

Abstract
This study aimed to evaluate the bioequivalence between test tablet dacomitinib and reference product Vizimpro® under fasting and fed conditions and assess their pharmacokinetic (PK) and safety profiles for gaining marketing approval of the new generic drug. A single‐center, randomized, open‐label, single‐dose, two‐treatment, two‐period, crossover bioequivalence study was conducted in healthy Chinese subjects. Eligible healthy subjects randomly received a single 45 mg dose of test or reference formulations with an administration sequence of test tablet (T), reference tablet (R), or (RT), under both fasting and fed conditions, and each single administration was followed by a 21‐day washout period. Plasma concentrations and corresponding non‐compartmental PK parameters of dacomitinib were determined. The 90% confidence intervals of the geometric mean ratio (GMR) (test/reference) for Cmax, AUC0–t , and AUC0–∞, respectively, were 97.75%–119.99%, 101.00%–115.09%, and 100.27%–113.90% under fasting conditions and 95.20%–104.94%, 97.24%–102.23%, and 97.27%–101.88% under fed conditions, which were within the limits of 80%–125%. Under fasting and fed conditions, the PK characteristics of the test dacomitinib tablet and reference Vizimpro® were comparable; the two formulations of dacomitinib were demonstrated to be bioequivalent and well‐tolerated in healthy Chinese volunteers.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
There are limited pharmacokinetic (PK) data for dacomitinib (Vizimpro®) in Chinese volunteers. The original drug is too expensive for most patients with cancer, and there is an urgent need to exploit generic drugs to provide the maximum accessibility of treatments for patients and relieve the socioeconomic burden in China.
WHAT QUESTION DID THIS STUDY ADDRESS?
The study evaluated the bioequivalence between 45 mg test dacomitinib tablet and 45 mg reference product Vizimpro® under fasted and fed conditions and assessed their PK and safety profiles for the marketing approval of the new generic drug.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
The test dacomitinib and reference product Vizimpro® were bioequivalent and well‐tolerated in healthy Chinese volunteers under fasting and fed conditions.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
Test dacomitinib tablets are comparable to reference Vizimpro® products with or without food intake, thus the generic drug could be an alternative to the expensive original drug in clinical practice. These results may provide a critical reference for the development of homogeneous generic drugs.
INTRODUCTION
Lung cancer has a high incidence rate and is the primary cause of cancer‐related deaths worldwide. Notwithstanding, non‐small cell lung cancer (NSCLC) accounts for approximately 85% of lung cancer cases. 1 , 2 Epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) have been approved as basic therapeutic agents for NSCLC with EGFR genetic mutation. 3 , 4 , 5 , 6 In contrast to first‐generation EGFR TKIs, second‐generation EGFR TKIs target EGFR selectively and are irreversible inhibitors sheltering activated mutations in the epidermal growth factor receptor 2 genes, such as human epidermal growth factor receptor‐1 (HER1), human epidermal growth factor receptor‐2 (HER2), and human epidermal growth factor receptor‐4 (HER4), belonging to the ErbB family, also known as the EGF receptor family or type I receptor family. 7 , 8 , 9 , 10 Dacomitinib, as a first‐line therapy for NSCLC patients with EGFR mutation positivity, has been demonstrated to significantly improve progression‐free survival (PFS) and overall survival (OS) of patients, as compared with gefitinib, which is a first‐generation EGFR TKI considered superior to standard chemotherapy. 11 , 12 , 13 , 14
Dacomitinib is an irreversible ErBb inhibitor that has been approved as first‐line therapy for NSCLC patients with EGFR‐positive mutation as first‐line therapy measurement. 7 , 8 , 15 In September 2018, dacomitinib received its first US Food and Drug Administration (FDA) global approval for use as the first‐line treatment of patients with metastatic NSCLC with EGFR exon 19 deletion or exon 21 L858R substitution mutations, as detected by an FDA‐approved test. 16 Japan, Europe, and China have also approved its marketing. In China, the National Medical Products Administration (NMPA) approved dacomitinib as a monotherapy for the first‐line treatment of adult patients with locally advanced or metastatic NSCLC with EGFR exon 19 deletion or exon 21 L858R substitution mutations in May 2019.
Dacomitinib (Vizimpro®) is available as 15, 30, and 45 mg tablets. Dacomitinib (45 mg) is the recommended dose for 1 day, and the physician modulates the dose based on tolerance until disease progression or intolerability. Previous studies have shown that food intake has no significant effect on its bioavailability; thus, it can be taken with or without food. 17 , 18 Dacomitinib has a rapid absorption and has 80% high bioavailability after oral administration. 19 The median T max was approximately 6 h (2–24 h) after the administration of 45 mg dacomitinib. 17 , 19 Blood drug concentrations stabilized within 14 days. 17 The large apparent volume of 1889 L indicated that the drug was distributed in the tissue extensively. 17 In vitro, human plasma proteins bound nearly 98% dacomitinib. The mean t 1/2 of dacomitinib was approximately 70 h, and the apparent plasma clearance was 24.9 L/h. 17 Most (79%) of the radiolabeled dacomitinib was eliminated by means of the fecal route, with the remaining portion being detected as unchanged dacomitinib. 20
Easton Biopharmaceuticals (Chengdu, China) has developed generic dacomitinib tablets. In line with the NMPA guidelines, we designed this study to evaluate the bioequivalence of test dacomitinib and reference Vizimpro®, and also evaluated dacomitinib's tolerability and pharmacokinetic (PK) characteristics. This study provides further support for the marketing authorization of generic drugs in China.
METHODS
Populations
Eligible subjects were aged 18–45 years with a body mass index (BMI; weight [kg]/height [m2]) ranging from 19.0 to 26.0 kg/m2 (including boundary values). Volunteers were in good physical condition with no abnormalities of clinical significance based on the physical examination, vital signs, 12‐lead electrocardiogram, chest X‐ray, and clinical laboratory examination results during the screening period; subjects had no plans to give birth or undergo sperm/egg donation and voluntarily used an effective physical contraceptive method. They also understood the study objectives and gave written informed consent.
Exclusion criteria: currently suffering from circulatory system, endocrine, nervous, digestive, respiratory system, hematology, immunology, urology, psychiatry, metabolic abnormalities, or any other clinically serious disease or any other disease that could influence the study implementation; history of solar dermatitis or allergies to ultraviolet (UV) rays or dacomitinib or its preparations or history of allergy; use of vaccine within 1 month, use of any prescription or non‐prescription medications in the previous 14 days, any drug–drug interactions with dacomitinib within 4 weeks of administration; presence of alcoholism, smoking addiction, or drug abuse; history of major surgery or having suffered serious external injury; use of special food (e.g., tea, other drink, coffee, etc.) for 48 h before registration; clinically significant abnormal results for HIV, hepatitis B, or hepatitis C; pregnant or lactating; participating in other clinical trials within 90 days prior to registration; and unable to adapt to dietary requirements.
