

Abstract
Background and objective
The ability to inhibit aggregation has been demonstrated with synthetically derived ginsenoside compounds G-Rp (1, 3, and 4) and ginsenosides naturally found in Panax ginseng 20(S)-Rg3, Rg6, F4, and Ro. Among these compounds, Rk3 (G-Rk3) from Panax ginseng needs to be further explored in order to reveal the mechanisms of action during inhibition.
Methodology
Our study focused to investigate the action of G-Rk3 on agonist-stimulated human platelet aggregation, inhibition of platelet signaling molecules such as fibrinogen binding with integrin αIIbβ3 using flow cytometry, intracellular calcium mobilization, dense granule secretion, and thromboxane B2 secretion. In addition, we checked the regulation of phosphorylation on PI3K/MAPK pathway, and thrombin-induced clot retraction was also observed in platelets rich plasma.
Key Results
G-Rk3 significantly increased amounts of cyclic adenosine monophosphate (cAMP) and led to significant phosphorylation of cAMP-dependent kinase substrates vasodilator-stimulated phosphoprotein (VASP) and inositol 1,4,5-trisphosphate receptor (IP3R). In the presence of G-Rk3, dense tubular system Ca2+ was inhibited, and platelet activity was lowered by inactivating the integrin αIIb/β3 and reducing the binding of fibrinogen. Furthermore, the effect of G-Rk3 extended to the inhibition of MAPK and PI3K/Akt phosphorylation resulting in the reduced secretion of intracellular granules and reduced production of TXA2. Lastly, G-Rk3 inhibited platelet aggregation and thrombus formation via fibrin clot.
Conclusions and implications
These results suggest that when dealing with cardiovascular diseases brought upon by faulty aggregation among platelets or through the formation of a thrombus, the G-Rk3 compound can play a role as an effective prophylactic or therapeutic agent.
Keywords: G-Rk3, platelet aggregation, cyclic nucleotide, intracellular Ca2+, granule secretion, `
Graphical abstract
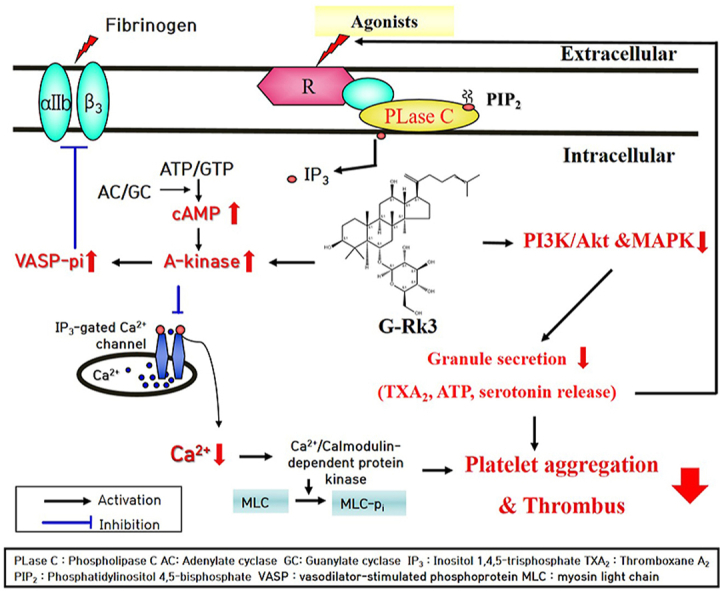
1. Introduction
Among various diseases, cardiovascular disease (CVD) is the leading cause of death worldwide, with 1 in 7 deaths from coronary heart disease and 1 in 9 deaths from heart failure in the United States alone [1]. After vascular injury has occurred, hemostasis relies on the proper functioning of platelets to allow clot formation and prevention of blood loss. Unfortunately, improper platelet activation and aggregation lead to the formation of unwanted blood clots, leading to common diseases of the cardiovascular system such as atherosclerosis, coronary artery disease, heart failure and stroke [2].
Treatment of thrombotic complications with pharmacological compounds has been shown to act through inhibition of platelets, and current CVD prevention and treatment exploits these advantages, but unfortunately comes with major complications and side effects, so better alternatives are needed [3]. One method that is proving promising in the treatment of thrombosis and disorders associated with platelet aggregation is the use of natural bioactive compounds [4]. Compounds found in the traditional diet of the Mediterranean and plants traditionally used for medicinal purposes have shown antiplatelet and cardioprotective properties in CVD prevention [[5], [6], [7]].
During normal blood circulation, endothelial cells in the vascular tissue secrete prostaglandin I2 and nitric oxide, which initiate the production of cAMP and cGMP in platelets. Production of cAMP activates protein kinase A (PKA) and production of cGMP activates protein kinase G (PKG). Both kinases phosphorylate vasodilator-stimulated phosphoprotein (VASP) and inositol 1, 4, 5-triphosphate receptor (IP3R) [8]. Phosphorylation of the IP3R inhibits the recruitment of Ca2+ from the dense tubular system into the cytoplasm [9]. VASP, an important PKG and PKA substrate, is involved in the regulation of αIIb/β3 and actin filaments [10,11].
The secretion of platelet granules containing serotonin and ATP is an important step during aggregation and is involved in the phosphorylation of mitogen-activated protein kinases (MAPKs) and PI3K/Akt phosphoproteins [12]. MAPKs are a group of kinases consisting of p38 MAPK, c-Jun N-terminal kinase (JNK), and extracellular signal-regulated kinase (ERK), which play a role in intracellular signaling during hemostasis and clot formation [13]. MAPKs are present in human platelets and play an important role in stimulating platelet granule secretion by acting as signaling molecules [[14], [15], [16], [17], [18]]. In addition, MAPK triggers phosphorylation of cPLA2 found in cell membranes, leading to an increase in TXA2, resulting in more platelet activation and aggregation [19,20]. The PI3K/Akt pathway is involved in dense granule secretion, platelet regulation and aggregation [21].
