a considerably more favorable binding affinity when compared to 1-methylchrysene-PTEN. The energy constituents for 3-methyl-1,8,9-anthracenetriol-PTK2 included Van der Waals interactions (VDWAALS) at − 27.58 kcal/mol, Electrostatic interactions (EEL) at − 9.75 kcal/mol, electrostatic solvation energy (EGB) at 19.3 kcal/mol, Surface area-based polar solvation energy (ESURF) at − 3.89 kcal/mol, the gas phase component of solvation energy (GGAS) at − 37.33 kcal/mol, and Solvation energy in the solvent (GSOLV) at 15.41 kcal/mol. These components, when combined, yielded a resultant binding free energy of − 21.92 kcal/mol for 3-methyl-1,8,9-anthracenetriol-PTK2 (Fig. 6b).
Table 3.
Net binding free energies of both the complexes (kcal/mol)
| Complex | ΔG complex | ΔG receptor | ΔG ligand | ΔG total |
|---|---|---|---|---|
| 1-methylchrysene-PTEN | − 5523.18 ± 62.47 | − 5599.08 ± 61.62 | 86.55 ± 4.01 | − 10.65 ± 6.91 |
| 3-methyl-1,8,9-anthracenetriol-PTK2 | − 4774.56 ± 58.13 | − 4776.51 ± 57.75 | 23.87 ± 3.71 | − 21.92 ± 2.24 |
Fig. 6.


a Energetic components of 1-methylchrysene with Human Phosphatase and Tensin homolog (PTEN), b energetic components of 3-methyl-1,8,9-anthracenetriol with Focal Adhesion Kinase 1 (PTK2), c BFE trajectory of 1-methylchrysene-PTEN, d BFE trajectory of 1-3-methyl-1,8,9-anthracenetriol-PTK2
In the binding free energy trajectories analysis, 3-methyl-1,8,9-anthracenetriol-PTK2 showed a more favorable average BFE of − 21.9 kcal/mol, while the 1-methylchrysene-PTEN exhibited a less favorable BFE trajectory of − 10.6 kcal/mol (Fig. 6c, d). The BFE trajectory for 3-methyl-1,8,9-anthracenetriol-PTK2 demonstrated a relatively stable binding energy with a lower standard deviation (STD) of 2.24 kcal/mol and a narrower range between − 32.6 and − 9.6 kcal/mol. In contrast, the 1-methylchrysene-PTEN complex displayed a wider BFE trajectory range between − 30.1 and 2.8 kcal/mol, accompanied by a higher SD of 6.91 kcal/mol, indicating more variability in its binding energy over time. This suggested that the 3-methyl-1,8,9-anthracenetriol-PTK2 complex generally maintained a more stable and favorable binding energy profile compared to the 1-methylchrysene-PTEN complex.
The findings of binding free energies reveal that the 1-methylchrysene-PTEN complex exhibits ΔG complex, ΔG receptor, ΔG ligand, and ΔG total values of − 5523.18, − 5599.08, 86.55, and − 10.65 kcal/mol, respectively. Similarly, for the 3-methyl-1,8,9-anthracenetriol-PTK2 complex, the ΔG complex, ΔG receptor, ΔG ligand, and ΔG total are − 4774.56, − 4776.51, 23.87, and − 21.92 kcal/mol, respectively. The values reflect the energetically favorable nature of the complexes, with negative ΔG complex and ΔG total, while positive ΔG receptor and ΔG ligand signify their individual energy contributions to binding. The larger negative ΔG Total for the 1-3-methyl-1,8,9-anthracenetriol-PTK2 complex suggest a stronger binding interaction compared to the methylchrysene-PTEN complex. Table 3 illustrates the binding free energies of both complexes along with their respective standard deviations.
Drug-likeness and ADMET profiling
The 1-methylchrysene and 3-methyl-1,8,9-anthracenetriol reveals several differences in their physicochemical properties, lipophilicity, water solubility, pharmacokinetics, druglikeness, and medicinal chemistry (Table 4). In terms of physicochemical properties, these compounds differ in their molecular formulas and molecular weights. 1-methylchrysene has a higher molecular weight (242.31) compared to 3-methyl-1,8,9-anthracenetriol (240.25). Furthermore, they have different numbers of heavy atoms, with 1-methylchrysene having 19 heavy atoms and 3-methyl-1,8,9-anthracenetriol having 18. Regarding lipophilicity, which indicates the compound’s ability to dissolve in lipids, the two compounds exhibit variations in their log octanol/water partition coefficients (log Po/w). 1-methylchrysene has higher log Po/w values across different methods (iLOGP, XLOGP3, SILICOS-IT) compared to 3-methyl-1,8,9-anthracenetriol. This suggests that 1-methylchrysene has a higher tendency to dissolve in lipid-based environments. In terms of water solubility, 1-methylchrysene is classified as poorly soluble, as indicated by its low log water solubility (Log S) value of − 6.03. On the other hand, 3-methyl-1,8,9-anthracenetriol exhibits moderate solubility with a log S value of − 4.43.
