

Abstract
Airway remodeling is a major feature of asthma. Interleukin (IL)-36γ is significantly upregulated and promotes airway hyper-responsiveness (AHR) in asthma, but its role in airway remodeling is unknown. Here, we aimed to investigate the role of IL-36γ in airway remodeling, and whether IL-38 can alleviate airway remodeling in chronic asthma by blocking the effects of IL-36γ. IL-36γ was quantified in mice inhaled with house dust mite (HDM). Extracellular matrix (ECM) deposition in lung tissues and AHR were assessed following IL-36γ administration to mice. Airway inflammation, AHR, and remodeling were evaluated after IL-38 or blocking IL-36 receptor (IL-36R) treatment in asthmatic mice. The effects of lung fibroblasts stimulated with IL-36γ and IL-38 were quantified in vitro. Increased expression of IL-36γ was detected in lung tissues of HDM-induced asthmatic mice. The intratracheal instillation of IL-36γ to mice significantly enhanced the ECM deposition, AHR, and the number of activated lung fibroblasts around the airways. IL-38 or blocking IL-36R treated asthmatic mice showed a significant alleviation in the airway inflammation, AHR, airway remodeling, and number of activated fibroblasts around airways as compared with the HDM group. In vitro, IL-36γ promoted the activation and migration of human lung fibroblasts (HFL-1). The administration of IL-38 can counteract these biological processes induced by IL-36γ in HFL-1cells. The results indicated that IL-38 can mitigate airway remodeling by blocking the profibrotic effects of IL-36γ in chronic asthma. IL-36γ may be a new therapeutic target, and IL-38 is a potential candidate agent for inhibiting airway remodeling in asthma.
Keywords: asthma, airway remodeling, interleukin-36γ, interleukin-38, lung fibroblasts
IL-36γ was upregulated in the lungs of chronic asthma mice, and it contributed to the airway remodeling. IL-38 can alleviate the airway remodeling in asthma by regulating the effect of IL-36γ.
Graphical Abstract
Graphical Abstract.
Introduction
Asthma is characterized as a heterogeneous chronic inflammatory airway disease that an estimated 300 million people suffer from it around the world, and it costs billions of dollars in medical expenses and causes 250 000 annual fatalities [1, 2]. A variety of cells are involved in the process of asthma and cause airway inflammation, airway hyper-reactivity (AHR), airway remodeling, and other pathological changes [3]. A long course of inflammation may lead to a suite of structural changes in the airway called airway remodeling. Airway remodeling is the pathological basics of fixed airflow restriction in asthma patients. In the past, persistent inflammation was believed to be the cause of airway remodeling. Severe asthma patients experience irreversible or partially reversible airflow limitation and endangering their lives. However, there is accumulating evidence that airway remodeling occurs concurrently with inflammation [4, 5]. Current therapeutic about airway inflammation do little for airway remodeling.
Interleukin (IL)-36 cytokines belong to IL-1 family and consist of three agonists IL-36 alpha (IL-36α), IL-36beta (IL-36β), and IL-36 gamma (IL-36γ) and the natural antagonist IL-36 receptor antagonist (IL-36Ra) and IL-38 [6]. IL-36 receptor agonists genes are localized to human and mouse chromosome 2 [7]. IL-36 receptor agonists exert the pro-inflammatory function by binding to the heterodimeric receptor compound of IL-36R and IL-1-receptor-affiliated protein IL-1RAcP1 and stimulating downstream signaling cascade including MyD88 adaptor protein complex, NF-kB, and MAPK pathways to promote inflammation [8]. Recently, a series of studies showed that IL-36α, IL-36β, and IL-36γ participate in allergic disease and were significantly upregulated, such as allergic rhinitis (AR), especially with asthma and allergic contact dermatitis (ACD) [9–12]. IL-36γ has been found to be upregulated in primary bronchial epithelial cells from asthma patients and is further increased upon infection with rhinovirus [13]. The mRNA level and the protein concentration of IL-36γ are both increased in the lung tissues of HDM-induced mouse asthma mode [7]. Several pieces of evidence have shown that IL-36 has a promoting effect on tissue fibrosis. For example, Zhang et al. demonstrated that IL-36γ promotes HFL-1 cells releasing IL-6 and CXCL8 [14]. Scheibe et al. have indicated that IL-36 ligands can stimulate colonic fibroblasts releasing the type IV collagen and inhibiting IL-36R can reduce the tissue fibrosis in the chronic intestinal inflammation mice [15]. But the relationship between IL-36γ and airway remodeling in asthma is still unknown.
IL-38 is a 17–18 kDa protein which belongs to IL-1 family and shares 40% sequence similar to IL-1Ra and IL-36Ra. Thus, it is thought to be a typical antagonist of the IL-1 family along with IL-1Ra and IL-36Ra [16]. Three candidate receptors, IL-36R, IL-1R1, and IL-1 receptor accessory protein-like 1 (IL-1RAPL1), have been proposed for IL-38 and IL-38 exerts anti-inflammatory activities by binding to these receptors and blocking intracellular signaling [17–19]. The protective effect of IL-38 in immune diseases has been widely studied, such as RA [20], SLE [21], psoriasis [22], and inflammatory bowel disease [23]. The mRNA level of IL-38 was decreased in the lung tissues of ovalbumin-induced mice [24]. Sun et al. have shown that airway inflammation, AHR, and Th2 cytokines are reduced in asthmatic mice upon the injection with IL-38 [25]. However, the effect of IL-38 in allergic asthma remains largely unknown, including the potential on airway remodeling.
In this study, we aimed to investigate the effect of IL-36γ on airway remodeling in experimental mice models of asthma and in vitro, and then further elucidated whether IL-38 could inhibit airway remodeling in HDM-induced mice by blocking the effect of IL-36γ.
Materials and methods
Animals
SPF BALB/c mice (female, 6–8 weeks) were purchased from Guangdong Yao Kang Biotechnology Co. Ltd (Guangdong, China) and allowed to acclimate for 1 week. All mice were placed in a specific sterile level (SPF) environment with temperature 23 ± 1°C, humidity 60 ± 5%, and light–dark cycles of 12 hours. The protocol was approved by the Committee on the Ethics of Animal Experiments of the Third Affiliated Hospital of Sun Yat-Sen University (number [2019]02-148-01).
HDM-induced asthma in mice
Twenty-five micrograms of house dust mite (cat. No. XPB82D3A2.5, Greer, USA) protein were resuspended in 25 µl phosphate-buffered saline (PBS) at a concentration of 1.0 mg/ml. Moreover, mice in the control group were injected with 25 μl of PBS meanwhile as a negative control group. All mice were euthanized 24 hours after last induced.
Intratracheal IL-36γ induction
One microgram of mouse recombinant IL-36γ was dissolved in 20 μl PBS and intratracheally instilled into mouse lungs for 2 weeks at a total of three times a week. The control group were received 20 μl of PBS. Lung tissues were collected for analysis 24 hours after the last exposure.
Airway hyper-responsiveness measurement
Invasive lung function assessment of mice was operated by using a BuxcoÒ FinePointe™ RC system (DSI, St. Paul, MN, USA). In brief, mice were anesthetized with 1% pentobarbital sodium (70 mg/kg, intraperitoneal injection) 24 hours after the last HDM induction. Mice were endotracheally intubated with a 24-gauge cannula and connected to a ventilator at a rate of 120 breaths per minute. Then, the airway resistance (RL, cm H2O s/ml) of mice to 20 μl PBS or different doses of inhaled methacholine (Mch, 6.25, 12.5, 25, and 50 mg/ml, Sigma–Aldrich) were recorded for 3 minutes at each dose [26].
Bronchoalveolar lavage fluid analyses
Bronchoalveolar lavage fluid (BALF) was obtained by endotracheal intubation after AHR measurement. After lysis of RBCs, the total number of inflammatory cells in BALF was determined using a hemocytometer. For the differential counts, the cells (5 × 104 cells/slide) in the BALF were centrifuged onto slides. Giemsa staining was used for differential cell counting. At least 400 cells per sample were examined.
Cytokines quantification by ELISA
The concentrations of IL-36γ, IL-38, IL-5, IL-6, IL-13, IL-17, TSLP, active TGF-β1, and TNF-α were measured by ELISA kit according to the manufacturer’s specifications.
