

Abstract
Autophagy is a cellular process with important functions that drive neurodegenerative diseases and cancers. Lysosomal hyperacidification is a hallmark of autophagy. Lysosomal pH is currently measured by fluorescent probes in cell culture, but existing methods do not allow for quantitative, transient, or in vivo measurements. Here, we developed near-infrared optical nanosensors using organic color centers (covalent sp3 defects on carbon nanotubes) to measure autophagy-mediated endolysosomal hyperacidification in live cells and in vivo. The nanosensors localize to the lysosomes, where the emission band shifts in response to local pH, enabling spatial, dynamic, and quantitative mapping of subtle changes in lysosomal pH. Using the sensor, we observed cellular and intratumoral hyperacidification upon administration of mTORC1 and V-ATPase modulators, revealing that lysosomal acidification mirrors the dynamics of S6K dephosphorylation and LC3B lipidation while diverging from p62 degradation. This sensor enables the transient and in vivo monitoring of the autophagy-lysosomal pathway.
Introduction
Lysosomes have emerged as essential signalling centres that govern cell growth, division, and differentiation, as well as catabolic activity.1 Dysregulated lysosomal function affects cell survival, invasion, immune evasion, and drug resistance. Lysosomes play a large role in autophagy, a process that controls cellular homeostasis and the progression of many diseases. The impact of autophagy on cancers is heterogeneous.2,3 While autophagy increases immunosurveillance that resses tumor initiation, it also sustains cellular homeostasis and prevents chronic cellular damage in tumors. In ovarian carcinoma, for example, autophagy-related proteins are downregulated, and autophagy activation can result in the death of these cancer cells.4,5
Autophagic processes within tissues and whole organisms exhibit substantial complexities beyond those found in cell culture studies.3,6,7 For instance, autophagy can be activated by hypoxic stress in the tumor microenvironment.8 The role of autophagy differs throughout tumor development in response to dynamic changes in stresses such as nutrient deprivation, hypoxia, and growth factor depletion in the microenvironment at different stages of progression.9,10 Autophagy-mediated degradation of immune signaling proteins helps tumors escape from both adaptive and innate immune system surveillance.11 Thus, elucidation of the role of autophagy on cell signaling, dynamically and in vivo, would facilitate greater understanding of cancer progression and other disease processes. Autophagic flux is typically assessed by biochemical assays or fluorescent protein-tagging of autophagy-related ubiquitin like protein ATG8 family members such as LC3B, autophagy substrates such as p62/SQSTM1, or other organellar markers in cells.12 Assessments of autophagy and related processes in organisms generally require tissue acquisition at selected endpoints7 or deletion of genes essential for the ATG8 conjugation pathway, such as ATG7 or ATG5.13,14 The knockout of ATG7 or ATG5 in mice causes defect in the autophagosome formation, thus, has been used to understand the physiological roles of autophagy. However, a permanent depletion of the autophagic pathway leads to neonatal lethality, organ failure, and neurological dysfunction.15,16 Alternatively, transgenic mice with systemic expression of GFP-tagged LC3B were used to monitor autophagy in vivo.14 Therefore, methods enabling dynamic and in vivo measurements of autophagy, that do not require alterations in host tissues, would provide important complementarity to facilitate mechanistic investigations and experimental therapeutics studies.
During the course of (macro)autophagy, a fusion event between lysosomes and autophagosomes exposes acid hydrolases to the autophagic cargoes for degradation. Acidic pH, generated by vacuolar H+ ATPase (V-ATPase), is essential to facilitate lysosomal proteolysis and recycle damaged lysosomes, enhancing autophagic efficiency and lysosomal biogenesis. Lysosomal hyperacidification, subtle reductions in pH below the normal acidic levels of late endosomes/lysosomes, is regarded as a hallmark of autophagy. The mechanism of lysosomal hyperacidification is not fully understood.17 There is a dearth of robust tools to quantify lysosomal pH in live cells and in vivo, however.18 Existing lysosomal pH reporters include pH-sensitive microelectrodes,19 organic dyes,20,21 synthetic nanoparticles,22 and pH-sensitive fluorescent proteins23. Intracellular pH recordings using microelectrode tips are considered label-free and enable high temporal resolution. However, the spatial resolution is limited by the size of the microelectrode tip, and the measurement may disrupt cellular processes due to membrane puncturing. On the other hand, the most common method, fluorescent dyes, exhibit a time-dependent alkalizing effect on lysosomes,20,24 making temporal tracking of lysosomal pH, and in vivo use, challenging. Also, most optical probes are excited and emit at visible (380–700 nm) and first near-infrared (NIR-I, 700–900 nm) wavelengths where they exhibit phototoxicity25,26 and shallow tissue penetration due to significant tissue absorption and light scattering, and overlap with autofluorescence, limiting their use for in vivo imaging.
Semiconducting single-walled carbon nanotubes (SWCNTs) exhibit electronic and optical properties that enable intracellular and in vivo imaging and sensing. SWCNT fluorescence exhibits narrow bandwidths (35–80 meV), allowing sensitive and precise quantification of local environments via quenching and solvatochromic responses and modulation of emission from quantum defect sites.27,28 SWCNTs fluoresce in the near-infrared window (NIR-II, 900–1700 nm),29 where emission can penetrate living tissues to distances in the centimetre range with minimal light scattering and tissue absorbance.30 Tissue autofluorescence drastically decreases at NIR-II wavelengths, improving contrast for in vivo imaging. SWCNT fluorescence () is highly photostable31 and can be imaged in live cells, tissues, and animals over long periods.31–33 Non-covalent encapsulation with polymers, including short oligonucleotides, facilitates aqueous suspension and confers colloidal stability and biocompatibility in biological systems.34 Organic color centers (OCCs) are molecularly tunable quantum defects on SWCNTs35 that harvest mobile excitons to produce distinct, bright fluorescence bands at longer wavelengths from the peak. The fluorescence of the OCC-functionalized-SWCNTs (OCC-SWCNTs) introduces new biochemical sensitivities to SWCNTs determined by the chemical nature of the defect, making OCCs the molecular focal points for local environmental responses.36
Herein, we developed an optical nanosensor to measure autophagy-mediated lysosomal hyperacidification events, quantitatively and temporally in live cells and in vivo. The sensors are composed of the N,N-diethyl-4-aminoaryl OCC modification of a SWCNT, encapsulated with an oligonucleotide. The nanosensor localized to the endolysosomal lumen where the emission of the OCCs responded via quantifiable wavelength shifts of the band in response to local pH, while the band remained largely stable, functioning as a reference. The nanosensor measured lysosomal pH changes upon treatment with V-ATPase targeting drugs and autophagy modulators in real-time. Using near-infrared hyperspectral imaging, the nanosensor enabled the acquisition of quantitative maps of endolysosomal pH spatially, allowing direct comparisons between different cell types and experiments. We found that the nanosensor enables transient, spatial, and non-invasive monitoring of pH, including subtle autophagy-mediated hyperacidification, within tumors in live mice. Monitoring the intratumorally-injected nanosensors enabled dynamic tracking of lysosomal pH upon pharmacologic modulation. The sensor revealed that lysosomal acidification mirrors dephosphorylation of S6 and lipidation of LC3B, exhibiting a sharp recovery after perturbation, while acidification diverged substantially from p62 degradation. These results suggest that pH temporally tracks mTORC1 inhibition in the tumor microenvironment. This sensor technology constitutes a quantitative tool for discovery and preclinical investigations of lysosomal biology, autophagy, and experimental therapeutics.