Study drug
Dacomitinib (45 mg/tablet) was obtained from Easton Biopharmaceuticals (Chengdu, China; batch number: P220601; expiry date: May 2025). The key to product preparation was crystallization of the active pharmaceutical ingredients (APIs). The reference preparation (Vizimpro®, 45 mg/tablet) was produced by Pfizer Europe MA EEIG, and provided by Easton Biopharmaceuticals (batch number EP3158, expiry date February 2023). All subjects underwent a single oral test or reference administration according to the sequence of the dosing randomization table during periods I and II.
Study design
This study included fasting and fed clinical trials, both of which were designed as single‐center, randomized, open‐label, single‐dose, two‐period, two‐treatment crossover bioequivalents. Eligible volunteers were enrolled at pre‐dose day and were randomized to receive T (test) or R (reference) administration in period I and to receive R or T administration in period II in reverse according to a sequence of dosing randomization scheme. A 21‐day washout period was set according to a t 1/2 of ~70 h. SAS (version 9.4) software was used to generate the randomization scheme in a 1:1 randomized design. A single‐dose test or reference product was orally taken with 240 mL warm water after at least a 10 h overnight fast for subjects following fasting conditions. The subjects in the fed study received a standard high‐fat breakfast comprising approximately 800–1000 kcal (protein ~150, carbohydrates ~250, and fat ~500–600 kcal, respectively) within 0.5 min of the pre‐dose and then carried out the same procedure. The researchers confirmed that the participants had swallowed the drugs. Water was prohibited for the subjects for 1 h before and after administration. At 4 h and 10 h after administration, the subjects received regular standardized meals. Safety evaluations were conducted throughout the study.
Sample size
According to previous studies, dacomitinib shows a low degree of variation. It was assumed that the GMR of T/R was 92% and intra‐coefficient of variation (CV) was 19%; therefore, a total of 48 subjects were required to establish the bioequivalence limits of 80.00%–125.00% between T and R, with 80% power at a significance level of 5%. Considering ~20% dropout and/or withdrawal, 60 patients were sufficient to meet statistical requirements.
Ethics statement
According to the Declaration of Helsinki and the principles of Good Clinical Practice, the study was reviewed and approved by the Institutional Ethics Committee of Chengdu Fifth People's Hospital (The Second Clinical Medical College, Affiliated Fifth People's Hospital of Chengdu University of Traditional Chinese Medicine, Chengdu, China; Approval No. 2022‐014(drug)‐01). Written informed consent was obtained from all the participants before conducting the study, and the study was registered in the Chinese Clinical Trial Registry (No. ChiCTR20222221).
Sample collection and handling
Blood samples were collected to assess the bioequivalence between T and R. The venous blood samples were collected in pre‐cooled heparin sodium anticoagulant vacuum sampling vessels from patients' forearm veins within 1 h at pre‐dose and at 1.0, 2.0, 4.0–9.0 (at intervals of 0.5 h each), 10.0, 12.0, 15.0, 24.0, 48.0, 72.0, 96.0, 120.0, 144.0, 192.0, and 240.0 h after a single oral dose. The blood samples were gently reversed and mixed five to eight times, and then placed in a low‐temperature (2–8°C) centrifuge (set at 4°C) and spun at 1700 g for 10 min, within 1 h of collection. Plasma samples were divided into test and backup tubes labeled with identification stickers. Each test tube contained ~0.8 mL of plasma sample for analysis, and the remaining plasma was stored in another tube as backup. The plasma sample volume did not exceed two‐thirds of the frozen tube volume. When the plasma volume was insufficient, the test tube capacity was maintained. All whole blood samples were temporarily stored in an ice bath after collection, and the plasma after separation was temporarily stored upright in ice bath conditions and transferred to a −80°C (−60°C to −90°C) refrigerator within 2 h of centrifugation until it was shipped to the analysis laboratory (Chengdu Finelyse Pharmaceutical Technology Co., Ltd., Chengdu, China). Backup tubes were kept upright in a −80°C refrigerator in the research institution to ensure that they were available when reanalysis was necessary.
Plasma concentration analysis
Plasma samples were analyzed at Chengdu Finelyse Pharmaceutical Technology Co., Ltd. Plasma concentrations of test dacomitinib tablet and reference product Vizimpro® were analyzed using high‐performance liquid chromatography–tandem mass spectrometry (LC–MS/MS). Validation parameters included stock check, selectivity, carryover, intra‐lot and inter‐lot precision and accuracy, dilution accuracy, batch capacity test, standard curve, matrix effect, recovery, durability and stability, and accompanying quality control and system suitable test. All the results met with regulatory requirements. The method was developed by Chengdu Finelyse Pharmaceutical Technology Co., Ltd. The linear range of the calibration curve for dacomitinib was 0.5–50 ng/mL, including 0.5000, 1.000, 4.000, 10.00, 25.00, 40.00, and 50.00 ng/mL, the lower limit of quantitation (LLOQ) was 0.5 ng/mL, the linearity acceptance criteria was set as R‐squared value ≥0.9800, and weighting was 1/C2. The quality control samples consisted of 1.50 (QC.1), 6.00 (QC.2), 20.00 (QC.3), 37.50 (QC.4), and 75.00 ng/mL (QC.DIL) with a dilution ratio of 4. For sample concentrations below the lower limit of quantification, a value of “0” was assigned for the time points before T max and recorded as missing data after T max during PK analysis.
For the analysis, 100 μL of the plasma samples and 50 μL of the internal standard (Dacomitinib‐d10, 50 ng/mL) solution were transferred to a 96‐hole plate, then vortexed for 30 s, and 250 μL of acetonitrile was added to the 96‐hole plate. After being vortexed for 3 min and centrifuged at 4000 rpm for 10 min at 4°C, 200 μL of the supernatant was transferred to a clean 96‐hole plate, and 100 μL of ultrapure water was added to it and mixed up. Then, after being vortexed for 1 min, 10 μL volumes were injected into the LC–MS/MS, and a 3‐min run time set up.