Determination of lung hydroxyproline
For determination of hydroxyproline (HYP) contents, 50 mg wet tissue from the frozen lung of mice was hydrolyzed and boiled for 20 minutes. After adjusting the pH to 6.5, the HYP levels in individual hydrolysates were determined using the HYP Test Kit according to the manufacturer’s instruction (Nanjing Jiancheng Bioengineering Institute, China). Briefly, each hydrolysate sample was reacted with activated carbon (20 mg), and after centrifuging, the absorbance of the supernatant was measured at 550 nm by ultraviolet spectrophotometer. The data were expressed as micrograms of HYP per milligram of lung tissue.
Lung histopathology
The lung tissues were fixed in 4% paraformaldehyde and then embedded in paraffin fixation. The paraffin sections (5 μm) were stained with hematoxylin and eosin (H&E), periodic acid-Schiff (PAS), Masson trichrome, and toluidine blue to evaluate the airway inflammation, the production of mucus, collagen deposition, and mast cells recruitment in mice lung tissues. The airway inflammation score and airway wall thickness were quantified according to previously published methods [27]. The quantification of PAS staining and Masson trichrome staining was performed by ImageJ software. Mast cells were counted at least in three fields per section.
Flow cytometric analyses
The lungs tissues were minced and subsequently incubated with shaking for 30 minutes at 37°C with the collagenase type IV solution. After red blood cell lysis, cell suspensions were filtered through a 70 μm cell strainer. The Fc-gamma receptors were blocked with an anti-CD16/32 (clone: W17231A, BioLegend, USA) antibody for 30 minutes to avoid nonspecific staining. The cells were then stained with anti-mouse CD3 (clone: 145-2c11, BioLegend) and anti-mouse CD4 (clone:RM4-5, BioLegend) antibodies for cell surface staining. Next, the cells were fixed with the Intracellular Fixation and Permeabilization kit at 4℃ for 1 hour and then stained with anti-mouse interferon (IFN)-γ (clone: XMG1.2, BioLegend) and anti-mouse IL-4 (clone: 11B11, BioLegend) antibodies for 30 minutes in the dark. Stained cells were analyzed using a flow cytometer. Data were analyzed using FlowJo software.
Immunohistochemistry
Antigenic repair was performed on the 5 μm paraffin sections, and endogenous peroxidase was wiped off. The IL-36γ (1:100, Affinity, China), α-SMA (1:200, Cell Signaling Company, USA), Fibronectin (1:200, Santa Cruz Company, USA), CD31 (1:40, Abcam, USA), and p-ERK (1:200, Cell Signaling Company) antibodies were incubated overnight at 4℃. Next day, the tissue sections were incubated with goat anti-rabbit antibody for 1 hour at room temperature, and then incubated with DAB for 5 minutes. Finally, the sections were dehydrated with a grade alcohol series as well as transparency with xylene, and photographed under the microscope. Protein expression was evaluated using ImageJ software.
Immunofluorescence
The lung tissue paraffin sections or HFL-1 cells were fixed with 4% paraformaldehyde and then permeated with 0.5% Triton X-100 (Solaibio, China) for 30 minutes. The paraffin sections were incubated with the antibodies of α-SMA (1:100, Cell Signaling Company), Vimentin (1:100, Cell Signaling Company), Collagen I (1:250 Santa Cruz), and Fibronectin (1:250, Santa Cruz) at 4°C overnight. The following day, the tissue sections were stained with Alexa Fluor goat anti-rabbit and DAPI in the dark. Finally, the sections were photographed with a fluorescence microscope (Nikon, Japan).
RNA isolation and quantitative real-time PCR
Total RNA of lung tissues and cells was extracted with Trizol reagent (CWBIO, China) and reversely transcribed into cDNA using Prime Script RT Reagent Kit (TaKaRa, Japan). The Light Cycler 480 System and ChamQ SYBR Green PCR kit (Vazyme, China) were used for real-time quantitative PCR. The relative expression of mRNA was normalized and evaluated by the 2−ΔΔCt method. Primer sequences are provided in Supplementary Table S1.
Cell culture and treatment
A normal human lung fibroblast cell line (HFL-1) was obtained (Procell, Wuhan, China) and cultured in Ham’s F-12K containing 10% FBS and stimulated with rhIL-36γ and rhIL-38.
Isolation of total protein and western blot
Cultured HFL-1 cells were washed with PBS and were subjected to protein extraction by using a lysis buffer (KeyGEN, China). A BCA protein assay kit (Ding guo, China) was used to measure the concentration of protein. The samples were loaded with sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto a polyvinylidene fluoride membrane (Millipore Company, USA). The membranes were blocked with 5% skimmed milk for 1 hour. The primary antibodies Fibronectin (1:500, Santa Cruz Company), Collagen I (1:500, Santa Cruz Company), α-SMA (1:1000, Cell Signaling Company), p-ERK (1:1000, Cell Signaling Company), t-ERK (1:1000, Cell Signaling Company), and GAPDH (1:2000, Cell Signaling Company) were incubated at 4°C overnight after washing with TBST solution. The PVDF membranes were washed with TBST and then incubated with HRP-conjugated goat anti-rabbit IgG for 1 hour. The PVDF membranes were washed three times with TBST and then visualized using an enhanced chemiluminescence reagent.
Wound healing and transwell assay
Cell migration ability was evaluated by using wound healing assay and transwell assay. A horizontal line was drawn with the pipette tip at the bottom behind the 6-well plate. 1 × 105 cells in each group were seeded into the six-well plate and incubated with PBS, rhIL-36γ, and rhIL-38 for 24 hours. The cells were cultured with serum-free medium and were treated with PBS, rhIL-36γ, and rhIL-38, respectively. Images of six randomly fields in each group were captured using a microscope at 40× magnification (0 and 24 hours) [28].
1 × 104 cells were suspended in 100 μl serum-free medium and cultured in the upper transwell chamber with an 8 μm pore polycarbonate membrane filter. 600 μl medium containing 10% FBS was placed in the bottom chamber. PBS and rhIL-36γ were added in the bottom chamber, respectively, incubated with or without rhIL-38. After 24 hours, the migrated cells on the membrane filter were fixed with 4% paraformaldehyde for 20 minutes, stained with 0.1% crystal violet for 30 minutes. After washing and drying, the migrated cells were photographed in five random fields using a 100× inverted microscope [28].
Statistical methods
All statistical analyses were performed by using GraphPad Prism 8. Unpaired Student’s t-test was used to analyze the differences between the two groups. One-way ANOVA was used to compare multiple groups to the designated control group. Data were expressed as mean ± standard deviation. Results were considered statistically significant at P < 0.05.
Results
IL-36γ was significantly upregulated and IL-38 was downregulated in the lungs of HDM-induced asthma mice
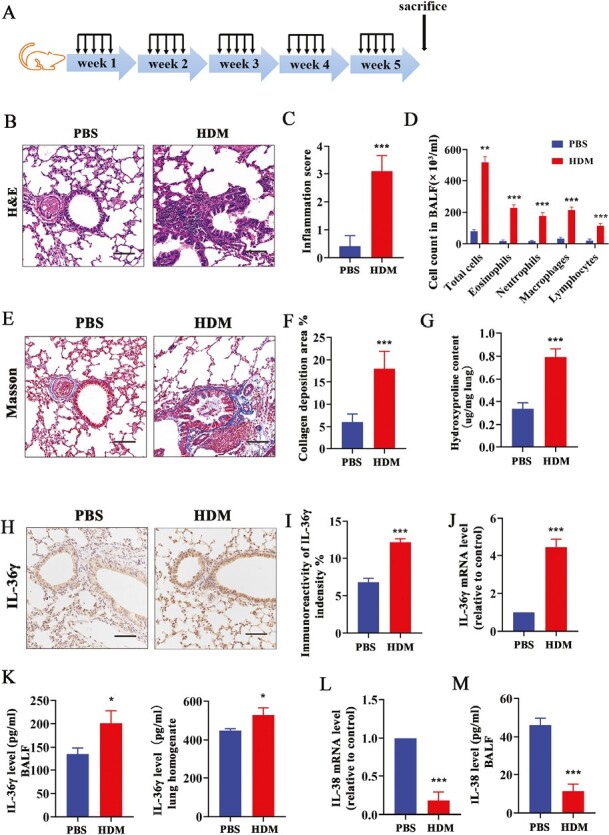
IL-36 receptor agonist IL-36γ gene, a quantitative trait locus on mouse chromosome 2, is associated with allergen-induced bronchial hyper-responsiveness [7]. To investigate whether IL-36γ was upregulated and participate in the process of chronic asthma, we successfully established a mice model of chronic asthma according to the protocol of our previous research (Fig. 1A) [29] and analyzed. Continuous induction with HDM elicited inflammatory cells infiltration (Fig 1B–D; Supplementary Fig. S1) and collagen deposition around the airways in the lungs of mice (Fig. 1E–G). Immunohistochemical staining of lung tissue sections revealed that the expression of IL-36γ was significantly increased in the HDM group as compared with the PBS control (Fig. 1H and I). The mRNA level of IL-36γ was also upregulated in the lung tissues of mice in HDM group (Fig. 1J). Meanwhile, the concentration of IL-36γ was remarkably increased in BALF and lung homogenate of chronic asthma mice as compared to that in PBS group, which were the same with the previous research (Fig. 1K) [7]. In addition, we found that both mRNA level of IL-38 in the lung tissues (Fig. 1L) and the concentration of IL-38 in BALF were downregulated in the HDM group mice compared with the PBS group (Fig. 1M).