Table 4.
Complete pharmacokinetics profiles for compounds
| Parameters | 1-methylchrysene | 3-methyl-1,8,9-anthracenetriol |
|---|---|---|
| Physicochemical properties | ||
| Formula | C19H14 | 242.31 |
| Molecular weight | C15H12O3 | 240.25 |
| SMILES | CC1=C2C=CC3=C(C2=CC=C1)C=CC4=CC=CC=C43 | CC1=CC2=C(C(=C1)O)C(=C3C(=C2)C=CC=C3O)O |
| Num. heavy atoms | 19 | 18 |
| Num. rotatable bonds | 0 | 0 |
| Num. H-bond acceptors | 0 | 3 |
| Num. H-bond donors | 0 | 3 |
| Molar refractivity | 83.93 | 72.49 |
| Lipophilicity | ||
| Log Po/w (iLOGP) | 2.91 | 2.3 |
| Log Po/w (XLOGP3) | 6.33 | 4.01 |
| Log Po/w (SILICOS-IT) | 5.58 | 3.02 |
| Consensus Log Po/w | 5.29 | 3.04 |
| Water solubility | ||
| Log S (ESOL) | − 6.03 | − 4.43 |
| Solubility | Poorly soluble | Moderately soluble |
| Pharmacokinetics | ||
| GI absorption | Low | High |
| BBB permeant | No | Yes |
| P-gp substrate | No | No |
| CYP1A2 inhibitor | Yes | Yes |
| CYP2C19 inhibitor | Yes | No |
| CYP2C9 inhibitor | No | Yes |
| CYP2D6 inhibitor | No | Yes |
| CYP3A4 inhibitor | No | No |
| Log Kp (skin permeation) | − 3.28 cm/s | − 4.92 cm/s |
| Toxicity | ||
| SkinSen | 0.955 | 0.954 |
| Carcinogenicity | 0.897 | 0.716 |
| Respiratory | 0.766 | 0.771 |
| Druglikeness | ||
| Lipinski | Yes; 1 violation | Yes; 0 violation |
| Ghose | Yes | Yes |
| Veber | Yes | Yes |
| Egan | Yes | Yes |
| Muegge | No | Yes |
| Bioavailability score | 0.55 | 0.55 |
| Medicinal chemistry | ||
| Leadlikeness | No; 2 violations: MW < 250, XLOGP3 > 3.5 | No; 2 violations: MW < 250, XLOGP3 > 3.5 |
| Synthetic accessibility | 1.00 | 1.12 |
In the context of pharmacokinetics, the compounds differ in their gastrointestinal (GI) absorption and blood–brain barrier (BBB) permeability. 1-methylchrysene shows low GI absorption, indicating limited absorption from the gastrointestinal tract, while 3-methyl-1,8,9-anthracenetriol exhibits high GI absorption, suggesting efficient absorption. Additionally, 3-methyl-1,8,9-anthracenetriol is BBB permeant, indicating its ability to cross the blood-brain barrier, whereas 1-methylchrysene is not. Examining druglikeness parameters, both compounds generally fulfill various criteria. They have favorable scores in Lipinski, Ghose, Veber, and Egan rules, which are guidelines used to assess drug-like properties. However, 1-methylchrysene violates one Lipinski rule, specifically regarding molecular weight and XLOGP3 exceeding certain thresholds. In terms of medicinal chemistry, the compounds differ in terms of leadlikeness and synthetic accessibility. Both the phytochemicals have good synthetic accessibility scores, indicating their feasibility for synthesis.
Discussion
Determining the therapeutic efficacy or mechanism of action of plant fractions is a difficult process, particularly for each drug-target combination. On the other hand, medicinal plants often contain multiple bioactive compounds that can act synergistically to produce therapeutic effects. Traditional use of medicinal plants encompasses a wide range of practices, including the use of plant parts like leaves, roots, bark, flowers, and fruits, as well as their extracts, decoctions, infusions, and oils. From the published literature study, we have found that S. singueana is used traditionally to treat cancer (Sobeh et al. 2017). Studying the interactions and synergistic effects of compounds within S. singueana can lead to the development of combination therapies or novel drug formulations. In this study, a total of 38 active compounds from published literature and 2 target genes were selected through PPI network analysis on the basis of the highest degree and further proceeded for GO and KEGG enrichment analyses of the therapeutic targets. According to the GO analysis, the therapeutic targets displayed strong correlations with BP (e.g., negative regulation of organ growth, regulation of focal adhesion assembly, and positive regulation of proteolysis involved in the protein catabolic process), CC (e.g. cytoplasmic side of the plasma membrane,) and MF terms (e.g., protein phosphatase activity, and phosphatidylinositol-3-Phosphate phosphatase activity.). In the KEGG pathway analysis, the targets belong to PI3K/Akt signaling pathways and the VEGF signaling pathway. It is well known that the frequently activated PI3K/Akt pathway promotes PDAC aggressiveness. It controls a variety of cellular processes, including angiogenesis, cell migration, and apoptosis (Mehra et al. 2021). Reduced overall survival in Pancreatic cancer is significantly correlated with the activation of the PI3K/AKT pathway (Zheng et al. 2022).