Results
OCC-DNA optical response to lysosomal pH
We developed pH-sensitive OCC-DNA complexes (Fig. 1a) and assessed their optical response. We covalently functionalized (6,5)-SWCNTs with an N,N-diethyl-4-aminobenzene diazonium salt in aqueous solution.36 Upon successful functionalization, the fluorescence spectrum of OCC-SWCNTs exhibited a pair of emission peaks: The nanotube host emission () centered at 993 nm, and the “OCC peak” at 1149 nm (Supplementary Fig. 1). The peak traced the titration curve of the aminobenzene group, shifting by 12 nm, while wavelength was nearly unchanged (< 3 nm) in the pH range of 3–8 (Fig. 1b). The emission response is attributed to modulation of electronic resonance and inductive effects resulting from protonation of the amine group of the OCC.37
Fig. 1 |. Synthesis of pH-responsive OCC-DNA complexes.
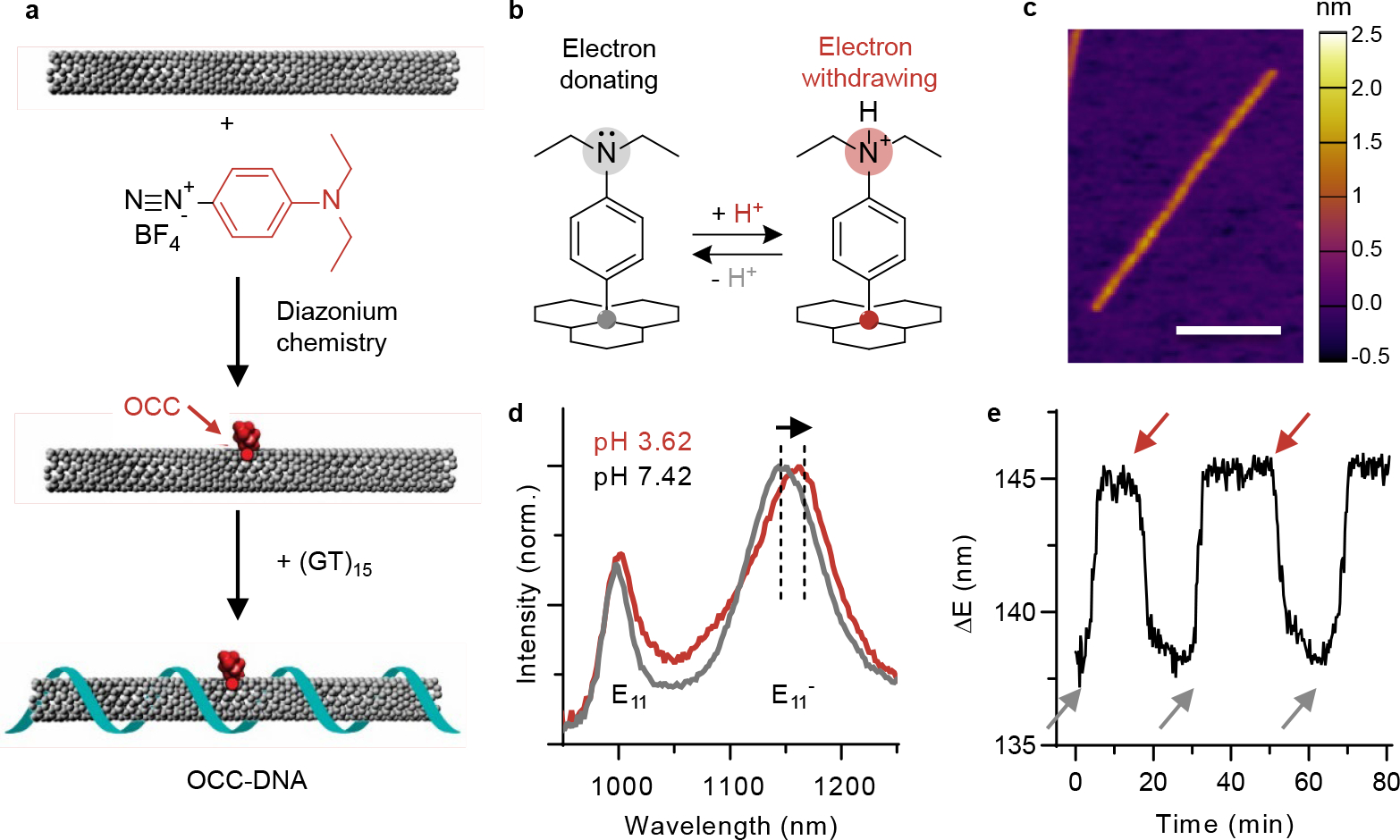
a, Schematic of the synthesis of OCC-DNA complexes. b, Schematic of the molecular mechanism of response that involves mechanism - protonation and deprotonation of the N,N-diethylamino moieties group on the aryl OCC. c, AFM image of OCC-DNA complex. Scale bar = 100 nm. Color map denotes height. d, Representative near-infrared fluorescence spectra of OCC-DNA complexes in PBS at pH 3.14 (red) and pH 7.02 (gray) at 575 nm excitation. The arrow indicates the redshift of centre wavelength in acidic buffer solution. e, Dynamics of OCC-DNA response and reversal in buffer at pH = 7.09 (gray arrows) and 3.32 (red arrows) at 575 nm excitation. The spectral shift is as measured from the emission peak centre wavelengths.
We wrapped the OCC-SWCNTs with a single-stranded oligonucleotide with the sequence (GT)15, known to confer colloidal stability and biocompatibility even in the lysosome38 (see Methods and Supplementary Fig. 2). Successful DNA wrapping was confirmed by periodic DNA pattern along the SWCNT axis, observed via atomic force microscopy (AFM, Fig. 1c). Both the optical and chemical properties (quantum yield and pH responsivity) of OCC-SWCNTs were preserved after the DNA wrapping process (Supplementary Fig. 3).