Ginsenosides found in Panax ginseng have demonstrated various biological reactions with major essential components of red and white ginseng being ginsenoside-Rb1, -Rc, -Rd, -Re, and -Rg1 and minor essential components being ginsenoside-Rg5, -Rk1, -Rg6 and -F4 [22,23]. G-Rk3 has been shown to have an anti-cancer effect on lung cancer, inhibited inflammation and apoptosis in liver damage, and protective effect on cisplatin-induced kidney damage [[24], [25], [26]]. G-Rk3 has been shown to have anticancer effects against lung cancer, inhibition of inflammation and apoptosis in liver damage, and protective effects against cisplatin-induced kidney damage [[24], [25], [26]]. However, due to the lack of studies investigating the mechanism of anti-platelet by G-Rk3, we investigated the regulatory action of G-Rk3 on human platelet aggregation.
2. Materials and methods
2.1. Materials
The antibodies and western blotting lysis buffers were ordered from Cell Signaling in Beverly, Massachusetts (USA). The ginsenoside Rk3, or G-Rk3, was ordered from Ambo Institue in Dajeon, (South Korea). The platelet inducers, collagen, U46619, and thrombin, were ordered from Chrono-Log Corporation in Havertown, Pennsylvania (USA). The Fura 2-AM and fibrinogen Alexa Fluor 488 conjugates were both ordered from Invitrogen Molecular Probes in Eugene, Oregon (USA). The enzyme immunoassays for TXB2, serotonin, ATP, and cyclic nucleotides (cAMP/cGMP) were all ordered at Cayman Chemical Co. in Ann Arbor, Michigan (USA). The polyvinylidene difluoride membrane and enhanced chemiluminescence solution were both ordered at Thermo Fisher Scientific in Gangnam-gu, Seoul (South Korea). All other reagents not listed above were ordered from Sigma Chemical Corporation in St. Louis, Missouri (USA).
2.2. Human washed platelets and preparation
The proven and tested method previously performed allowed for proper preparation of the platelets [27]. The human plasma-rich plasma (PRP) was ordered from the Korean Red Cross Blood Center in Suwon (South Korea). To allow for collection of platelets, the PRP needed to be centrifuged for a total of 10 minutes at a speed of 1,300 × g. Following the collection, a wash was performed twice. The buffer used during the wash consisted of 2.7 mM KCl, 138 mM NaCl, 12 mM NaHCO3, 0.36 mM NaH2PO4, 1 mM Na2EDTA, and 5.5 mM glucose and a pH of 6.9. After the wash is complete, the platelets are to be suspended. The buffer used to suspend the platelets consisted of 2.7 mM KCl, 138 mM NaCl, 12 mM NaHCO3, 0.49 mM MgCl2, 0.36 mM NaH2PO4, 5.5 mM glucose, 0.25% gelatin and a pH of 7.4. The suspended platelets came to a final concentration of 108 cells/mL. To ensure that unnecessary platelet aggregation was avoided, a temperature of 25°C was maintained for all procedures. For the use of all human material, proper approval was acquired from the Institutional Review Board (1041479-HR-202110-002) at Namseoul University (South Korea).
2.3. Platelet aggregation measurement
The suspended platelets at a concentration of 108 cells/mL were incubated while different doses of G-Rk3 were added. The temperature for incubation was set to 37°C and a time limit of 3 minutes. Following this, 2 mM CaCl2 was introduced and then, for stimulation, collagen was added. The stimulation took place over a 5-minute time period. Using the aggregometer from Chrono-Log Co in Havertown, Pennsylvania (USA), a stirring speed was set to 1,000 rpm. Any increase in the transmittance of light was used to calculate the aggregation rate. For a reference value, a suspension buffer of 0% (permeability) was used. Finally, to dissolve the G-Rk3, dimethyl sulfoxide (DMSO) with a 0.1% concentration was administered (same dose used for all tests).
2.4. Cytotoxicity measurement
Any cytotoxicity could be confirmed with the presence of lactate dehydrogenase (LDH) from the cytoplasm. The suspended platelets at a concentration of 108 cells/mL needed to be incubated. The incubation took place over 2 hours and at room temperature. During the incubation, different G-Rk3 doses were added. Following that, the solution of platelets was centrifuged with the speed set to 12,000 × g and time duration of 2 minutes. The resulting supernatant was analyzed with an LDH EIA kit and the synergy HT multi-reader from BioTek Instruments in Winooski, Vermont (USA).
2.5. Cyclic nucleotides (cAMP and cGMP) production measurement
With concentration of 108 cells/mL, the suspended platelets had different doses of G-Rk3 added while being incubated. The temperature for incubation was set at 37°C and continued for a duration of 3 minutes. Following the incubation, 2 mM CaCl2 was administered along with the addition of collagen to initiate stimulation. This took place for a duration of 5 minutes and then 1 M HCl of was added. The HCL was used to terminate the reaction. The cyclic nucleotides were analyzed using the EIA kit (cAMP/cGMP) and the Synergy HT Multi-Reader from BioTek Instruments in Winooski, Vermont (USA).
