

Abstract
Genome-wide association studies (GWAS) revolutionized our understanding of common genetic variation and its impact on common human disease and traits. Developed and adopted in the mid-2000s, GWAS led to searchable genotype–phenotype catalogs and genome-wide datasets available for further data mining and analysis for the eventual development of translational applications. The GWAS revolution was swift and specific, including almost exclusively populations of European descent to the neglect of the majority of the world’s genetic diversity. In this narrative review, we recount the GWAS landscape of the early years that established a genotype–phenotype catalog that is now universally understood to be inadequate for a complete understanding of complex human genetics. We then describe approaches taken to augment the genotype–phenotype catalog, including the study populations, collaborative consortia, and study design approaches aimed to generalize and then ultimately discover genome-wide associations in non-European descent populations. The collaborations and data resources established in the efforts to diversify genomic findings undoubtedly provide the foundations of the next chapters of genetic association studies with the advent of budget-friendly whole-genome sequencing.
Keywords: diversity, genome-wide association studies, cohorts, consortia, precision medicine, polygenic risk scores, generalizability
INTRODUCTION
In recent years, genetic association studies have uncovered information on the genetic basis of disease for thousands of phenotypes (1). While early genetic studies consisted of smaller sample sizes and focused on a single phenotype, efforts soon after the first genome-wide association studies (GWAS) focused on aggregating genotype and phenotype data from hundreds of thousands of study participants via large consortia (Figure 1). These analyses focused on larger samples to ensure sufficient statistical power to detect genetic associations. As a result of past (2–11) and ongoing (12–14) genomic discovery efforts, genetic associations continue to be identified even for the most well-studied phenotypes (Figure 2), revealing the underlying genetic architecture of and estimated heritabilities for important human clinical outcomes and traits.
Figure 1.
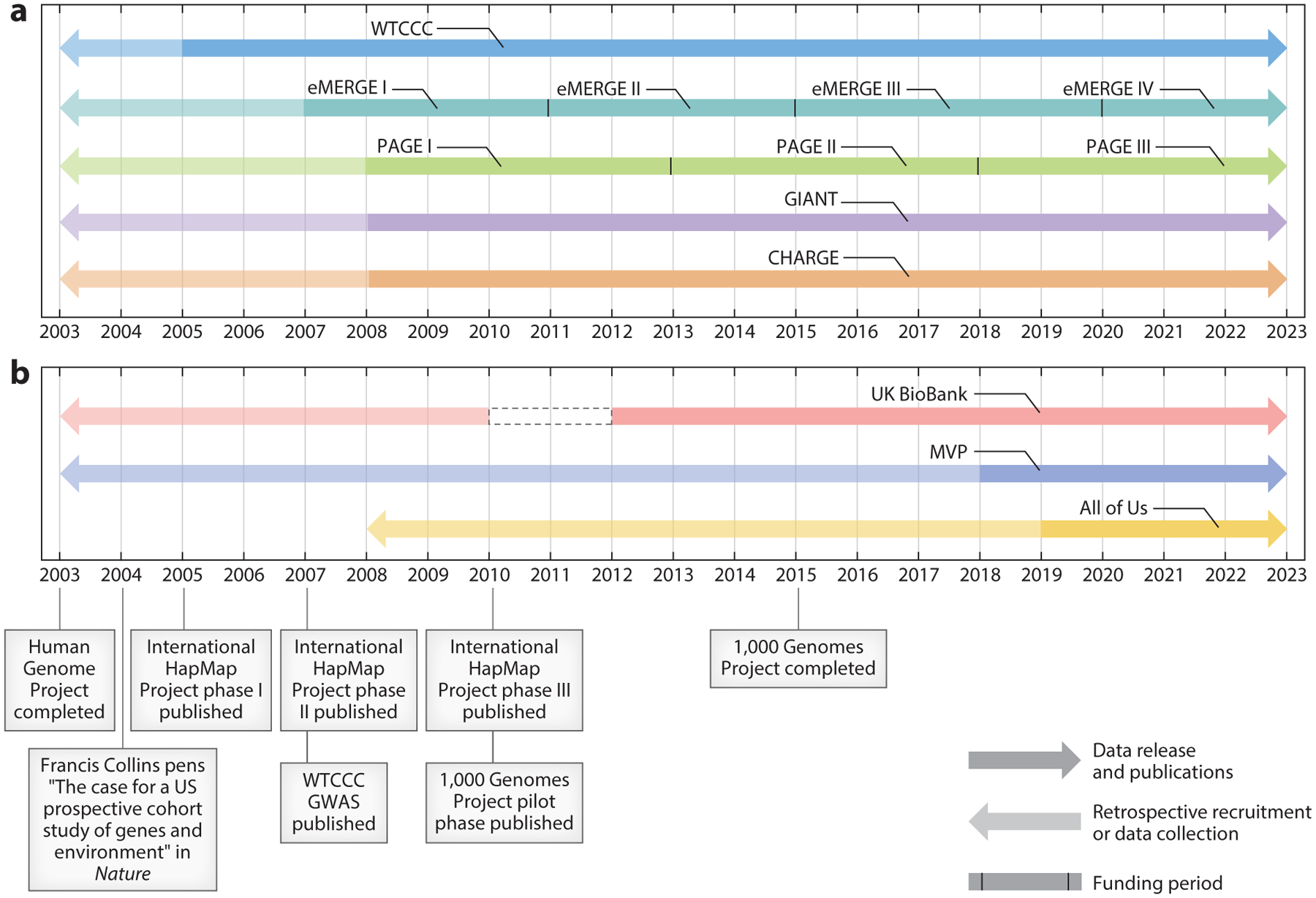
A timeline of the complicated evolution of GWAS consortia. This timeline is a snapshot of the formation of (a) select consortia that serve as the foundation of many contemporary GWAS and (b) newer large, prospective studies with genome-wide data that are fueling the next generation of GWAS consortia. The years on the x-axis represent a 20-year time frame within which we highlight some of the major pre-GWAS accomplishments that enabled the first and now commonplace GWAS. (a) The faded arrows pointing retrospectively represent studies within consortia that recruited participants or collected data prior to the years on the timeline (i.e., the 1958 British Birth Cohort). The black dividing markers represent the NIH funding periods for each iteration of the eMERGE and PAGE studies. While WTCCC is no longer aggregating new data from study investigators, we present this consortium as active since this dataset is one of many in the largest currently active consortium. Unlike WTCCC, GIANT and CHARGE are actively acquiring data to increase diversity and sample size. As such the forward arrows represent both the inclusion of new data and the use of these data in present-day GWAS. Similar to the forward pointing arrows in panel a, those in panel b also represent both data that are used in GWAS today and studies that are actively recruiting or collecting data. Abbreviations: CHARGE, Cohorts for Heart and Aging Research in Genomic Epidemiology; eMERGE, Electronic Medical Records and Genomics; GIANT, Genetic Investigation of Anthropometric Traits; GWAS, genome-wide association study; NIH, National Institutes of Health; PAGE, Population Architecture using Genomics and Epidemiology; WTCCC, Wellcome Trust Case Control Consortium. Figure adapted from images created with BioRender.com.
Figure 2.
