

Abstract
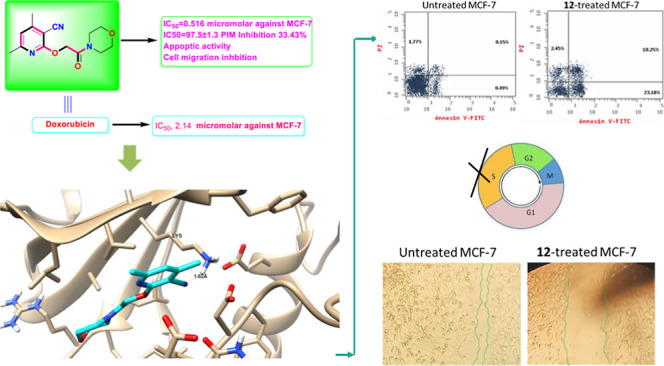
2-((3-Cyano-4,6-dimethylpyridin-2-yl)oxy)acetohydrazide 1 was used as the precursor for the synthesis of 5-thioxo-1,3,4-oxadiazol-2-yl)methoxy)nicotinonitrile 2. The latter was alkylated with different alkylating agents to produce the S-alkylated products 3–6. Galactosylation of 5-thioxo-1,3,4-oxadiazol-2-yl)methoxy)nicotinonitrile 2 produces a mixture of S- and N-galactosides 8 and 9. The hydrazide 1 is converted to azide 10, coupled with glycine methyl ester hydrochloride and a set of amines to produce the target coupled amides 11–15. New compounds were assigned using NMR and elemental analysis. Compound 12 had potent cytotoxicity with IC50 values of 0.5 and 5.27 μM against MCF-7 and HepG2 cell lines compared with doxorubicin, which displayed the following IC50: 2.14 and 2.48 μM for the mentioned cell lines, respectively. Regarding the molecular target, compound 12 exhibited potent PIM-1 inhibition activity with 97.5% with an IC50 value of 14.3 nM compared to Staurosporine (96.8%, IC50 = 16.7 nM). Moreover, compound 12 significantly activated apoptotic cell death in MCF-7 cells, increasing the cell population by total apoptosis by 33.43% (23.18% for early apoptosis and 10.25% for late apoptosis) compared to the untreated control group (0.64%), and arresting the cell cycle at S-phase by 36.02% compared to control 29.12%. Besides, compound 12 caused tumor inhibition by 42.1% in solid tumors in the SEC-bearing mice. Results disclosed that compound 12 significantly impeded cell migration and cell proliferation by interfering with PIM-1 enzymatic activity via considerable apoptosis-induction, which made it an attractive lead compound for the development of chemotherapeutics to treat breast cancer.
Introduction
Due to its extensive and complex etiology, cancer poses a serious threat to human health.1 Being the third most common cause of cancer death and the sixth most prevalent cancer type, liver cancer is a problem for the world’s health. Furthermore, 14% of all cancer-related deaths among women are caused by breast cancer, which is the most prevalent cancer in the world.2,3 Drug resistance continues to be one of the biggest obstacles in treating breast cancer, despite the recent identification of numerous potentially novel treatments that have shown significant therapeutic success.4 In contrast, 75–85% of all instances of liver cancer are hepatocellular carcinoma (HCC). Despite the incidence of HCC, few drugs are available for clinical therapy, especially in the advanced stages; consequently, major efforts should be made.2,5
In addition to signaling pathways associated with malignancies, PIM kinases are crucial for many biological processes, such as cell differentiation, proliferation, and programmed cell death (apoptosis).6,7 The original gene PIM-1, which serves as the proviral insertion site for the Moloney murine leukemia virus, is where the name PIM originates.8,9 High levels of PIM expression have been related in several studies to human epithelial and hematologic cancers, making them promising therapeutic targets.10,11 Because PIM-1 kinase has a distinctive active site, it is simpler to develop small compounds that function as PIM-1 inhibitors.12,13 PIM-1 elevations have been linked to the development and progression of cancer.14,15 PIM-1 kinase was highly expressed in several tumor types, including breast, prostate, hepatic, colon, and pancreatic malignancies.16−18 Consequently, it is a promising strategy to stop the spread of cancer by inhibiting PIM-1 using small-molecule drugs.19
Since many pharmaceutically and physiologically active substances, including etoricoxib, nifedipine, amlodipine, and clevidipine,20,21 include the pyridine motif, some anticancer medications, including Veliparib, Niraparib, Olaparib, and Rucaparib, were revealed to have essential pharmacophoric groups due to the presence of an amide group, which functions as a hydrogen bond donor for the interaction with biological targets.22,23 Compound 6-phenyl-N-(pyridin-3-yl)picolinamide (LGB321), the most powerful PIM inhibitor identified to date, has pyridine, amine, and amide groups in its structure that are required for hydrogen bond interactions with the residues of PIM-1’s ATP-binding site.24,25 While 2-(3-cyano-4,6-diphenylpyridin-2-yloxy)acetohydrazide I was said to have a potent effect that significantly inhibited HepG2 cell reproduction and cell migration by interfering with PIM-1 enzymatic activity26 (Figure 1). Moreover, by inhibiting PIM-1 enzymatic activity, 6-(4-(benzyloxy)phenyl)-4-(4-(dimethylamino)phenyl)-2-ethoxynicotinonitrile II demonstrated a strong action that drastically decreased PC3 cell reproduction and migration.27 Compound III showed roughly equal activity against the HepG2 cell line as the reference drug in 2023 (IC50 of 0.0486 M).28 Compound IV showed the best antiproliferative activities with an IC50 value of 1.18 μM against MCF-7, better than lapatinib as a reference standard (IC50 = 4.69 μM).29 Compounds V and VI were found to be the most active congeners against MCF-7 cells (IC50 = 0.22 and 1.88 μM after 48 h treatment; 0.11 and 0.80 μM after 72 h treatment, respectively), with increased activity compared to the reference drug doxorubicin (IC50 = 1.93 μM).30
Figure 1.
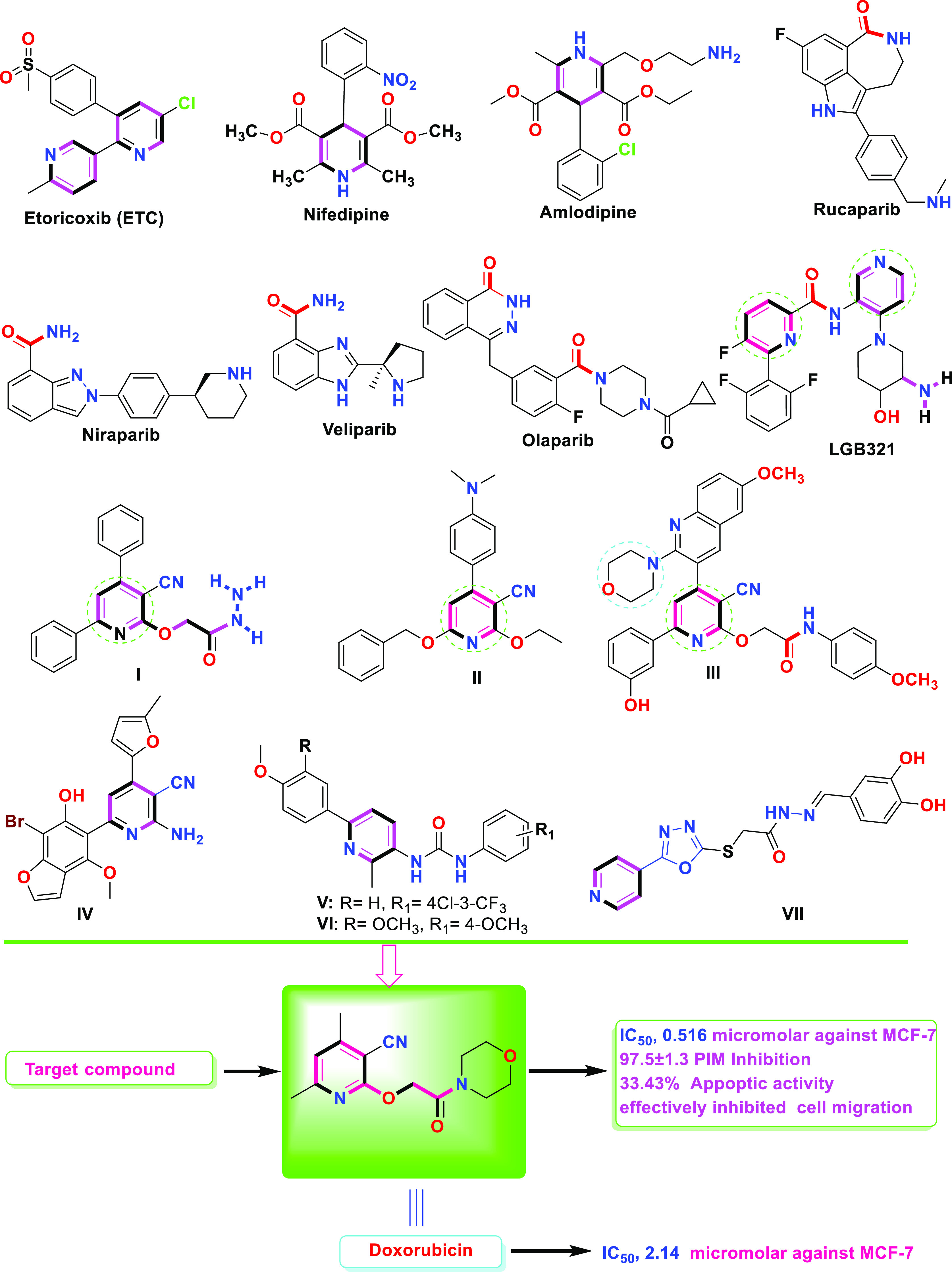
Structures of selected FDA-approved drugs and bioactive lead compounds containing a pyridine moiety.