The binding affinity and interactions between these compounds and the targeted proteins were evaluated. Among the S. singueana compounds, 1-methylchrysene and 9-benzylidenexanthene showed significant binding affinity against the 5BZX protein, while 1-methylchrysene, 6-dehydroestradiol, and 3-methyl-1,8,9-anthracenetriol exhibited noteworthy binding affinity with the 3BZ3 protein. These compounds demonstrated stronger binding affinity than the positive control drug, Belzutifan. The findings suggest the potential of these compounds as therapeutic agents, although further studies are needed to confirm their efficacy. These results align with previous studies that have highlighted the multi-targeting potential of phytochemicals and the synergistic effects of compounds derived from medicinal plants (Antony et al. 2020; Qi et al. 2021). The presence of multiple bioactive compounds in medicinal plants offers the opportunity for enhanced therapeutic effects through synergistic interactions (Shi et al. 2022). Furthermore, studies focusing on the binding affinity and interactions between compounds and target proteins have provided valuable insights into the development of novel therapeutic agents (Qi et al. 2021). Overall, the findings of this study contribute to the growing body of evidence supporting the potential of plant-derived compounds as multi-targeting inhibitors for therapeutic applications.
The molecular dynamics simulations were conducted to investigate the binding characteristics and stability of two compound-target dockings: 3-methyl-1,8,9-anthracenetriol with Focal Adhesion Kinase 1 (PTK2) and 1-methylchrysene with Human Phosphatase and Tensin homolog (PTEN). In this study, we performed 1000 picosecond NVT simulations on protein-ligand complexes at both 300 and 310 K. Notably, the simulations at 300 K exhibited increased stability and a reduced standard deviation compared to those at 310 K, emphasizing the temperature’s crucial role in system dynamics. The choice of 300 K aligns with established conventions in the field, as seen in prior studies involving PTEN and Focal Adhesion Kinase 1 (PTK2), ensuring comparability across research (Khan et al. 2017; Shi et al. 2022). In a similar study, the phytochemical components of Bauhinia variegata were analyzed to identify potential candidate inhibitors for the SARS-CoV-2 Mpro through molecular dynamics simulations conducted at a temperature of 300 K (More-Adate et al. 2022). This temperature choice enhances the stability of the complex, minimizes fluctuations, and underscores the importance of adhering to standards for meaningful data interpretation and inter-study comparisons, ultimately contributing to a more comprehensive understanding of the protein-ligand complex’s behavior.
The observed stability of the 3-methyl-1,8,9-anthracenetriol-PTK2 complex throughout the simulation is consistent with similar studies involving other ligands and protein targets (Ahmad et al. 2022; Fang et al. 2018). Similarly, Shi et al. (2022) performed MD simulations on the binding of a different compound to PTK2 and reported stable RMSD values over the simulation time (Shi et al. 2022). These findings indicate that stable RMSD values are indicative of strong and stable binding interaction. In contrast, the larger fluctuations in RMSD observed in the 1-methylchrysene-PTEN complex align with studies that have reported less stable binding interactions. The complex formed by 1-methylchrysene and PTEN appears to be unstable, as indicated by elevated RMSD, high RMSF, a large radius of gyration, high SASA, and binding free energy values. This instability may be attributed to weak binding interactions between 1-methylchrysene and PTEN, resulting in loose conjugation, or fluctuations in the conformation of 1-methylchrysene within the PTEN binding site. As a result, the complex lacks strong binding and experiences dynamic fluctuations, which contribute to its instability. Similarly, Jang et al. (2021) investigated the binding of various ligands to PTEN using MD simulations and observed significant RMSD fluctuations, indicating a weaker and less stable binding (Jang et al. 2021). The stability analysis of the 3-methyl-1,8,9-anthracenetriol-PTK2 complex aligns with studies emphasizing the significance of early stabilization in ligand binding (Bijian et al. 2022; Zhou et al. 2020). The stability analysis results obtained in our study also align with Zhou et al. (2020), who observed rapid attainment of stable binding conformations in a different ligand-PTK2 system, indicating early stabilization. Our findings support the notion that ligands with lower binding stability may induce larger conformational changes in the target protein.