Figure 1.
The expression of IL-36γ and IL-38 in HDM-induced chronic asthmatic mice. (A) The protocol of chronic asthma animal experimental model (n = 6 per group). (B and C) Representative histopathological images and quantification of the lung tissues in mice were performed with H&E staining. Scale bar = 50 μm (×200). (D) The total and differential inflammatory cell counts in BALF were determined by Giemsa staining. (E and F) Representative histopathological images and quantification of the lung tissues in mice were performed with Masson staining. Scale bar = 50 μm (×200). (G) The HYP level in lung of each group mice was measured by HYP assay kit. (H and I) Representative immunoreactivity images and quantification of IL-36γ staining in lung tissues from each group. (J) The mRNA level and (K) concentration of IL-36γ in lung tissue and BALF in each group mice. (L) The mRNA level and (M) concentration of IL-38 in lung tissue in each group mice. Bar diagrams and data are presented as the mean ± standard deviation (SD). * vs. PBS. * P < 0.05; **P < 0.01; ***P < 0.001
Continues exposure of IL-36γ increased extracellular matrix deposition and AHR in chronic asthma mice
Increased deposition of extracellular matrix (ECM) is the main characteristic of airway remodeling [30]. In order to assess whether IL-36γ can contribute to airway remodeling, intra-nasal IL-36γ was administrated to BALB/C mice for 2 weeks at three times a week (Fig. 2A). Pathological staining results showed that the inflammatory cells infiltration around the airway and inflammation score were significantly increased after administration of IL-36γ at week 2 as compared with the PBS group and at week 1 (Fig. 2B and E). The collagen deposition level around the airway (Fig. 2C, F, and G) and the mRNA level of elastin (Fig. 2H) were remarkably upregulated after IL-36γ treatment compared to the PBS group. At the same time, the expression of Fibronectin was also obviously enhanced after 2 weeks of IL-36γ treatment by immunohistochemistry analysis (Fig. 2D and I). In addition, AHR was drastically increased after exposure of IL-36γ compared to the control group and was significantly higher at week 2 than week 1 (Fig. 2J).
Figure 2.
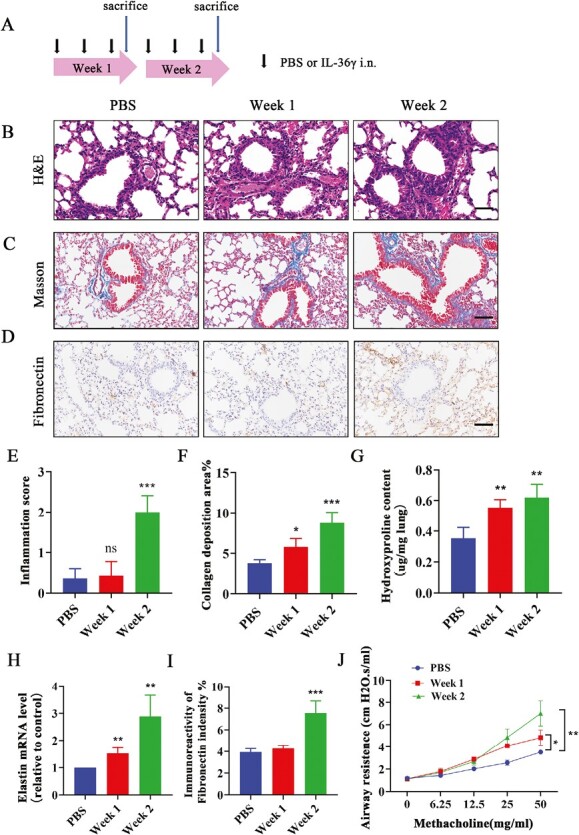
The effect of IL-36γ stimulation to airway inflammation, AHR and ECM deposition in vivo. (A) Intranasal administration of IL-36γ to mice for 2 weeks (n = 6 per group). (B and C) Representative histopathological images of airway and lung tissues from each group mice were performed with H&E staining and Masson staining. Scale bar = 50 μm (×200). (D) Representative immunohistochemical images of Fibronectin in mice lung tissues from each group. Scale bar = 50 μm (×200). Quantifications of the lung tissues were performed with H&E staining (E) and Masson staining (F). (G) The HYP level in lung of each group mice was measured by HYP assay kit. (H) Quantification of elastin mRNA level in the mice lung tissues from each group. (I) Quantification of Fibronectin expression in the lung of each group mice. (J) Airway resistance response to various doses of methacholine was tested within 24 hours after the final IL-36γ stimulated. Bar diagrams and data are presented as the mean ± standard deviation (SD). * vs. PBS. *P < 0.05; **P < 0.01; ***P < 0.001; ns, no significant differences
Fibroblasts are key structural cells involved in airway remodeling in asthma. Activated fibroblasts can induce sub-epithelial fibrosis and produce large amounts of ECM [31]. In this study, we also found that the number of α-SMA and vimentin double positive activated lung fibroblasts around the airway was increased after IL-36γ stimulated as compared with the PBS group by immunofluorescence assay (Fig. 3A and B). The mRNA level of α-SMA and vimentin in the lung tissues was also upregulated after IL-36γ-administrated than the PBS group (Fig. 3C and D). All these findings suggested that IL-36γ may play an important role in airway remodeling.
Figure 3.
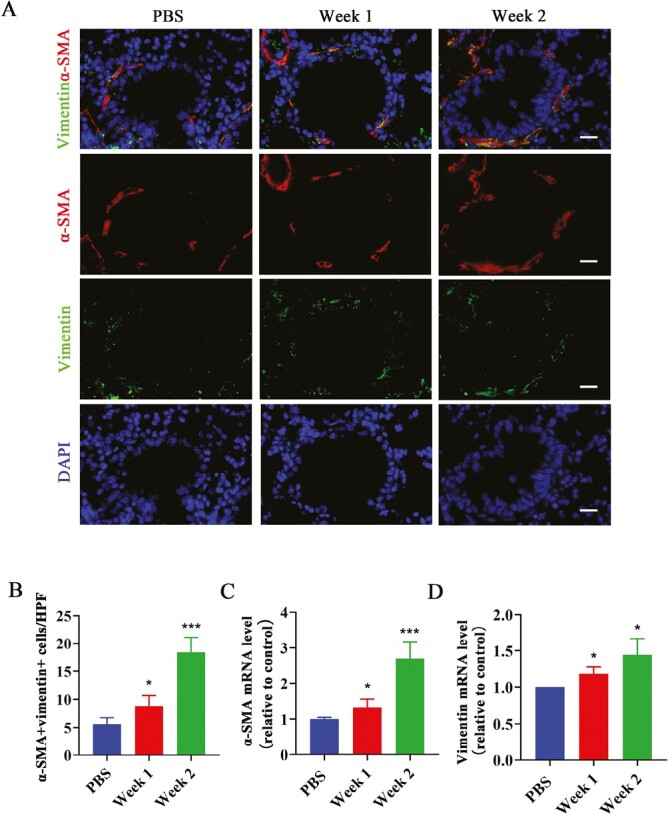
The effect of IL-36γ on the activation of lung fibroblast around airway. (A and B) Representative immunofluorescence images and quantification of α-SMA and vimentin double-positive activated fibroblast cells around the airways in each group. Scale bar = 20 μm (×400). (C and D) Quantification of α-SMA and vimentin mRNA level in the mice lung tissues from each group. Bar diagrams and data are presented as the mean ± standard deviation (SD). * vs. PBS. *P < 0.05; ***P < 0.001
IL-36γ promoted the activation of HFL-1 cells by ERK1/2 signaling pathway
Activated lung fibroblasts can produce amount of ECM which is a characteristic of airway remodeling [31]. Other research has demonstrated that IL-36 receptor agonists can create colonic fibroblasts activation by stimulating IL-36R [15]. To investigate the effect of IL-36γ on lung fibroblasts, rhIL-36γ was added to HFL-1 cells and analyzed. Cells were treated with 100 ng/ml IL-36γ [14]. As shown in Fig. 4A, the mRNA level of Fibronectin, Collagen I, and α-SMA was significantly increased in IL-36γ treated group as compared with the PBS group. Likewise, the protein expression of these three markers was also confirmed by immunofluorescence staining (Fig. 4B). Simultaneously, we found that IL-36γ enhanced the expression of Fibronectin, Collagen I, and α-SMA with a time-dependent manner by western blot (Fig. 4C).