2.6. Intracellular Ca2+ mobilization measurement
Fura 2-AM and PRP were incubated. An amount of 5 μM of Fura-2AM was used and the temperature was set at 37°C for a duration of 60 minutes. The platelet suspension was prepared using the steps described earlier. Following, the platelets were washed and incubated with the addition of 2 mM CaCl2. The temperature was set to 37°C and the duration was 3 minutes. Then collagen was used for stimulation at a duration of 5 minutes. To measure the Fura 2 fluorescence, a spectrophotometer (SFM 25, USA) from BioTek Instruments in Winooski, Vermont (USA) was used. The excitation wavelength began at 340 nm and progressed every 0.5 seconds to 380 nm. The emission wavelength was set to 510nm. The equation established by Grynkiewicz was used to calculate the mobilized Ca2+ [28].
2.7. Fibrinogen binding measurement
A treatment using 2 mM CaCl2 was applied to 30 μg/mL of Alexa Fluor 488-human fibrinogen and the suspended human platelets at a concentration of 108 cells/mL. Following this, collagen was administered and present for a duration of 5 minutes. In order to terminate the reaction, phosphate-buffered saline (PBS, pH 7.4) with 0.5% paraformaldehyde was applied, and for the accuracy of the experiment, we performed the experiment while blocking light. To assess fibrinogen binding, we used a flow cytometer (FACS) from BD Biosciences in San Jose, California (USA). Cell-Quest software was also from BD Biosciences.
2.8. TXB2 production measurement
The suspended platelets at a concentration of 108 cells/mL were incubated with different doses of G-Rk3. The incubation temperature was set at 37°C and for a duration of 3 minutes. Following this, 2 mM CaCl2 was administered along with collagen to promote stimulation. This occurred at a duration of 5 minutes. Since TXB2 is produced from the metabolism of TXA2, the EIA KIT (TXB2) and the Synergy HT Multi-Reader from BioTek Instruments in Winooski, Vermont (USA) were used to analyze the production of TXB2.
2.9. ATP and serotonin release measurement
The suspended platelets at a concentration of 108 cells/mL were incubated with different doses of G-Rk3. The incubation temperature was set at 37°C and for a duration of 3 minutes. Following this, 2 mM CaCl2 was administered along with collagen to promote stimulation. This occurred at a duration of 5 minutes. To terminate the reaction, 2 mM EDTA of ice-cold water was administered. The result was centrifuged to collect the released serotonin and ATP in the upper layer. Then the EIA kit (serotonin and ATP) and the Synergy HT Multi-Reader from BioTek Instruments in Winooski, Vermont (USA) were used to analyze the production of serotonin and ATP.
2.10. Western immunoblotting measurement
A 1x lysis buffer was administered to halt the reaction. From the lysed platelets, the concentration of proteins was assessed using a BCA protein kit from Pierce Biotechnology in Rockford, IL (USA). Then 20 μg of protein was isolated and transferred using a 4-20% SDS-PAGE and PVDF membrane, respectively. Treatment occurred with primary antibody at 1:1000 dilution factor. Treatment occurred with secondary antibody at 1:2000 dilution factor. ECL reagent from Thermo Fisher Scientific in Gangnam-gu, Seoul (South Korea) allowed for proper visualization.
2.11. Platelet-mediated fibrin clot formation measurement
An amount of 2 mM CaCl2 was added to 500 μL of PRP and then 0.05 U/mL of thrombin was administered to cause stimulation. A polyethylene tube was chosen in order to avoid any sticking. The temperature was set to 37°C and a duration of 15 minutes. All fibrin-based clots were photographed with a digital camera and ImageJ software (v. 1.46) from the National Institutes of Health (USA) allowed for calculation of the coagulation area.
2.12. Statistical analysis
To represent all the results of the experiment, the standard deviation (mean ±) was used. Results were statistically significant when P < 0.05 using Student's t-test or ANOVA. If the group means had any significant differences in terms of analysis of variance, they were further examined with Scheffe's method. Statistical analysis was performed using SPSS 21.0.0.0 software (SPSS, Chicago, IL, USA), and p < 0.05 was considered statistically significant.
3. Results
3.1. Actions of G-Rk3 on human platelet aggregation and cytotoxicity
Collagen (2.5 μg/mL), U46619 (200 nM), and thrombin (0.05 U/mL) initiated aggregation among human platelets (Fig. 1A, B, 1C). The aggregation values came to 90.8% with collagen, 91% with U46619, and 90.3% with thrombin. With the addition of G-Rk3, the strongest inhibitory effect on platelet aggregation was observed in platelets induced with collagen. The percent of aggregation inhibition was positively correlated with the doses of G-Rk3 (45 μM/19.0%, 60 μM/44.0%, 90 μM/81.2%, and 120 μM/96.5%) (Fig. 1A). Equally important, G-Rk3 displayed no cytotoxic activity (Fig. 1D). Taken together, this shows G-Rk3 has a strong ability to inhibit platelet aggregation without causing unnecessary cytotoxicity.
Fig. 1.

G-Rk3's effect on agonists-induced human platelet aggregation. (A) G-Rk3's effect on collagen-induced human platelet aggregation. (B) G-Rk3's effect on thrombin-induced human platelet aggregation. (C) G-Rk3's effect on U46619-induced human platelet aggregation. (D) Cytotoxicity of G-Rk3 on human platelets. The results are shown as mean ± SD (n = 4). ∗P < 0.05, ∗∗P < 0.001 in comparison to the collagen-induced platelets.
3.2. Actions of G-Rk3 on the cyclic nucleotides production, intracellular Ca2+ mobilization, and IP3R phosphorylation
The cyclic nucleotides cGMP and cAMP lower the rate of platelet activation and for this reason, the ability of G-Rk3 to increase cyclic nucleotide (cGMP/cAMP) was investigated. There was a positive correlation between the dose of G-Rk3 and cAMP production. A significant increase in cGMP was not observed (data not shown). The results demonstrate that G-Rk3 is able to inhibit the activation of platelets by significantly increasing the production of cAMP (but not cGMP) in collagen-induced platelets.