The analysis of residue flexibility using RMSF is consistent with previous research on protein-ligand interactions (Sharma et al. 2022). Fang et al. (2018) conducted MD simulations on various ligand-target complexes and found that ligands inducing higher flexibility in the target protein exhibited weaker binding (Fang et al. 2018). In this study, greater flexibility of residues in PTK2 bound with 3-methyl-1,8,9-anthracenetriol compared to PTEN bound with 1-methylchrysene, indicating a potentially stronger binding interaction between 3-methyl-1,8,9-anthracenetriol and PTK2. The analysis of the radius of gyration is consistent with studies investigating the compactness of protein-ligand complexes (Kamath et al. 2022; Rai et al. 2022). Similarly, Guo et al. (2022) performed MD simulations on various ligand–protein complexes and observed that ligands with higher compactness in the binding pocket exhibited stronger binding affinity (Guo et al. 2022). The SASA analysis, which assesses the surface area accessible to solvent molecules, corresponds with studies investigating the tightness of protein–ligand interfaces (Al-Wahaibi et al. 2022; Khan et al. 2020). Likewise, Somadder et al. (2023) investigated the binding of different ligands to protein targets and found that ligands with lower SASA values formed more tightly bound interfaces (Somadder et al. 2023). Our results align with these findings, as we observed lower SASA values in the 3-methyl-1,8,9-anthracenetriol-PTK2 complex compared to the 1-methylchrysene-PTEN complex, indicating a more tightly bound interface in the former. Moreover, loose conjugation of 1-methylchrysene with PTEN can be attributed to several factors, including a high root mean square deviation (RMSD), fewer hydrogen bonds, and lower free binding energy.
The present study showed that the 3-methyl-1,8,9-anthracenetriol-PTK2 complex is more favourable for drug discovery being of having higher occurrence of both stable and intermittent hydrogen bond formations in this complex compared to the 1-methylchrysene-PTEN complex. Stable hydrogen bonds are indicative of strong and consistent molecular interactions, which are essential for drug-target binding and stability (Shi et al. 2022). Additionally, intermittent hydrogen bonds contribute to conformational flexibility, enabling the complex to adapt and respond to its environment (Arnittali et al. 2019). Therefore, the 3-methyl-1,8,9-anthracenetriol-PTK2 complex, with its greater number of stable and intermittent hydrogen bonds, holds promise as a potential drug target for further investigation and drug discovery efforts. These results highlight the importance of considering structural stability and interaction energy in the evaluation of ligand-target complexes and support the potential of 3-methyl-1,8,9-anthracenetriol as a candidate for drug development in the field of pancreatic cancer therapeutics.
Binding free energy calculations are indispensable in computational drug discovery, offering profound insights into the thermodynamics and energetics of molecular interactions. These calculations are vital for assessing the binding affinity of ligands to target proteins, illuminating the mechanisms of binding, guiding drug design and optimization, and streamlining virtual screening efforts (Pandya et al. 2020). In the present study, the 3-methyl-1,8,9-anthracenetriol-PTK2 complex exhibited a notably more favourable binding affinity, with a substantially lower binding free energy (ΔG) of − 21.92 kcal/mol, in contrast to the 1-methylchrysene-PTEN complex, which had a less favourable ΔG of − 10.65 kcal/mol. The disparity could be attributed to the energetic components in each complex. In the 1-methylchrysene-PTEN complex, electrostatic solvation energy (EGB) dominated, which countered the stronger van der Waals interactions (VDWAALS), resulting in a relatively weak binding affinity. In contrast, the 3-methyl-1,8,9-anthracenetriol-PTK2 complex benefited from more substantial van der Waals interactions, leading to a stable and energetically favourable binding. In a comparable investigation, the employment of the molecular mechanics generalized Born surface area method to evaluate the binding free energy of the aminopyrimidine group in VS-4718/FAK demonstrated concurrence with the current study’s findings (Shi et al. 2022). Another study identified Focal Adhesion Kinase PTK2 and Integrin αIIbβ3 as essential components with potential therapeutic value, demonstrating the promise of CIB1-interfering peptides designed to bind to Integrin Chain αIIb, aligning with the finding of current study (Huang et al. 2022a).
In this study, the physicochemical properties, pharmacokinetics, druglikeness, and medicinal chemistry of 1-methylchrysene and 3-methyl-1,8,9-anthracenetriol, revealing differences in their molecular weight, lipophilicity, water solubility, GI absorption, BBB permeability, and druglikeness compared for both the potential drug candidates. These findings are in line with previous research on the importance of considering such properties in drug design (Isvoran et al. 2017; Riyadi et al. 2021). The variations in water solubility highlight the need for improved formulation and delivery strategies to enhance bioavailability and clinical effectiveness. Likewise, the differences in GI absorption and BBB permeability provide insights into the compounds’ pharmacokinetic behaviour, aiding in target-specific drug distribution (Di et al. 2020). The evaluation of druglikeness parameters and medicinal chemistry aligns with current drug discovery trends, emphasizing the assessment of favorable pharmacological profiles and leadlikeness criteria (Apostolov et al. 2020). These aspects play a crucial role in identifying potential therapeutic candidates.