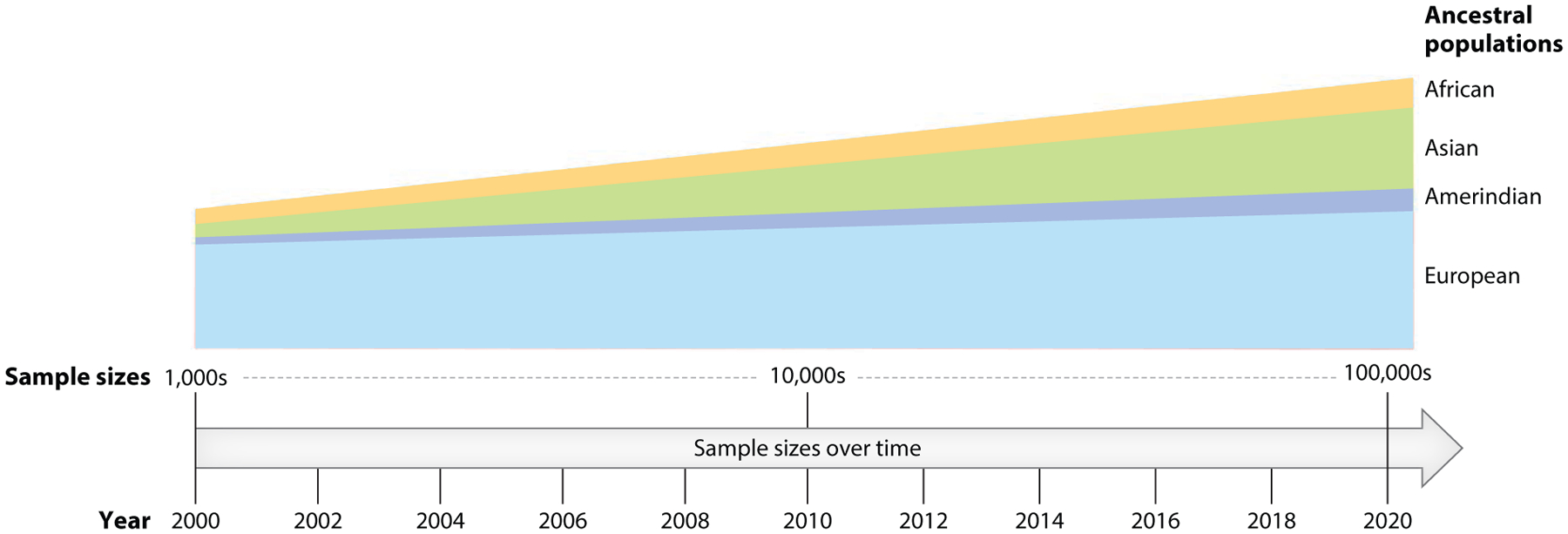
Genome-wide association study (GWAS) consortia population distribution from the early 2000s to the 2020s. Historically, GWAS have been made up of an overwhelming majority of individuals of European ancestry. Therefore, it is unsurprising that the early consortia-powered GWAS lack ancestral diversity. While recent efforts to purposely oversample populations historically underrepresented in research are underway, equitable healthcare can only be achieved if these efforts are more widespread. A 2019 commentary summarized the ancestral distribution of GWAS studies and individuals within studies, finding that while approximately half (48%) of research studies contain data from non-European participants, nearly 79% of the study samples are participants of European ancestry, 10% are Asian, 2% are African, and 1% are Hispanic or Latin American (20). This lack of diverse representation exacerbates health disparities and hinders our understanding of the role of genetic ancestry in disease etiology. Figure adapted from images created with BioRender.com.
From their inception, GWAS have consisted of predominantly European-descent individuals. A consistent lack of diverse ancestral representation in these studies has led to an incomplete understanding of the genetic architecture of phenotypes, resulting in limited opportunities to apply these data to at-risk individuals of non-European ancestry (15). This disparate representation in genome-wide studies has the potential to exacerbate health care inequities for historically underrepresented groups in human genetics and genomics research. It has been well demonstrated that ancestrally diverse GWAS expand gene discovery (16) and improve risk estimation via polygenic risk scores (17, 18), which leads to better cross-population utility of results (19). Increased genetic diversity allows for better characterization of the underlying genetic architecture of complex polygenic traits beyond the group in which genetic architecture is examined (20).
The objective of conducting GWAS is to identify genetic variants associated with a phenotype of interest (21). For complex polygenic traits such as height or blood pressure, GWAS may return many statistically significant associations for genetic variants with varying effect sizes. Larger sample sizes enable identification of genetic associations with small effect sizes, offering finer granularity in the understanding of all the genetic variants relevant for the outcome or trait of interest. This is especially important in the context of complex polygenic diseases, as many genetic loci with varying effect sizes are involved in the risk of disease development and progression. As GWAS typically generate hypotheses, results are then further explored in subsequent fine-mapping analyses and functional in silico or in vivo studies to better define causal variants and the biological and molecular processes that they impact. Compared to linkage approaches, whose sample sizes range from a large multigenerational extended family to smaller families or affected sibpairs (22), typical, contemporary GWAS, whether case–control studies or studies of quantitative traits, are conducted by analyzing DNA samples from thousands of unrelated individuals. While GWAS can be conducted using a parent–offspring study design, it is more difficult to ascertain sufficient numbers of trios compared with the easier enrollment of unrelated individuals drawn from a general or clinical population. Additionally, GWAS conducted in trios require that more study participants be genotyped or sequenced compared with the study design using unrelated individuals.
While modern mega GWAS statistically allow for inclusion of individuals from diverse geographic and ancestral backgrounds in both discovery and fine-mapping efforts, resources to enable these study designs have often been insufficient. As early as the dawn of GWAS (23), the reliance on existing cohorts with biospecimens had the effect of passively excluding groups historically underrepresented in biomedical research. Now, GWAS and genomic discovery in general remain dominated by DNA samples and genetic variation from European-descent participants (20) (Figure 3). Given this landscape, this review focuses on two scientific approaches designed to address persistent inequities in human genetics and genomics research, neither of which is exclusive of the other. We first describe approaches designed to generalize GWAS-identified variants in existing ancestrally diverse populations, noting historic European-only GWAS, major milestones, and lessons learned, including the need to develop more diverse study cohorts for genomic discovery. We next summarize ongoing efforts to build diverse, inclusive cohorts to amplify representation in genetic studies.
Figure 3.
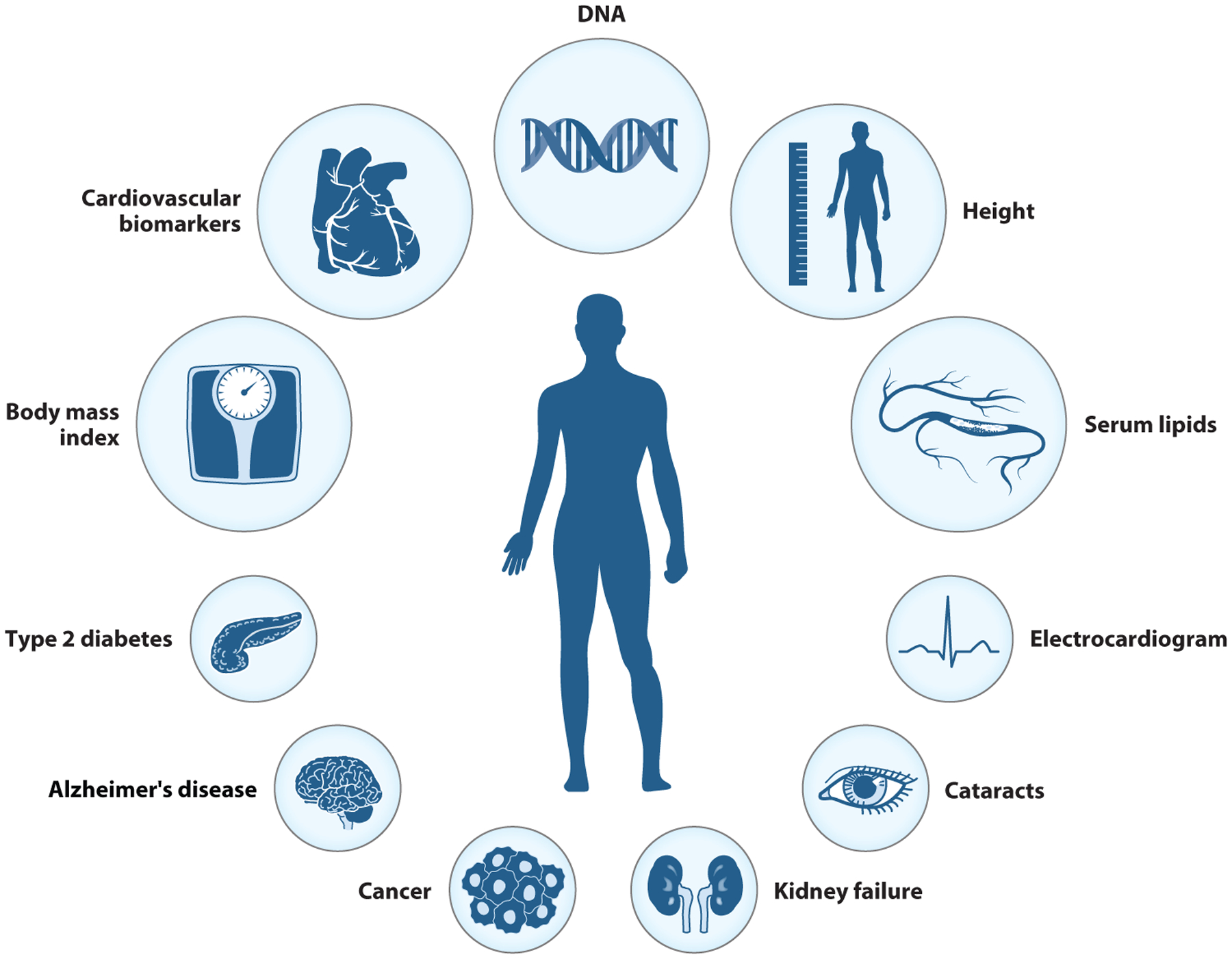
The phenotypes in this figure represent commonly measured phenotypes in the described genome-wide association study consortia. The larger circles represent those phenotypes for which data have been collected for a large number of participants (sample sizes in the hundreds of thousands to millions). These include body mass index, cardiovascular/inflammatory biomarkers (C-reactive protein and erythrocyte sedimentation rate), height, and serum lipids (LDL-C, HDL-C, triglycerides, and total cholesterol). Data for these phenotypes come from a majority of the studies described in this narrative review (e.g., GIANT, CHARGE, PAGE, eMERGE, and WTCCC). Compared to the larger circles, the smaller circles represent those phenotypes that are well-described in comparatively smaller consortia (sample sizes range from thousands to tens of thousands), such as ADGC, PRACTICAL, eMERGE, and DIAGRAM. These include type 2 diabetes, Alzheimer’s disease, cancer (breast and prostate), kidney failure, cataracts, and electrocardiographic traits. Abbreviations: ADGC, Alzheimer’s Disease Genetics Consortium; CHARGE, Cohorts for Heart and Aging Research in Genomic Epidemiology; DIAGRAM, Diabetes Genetics Replication and Meta-Analysis Consortium; eMERGE, Electronic Medical Records and Genomics; GIANT, Genetic Investigation of Anthropometric Traits; HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol; PAGE, Population Architecture using Genomics and Epidemiology; PRACTICAL, Prostate Cancer Association Group to Investigate Cancer Associated Alterations in the Genome; WDCCC, Wellcome Trust Case Control Consortium. Figure adapted from images created with BioRender.com.