The mobilization of intracellular Ca2+ is critical for the proper functioning of platelet activation and the effect of G-Rk3 on the mobilization of calcium was investigated. When exposed to collagen, intracellular calcium showed an increase from 100.2 ± 0.5 nM to 600.5 ± 7.5 nM. Once G-Rk3 was introduced at varying doses (45∼120 μM), an inverse relationship was observed between the dose of G-Rk3 and levels of calcium (Fig. 2B). The protein IP3R also contributes to the levels of intracellular calcium and therefore, the effect of G-Rk3 on IP3R phosphorylation was investigated. Here as well, G-Rk3 was introduced at varying doses (45∼120 μM) and a positive correlation was seen between G-Rk3 dose and IP3R phosphorylation. All concentrations above 45 μM were deemed significant. Based on these results, the ability of G-Rk3 to reduce intracellular calcium concentration is through increased phosphorylation of IP3R.
Fig. 2.

G-Rk3's effect on cAMP production, intracellular Ca2+ mobilization, and related phophoprotein. (A) G-Rk3's effect on cAMP production. (B) Effects of G-Rk3 on intracellular Ca2+ mobilization. (C) Effects of G-Rk3 on IP3R phosphorylation. (D) G-Rk3's effect on VASP phosphorylation. The results are shown as mean ± SD (n = 4). aP < 0.05 in comparison to no-stimulated platelets, ∗P < 0.05, ∗∗P < 0.001 in comparison to the collagen -induced platelets.
3.3. Actions of G-Rk3 on VASP phosphorylation and fibrinogen binding
Based on the fact that exposure to G-Rk3 with collagen-induced platelets caused an increase in cAMP, the effect of G-Rk3 on the cAMP/cGMP-dependent VASP was investigated. The phosphorylation of Ser157 of cAMP-dependent VASP was found to be significantly increased in the presence of G-Rk3 while there was no significant change in the phosphorylation of Ser239 of cGMP-dependent VASP (Fig. 2D). More specifically, doses of more than 60 μM were calculated as significant. This proves that the increase in cAMP from G-Rk3 leads to a significant increase in the phosphorylation of Ser157 on cAMP-dependent VASP.
Due to the ability of G-Rk3 to increase the production of cAMP and the phosphorylation of Ser157 cAMP-dependent VASP, the effect of G-Rk3 on fibrinogen binding to αIIb/β3 was further investigated. In the presence of collagen, fibrinogen/αIIb/β3 binding increased to 92.1 ± 1.4% (Fig. 3A and b, 3B). However, once exposed to G-Rk3, the binding of fibrinogen/αIIb/β3 was inhibited and showed a positive correlation between the dose of G-Rk3 and the amount of inhibition. Among the attempted doses, 120 μM of G-Rk3 had a confirmed rate of inhibition of 93.3% (Fig. 3A–f, 3B).
Fig. 3.

G-Rk3's effects on the binding of fibrinogen. (A) The histograms of flow cytometry on the binding of fibrinogen. a, Intact platelets(base); b, collagen (2.5 μg/mL); c, collagen (2.5 μg/mL) + G-Rk3 (45 μM); d, collagen (2.5 μg/mL) + G-Rk3 (60 μM); e, collagen (2.5 μg/mL) + G-Rk3 (90 μM); f, collagen (2.5 μg/mL) + G-Rk3 (120 μM). (B) G-Rk3's effect on the binding (%) of fibrinogen induced by collagen.The results are shown as mean ± SD (n = 4). ap < 0.05 in comparison to no-stimulated platelets, ∗p < 0.05, ∗∗p < 0.001 in comparison to the collagen-induced platelets.
3.4. Actions of G-Rk3 on phosphorylation of PI3K/Akt and MAPK
The phosphoprotein PI3K/Akt is involved in the release of platelet granules and thus the effect of G-Rk3 on PI3K/Akt phosphorylation was investigated. When exposed to collagen, both PI3K and Akt showed a significant increase in phosphorylation. Yet, in the presence of G-Rk3, the phosphorylation from collagen exposure was significantly decreased (Fig. 4A). This confirms that phosphorylation of PI3K/Akt induced by collagen is inhibited due to G-Rk3.
Fig. 4.

G-Rk3's effect on the phosphorylation of PI3K/Akt and MAPK. (A) G-Rk3's effect on PI3K/Akt phosphorylation. (B) G-Rk3's effect on MAPK phosphorylation. The results are shown as mean ± SD (n = 4). ap < 0.05 in comparison to no-stimulated platelets, ∗p < 0.05, ∗∗p < 0.001 in comparison to the collagen-induced platelets.
The MAPK proteins, named p38, ERK, and JNK are involved in platelet granule release and the production of TXA2. Therefore, the effect of G-Rk3 and the phosphorylation of MAPK was investigated. With all 3 MAPK proteins (p38, ERK, and JNK), phosphorylation was significantly increased in the presence of collagen when compared to intact cells. However, the phosphorylation of the same MAPK proteins from collagen was inhibited significantly in the presence of G-Rk3 (Fig. 4B). This confirms the ability of G-Rk3 to regulate platelet aggregation by inhibiting the phosphorylation of ERK, JNK, and p38 (MAPK proteins).