Hybridization of the 1,3,4-oxadiazole moiety with other heterocyclic pharmacophores is a promising approach to overcome various disadvantages of current anticancer drugs, such as drug resistance, toxicity, and other side effects. Thus, the hybridization of 1,3,4-oxadiazole with pyridine may also lead to the development of new anticancer drugs with multiple action mechanisms. Zhang et al.31 reported that the anticancer activity of 1,3,4-oxadiazole-pyridine hybrid VII against four cancer cell lines (HepG2, MCF-7, SW1116, and BGC823) exhibited more potent activity (IC50 = 0.76–12.21 μM) than the positive control (5-Fluorouracil, IC50 = 5.26–9.79 μM).
As part of our continuous efforts to synthesize and discover novel anticancer drugs, we created stepwise biological work packages that involve cytotoxic screening and apoptosis induction.32−38 In this work, novel pyridine compounds are designed and synthesized, and their antibreast and antiliver cancer properties are assessed.
Results and Discussion
The synthetic strategy was as follows: 2-((3-cyano-4,6-dimethylpyridin-2-yl)oxy)acetohydrazide 1 reacted with carbon disulfide and potassium hydroxide in ethanol under reflux conditions to afford 4,6-dimethyl-2-((5-thioxo-4,5-dihydro-1,3,4-oxadiazol-2-yl)methoxy)nicotinonitrile 2. The 1,3,4-oxadiazole-thione moiety in 2 alkylated with a set of alkylating agents, including ethyl chloroacetate, benzyl chloride, allyl bromide, and amyl bromide, in the presence of potassium carbonate in acetone gave the S-alkylated products 3-4-6 respectively (Scheme 1).
Scheme 1. Alkylation of 5-Thioxo-1,3,4-oxadiazol-2-yl)methoxy)nicotinonitrile 2.
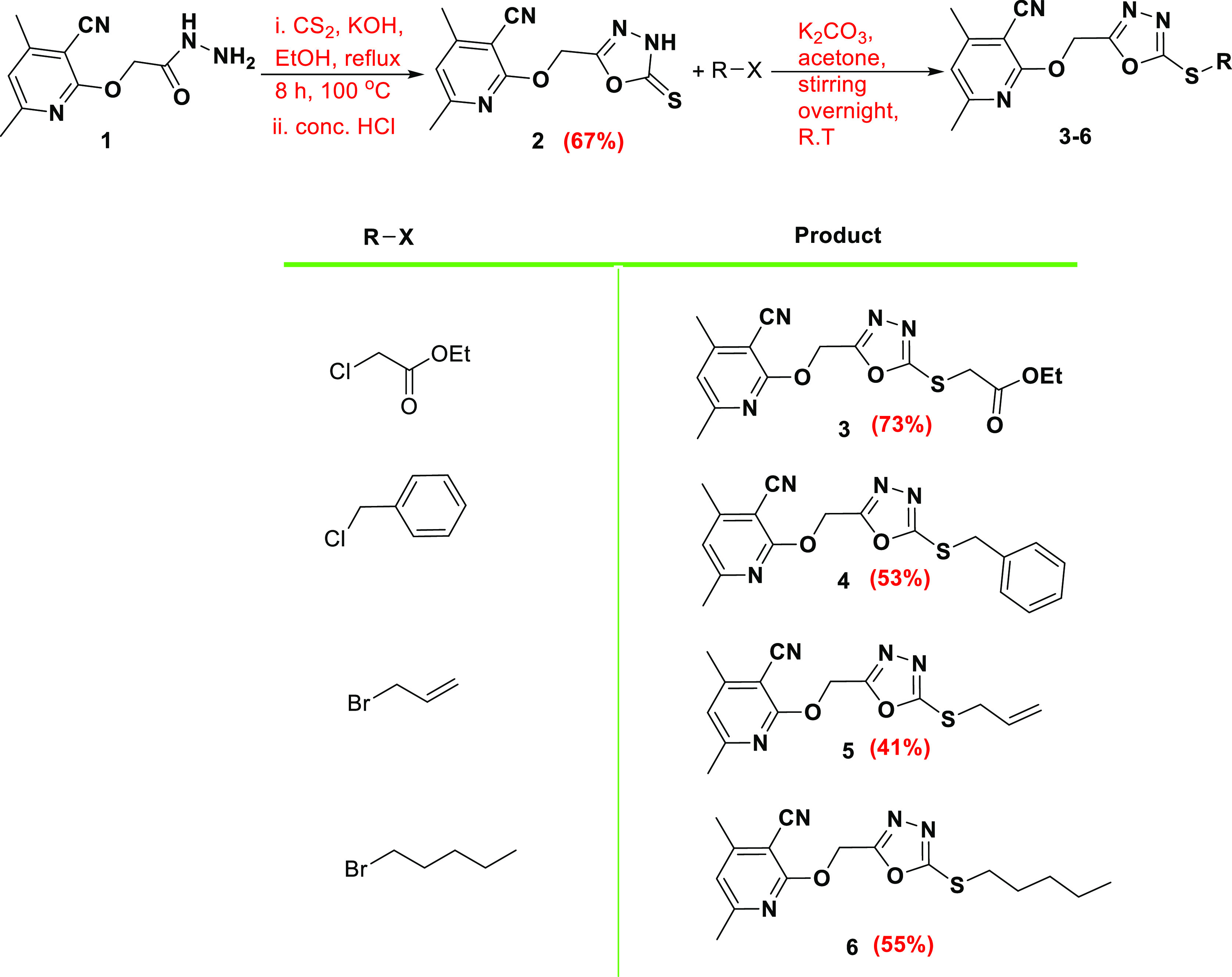
The 1H NMR of 2 showed the two methyl group proton signals at 2.42 and 2.44 ppm, the OCH2 signal appeared at 5.55 ppm, the aromatic CH proton was found at 7.09 ppm, whereas the oxadiazole NH proton was detected at 14.69 ppm. The 13C NMR displayed the two methyl group carbons at 20.11 ppm and 24.51 ppm, the OCH2 carbon appeared at 58.05 ppm, while the thiocarbonyl carbon (C=S) was found at 178.38 ppm, which confirmed the thione form. S-alkylation was established based on the 13C NMR signal of the methylene carbon attached to sulfur since the S-CH2 signal values in ppm were 34.25 (3), 36.34 (4), 35.13 (5), and 32.44 (6).
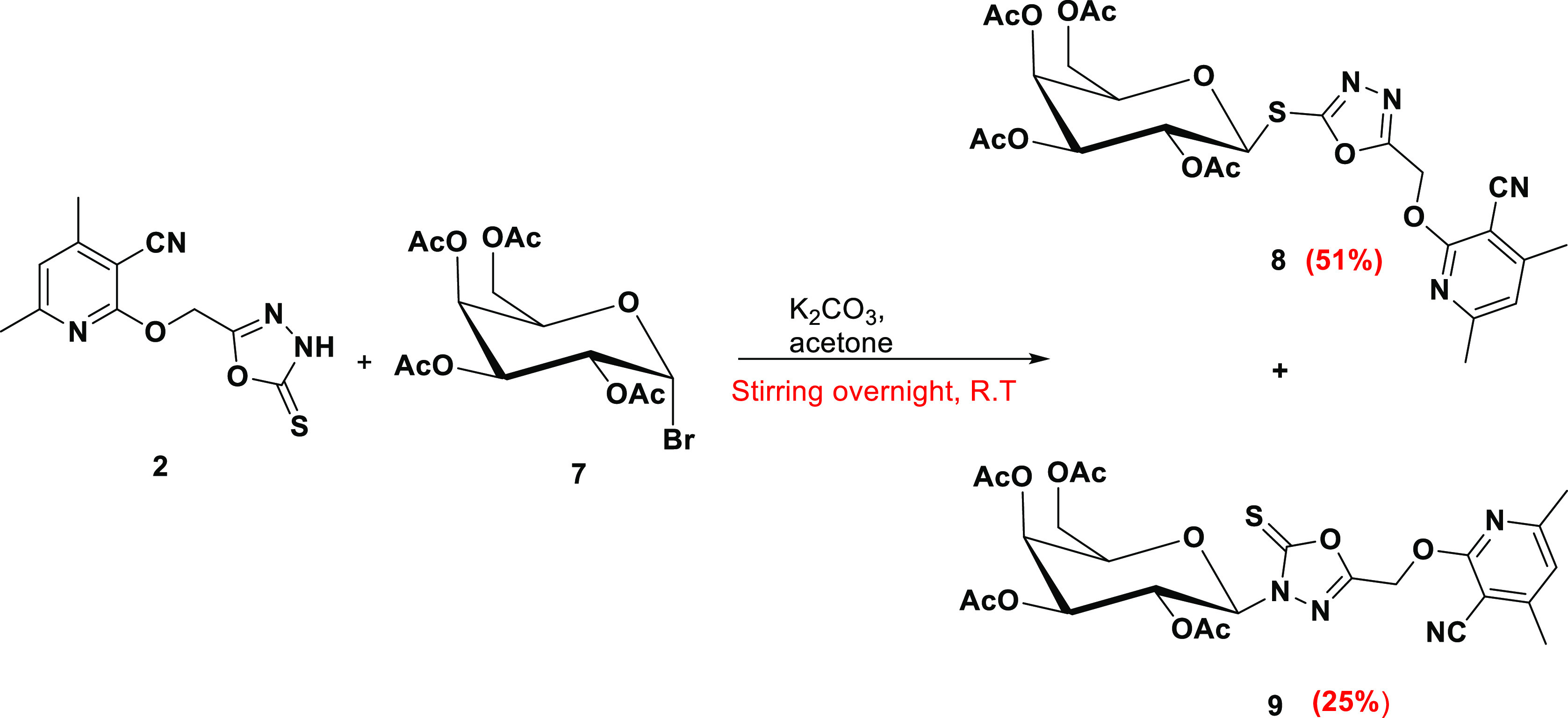
Compound 5-thioxo-1,3,4-oxadiazol-2-yl)methoxy)nicotinonitrile 2 was galactosylated with acetobromogalactose 7 under the same conditions to afford a mixture of S-galactoside 8 and N-galactoside 9, which were separated using silica-column chromatography (Scheme 2).