Data acquisition and integration were performed using Analyst 1.6.3 (Applied Biosystems, USA) and the corresponding plasma concentrations were calculated using Watson LIMS 7.5 sp1 (Thermo Fisher Corporation, USA).
Dacomitinib was detected by positive ionization in multiple reaction monitoring modes using an AB SCIEX API 4000 LC–MS. The typical ion source parameters were: declustering potential 105 V, curtain gas 20 psi, ion source gas1 30 psi, gas2 40 psi, collision energy 45 eV, and temperature 550°C. The mass transitions of m/z 470.2/319.2 and 480.2/319.2 were used to quantify dacomitinib and dacomitinib‐d10, respectively. A Phenomenex Kinetex C18 column (2.1 mm ID × 50 mm L, 5 μm) was used as the analytical column. The mobile phases included mobile phase A: 25% ammonia liquor, LC–MS: ultrapure water (1:2000, v/v), and mobile phase B: methanol: acetonitrile (1:1, v/v). A gradient elution procedure was used to separate the components.
PK statistical analysis
PK evaluations contained plasma concentrations and PK parameters of test dacomitinib tablet and reference product Vizimpro®. The primary PK parameters were as follows: Cmax, the maximum plasma drug concentration; AUC0–t, area under the plasma concentration–time curve (AUC) from time 0 to the last measurable concentration, calculated using the log‐linear trapezoid method: AUC(i,i + 1) = (T i + 1 − T i )(C i + C i + 1)/2, where AUC0–t is the sum of all AUC(i,i + 1); AUC0–∞, AUC from time 0 to infinity, calculated using AUC0–t + C t /λz, where C t is the last quantifiable concentration; T max, peak time of plasma drug concentration reached at Cmax; λ z, terminal elimination rate (i.e., the slope at the end of a semi‐logarithmic time curve, calculated using linear regression); t 1/2, time of elimination half‐life, calculated by ln2/λ z ; AUC_%Extrap, percentage of AUC derived after extrapolation, calculated using (AUC0–∞ to AUC0–t )/AUC0–∞ × 100%; and if AUC_%Extrap >20%, the AUC0–∞ was considered unreliable; Vz/F, apparent volume of distribution; and CL/F, apparent clearance. We performed non‐compartmental analysis to calculate plasma drug concentration–time data by WinNonlin (Pharsight Corporation 8.3). A mixed‐effect model was used to analyze the main log‐transformed PK parameters (AUC0–∞, AUC0–t and Cmax). In the model, the sequence, period, and formulation were fixed effects and the subjects (sequences) were random effects. Bioequivalence was assessed using 90% CIs of the GMR of the T/R. The two formulations were considered to be bioequivalent if the values fell within the limits of 80.00%–125.00%. T max was analyzed using a non‐parametric test analysis (Wilcoxon signed‐rank test). PK parameter calculations were performed using WinNonlin and the remaining analyses were performed using SAS 9.4.
Safety assessment
To assess the safety of dacomitinib tablets, vital signs (blood pressure, pulse, and body temperature), physical examinations, chest radiography, 12‐lead electrocardiograms, and clinical laboratory tests (including chemistry, hematology, blood coagulation, urinalysis, immunology, blood lipid tests, and blood pregnancy tests [for women of childbearing age only]) were performed at regular intervals throughout the study. Vital signs were measured at pre‐dose (within 1 h) and 2.0, 4.0, 8.0, 12.0, 24.0, 48.0, 72.0, 96.0, 120.0, 144.0, 192.0, and 240.0 h after drug administration in each period. Physical examinations, 12‐lead electrocardiography, and clinical laboratory tests were performed for each subject during the screening and follow‐up periods, as planned. In addition, from the first dose to the end of the study, all adverse events (AEs), treatment‐emergent adverse events (TEAEs), serious treatment‐emergent adverse events (STEAEs), drug‐related TEAEs, TEAEs that caused subjects to withdraw from the trial, and TEAEs associated with the study drug that caused subjects to withdraw from the trial were reported and classified on the basis of the System Organ Class (SOC) and Preferred Term (PT). National Cancer Institute (NCI) Common Terminology Criteria Adverse Events (CTCAE) Ver. 5.0 was used to determine the corresponding severity.
RESULTS
Subjects’ disposition and baseline characteristics
77 subjects were screened for the fasting group, and 104 subjects were screened for the fed group. Ultimately, 30 eligible subjects were randomized to the two‐part study; 28 subjects in the fasted study and 29 subjects in the fed study completed the trial, and all randomized subjects were included in the bioequivalence, PK, and safety analysis sets. The baseline characteristics of the patients are summarized in Table 1. The majority (85%) of subjects were male in the two‐part study. The mean ± SD ages were 27.20 ± 6.31 years for the fasting study and 25.73 ± 4.60 years for the fed study, and the mean ± SD BMI were 21.80 ± 1.81 kg/m2 and 21.72 ± 1.72 kg/m2, respectively.
TABLE 1.
Demographic and baseline characteristics.
| Characteristic | Fasting condition (n = 30) | Fed condition (n = 30) |
|---|---|---|
| Age (years) | 27.20 ± 6.31 | 25.73 ± 4.60 |
| Gender, n (%) | ||
| Male | 25 (83.3) | 26 (86.7) |
| Female | 5 (16.7) | 4 (13.3) |
| Height (cm) | 166.83 ± 8.66 | 167.20 ± 7.44 |
| Weight (kg) | 60.84 ± 8.61 | 60.83 ± 7.38 |
| Body mass index (kg/m2) | 21.80 ± 1.81 | 21.72 ± 1.72 |
PK and bioequivalence
As shown in Figure 1, the trends of the mean plasma concentration–time curves for the two formulations were similar under fasting conditions, and separate semi‐logarithmic plots of plasma concentrations versus time in the fasted state are shown in Figure S1. The PK parameters of the subjects after a single oral dose of the test and reference drugs under fasting conditions are summarized in Table 2. After oral administration of dacomitinib under fasting conditions, the mean Cmax value was 37.58 ± 11.03 ng/mL and this was reached within 4.5–10 h; the mean AUC0–t and AUC0–∞ values were 1709.51 ± 387.14 and 1851.27 ± 467.85 ng·h/mL, respectively; and the mean t 1/2 was 62.95 ± 16.58 h. After receiving the reference products, the mean Cmax value was 35.01 ± 11.23 ng/mL and this was reached within 4.5–48 h; the mean AUC0–t and AUC0–∞ values were 1607.10 ± 452.92 and 1762.55 ± 541.14 ng·h/mL, respectively; and the mean t 1/2 was 67.10 ± 22.57 h. The PK properties of the two formulations were comparable in the fasting study.