LARGE GWAS WERE (AND ARE) CONDUCTED PRIMARILY IN EUROPEAN-DESCENT POPULATIONS
Historic GWAS laid the foundation for the study design, quality control, and now rote statistical methods for future discovery efforts. These early GWAS also generated data and findings that prompted the first observations that the study of homogeneous populations would not be sufficient. As mentioned above, nearly 20 years ago at its inception (see the sidebar titled The Early History of GWAS) large GWAS were (and to some extent still are) primarily conducted in European-descent populations (20). As an example, established in 2005, the Wellcome Trust Case Control Consortium (WTCCC) is one of the earliest collaborative efforts designed to understand genetic variation of human disease with the intent of providing opportunity for large-scale GWAS (24). The initial major WTCCC GWAS included 2,000 cases, each for seven human diseases/outcomes, and 3,000 shared controls drawn from the 1958 British Birth Cohort (24). This and other early WTCCC GWAS identified thousands of putative candidate loci for breast cancer (25), coronary artery disease (24), multiple sclerosis (25), malaria (26), and tuberculosis (27).
THE EARLY HISTORY OF GWAS
In 2003, the initial iteration of the 13-year Human Genome Project was announced (118). While monumental, data from the Human Genome Project alone were not sufficient for the understanding of how sequence variation impacts complex human diseases. Genotype–phenotype studies for common human diseases would require large prospective cohorts, as advocated for by the then-director of the NIH’s (National Institutes of Health) National Human Genome Research Institute, Francis Collins, in 2004 (119). Also required would be a catalog of genetic variation and an understanding of the patterns of variation and linkage disequilibrium in human populations. To supply these data, the International HapMap Project was formed in late 2002, and in 2005 the project published data in three ancestral populations from phase I, making large-scale human genotype patterns widely available for the first time (5). Also in 2005, an early GWAS was published for age-related macular degeneration describing a significant association between common variation in CFH with what is now recognized to be an unusually large genetic effect (76) (not shown in the figure). In 2006, recruitment started for the UK Biobank. While the WTCCC data were first released in 2005, it was not until 2007 that the GWAS from this effort was published. The 2007 WTCCC GWAS set the precedent for future GWAS by modeling the importance of nontrivial components such as large sample size, discovery, and replication cohorts, as well as multiple-testing correction (24). The same year, phase II of the HapMap project was published characterizing over 3.1 million SNPs (single-nucleotide polymorphisms) (6). By 2010, phase III of the HapMap project was finished (7), while the pilot phase of the 1000 Genomes Project was first published, describing genetic variation yields from the newer next-generation sequencing technologies (8). Although recruitment for the UK Biobank was completed in 2010, early data in the form of surveys were not released until two years later in 2012. By this point, recruitment for the Million Veteran Program had already been underway for a year. The 1000 Genomes Project was completed in 2015 characterizing over 88 million SNPs across 26 ancestral populations (10). Three years later, recruitment began for the NIH’s All of Us research program.
Following the establishment and success of the WTCCC, other cohort study collaborations arose exploring additional polygenic traits in European-descent populations. Phenotype-driven consortia such as the GIANT (Genetic Investigation of Anthropometric Traits) consortium focused on common human traits measured in most epidemiologic studies or data resources such as body mass index (BMI) (28, 29), height (30–33), and obesity (28). The GIANT consortium began as modest collaborations accessing study populations from Finland and Sardinia (30), later expanding to add other European epidemiologic studies with DNA samples linked to anthropometric traits of interest such as the KORA (Cooperative Health Research in the Region Augsburg) cohort study (28, 31). Starting with an initial sample size of ~6,600, the incorporation of additional collaborative studies over approximately four to five years quickly resulted in the largest GWAS sample size at the time with ~250,000 participants (33) (Figure 4).
Figure 4.
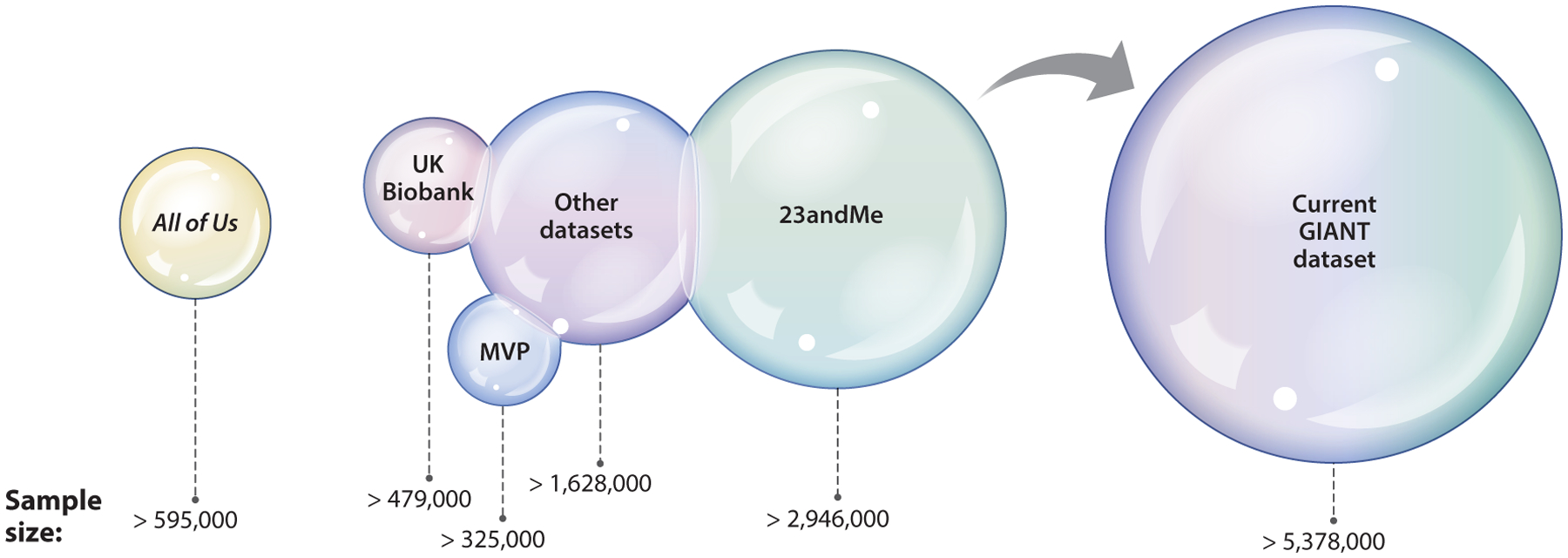
The GIANT (Genetic Investigation of Anthropometric Traits) consortium of consortia is arguably the largest consortium to date. The current iteration of GIANT is a conglomeration of more than 200 distinct studies or cohorts, which, with the recent incorporation of data from 23andMe, Million Veteran Program (MVP), and UK Biobank, has increased in sample size to over 5.38 million participants. A genome-wide study with this sample size has identified more than 12,000 SNPs (single-nucleotide polymorphisms) associated with height (46). As GIANT continues to grow, we expect that other larger cohorts will also be incorporated in the future as it moves toward completing the genetic map for human height. Figure adapted from images created with BioRender.com.
Like the anthropometric traits in the GIANT consortium, other commonly measured quantitative traits such as lipid traits, low-density lipoprotein cholesterol (LDL-C), high-density lipoprotein cholesterol (HDL-C), triglycerides, and total cholesterol enjoyed early GWAS success and subsequent consortium branding to amass large data resources for genomic discovery. In 2008, a GWAS meta-analysis of nearly 12,000 European-descent individuals revealed several genetic variants strongly associated with lipid traits (34). Two years later, that sample size grew exponentially to more than 100,000 European-descent individuals, resulting in nearly 100 significantly associated loci after genome-wide multiple testing correction (35). These early consortium efforts were formalized into what is now known as the Global Lipids Genetics Consortium (35).