The optical properties of the resulting OCC-DNA complexes were characterized by fluorescence spectroscopy. Upon wrapping the OCC-SWCNTs with ss(GT)15, the and emission peaks red-shifted by 6 nm, consistent with a shift in the absorbance band (Supplementary Fig. 4). We assessed the response of the OCC-DNA complexes to pH by observing the emission peak wavelength shift in various buffer/media conditions with different pH values (Fig. 1d, Extended Data Fig. 1). The variation of shifts in triplicate measurements was small. The maximum variation between repeat measurements of the same sample was less than 0.1 nm, and small spectral shifts of less than 0.5 nm were easily identifiable. Multivalent metal ions, lipoproteins, and small metabolites slightly shifted the OCC-DNA fluorescence wavelengths, but the pH sensitivity in the presence of these interferents remained consistent. Exposure to high concentrations of reactive oxygen species (ROS), including hydrogen peroxide, superoxide radical anion, or hydroxy radical, irreversibly inactivated the OCC-DNA complexes. This is attributed to the fact that 4-N,N-diethylaminoaryl OCC reacts with ROS to form tertiary amine oxide that does not significantly modulate the electronic energy level of the defect state upon protonation. In 20% fetal bovine serum (FBS), both and emission peaks red-shifted approximately 1–2 nm, comprising a change in the baseline of the sensor but without changing the dynamic range of the pH response. This phenomenon is attributed to the exposure of the SWCNT to the electrostatic charges of proteins upon their adsorption to the SWCNT sidewall.39
The pH-responsive dynamic range of the OCC-DNA fluorescence depended on the protein composition and density (Extended Data Fig. 2), but the emission wavelength still responded quantitatively to solution pH within the physiologic pH range (pH 3–7.4). While peak wavelength exhibited < 3 nm shifts upon varying pH from 7.4 to 3.5, the wavelengths red-shifted as high as 12 nm in protein-free buffer solutions. The dynamic range and wavelength shifts were independent of solution viscosity (Extended Data Fig. 3). To account for these protein-related effects, we used the emission peak wavelength as an internal reference of the response. We thus reported the relative spectral shift of the OCC band, as the ultimate readout of pH response.
We then assessed the kinetics and reversibility of the OCC-DNA responses in PBS at pH values of 3.32 and 7.09 (Fig. 1e). The OCC-DNA complexes were loaded into a semipermeable polyvinylidene fluoride membrane capillary with a 500 kDa cutoff. The membrane was immersed in the buffer solution, and the solution pH was modulated by alternatively exchanging acidic and basic buffer solutions every 20 minutes. The OCC-DNA emission through the membrane was continuously monitored. The exhibited immediate and reversible changes upon pH modulation.
To assess long-term functionality in biological conditions, we incubated OCC-DNA complexes in 20% FBS at 37°C for a week and compared the responsivity with fresh complexes (Supplementary Fig. 5). Although the intensity reduced by >80%, the pH sensitivity and peak wavelengths remained the same. The results suggest that the pH-responsive amine group of the OCCs does not easily degrade in biological environments.
To investigate the biocompatibility of the OCC-DNA complexes, we assessed dose-dependent cell viability. We used a human ovarian cancer cell line, SKOV3, to test the cell viability at a range of OCC-DNA concentrations (0–0.08 mg/L, Fig. 2a). The OCC-DNA complexes, incubated at up to 0.05 mg/L for three days, did not affect cell viability. We used 0.0015–0.01mg/L as the working concentrations based on signal intensity in cells (Fig. 2b). At this concentration range, we observed OCC-DNA emission from individual puncta of the live cells in the hyperspectral imaging cube, suggesting that it is possible to obtain spatially-resolved pH information. We did not observe rotational or tumbling motions of individual OCC-DNA complexes in the NIR movies of OCC-DNA complexes in live cells, suggesting that each punctum likely contained more than one OCC-DNA (Supplementary Video 1). We tested cell viability at the highest working concentration (0.01 mg/L) in several more cell lines, including human epithelial ovarian carcinoma (OVCAR3 and OVCAR8), human epithelial adenocarcinoma (HeLa), human epithelial kidney (HEK293T), mouse embryonic fibroblast (MEF), and mouse prostate cancer (RM1 and Myc-CaP) cells, and no cytotoxicity was observed (Extended Data Fig. 4). We also confirmed that 0.01 mg/L of OCC-DNA treatment did not induce NLRP3 inflammasome signaling (Supplementary Fig. 6).
Fig. 2 |. OCC-DNA complexes respond to endolysosomal pH.

a, Viability of SKOV3 cells at increasing concentrations of OCC-DNAs, measured via CellTiter-Glo 2.0, after 72 hours of incubation. N=3. The data are presented as mean values with error bars as standard deviation. The experiment was performed at least three times with comparable results. b, Overlay of near-infrared emission of the OCC-DNA complexes from individual puncta of the live cells at 730 nm excitation and the transmitted light image. Arrows mark the 12 cells used for the OCC-DNA response analysis in Fig. e. Scale bar is 50 μm. c, Representative confocal microscopy images of Cy5-labelled OCC-SWCNT complexes (red) and LysoTracker Green (green) lysosomal imaging dye in live SKOV3 cells. Scale bar is 50 μm. d, OCC-DNA response in SKOV3 cells modulated by HEPES or MES buffer with monensin, measured under 730 nm excitation. Data are presented as mean values with error bars and error bands as standard deviation. N=25. e, OCC-DNA response in each cell upon exposure to buffer solutions of varying pHs with monensin. Each line denotes emission response obtained from 12 individual cells. Gray error band indicates the standard deviation of the sensor responses.
We then investigated where the OCC-DNA complexes localized within live cells. We encapsulated OCC-SWCNTs with Cyanine5 (Cy5)-labelled-ss(GT)15 and treated the cells with 0.01 mg/L of the OCC-DNAs for 16 hours. In the confocal microscopy imaging, we observed that the Cy5 emission colocalized with the fluorescence of LysoTracker Green, which stains acidic compartments in cells (Fig. 2c). Quantitative colocalization analysis found a Manders’ overlap coefficient between Cy5 and LysoTracker Green of 0.893 (±0.036), and the fraction of Cy5 emission overlapping LysoTracker emission was 0.988. These data suggest that the overwhelming majority of OCC-DNA complexes were localized to the lysosomes. Based on the similar colocalization behaviour of OCC-DNAs to un-derivatized DNA-SWCNTs (described extensively in Jena et al.32), we concluded that the OCC-DNA complexes behaved similarly to DNA-SWCNTs with respect to localization to the endolysosomal lumen. A significant reduction in cellular uptake of OCC-DNAs incubated at 4°C, as compared to 37°C, suggests an energy-dependent, endocytic internalization mechanism (Supplementary Fig. 7). These similarities are anticipated based on the minor changes in the overall nanotube surface chemistry by the minimal functionalization with OCCs (one defect per 20 nm of SWCNT length).