FIGURE 1.
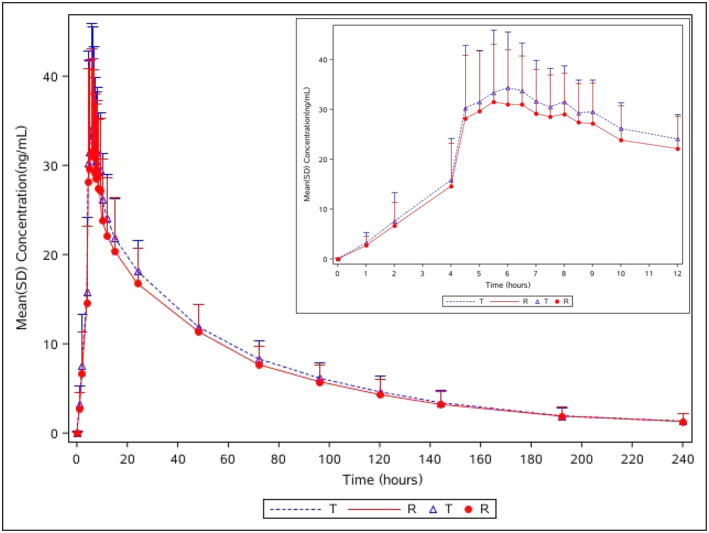
Plasma concentration versus time curves for dacomitinib and Vizimpro® under fasting conditions; n = 30 for dacomitinib (T), n = 29 for Vizimpro® (R). The inset enlargement represents details of the 0–12 h period. Data are presented as mean ± standard deviation (SD). R, reference tablet; T, test tablet.
TABLE 2.
Summary of pharmacokinetic parameters of dacomitinib and Vizimpro® under fasting and fed conditions.
| PK parameter | Fasting condition | Fed condition | ||
|---|---|---|---|---|
| Test (n = 30) | Reference (N = 29) | Test (N = 29) | Reference (N = 30) | |
| T max (h) | 6.00 (4.5, 10) | 6.00 (4.5, 48) | 5.00 (4.5, 12) | 5.00 (4.5, 12) |
| Cmax (ng/mL) | 37.58 ± 11.03 | 35.01 ± 11.23 | 35.47 ± 8.79 | 35.50 ± 7.81 |
| AUC0–t (ng∙h/mL) | 1709.51 ± 387.14 | 1607.10 ± 452.92 | 1769.02 ± 388.25 | 1790.58 ± 377.98 |
| AUC0–∞ (ng∙h/mL) | 1851.27 ± 467.85 | 1762.55 ± 541.14 | 1901.05 ± 438.31 | 1927.24 ± 417.64 |
| λ z (h–1) | 0.0117 ± 0.0030 | 0.0114 ± 0.0036 | 0.0119 ± 0.0031 | 0.0115 ± 0.0030 |
| t 1/2 (h) | 62.95 ± 16.58 | 67.10 ± 22.57 | 61.70 ± 15.04 | 63.95 ± 16.82 |
| AUC_%Extrap (%) | 7.09 ± 4.38 | 8.14 ± 5.32 | 6.68 ± 3.41 | 6.90 ± 3.63 |
| Vz/F (L) (CV%) | 2206.28 (26.62) | 2465.86 (32.66) | 2097.34 (26.01) | 2134.01 (28.63) |
| CL/F (L/h) (CV%) | 25.09 (23.64) | 26.76 (34.59) | 24.26 (22.85) | 23.88 (21.76) |
Note: Vz/F and CL/F values are geometric mean (CV%); T max values are median (minimum, maximum); the remaining values are arithmetic mean ± standard deviation (SD).
Abbreviations: AUC0–t , area under the concentration–time curve from time 0 to the last observed non‐zero concentration calculated by the linear trapezoidal method; AUC0–∞, area under the concentration–time curve from time 0 extrapolated to infinity; AUC_%Extrap, the percentage of the AUC derived after extrapolation; Cmax, maximum plasma concentration; CL/F: apparent clearance; CV, coefficient of variation; λ z : the terminal elimination rate; PK, pharmacokinetic; t 1/2, terminal half‐life; T max, time to maximum plasma concentration; Vz/F: apparent volume of distribution.
The PK bioequivalence assessments under fasting conditions are summarized in Table 3. The 90% CIs of the GMR of the T/R were 97.75%–119.99% for Cmax, 101.00%–115.09% for AUC0–t , and 100.27%–113.90% for AUC0–∞, respectively. The values of these PK parameters fell within the limits of 80.00%–125.00%, which were considered to be bioequivalent for the two formulations under fasting conditions. The coefficients of intra‐subject variation for Cmax, AUC0–t , and AUC0–∞ were 23.28%, 14.44%, and 14.08%, respectively, which implied that dacomitinib is not a highly variable drug according to these PK results. The power of Cmax, AUC0–t , and AUC0–∞ were 75.07%, 98.27%, 99.26%, respectively, which demonstrated that the sample size met statistical requirements.
TABLE 3.
Statistical comparison of bioequivalence parameters of dacomitinib and Vizimpro®.
| PK parameter | Fasting condition | Fed condition | ||||||
|---|---|---|---|---|---|---|---|---|
| GMR (%) | 90% CI | Intra‐subject CV (%) | Power (%) | GMR (%) | 90% CI | Intra‐subject CV (%) | Power (%) | |
| Cmax (ng/mL) | 108.30 | 97.75–119.99 | 23.28 | 75.07 | 99.95 | 95.20–104.94 | 10.94 | >99.99 |
| AUC0–t (ng∙h/mL) | 107.82 | 101.00–115.09 | 14.44 | 98.27 | 99.71 | 97.24–102.23 | 5.60 | >99.99 |
| AUC0–∞ (ng∙h/mL) | 106.86 | 100.27–113.90 | 14.08 | 99.26 | 99.55 | 97.27–101.88 | 5.17 | >99.99 |
Abbreviations: AUC0–t , area under the concentration–time curve from time 0 to the last observed non‐zero concentration calculated by the linear trapezoidal method; AUC0–∞, area under the concentration–time curve from time 0 extrapolated to infinity; Cmax, maximum plasma concentration; CI, confidence interval; CV, coefficient of variation; GMR, geometric mean ratio; PK, pharmacokinetic.