In summary, the data reveals a striking contrast between these two complexes. 3-methyl-1,8,9-anthracenetriol exhibits a much stronger and more stable binding affinity for the PTK2 receptor compared to 1-methylchrysene’s interaction with PTEN. Moreover, significant difference in binding affinities can be attributed to the substantial variations in the energy components contributing to the total binding free energy for each complex, with 3-methyl-1,8,9-anthracenetriol-PTK2 displaying more favourable energetic contributions. Based on the results of the molecular dynamics simulations, it can be inferred that 3-methyl-1,8,9-anthracenetriol has the potential to act as a potent inhibitor of Focal Adhesion Kinase 1 (FAK1) and could serve as a promising lead compound for the development of novel therapeutics for pancreatic cancer. The comprehensive analysis of various parameters including RMSD, RMSF, the radius of gyration, potential energy, and the solvent-accessible surface area provides valuable insights into the structural stability and interaction energy of the protein-ligand complex.
Conclusion
The comprehensive analysis of the compounds derived from S. singueana highlights their potential as multi-targeting inhibitors in the treatment of pancreatic cancer. The enrichment analysis revealed significant associations with key biological processes and molecular functions, shedding light on the underlying mechanisms of action. Moreover, the identification of PTK2 and PTEN as highly involved genes in various cellular processes and pathways emphasizes their significance as therapeutic targets. The molecular docking results revealed that 1-methylchrysene and 9-benzylidenexanthene exhibited the highest binding affinities against the targeted protein 5BZX. Additionally, 1-methylchrysene, 6-dehydroestradiol, and 3-methyl-1,8,9-anthracenetriol showed noteworthy binding affinities with the protein 3BZ3, outperforming the positive control drug Belzutifan. These findings support the potential of these compounds as multi-targeting inhibitors for pancreatic cancer treatment. Further analysis using molecular dynamics simulations provided insights into the stability and interaction dynamics of the ligand-target complexes. The consistent and stable binding interaction observed in the 3-methyl-1,8,9-anthracenetriol-PTK2 complex suggests its potential as a potent inhibitor of Focal Adhesion Kinase 1. In contrast, the fluctuations in the 1-methylchrysene-PTEN complex indicate a less stable binding interaction. Moreover, 3-methyl-1,8,9-anthracenetriol-PTK2 complex showed significantly stronger binding affinity and a considerably lower binding free energy (ΔG) compared to the 1-methylchrysene-PTEN complex, which exhibited a less favourable ΔG value. Although, both compounds show promise in drug development based on their druglikeness and synthetic accessibility. These findings contribute to the growing body of knowledge regarding the multi-target effects of S. singueana and its potential as a valuable treatment option for pancreatic cancer. While this study employed bioinformatics approaches, additional in-vivo, and in-vitro studies are necessary to validate the efficacy and therapeutic potential of the identified compounds. Continued research and exploration are essential to translate these findings into clinical applications and improve patient outcomes.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary material 1 (DOCX 220.5 kb)
Acknowledgements
The authors would like to express their gratitude to the revered Swami Ramdev for his inspiration. Further, the authors are thankful to the Ministry of AYUSH under Grant-In-Aid for the Establishment of the Centre of Excellence of Renovation and Upgradation of Patanjali Ayurveda Hospital, Haridwar.
Author contributions
AB: Conceptualization; DS: Data curation, Methodology, Writing—Original draft preparation; MT: Investigation; VA: Writing—Review and editing; AD: Data visualization, Formal analysis.
Funding
This research was supported by the Ministry of Ayush, Government of India, under Ayurswasthya Yojana.
Data availability
Data will be made available on request.