3.5. Actions of G-Rk3 on granule secretion, thromboxane B2 and cPLA2 dephosphorylation
The release of platelet granules (ATP and serotonin) is involved in the aggregation of platelets and so the ability of G-Rk3 to affect granule release was investigated. When exposed to 2.5 μg/mL collagen, the release of ATP increased to 9.2 ± 0.2 μM compared to intact cells at 1.1 ± 0.1 μM (Fig. 5A). Yet, the presence of G-Rk3 at various doses (45, 60, 90, and 120 μM) suppressed that increase significantly. As for serotonin release, when exposed to 2.5 μg/mL collagen, the release increased to 125.8 ± 6.2 ng/108 cells from 16.3 ± 2.1 ng/108 cells in intact cells. Just as before, the addition of G-Rk3 suppressed the release of serotonin from collagen exposure. A positive correlation was seen between dose and inhibition with 45 μM at 96.5 ± 8.4 ng/108, 60 μM at 76.5 ± 2.16 ng/108, 90 μM at 69.0 ± 2.9 ng/108, and 120 μM at 43.9 ± 7.5 ng/108 cells (Fig. 5B). Taken together, G-Rk3 is able to significantly inhibit granule release through ATP and serotonin.
Fig. 5.

G-Rk3's effect on the secretion of granules, TXA2 production and cPLA2 phosphorylation. (A) G-Rk3's effect on ATP release. (B) G-Rk3's effect on the release of serotonin. (C) G-Rk3's effect on TXA2 production. (D) G-Rk3's effect on cPLA2 phosphorylation. The results are shown as mean ± SD (n = 4). aP < 0.05 in comparison to no-stimulated platelets, ∗P < 0.05, ∗∗P < 0.001 in comparison to the collagen -induced platelets.
TXA2 is known as an autacoid and is responsible for increasing the aggregation of platelets and so the ability of G-Rk3 to affect TXA2 was investigated. The levels of TXA2 (based on its metabolite TXB2) increased greatly from 1.2 ± 0.3 ng/108 platelets (intact platelets) to 62.5 ± 2.1 ng/108 platelets (2.5 μg/mL collagen) (Fig. 5C). With the addition of G-Rk3, the increase in TXA2 seen from collagen was lowered to 5.4 ± 0.5 ng/108 platelets. A dose of 120 μM showed a remarkable suppression of 98% (Fig. 5D).
The release of arachidonic acid into the cytosol from its initial position in the membrane is regulated by cPLA2 and we examined the ability of G-Rk3 to affect cPLA2 phosphorylation. The release of intracellular calcium leads to the activation of cPLA2 and initiates its movement to the membrane. Full enzyme activity takes place once Ser505 becomes phosphorylated. The introduction of collagen led to the phosphorylation of cPLA2. However, as expected, the presence of G-Rk3 inhibited cPLA2 phosphorylation and showed a positive correlation between G-Rk3 dose and inhibition level.
3.6. Actions of G-Rk3 on platelet-mediated fibrin clot formation
The formation of a fibrin clot stems from the initiation of platelet inducers causing activation and aggregation and eventually leading to the transduction of signals from the external pathway. Based on this knowledge, we investigated the ability of G-Rk3 to affect fibrin-based clot formation from exposure to thrombin. Once exposed to thrombin, a fibrin clot was readily formed (Fig. 6A), but with the addition of various doses of G-Rk3, the fibrin clot formation was inhibited. There was a positive correlation between the G-Rk3 dose and inhibition rates with 60 μM at 17.7%, 90 μM at 54.7%, and 120 μM at 97.1% (Fig. 6B). Give this data, G-Rk3 shows a clear ability to inhibit fibrin clot and thrombus formation.
Fig. 6.

G-Rk3's effect on platelet-mediated fibrin clot formation. (A) G-Rk3's effect on thrombin-retracted fibrin clot photographs. (B) G-Rk3's effect on thrombin-retracted fibrin clot area. The results are shown as mean ± SD (n = 4). ap < 0.05 in comparison to no-stimulated platelets, ∗p < 0.05, ∗∗p < 0.001 in comparison to the thrombin-induced platelet.
4. Discussion
Two products, diacylglycerol (DG) and inositol 1,4,5-triphosphate (IP3), stem from phosphatidylinositol 4,5-bisphosphate (PIP2) on the platelet membrane. This reaction takes place due to phospholipase C-γ2 (PLC- γ2), IP3 initiates the flow of calcium (Ca2+) from the dense tubular system directly into the cytosol [29]. The availability of calcium phosphorylates myosin light chain and pleckstrin in the process of platelet aggregation [30].
On the other hand, intracellular calcium decreases in the presence of both cyclic nucleotides, cAMP and cGMP. These two cyclic nucleotides also activate the cyclic-dependent protein kinases, protein kinase A (PKA) and protein kinase G (PKG) which inhibit platelet aggregation [31]. This study demonstrated that G-Rk3 significantly increased the production of the cyclic nucleotide cAMP, and G-Rk3 was able to inhibit the release of intracellular calcium [Ca2+]i. The increase in cAMP results in the activation of PKA causing a series of substrates to become phosphorylated. Among these substrates is inositol 1,4,5-trisphosphate receptor (IP3R) [8]. Phosphorylation of IP3R was increased with each dose of G-Rk3 as can be seen Fig. 3B. Therefore, the activation of the cAMP/PKA/IP3R pathway and reduction in intracellular calcium is clearly due to the addition of the G-Rk3 compound. The increase in cAMP production from G-Rk3 also facilitated the phosphorylation of vasodilator-stimulated phosphoprotein (VASP) via PKA activation. Vasodilator-stimulated phosphoprotein (VASP) is a critical substrate for both PKA and PKG, and aids in the regulation of platelet activation by controlling platelet secretion and adhesion properties. The phosphorylation of this protein leads to aggregation inhibition among platelets through the inhibition of integrin αIIb/β3 activation [32,33].