Scheme 2. Galactosylation of 2 Produced S- and N-Galactosides.
In addition, 2-((3-cyano-4,6-dimethylpyridin-2-yl)oxy)acetohydrazide 1 is converted to azide 10 by the reaction with sodium nitrite and HCl under low temperature (−5 °C). The azide 10 is coupled with glycine methyl ester hydrochloride via the azide coupling method in the presence of Et3N to give the glycinated compound 11. Moreover, the azide 10 is coupled with a set of amines, including morpholine, pipridine, benzylamine, and allylamine, in the presence of Et3N to yield the amine-coupled products 12–15, respectively (Scheme 3). The azide compound 10 missed any NH signal and showed the carbonyl carbon (C=O) signal at 176.17 ppm. The glycinated compound 11 showed the protons of the methoxy group proton at 3.64 ppm, the two-methylene groups detected at 3.88 and 4.92 ppm, and the NH appeared at 8.22 ppm. The carbonyl carbons were found at 168.38 and 170.51 ppm. The coupled amines showed new characteristic signals. Compounds 14 and 15 displayed NH at 8.45 and 8.21 ppm, respectively. The coupled amines 12–15 displayed the carbonyl carbons of the amide group at 165.97 ppm (12), 165.23 ppm (13), 167.83 ppm (14), and 167.42 ppm (15).
Scheme 3. Azide Coupling of 10 with Glycine Methyl Ester Hydrochloride and Amines.

Biology
Cytotoxicity Using the MTT Assay
The synthesized compounds were tested for their cytotoxicity against breast MCF-7 and liver HepG2 cancer cells. Compounds demonstrated good cytotoxicity with a promising percentage of cell death at the maximum concentration [100 μM], as shown in Table 1. Compounds 11 and 12 revealed interesting IC50 values against MCF-7 (0.73 and 0.5 μM, respectively), whereas compounds 6 and 12 showed the following IC50 against HepG2 cell lines (5.27 and 6.6 μM, respectively), as shown in Table 2. The percentages of cell growth inhibition of MCF-7 and HepG2 cancer cells and MCF-10A normal cells against the concentration of tested compounds are summarized in Figure 2.
Table 1. Percentage of Cell Growth Inhibition of the Tested Compounds against MCF-7 and HepG2 Lines Using the MTT Assay.
| code | % of cell inhibition ± SD at [100
μM] |
|
|---|---|---|
| MCF7 | HepG2 | |
| 2 | 41.92 ± 1.9 | 43.49 ± 1.8 |
| 3 | 55.07 ± 2.1 | 57.79 ± 1.9 |
| 4 | 72.13 ± 2.9 | 75.85 ± 2.3 |
| 5 | 46.96 ± 1.4 | 68.98 ± 2.1 |
| 6 | 78.25 ± 2.2 | 79.25 ± 2.2 |
| 8 | 59.75 ± 1.7 | 57.84 ± 1.6 |
| 11 | 78.59 ± 2.3 | 68.77 ± 1.6 |
| 12 | 84.20 ± 2.6 | 85.13 ± 2.1 |
| 13 | 64.56 ± 1.8 | 56.36 ± 1.4 |
| 14 | 48.76 ± 1.2 | 35.55 ± 1.1 |
| doxorubicin | 90.20 ± 2.1 | 79.13 ± 2.3 |
Table 2. Cytotoxic IC50 Values of the Promising Compounds against MCF7 and HepG2 Cell Lines Using the MTT Assay.
| code | IC50 ± SD [μM]a |
||
|---|---|---|---|
| MCF7 | HepG2 | MCF-10A | |
| 6 | NT | 6.64 ± 0.28 | NT |
| 11 | 0.73 ± 0.04 | NT | NT |
| 12 | 0.51 ± 0.02 | 5.27 ± 0.2 | 52.85 ± 2.1 |
| doxorubicin | 2.14 ± 0.1 | 2.48 ± 0.1 | 15.75 ± 0.63 |
Values are expressed as the mean ± SD of three independent triplets (n = 3). Doxorubicin is a reference drug. NT: non-Tested. IC50 values were calculated using the dose–response curve using EXCEl.
Figure 2.
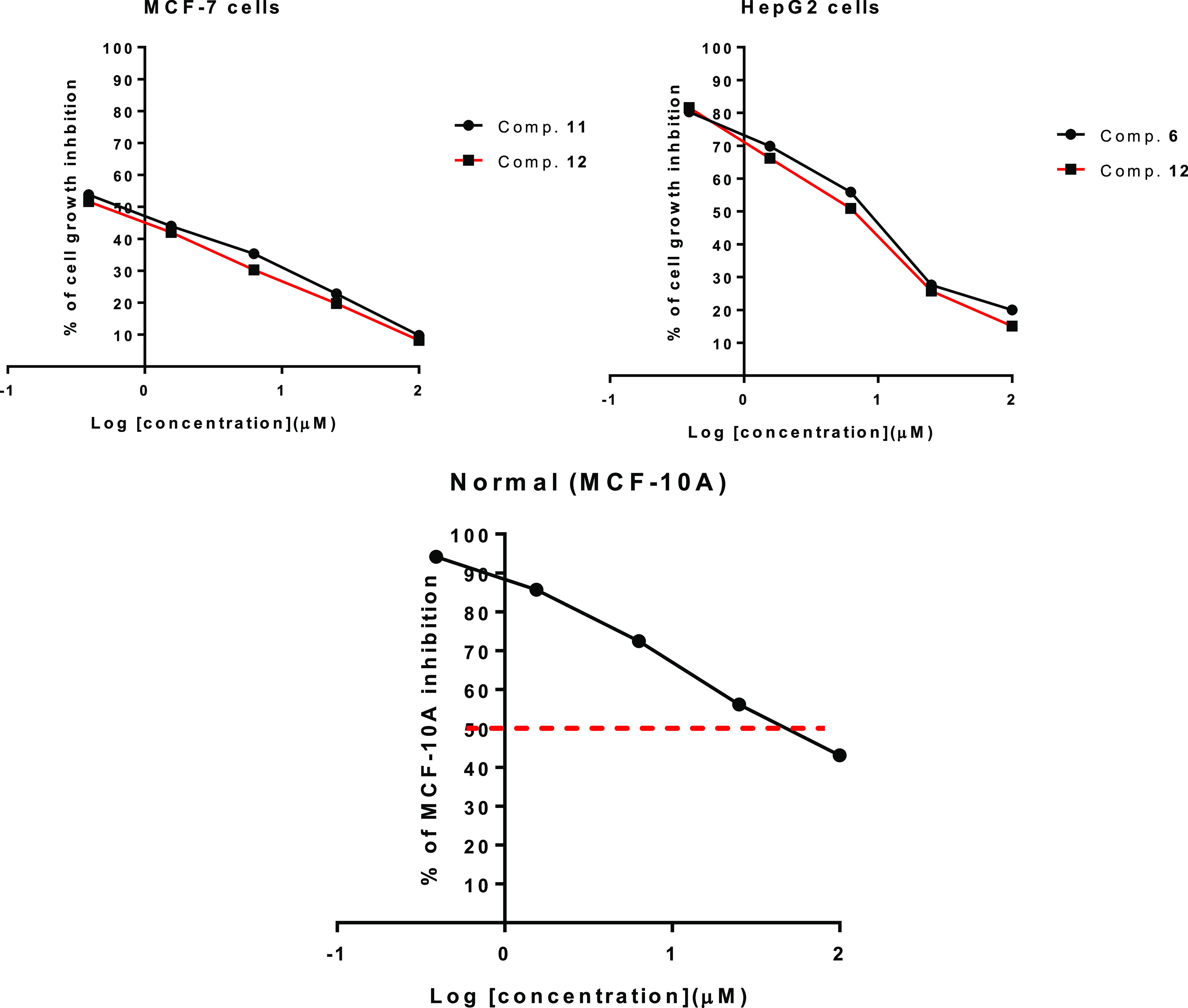
Dose–response curve for the tested compounds 6, 11, and 12 against cancer and normal cells using the MTT assay.
PIM-1 Kinase Inhibitory Assay
To highlight their effective molecular target, compounds 6, 11–13, with the highest cytotoxic activity against MCF-7 cells, were evaluated against PIM-1 inhibitory activities. As shown in Table 3, the investigated compounds displayed promising PIM-1 inhibitory capabilities with IC50 values of 19.4, 42.3, 14.3, and 19.8 nM (percentage of enzyme inhibition by 90.1–97.5%) when compared to Staurosporine (IC50 = 16.7 nM, 96.8%). As a result, compound 12 was studied further for apoptotic cell death in MCF-7 cells (Table 3).
Table 3. IC50 Values of VEGFR2 Kinase Activities of the Tested Compounds.
| compound | PIM-1 kinase |
|
|---|---|---|
| % of kinase inhibition | IC50 [nM] ± SDa | |
| 6 | 94.3 ± 1.3 | 19.4 ± 0.7 |
| 11 | 90.1 ± 2.7 | 42.3 ± 1.1 |
| 12 | 97.5 ± 1.3 | 14.3 ± 0.6 |
| 13 | 94.9 ± 2.9 | 19.8 ± 1.1 |
| staurosporine | 96.8 ± 2.1 | 16.7 |
Values are expressed as the average of three independent replicates. “IC50 values were calculated using the sigmoidal non-linear regression curve fit of percentage inhibition against five concentrations of each compound”.