No clear significance was observed for the T max between the two formulations (p = 0.7281) using the non‐parametric Wilcoxon signed‐rank test, which also demonstrated the bioequivalence of the fasting study. As shown in Table 4, there were no period, sequence, or formulation effects observed for lnCmax, lnAUC0–t , and lnAUC0–∞, Subject*sequence had a clear significance (p < 0.05) for lnCmax, lnAUC0–t , and lnAUC0–∞, whereas the significance difference did not affect the bioequivalence evaluation in the fasting study.
TABLE 4.
ANOVA p‐values for dacomitinib and Vizimpro®.
| Parameter | Fasting condition | Fed condition | ||||
|---|---|---|---|---|---|---|
| lnCmax | lnAUC0–t | lnAUC0–∞ | lnCmax | lnAUC0–t | lnAUC0–∞ | |
| Sequence | 0.9618 | 0.7303 | 0.8402 | 0.1065 | 0.0956 | 0.0904 |
| Period | 0.5010 | 0.1940 | 0.2702 | 0.1261 | <0.0001 | <0.0001 |
| Formulation | 0.2403 | 0.0606 | 0.0842 | 0.9994 | 0.8757 | 0.7700 |
| Subject*sequence | 0.0002 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 |
Abbreviations: AUC0–t , area under the concentration–time curve from time 0 to the last observed non‐zero concentration calculated by the linear trapezoidal method; AUC0–∞, area under the concentration–time curve from time 0 extrapolated to infinity; Cmax, maximum plasma concentration.
As shown in Figure 2, the trends of the mean plasma concentration–time curves of dacomitinib and Vizimpro® were similar under fed condition. Separate semi‐logarithmic plots of plasma concentrations versus time in fed state are shown in Figure S2. PK parameters are listed in Table 2. After receiving test or reference products, the mean Cmax values were 35.47 ± 8.79 and 35.50 ± 7.81 ng/mL, respectively, and these values were reached for both formulations within 4.5–12 h. The mean AUC0–t values for dacomitinib and Vizimpro® were 1769.02 ± 388.25 and 1790.58 ± 377.98 ng·h/mL, respectively; and the mean AUC0–∞ values for dacomitinib and Vizimpro® were 1901.05 ± 438.31 and 1927.24 ± 417.64 ng·h/mL, respectively. The mean t 1/2 for dacomitinib and Vizimpro® were 61.70 ± 15.04 and 63.95 ± 16.82 h, respectively. The PK properties of both the test and reference formulations were comparable under fed conditions.
FIGURE 2.
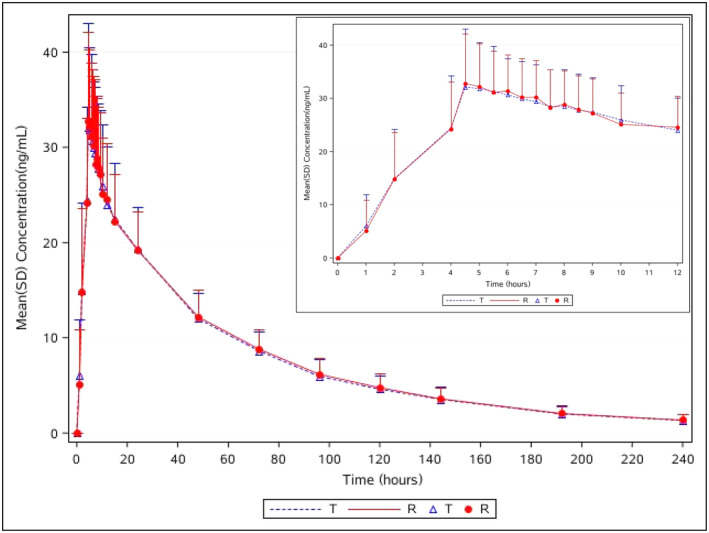
Plasma concentration versus time curves for dacomitinib and Vizimpro® under fed conditions; n = 29 for dacomitinib (T), n = 30 for Vizimpro® (R). The inset enlargement represents details of the 0–12 h period. Data are presented as mean ± standard deviation (SD). R, reference tablet; T, test tablet.
The PK bioequivalence assessments under fed conditions are summarized in Table 3. The 90% CIs of GMR of T/R were 95.20%–104.94% for Cmax, 97.24%–102.23% for AUC0–t , and 97.27%–101.88% for AUC0–∞, respectively. The PK parameter values fell within the limits of 80.00%–125.00%, which implied that the two formulations were bioequivalent. The coefficients of intra‐subject variation for Cmax, AUC0–t , and AUC0–∞ were 10.94%, 5.60%, and 5.17%, respectively. The power of Cmax, AUC0–t , and AUC0–∞ were all above 99.99%, which demonstrated that the sample size met statistical requirements. No statistically significant difference was observed in T max (p = 0.2059), according to the non‐parametric Wilcoxon signed‐rank test, which demonstrated the bioequivalence of the fed study. As shown in Table 4, under fed conditions, no significant effect of lnCmax was observed in sequence, period, and formulation (all p > 0.05), Subject*sequence had a significant effect on lnCmax, lnAUC0–t , and lnAUC0–∞ (p < 0.05), and period had a significant effect on lnAUC0–t and lnAUC0–∞. However, this significant difference did not affect bioequivalence evaluation in the fed study.
Safety
The AEs are summarized in Table 5. Of the 30 subjects under fasting conditions, 21 (70%, 21/30) experienced 41 TEAEs, of which 18 TEAEs occurred in 11 (36.7%, 11/30) subjects after receiving the test product and 23 occurred in 13 (44.8%, 13/29) subjects after reference product administration. A total of 36 drug‐related TEAEs occurred in 21 subjects, of which 15 drug‐related TEAEs occurred in 10 (33.3%, 10/30) subjects after test preparation and 21 drug‐related TEAEs occurred in 12 (41.4%, 12/29) subjects after reference preparation. All the TEAEs were grade 1 in severity except for one AE (gallstones with acute cholecystitis; 3.3%, 1/30) grade 2 after test product, and one AE (epidermal cyst of scrotum; 3.4%, 1/29) grade 2 after reference product, respectively. All the TEAEs were monitored until recovery or remission.
TABLE 5.