Figure 4.
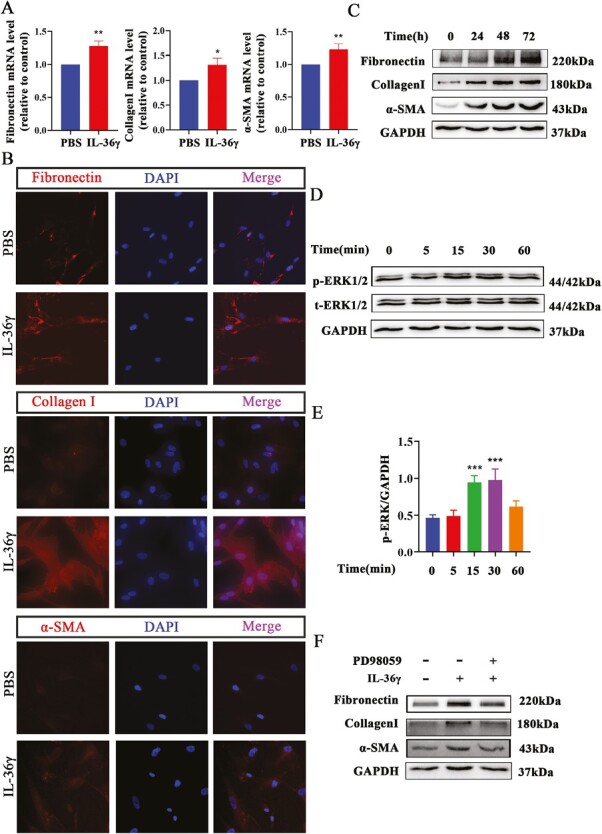
IL-36γ induced the activation of HFL-1 cells by ERK1/2 pathway. (A) Quantification of Fibronectin, Collagen I, and α-SMA mRNA level in HFL-1 cells treated with 100 ng/ml IL-36γ by RT–qPCR. (B) Representative immunofluorescence images of Fibronectin, Collagen I, and α-SMA protein expression in HFL-1 cells treated with 100 ng/ml IL-36γ. (C) Western blot analysis of Fibronectin, Collagen I, and α-SMA protein expression in HFL-1 cells treated with 100 ng/ml IL-36γ at different time. (D and E) Representative immunoblot analysis and quantification of the activation of ERK1/2 signaling pathway in HFL-1 cells treated with100 ng/ml IL-36γ at different time. (F) Representative immunoblot analysis of Fibronectin, Collagen I, and α-SMA in HFL-1 cells exposed to IL-36γ with or without PD98059 treated. Bar diagrams and data are presented as the mean ± standard deviation (SD). * vs. PBS group. *P < 0.05; **P < 0.01; ***P < 0.001
It has been verified that IL-36 receptor agonists trigger downstream signaling cascade including MyD88 adaptor protein complex, NF-kB, and MAPK by binding to the heterodimeric receptor compound of IL-36R and IL-1 receptor-affiliated protein IL-1RAcP1 [7]. Previous studies have reported that phospho (p)-ERK1/2 is involved in the proliferation and activation of airway smooth muscle cells (ASMCs) and regulate airway remodeling of asthma [32]. But whether ERK1/2 signaling pathway participated in the activation of fibroblasts induced by IL-36γ is unknown. In this study, we found that IL-36γ can induce ERK1/2 phosphorylation in a time-dependent manner (Fig. 4D and E). PD98059 (a specific inhibitor of ERK1/2) can inhibit the expression of Fibronectin, Collagen I, and α-SMA in HFL-1 cells induced by IL-36γ (Fig. 4F). In general, we considered that IL-36γ regulates the activation of lung fibroblasts via an ERK1/2-dependent mechanism.
IL-38 counteracted the activation and migration of lung fibroblasts induced by IL-36γ
IL-38 is considered to be a partial receptor antagonist of IL-36 and can exert its anti-inflammatory action by binding to IL-36 receptor [17]. It has been indicated that IL-38 can inhibit peripheral blood mononuclear cells (PBMCs) secreting IL-8 stimulated by IL-36γ [17]. To verify whether IL-38 can block the effect of IL-36γ on lung fibroblasts, IL-38 was treated to HFL-1 cells before IL-36γ stimulated. It showed that IL-38 not only remarkably decreased the elevated phosphorylation level of ERK1/2, similar to PD98059 (Fig. 5A and B), but also inhibited the expression of Fibronectin, Collagen I, and α-SMA induced by IL-36γ in HFL-1 cells (Fig. 5C and D). Other than blocking the activation of fibroblasts, we also found that there is an enhancement in the migratory ability of HFL-1 cells after IL-36γ stimulated through wound healing and transwell assays, and pretreatment of IL-38 can reverse this effect (Fig. 5E and F). Collectively, all these data supported the conjecture that IL-38 could restrain IL-36γ-induced activation and migration of HFL-1 cells.
Figure 5.
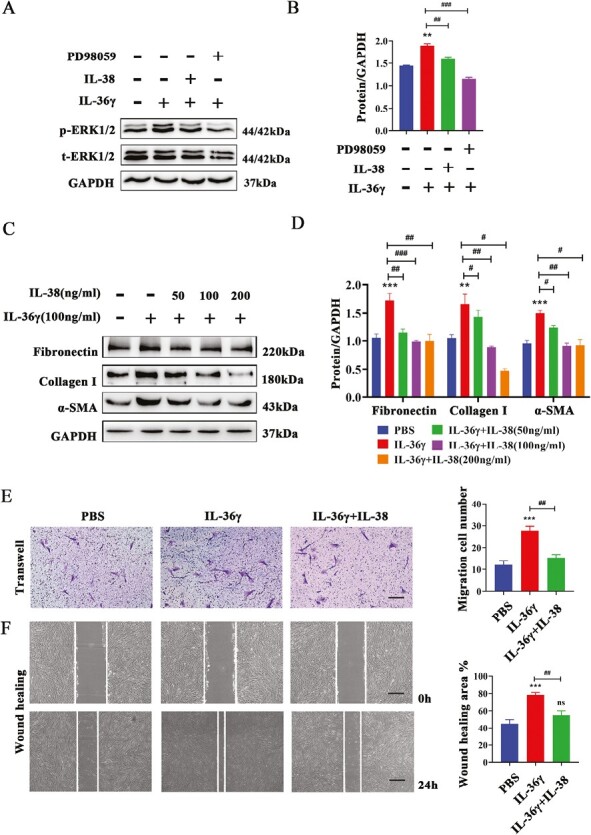
IL-38 inhibited IL-36γ-induced activation and migration in HFL-1 cells. (A and B) Western blotting analysis and quantification of the ERK1/2 pathway treated with IL-36γ with or without IL-38 or PD98059. (C and D) The protein expression of Fibronectin, Collagen I, and α-SMA in HFL-1 cells treated with IL-36γ, with or without different dose of IL-38 (50–200 ng/ml) by immunoblots analysis. (E and F) Representative images and quantification of cells’ migrated ability induced by IL-36γ, with or without IL-38 through transwell assay (scale bar = 50 μm (×100)) and wound healing assay (scale bar = 20 μm (×40)). Bar diagrams and data are presented as the mean ± standard deviation (SD). * vs. PBS group; # vs. IL-36γ group. *,#P < 0.05; **,##P < 0.01; ***,###P < 0.001. Abbreviation: ns: no significant differences
IL-38 or blocking IL-36R ameliorates airway inflammation and AHR in HDM-induced asthma mice
To further confirm the effect of IL-38 on asthmatic airway inflammation and AHR in vivo, we used an HDM-induced asthma model in BALB/c mice, which could better mimic the key characteristics of asthma [25]. Mice were randomly divided into six groups: PBS group (n = 6), HDM group (n = 6), HDM + IL-36Ra group (n = 6), HDM + IL-38 group (n = 6), HDM + IL-36γ group (n = 6), and HDM + IL-36γ + IL-38 group (n = 6). The protocol of establishing the asthmatic model is shown in Fig. 6A. For intervention experiments, mice were intraperitoneally injected with 1 μg recombinant murine IL-38 (aa3–152) (Adipogen Life Sciences, USA) PBS solution and 1 μg recombinant murine IL-36Ra (PeproTech, USA) PBS solution 30 minutes before HDM exposure beginning at the third week, three times a week accompanied with 1 µg recombinant murine IL-36γ (NovoProtein, China) PBS solution intratracheally instilled into lungs. Our data showed that both IL-38 and IL-36Ra treatment can remarkably inhibit peribronchial inflammatory cells infiltration in the lung and the thickness of bronchial wall compared with experimental allergic asthma mice (Fig. 6B–D). Meanwhile, AHR in response to methacholine was also decreased after the administration of IL-38 or IL-36Ra (Fig. 6E). Moreover, the number of inflammatory cells (Fig. 6F; Supplementary Fig. S2) and the concentrations of inflammatory cytokines including IL-5, IL-13, IL-6, IL-17, TSLP, active TGF-β1, and TNF-α in the BALF were observed to decrease after IL-38 or IL-36Ra treated as compared with the HDM group (Fig. 6G). The increased recruited mast cells (Supplementary Fig. S3) and proportion of Th2 lymphocytes (Fig. 6H) in the lung tissues of HDM group were also significantly attenuated after IL-38 or IL-36Ra treated. In addition, we found that inflammation cells and Th2 lymphocytes infiltration, the thickness of bronchial wall, and AHR were obviously enhanced in the mice from HDM + IL-36γ group accompanied with the increased concentrations of cytokines in BALF. After treating with IL-38, all these changes were reversed.