Another well-studied common quantitative and polygenic trait is blood pressure. In 2009, two GWASs for blood pressure conducted in 25,000 European-descent participants each identified 13 associated genetic variants (36, 37). A decade later with now 200,000 European-descent participants, The International Consortium for Blood Pressure GWAS added an additional 16 associated loci (38).
In comparison to quantitative traits, very large GWAS for diseases of interest such as type 2 diabetes (T2D) were slower to organize since these phenotypes require more effort to measure and consequently are less ubiquitous in data resources linked to DNA samples. T2D GWAS debuted in 2007 with a genome-wide study in ~1,100 Finish cases and controls (34), followed relatively quickly with a genome-wide meta-analysis of more than 10,000 individuals of European descent (35). The efforts to assemble datasets for the meta-analysis led to the formation of MAGIC (Meta-Analyses of Glucose and Insulin-related traits Consortium) and DIAGRAM (Diabetes Genetics Replication and Meta-analysis Consortium) (39). By 2017, DIAGRAM amassed almost 27,000 T2D cases of European descent, culminating in 128 statistically significant genetic associations involving 113 loci (40).
MANY EUROPEAN-DESCENT COHORTS WITH GENOME-WIDE DATA ARE AVAILABLE AND USED IN VARIOUS GWAS
The WTCCC and early GWAS of anthropometric traits, lipid traits, blood pressure, and T2D capitalized on the availability of existing cohorts or case–control studies, the majority of which were limited to European-descent populations. This trend continued after GWAS was widely adopted as the study design of choice, leading to the genotyping and incorporation of many European-descent cohorts into meta-analyses or consortium-style genome-wide analyses. Examples of these cohorts include the Framingham Heart Study (FHS) (41), the Helsinki Birth Cohort Study (42), the Nurses’ Health Study (43), the Rotterdam Study (44), and the 1958 National Child Development Study (45) (also known as the 1958 British Birth Cohort). While adequately powered GWAS became possible with the availability of these data, their inclusion in the ever-growing GWAS cohort sample sizes created a genotype–phenotype catalog almost exclusively containing data from European-descent populations.
CONTEMPORARY GWAS AND CONSORTIA ARE MORE DIVERSE BUT STILL DOMINATED BY EUROPEAN-DESCENT DATA
More recent consortia like GIANT (46), the Global Lipids Genetics Consortium (47), and DIAGRAM (48) tout larger sample sizes but have made little improvements in proportional diversity, as most consortia now include the genome-wide data available in the UK Biobank (49, 50) The UK Biobank is a large, prospective cohort of ~500,000 adults of 40–69 years of age at the time of ascertainment (50). This large prospective cohort collects health, lifestyle, and behavior data through a variety of mechanisms including direct measurement, questionnaires or surveys, and linkage to electronic health records. While most participants are of “white British ancestry,” roughly 78,000 individuals are of “nonwhite British ancestry.” Global ancestry estimates suggest that the majority of “nonwhite British ancestry” participants are of European descent (n = 50,685), with the remaining being of African (n = 6,653), South Asian (n = 2,782), and East Asian (n = 2,364) descent (51). The UK Biobank also has genome-wide genotype data available and is now generating and releasing whole-exome and whole-genome sequencing data. The UK Biobank is somewhat unique in its ease of access for research (52), making this mostly European-descent data resource an attractive and realistic cohort to include in any ongoing genome-wide consortium effort.
DIVERSE COHORTS WITH GENOME-WIDE DATA ARE A RECOGNIZED NEED BUT ARE STILL COMPARATIVELY SMALL
The demand to fuel continuing consortia growth for genomic discovery has highlighted the need for additional independent genotype–phenotype data not yet subsumed by past consortia analyses. The demand coupled with the recognized need for diversity has also led to an appreciation for already established cohorts and the establishment of new data resources, including biobanks in clinical populations linked to electronic health records. Examples of already established but now greatly appreciated cohorts include the Multiethnic Cohort (MEC) (53), Women’s Health Initiative (WHI) (54), and the Jackson Heart Study (55). These cohorts have sizable African American/Black and Native Hawaiian/Pacific Islander subgroups with genome-wide data. The Hispanic Community Health Study/Study of Latinos (HCHS/SOL), which has 16,000 adult participants representing several groups under the broad umbrella term “Hispanic” (56), is an example newer cohort specifically established to fill the underrepresentation void for this heterogeneous and highly admixed sample in biomedical research.
Prospective cohort studies are the gold-standard study design for GWAS because they minimize biases and establish causality between a suspected risk factor or exposure and the outcome of interest (57). However, cohorts with sufficient sample sizes for genome-wide studies can take years to decades to assemble. To accelerate the availability of data resources available for research, several medical centers have established biobanks that leverage patient biospecimens and the real-world clinical data collected in outpatient settings. Today, various biobanks are linked to electronic health records available in diverse clinical populations, such as Mount Sinai’s BioMe (58), Vanderbilt University Medical Center’s BioVU (59), Northwestern University’s NUGene (60), Kaiser Permanente’s Resource for Genetic Epidemiology Research on Aging (61), and the University of Pennsylvania’s Penn Medicine BioBank (62). Although outside the scope of this review, it should be noted that studies using health data linked to biobanks are associated with many challenges and limitations compared with studies using a traditional cohort design (55). The extent of bias and data missingness will vary depending on the patient population sampled (63).
GENERALIZING GENOTYPE–PHENOTYPE ASSOCIATIONS FROM EUROPEAN TO DIVERSE POPULATIONS
Despite the emergence of new, independent, and diverse data resources for genome-wide studies, individually, sample sizes of these newer studies remain small compared to previous large, European-descent sample sizes represented in consortia-based studies. As we describe above, GWAS began with cohorts and case–control studies drawn from populations of European and built upon their initial success with additional populations of European descent. Existing cohorts from non-European participants were comparatively smaller and fewer, and new cohorts have been slower to mobilize to contribute to genomic discovery. In parallel to cohort and resource building to deliver diversity to GWAS, there has been increased interest in cataloging the replication or generalization of associations identified in GWAS with cohorts of European descent as meaningful data for non-European-descent populations.
Generalization studies hypothesize that a genetic variant identified as associated with a phenotype of interest in European populations is also associated, with similar effect sizes and in the same direction, with the phenotype in non-European populations. To maximize power, rather than genotyping and testing millions of SNPs (single-nucleotide polymorphisms) for associations, a generalization study tests associations between the outcome of interest and a limited list of genetic variants based on an in-depth literature review and a search for previously associated variants in the GWAS Catalog (https://www.ebi.ac.uk/gwas/). An advantage of this more focused study design is that fewer statistical tests are conducted, ultimately allowing for less stringent significance thresholds. In this context, moderately sized cohorts or data resources that characterize most non-European datasets have sufficient power to distinguish between genetic associations that are population specific and those that are universal.
PAGE I
One of the earliest examples of generalization of GWAS-identified variants is the PAGE (Population Architecture using Genomics and Epidemiology) study. Started in 2008, PAGE is a collaborative effort funded by the National Human Genome Research Institute (NHGRI) to investigate the association between genetic variants and complex diseases using ancestrally diverse populations (65). The first phase of the PAGE study (PAGE I) consisted of four research groups or consortia accessing diverse population-based cohorts or cross-sectional studies: the EAGLE (Epidemiological Architecture for Genes Linked to Environment) study, accessing the National Health and Nutrition Examination Surveys (55); MEC (53); WHI (54); and CALiCo (Causal Variants Across the Life Course), which is itself a consortium of cardiovascular disease cohort studies, including the Strong Heart Study (66, 67), the Cardiovascular Health Study (CHS) (68), the Atherosclerosis Risk in Communities Study (ARIC) (69), the Coronary Artery Risk Development in Young Adults (CARDIA) (70), and HCHS/SOL (71). Of the more than 120,000 participants in PAGE I, less than half (47%) were of European descent. The majority (53%) of participants represented five self-identified non-European groups from the United States: African Americans, Hispanics, East Asians, Native Hawaiians, and American Indians.
The PAGE I study conducted several notable generalization studies for a variety of phenotypes from European-descent GWAS (71–75). In one such study, PAGE I investigators examined variants previously found in European-descent GWAS to be associated with age-related macular degeneration (AMD) in their diverse populations, including the highly significant missense mutation CFH rs1061170, which is presumably the causal variant in linkage disequilibrium with the original genome-wide finding among participants self-described as non-Hispanic White (76). Of the genetic variants tested, none were significantly associated with AMD in African Americans or Mexican Americans in PAGE I, despite sufficient statistical power to detect associations with large effect sizes, contrary to what would be expected based on European-descent results for CFH rs1061170 and AMD. These data demonstrate that population differences such as linkage disequilibrium and population-specific associations can affect even the most well-studied phenotypes of early European-descent GWAS such as AMD, whose association with CFH rs1061170 is one of the strongest and most replicable in European-descent GWAS genotype–phenotype associations for complex human diseases apart from Alzheimer’s disease and the gene APOE (77).