Summary of adverse effects occurring under fasting and fed conditions.
| TEAEs | Fasting condition | Fed condition | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Test (N = 30) | Reference (N = 29) | Total (N = 30) | Test (N = 29) | Reference (N = 30) | Total (N = 30) | |||||||
| AE count | N (%) | AE count | N (%) | AE count | N (%) | AE count | N (%) | AE count | N (%) | AE count | N (%) | |
| AEs | 18 | 11 (36.7) | 23 | 13 (44.8) | 41 | 21 (70.0) | 38 | 13 (44.8) | 22 | 10 (33.3) | 60 | 18 (60.0) |
| Adverse reaction | 15 | 10 (33.3) | 21 | 12 (41.4) | 36 | 21 (70.0) | 37 | 12 (41.4) | 19 | 10 (33.3) | 56 | 17 (56.7) |
| Serious AE | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) |
| Serious adverse reaction | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) |
| AEs that led to withdrawal | 1 | 1 (3.3) | 0 | 0 (0) | 1 | 1 (3.3) | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) |
| Adverse reaction that that led to withdrawal | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) |
| Results of inspection | 11 | 5 (16.7) | 9 | 4 (13.8) | 20 | 8 (26.7) | 29 | 8 (27.6) | 10 | 5 (16.7) | 39 | 10 (33.3) |
| Heart rate elevated | 6 | 1 (3.3) | 6 | 1 (3.4) | 12 | 1 (3.3) | 12 | 3 (10.3) | 4 | 1 (3.3) | 16 | 3 (10.0) |
| BP elevated | 10 | 3 (10.3) | 3 | 2 (6.7) | 13 | 3 (10.0) | ||||||
| BP reduced | 3 | 2 (6.9) | 0 | 0 (0) | 3 | 2 (6.7) | ||||||
| Urine leukocyte positive | 2 | 2 (6.7) | 2 | 2 (6.9) | 4 | 4 (13.3) | 2 | 2 (6.9) | 0 | 0 (0) | 2 | 2 (6.7) |
| Urine erythrocyte positive | 1 | 1 (3.3) | 0 | 0 (0) | 1 | 1 (3.3) | 0 | 0 (0) | 1 | 1 (3.3) | 1 | 1 (3.3) |
| Urine occult blood positive | 1 | 1 (3.3) | 0 | 0 (0) | 1 | 1 (3.3) | ||||||
| Blood uric acid elevated | 1 | 1 (3.4) | 0 | 0 (0) | 1 | 1 (3.3) | ||||||
| AST increased | 1 | 1 (3.3) | 0 | 0 (0) | 1 | 1 (3.3) | ||||||
| ALT increased | 0 | 0 (0) | 1 | 1 (3.4) | 1 | 1 (3.3) | 1 | 1 (3.4) | 1 | 1 (3.3) | 2 | 2 (6.7) |
| γ‐GT increased | 0 | 0 (0) | 1 | 1 (3.3) | 1 | 1 (3.3) | ||||||
| Gastrointestinal diseases | 4 | 4 (13.3) | 8 | 8 (27.6) | 12 | 12 (40.0) | 7 | 7 (24.1) | 5 | 4 (13.3) | 12 | 10 (33.3) |
| Dental ulcer | 3 | 3 (10.0) | 8 | 8 (27.6) | 11 | 11 (36.7) | 6 | 6 (20.7) | 3 | 3 (10.0) | 9 | 8 (26.7) |
| Diarrhea | 1 | 1 (3.3) | 0 | 0 (0) | 1 | 1 (3.3) | 0 | 0 (0) | 2 | 2 (6.7) | 2 | 2 (6.7) |
| Gingival pain | 1 | 1 (3.4) | 0 | 0 (0) | 1 | 1 (3.3) | ||||||
| Diseases of the skin and subcutaneous tissue | 1 | 1 (3.3) | 2 | 2 (6.9) | 3 | 3 (10.0) | 0 | 0 (0) | 1 | 1 (3.3) | 1 | 1 (3.3) |
| Rash | 1 | 1 (3.3) | 2 | 2 (6.9) | 3 | 3 (10.0) | 0 | 0 (0) | 1 | 1 (3.3) | 1 | 1 (3.3) |
| Respiratory, chest, and mediastinal diseases | 0 | 0 (0) | 3 | 3 (10.3) | 3 | 3 (10.0) | 0 | 0 (0) | 3 | 3 (10.0) | 3 | 3 (10.0) |
| Oropharyngeal pain | 0 | 0 (0) | 3 | 3 (10.3) | 3 | 3 (10.0) | 0 | 0 (0) | 1 | 1 (3.3) | 1 | 1 (3.3) |
| Epistaxis | 0 | 0 (0) | 1 | 1 (3.3) | 1 | 1 (3.3) | ||||||
| Throat irritation | 0 | 0 (0) | 1 | 1 (3.3) | 1 | 1 (3.3) | ||||||
| Diseases of the hepatobiliary system | 1 | 1 (3.3) | 0 | 0 (0) | 1 | 1 (3.3) | ||||||
| Acute cholecystitis | 1 | 1 (3.3) | 0 | 0 (0) | 1 | 1 (3.3) | ||||||
| Diseases of the reproductive system and breast | 0 | 0 (0) | 1 | 1 (3.4) | 1 | 1 (3.3) | ||||||
| Scrotal cyst | 0 | 0 (0) | 1 | 1 (3.4) | 1 | 1 (3.3) | ||||||
| Infectious and infective diseases | 1 | 1 (3.3) | 0 | 0 (0) | 1 | 1 (3.3) | 2 | 2 (6.9) | 2 | 2 (6.7) | 4 | 3 (10.0) |
| URTI | 1 | 1 (3.3) | 0 | 0 (0) | 1 | 1 (3.3) | 2 | 2 (6.9) | 1 | 1 (3.3) | 3 | 3 (10.0) |
| Bacterial tonsillitis | 0 | 0 (0) | 1 | 1 (3.3) | 1 | 1 (3.3) | ||||||
| Ocular organ disease | 0 | 0 (0) | 1 | 1 (3.3) | 1 | 1 (3.3) | ||||||
| Conjunctival ulcer | 0 | 0 (0) | 1 | 1 (3.3) | 1 | 1 (3.3) | ||||||
Abbreviations: AE, adverse event; ALT, alanine aminotransferase; AST, aspartate aminotransferase; BP, blood pressure; γ‐GT, gamma‐glutamyl transferase; TEAE, treatment‐emergent adverse event; URTI, upper respiratory tract infection.
In the fed trial, 30 subjects were included in the safety analysis. A total of 60 TEAEs occurred in 18 (60%, 18/30) subjects, of which 38 TEAEs occurred in 13 (44.8%, 13/29) subjects after receiving the test drug and 22 occurred in 10 (33.3%, 10/30) subjects who had received the reference drug. Of the reported TEAEs, only one (acute suppurative tonsillitis) was grade 2 in severity after the reference administration, and the others were mild. Some 56 drug‐related TEAEs occurred in 17 subjects, of which 37 drug‐related TEAEs occurred in 12 (41.4%, 12/29) subjects after test preparation and 19 drug‐related TEAEs occurred in 10 (33.3%, 10/30) subjects after reference preparation. All the TEAEs were monitored until recovery.