Based on the results of this study, the addition of G-Rk3 led to the inhibition of fibrinogen and αIIb/β3 binding. This may be due to the ability of G-Rk3 to initiate the cAMP/PKA/VASP pathway which ultimately leads to phosphorylation of VASP Ser157 and restricts the binding of fibrinogen and αIIb/β3. Future studies will be needed in order to reveal the ways in which cAMP is being increased through the addition of G-Rk3. One possible avenue to consider is that the cyclic nucleotides (cAMP and cGMP) are dependent on the activation of cyclase (adenylyl/guanylyl) and phosphodiesterase (PDE) [34]. When PDE is inhibited during platelet aggregation, cyclic nucleotides do increase and PDE inhibitors are successful when treating thrombosis [35]. Many common PDE inhibitors such as cilostazol, dipyridamole, and triple rusal are used in clinical settings to achieve increased production of cyclic nucleotides [36,37]. Therefore, there is reason to believe that G-Rk3 could provide similar therapeutic benefits.
In addition, the PI3K/Akt pathway and mitogen-activated protein kinase (MAPK) are vital phosphoproteins involved in the function of platelets such as activation, aggregation, and secretion [20,21]. According to Mei-Chi et al, phosphorylation of MAPKs such as p38 is also known to be important for the formation of TXA2, which leads to arachidonic acid release (precursor of TXA2) and platelet aggregation via phosphorylation of cyclic PLA2 [38]. TXA2 production acts as a potent autacoid that induces further platelet activation and aggregation, and is an important indicator when evaluating substances or components involved in inhibition of platelet activity [38].
In this study, G-Rk3 showed a significant concentration-dependent inhibitory effect on collagen-induced platelet aggregation. In addition, G-Rk3 decreased the phosphorylation of cyclic PLA2 in a concentration-dependent manner, suppressed the strongly increased TXA2 production into collagen, and strongly decreased the release of serotonin and ATP (intracellular granule secretion indicators). In addition, PI3K/Akt and MAPK phosphorylation were significantly inhibited by G-Rk3. G-Rk3 inhibits phosphoproteins (PI3K/Akt and MAPK) phosphorylation, reduces TXA2 production, and inhibits platelet aggregation through secretion of intracellular granules which were serotonin and ATP.
In addition, increased signaling mediated from integrin αIIb/β3 and release of granules normally alters the cytoskeleton of platelets, altering aggregation of platelets and the formation of a thrombus, which is the most important step in repairing damaged blood vessels. At this point, activated platelets accumulate and clots develop from the association of fibrin and platelets. Once a fibrin clot forms, the contraction process occurs for anywhere to 30 to 60 minutes, finally resulting in a clot plug. Contractile formation of fibrin clots is highly dependent on fibrin-one and αIIb/β3 interactions, and inhibitors of αIIb/β3 activity can slow or stop thrombus formation [39]. Coagulation and platelet αIIb/β3 activation can be induced by thrombin to increase the ability of fibrinogen to bind αIIb/β3, leading to clot formation. In this study, the antiplatelet effect of G-Rk3 inhibited thrombin-induced fibrin coagulation in a concentration-dependent manner, and this result shows that G-Rk3 has the potential as a powerful compound with antiplatelet effects that can slow or stop thrombus formation. Previous ginseng studies have reported that synthetic compounds such as 20(S)-Rg3, Ro and G-Rp1, G-Rp3 and G-Rp4, as well as trace amounts of ginsenosides, F4 and Rg6, have antiplatelet effects [22,23]. In addition, we investigated the antiplatelet mechanism of Rk1, which has a structure resembling Rk3, and revealed that Rk1 elicits Ca2+ by elevating cAMP, and inhibits platelet-mediated thrombosis by downregulating granule release and αIIb/β3 activation [40]. Through this study, it was confirmed that Rk3 also has an antiplatelet effect through a mechanism similar to that of Rk1.
In conclusion, G-Rk3 increases cAMP in human platelets, induces IP3R and VASP phosphorylation, and significantly inhibits Ca2+ recruitment and cytoplasmic activation of integrin αIIb/β3. Furthermore, G-Rk3 exhibits an antiplatelet ability by reducing TXA2 production and granule release by controlling phosphorylation of phosphoproteins acting on signal transduction such as PI3K/Akt and MAPK. Ultimately, G-Rk3 significantly inhibits the formation of thrombin-induced fibrin clots.