Figure 6.

IL-38 or blocking IL-36R alleviated airway inflammation and AHR in asthma mice model. (A) Schematic diagram of the experimental protocol for the chronic asthma (n = 6 mice per group). (B) Representative histopathological image of lung tissues was performed with H&E staining. Scale bar = 50 μm (×200). Quantification was performed with H&E staining (C and D) in each group. (E) Airway resistance response to methacholine (0–50 mg/ml) was determined within 24 hours after the final HDM induced. (F) The total and differential inflammatory cell counts in the BALF from each group were determined by Giemsa staining. (G) Analysis of the concentrations of IL-5, IL-6, IL-17, IL-13, TSLP, active TGF-β1, and TNF-α in BALF from each group. (H) The frequencies of Th1 (CD4+ IFN-γ+) and Th2 (CD4+ IL-4+) cells in lung tissue were analyzed by using flow cytometry. Bar diagrams and data are presented as the mean ± standard deviation (SD). * vs. PBS group; # vs. the indicated group. *,#P < 0.05; **,##P < 0.01; ***,###P < 0.001
IL-38 or blocking IL-36R alleviated airway remodeling in HDM-induced chronic asthma mice
To better understand the IL-38 potential on airway remodeling, we analyze the airway remodeling markers by histopathology. PAS-stained and Masson trichrome-stained sections demonstrated remarkable goblet cell hyperplasia and high collagen deposition around the bronchioles in the HDM group. The HYP content and mRNA level of elastin of lung tissues were also upregulated in asthmatic mice. These changes could be partly inhibited after the administration of IL-38 and IL-36Ra (Fig. 7A–F). Immunohistochemical staining of tissue sections revealed that around the bronchia, the expressions of α-SMA (Fig. 7G and J) and Fibronectin (Fig. 7H and K) were significantly increased in HDM group as compared with the PBS control. IL-38 and IL-36Ra treatment can dramatically inhibit the expression of these two markers. Meanwhile, immunohistochemistry staining for CD31 showed that the HDM-induced mice exhibited increased area of microvessels in the sub-epithelium when compared with the control groups, while IL-38 or IL-36Ra treatment stunted these increases (Supplementary Fig. S4). In addition, IL-38 can inhibit the expression of p-ERK1/2 which was upregulated in HDM group, and it was similar to the treatment with IL-36Ra (Fig. 7I and L). In HDM + IL-36γ group, all the above pathogeny structure were remarkably upregulated while they can be attenuated after the treatment of IL-38.
Figure 7.
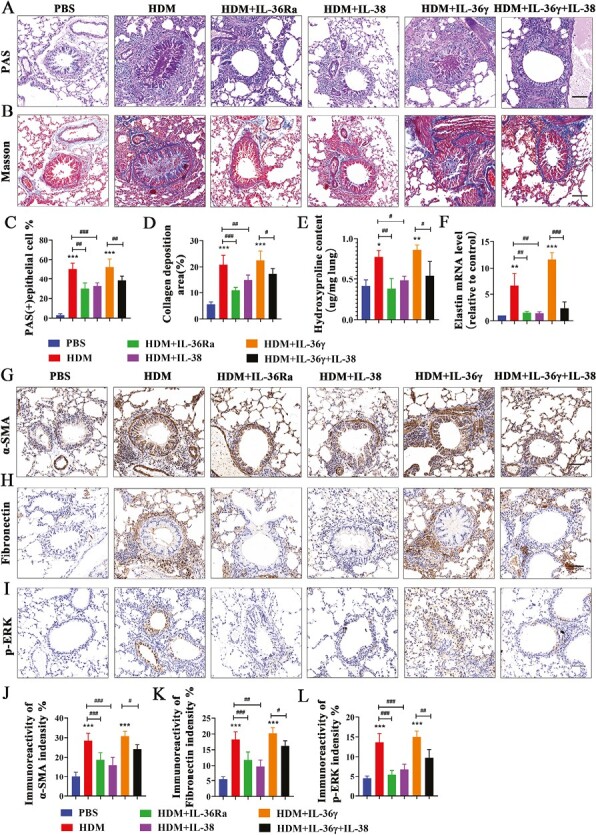
IL-38 or blocking IL-36R alleviated airway remodeling and the expression of p-ERK1/2 in an HDM-induced asthma murine model. (A and B) Representative images of PAS staining and Masson staining in lung tissues from each group. Scale bar = 50 μm (×200). (C and D) Quantification of PAS staining and Masson staining in each group. (E) The HYP content in lung of each group mice was measured by HYP assay kit. (F) Quantification of elastin mRNA level in the mice lung tissues of each group. (G–I) Representative immunoreactivity images of α-SMA, Fibronectin and p-ERK1/2 in lung tissues from each group. Scale bar = 50 μm (×200). (J–L) Quantification of the expression of α-SMA, Fibronectin , and p-ERK1/2 in each group. Bar diagrams and data are presented as the mean ± standard deviation (SD). * vs. PBS group; # vs. the indicated group. *,#P < 0.05; **,##P < 0.01; ***,###P < 0.001
At the same time, the number of α-SMA and vimentin double-positive lung fibroblasts around the airway was remarkably increased in the HDM group compared with the PBS group, while IL-38 or IL-36Ra treatment group showed a decrease in the number by immunofluorescence assay (Fig. 8A and B). The mRNA level of α-SMA and vimentin in the lung tissues of mice in the HDM group was also increased and diminished after IL-38 or IL-36Ra treatment (Fig. 8C and D). Likewise, IL-36γ co-administration HDM increased the number of activated fibroblasts around the airways and treatment with IL-38 can recover these changes.
Figure 8.