Under both fasting and fed conditions, no deaths, serious TEAEs, or serious drug‐related TEAEs occurred during the study, and only one subject withdrew from the study due to an AE (gallstones with acute cholecystitis) after receiving the test preparation under fasting conditions. Overall, both test and reference formulations were well‐tolerated.
DISCUSSION
It is well known that original drugs are too expensive for most patients with cancer 21 ; thus, generic drugs provide the maximum accessibility of treatments for patients, relieve the social economic burden.
In September 2018, dacomitinib was approved as a first‐line treatment for patients with metastatic NSCLC with EGFR exon 19 deletions or exon 21 L858R substitution mutations in the USA. 16 However, there are limited published PK data on dacomitinib, particularly for Chinese patients. We performed a randomized, open‐label, single‐dose, two‐way, crossover trial in healthy Chinese subjects and demonstrated the bioequivalence of the test and reference formulations under fasting and fed conditions. Consistent with the 90% CI of the GMR, both fell within the prescriptive limits of 80%–125%, values that abide by the NMPA supervision requirement for bioequivalence of generic drugs. 22
The median T max of the two formulations under fasting conditions were 6.0 h (range: 4.5–10 h) and 6.0 h (range: 4.5–48 h), respectively, and under fed conditions were both 5.00 h (range: 4.5–12 h), which were similar to the range of approximately 6 h (2–24 h) demonstrated in previous studies. 17 We set the washout period at 21 days according to the mean t 1/2 of ~70 h, which was more than seven times the t 1/2 to fulfil regulatory requirements. 21 The mean AUC_%Extrap for the test and reference formulations under fasting and fed conditions were all <20%, indicating that the sampling time (240 h) was long enough to ensure absorption. In the current study, there was no significant difference observed for t 1/2, Cmax, and AUC0–∞ between the test and reference formulations under fasting and fed conditions, and the food intake had no effect on the main PK parameters, which were the same as previous results. 17 , 18 According to previous study results, 17 the geometric mean large apparent volume of distribution of dacomitinib (Vss) was 1889 L, and the geometric mean apparent plasma clearance was 24.9 L/h in cancer patients. In the present study, under fasting and fed conditions, Vz/F was larger than that in previous data, which may indicate that the drug undergoes more extensive tissue penetration in Chinese subjects. The CL/F was slightly larger than that in previous data, which may be related to the study population and health conditions.
Both in the fasting and fed trials, the subject*sequence had a significant effect on lnCmax, ln AUC0–t and, ln AUC0–∞ (all p < 0.05). In the mixed‐effect model of dacomitinib Cmax, AUC0–t , and AUC0–∞ after ln transformation, their influence was analyzed with sequence, formulation, and period as fixed effects and subject*sequence as a random effect. First, the reasons for the statistical difference in the subject*sequence may be the individual constitutions and differences in drug absorption of each subject. Moreover, 30 subjects were included in the analysis of fasting and fed states, and the sum of the squares of deviation was large, which made it easy to draw a conclusion regarding the statistical difference. Second, in the equivalence evaluation, the influence of subject*sequence was considered in the ANOVA, and the results of the 90% CIs of the GMR of major PK parameters for the tested preparation and the reference preparations were still in the range 80.00%–125.00%. In addition, the period had a clear difference on lnAUC0–t and lnAUC0–∞ in the fed trial, while no statistical difference was found in sequence, indicating that the period difference between the test preparation and the reference preparation was consistent, and the point estimation of the difference between the two preparations could not cause systematic bias. However, this did not affect the results. There were no statistically significant differences between sequences, indicating that the residual effects were equal. In summary, significant differences had no effect on the bioequivalence evaluation in this study.
In the fasting trial, a sensitivity analysis was conducted after removing the relative data of three subjects, and the 90% CIs of the GMR of test to reference preparation of AUC0–∞ was 108.06% (100.67%–116.00%) between 80% and 125%, which also met with the bioequivalence evaluation criteria. In the fed trial, the sensitivity analysis after eliminating one subject's relative data showed that the 90% CIs of the GMRs of AUC0–t and AUC0–∞ were 99.70% (97.15%–102.33%) and 99.67% (97.31%–102.10%), respectively, both falling within 80%–125%, which also met with the evaluation criteria of bioequivalence.
According to the previous data, 19 the most common TEAEs reported previously (occurring in ≥30%) were diarrhea, paronychia, dermatitis acneiform, stomatitis, and decreased appetite. 17 In the fasting trial, the most commonly observed AEs (≥10%) were urine leukocyte positive (13.3%), dental ulcer (36.7%), rash (10.0%) occurring in single oral test or reference formulation, and oropharyngeal pain (10.0%) only occurring in reference formulation administration. In the fed trial, the most commonly observed AEs were elevated heart rate (10.0%), elevated blood pressure (10.0%), dental ulcers (26.7%), and upper respiratory tract infections (10.0%). Overall, the vast majority of the observed and volunteered AEs in our study were reported in the drug specifications.
In conclusion, this study validated the comparable PK bioequivalence of the test and reference products under fasting and fed conditions. The two formulations were well‐tolerated, and no serious TEAEs occurred during the study. Generic drug preparations could be an alternative to expensive original drugs in clinical practice. These results may provide a critical reference for the development of homogeneous generic drugs.
AUTHOR CONTRIBUTIONS
X.Y., J.L., T.Z., and Q.X. wrote the manuscript. W.Z. designed the research. Y.C. analyzed the data. W.H. performed the research.
FUNDING INFORMATION
The study was funded by Easton Biopharmaceuticals (Chengdu, China).
CONFLICT OF INTEREST STATEMENT
Wei Zhang is a paid employee of Easton Biopharmaceuticals (Chengdu, China). All the other authors declared no competing interests for this work.
Supporting information
Figure S1
Figure S2
ACKNOWLEDGMENTS
The authors are grateful for the devotion of the volunteers and the work of the researchers conducting the study, which was funded by Easton Biopharmaceuticals (Chengdu, China). The sponsor designed the research, and Shanghai Renzhi Data Technology Co., Ltd. analyzed the data. The analytical method was provided by Chengdu Finelyse Pharmaceutical Technology Co., Ltd. (Chengdu, China), and all the authors revised the manuscript.