To investigate lysosomal integrity in the presence of OCC-DNA complexes, we assessed cell viability with a lysosomotropic agent, L-Leucyl-L-Leucine methyl ester (LLOMe). No significant changes in LLOMe-dose-dependent viability were observed between control and OCC-DNA-treated cells (Supplementary Fig. 8). In addition, the treatment of OCC-DNA complexes did not induce NLRP3 inflammation–downstream signalling of cathepsin release to cytosol by rupture and permeabilization of lysosomal membranes40 (Supplementary Fig. 6). The median length of the OCC-DNA complexes was 252 nm (Supplementary Fig. 9), and lysosomes measure 0.1–1.2 μm in diameter.41,42 However, endocytosis pathways can preferentially take up SWCNTs of certain sizes and largely reject particles that are too large for endosomes.43 The results also agree with previous studies with DNA-SWCNTs32, showing that they do not perturb the endolysosomal limiting membrane.
We then studied the effects of the OCC-DNAs on lysosomal function. We used the dye quenched-bovine serum albumin (DQ-BSA) assay, which measures lysosomal protein degradation activity. The assay confirmed that the OCC-DNA treatment did not alter lysosomal enzymatic function (Supplementary Fig. 10).
We measured the pH-induced peak shifts of the OCC-DNAs in live cells using near-infrared fluorescence spectroscopy (Supplementary Fig. 11). We treated SKOV3 cells with OCC-DNAs overnight and replaced with fresh media 4 hours before the spectroscopy measurement. The cells were then incubated in HEPES or MES buffer with 10 μM of monensin (an ionophore that permeabilizes the lysosomal membrane and equilibrates the pH throughout the cell) in the pH range of 3–7.4 for 10 minutes before acquiring fluorescence spectra of ensemble emission of the OCC-DNA complexes within the live cells at 575 nm excitation (Fig. 2d). A gradual increase in was observed as the cells were exposed to acidic buffer solutions. We also tracked the sensor response at the individual cell level at varying buffer pH conditions using hyperspectral near-infrared fluorescence microscopy44. The emission analysis showed that the shift and the dynamic range of pH responsivity were consistent with the spectral measurements of Fig. 2d (Fig. 2e).
To evaluate the pH response of OCC-DNAs on permeabilized lysosomes, we obtained OCC-DNA responses upon 2-hour treatment of 10mM LLOMe in SKOV3 cells (Supplementary Fig. 12). The pH responsivity in the presence/absence of LLOMe remained the same. The lysosomal pH basified by 1.30 pH units in OCC-DNA/LLOMe co-treated cells, suggesting that permeabilization by LLOMe basifies lysosomes close to cytosolic pH, and treatment with OCC-DNA complexes alone maintains the lysosomal pH in the expected range45.
We examined the OCC-DNA response to pH across different cell lines. We found that, although the wavelength shifts were qualitatively consistent, the dynamic range differed to some extent by cell type (Extended Data Fig. 5). Therefore, a specific titration curve for each cell line was used to interpolate the estimated lysosomal pH in all live cell imaging and screening experiments. Based on the above experiments, we concluded that the OCC-DNA complexes constituted a sensor with the parameters defined by these experiments. We thus called the complexes a “sensor” or “nanosensor(s)” in the following studies.
pH response to modulation of V-ATPase and mTORC1 activity
To measure lysosomal pH under pharmacologic perturbations, we assessed the nanosensor response in cells treated with V-ATPase modulators bafilomycin A1 (Baf A1) and EN6. V-ATPase is a proton pump directly responsible for maintaining acidic pH in lysosomes. Baf A1 is a V-ATPase inhibitor that increases the lysosomal pH and is commonly used to block the fusion between autophagosomes and lysosomes.46,47 EN6 covalently binds the ATPV1A subunit, which decouples the V-ATPase from Ragulator complex, a pentameric protein complex that recruits mTORC1 to the lysosomal surface for activation. This leads to the inhibition of mTORC1 signaling and additionally increases the catalytic activity of V-ATPase.48 V-ATPase plays a vital role in Rag-mTORC1-related autophagy activation.17 Rag family proteins are direct recruiters for mTORC1 on the lysosome surface and control the activation state of mTORC1.17 We first confirmed that the nanosensors remained inside the lysosomes regardless of the drug treatment and can report drug-induced endolysosomal pH changes (Supplementary Fig. 13). Colocalization analysis indicates that the average fraction of nanosensor emission overlapping emission from the lysosomal dye, LysoTracker Green does not significantly varies between treatment conditions.
We acquired the sensor responses within SKOV3 cells under 100 nM Baf A1 and 100 μM EN6 treatments using near-infrared hyperspectral microscopy. The sensor emission spectra from individual cells showed blue-shifted peak wavelengths for Baf A1 and red-shifted peak wavelengths for EN6, compared to the control (Fig. 3a). The spatially-resolved emission from the nanosensors facilitated the production of quantitative, live-cell maps of lysosomal pH (Fig. 3b). In SKOV3 cells, the mean of the distribution decreased by 0.6 nm under Baf A1 treatment and increased 0.5 nm upon EN6 treatment, indicating lysosomal basification from pH 5.15 to pH 6.15 and acidification to pH 4.35 in each treatment condition, respectively (Fig. 3c).
Fig. 3 |. Nanosensor response under modulation of V-ATPase in live cells.

a, Near-infrared fluorescence emission spectra of regions of interest (ROIs) in the images in panel b. Each ROI is marked with dashed circle. b, Maps of the nanosensor pH response in SKOV3 cells, as measured by near-infrared hyperspectral microscopy at 730 nm excitation, overlaid onto brightfield images 4 hours after the introduction of DMSO (control), EN6 (100 μM), or Baf A1 (100 nM). Scale bar is 50 μm. c, Histogram of from all pixels with sensor emission from the hyperspectral images of cells in Fig. b. d, Lysosomal pH changes estimated from values from fluorescence spectroscopy measurements after 4-hour drug treatment in 7 different cell lines. N = 24, 10, 22, 19, 24, 5, and 5 spectra acquired for each condition, from the left to right cell lines. The bars are presented as mean values with error bars as standard deviation.