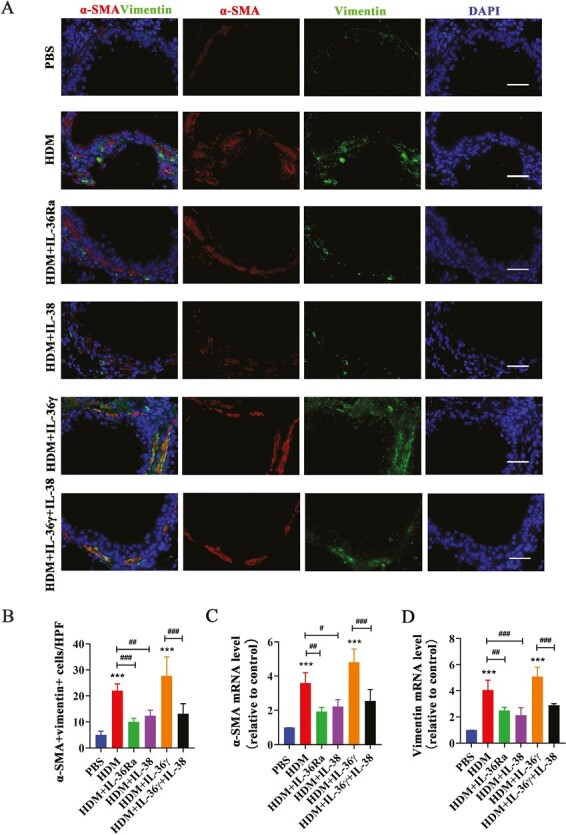
IL-38 or blocking IL-36R inhibits activation of lung fibroblast around airway in asthma mice. (A and B) Representative immunofluorescence images and quantification of α-SMA and vimentin double positive cells around the airways in the lung tissues from each group. Scale bar = 50 μm (×400). (C and D) Quantification of α-SMA and vimentin mRNA level in the mice lung tissues each group. Bar diagrams and data are presented as the mean ± standard deviation (SD). * vs. PBS group; # vs. the indicated group. *,#P < 0.05; **,##P < 0.01; ***,###P < 0.001
Discussion
Asthma is a chronic inflammatory disease of the airways. Airway remodeling is considered to be an important and independent pathologic feature of asthma. The main features include destruction of epithelial cells, abnormal deposition of ECM, airway smooth muscle hyperplasia/hypertrophy, and angiopoiesis [33]. Any of these pathologies can occur alone or in combination during the process of asthma [34]. In our research, we found that in vivo, IL-36γ was upregulated in chronic asthma mice lung tissues and continues intratracheally administration of IL-36γ can induce ECM deposition, AHR, and increased number of activated fibroblasts around mice airways. IL-38 or blocking IL-36R treatment can alleviate the airway inflammation, AHR, airway remodeling, and the increased number of activated lung fibroblasts in chronic asthmatic mice. Meanwhile, in vitro, IL-36γ can promote the activation and migration of lung fibroblasts, which can be inhibited by IL-38. All these evidences can provide that IL-36γ may participate in asthma airway remodeling by upregulating profibrotic function on lung fibroblasts, and IL-38 can alleviate the airway remodeling by inhibiting this effect of IL-36γ.
Activated fibroblasts in the airways of asthmatic patients can produce large amounts of ECM and abnormal accumulation of ECM results in changes of tissue structure and function which contribute to airway remodeling and airflow limitation [35–37]. In this study, we revealed that intratracheal administration of IL-36γ to mice can promote deposition of collagen, upregulate the mRNA level of elastin and expression of Fibronectin, and increase the number of activated fibroblasts around the airway in vivo. It also enhanced AHR compared with the controls. Treatment with IL-36Ra can alleviate AHR and airway remodeling in chronic asthma mice. In vitro, IL-36γ can upregulate the expression of Fibronectin, Collagen I, and α-SMA in HFL-1 cells through ERK1/2 phosphorylation pathway which is involved in lung fibroblasts to myofibroblasts transition [38] and highly activated in chronic asthma model [39]. Meanwhile, IL-36γ also enhance the migratory ability of HFL-1 cells. All these evidences hint that IL-36γ may contribute to airway remodeling in HDM-induced asthma mice. In fact, many studies have confirmed that the IL-36 cytokines are associated with tissue remodeling in other diseases. For example, it has been demonstrated that IL-36 receptor agonists can activate intestinal fibroblasts and epithelial cells [15, 40]. Strong expression of IL-36α was found in samples from IBD patients with intestinal fibrous stenosis and correlated with the severity of inflammation [15]. IL-36R KO mice had a lower fibrosis score and reduced submucosal thickening in mice with inflammatory bowel disease, along with a reduction in the number of activated fibroblasts compared with WT mice [15]. Chi et al. confirmed that compared with WT mice, IL-36 receptor agonist IL-36α was upregulated in UUO mice, and IL-36R KO mice exhibited ameliorative renal function and inhibited fibrosis [41]. In addition, it has been found that IL -36γ and IL-36α were highly expressed in serum of IPF patients, and the increased levels are correlated with disease severity [42]. Collectively, connecting with our findings suggested that IL-36γ, as one of IL-36 receptor agonists, may be a potential target for airway remodeling in asthma.
IL-38 is an IL-1 family anti-inflammatory factor that can exert its function by binding the receptors, IL-36R, IL-1R1, or IL-1RAPL1 [17–19]. Sun et al. have demonstrated that IL-38 can alleviate airway obstruction, reduced eosinophil aggregation and proliferation of Th2 and Th7 cells, and promoted Treg-cell differentiation while inhibiting the release of Th2-type cytokines in asthma mice [25]. However, there was no evidence that IL-38 alleviates airway remodeling in chronic asthma. But the relation between IL-38 and tissues remodeling has been verified in other diseases. For example, IL-38 can ameliorate the pathological manifestations of bleomycin-induced idiopathic pulmonary fibrosis [43]. Wei et al.’s research showed that IL-38 may protect against ventricular remodeling after MI by regulating the function of dendritic cells (DCs) [44]. Shi et al. have indicated that different concentrations of IL-38 exert anti-inflammatory and antifibrotic effects in Thyroid-Associated Ophthalmopathy in vitro [45]. In this study, we have verified that IL-38 can inhibit the activation and migration of lung fibroblast induced by IL-36γ in vitro, and in vivo, with the treatment of IL-38, the airway inflammation, hyper-responsiveness, and remodeling in chronic asthma mice were significantly alleviated, similar to treatment of IL-36Ra. The high expression of p-ERK1/2 in the lung tissues of asthmatic mice was inhibited after IL-38 treatment. The number of activated fibroblasts around airway was also decreased after IL-38 treated compared with the asthma group. Collectively, all these data indicated that IL-38 has a promising therapeutic effect on chronic asthma airway remodeling, and the mechanism may be related to blocking the profibrotic effect of IL-36γ.
In this present study, we also tested the mRNA level of IL-36α and IL-36β in the mice lung tissues. IL-36α mRNA level was mildly elevated, and no IL-36β mRNA was detected in asthmatic mice lungs (data not shown). Although it has demonstrated the fibrotic effect of IL-36α on renal fibrosis and inflammatory bowel disease, the role of IL-36α on chronic asthma airway remodeling is worth to further in-depth research. Moreover, animal models can only mimic partial characteristics of human disease, and we need to collect more tissue specimens from asthma patients in subsequent studies to further analyze the relationship between IL-36γ and airway remodeling in asthma.
Conclusion
In summary, we demonstrated that IL-36γ may participate in promoting airway remodeling in chronic asthma by upregulating profibrotic functions of lung fibroblasts. Furthermore, we also revealed that IL-38 has a protective role on airway remodeling in chronic asthma through inhibiting the profibrotic effect of IL-36γ. IL-38 may be a promising target for the treatment of chronic asthma.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgement
We sincerely thank Tian-Yun Lan (Central Laboratory, The Third Affiliated Hospital of Sun Yat-Sen University, Guangzhou, China) for assistance with flow cytometry analysis.
Glossary
Abbreviations:
- AHR
airway hyper-responsiveness
- α-SMA
alpha smooth muscle actin
- BALF
bronchoalveolar lavage fluid
- DAPI
4,6-diamidino-2-phenylindole
- ELISA
enzyme-linked immunosorbent assay
- FBS
fetal bovine serum
- HDM
house dust mite
- IL
interleukin
- IL-36R
interleukin-36 receptor
- IL-36Ra
interleukin-36 receptor antagonist
Contributor Information
Min Zhang, Department of Pulmonary and Critical Care Medicine, The Third Affiliated Hospital of Sun Yat-Sen University, Institute of Respiratory Disease of Sun Yat-Sen University, Guangzhou, Guangdong, China.
Jian-Xia Zhou, Department of Pulmonary and Critical Care Medicine, The Third Affiliated Hospital of Sun Yat-Sen University, Institute of Respiratory Disease of Sun Yat-Sen University, Guangzhou, Guangdong, China.
Chu-Qin Huang, State Key Laboratory of Respiratory Disease, National Clinical Research Center for Respiratory Disease, Guangzhou Institute of Respiratory Health, The First Affiliated Hospital of Guangzhou Medical University, Guangzhou, Guangdong, China.