Apoptotic Investigation
Annexin V/PI Staining with Cell Cycle Analysis
To investigate the apoptotic activity of compound 12 (IC50 = 1.62 M, 48 h), flow cytometric evaluation of Annexin V/PI staining was utilized to examine apoptotic cell death in untreated and treated MCF-7 cells. Figure 3 shows that compound 12 dramatically increased apoptotic cell death in MCF-7 cells, increasing total apoptosis by 33.43% (23.18% for early apoptosis and 10.25% for late apoptosis) compared to the untreated control group (0.64%). As a result, 12-treatment increased apoptosis by 52.2 fold.
Figure 3.

Apoptosis/necrosis assessment using Annexin-V/Propidium Iodide staining of untreated and 12-treated MCF-7 cells with the IC50 value (IC50 = 1.62 μM, 48 h).
Following that, DNA flow cytometry was utilized to determine the cell population in each cell phase after treatment with a cytotoxic agent. As shown in Figure 4, compound 12 administration raised the S-phase cell population by 36.02% compared to the control by 29.12%, while cells in other phases declined or increased insignificantly. As a result, chemical 12 caused apoptosis in MCF-7 cells, halting cell proliferation at the S-phase.
Figure 4.

Percentage of cell population at each cell cycle G1, S, and G2/M in untreated and 12-treated MCF-7 cells with the IC50 value (IC50 = 1.62 μM, 48 h) using DNA content-flow cytometry-aided cell cycle analysis.
Autophagy Assessment of 12-Treatment
The investigated chemical 12 caused considerable autophagic cell death, activating autophagic cell death by 87.6 × 103 cells compared to 32.8 × 103 for the control, demonstrating compound 12’s autophagy activity and apoptosis-induction as a dual activity (Figure 5).
Figure 5.
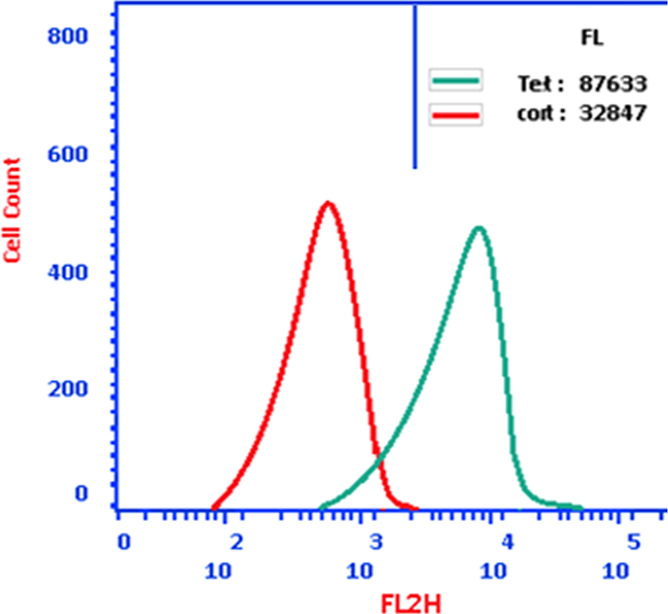
Using the acridine orange lysosomal dye and the flow cytometric analysis, autophagic cell death in MCF-7 treated with compound 12 (IC50 = 1.62 M, 48 h) was assessed. The green curve represents cells treated with compound 12; the red curve represents cells under control (untreated).
Effect of 12 Treatment against Cell Migration Using Wound-Healing Activity
As shown in Table 4 and Figure 6, the wounded area between cell layers following a scratch was partially filled by migrating MCF-7 control cells (95.6% wound closure), while only 71.8% of the wound was closed after 48 h of 12-treatment.
Table 4. Cell Migration Parameters in the Untreated and 12-Treated MCF-7 Cells.
| samples | % closure | total area | migrated cell area | time | length (mm) | L1 (mm) | L2 (mm) | L3 (mm) | average L of migration (mm) |
|---|---|---|---|---|---|---|---|---|---|
| 12-treated MCF-7 | 71.85 | 0.225 | 0.161 | 72 h | 0.25 | 0.33 | 0.32 | 0.32 | 0.32 |
| Cont.MCF-7 | 95.55 | 0.225 | 0.215 | 0.25 | 0.44 | 0.41 | 0.44 | 0.43 |
Figure 6.

As determined by the wound-healing experiment, migration of MCF-7 cells treated with 12 for 72 h was seen under a light microscope.
In Vivo (SEC-Bearing Mice)
A solid Ehrlich carcinoma cell was implanted, and compound 12 was injected intraperitoneally (IP) throughout the experiment to confirm its anticancer efficacy. Table 5 summarizes the outcomes of the tumor measurement experiments.
Table 5. Antitumor Parameters in Normal, Untreated, and Treated SEC-Bearing Micea.
| treatments |
||||
|---|---|---|---|---|
| parameter | SEC control | SEC + 12 | SEC + dox | |
| tumor potentiality | tumor weight (mg) | 198.5 ± 10.6 | 72.8 ± 1.74 | 63.4 ± 1.23 |
| tumor volume (mm3) | 274.8 ± 16.6 | 159.2 ± 10.1 | 112.3 ± 9.2 | |
| tumor inhibition ratio (TIR %) | 42.1 ± 1.0 | 59.14 ± 1.01 | ||
“Mean ± SD values of mice in each group (n = 6)”. “* Values are significantly different (P ≤ 0.05) between the SEC control and normal group”, while “# values are significantly different (P ≤ 0.05) between treated SEC and SEC control mice using the unpaired test in GraphPad prism”. TIR % = C – T/C × 100.
As a result, tumor proliferation revealed an increase in solid tumor mass of approximately 198.5 mg related to tumor potential. Following treatment with 12 and doxorubicin, the solid tumor mass decreased to 72.8 mg and 63.4 mg, respectively. As a result, treatments with 12 considerably reduced tumor volume from 274.8 mm3 in the untreated control to 159.2 mm3 and greatly decreased tumor proliferation by 42.1%. It reduced tumor volume to 112.3 mm3 and suppressed tumor development by 59.1% compared to doxorubicin.
Molecular Docking
The molecular docking study validated the binding affinity of the promising active compound 12 toward the PIM-1 active site. Docking results highlighted its binding mode with a binding energy of −17.38 Kcal/mol, and it formed a good binding interaction with Lys67 as an H-bond acceptor through the nitrile group. Besides the H-bond interactions, compound 12 made lipophilic interactions with the nonpolar amino acids, including Phe49, Ile104, Ile185, Leu174, Leu120, Ala65, and Val52. As seen in Figure 7, with the superimposition of compound 12 with the cocrystallized ligand, it exhibited the same binding mode with binding interactions.
Figure 7.

Binding disposition as surface presentation (A) and ligand–receptor interaction (B) of the docked compound 12 toward the PIM-1 kinase active site. 3D images were generated using Chimera-UCSF software.
Experimental Part
Chemistry
General Procedures
The values for the melting points are uncorrected and were determined in open capillaries using Temp-melt II melting point equipment. On silica gel 60 (230–400 mesh ASTM), flash chromatography was carried out. On silica gel 60 F254 aluminum plates (E. Merck, layer thickness 0.2 mm), thin layer chromatography was performed. The spots were found using a UV lamp. Using DMSO-d6 and CDCl3 as solvents, the 1H and 13C NMR spectra were captured on Bruker instruments at 400 MHz for 1H NMR and 101 MHz for 13C NMR, respectively. Using KBr and a Perking Elmer 1430 ratio recording infrared spectrophotometer, Bruker’s Fourier transform infrared spectrophotometry was used to record the IR spectra, and Flash EA-1112 equipment was used to do the CHNS-microanalysis.
Synthesis of 2-((3-Cyano-4,6-dimethylpyridin-2-yl)oxy)acetohydrazide (1)
Procedures as mentioned in Keshk39 Yield: 70%, mp 196 °C (Lit.39 198 °C); 1H NMR (DMSO-d6, 400 MHz): δ 2.39 (s, 3H, CH3), 2.43 (s, 3H, CH3), 4.25 (s, 2H, NH2), 4.84 (s, 2H, OCH2), 6.99 (s, 1H, Hpy), 9.20 (s, 1H. NH); IR (KBr/cm–1): 3322, 3266 (NH, NH2), 2963 (C–H aliphatic), 2222 (CN), 1688 (C=O amide), 1605 (C=C); Elemental analysis Calcd for [C10H12N4O2]: C, 54.54; H, 5.49; N, 25.44 Found C, 54.44; H, 5.55; N, 25.3.
Synthesis of 4,6-Dimethyl-2-((5-thioxo-4,5-dihydro-1,3,4-oxadiazol-2-yl)methoxy)nicotinonitrile (2)
Carbon disufide (5 mmol) was added to a solution of hydrazide 1 (1.0 mmol) in ethanol (30 mL) that contained KOH (1.5 mmol), and the reaction mixture was refluxed for 8 h. After being allowed to reach room temperature, the mixture was acidified with concentrated HCl. In order to get 2 as yellow crystals, the precipitate that had developed was filtered out, washed with water, dried, and recrystallized from absolute ethanol.
Yield: 67%, mp 146 °C. 1H NMR (DMSO-d6, 400 MHz): δ 2.42 (s, 3H, CH3), 2.44 (s, 3H, CH3), 5.55 (s, 2H, OCH2), 7.09 (s, 1H, Hpy), 14.69 (s, 1H, NH); 13C NMR (DMSO-d6, 101 MHz): δ 20.11 (CH3), 24.51 (CH3), 58.05 (OCH2), 93.57, 114.76, 119.67, 155.99, 159.89, 160.94, 161.92, 178.38 (C=S); IR (KBr/cm–1): 3335 (NH), 3096 (C–H aromatic), 2947 (C–H aliphatic), 2224 (CN), 1606 (C=C); Elemental analysis Calcd for [C11H10N4O2S]: C, 50.37; H, 3.84; N, 21.36; S, 12.23 Found C, 50.34; H, 3.87; N, 21.42; S, 12.33.