Via near-infrared fluorescence spectroscopy of wells in a 96-well plate, we measured the ensemble fluorescence emission of the sensors from within adhered cells. After 4-hour treatment with 100 μM of EN6, we observed a 0.8 nm increase in , corresponding to lysosomal acidification from pH 4.93 to 3.38 (Fig. 3d). Conversely, 100 nM Baf A1 resulted in a 1.22 nm attenuation of , corresponding to the lysosomal basification to pH 6.51. Using the DQ-BSA assay, we found elevated and reduced enzymatic activities of lysosomes upon EN6 and Baf A1 treatments, respectively (Supplementary Fig. 14), as expected.49,50 We also measured the sensor emission in 6 additional cell lines under 4-hour treatments of EN6 and Baf A1. The and lysosomal pH responses were qualitatively consistent, further validating the sensor functionality in live cells.
We used the nanosensor to dynamically measure pH changes upon pharmacologic perturbation. Continuous monitoring of the sensor emission resulted in transient quantification of lysosomal pH in SKOV3 cells (Fig. 4a). Baf A1 caused immediate lysosomal basification, which equilibrated before the first data point at 15 minutes, consistent with the known mechanism of action.46,47 The introduction of EN6 to the media resulted in acidification of the lysosomes, which began within minutes. This rapid response suggests that EN6 induces lysosomal pH changes in these cells due to the direct modulation of catalytic efficiency of V-ATPase rather than the modulation of V-ATPase expression levels by the mTORC1 inhibition and TFEB translocation, which only becomes evident after several hours.51
Fig. 4 |. Dynamic response to V-ATPase and mTORC modulation in live cells.
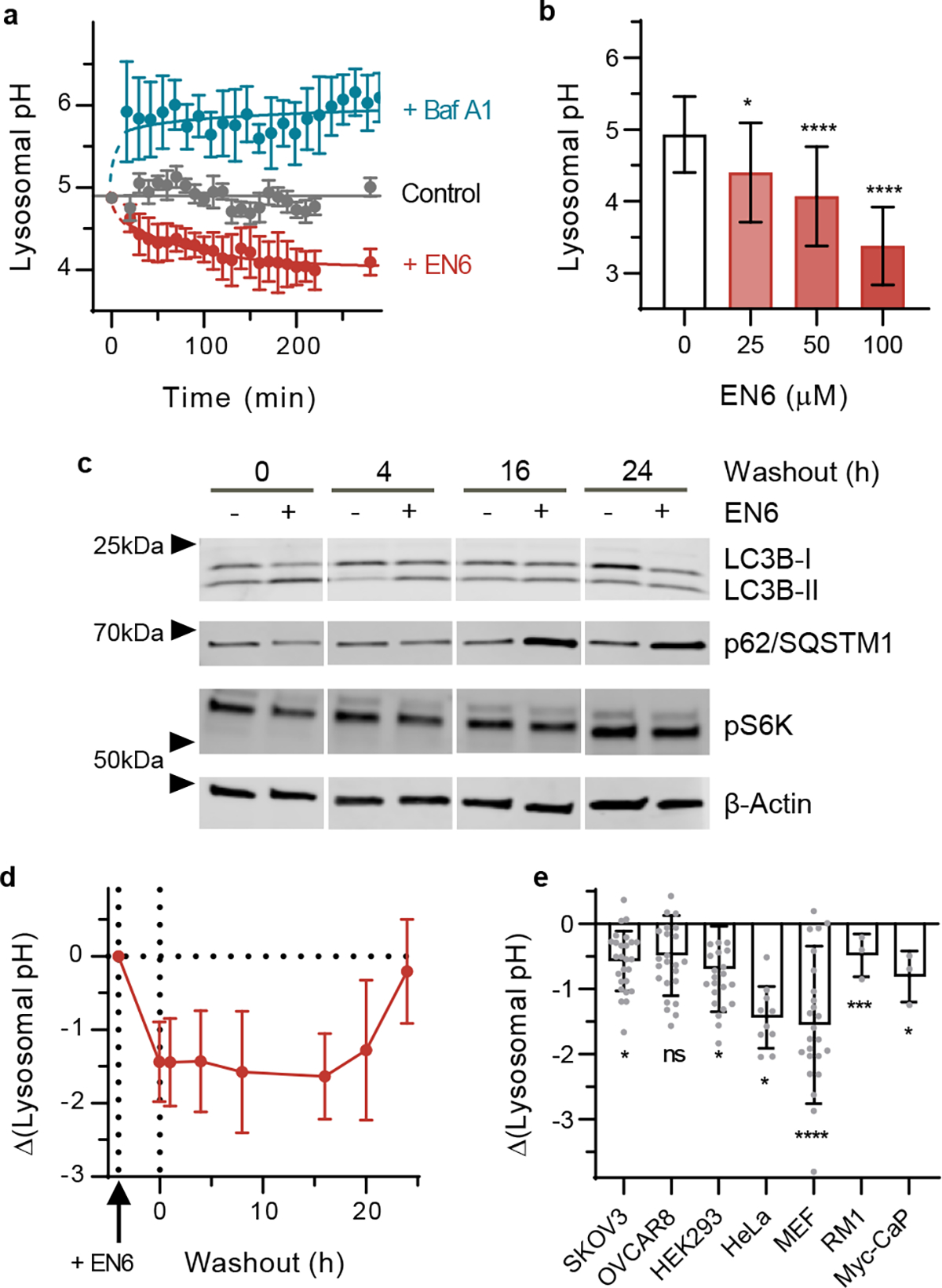
a, Time-dependent lysosomal pH in response to EN6 (red), Baf A1 (blue) or control (black) treatment conditions in SKOV3 cells. N=3 spectra. b, Dose-dependent lysosomal pH response in SKOV3 cells upon 4 hours of treatment with EN6. * = 0.0147, **** < 0.0001. N=20 for DMSO and 25 for EN6 treatments. c, Time-dependent expression of autophagy and mTORC signalling effectors in SKOV3 cells upon introduction and wash-out of EN6. Western blotting was performed at least three times with comparable results. d, Time-dependent pH response of nanosensors in SKOV3 cells to 100 μM EN6 treatment for 4 hours and wash-out. N=25. e, Change in lysosomal pH upon 4-hour treatment of 250 nM torin 1, with respect to DMSO control. N = 26, 25, 24, 10, 26, 3, and 3, from left to right cell lines. *: 0.005< p <0.05, ***: p = 0.0005, ****: p < 0.0001 All data are presented as mean values with error bars as standard deviation (a,b,d,e), and statistical significance was analysed using a one-way ANOVA followed by Dunnett’s multiple comparison test (b,e). All experiments were performed at least three times with comparable results.