Yang X, Li J, Zhang T, et al. Bioequivalence study of dacomitinib and Vizimpro® in healthy Chinese volunteers under fasting and fed conditions: A randomized, open‐label, single‐dose, crossover trial. Clin Transl Sci. 2023;16:2591‐2603. doi: 10.1111/cts.13653
REFERENCES
- 1. Le XN, Nilsson M, Goldman J, et al. Dual EGFR/VEGF pathway inhibition: a promising strategy for patients with EGFR mutant NSCLC. J Thorac Oncol. 2021;16(2):205‐215. [DOI] [PubMed] [Google Scholar]
- 2. Siegel RL, Miller KD, Jemal A. Cancer statistics. CA Cancer J Clin. 2019;69(1):7‐34. [DOI] [PubMed] [Google Scholar]
- 3. Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non‐small‐cell lung cancer to gefitinib. N Engl J Med. 2004;350(21):2129‐2139. [DOI] [PubMed] [Google Scholar]
- 4. Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first‐line treatment for European patients with advanced EGFR mutation‐positive non‐small‐cell lung cancer (EURTAC): a multicentre, open‐label, randomised phase 3 trial. Lancet Oncol. 2012;13(3):239‐246. [DOI] [PubMed] [Google Scholar]
- 5. Zhou CC, Wu YL, Chen GY, et al. Erlotinib versus chemotherapy as first‐line treatment for patients with advanced EGFR mutation‐positive non‐small‐cell lung cancer (OPTIMAL, CTONG‐0802): a multicentre, open‐label, randomised, phase 3 study. Lancet Oncol. 2011;12(8):735‐742. [DOI] [PubMed] [Google Scholar]
- 6. Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non‐small‐cell lung cancer with mutated EGFR. N Engl J Med. 2010;362(25):2380‐2388. [DOI] [PubMed] [Google Scholar]
- 7. Gonzales AJ, Hook KE, Althaus IW, et al. Antitumor activity and pharmacokinetic properties of PF‐00299804, a second‐generation irreversible pan‐erbB receptor tyrosine kinase inhibitor. Mol Cancer Ther. 2008;7(7):1880‐1889. [DOI] [PubMed] [Google Scholar]
- 8. Engelman JA, Zejnullahu K, Gale CM, et al. PF00299804, an irreversible pan‐ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res. 2007;67(24):11924‐11932. [DOI] [PubMed] [Google Scholar]
- 9. Li D, Ambrogio L, Shimamura T, et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene. 2008;27(34):4702‐4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Solca F, Dahl G, Zoephel A, et al. Target binding properties and cellular activity of afatinib (BIBW 2992), an irreversible ErbB family blocker. J Pharmacol Exp Ther. 2012;343(2):342‐350. [DOI] [PubMed] [Google Scholar]
- 11. Wu YL, Cheng Y, Zhou X, et al. Dacomitinib versus gefitinib as first‐line treatment for patients with EGFR‐mutation‐positive non‐small‐cell lung cancer (ARCHER 1050): a randomised, open‐label, phase 3 trial. Lancet Oncol. 2017;18(11):1454‐1466. [DOI] [PubMed] [Google Scholar]
- 12. Mok TS, Cheng Y, Zhou XD, et al. Improvement in overall survival in a randomized study that compared dacomitinib with gefitinib in patients with advanced non‐small‐cell lung cancer and EGFR‐activating mutations. J Clin Oncol. 2018;36(22):2244‐2250. [DOI] [PubMed] [Google Scholar]
- 13. Mok TS, Cheng Y, Zhou XD, et al. Updated overall survival in a randomized study comparing dacomitinib with gefitinib as first‐line treatment in patients with advanced non‐small‐cell lung cancer and EGFR‐activating mutations. Drugs. 2021;81(2):257‐266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Passaro A, Mok T, Peters S, Popat S, Ahn MJ, de Marinis F. Recent advances on the role of EGFR TKIs in the management of NSCLC with uncommon, non‐exon 20 insertion EGFR mutations. J Thorac Oncol. 2021;16(5):764‐773. [DOI] [PubMed] [Google Scholar]
- 15. Zhang JY, Wang Y, Liu ZL, et al. Efficacy of dacomitinib in patients with EGFR‐mutated NSCLC and brain metastases. Thorac Cancer. 2021;12(24):3407‐3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pfizer. U.S . FDA approves VIZIMPRO® (dacomitinib) for the first‐line treatment of patients with EGFR‐mutated metastatic non‐small cell lung cancer. 2018. https://www.pfizer.com/news/press‐release/press‐release‐detail/u_s_fda_approves_vizimpro_dacomitinib_for_the_first_line_treatment_of_patients_with_egfr_mutated_metastatic_non_small_cell_lung_cancer. Accessed 07 Oct 2018. [DOI] [PubMed]
- 17. US FDA . VIZIMPRO® (dacomitinib) tablets, for oral use. 2018. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/211288s000lbl.pdf Accessed 07 Oct 2018.
- 18. Ruiz‐Garcia A, Masters JC, Mendes da Costa L, et al. Effect of food or proton pump inhibitor treatment on the bioavailability of dacomitinib in healthy volunteers. J Clin Pharmacol. 2016;56(2):223‐230. [DOI] [PubMed] [Google Scholar]
- 19. Matt S. Dacomitinib: first global approval. Drugs. 2018;78(18):1947‐1953. [DOI] [PubMed] [Google Scholar]
- 20. Bello CL, Smith E, Ruiz‐Garcia A, Ni G, Alvey C, Loi CM. A phase I, open label, mass balance study of [14C] dacomitinib (PF‐00299804) in healthy male volunteers. Cancer Chemother Pharmacol. 2013;72(2):379‐385. [DOI] [PubMed] [Google Scholar]
- 21. Javier AS, Vicente GB, Alfonso PC, et al. Dacomitinib in first‐line treatment of advanced EGFR‐mutated non‐small‐cell lung cancer: a cost–effectiveness analysis. J Comp Eff Res. 2021;10(4):325‐335. [DOI] [PubMed] [Google Scholar]
- 22. Drug Evaluation Center of the State Drug Administration . Technical guiding principles for human bioequivalence studies of chemical drug generics using pharmacokinetic parameters as endpoint evaluation indicators. https://www.cde.org.cn/zdyz/domesticinfopage?zdyzIdCODE=1e218f70d9b7c99c2663de9f6655bc5b Accessed 08 Mar 2016.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2