Kang-Ni Feng, Department of Pulmonary and Critical Care Medicine, The Third Affiliated Hospital of Sun Yat-Sen University, Institute of Respiratory Disease of Sun Yat-Sen University, Guangzhou, Guangdong, China.
Xiao-Ling Zou, Department of Pulmonary and Critical Care Medicine, The Third Affiliated Hospital of Sun Yat-Sen University, Institute of Respiratory Disease of Sun Yat-Sen University, Guangzhou, Guangdong, China.
Jie-Mei Cen, Department of Pulmonary and Critical Care Medicine, The Third Affiliated Hospital of Sun Yat-Sen University, Institute of Respiratory Disease of Sun Yat-Sen University, Guangzhou, Guangdong, China.
Ping Meng, Department of Pulmonary and Critical Care Medicine, The Third Affiliated Hospital of Sun Yat-Sen University, Institute of Respiratory Disease of Sun Yat-Sen University, Guangzhou, Guangdong, China.
Hong-Tao Li, Department of Pulmonary and Critical Care Medicine, The Third Affiliated Hospital of Sun Yat-Sen University, Institute of Respiratory Disease of Sun Yat-Sen University, Guangzhou, Guangdong, China.
Tian-Tuo Zhang, Department of Pulmonary and Critical Care Medicine, The Third Affiliated Hospital of Sun Yat-Sen University, Institute of Respiratory Disease of Sun Yat-Sen University, Guangzhou, Guangdong, China.
Ethics approval
The study was approved by the Ethics Committee of the Third Affiliated Hospital of Sun Yat-Sen University (number [2019]02-148-01). The authors confirm that the animal research adhere to the ARRIVE guidelines.
Conflict of interests
None declared.
Funding
This study was supported by grants from the National Natural Science Foundation of China (grant nos. 81970017, 81973984).
Data availability
All data included in this study are available upon request by contact with the corresponding author.
Author contributions
Conceptualization: M.Z. and T.Z. Data curation: M.Z., J.Z., and T.Z. Formal analysis: M.Z., J.Z., C.H., X.Z., and T.Z. Funding acquisition: T.Z. and H.L. Investigation: M.Z., J.Z., K.F., and P.M. Methodology: M.Z., J.Z., C.H., K.F., and J.C. Validation: T.Z. Project administration: T.Z. Writing—original draft preparation: M.Z. Visualization: X.Z. All authors have read and agreed to the published version of the manuscript.
References
- 1. Zhang FQ, Han XP, Zhang F, Ma X, Xiang D, Yang XM, et al. . Therapeutic efficacy of a co-blockade of IL-13 and IL-25 on airway inflammation and remodeling in a mouse model of asthma. Int Immunopharmacol 2017, 46, 133–40. doi: 10.1016/j.intimp.2017.03.005 [DOI] [PubMed] [Google Scholar]
- 2. GBD 2016 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1211–59. doi: 10.1016/S0140-6736(17)32154-2. Erratum in: Lancet. 2017 Oct 28;390(10106): e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lambrecht BN, Hammad H.. The immunology of asthma. Nat Immunol 2015, 16, 45–56. doi: 10.1038/ni.3049 [DOI] [PubMed] [Google Scholar]
- 4. Boulet LP. Airway remodeling in asthma: update on mechanisms and therapeutic approaches. Curr Opin Pulm Med Jan, 24, 56–62. doi: 10.1097/MCP.0000000000000441 [DOI] [PubMed] [Google Scholar]
- 5. Warner SM, Knight DA.. Airway modeling and remodeling in the pathogenesis of asthma. Curr Opin Allergy Clin Immunol 2008, 8, 44–8. doi: 10.1097/ACI.0b013e3282f3b5cb [DOI] [PubMed] [Google Scholar]
- 6. Towne JE, Renshaw BR, Douangpanya J, Lipsky BP, Shen M, Gabel CA, et al. . Interleukin-36 (IL-36) ligands require processing for full agonist (IL-36α, IL-36β, and IL-36γ) or antagonist (IL-36Ra) activity. J Biol Chem 2011, 286, 42594–602. doi: 10.1074/jbc.M111.267922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ramadas RA, Ewart SL, Medoff BD, LeVine AM.. Interleukin-1 family member 9 stimulates chemokine production and neutrophil influx in mouse lungs. Am J Respir Cell Mol Biol 2011, 44, 134–45. doi: 10.1165/rcmb.2009-0315OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Murrieta-Coxca JM, Rodríguez-Martínez S, Cancino-Diaz ME, Markert UR, Favaro RR, Morales-Prieto DM.. IL-36 cytokines: regulators of inflammatory responses and their emerging role in immunology of reproduction. Int J Mol Sci 2019, 20, 1649. doi: 10.3390/ijms20071649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Qin X, Liu M, Zhang S, Wang C, Zhang T.. The role of IL-36γ and its regulation in eosinophilic inflammation in allergic rhinitis. Cytokine 2019, 117, 84–90. doi: 10.1016/j.cyto.2019.02.008 [DOI] [PubMed] [Google Scholar]
- 10. Qin X, Zhang T, Wang C, Li H, Liu M, Sun Y.. IL-36α contributes to enhanced T helper 17 type responses in allergic rhinitis. Cytokine 2020, 128, 154992. doi: 10.1016/j.cyto.2020.154992 [DOI] [PubMed] [Google Scholar]
- 11. Wang H, Li ZY, Jiang WX, Liao B, Zhai GT, Wang N, et al. . The activation and function of IL-36γ in neutrophilic inflammation in chronic rhinosinusitis. J Allergy Clin Immunol 2018, 141, 1646–58. doi: 10.1016/j.jaci.2017.12.972 [DOI] [PubMed] [Google Scholar]
- 12. Mattii M, Ayala F, Balato N, Filotico R, Lembo S, Schiattarella M, et al. . The balance between pro- and anti-inflammatory cytokines is crucial in human allergic contact dermatitis pathogenesis: the role of IL-1 family members. Exp Dermatol 2013, 22, 813–9. doi: 10.1111/exd.12272 [DOI] [PubMed] [Google Scholar]
- 13. Bochkov YA, Hanson KM, Keles S, Brockman-Schneider RA, Jarjour NN, Gern JE.. Rhinovirus-induced modulation of gene expression in bronchial epithelial cells from subjects with asthma. Mucosal Immunol 2010, 3, 69–80. doi: 10.1038/mi.2009.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang J, Yin Y, Lin X, Yan X, Xia Y, Zhang L, et al. . IL-36 induces cytokine IL-6 and chemokine CXCL8 expression in human lung tissue cells: implications for pulmonary inflammatory responses. Cytokine 2017, 99, 114–23. doi: 10.1016/j.cyto.2017.08.022 [DOI] [PubMed] [Google Scholar]
- 15. Scheibe K, Kersten C, Schmied A, Vieth M, Primbs T, Carlé B, et al. . Inhibiting interleukin 36 receptor signaling reduces fibrosis in mice with chronic intestinal inflammation. Gastroenterology 2019, 156, 1082–1097.e11. doi: 10.1053/j.gastro.2018.11.029 [DOI] [PubMed] [Google Scholar]
- 16. Bensen JT, Dawson PA, Mychaleckyj JC, Bowden DW.. Identification of a novel human cytokine gene in the interleukin gene cluster on chromosome 2q12-14. J Interferon Cytokine Res 2001, 21, 899–904. doi: 10.1089/107999001753289505 [DOI] [PubMed] [Google Scholar]
- 17. van de Veerdonk FL, Stoeckman AK, Wu G, Boeckermann AN, Azam T, Netea MG, et al. . IL-38 binds to the IL-36 receptor and has biological effects on immune cells similar to IL-36 receptor antagonist. Proc Natl Acad Sci U S A 2012, 109, 3001–5. doi: 10.1073/pnas.1121534109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mora J, Schlemmer A, Wittig I, Richter F, Putyrski M, Frank A-C, et al. . Interleukin-38 is released from apoptotic cells to limit inflammatory macrophage responses. J Mol Cell Biol 2016, 8, 426–38. doi: 10.1093/jmcb/mjw006 [DOI] [PubMed] [Google Scholar]
- 19. Yuan XL, Li Y, Pan XH, Zhou M, Gao QY, Li MC.. Production of recombinant human interleukin-38 and its inhibitory effect on the expression of proinflammatory cytokines in THP-1 cells. Mol Biol 2016, 50, 405–11. doi: 10.1134/s0026893316030134 [DOI] [PubMed] [Google Scholar]