The inability to generalize or replicate GWAS-identified variants from European-descent populations was a theme of PAGE I. Similar to the AMD example, in a PAGE I EAGLE substudy, none of the tested MYH9 variants were associated with chronic kidney disease in non-Hispanic Blacks (74). Furthermore, none of the MYH9 variants showed consistent direction of effect across the three groups tested, which included non-Hispanic Whites, non-Hispanic Blacks, and Mexican Americans (74). The lack of associations was surprising given that the MYH9 variants are in strong linkage disequilibrium with APOL1 variants (78), both of which have been strongly associated with kidney diseases in African-descent participants but not European-descent participants (79). The lack of association could be due to the combination of heterogeneous kidney diseases in the tested populations, misspecification of genetic models, and differences in effect sizes compared with the original literature. In support of the different effect size explanation, a PAGE I reexamination of generalization study results for five common diseases and traits including BMI, T2D, and lipid levels demonstrated that although many of the variants tested were associated regardless of significance threshold in the same direction, the effect sizes varied when comparing European Americans to non-European Americans, especially in African Americans, where the effect sizes were smaller compared with European descent populations in PAGE I (80). The dilution or heterogeneity of effect sizes may be due to differences in linkage disequilibrium, where the tested variant tags the causal variant in Europeans but does so imperfectly or not at all in other populations (80).
FROM GENERALIZATION TO DISCOVERY IN CONSORTIA WITH DIVERSE POPULATIONS
The PAGE study was one of the largest consortia at the time focused on generalization of GWAS-identified variants; it subsequently shifted its focus to discovery efforts using the Metabochip (81) and then other genome-wide array data as part of PAGE II (82). Other consortia contemporary to PAGE I also contributed to knowledge of generalization and population-specific associations and conducted some of the first albeit underpowered GWAS for several outcomes and traits in non-European-descent populations. One such consortium is the eMERGE (Electronic Medical Records and Genomics) network, formed and supported in 2007 by the NHGRI (58, 60, 83). Now in its fourth iteration, the eMERGE network was initially a consortium of five biobanks, each focused on an outcome or clinical trait of interest for GWAS. The first two cycles of the eMERGE network examined the extent to which variants associated with electrocardiographic traits in European-descent GWAS were generalizable to non-European-descent populations (84). In parallel, the eMERGE network conducted GWAS in African American participants and patients for red blood cell traits (85), lipid levels (HDL-C and LDL-C) (86, 87), atrioventricular conduction (88), and resistant hypertension (89), among other traits. While the majority of these African-descent GWAS were statistically underpowered, these data were used in subsequent GWAS and meta-analysis as part of larger consortia of consortia (90), demonstrating the usefulness of generating these data that are often left out of studies due to lack of statistical power (91).
Like the PAGE study and the eMERGE network, the CHARGE (Cohorts for Heart and Aging Research in Genomic Epidemiology) consortium (92), which comprised ten prospective cohorts, began conducting genome-wide studies in multiple populations. CHARGE included the AGES (Age, Gene/Environment Susceptibility)–Reykjavik study (94), ARIC (69), CHS (68), FHS (offspring and Gen3) (41), the Rotterdam Study (44), CARDIA (70), the HABC (Health, Aging, and Body Composition) study, and the MESA (Multi-Ethnic Study of Atherosclerosis) study (95). While these studies mainly consist of European-descent individuals, the consortium was able to capture information from a large number of African-descent individuals. As a result of this diverse, multicohort collaboration, a GWAS for an electrocardiographic trait (QRS duration) was conducted in African Americans (96), resulting in two novel loci associated with QRS width. CHARGE investigators also described the transferability or generalization of previously identified loci from the European-descent cohorts to the African American cohorts.
LIMITATIONS OF GENERALIZING GWAS-IDENTIFIED VARIANTS TO DIVERSE POPULATIONS
While restricting tests of association to variants or gene/gene regions identified in previous European-descent GWAS preserves statistical power for small and moderately sized diverse cohorts, this approach has some notable limitations. A major limitation is the assumption that the GWAS-identified variant, also known as the index variant, is either the causal variant or in linkage disequilibrium with the causal variant. Differences in linkage disequilibrium and the impact of these differences on generalization were noted soon after GWAS studies were first published in the mid-2000s (97). GWAS-identified variants may also differ in frequency across populations, affecting both statistical power and linkage disequilibrium. At its most extreme, observed allele frequency differences include population-specific variants or genes like APOL1 (79), which are common in African-descent populations but rare or absent in European-descent populations. African-descent populations are the genomically most diverse populations in the world (98), and genetic association studies limited to European findings can only characterize variants shared across populations, which are much fewer than those specific to certain populations (10). Complicating the variant and linkage disequilibrium landscape is admixture, a prominent feature of genomes for many present-day populations with complex, recent migratory histories (99, 100). In this context, alternative or adjuvant approaches to GWAS such as admixture mapping, which leverages allele frequency differences between ancestral haplotypes to identify index variants associated with the phenotype of interest (101, 102), may be of use, as described in the next section.
STATISTICALLY POWERED GENOMIC DISCOVERY IN DIVERSE POPULATIONS
Statistically, genomic discovery depends on a well-powered genome-wide array, sequencing association, or admixture study. Toward the latter, based on the assumption that ancestry influences genetic architecture, admixture mapping is a robust statistical approach for delineating genetic risk for disease in recently (approximately 20–30 generations) admixed populations. Specifically, in two-way admixture analysis (101, 102), regions of the genome of differing frequencies between parental populations are chosen for further investigation. These regions are then compared based on differential distribution among cases and controls. Index loci are then identified and further explored for a possible role in disease etiology or tested for association with the putative causal variant.
As described above, various cohorts and consortia were formed to conduct generalization and replication studies of early GWAS studies, but it was not until recent years that resources were available to conduct properly powered trans-population and non-European-specific genomic discovery studies. Innovative statistical methods were first developed in early consortia-led GWAS. This has served as model for the subsequent development of large-scale population specific and trans-population GWAS (103). These methods include using summary statistics and the application of meta-analysis approaches (for example, fixed versus random effects) (104). The use of summary statistics as opposed to individual-level data is an attractive approach because it allows for the inclusion of datasets subject to otherwise restricted access without the loss of statistical power (105). Meta-analysis allowing for random effects provides an opportunity to examine heterogeneity likely observed when GWAS include multiple cohorts from diverse populations.
Trans-population meta-analyses, like the previously described early GWAS of QRS duration by the CHARGE consortium (96), are also now possible thanks to the establishment and continued growth of these diverse consortia. Genome-wide consortia from several years ago included cohorts that represented only a few countries, with the largest contributions coming from the United States and the United Kingdom. Multicountry genomic resources such as the 1000 Genomes Project (10) dataset were limited to genome-wide genotyping or sequencing for population genetics research but were too small and lacked phenotype data for genetic association studies. Several cohorts and biobanks outside of the United States and the United Kingdom, such as the Biobank Japan (106), H3Africa (Humans, Heredity, and Health in Africa) (107), and INMEGEN (Mexico National Institute of Genomic Medicine) (108), are now being considered for worldwide consortia efforts for genomic discovery. The latest iterations of consortia of consortia, like the Global Biobank Meta-analysis Initiative (GBMI) (109, 110), leverages the availability of worldwide biobanks. GBMI consists of 24 biobanks across the world with more than 2.2 million individuals recruited through both population-based and hospital-based approaches. This collaboration spans five continents including Europe, Asia, North America, Australia, and Africa. Despite the fact that more than half of participants reside in Europe, this is a geographically and ancestrally diverse genomic resource. Another impressive global-scale consortium is the new COVID-19 Host Genetics Initiative (111). At release 6 (June 2021), the COVID-19 Host Genetics Initiative included data from 54 studies conducted by ~3,000 scientists worldwide.
The global datasets of GBMI and the COVID-19 Host Genetics Initiative are examples of general approaches being taken to develop new data resources for GWAS. These new data resources can be disease-agnostic or disease-centric consortia. Although first developed around specific outcomes of interest, the linkage of electronic health records makes the eMERGE network disease agnostic. Other examples of diverse disease-agnostic consortia, cohorts, or companies are PAGE II, the Million Veteran Program (MVP) (112), and the 23andMe Research Innovation Collaborations Program. MVP is a longitudinal cohort study conducted by the Department of Veterans Affairs healthcare system. MVP participants are US veterans who consent to donate biospecimens and their electronic health records for research. Participating veterans also take questionnaires designed to collect data on health, lifestyle, behaviors, and exposures. MVP genome-wide data include genome-wide array and sequencing data. The MVP currently has more than 900,000 participants, and as the name implies, the MVP intends to recruit approximately one million participants. While the MVP is one of the largest US biobanks, 23andMe eclipses it with more than 10 million customers, 80% of whom have consented to share their genome-wide data for research (113). Disease-centric diverse population consortia include the PRACTICAL (Prostate Cancer Association Group to Investigate Cancer Associated Alterations in the Genome) consortium (114) and ADGC (Alzheimer’s Diseases Genetics Consortium) (115).