References
- 1.Mozaffarian D., Benjamin E.J., Go A.S., Arnett D.K., Blaha M.J., Cushman M., Das S.R., de Ferranti S., Després J.P., Fullerton H.J., et al. Heart disease and stroke statistics-2016 update a report from the American Heart Association. Circulation. 2016;133:e38–e48. doi: 10.1161/CIR.0000000000000350. [DOI] [PubMed] [Google Scholar]
- 2.Andrews R.K., Berndt M.C. Platelet physiology and thrombosis. Thromb Res. 2004;114:447–453. doi: 10.1016/j.thromres.2004.07.020. [DOI] [PubMed] [Google Scholar]
- 3.Barrett N.E., Holbrook L., Jones S., Kaiser W.J., Moraes L.A., Rana R., Wright B., et al. Future innovations in anti-platelet therapies. Br J Pharmacol. 2008;154:918–939. doi: 10.1038/bjp.2008.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Badimon L., Vilahur G., Padro T. Nutraceuticals and atherosclerosis: human trials. Cardiovasc Ther. 2010;28:202–215. doi: 10.1111/j.1755-5922.2010.00189.x. [DOI] [PubMed] [Google Scholar]
- 5.Estruch R., Ros E., Salas-Salvado J., et al. Primary prevention of cardiovascular disease with a Mediterranean diet. N Engl J Med. 2013;368:1279–1290. doi: 10.1056/NEJMoa1200303. [DOI] [PubMed] [Google Scholar]
- 6.Irfan M., Kwon T.H., Yun B.S., Park N.H., Rhee M.H. Eisenia bicyclis (brown alga) modulates platelet function and inhibits thrombus formation via impaired P2Y12 receptor signaling pathway. Phytomedicine. 2018;40:79–87. doi: 10.1016/j.phymed.2018.01.003. [DOI] [PubMed] [Google Scholar]
- 7.Rastogi S., Pandey M.M., Rawat A. Traditional herbs: a remedy for cardiovascular disorders. Phytomedicine. 2016;23:1082–1089. doi: 10.1016/j.phymed.2015.10.012. [DOI] [PubMed] [Google Scholar]
- 8.Schwarz U.R., Walter U., Eigenthaler M. Taming platelets with cyclic nucleotides. Biochem Pharmacol. 2001;62:1153–1161. doi: 10.1016/s0006-2952(01)00760-2. [DOI] [PubMed] [Google Scholar]
- 9.Cavallini L., Coassin M., Borean A., Alexandre A. Prostacyclin and sodium nitroprusside inhibit the activity of the platelet inositol 1,4,5-trisphosphate receptor and promote its phosphorylation. J Biol Chem. 1996;271:5545–5551. doi: 10.1074/jbc.271.10.5545. [DOI] [PubMed] [Google Scholar]
- 10.Laurent V., Loisel T.P., Harbeck B., Wehman A., Gröbe L., Jockusch B.M., et al. Carlier M.F. Role of proteins of the Ena/VASP family in actin-based motility of Listeria monocytogenes. J Cell Biol. 1999;144:1245–1258. doi: 10.1083/jcb.144.6.1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sudo T., Ito H., Kimura Y. Phosphorylation of the vasodilator-stimulated phosphoprotein (VASP) by the anti-platelet drug, cilostazol, in platelets. Platelets. 2003;14:381–390. doi: 10.1080/09537100310001598819. [DOI] [PubMed] [Google Scholar]
- 12.Irfan M., Jeong D., Kwon H.W., Shin J.H., Park S.J., Kwak D., et al. Rhee M.H.Ginsenoside-Rp3 inhibits platelet activation and thrombus formation by regulating MAPK and cyclic nucleotide signaling. Vascul Pharmacol. 2018;109:45–55. doi: 10.1016/j.vph.2018.06.002. [DOI] [PubMed] [Google Scholar]
- 13.Adam F., Kauskot A., Rosa J.P., Bryckaert M. Mitogen activated protein kinases in hemostasis and thrombosis. J Thromb Haemost. 2008;6:2007–2016. doi: 10.1111/j.1538-7836.2008.03169.x. [DOI] [PubMed] [Google Scholar]
- 14.Bugaud F., Nadal-Wollbold F., Lévy-Toledano S., Rosa J.P., Bryckaert M. Regulation of c-jun-NH2 terminal kinase and extracellular-signal regulated kinase in human platelets. Blood. 1990;94:3800–3805. [PubMed] [Google Scholar]
- 15.Kramer R.M., Roberts E.F., Strifler B.A., Johnstone E.M. Thrombin induces activation of p38 MAP kinase in human platelets. J Biol Chem. 1995;270:27395–27398. doi: 10.1074/jbc.270.46.27395. [DOI] [PubMed] [Google Scholar]
- 16.Nadal-Wollbold F., Pawlowski M., Lévy-Toledano S., Berrou E., Rosa J.P., Bryckaert M. Platelet ERK2 activation by thrombin is dependent on calcium and conventional protein kinases C but not Raf-1 or B-Raf. FEBS Lett. 2002;531:475–482. doi: 10.1016/s0014-5793(02)03587-1. [DOI] [PubMed] [Google Scholar]
- 17.Michelson A.D. Antiplatelet therapies for the treatment of cardiovascular disease. Nat Rev Drug Discov. 2010;9:154–169. doi: 10.1038/nrd2957. [DOI] [PubMed] [Google Scholar]
- 18.Flevaris P., Li Z., Zhang G., Zheng Y., Liu J., Du X. Two distinct roles of mitogen-activated protein kinases in platelets and a novel Rac1-MAPK-dependent integrin outside-in retractile signaling pathway. Blood. 2009;113:893–901. doi: 10.1182/blood-2008-05-155978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kramer R.M., Roberts E.F., Um S.L., Börsch-Haubold A.G., Watson S.P., Fisher M.J. Jakubowski J.A.p38 mitogenactivated protein kinase phosphorylates cytosolic phospholipase A2 (cPLA2) in thrombin-stimulated platelets. Evidence that proline-directed phosphorylation is not required for mobilization of arachidonic acid by cPLA2. J Biol Chem. 1996;271:27723–27729. doi: 10.1074/jbc.271.44.27723. [DOI] [PubMed] [Google Scholar]
- 20.McNicol A., Shibou T.S. Translocation and phosphorylation of cytosolic phospholipase A2 in activated platelets. Thromb Res. 1998;92:19–26. doi: 10.1016/s0049-3848(98)00097-8. [DOI] [PubMed] [Google Scholar]