General Procedure for the Preparation of Compounds (3–6 and 8,9)
To a solution of oxadiazole-thione 2 (1.0 mmol) in acetone (20 mL), potassium carbonate (1.2 mmol) was added and stirred for 1 h; then the appropriate alkyl or galactosyl bromide (1.1 mmol) was added. The reaction mixture was stirred at room temperature overnight. The excess solvent evaporated, and cold water was added. The formed products were filtered off, dried, and purified by crystallization from methanol in the case of products 3–6 or using column chromatography (ethyl acetate/petroleum ether 1:2) in the cases of 8 and 9.
Ethyl 2-((5-(((3-Cyano-4,6-dimethylpyridin-2-yl)oxy)methyl)-1,3,4-oxadiazol-2-yl)thio)acetate (3)
Yield: 73%, mp 85 °C. 1H NMR (DMSO-d6, 400 MHz): δ 1.18 (t, 3H, J = 7.2 Hz, CH3), 2.41 (s, 3H, CH3), 2.43 (s, 3H, CH3), 4.10 (q, 2H, CH2), 4.23 (s, 2H, SCH2), 5.69 (s, 2H, OCH2), 7.07 (s, 1H, Hpy); 13C NMR (DMSO-d6, 101 MHz): δ 14.38 (CH3), 20.08 (CH3), 24.47 (CH3), 34.25 (SCH2), 57.93 (OCH2), 62.06 (OCH2), 93.57, 114.73, 119.55, 155.89, 160.93, 162.00, 164.05, 164.27, 167.93 (C=O); IR (KBr) cm–1: 2987 (C–H aliphatic), 2224 (CN), 1743 (C=Oester), 1602 (C=C); Elemental analysis Calcd for [C15H16N4O4S]: C, 51.71; H, 4.63; N, 16.08; S, 9.20 Found C, 51.76; H, 4.58; N, 16.18; S, 9.30.
2-((5-(Benzylthio)-1,3,4-oxadiazol-2-yl)methoxy)-4,6-dimethylnicotinonitrile (4)
Yield: 53%, mp 68 °C. 1H NMR (DMSO-d6, 400 MHz): δ 2.39 (s, 3H, CH3), 2.43 (s, 3H, CH3), 4.51 (s, 2H, SCH2), 5.69 (s, 2H, OCH2), 7.04 (s, 1H, Hpy), 7.27–7.42 (m, 5H, Hph); 13C NMR (DMSO-d6, 101 MHz): δ 20.07 (CH3), 24.47 (CH3), 36.34 (SCH2), 57.96 (OCH2), 93.58, 114.74, 119.22, 128.24, 128.41, 128.81, 129.01, 129.24, 136.85, 155.85, 160.90, 162.00, 164.04, 164.53; IR (KBr/cm–1): 3090 (C–H aromatic), 2979 (C–H aliphatic), 2221 (CN), 1604 (C=C); Elemental analysis Calcd for [C18H16N4O2S, 352.41]: C, 61.35; H, 4.58; N, 15.90; S, 9.10 Found C, 61.44; H, 4.63; N, 15.86; S, 9.15.
2-((5-(Allylthio)-1,3,4-oxadiazol-2-yl)methoxy)-4,6-dimethylnicotinonitrile (5)
Yield: 41%, mp 190 °C. 1H NMR (DMSO-d6, 400 MHz): δ 2.42 (s, 3H, CH3), 2.45 (s, 3H, CH3), 3.89 (d, 2H, J = 6.4 Hz, SCH2), 5.14 (d, 1H, J = 10.0 Hz, CH=CHcisH), 5.30 (d, 1H, J = 16.8 Hz, CH=CHHtrans), 5.70 (s, 2H, OCH2), 5.92–5.98 (m, 1H, CH=CH2), 7.06 (s, 1H, Hpy); 13C NMR (DMSO-d6, 101 MHz): δ 19.97 (CH3), 24.37 (CH3), 35.13 (SCH2), 57.94 (OCH2), 93.54, 114.73, 119.56, 119.78, 132.83, 155.92, 161.04, 162.02, 164.12, 164.46; IR (KBr/cm–1): 3086 (C–H aromatic), 2968 (C–H aliphatic), 2224 (CN), 1641 (C=N), 1596 (C=C); Elemental analysis Calcd for [C14H14N4O2S]: C, 55.61; H, 4.67; N, 18.53; S, 10.61 Found C, 55.67; H, 4.47; N, 18.39; S, 10.64.
4,6-Dimethyl-2-((5-(pentylthio)-1,3,4-oxadiazol-2-yl)methoxy)nicotinonitrile (6)
Yield: 55%, mp 170 °C. 1H NMR (DMSO-d6, 400 MHz): δ 0.86 (t, 3H, J = 7.0 Hz, CH3), 1.26–1.38 (m, 4H, 2CH2), 1.67–1.74 (m, 2H, CH2), 2.41 (s, 3H, CH3), 2.44 (s, 3H, CH3), 3.21 (t, 2H, J = 7.4 Hz, SCH2), 5.69 (s, 2H, OCH2), 7.08 (s, 1H, Hpy); 13C NMR (DMSO-d6, 101 MHZ): δ 14.23 (CH3), 20.08 (CH3), 21.99 (CH2), 24.49 (CH3), 29.09 (CH2), 30.42 (CH2), 32.44 (SCH2), 58.03 (OCH2), 93.58, 114.76, 119.54, 155.88, 160.91, 162.05, 163.83, 165.10; IR (KBr/cm–1): 3030 (C–H aromatic), 2956 (C–H aliphatic), 2224 (CN), 1604 (C=C); Elemental analysis Calcd for [C16H20N4O2S]: C, 57.81; H, 6.06; N, 16.85; S, 9.65 Found C, 57.87; H, 6.16; N, 16.88; S, 9.58.
2- ((5-(2,3,4,6-Tetra-O-acetyl-α-D-galactopyranosylsulfanyl)-1,3,4-oxadiazol-2-yl)methoxy)-4,6-dimethylpyridine-3-carbonitrile (8)
Yield: 51%, mp 145 °C. 1H NMR (DMSO-d6, 400 MHz): δ 1.95, 1.97, 2.05, 2.14 (4s, 12H, 4CH3CO), 2.41 (s, 3H, CH3), 2.45 (s, 3H, CH3), 3.99–4.06 (m, 2H, H-6Gal), 4.37–4.40 (m, 1H, H-5Gal), 5.23 (dd, 1H, J = 9.6, J = 10 Hz, H-2Gal), 5.34–5.40 (m, 2H, H-3Gal, H-4Gal), 5.71(d, 1H, J = 10 Hz, H-1Gal), 5.74 (s, 2H, OCH2), 7.09 (s, 1H, Hpy); 13C NMR (DMSO-d6, 101 MHz): δ 20.10, 20.79, 20.82, 20.84, 20.88, 24.47, 58.01, 61.73, 67.72, 71.02, 74.72, 82.99, 93.60, 114.76, 119.61, 155.95, 160.95, 161.24, 162.00, 164.92, 169.86, 169.99, 170.28, 170.40; IR (KBr/cm–1): 3038 (C–H aromatic), 2959 (C–H aliphatic), 2224 (CN), 1739 (C=Oester), 1603 (C=C); Elemental analysis Calcd for [C25H28N4O11S]: C, 50.67; H, 4.76; N, 9.45; S, 5.41 Found C, 50.47; H, 4.73; N, 9.49; S, 5.36.
3- ((5-(2,3,4,6-Tetra-O-acetyl-α-D-galactopyranosyl)-1,3,4-oxadiazol-2-ylthione)methoxy)-4,6-dimethylpyridine-3-carbonitrile (9)
Yield: 25%, oily. 1H NMR (CDCl3, 400 MHz): δ 1.98, 2.02, 2.08, 2.17 (4s, 12H, 4CH3CO), 2.23 (s, 3H, CH3), 2.47 (s, 3H, CH3), 4.16–4.22 (m, 3H, H-5Gal, H-6Gal, H-6̀Gal), 5.25 (dd, 1H, J = 3.2, J = 10.4 Hz, H-3Gal), 5.52 (d, 3H, J = 3.2 Hz, H-4Gal, OCH2), 5.65 (dd, 1H, J = 9.6 Hz, H-2Gal), 5.89 (d, 1H, J = 9.2 Hz, H-1Gal), 6.82 (s, 1H, HPy); 13C NMR (CDCl3, 101 MHz): δ 20.10, 20.47 (CH3), 20.63, 24.25, 57.17, 61.18, 66.78, 66.86, 71.25, 73.72, 83.52 (C-1Gal), 93.93, 114.29, 119.11, 154.88, 158.04, 160.49, 161.58, 168.87, 169.13, 170.05, 170.33; Elemental analysis Calcd for [C25H28N4O11S]: C, 50.67; H, 4.76; N, 9.45; S, 5.41 Found C, 50.47; H, 4.73; N, 9.49; S, 5.36.
General Procedure for Azide-Coupling of Amino Acid Methyl Ester and Amines
A cold solution (0 °C) of sodium nitrite (0.14 g, 2.0 mmol) in water (6 mL) was added to a cold solution (−5 °C) of hydrazide 1 (1.6 mmol) in acetic acid (12 mL), hydrochloric acid (5 M, 6 mL), and water (50 mL). Following 30 min of stirring at the same temperature, the formed azide was removed using cold ethyl acetate and rinsed with a cold solution of NaHCO3 (5%) and water. In the following stage, azide 10 was employed without further purification after drying over anhydrous sodium sulfate.