Here we make special mention of All of Us Research Program of the NIH (National Institutes of Health) (116, 117). The United States does not have a national cohort nor does it have a national healthcare system. The NIH established All of Us to provide the scientific community with data resources that include US populations or groups historically underrepresented in biomedical research. All of Us is reminiscent of the UK Biobank in that participants can consent to include health data from their electronic health records as well as health data directly measured from exams, biospecimens, or questionnaires. All of Us deviates from the UK Biobank in oversampling by self-identified non-European race/ethnicity as well as geography, socioeconomic status, age, disability, and other dimensions of diversity. All of Us enrolls participants primarily through healthcare provider organizations but also allows for volunteer participants outside of the healthcare system. Similar to the UK Biobank, All of Us promises ease of data access to better ensure properly powered genomic discovery studies will be conducted sooner rather than later for diverse populations.
NEXT STEPS
To better understand the underlying genetic architecture of complex disease, more effort must be made toward global inclusion at every step of the research process, from study design to analyses. At the recruitment stage, more attention should be paid toward recruitment for bigger and more ancestrally and geographically diverse cohorts. This recruitment effort will likely require expansion beyond the Eurocentric recruitment methods applied to date. Along with the use of larger sample sizes, future studies will be able to conduct whole-genome sequencing at the scale now enjoyed by budget-friendly genome-wide arrays. As the cost of whole-genome sequencing decreases, more population-specific variation data associated with phenotypes will be available. These data will contribute to our complete understanding of the population-shared and population-unique genomic basis of complex human diseases and traits, ultimately informing translational applications emerging from the currently incomplete and Eurocentric databases established almost two decades ago.
ACKNOWLEDGMENTS
We would like to acknowledge Shiying Liu for her help with creating and editing figures for this manuscript. L.A.C. is supported by the National Institutes of Health (NIH) grant T32 HL007567 from the National Heart, Lung, and Blood Institute. This work was supported by the Cleveland Institute for Computational Biology; the Clinical and Translational Science Collaborative of Cleveland, which is funded by the NIH; the National Center for Advancing Translational Science; the Clinical and Translational Science Award grant UL1TR002548; and NIH grant R01 EY033829-01.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Sollis E, Mosaku A, Abid A, Buniello A, Cerezo M, et al. 2023. The NHGRI-EBI GWAS Catalog: knowledgebase and deposition resource. Nucleic Acids Res. 51(D1):D977–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, et al. 2001. Initial sequencing and analysis of the human genome. Nature 409(6822):860–921 [DOI] [PubMed] [Google Scholar]
- 3.Nurk S, Koren S, Rhie A, Rautiainen M, Bzikadze AV, et al. 2022. The complete sequence of a human genome. Science 376(6588):44–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Int. HapMap Consort. 2003. The International HapMap Project. Nature 426(6968):789–96 [DOI] [PubMed] [Google Scholar]
- 5.Altshuler D, Donnelly P, Int. HapMap Consort. 2005. A haplotype map of the human genome. Nature 437(7063):1299–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, et al. 2007. A second generation human haplotype map of over 3.1 million SNPs. Nature 449(7164):851–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Int. HapMap 3 Consort., Altshuler DM, Gibbs RA, Peltonen L, Altshuler DM, et al. 2010. Integrating common and rare genetic variation in diverse human populations. Nature 467(7311):52–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.1000 Genomes Proj. Consort., Abecasis GR, Altshuler D, Auton A, Brooks LD, et al. 2010. A map of human genome variation from population-scale sequencing. Nature 467(7319):1061–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.1000 Genomes Project Consort., Abecasis GR, Auton A, Brooks LD, DePristo MA, et al. 2012. An integrated map of genetic variation from 1,092 human genomes. Nature 491(7422):56–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.1000 Genomes Project Consort., Auton A, Brooks LD, Durbin RM, Garrison EP, et al. 2015. A global reference for human genetic variation. Nature 526(7571):68–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, et al. 2015. An integrated map of structural variation in 2,504 human genomes. Nature 526(7571):75–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang T, Antonacci-Fulton L, Howe K, Lawson HA, Lucas JK, et al. 2022. The Human Pangenome Project: a global resource to map genomic diversity. Nature 604(7906):437–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taliun D, Harris DN, Kessler MD, Carlson J, Szpiech ZA, et al. 2021. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 590(7845):290–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, et al. 2016. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536(7616):285–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martin AR, Kanai M, Kamatani Y, Okada Y, Neale BM, Daly MJ. 2019. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat. Genet 51(4):584–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wojcik GL, Graff M, Nishimura KK, Tao R, Haessler J, et al. 2019. Genetic analyses of diverse populations improves discovery for complex traits. Nature 570(7762):514–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Graham BE, Plotkin B, Muglia L, Moore JH, Williams SM. 2021. Estimating prevalence of human traits among populations from polygenic risk scores. Hum. Genom 15:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Y, Tsuo K, Kanai M, Neale BM, Martin AR. 2022. Challenges and opportunities for developing more generalizable polygenic risk scores. Annu. Rev. Biomed. Data Sci 5:293–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hindorff LA, Bonham VL, Brody LC, Ginoza MEC, Hutter CM, et al. 2018. Prioritizing diversity in human genomics research. Nat. Rev. Genet 19(3):175–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sirugo G, Williams SM, Tishkoff SA. 2019. The missing diversity in human genetic studies. Cell 177(1):26–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scott WK, Ritchie MD. 2022. Genetic Analysis of Complex Disease. Hoboken, NJ: Wiley-Blackwell. 3rd ed. [Google Scholar]
- 22.Borecki IB, Province MA. 2008. Linkage and association: basic concepts. Adv, Genet. 60:51–74 [DOI] [PubMed] [Google Scholar]
- 23.Collins FS, Manolio TA. 2007. Merging and emerging cohorts: necessary but not sufficient. Nature 445(7125):259. [DOI] [PubMed] [Google Scholar]
- 24.Wellcome Trust Case Control Consort. 2007. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447(7145):661–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burton PR, Clayton DG, Cardon LR, Craddock N, Deloukas P, et al. 2007. Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat. Genet 39(11):1329–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jallow M, Teo YY, Small KS, Rockett KA, Deloukas P, et al. 2009. Genome-wide and fine-resolution association analysis of malaria in West Africa. Nat. Genet 41(6):657–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thye T, Vannberg FO, Wong SH, Owusu-Dabo E, Osei I, et al. 2010. Genome-wide association analyses identifies a susceptibility locus for tuberculosis on chromosome 18q11.2. Nat. Genet 42(9):739–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Loos RJF, Lindgren CM, Li S, Wheeler E, Zhao JH, et al. 2008. Common variants near MC4R are associated with fat mass, weight and risk of obesity. Nat. Genet 40(6):768–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Speliotes EK, Willer CJ, Berndt SI, Monda KL, Thorleifsson G, et al. 2010. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat. Genet 42(11):937–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanna S, Jackson AU, Nagaraja R, Willer CJ, Chen W-M, et al. 2008. Common variants in the GDF5-UQCC region are associated with variation in human height. Nat. Genet 40(2):198–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lettre G, Jackson AU, Gieger C, Schumacher FR, Berndt SI, et al. 2008. Identification of ten loci associated with height highlights new biological pathways in human growth. Nat. Genet 40(5):584–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lango Allen H, Estrada K, Lettre G, Berndt SI, Weedon MN, et al. 2010. Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature 467(7317):832–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wood AR, Esko T, Yang J, Vedantam S, Pers TH, et al. 2014. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat. Genet 46(11):1173–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Willer CJ, Sanna S, Jackson AU, Scuteri A, Bonnycastle LL, et al. 2008. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat. Genet 40(2):161–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, et al. 2010. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 466(7307):707–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Newton-Cheh C, Johnson T, Gateva V, Tobin MD, Bochud M, et al. 2009. Genome-wide association study identifies eight loci associated with blood pressure. Nat. Genet 41(6):666–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Levy D, Ehret GB, Rice K, Verwoert GC, Launer LJ, et al. 2009. Genome-wide association study of blood pressure and hypertension. Nat. Genet 41(6):677–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Int. Consort. Blood Pressure Genome-Wide Assoc. Studies, Ehret GB, Munroe PB, Rice KM, Bochud M, et al. 2011. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature 478(7367):103–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dupuis J, Langenberg C, Prokopenko I, Saxena R, Soranzo N, et al. 2010. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat. Genet 42(2):105–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scott RA, Scott LJ, Mägi R, Marullo L, Gaulton KJ, et al. 2017. An expanded genome-wide association study of type 2 diabetes in Europeans. Diabetes 66(11):2888–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Andersson C, Johnson AD, Benjamin EJ, Levy D, Vasan RS. 2019. 70-year legacy of the Framingham Heart Study. Nat. Rev. Cardiol 16(11):687–98 [DOI] [PubMed] [Google Scholar]