We studied the relationship between EN6-induced lysosomal acidification and autophagy flux in SKOV3 cells. A dose-dependent increase in autophagy activation was observed upon 4-hour treatment with EN6 via Western blot (Supplementary Fig. 15). Upon increasing concentrations of EN6, the level of phosphorylated canonical substrate S6 kinase 1 (pS6K) gradually decreased, indicating the inactivation of mTORC1 signaling. The number of autophagosomes and autophagic flux increased, evidenced by the increased levels of conjugated LC3B-II and degradation of p62/SQSTM1 (sequestosome 1). We observed dose-dependent lysosomal acidification as measured by the nanosensor (Fig. 4b). The pH response was consistent with reports of lysosomal acidification concomitant with autophagy activation.48
We further interrogated the dynamics of the V-ATPase-specific autophagy activator, EN6, in SKOV3 cells. We conducted a washout experiment to assess both EN6-induced autophagy activation and lysosomal pH modulation and reversal. The cells were exposed to 100 μM of EN6 for 4 hours, then washed out and grown in fresh media free of EN6. Autophagy markers were measured after washout. We observed that LC3B-II and p-S6K returned to the untreated levels after 16 hours, while V-ATPase expression did not change in response to the drug. (Fig. 4c and Supplementary Fig. 16). In parallel, we measured the nanosensor emission to estimate the lysosomal pH at each time point (Fig. 4d). The lysosomes maintained a pH of approximately 3.75 up to 16 hours, before starting to recover the basal lysosomal pH of 4.93, which fully returned to baseline levels 24 hours post-washout. The prolonged effects of lysosomal acidification and autophagy activation were attributed to the irreversible nature of the covalent modulator on the target. Additionally, even 16 hours after washout, we observed persistent effects of the EN6. We surmise that EN6-bound V-ATPase gradually turned over in 16 hours; newly synthesized V-ATPases started to restore the lysosomal pH, and mTORC1 was recruited back to the lysosomes. We also observed that the levels of p62/SQSTM1 decreased after EN6 washout and were restored after 4 hours but then increased after 16 hours, likely due to the feedback effects on TFEB.52 TFEB directly regulates p62/SQSTM1, whose expression level increases similar to cells under prolonged starvation since covalently bound-EN6 continued to inhibit mTORC1 even after washout.
We investigated lysosomal pH modulation due to mTORC1/2 inhibition by torin 1. Torin 1 is a potent autophagy activator that directly competes with the ATP binding pocket in mTORC1/2.53 We observed lysosomal acidification within 4 hours of torin 1 treatment at a concentration of 250 nM, where the degree of acidification was cell-type dependent (Fig. 4e). Consistent with the previous reports, torin 1 acidified the lysosomal pH in HeLa and MEF cells. The prostate cancer cell lines, Myc-CaP and RM1, human ovarian cancer cells, SKOV3 and OVCAR8, and HEK293T cells exhibited relatively small decreases in lysosomal pH. Concomitantly, autophagy markers confirmed autophagy activation upon torin 1 treatment (Supplementary Fig. 17). The results suggest that divergent responses of lysosomal activity upon autophagy activation may explain the heterogeneous sensitivities of autophagic processes in different cell types and the time course of autophagy can be monitored by the nanosensors.
We assessed inhibitor-mediated modulation of the nanosensor response. The emission wavelength shifted, but the dynamic range remained the same, when cells were treated with torin 1 but not with the other inhibitors (Extended Data Fig. 6). We thus applied a new calibration curve to correct the lysosomal pH change upon torin 1 treatment. We hypothesized that the torin 1-mediated OCC-DNA response derived from autophagy activation-induced accumulation of proteins in the lysosome, which influenced the local electrostatic environment in the lysosomes, and thus the optical bandgap of SWCNTs39, as described heretofore (Extended Data Fig. 2). We thus assessed torin 1-induced shifting of the nanosensor response in autophagy-defective ATG7−/− HEK293T cells (Extended Data Fig. 7). We found that knocking out ATG7 abrogated the differences in OCC-DNA response. As ATG7 knockout prevents mTORC1/2 inhibition-mediated autophagy activation and concomitant changes in lysosomal composition1,17, the results suggest that a large influx of cargo proteins can cause modulation of the OCC-DNA wavelength.
In vivo, quantitative, dynamic imaging of pH response
We investigated the nanosensor in vivo using a xenograft model of ovarian cancer. Nanosensors were injected intratumorally into SKOV3 tumors when they reached 120 mm3 (Fig. 5a). Emission spectra of the sensors were obtained starting 24 h after injection using a near-infrared hyperspectral mouse imager (Supplementary Fig. 18). Strong nanosensor fluorescence was detected within tumors (Fig. 5b), which gradually decreased by 35% after three days (Extended Data Fig. 8). We note that the intensity attenuation was not statistically significant during the measurements. Immunofluorescence imaging of lysosome-associated membrane glycoprotein 1 (LAMP1) was conducted in Cy5-labeled, nanosensor-injected tumor slices (Fig. 5c). Adjacent, 5-micron sections were required for tissue imaging, due to the need to protect the labeled nanosensor emission during the staining protocol. Quantitative analysis revealed that the fraction of Cy5 (nanosensor) emission co-localized with LAMP1 signal was 0.715. A small fraction of Cy5 signal (0.083) was adjacent to LAMP1 emission but not overlapping. Cy5 co-localized with DAPI to a small degree (0.210), and a minor fraction of LAMP1 emission overlapped with DAPI (0.202) as well. We believe that the 5-micron thickness of the slices led to unexpected colocalization between DAPI and LAMP1/Cy5 and the limited z-resolution, which were not of concern in the live cell colocalization study (Fig. 2c). We did not find significant Cy5 emission in the extratumoral space. We, therefore, conclude that the nanosensors localized largely to late endosomes or lysosomes in the tumor cells in vivo. The haematoxylin and eosin (H&E)-stained tissues were assessed by a trained pathologist; no signs of injury or other abnormalities were found in the tumor tissues upon intratumoral injection of the sensor (Supplementary Fig. 19).
Fig. 5 |. In vivo nanosensor response to V-ATPase and mTORC modulation.