- 20. Boutet MA, Najm A, Bart G, Brion R, Touchais S, Trichet V, et al. . IL-38 overexpression induces anti-inflammatory effects in mice arthritis models and in human macrophages in vitro. Ann Rheum Dis 2017, 76, 1304–12. doi: 10.1136/annrheumdis-2016-210630 [DOI] [PubMed] [Google Scholar]
- 21. Xu WD, Su LC, Fu L, Lan YY, Liu XY, Huang Q, et al. . IL-38, a potential therapeutic agent for lupus, inhibits lupus progression. Inflamm Res 2022, 71, 963–75. doi: 10.1007/s00011-022-01581-3 [DOI] [PubMed] [Google Scholar]
- 22. Mercurio L, Morelli M, Scarponi C, Eisenmesser EZ, Doti N, Pagnanelli G, et al. . IL-38 has an anti-inflammatory action in psoriasis and its expression correlates with disease severity and therapeutic response to anti-IL-17A treatment. Cell Death Dis 2018, 9, 1104. doi: 10.1038/s41419-018-1143-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ohno M, Imai T, Chatani M, Nishida A, Inatomi O, Kawahara M, et al. . The anti-inflammatory and protective role of interleukin-38 in inflammatory bowel disease. J Clin Biochem Nutr 2022, 70, 64–71. doi: 10.3164/jcbn.21-104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wu Z, Mehrabi Nasab E, Arora P, Athari SS.. Study effect of probiotics and prebiotics on treatment of OVA-LPS-induced of allergic asthma inflammation and pneumonia by regulating the TLR4/NF-kB signaling pathway. J Transl Med 2022, 20, 130. doi: 10.1186/s12967-022-03337-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sun X, Hou T, Cheung E, Iu TN, Tam VW, Chu IM, et al. . Anti-inflammatory mechanisms of the novel cytokine interleukin-38 in allergic asthma. Cell Mol Immunol 2020, 17, 631–46. doi: 10.1038/s41423-019-0300-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Feng KN, Meng P, Zhang M, Zou XL, Li S, Huang CQ, et al. . IL-24 contributes to neutrophilic asthma in an IL-17A-dependent manner and is suppressed by IL-37. Allergy Asthma Immunol Res 2022 Sep, 14, 505–27. doi: 10.4168/aair.2022.14.5.505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen J, Zhou H, Wang J, Zhang B, Liu F, Huang J, et al. . Therapeutic effects of resveratrol in a mouse model of HDM-induced allergic asthma. Int Immunopharmacol 2015, 25, 43–8. doi: 10.1016/j.intimp.2015.01.013 [DOI] [PubMed] [Google Scholar]
- 28. He H, Cao L, Wang Z, Wang Z, Miao J, Li XM, et al. . Sinomenine relieves airway remodeling by inhibiting epithelial-mesenchymal transition through downregulating TGF-β1 and Smad3 expression in vitro and in vivo. Front Immunol 2021, 12, 736479. doi: 10.3389/fimmu.2021.736479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Feng KN, Meng P, Zou XL, Zhang M, Li HK, Yang HL, et al. . IL-37 protects against airway remodeling by reversing bronchial epithelial-mesenchymal transition via IL-24 signaling pathway in chronic asthma. Respir Res 2022, 23, 244. doi: 10.1186/s12931-022-02167-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Olczyk P, Mencner L, Komosinska-Vassev K.. The role of the extracellular matrix components in cutaneous wound healing. Biomed Res Int 2014, 2014, 747584. doi: 10.1155/2014/747584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rosethorne EM, Charlton SJ.. Airway remodeling disease: primary human structural cells and phenotypic and pathway assays to identify targets with potential to prevent or reverse remodeling. J Exp Pharmacol 2018, 10, 75–85. doi: 10.2147/JEP.S159124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dai Y, Li F, Wu L, Wang R, Li P, Yan S, et al. . Roxithromycin treatment inhibits TGF-β1-induced activation of ERK and AKT and down-regulation of caveolin-1 in rat airway smooth muscle cells. Respir Res 2014, 15, 96. doi: 10.1186/s12931-014-0096-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Johnson MT, Xin P, Benson JC, Pathak T, Walter V, Emrich SM, et al. . STIM1 is a core trigger of airway smooth muscle remodeling and hyperresponsiveness in asthma. Proc Natl Acad Sci U S A 2022, 119, e2114557118. doi: 10.1073/pnas.2114557118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fang L, Sun Q, Roth M.. Immunologic and non-immunologic mechanisms leading to airway remodeling in asthma. Int J Mol Sci 2020, 21, 757. doi: 10.3390/ijms21030757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mostaço-Guidolin LB, Osei ET, Ullah J, Hajimohammadi S, Fouadi M, Li X, et al. . Defective Fibrillar Collagen Organization by fibroblasts contributes to airway remodeling in asthma. Am J Respir Crit Care Med 2019, 200, 431–43. doi: 10.1164/rccm.201810-1855OC [DOI] [PubMed] [Google Scholar]
- 36. Reeves SR, Kolstad T, Lien TY, Elliott M, Ziegler SF, Wight TN, et al. . Asthmatic airway epithelial cells differentially regulate fibroblast expression of extracellular matrix components. J Allergy Clin Immunol 2014, 134, 663–670.e1. doi: 10.1016/j.jaci.2014.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fang CL, Yin LJ, Sharma S, Kierstein S, Wu HF, Eid G, et al. . Resistin-like molecule-β (RELM-β) targets airways fibroblasts to effect remodelling in asthma: from mouse to man. Clin Exp Allergy 2015, 45, 940–52. doi: 10.1111/cea.12481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu H, Zhang X, Shao Y, Lin X, Dong F, Liu X.. Danshensu alleviates bleomycin-induced pulmonary fibrosis by inhibiting lung fibroblast-to-myofibroblast transition via the MEK/ERK signaling pathway. Bioengineered 2021, 12, 3113–24. doi: 10.1080/21655979.2021.1944020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Xie M, Liu XS, Xu YJ, Zhang ZX, Bai J, Ni W, et al. . ERK1/2 signaling pathway modulates the airway smooth muscle cell phenotype in the rat model of chronic asthma. Respiration 2007, 74, 680–90. doi: 10.1159/000108783 [DOI] [PubMed] [Google Scholar]
- 40. Scheibe K, Backert I, Wirtz S, Hueber A, Schett G, Vieth M, et al. . IL-36R signalling activates intestinal epithelial cells and fibroblasts and promotes mucosal healing in vivo. Gut 2017, 66, 823–38. doi: 10.1136/gutjnl-2015-310374 [DOI] [PubMed] [Google Scholar]
- 41. Chi HH, Hua KF, Lin YC, Chu CL, Hsieh CY, Hsu YJ, et al. . IL-36 signaling facilitates activation of the NLRP3 inflammasome and IL-23/IL-17 axis in renal inflammation and fibrosis. J Am Soc Nephrol 2017, 28, 2022–37. doi: 10.1681/ASN.2016080840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang Q, Guo L, Song X, Lv C, Tang P, Li Y, et al. . Serum IL-36 cytokines levels in idiopathic pulmonary fibrosis and connective tissue disease-associated interstitial lung diseases. Clin Chim Acta 2022, 530, 8–12. doi: 10.1016/j.cca.2022.02.015 [DOI] [PubMed] [Google Scholar]
- 43. Xu Z, Yuan X, Gao Q, Li Y, Li M.. Interleukin-38 overexpression prevents bleomycin-induced mouse pulmonary fibrosis. Naunyn Schmiedebergs Arch Pharmacol 2021, 394, 391–9. doi: 10.1007/s00210-020-01920-3 [DOI] [PubMed] [Google Scholar]
- 44. Wei Y, Lan Y, Zhong Y, Yu K, Xu W, Zhu R, et al. . Interleukin-38 alleviates cardiac remodelling after myocardial infarction. J Cell Mol Med 2020, 24, 371–84. doi: 10.1111/jcmm.14741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shi L, Ye H, Huang J, Li Y, Wang X, Xu Z, et al. . IL-38 exerts anti-inflammatory and antifibrotic effects in thyroid-associated ophthalmopathy. J Clin Endocrinol Metab 2021, 106, e3125–42. doi: 10.1210/clinem/dgab154 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data included in this study are available upon request by contact with the corresponding author.