For 20 min at 0 °C, glycine methyl ester hydrochloride or the corresponding amine (1.8 mmol) was agitated in 50 mL of ethyl acetate that also included 0.2 mL of triethyl amine. The produced triethyl amine hydrochloride was filtered out, and the filtrate was then added to the cold solution of azide 10 that had already been made. The mixture was then kept at ambient temperature for an additional 12 h after spending the first 12 h in the refrigerator. The reaction mixture was rinsed with a water and NaHCO3 (5%) solution before being dried on anhydrous sodium sulfate. The solvent was evaporated under a vacuum, and the leftover mixture of ethyl acetate and petroleum ether was recrystallized to produce the desired product.
2-((3-Cyano-4,6-dimethylpyridin-2-yl)oxy)acetyl Azide (10)
Yield: 77%, mp 127 °C. 1H NMR (DMSO-d6, 400 MHz): δ 2.40 (s, 3H, CH3), 2.45 (s, 3H, CH3), 5.11 (s, 2H, OCH2), 7.07 (s, 1H, Hpy); 13C NMR (DMSO-d6, 101 MHz): δ 20.08 (CH3), 24.52 (CH3), 65.20 (OCH2), 93.35, 114.84, 119.45, 155.84, 160.85, 162.19, 176.17 (C=O); IR (KBr/cm–1): 3003 (C–H aromatic), 2957 (C–H aliphatic), 2222 (CN), 1661 (C=O), 1601 (C=C); Elemental analysis Calcd for [C10H9N5O2]: C, 51.95; H, 3.92; N, 30.29 Found C, 52.02; H, 3.82; N, 30.36.
Methyl (2-((3-Cyano-4,6-dimethylpyridin-2-yl)oxy)acetyl)glycinate (11)
Yield: 50%, mp 126 °C. 1H NMR (DMSO-d6, 400 MHz): δ 2.41 (s, 3H, CH3), 2.44 (s, 3H, CH3), 3.64 (s, 3H, OCH3), 3.88 (d, 2H, J = 4.4 Hz, NHCH2), 4.92 (s, 2H, OCH2), 7.00 (s, 1H, Hpy), 8.33 (s, 1H, NH); 13C NMR (DMSO-d6, 101 MHz): δ 19.96 (CH3), 24.46 (CH3), 40.81 (CH2NH), 52.21 (OCH3), 64.65 (OCH2), 93.59, 115.19, 118.93, 155.33, 160.85, 162.75, 168.38 (C=OAmide), 170.51 (C=OEster); IR (KBr/cm–1): 3299 (NH), 3088 (C–H aromatic), 2947 (C–H aliphatic), 2220 (CN), 1740 (C=Oester), 1658 (C=Oamide), 1601 (C=C); Elemental analysis Calcd for[C13H15N3O4]: C, 56.31; H, 5.45; N, 15.15 Found C, 56.44; H, 5.32; N, 15.05.
4,6-Dimethyl-2-(2-morpholino-2-oxoethoxy)nicotinonitrile (12)
Yield: 66%, mp 131 °C. 1H NMR (DMSO-d6, 400 MHz): δ 2.39 (s, 3H, CH3), 2.43 (s, 3H, CH3), 3.47 (s, 4H, 2CH2), 3.62 (s, 4H, 2OCH2), 5.17 (s, 2H, OCH2), 6.97 (s, 1H, Hpy); 13C NMR (DMSO-d6, 101 MHz): δ 19.94 (CH3), 24.55 (CH3), 45.21 (2CH2N), 63.65 (OCH2), 66.53 (2CH2O), 93.49, 115.13, 118.66, 155.22, 160.68, 163.17, 165.97 (C=OAmide); IR (KBr/cm–1): 2973 (C–H aliphatic), 2220 (CN), 1647 (C=Oamide), 1601 (C=C); Elemental analysis Calcd for [C14H17N3O3]: C, 61.08; H, 6.22; N, 15.26 Found C, 61.18; H, 6.18; N, 15.36.
4,6-Dimethyl-2-(2-oxo-2-(piperidin-1-yl)ethoxy)nicotinonitrile (13)
Yield: 55%, mp 116 °C. 1H NMR (DMSO-d6, 400 MHz): δ 1.48–1.62 (m, 6H, 3CH2), 2.38 (s, 3H, CH3), 2.43 (s, 3H, CH3), 3.41 (s, 4H, 2CH2), 5.15 (s, 2H, OCH2), 6.96 (s, 1H, Hpy); 13C NMR (DMSO-d6, 101 MHz): δ 19.98 (CH3), 24.44 (CH3), 24.57, 25.76, 26.34, 42.86, 45.57, 63.72, 93.34, 115.29, 118.57, 155.21, 160.67, 163.28, 165.23 (C=OAmide); IR (KBr/cm–1): 3018 (C–H aromatic), 2996 (C–H aliphatic), 2219 (CN), 1659 (C=O amide), 1599 (C=C); Elemental analysis Calcd for [C15H19N3O2]: C, 65.91; H, 7.01; N, 15.37 Found C, 65.97; H, 6.98; N, 15.46.
N-Benzyl-2-((3-cyano-4,6-dimethylpyridin-2-yl)oxy)acetamide (14)
Yield: 45%, mp 121 °C. 1H NMR (DMSO-d6, 400 MHz): δ 2.39 (s, 3H, CH3), 2.43 (s, 3H, CH3), 4.32 (d, 2H, J = 4.8 Hz, CH2NH), 4.91 (s, 2H, OCH2), 6.99 (s, 1H, Hpy), 7.25–7.32 (m, 5Hph), 8.45 (s, 1H, NH); 13C NMR (DMSO-d6, 101 MHz): δ 19.95 (CH3), 24.43 (CH3), 42.23 (CH2NH), 65.21 (OCH2), 93.79, 115.17, 118.83, 127.18, 127.42, 128.61, 139.70, 155.21, 160.70, 163.02, 167.83 (C=OAmide); IR (KBr/cm–1): 3290 (NH), 3031 (C–H aromatic), 2961 (C–H aliphatic), 2224 (CN), 1649 (C=Oamide), 1596 (C=C); Elemental analysis Calcd for [C17H17N3O2]: C, 69.14; H, 5.80; N, 14.23 Found C, 69.04; H, 5.88; N, 14.13.
N-Allyl-2-((3-cyano-4,6-dimethylpyridin-2-yl)oxy)acetamide (15)
Yield: 80%, mp 139 °C. 1H NMR (DMSO-d6, 400 MHz): δ 2.38 (s, 3H, CH3), 2.43 (s, 3H, CH3), 3.72–3.75 (m, 2H, NHCH2), 4.87 (s, 2H, OCH2), 5.06 (dd, 1H, J = 10.4 Hz, J = 1.6 Hz, CH=CHcisH), 5.16 (dd, 1H, J = 17.2 Hz, J = 1.6 Hz, CH=CHHtrans), 5.75–5.84 (m, 1H, CH=CH2), 6.99 (s, 1H, Hpy), 8.21 (d, 1H, J = 5.2 Hz, NH); 13C NMR (DMSO-d6, 101 MHz): δ 19.99 (CH3), 24.50 (CH3), 41.37 (CH2NH), 65.04 (OCH2), 93.78, 115.18, 115.32, 118.78, 135.53, 155.16, 160.62, 163.01, 167.42 (C=Oamide); Elemental analysis Calcd for [C13H15N3O2]: C, 63.66; H, 6.16; N, 17.13 Found C, 63.55; H, 6.20; N, 17.03.
Material and Methods
Biology
Cytotoxicity
The breast cancer (MCF-7) and liver (HepG2) cells, which were obtained and grown in RPMI-1640 medium l-glutamine (Lonza Verviers SPRL, Belgium, cat#12-604F), were provided by the National Research Institute in Egypt. Both cell lines received 10% fetal bovine serum (FBS; Sigma-Aldrich, MO, USA) and 1% penicillin–streptomycin (Lonza, Belgium). On the second day, cells were exposed to the chemicals in concentrations of (0.01, 0.1, 1, 10, and 100 M). Cell viability was assessed using the MTT solution (Promega, USA) after 48 h.40 The plate was incubated for 3 h after each well had been injected with 20 μL of MTT dye. The viability was calculated with respect to the control, and GraphPad Prism 7 was used to determine the IC50 values.
PIM-1 Kinase Inhibitory Assay
The PIM-1 kinase test was done using an ELISA kit in accordance with manufacturer instructions (Kit #7573). To assess the inhibitory potency of drugs 6, 11, 12, and 13 against the PIM-1 kinase activity, kinase inhibitory tests were carried out. The following calculation was used to compute the proportion in which chemicals inhibited autophosphorylation: 100-[(A treated)/(A control)-control] Using the GraphPad prism7 program, the IC50 was calculated using the curves of the percentage inhibition of five concentrations of each chemical.41,42
Investigation of Apoptosis
Annexin V/PI Staining and Cell Cycle Analysis
3–105 MCF-7 cells were added to 6-well culture plates, which were then placed in the incubator for the night. Following that, cells were treated for 48 h to compound 12 at its IC50 levels. Following that, PBS was rinsed with ice-cold water before cells and media supernatants were gathered. The cells were then treated with ″Annexin V-FITC solution (1:100) and propidium iodide (PI)″ at a concentration of 10 g/mL for 30 min in the dark after being suspended in 100 L of annexin binding buffer solution, which is composed of 25 mM CaCl2, 1.4 M NaCl, and 0.1 M Hepes/NaOH, pH 7.4. Then, labeled cells were collected using the Cytoflex FACS system. The data were assessed using the cytExpert program.43
Autophagy Evaluation Using Acridine Orange Quantitative Assessment
Autophagic cell death is measured by flow cytometric analysis and an acridine orange lysosomal stain. After being exposed to the test substance for 48 h, MCF-7 cells (105 cells) were extracted by trypsinization and washed twice with ice-cold PBS (pH 7.4). The cells were stained with acridine orange (10 mM) and incubated at 37 °C in the dark for 30 min. After staining, cells were injected into the ACEA NovoCyte flow cytometer (ACEA Biosciences Inc., San Diego, CA, USA), and a FL1 signal detector (lex/em 488/530 mM) was used to assess the fluorescent signals from acridine orange. After 12,000 events were recorded for each sample, ACEA NovoExpress software (ACEA Biosciences Inc., San Diego, CA, USA) was used to calculate the net fluorescence intensities (NFI) for each sample.