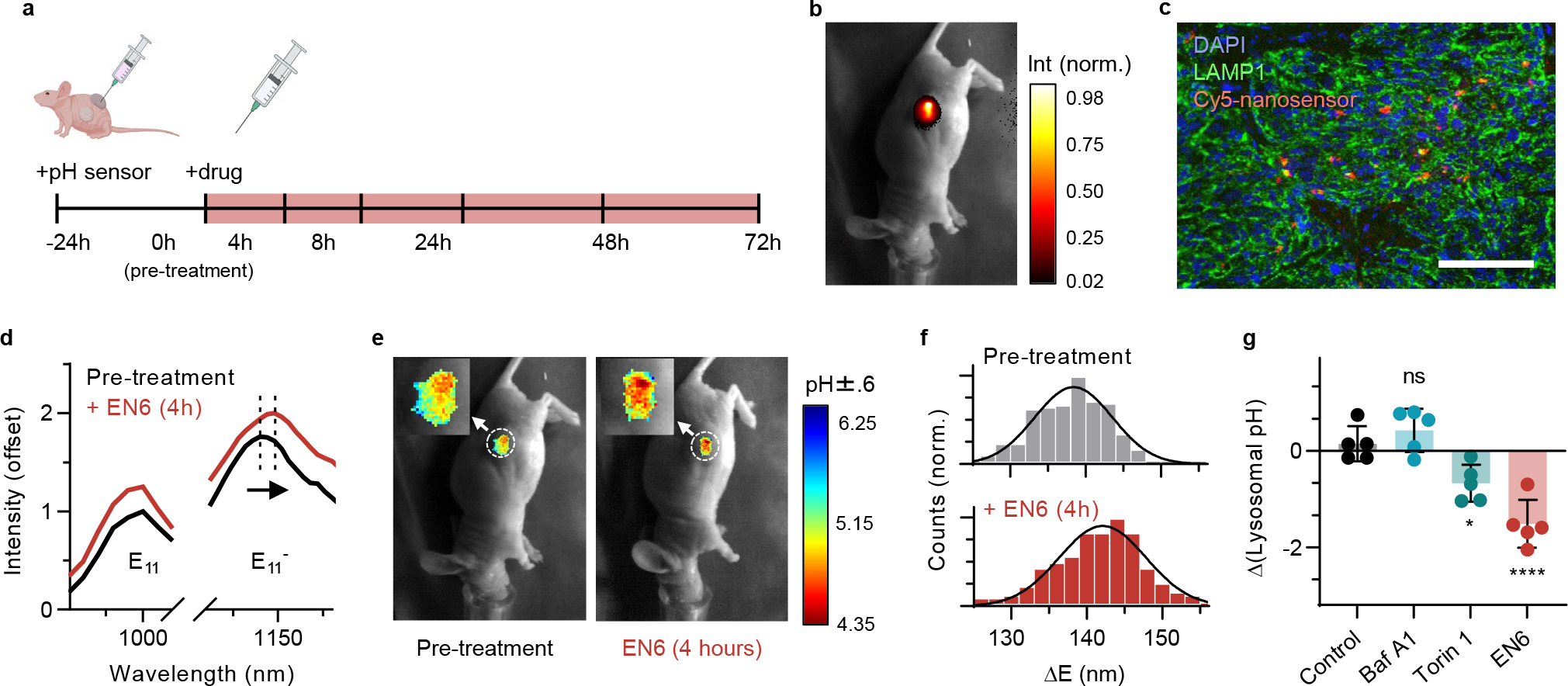
a, Experimental scheme used in Fig. 5–6. b, Overlay of transmitted light and near-infrared fluorescence emission of the nanosensors in vivo in a mouse with SKOV3 tumour 24 hours after the sensor injection (30 μL of the nanosensors in PBS, 0.1 mg/L). c, Overlay of the Cy5-labeld nanosensors (red), LAMP1 (green), and DAPI (blue) from a tumour slice 24 hours after the sensor injection. Scale bar is 100 μm. d, Averaged sensor emission from the mouse tumour before and after EN6 treatment at 808 nm excitation. The selected ROIs are marked with dashed circle in Fig. e. e, Overlay of transmitted light and hyperspectral wavelength analysis images in vivo before (left) and after EN6 treatment (right) in the same mouse as in panel b. f, Histogram of from all pixels with sensor emission from the hyperspectral images of solid tumours in Fig. d. g, Lysosomal pH change 4 hour after the drug treatment. N=5 biological replicates per group. Statistical significance was analysed using a one-way ANOVA followed by Dunnett’s multiple comparison test. *: p= 0.0206, ****: p < 0.001. Data are presented as mean values with error bars as standard deviation.
We measured the intratumoral pH response of the nanosensor to autophagy modulators in vivo. Mice were randomized into four treatment groups 24 h after nanosensor injection. Mice were administered either EN6 (50 mg/kg), Baf A1 (0.125 mg/kg), torin 1 (20 mg/kg), or vehicle (saline:DMSO:polyethylene glycol 400, v:v:v = 6:1:1) intraperitoneally. The sensor response from the solid tumors was monitored for 72 hours. The emission wavelength remained stable over the experiments for the control group, denoting no significant pH changes. The in vivo spectra showed distinct red shifts in the EN6 treatment group (Fig. 5d). We employed the in vitro calibration curve to estimate the actual lysosomal pH from values in vivo. Due to the low spectral resolution in the peak (10 nm), the variation in estimated pH was larger than in the cell experiments (|max error| < pH 0.6 unit), but the sensor response was qualitatively consistent with the results of the live cell experiments (Fig. 5e). Within 4 hours after the EN6 treatment, the nanosensors reported statistically significant red-shifting in wavelength and increased (Fig. 5f), indicating lysosomal acidification in the tumor from pH 5.15 to 4.35 (Fig. 5g).
We measured the intratumoral dynamics of lysosomal pH modulation in vivo. Lysosomes of the SKOV3 xenograft tumor cells acidified within 4 h and returned to normal levels 24 h after EN6 treatment (Fig. 6a). Torin 1 treatment elicited a smaller but significant lysosomal acidification event (Fig. 6b). Lysosomal pH remained low for 24 hours before starting to recover. Baf A1 treatment elicited the opposite change in emission wavelength as expected, although the change was not statistically significant. We also assessed the effectors of the drugs in the tumors. EN6 and torin 1 treatments both inhibited mTORC1 signalling as demonstrated by reduced phosphorylation of S6 in the tumor tissues (Fig. 6c), whereas autophagy was activated, evinced by reduced p62/SQSTM1 levels (Fig. 6d) in immunohistochemical stains (Supplementary Fig. 20–22). The timeframe of pS6 reduction and reversal (and thus, mTORC1 activity) closely followed that of lysosomal acidification. However, p62/SQSTM1 levels decreased within 4 hours and remained low over 72 hours. The dynamics of the lysosomal pH in vivo was consistent with the in vitro washout experiment of EN6 (Fig. 4c,d). The onset of lysosomal acidification mirrored the attenuation of pS6, an indication of reduced mTORC1 activity. Conversely, lysosomal pH dynamics diverged from downstream autophagy signaling (p62/SQSTM1).
Fig. 6 |. In vivo dynamic monitoring of autophagy induction.