Declarations
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this research article.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Adedoyin B, Muhammad A, Dangoggo SM, Rabah A, Sharples G, Nahar L, Sarker S. Chemical composition and bioactivity of the essential oil of Cassia singueana flowers growing in Nigeria. Pharm Biomed Res. 2019;5:1–7. doi: 10.18502/pbr.v5i3.2110. [DOI] [Google Scholar]
- Adler DG (2021) Pancreatic adenocarcinoma. In: Atlas of endoscopic ultrasonography, 2nd edn. pp 104–107. 10.1002/9781119522997.ch22
- Adzu B, Abbah J, Vongtau H, Gamaniel K. Studies on the use of Cassia singueana in malaria ethnopharmacy. J Ethnopharmacol. 2003;88:261–267. doi: 10.1016/S0378-8741(03)00257-5. [DOI] [PubMed] [Google Scholar]
- Ahmad S, Sayeed S, Bano N, Sheikh K, Raza K. In-silico analysis reveals auinic acid as a multitargeted inhibitor against cervical cancer. J Biomol Struct Dyn. 2022 doi: 10.1080/07391102.2022.2146202. [DOI] [PubMed] [Google Scholar]
- Al-Wahaibi LH, Mary YS, Shyma Mary Y, Al-Mutairi AA, Hassan HM, El-Emam AA, Yadav R. Investigation of the electronic properties of solvents (water, benzene, methanol) using IEFPCM model, spectroscopic investigation with docking and MD simulations of a thiadiazole derivative with anti-tumor activities. J Mol Liq. 2022;348:118061. doi: 10.1016/j.molliq.2021.118061. [DOI] [Google Scholar]
- Antony P, Baby B, Homedi A, Al Halabi Z, Ali W, Vijayan A. Polypharmacological potential of natural compounds against prostate cancer explored using molecular docking and molecular dynamics simulations. Int J Comput Biol Drug Des. 2020;13:181–199. doi: 10.1504/IJCBDD.2020.107314. [DOI] [Google Scholar]
- Apostolov S, Mijin D, Petrović S, Vastag G. In silico approach in the assessment of chromatographic parameters as descriptors of diphenylacetamides’ biological/pharmacological profile. J Liq Chromatogr Relat Technol. 2020;43:852–862. doi: 10.1080/10826076.2020.1835672. [DOI] [Google Scholar]
- Arnittali M, Rissanou AN, Harmandaris V (2019) Structure of biomolecules through molecular dynamics simulations. In: Procedia computer science, pp 69–78. 10.1016/j.procs.2019.08.181
- Bijian K, Wernic D, Nivedha AK, Su J, Lim FPL, Miron CE, Amzil H, Moitessier N, Alaoui-Jamali MA, Novel Aurora A, Protein Kinase C. (α, β1, β2, and θ) multitarget inhibitors: impact of selenium atoms on the potency and selectivity. J Med Chem. 2022;65:3134–3150. doi: 10.1021/acs.jmedchem.1c01031. [DOI] [PubMed] [Google Scholar]
- Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di L, Artursson P, Avdeef A, Benet LZ, Houston JB, Kansy M, Kerns EH, Lennernäs H, Smith DA, Sugano K. The critical role of passive permeability in designing successful drugs. ChemMedChem. 2020;15:1862–1874. doi: 10.1002/cmdc.202000419. [DOI] [PubMed] [Google Scholar]
- Fang Y, Wang D, Xu X, Dava G, Liu J, Li X, Xue Q, Wang H, Zhang J, Zhang H. Preparation,: in vitro and in vivo evaluation, and molecular dynamics (MD) simulation studies of novel F-18 labeled tumor imaging agents targeting focal adhesion kinase (FAK) RSC Adv. 2018;8:10333–10345. doi: 10.1039/c8ra00652k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaidhani RH, Balasubramaniam G. An epidemiological review of pancreatic cancer with special reference to India. Indian J Med Sci. 2021;73:99–109. doi: 10.25259/ijms_92_2020. [DOI] [Google Scholar]
- Gidado A, Boonpisuttinant K, Kanjanawongwanich S. Anti-cancer and anti-oxidative activities of Nigerian traditional medicinal plants/recipes. J Complement Med Res. 2019;10:200. doi: 10.5455/jcmr.20190731050619. [DOI] [Google Scholar]
- Guo H, Wang Z, Wang B, Zhang Y, Meng H, Sui H. Molecular dynamics simulations of oil recovery from dolomite slit nanopores enhanced by CO2 and N2 injection. Adv Geo-Energy Res. 2022;6:306–313. doi: 10.46690/ager.2022.04.05. [DOI] [Google Scholar]
- Huang J, Jooss NJ, Fernández DI, Sickmann A, García Á, Wichapong K, Dijkgraaf I, Heemskerk JWM. Roles of focal adhesion kinase PTK2 and integrin αIIbβ3 signaling in collagen- and GPVI-dependent thrombus formation under shear. Int J Mol Sci. 2022 doi: 10.3390/ijms23158688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Zhang G, Tang TY, Gao X, Liang TB. Personalized pancreatic cancer therapy: from the perspective of mRNA vaccine. Mil Med Res. 2022 doi: 10.1186/s40779-022-00416-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim MAA, Badr EAA, Abdelrahman AHM, Almansour NM, Mekhemer GAH, Shawky AM, Moustafa MF, Atia MAM. Silico targeting human multidrug transporter ABCG2 in breast cancer: database screening, molecular docking, and molecular dynamics study. Mol Inf. 2022;41:2060039. doi: 10.1002/minf.202060039. [DOI] [PubMed] [Google Scholar]