Wound-Healing Assay (Scratch Assay)
Research from the past has referenced the wound-healing test.45 MCF-7 cells (4 105/well) were added to 6-well plates containing starvation medium and then incubated at 37 °C for an overnight period. When it was determined that the cells had adhered to the well and that cell confluence had reached 90% the next day, a scratch of the cell monolayer was created using a sterile 1 mL pipet tip. To remove the cells from the plates, the cells were cleansed with starvation media. The medium was replaced with PBS right away after 48 h, the wound gap was inspected, and cells (both control and treated) were photographed using a digital camera mounted to an Olympus microscope. Measurements were made for the wound closure area.
In Vivo
The Suez Canal University Research Ethics Committee validated the experimental procedure (Approval number REC220/2023, Faculty of Science, Suez Canal University). Detailed methodology is provided in the supplementary file.
Molecular Docking
Maestro was used to construct, optimize, and energetically favor ligand structures. The X-ray crystallographic structure of PIM-1 kinase (PDB ID: 2OBJ)44 was subjected to a molecular docking investigation using the AutoDock Vina software, followed by the Chimera-UCSF software after routine work.
Conclusions
Compound 2 as 5-thioxo-1,3,4-oxadiazol-2-yl)methoxy)nicotinonitrile was synthesized and coupled with alkyl halides to produce new compounds. The azide-coupling method used for coupling the azide 10 with glysine methyl ester and a set of amines. Compound 12 had potent cytotoxicity with IC50 values of 0.5 and 5.27 μM against MCF-7 and HepG2, respectively. For molecular target, compound 12 exhibited potent PIM-1 inhibition activity with a 97.5% IC50 value of 14.3 nM compared to Staurosporine (96.8%, IC50 = 16.7 nM). Compound 12 significantly activated apoptotic cell death in MCF-7 cells, increasing the cell population by total apoptosis by 33.43% (23.18% for early apoptosis and 10.25% for late apoptosis) compared to the untreated control group (0.64%), and arresting the cell cycle at S-phase by 36.02% compared to control 29.12%. Accordingly, compound 12 induced potent cytotoxicity against MCF-7 cells through PIM-1 inhibition with apoptosis-induction, and it can be developed as a chemotherapeutic antibreast cancer agent.
Acknowledgments
This paper is based upon work supported by the Science, Technology & Innovation Funding Authority (STDF) under grant no. (project ID: 45096).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.3c06700.
1H-, 13C-NMR spectra for all compounds and in vivo assay additional experimental details (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Wu S.; Zhu W.; Thompson P.; Hannun Y. A. Evaluating intrinsic and non-intrinsic cancer risk factors. Nat. Commun. 2018, 9, 3490. 10.1038/s41467-018-05467-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung H.; Ferlay J.; Siegel R. L.; Laversanne M.; Soerjomataram I.; Jemal A.; Bray F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. 2021, 71, 209–249. 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- Presti D.; Quaquarini E. The PI3K/AKT/mTOR and CDK4/6 Pathways in Endocrine Resistant HR+/HER2- Metastatic Breast Cancer: Biological Mechanisms and New Treatments. Cancers 2019, 11, 1242. 10.3390/cancers11091242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lainetti P. d. F.; Leis-Filho A. F.; Laufer-Amorim R.; Battazza A.; Fonseca-Alves C. E. Mechanisms of Resistance to Chemotherapy in Breast Cancer and Possible Targets in Drug Delivery Systems. Pharmaceutics 2020, 12, 1193. 10.3390/pharmaceutics12121193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X.-Y.; Wu K.-M.; He X.-X. Advances in drug development for hepatocellular carcinoma: clinical trials and potential therapeutic targets. J. Exp. Clin. Cancer Res. 2021, 40, 172. 10.1186/s13046-021-01968-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Lohuizen M.; Verbeek S.; Krimpenfort P.; Domen J.; Saris C.; Radaszkiewicz T.; Berns A. Predisposition to lymphomagenesis in pim-1 transgenic mice: Cooperation with c-myc and N-myc in murine leukemia virus-induced tumors. Cell 1989, 56, 673–682. 10.1016/0092-8674(89)90589-8. [DOI] [PubMed] [Google Scholar]
- Theo Cuypers H.; Selten G.; Quint W.; Zijlstra M.; Maandag E. R.; Boelens W.; van Wezenbeek P.; Melief C.; Berns A. Murine leukemia virus-induced T-cell lymphomagenesis: Integration of proviruses in a distinct chromosomal region. Cell 1984, 37, 141–150. 10.1016/0092-8674(84)90309-X. [DOI] [PubMed] [Google Scholar]
- Foulks J. M.; Carpenter K. J.; Luo B.; Xu Y.; Senina A.; Nix R.; Chan A.; Clifford A.; Wilkes M.; Vollmer D.; et al. A Small-Molecule Inhibitor of PIM Kinases as a Potential Treatment for Urothelial Carcinomas. Neoplasia 2014, 16, 403–412. 10.1016/j.neo.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abouzid K. A. M.; Al-Ansary G. H.; El-Naggar A. M. Eco-friendly synthesis of novel cyanopyridine derivatives and their anticancer and PIM-1 kinase inhibitory activities. Eur. J. Med. Chem. 2017, 134, 357–365. 10.1016/j.ejmech.2017.04.024. [DOI] [PubMed] [Google Scholar]
- Chen L. S.; Redkar S.; Taverna P.; Cortes J. E.; Gandhi V. Mechanisms of cytotoxicity to Pim kinase inhibitor, SGI-1776, in acute myeloid leukemia. Blood 2011, 118, 693–702. 10.1182/blood-2010-12-323022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeton E. K.; McEachern K.; Dillman K. S.; Palakurthi S.; Cao Y.; Grondine M. R.; Kaur S.; Wang S.; Chen Y.; Wu A.; et al. AZD1208, a potent and selective pan-Pim kinase inhibitor, demonstrates efficacy in preclinical models of acute myeloid leukemia. Blood 2014, 123, 905–913. 10.1182/blood-2013-04-495366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheney I. W.; Yan S.; Appleby T.; Walker H.; Vo T.; Yao N.; Hamatake R.; Hong Z.; Wu J. Z. Identification and structure–activity relationships of substituted pyridones as inhibitors of Pim-1 kinase. Bioorg. Med. Chem. Lett. 2007, 17, 1679–1683. 10.1016/j.bmcl.2006.12.086. [DOI] [PubMed] [Google Scholar]
- Abdelaziz M. E.; El-Miligy M. M. M.; Fahmy S. M.; Mahran M. A.; Hazzaa A. A. Design, synthesis and docking study of pyridine and thieno[2,3-b] pyridine derivatives as anticancer PIM-1 kinase inhibitors. Bioorg. Chem. 2018, 80, 674–692. 10.1016/j.bioorg.2018.07.024. [DOI] [PubMed] [Google Scholar]
- Asati V.; Mahapatra D. K.; Bharti S. K. PIM kinase inhibitors: Structural and pharmacological perspectives. Eur. J. Med. Chem. 2019, 172, 95–108. 10.1016/j.ejmech.2019.03.050. [DOI] [PubMed] [Google Scholar]
- Rizk O. H.; Teleb M.; Abu-Serie M. M.; Shaaban O. G. Dual VEGFR-2/PIM-1 kinase inhibition towards surmounting the resistance to antiangiogenic agents via hybrid pyridine and thienopyridine-based scaffolds: Design, synthesis and biological evaluation. Bioorg. Chem. 2019, 92, 103189. 10.1016/j.bioorg.2019.103189. [DOI] [PubMed] [Google Scholar]
- Farrag A. M.; Ibrahim M. H.; Mehany A. B. M.; Ismail M. M. F. New cyanopyridine-based scaffold as PIM-1 inhibitors and apoptotic inducers: Synthesis and SARs study. Bioorg. Chem. 2020, 105, 104378. 10.1016/j.bioorg.2020.104378. [DOI] [PubMed] [Google Scholar]
- Ismail M. M. F.; Farrag A. M.; Harras M. F.; Ibrahim M. H.; Mehany A. B. M. Apoptosis: A target for anticancer therapy with novel cyanopyridines. Bioorg. Chem. 2020, 94, 103481. 10.1016/j.bioorg.2019.103481. [DOI] [PubMed] [Google Scholar]
- Madheswaran T.; Kandasamy M.; Bose R. J.; Karuppagounder V. Current potential and challenges in the advances of liquid crystalline nanoparticles as drug delivery systems. Drug Discov. Today 2019, 24, 1405–1412. 10.1016/j.drudis.2019.05.004. [DOI] [PubMed] [Google Scholar]
- Abdelrahman F. E.; Elsayed I.; Gad M. K.; Badr A.; Mohamed M. I. Investigating the cubosomal ability for transnasal brain targeting: In vitro optimization, ex vivo permeation and in vivo biodistribution. Int. J. Pharm. 2015, 490, 281–291. 10.1016/j.ijpharm.2015.05.064. [DOI] [PubMed] [Google Scholar]