- 42.Barker DJP, Osmond C, Forsén TJ, Kajantie E, Eriksson JG. 2005. Trajectories of growth among children who have coronary events as adults. N. Engl. J. Med 353(17):1802–9 [DOI] [PubMed] [Google Scholar]
- 43.Bao Y, Bertoia ML, Lenart EB, Stampfer MJ, Willett WC, et al. 2016. Origin, methods, and evolution of the three nurses’ health studies. Am. J. Public Health 106(9):1573–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ikram MA, Brusselle G, Ghanbari M, Goedegebure A, Ikram MK, et al. 2020. Objectives, design and main findings until 2020 from the Rotterdam Study. Eur. J. Epidemiol 35(5):483–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Power C, Elliott J. 2006. Cohort profile: 1958 British birth cohort (National Child Development Study). Int. J. Epidemiol 35(1):34–41 [DOI] [PubMed] [Google Scholar]
- 46.Yengo L, Vedantam S, Marouli E, Sidorenko J, Bartell E, et al. 2022. A saturated map of common genetic variants associated with human height. Nature 610(7933):704–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Graham SE, Clarke SL, Wu K-HH, Kanoni S, Zajac GJM, et al. 2021. The power of genetic diversity in genome-wide association studies of lipids. Nature 600(7890):675–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.DIAbetes Genet. Replication Meta-anal. (DIAGRAM) Consort., Asian Genet. Epidemiol. Netw. Type 2 Diabetes (AGEN-T2D) Consort., South Asian Type 2 Diabetes (SAT2D) Consort., Mex. Am. Type 2 Diabetes (MAT2D) Consort., Type 2 Diabetes Genet. Explor. Next-gener. seq. multi-Ethnic Samples (T2D-GENES) Consort., et al. 2014. Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat. Genet 46(3):234–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ollier W, Sprosen T, Peakman T. 2005. UK Biobank: from concept to reality. Pharmacogenomics 6(6):639–46 [DOI] [PubMed] [Google Scholar]
- 50.Sudlow C, Gallacher J, Allen N, Beral V, Burton P, et al. 2015. UK Biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLOS Med. 12(3):e1001779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Constantinescu A-E, Mitchell RE, Zheng J, Bull CJ, Timpson NJ, et al. 2022. A framework for research into continental ancestry groups of the UK Biobank. Hum. Genom 16:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Conroy M, Sellors J, Effingham M, Littlejohns TJ, Boultwood C, et al. 2019. The advantages of UK Biobank’s open-access strategy for health research. J. Intern. Med 286(4):389–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kolonel LN, Henderson BE, Hankin JH, Nomura AMY, Wilkens LR, et al. 2000. A multiethnic cohort in Hawaii and Los Angeles: baseline characteristics. Am. J. Epidemiol 151(4):346–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Women’s Health Initiat. Study Group. 1998. Design of the Women’s Health Initiative clinical trial and observational study. Control. Clin. Trials 19(1):61–109 [DOI] [PubMed] [Google Scholar]
- 55.Crawford DC, Goodloe R, Farber-Eger E, Boston J, Pendergrass SA, et al. 2015. Leveraging epidemiologic and clinical collections for genomic studies of complex traits. Hum. Hered 79(3–4):137–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.LaVange LM, Kalsbeek WD, Sorlie PD, Avilés-Santa LM, Kaplan RC, et al. 2010. Sample design and cohort selection in the Hispanic Community Health Study/Study of Latinos. Ann. Epidemiol 20(8):642–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Manolio TA, Bailey-Wilson JE, Collins FS. 2006. Genes, environment and the value of prospective cohort studies. Nat. Rev. Genet 7(10):812–20 [DOI] [PubMed] [Google Scholar]
- 58.Gottesman O, Kuivaniemi H, Tromp G, Faucett WA, Li R, et al. 2013. The Electronic Medical Records and Genomics (eMERGE) Network: past, present, and future. Genet. Med 15(10):761–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Roden D, Pulley J, Basford M, Bernard G, Clayton E, et al. 2008. Development of a Large-scale de-identified DNA biobank to enable personalized medicine. Clin. Pharmacol. Ther 84(3):362–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McCarty CA, Chisholm RL, Chute CG, Kullo IJ, Jarvik GP, et al. 2011. The eMERGE Network: a consortium of biorepositories linked to electronic medical records data for conducting genomic studies. BMC Med. Genom 4:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kvale MN, Hesselson S, Hoffmann TJ, Cao Y, Chan D, et al. 2015. Genotyping informatics and quality control for 100,000 subjects in the Genetic Epidemiology Research on Adult Health and Aging (GERA) cohort. Genetics 200(4):1051–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Verma A, Damrauer SM, Naseer N, Weaver J, Kripke CM, et al. 2022. The Penn Medicine BioBank: towards a genomics-enabled learning healthcare system to accelerate precision medicine in a diverse population. J. Pers. Med 12(12):1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pendergrass SA, Crawford DC. 2019. Using electronic health records to generate phenotypes for research. Curr. Protoc. Hum. Genet 100(1):e80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Delete in proof.
- 65.Matise TC, Ambite JL, Buyske S, Carlson CS, Cole SA, et al. 2011. The next PAGE in understanding complex traits: design for the analysis of Population Architecture Using Genetics and Epidemiology (PAGE) study. Am. J. Epidemiol 174(7):849–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee ET, Welty TK, Fabsitz R, Cowan LD, Le NA, et al. 1990. The Strong Heart Study. A study of cardiovascular disease in American Indians: design and methods. Am. J. Epidemiol 132(6):1141–55 [DOI] [PubMed] [Google Scholar]
- 67.North KE, Howard BV, Welty TK, Best LG, Lee ET, et al. 2003. Genetic and environmental contributions to cardiovascular disease risk in American Indians: the Strong Heart Family Study. Am. J. Epidemiol 157(4):303–14 [DOI] [PubMed] [Google Scholar]
- 68.Fried LP, Borhani NO, Enright P, Furberg CD, Gardin JM, et al. 1991. The cardiovascular health study: design and rationale. Ann. Epidemiol 1(3):263–76 [DOI] [PubMed] [Google Scholar]
- 69.ARIC (Atheroscler. Risk Commun.) Investig. 1989. The Atherosclerosis Risk in Communities (ARIC) Study: design and objectives. Am. J. Epidemiol 129(4):687–702 [PubMed] [Google Scholar]
- 70.Hughes GH, Cutter G, Donahue R, Friedman GD, Hulley S, et al. 1987. Recruitment in the Coronary Artery Disease Risk Development in Young Adults (CARDIA) study. Control. Clin. Trials 8(4 Suppl.):68S–73S [DOI] [PubMed] [Google Scholar]
- 71.Haiman CA, Fesinmeyer MD, Spencer KL, Bůžková P, Voruganti VS, et al. 2012. Consistent directions of effect for established type 2 diabetes risk variants across populations. Diabetes 61(6):1642–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dumitrescu L, Carty CL, Taylor K, Schumacher FR, Hindorff LA, et al. 2011. Genetic determinants of lipid traits in diverse populations from the Population Architecture using Genomics and Epidemiology (PAGE) study. PLOS Genet. 7(6):e1002138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Restrepo NA, Spencer KL, Goodloe R, Garrett TA, Heiss G, et al. 2014. Genetic determinants of age-related macular degeneration in diverse populations from the PAGE study. Investig. Ophthalmol. Vis. Sci 55(10):6839–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bailey JNC, Wilson S, Brown-Gentry K, Goodloe R, Crawford DC. 2015. Kidney disease genetics and the importance of diversity in precision medicine. Pac. Symp. Biocomput 21:285–96 [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang L, Spencer KL, Voruganti VS, Jorgensen NW, Fornage M, et al. 2013. Association of functional polymorphism rs2231142 (Q141K) in the ABCG2 gene with serum uric acid and gout in 4 US populations: the PAGE Study. Am. J. Epidemiol 177(9):923–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Klein RJ, Zeiss C, Chew EY, Tsai J-Y, Sackler RS, et al. 2005. Complement factor H polymorphism in age-related macular degeneration. Science 308(5720):385–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rajabli F, Feliciano BE, Celis K, Hamilton-Nelson KL, Whitehead PL, et al. 2018. Ancestral origin of ApoE ε4 Alzheimer disease risk in Puerto Rican and African American populations. PLOS Genet. 