- 21.Chuang W.Y., Kung P.H., Kuo C.Y., Wu C.C. Sulforaphane prevents human platelet aggregation through inhibiting the phosphatidylinositol 3-kinase/Akt pathway. Thromb Haemost. 2013;109:1120–1130. doi: 10.1160/TH12-09-0636. [DOI] [PubMed] [Google Scholar]
- 22.Nam K.Y. The comparative understanding between red ginseng and white ginsengs, processed ginsengs (Panax ginseng CA Meyer) J Ginseng Res. 2005;29:1–18. [Google Scholar]
- 23.Ha Y.W., Lim S.S., Ha I.J., Na Y.C., Seo J.J., Shin H., Son S.H., Kim Y.S. Preparative isolation of four ginsenosides from Korean red ginseng (steam-treated Panax ginseng CA Meyer), by high-speed counter-current chromatography coupled with evaporative light scattering detection. J Chromatogr A. 2007;1151:37–44. doi: 10.1016/j.chroma.2007.01.038. [DOI] [PubMed] [Google Scholar]
- 24.Duan Z., Deng J., Dong Y., Zhu C., Li W., Fan D. Anticancer effects of ginsenoside Rk3 on non-small cell lung cancer cells: in vitro and in vivo. Food Funct. 2017;8:3723–3736. doi: 10.1039/c7fo00385d. [DOI] [PubMed] [Google Scholar]
- 25.Qu L., Zhu Y., Liu Y., Yang H., Zhu C., Ma P., Qu L., Deng J., Fan D. Protective effects of ginsenoside Rk3 against chronic alcohol-induced liver injury in mice through inhibition of inflammation, oxidative stress, and apoptosis. Food Chem Toxicol. 2019;126:277–284. doi: 10.1016/j.fct.2019.02.032. [DOI] [PubMed] [Google Scholar]
- 26.Baek S.H., Shin B.K., Kim N.J., Chang S.Y., Park J.H. Protective effect of ginsenosides Rk3 and Rh4 on cisplatin-induced acute kidney injury in vitro and in vivo. J Ginseng Res. 2017;41:233–239. doi: 10.1016/j.jgr.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee D.H. Inhibitory effects of isoscopoletin on thrombus formation via regulation of cyclic nucleotides in collagen-induced platelets. J Appl Biol Chem. 2020;63:235–241. [Google Scholar]
- 28.Grynkiewicz G., Poenie M., Tsien R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 29.Berridge M.J., Irvine R.F. Inositol phosphates and cell signalling. Nature. 1989;341:197–205. doi: 10.1038/341197a0. [DOI] [PubMed] [Google Scholar]
- 30.Nishikawa M., Tanaka T., Hidaka H. Ca2+ -calmodulin-dependent phosphorylation and platelet secretion. Nature. 1980;287:863–865. doi: 10.1038/287863a0. [DOI] [PubMed] [Google Scholar]
- 31.Kuo J.F., Andersson R.G., Wise B.C., Mackerlova L., Salomonsson I., Brackett N.L., Katoh N., Shoji M., Wrenn R.W. Calcium¬dependent protein kinase: widespread occurrence in various tissues and phyla of the animal kingdom and comparison of effects of phospholipid, calmodulin, and trifluoperazine. Proc Natl Acad Sci. 1980;77:7039–7043. doi: 10.1073/pnas.77.12.7039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wentworth J.K., Pula G., Poole A.W. Vasodilator-stimulated phosphoprotein (VASP) is phosphorylated on Ser157 by protein kinase C-dependent and-independent mechanisms in thrombin-stimulated human platelets. Biochem J. 2006;393:555–564. doi: 10.1042/BJ20050796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Napeñas J., Oost F.C., DeGroot A., Loven B., Hong C.H., Brennan M.T., Lockhart P.B., van Diermen D.E.Review of postoperative bleeding risk in dental patients on antiplatelet therapy. Oral Surg Oral Med Oral Pathol Oral Radiol. 2013;115:491–499. doi: 10.1016/j.oooo.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 34.Gao J., Tao J., Liang W., Zhao M., Du X., Cui S., Duan H., Kan B., Su X., Jiang Z. Identification and characterization of phosphodiesterases that specifically degrade 3’3’-cyclic GMP-AMP. Cell Res. 2015;25:539–550. doi: 10.1038/cr.2015.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haslam R.J., Dickinson N.T., Jang E.K. Cyclic nucleotides and phos- phodiesterases in platelets. Thromb Haemost. 1999;82:412–423. [PubMed] [Google Scholar]
- 36.Quinton T.M., Dean W.L. Cyclic AMP-dependent phosphorylation of the inositol-1,4,5-trisphosphate receptor inhibits Ca2+ release from platelet membranes. Biochemical and Biochem Biophys Res Commun. 1992;184:893–899. doi: 10.1016/0006-291x(92)90675-b. [DOI] [PubMed] [Google Scholar]
- 37.Menshikov M.Y.U., Ivanova K., Schaefer M., Drummer C., Gerzer R. Influence of the cGMP analog 8-PCPT-cGMP on agonist-induced increases in cytosolic ionized Ca2+ and on aggregation of human platelets. Eur J Pharmacol. 1993;245:281–284. doi: 10.1016/0922-4106(93)90108-l. [DOI] [PubMed] [Google Scholar]
- 38.Cipollone F., Patrignani P., Greco A., Panara M.R., Padovano R., Cuccurullo F., Patrono C., Rebuzzi A.G., Liuzzo G., Quaranta G., et al. Differential suppression of thromboxane biosynthesis by indobufen and aspirin in patients with unstable angina. Circulation. 1997;96:1109–1116. doi: 10.1161/01.cir.96.4.1109. [DOI] [PubMed] [Google Scholar]
- 39.Calderwood D.A. Integrin activation. J. Cell Sci. 2004;117:657–666. doi: 10.1242/jcs.01014. [DOI] [PubMed] [Google Scholar]
- 40.Shin J.H., Kwon H.W., Irfan M., Rhee M.H., Lee D.H. Ginsenoside Rk1 suppresses platelet mediated thrombus formation by downregulation of granule release and αIIbβ3 activation. J Ginseng Res. 2021;45:490–497. doi: 10.1016/j.jgr.2020.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]