- Isvoran A, Ciorsac AA, Ostafe V. ADME-Tox profiling of some low molecular weight water soluble chitosan derivatives. ADMET DMPK. 2017;5:192–200. doi: 10.5599/admet.5.3.423. [DOI] [Google Scholar]
- Jang H, Smith IN, Eng C, Nussinov R. The mechanism of full activation of tumor suppressor PTEN at the phosphoinositide-enriched membrane. iScience. 2021 doi: 10.1016/j.isci.2021.102438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath A, Laha A, Pandiyan S, Aswath S, Vatti AK, Dey P. Atomistic investigations of polymer-doxorubicin-CNT compatibility for targeted cancer treatment: a molecular dynamics study. J Mol Liq. 2022;348:118005. doi: 10.1016/j.molliq.2021.118005. [DOI] [Google Scholar]
- Khan I, Ansari IA, Singh P, Dass FP. Prediction of functionally significant single nucleotide polymorphisms in PTEN tumor suppressor gene: an in silico approach. Biotechnol Appl Biochem. 2017 doi: 10.1002/bab.1483. [DOI] [PubMed] [Google Scholar]
- Khan FI, Lai D, Anwer R, Azim I, Khan MKA. Identifying novel sphingosine kinase 1 inhibitors as therapeutics against breast cancer. J Enzyme Inhib Med Chem. 2020;35:172–186. doi: 10.1080/14756366.2019.1692828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein AP. Pancreatic cancer epidemiology: understanding the role of lifestyle and inherited risk factors. Nat Rev Gastroenterol Hepatol. 2021 doi: 10.1038/s41575-021-00457-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson SC, Permert J, Håkansson N, Näslund I, Bergkvist L, Wolk A. Overall obesity, abdominal adiposity, diabetes and cigarette smoking in relation to the risk of pancreatic cancer in two Swedish population-based cohorts. Br J Cancer. 2005;93:1310–1315. doi: 10.1038/sj.bjc.6602868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majeed A, Hussain W, Yasmin F, Akhtar A, Rasool N. Virtual screening of phytochemicals by targeting HR1 domain of SARS-CoV-2 S protein: molecular docking, molecular dynamics simulations, and DFT studies. Biomed Res Int. 2021 doi: 10.1155/2021/6661191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehra S, Deshpande N, Nagathihalli N. Targeting pi3k pathway in pancreatic ductal adenocarcinoma: rationale and progress. Cancers (Basel) 2021 doi: 10.3390/cancers13174434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesmar J, Fardoun MM, Abdallah R, Al Dhaheri Y, Yassine HM, Iratni R, Badran A, Eid AH, Baydoun E. Ziziphus nummularia attenuates the malignant phenotype of human pancreatic cancer cells: role of ros. Molecules. 2021;26:4295. doi: 10.3390/molecules26144295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BR, McGee TD, Swails JM, Homeyer N, Gohlke H, Roitberg AE. MMPBSA.py: an efficient program for end-state free energy calculations. J Chem Theory Comput. 2012;8:3314–3321. doi: 10.1021/ct300418h. [DOI] [PubMed] [Google Scholar]
- More-Adate P, Lokhande KB, Swamy KV, Nagar S, Baheti A. GC-MS profiling of Bauhinia variegata major phytoconstituents with computational identification of potential lead inhibitors of SARS-CoV-2 Mpro. Comput Biol Med. 2022;147:105679. doi: 10.1016/j.compbiomed.2022.105679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni D, Lu S, Zhang J. Emerging roles of allosteric modulators in the regulation of protein–protein interactions (PPIs): a new paradigm for PPI drug discovery. Med Res Rev. 2019 doi: 10.1002/med.21585. [DOI] [PubMed] [Google Scholar]
- Pandya PN, Kumar SP, Bhadresha K, Patel CN, Patel SK, Rawal RM, Mankad AU. Identification of promising compounds from curry tree with cyclooxygenase inhibitory potential using a combination of machine learning, molecular docking, dynamics simulations and binding free energy calculations. Mol Simul. 2020;46:812–822. doi: 10.1080/08927022.2020.1764552. [DOI] [Google Scholar]
- Qi Y, Li Y, Fang Y, Gao H, Qiang B, Wang S, Zhang H. Design, synthesis, biological evaluation, and molecular docking of 2,4-diaminopyrimidine derivatives targeting focal adhesion kinase as tumor radiotracers. Mol Pharm. 2021;18:1634–1642. doi: 10.1021/acs.molpharmaceut.0c01088. [DOI] [PubMed] [Google Scholar]
- Rai K, Kumbhar BV, Panda D, Kunwar A. Computational study of interactions of anti-cancer drug eribulin with human tubulin isotypes. Phys Chem Chem Phys. 2022;24:16694–16700. doi: 10.1039/d1cp04454k. [DOI] [PubMed] [Google Scholar]