14(12):e1007791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Genovese G, Tonna SJ, Knob AU, Appel GB, Katz A, et al. 2010. A risk allele for focal segmental glomerulosclerosis in African Americans is located within a region containing APOL1 and MYH9. Kidney Int. 78(7):698–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yusuf AA, Govender MA, Brandenburg J-T, Winkler CA. 2021. Kidney disease and APOL1. Hum. Mol. Genet 30(R1):R129–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Carlson CS, Matise TC, North KE, Haiman CA, Fesinmeyer MD, et al. 2013. Generalization and dilution of association results from European GWAS in populations of non-European ancestry: the PAGE study. PLOS Biol. 11(9):e1001661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Buyske S, Wu Y, Carty CL, Cheng I, Assimes TL, et al. 2012. Evaluation of the metabochip genotyping array in African Americans and implications for fine mapping of GWAS-identified loci: the PAGE study. PLOS ONE 7(4):e35651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hu Y, Graff M, Haessler J, Buyske S, Bien SA, et al. 2020. Minority-centric meta-analyses of blood lipid levels identify novel loci in the Population Architecture using Genomics and Epidemiology (PAGE) study. PLOS Genet. 16(3):e1008684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Crawford DC, Crosslin DR, Tromp G, Kullo IJ, Kuivaniemi H, et al. 2014. eMERGEing progress in genomics—the first seven years. Front. Genet 5:184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jeff JM, Ritchie MD, Denny JC, Kho AN, Ramirez AH, et al. 2013. Generalization of variants identified by genome-wide association studies for electrocardiographic traits in African Americans. Ann. Hum. Genet 77(4):321–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ding K, de Andrade M, Manolio TA, Crawford DC, Rasmussen-Torvik LJ, et al. 2013. Genetic variants that confer resistance to malaria are associated with red blood cell traits in African-Americans: an electronic medical record-based genome-wide association study. G3 3(7):1061–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Turner SD, Berg RL, Linneman JG, Peissig PL, Crawford DC, et al. 2011. Knowledge-driven multi-locus analysis reveals gene-gene interactions influencing HDL cholesterol level in two independent EMR-linked biobanks. PLOS ONE 6(5):e19586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rasmussen-Torvik LJ, Pacheco JA, Wilke RA, Thompson WK, Ritchie MD, et al. 2012. High density GWAS for LDL cholesterol in African Americans using electronic medical records reveals a strong protective variant in APOE. Clin. Transl. Sci 5(5):394–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Denny JC, Ritchie MD, Crawford DC, Schildcrout JS, Ramirez AH, et al. 2010. Identification of genomic predictors of atrioventricular conduction: using electronic medical records as a tool for genome science. Circulation 122(20):2016–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dumitrescu L, Ritchie MD, Denny JC, El Rouby NM, McDonough CW, et al. 2017. Genome-wide study of resistant hypertension identified from electronic health records. PLOS ONE 12(2):e0171745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ng MCY, Shriner D, Chen BH, Li J, Chen W-M, et al. 2014. Meta-analysis of genome-wide association studies in African Americans provides insights into the genetic architecture of type 2 diabetes. PLOS Genet. 10(8):e1004517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ben-Eghan C, Sun R, Hleap JS, Diaz-Papkovich A, Munter HM, et al. 2020. Don’t ignore genetic data from minority populations. Nature 585(7824):184–86 [DOI] [PubMed] [Google Scholar]
- 92.Psaty BM, O’Donnell CJ, Gudnason V, Lunetta KL, Folsom AR, et al. 2009. Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium. Circ. Cardiovasc. Genet 2(1):73–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Delete in proof.
- 94.Harris TB, Launer LJ, Eiriksdottir G, Kjartansson O, Jonsson PV, et al. 2007. Age, Gene/Environment Susceptibility–Reykjavik study: multidisciplinary applied phenomics. Am. J. Epidemiol 165(9):1076–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bild DE, Bluemke DA, Burke GL, Detrano R, Diez Roux AV, et al. 2002. Multi-ethnic study of atherosclerosis: objectives and design. Am. J. Epidemiol 156(9):871–81 [DOI] [PubMed] [Google Scholar]
- 96.Evans DS, Avery CL, Nalls MA, Li G, Barnard J, et al. 2016. Fine-mapping, novel loci identification, and SNP association transferability in a genome-wide association study of QRS duration in African Americans. Hum. Mol. Genet 25(19):4350–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Teo Y-Y, Small KS, Kwiatkowski DP. 2010. Methodological challenges of genome-wide association analysis in Africa. Nat. Rev. Genet 11(2):149–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pereira L, Mutesa L, Tindana P, Ramsay M. 2021. African genetic diversity and adaptation inform a precision medicine agenda. Nat. Rev. Genet 22(5):284–306 [DOI] [PubMed] [Google Scholar]
- 99.Homburger JR, Moreno-Estrada A, Gignoux CR, Nelson D, Sanchez E, et al. 2015. Genomic insights into the ancestry and demographic history of South America. PLOS Genet. 11(12):e1005602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bryc K, Durand EY, Macpherson JM, Reich D, Mountain JL. 2015. The genetic ancestry of African Americans, Latinos, and European Americans across the United States. Am. J. Hum. Genet 96(1):37–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Winkler CA, Nelson GW, Smith MW. 2010. Admixture mapping comes of age. Annu. Rev. Genom. Hum. Genet 11:65–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Seldin MF, Pasaniuc B, Price AL. 2011. New approaches to disease mapping in admixed populations. Nat. Rev. Genet 12(8):523–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Peterson RE, Kuchenbaecker K, Walters RK, Chen C-Y, Popejoy AB, et al. 2019. Genome-wide association studies in ancestrally diverse populations: opportunities, methods, pitfalls, and recommendations. Cell 179(3):589–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Evangelou E, Ioannidis JPA. 2013. Meta-analysis methods for genome-wide association studies and beyond. Nat. Rev. Genet 14(6):379–89 [DOI] [PubMed] [Google Scholar]
- 105.Lin DY, Zeng D. 2010. On the relative efficiency of using summary statistics versus individual-level data in meta-analysis. Biometrika 97(2):321–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nagai A, Hirata M, Kamatani Y, Muto K, Matsuda K, et al. 2017. Overview of the BioBank Japan Project: study design and profile. J. Epidemiol 27(3S):S2–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Choudhury A, Aron S, Botigué LR, Sengupta D, Botha G, et al. 2020. High-depth African genomes inform human migration and health. Nature 586(7831):741–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Moreno-Estrada A, Gignoux CR, Fernández-López JC, Zakharia F, Sikora M, et al. 2014. The genetics of Mexico recapitulates Native American substructure and affects biomedical traits. Science 344(6189):1280–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wojcik GL. 2022. By their powers combined, global initiative joins forces for genomic research. Cell 185(23):4256–58 [DOI] [PubMed] [Google Scholar]
- 110.Zhou W, Kanai M, Wu K-HH, Rasheed H, Tsuo K, et al. 2022. Global Biobank Meta-analysis Initiative: powering genetic discovery across human disease. Cell Genom. 2(10):100192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.COVID-19 Host Genet. Initiat. 2020. The COVID-19 Host Genetics Initiative, a global initiative to elucidate the role of host genetic factors in susceptibility and severity of the SARS-CoV-2 virus pandemic. Eur. J. Hum. Genet 28(6):715–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gaziano JM, Concato J, Brophy M, Fiore L, Pyarajan S, et al. 2016. Million Veteran Program: a mega-biobank to study genetic influences on health and disease. J. Clin. Epidemiol 70:214–23 [DOI] [PubMed] [Google Scholar]
- 113.Majumder MA, Guerrini CJ, McGuire AL. 2021. Direct-to-consumer genetic testing: value and risk. Annu. Rev. Med 72:151–66 [DOI] [PubMed] [Google Scholar]
- 114.Conti DV, Darst BF, Moss LC, Saunders EJ, Sheng X, et al. 2021. Trans-ancestry genome-wide association meta-analysis of prostate cancer identifies new susceptibility loci and informs genetic risk prediction. Nat. Genet 53:65–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kunkle BW, Schmidt M, Klein H-U, Naj AC, Hamilton-Nelson KL, et al. 2021. Novel Alzheimer Disease risk loci and pathways in African American individuals using the African genome resources panel: a meta-analysis. JAMA Neurol. 78(1):102–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.All of Us Res. Prog. Investig., Denny JC, Rutter JL, Goldstein DB, Philippakis A, et al. 2019. The “All of Us” Research Program. N. Engl. J. Med 381(7):668–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mayo KR, Basford MA, Carroll RJ, Dillion M, Fullen H, et al. 2023. The All of Us Data and Research Center: creating a secure, scalable, and sustainable ecosystem for biomedical research. Annu. Rev. Biomed. Data Sci 6. In press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Collins FS, Morgan M, Patrinos A. 2003. The Human Genome Project: lessons from large-scale biology. Science 300(5617):286–90 [DOI] [PubMed] [Google Scholar]
- 119.Collins FS. 2004. The case for a US prospective cohort study of genes and environment. Nature 429(6990):475–77 [DOI] [PubMed] [Google Scholar]