- Martina S. D.; Vesta K. S.; Ripley T. L. Etoricoxib: A Highly Selective COX-2 Inhibitor. Ann. Pharmacother 2005, 39, 854–862. 10.1345/aph.1E543. [DOI] [PubMed] [Google Scholar]
- Tatar S.; Atmaca S. Determination of amlodipine in human plasma by high-performance liquid chromatography with fluorescence detection. J. Chromatogr. B: Biomed. Sci. Appl. 2001, 758, 305–310. 10.1016/S0378-4347(01)00197-9. [DOI] [PubMed] [Google Scholar]
- Papeo G.; Posteri H.; Borghi D.; Busel A. A.; Caprera F.; Casale E.; Ciomei M.; Cirla A.; Corti E.; D’Anello M.; et al. Discovery of 2-[1-(4,4-Difluorocyclohexyl)piperidin-4-yl]-6-fluoro-3-oxo-2,3-dihydro-1 H -isoindole-4-carboxamide (NMS-P118): A Potent, Orally Available, and Highly Selective PARP-1 Inhibitor for Cancer Therapy. J. Med. Chem. 2015, 58, 6875–6898. 10.1021/acs.jmedchem.5b00680. [DOI] [PubMed] [Google Scholar]
- Bozorov K.; Zhao J. y.; Nie L. F.; Ma H.-R.; Bobakulov K.; Hu R.; Rustamova N.; Huang G.; Efferth T.; Aisa H. A. Synthesis and in vitro biological evaluation of novel diaminothiophene scaffolds as antitumor and anti-influenza virus agents. Part 2. RSC Adv. 2017, 7, 31417–31427. 10.1039/C7RA04808D. [DOI] [Google Scholar]
- Burger M. T.; Han W.; Lan J.; Nishiguchi G.; Bellamacina C.; Lindval M.; Atallah G.; Ding Y.; Mathur M.; McBride C.; et al. Structure Guided Optimization, in Vitro Activity, and in Vivo Activity of Pan-PIM Kinase Inhibitors. ACS Med. Chem. Lett. 2013, 4, 1193–1197. 10.1021/ml400307j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia P. D.; Langowski J. L.; Wang Y.; Chen M.; Castillo J.; Fanton C.; Ison M.; Zavorotinskaya T.; Dai Y.; Lu J.; et al. Pan-PIM Kinase Inhibition Provides a Novel Therapy for Treating Hematologic Cancers. Clin. Cancer Res. 2014, 20, 1834–1845. 10.1158/1078-0432.CCR-13-2062. [DOI] [PubMed] [Google Scholar]
- Boraei A. T. A.; Eltamany E. H.; Ali I. A. I.; Gebriel S. M.; Nafie M. S. Synthesis of new substituted pyridine derivatives as potent anti-liver cancer agents through apoptosis induction: In vitro, in vivo, and in silico integrated approaches. Bioorg. Chem. 2021, 111, 104877. 10.1016/j.bioorg.2021.104877. [DOI] [PubMed] [Google Scholar]
- Ibrahim M. H.; Harras M. F.; Mostafa S. K.; Mohyeldin S. M.; Al kamaly O.; Altwaijry N.; Sabour R. Development of novel cyanopyridines as PIM-1 kinase inhibitors with potent anti-prostate cancer activity: Synthesis, biological evaluation, nanoparticles formulation and molecular dynamics simulation. Bioorg. Chem. 2022, 129, 106122. 10.1016/j.bioorg.2022.106122. [DOI] [PubMed] [Google Scholar]
- El-Miligy M. M. M.; Abdelaziz M. E.; Fahmy S. M.; Ibrahim T. M.; Abu-Serie M. M.; Mahran M. A.; Hazzaa A. A. Discovery of new pyridine-quinoline hybrids as competitive and non-competitive PIM-1 kinase inhibitors with apoptosis induction and caspase 3/7 activation capabilities. J. Enzyme Inhib. Med. Chem. 2023, 38, 2152810. 10.1080/14756366.2022.2152810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelhafez O. M.; Ahmed E. Y.; Abdel Latif N. A.; Arafa R. K.; Abd Elmageed Z. Y.; Ali H. I. Design and molecular modeling of novel P38α MAPK inhibitors targeting breast cancer, synthesized from oxygen heterocyclic natural compounds. Bioorg. Med. Chem. 2019, 27, 1308–1319. 10.1016/j.bmc.2019.02.027. [DOI] [PubMed] [Google Scholar]
- El-Naggar M.; Almahli H.; Ibrahim H. S.; Eldehna W. M.; Abdel-Aziz H. A. Pyridine-Ureas as Potential Anticancer Agents: Synthesis and In Vitro Biological Evaluation. Molecules 2018, 23, 1459. 10.3390/molecules23061459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F.; Wang X.-L.; Shi J.; Wang S.-F.; Yin Y.; Yang Y.-S.; Zhang W. M.; Zhu H. L. Synthesis, molecular modeling and biological evaluation of N-benzylidene-2-((5-(pyridin-4-yl)-1,3,4-oxadiazol-2-yl)thio)acetohydrazide derivatives as potential anticancer agents. Bioorg. Med. Chem. 2014, 22, 468–477. 10.1016/j.bmc.2013.11.004. [DOI] [PubMed] [Google Scholar]
- Boraei A. T. A.; Ashour H. K.; El Tamany E. S. H.; Abdelmoaty N.; El-Falouji A. I.; Gomaa M. S. Design and synthesis of new phthalazine-based derivatives as potential EGFR inhibitors for the treatment of hepatocellular carcinoma. Bioorg. Chem. 2019, 85, 293–307. 10.1016/j.bioorg.2018.12.039. [DOI] [PubMed] [Google Scholar]
- Boraei A. T. A.; Gomaa M. S.; El Ashry E. S. H.; Duerkop A. Design, selective alkylation and X-ray crystal structure determination of dihydro-indolyl-1,2,4-triazole-3-thione and its 3-benzylsulfanyl analogue as potent anticancer agents. Eur. J. Med. Chem. 2017, 125, 360–371. 10.1016/j.ejmech.2016.09.046. [DOI] [PubMed] [Google Scholar]
- Boraei A. T. A.; Singh P. K.; Sechi M.; Satta S. Discovery of novel functionalized 1,2,4-triazoles as PARP-1 inhibitors in breast cancer: Design, synthesis and antitumor activity evaluation. Eur. J. Med. Chem. 2019, 182, 111621. 10.1016/j.ejmech.2019.111621. [DOI] [PubMed] [Google Scholar]
- Nafie M. S.; Amer A. M.; Mohamed A. K.; Tantawy E. S. Discovery of novel pyrazolo[3,4-b]pyridine scaffold-based derivatives as potential PIM-1 kinase inhibitors in breast cancer MCF-7 cells. Bioorg. Med. Chem. 2020, 28, 115828. 10.1016/j.bmc.2020.115828. [DOI] [PubMed] [Google Scholar]
- El-Gohary N. S.; Hawas S. S.; Gabr M. T.; Shaaban M. I.; El-Ashmawy M. B. New series of fused pyrazolopyridines: Synthesis, molecular modeling, antimicrobial, antiquorum-sensing and antitumor activities. Bioorg. Chem. 2019, 92, 103109. 10.1016/j.bioorg.2019.103109. [DOI] [PubMed] [Google Scholar]
- El-Gohary N. S.; Shaaban M. I. New pyrazolopyridine analogs: Synthesis, antimicrobial, antiquorum-sensing and antitumor screening. Eur. J. Med. Chem. 2018, 152, 126–136. 10.1016/j.ejmech.2018.04.025. [DOI] [PubMed] [Google Scholar]
- El-Gohary N. S.; Shaaban M. I. Design, synthesis, antimicrobial, antiquorum-sensing and antitumor evaluation of new series of pyrazolopyridine derivatives. Eur. J. Med. Chem. 2018, 157, 729–742. 10.1016/j.ejmech.2018.08.008. [DOI] [PubMed] [Google Scholar]
- Keshk R. M. Design and synthesis of new series of 3-cyanopyridine and pyrazolopyridine derivatives. J. Heterocycl. Chem. 2020, 57, 3384–3393. 10.1002/jhet.4058. [DOI] [Google Scholar]
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Nafie M. S.; Boraei A. T. A. Exploration of novel VEGFR2 tyrosine kinase inhibitors via design and synthesis of new alkylated indolyl-triazole Schiff bases for targeting breast cancer. Bioorg. Chem. 2022, 122, 105708. 10.1016/j.bioorg.2022.105708. [DOI] [PubMed] [Google Scholar]
- Elgawish M. S.; Nafie M. S.; Yassen A. S. A.; Yamada K.; Ghareb N. The design and synthesis of potent benzimidazole derivatives via scaffold hybridization and evaluating their antiproliferative and proapoptotic activity against breast and lung cancer cell lines. New J. Chem. 2022, 46, 4239–4256. 10.1039/D1NJ05655G. [DOI] [Google Scholar]
- Nafie M. S.; Elghazawy N. H.; Owf S. M.; Arafa K.; Abdel-Rahman M. A.; Arafa R. K. Control of ER-positive breast cancer by ERα expression inhibition, apoptosis induction, cell cycle arrest using semisynthetic isoeugenol derivatives. Chem. Biol. Interact. 2022, 351, 109753. 10.1016/j.cbi.2021.109753. [DOI] [PubMed] [Google Scholar]
- Turner D. P.; Moussa O.; Sauane M.; Fisher P. B.; Watson D. K. Prostate-derived ETS factor is a mediator of metastatic potential through the inhibition of migration and invasion in breast cancer. Cancer Res. 2007, 67 (4), 1618–1625. 10.1158/0008-5472.CAN-06-2913. [DOI] [PubMed] [Google Scholar]
- Rao X.; Huang X.; Zhou Z.; Lin X. An improvement of the 2̂(−delta delta CT) method for quantitative real-time polymerase chain reaction data analysis. Biostat. Bioinforma. Biomath. 2013, 3, 71–85. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.