- Reddy PS, Lokhande KB, Nagar S, Reddy VD, Murthy PS, Swamy KV. Molecular modeling, docking, dynamics and simulation of gefitinib and its derivatives with EGFR in non-small cell lung cancer. Curr Comput Aided Drug Des. 2018;14:246–252. doi: 10.2174/1573409914666180228111433. [DOI] [PubMed] [Google Scholar]
- Riyadi PH, Romadhon, Sari ID, Kurniasih RA, Agustini TW, Swastawati F, Herawati VE, Tanod WA (2021) SwissADME predictions of pharmacokinetics and drug-likeness properties of small molecules present in Spirulina platensis. In: IOP conference series: earth and environmental science, p 012021. 10.1088/1755-1315/890/1/012021
- Salo-Ahen OMH, Alanko I, Bhadane R, Alexandre AM, Honorato RV, Hossain S, Juffer AH, Kabedev A, Lahtela-Kakkonen M, Larsen AS, Lescrinier E, Marimuthu P, Mirza MU, Mustafa G, Nunes-Alves A, Pantsar T, Saadabadi A, Singaravelu K, Vanmeert M. Molecular dynamics simulations in drug discovery and pharmaceutical development. Processes. 2021 doi: 10.3390/pr9010071. [DOI] [Google Scholar]
- Sharma A, Sinha S, Rathaur P, Vora J, Jha PC, Johar K, Rawal RM, Shrivastava N. Reckoning apigenin and kaempferol as a potential multi-targeted inhibitor of EGFR/HER2-MEK pathway of metastatic colorectal cancer identified using rigorous computational workflow. Mol Divers. 2022;26:3337–3356. doi: 10.1007/s11030-022-10396-7. [DOI] [PubMed] [Google Scholar]
- Shi M, Chen T, Wei S, Zhao C, Zhang X, Li X, Tang X, Liu Y, Yang Z, Chen L. Molecular docking, molecular dynamics simulations, and free energy calculation insights into the binding mechanism between VS-4718 and focal adhesion kinase. ACS Omega. 2022;7:32442–32456. doi: 10.1021/acsomega.2c03951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- Sobeh M, Mahmoud MF, Hasan RA, Cheng H, El-Shazly AM, Wink M. Senna singueana: antioxidant, hepatoprotective, antiapoptotic properties and phytochemical profiling of a methanol bark extract. Molecules. 2017;22:1502. doi: 10.3390/molecules22091502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somadder PD, Hossain MA, Ahsan A, Sultana T, Soikot SH, Rahman MM, Ibrahim SM, Ahmed K, Bui FM. Drug repurposing and systems biology approaches of enzastaurin can target potential biomarkers and critical pathways in colorectal cancer. Comput Biol Med. 2023;155:106630. doi: 10.1016/j.compbiomed.2023.106630. [DOI] [PubMed] [Google Scholar]
- Strobel O, Neoptolemos J, Jäger D, Büchler MW. Optimizing the outcomes of pancreatic cancer surgery. Nat Rev Clin Oncol. 2019 doi: 10.1038/s41571-018-0112-1. [DOI] [PubMed] [Google Scholar]
- Sun S, Kim MJ, Dibwe DF, Omar AM, Athikomkulchai S, Phrutivorapongkul A, Okada T, Tsuge K, Toyooka N, Awale S. Anti-austerity activity of Thai medicinal plants: chemical constituents and anti-pancreatic cancer activities of Kaempferia parviflora. Plants. 2021;10:1–12. doi: 10.3390/plants10020229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, Jensen LJ, Von Mering C. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47:D607–D613. doi: 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2009 doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdés-Tresanco MS, Valdés-Tresanco ME, Valiente PA, Moreno E. Gmx_MMPBSA: a new tool to perform end-state free energy calculations with GROMACS. J Chem Theory Comput. 2021;17:6281–6291. doi: 10.1021/acs.jctc.1c00645. [DOI] [PubMed] [Google Scholar]
- Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJC. GROMACS: fast, flexible, and free. J Comput Chem. 2005 doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- Webb B, Sali A. Comparative protein structure modeling using MODELLER. Curr Protoc Bioinforma. 2016;54:5–6. doi: 10.1002/cpbi.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong G, Wu Z, Yi J, Fu L, Yang Z, Hsieh C, Yin M, Zeng X, Wu C, Lu A, Chen X, Hou T, Cao D. ADMETlab 2.0: an integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021;49:W5–W14. doi: 10.1093/nar/gkab255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Ji S, Li X, Feng Q. Active ingredients and molecular targets of Taraxacum mongolicum against hepatocellular carcinoma: network pharmacology, molecular docking, and molecular dynamics simulation analysis. PeerJ. 2022;10:e13737. doi: 10.7717/peerj.13737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Zhou H, Ponzoni L, Luo A, Zhu R, He M, Huang Y, Guan KL, Bahar I, Liu Z, Wan Y. EIF3H orchestrates hippo pathway-mediated oncogenesis via catalytic control of YAP stability. Cancer Res. 2020;80:2550–2563. doi: 10.1158/0008-5472.CAN-19-3718. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material 1 (DOCX 220.5 kb)
Data Availability Statement
Data will be made available on request.