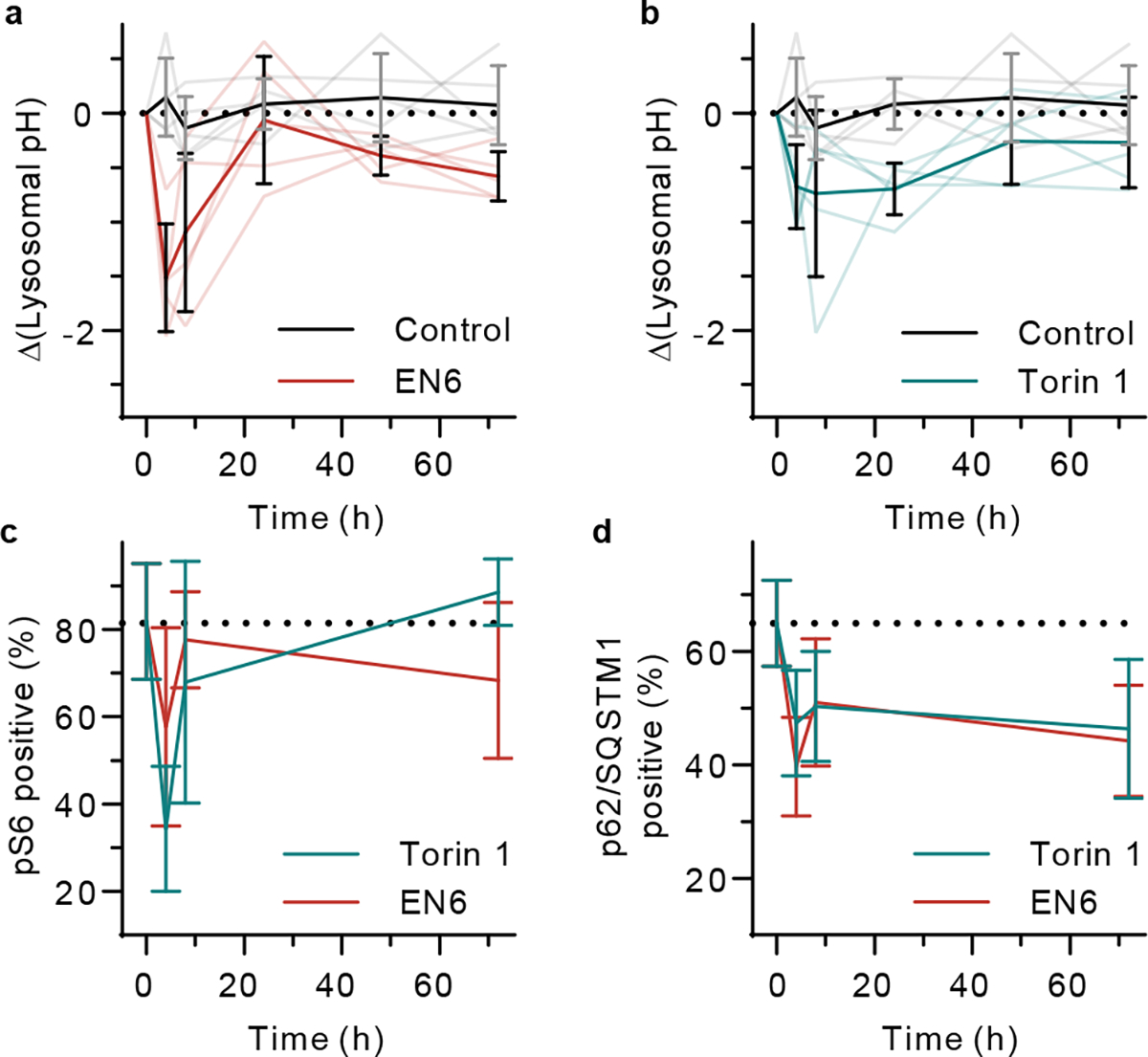
Time-dependent lysosomal pH in control versus a, EN6 (50 mg/kg) and b, Torin 1 (20 mg/kg) treatment groups. Opaque lines denote mean values and transparent lines denote individual datasets. N=5 biological replicates per group. Quantitative histology image analysis of c, phosphorylated S6 and d, p62/SQSTM1 positive cells with EN6 or Torin 1 treatment groups. DMSO control is considered as 0h treatment. N=11 immunohistochemistry images for 5 biological replicates per group. All data are presented as mean values with error bars as standard deviation (a-d).
Discussion
We developed an optical nanosensor that enabled quantitative and dynamic monitoring of endolysosomal pH in live cells and in vivo. The nanosensor reported lysosomal pH changes induced by modulation of the catalytic activity of V-ATPase and mTORC1 inhibition in various cell lines. Using the method, we found sustained pH dysregulation induced by covalent targeting of V-ATPase in live cells and provided dynamic measurements of intratumoral lysosomal pH in vivo. We also found that autophagy modulators induced pronounced differences in lysosomal acidification, depending on the cell type. The results suggest that pH dysregulation may correspond with heterogeneous responses of autophagy activation among cancers, and certain autophagic vulnerabilities in cancer cells may result from lysosomal dysregulation. Further investigations, regarding the signaling pathways regulating autophagy-related lysosomal hyperacidification and the degree and causes of any divergence from the canonical acidification behavior among cancer cells, are warranted.17
Experiments in live cells revealed that lysosomal pH dynamics mirrored dephosphorylation of S6K and lipidation of LC3B upon autophagy activation, but that pH desynchronized with p62/SQSTM1 degradation. In vivo, lysosomal pH was concomitant with pS6 but again diverged from p62/SQSTM1. The results suggest that lysosomal hyper-acidification, a hallmark of autophagy, is an indicator of mTORC1 inhibition.
We found that this method can dynamically assess autophagy-associated processes by monitoring lysosomal hyperacidification in vivo, addressing certain limitations of current autophagy assays.7,12 Molecular markers such as p62/SQSTM1 and LC3B for autophagy monitoring, through either immunoblotting or fluorescent protein tags, currently provide the most direct measurements of autophagic flux. However, the dynamic process of autophagy may not be fully captured by static imaging.7 For instance, enhanced autophagic flux facilitates the degradation of p62/SQSTM1 but also upregulates the expression through TFEB and autophagy-derived amino acids.54 Hydrolase activity assays like DQ-BSA provide a high-throughput readout for autophagic screenings. However, different hydrolases perform optimally under different pH values, and whether more efficient cleavage of a universal fluorescent marker is directly related to autophagy flux is still in question. In vivo applications using these bioassays are even more challenging due to the limitations of using these probes in living tissues. We found that the nanosensor can monitor the fluctuation of the tumor autophagy (or lysosomal) activity to facilitate investigation without the need for permanent alteration of the autophagy signaling in host and/or tumor, complementing studies using transgenic mouse models.
We provide an assessment of the limitations of this technology. The nanosensors currently only measure pH within the endolysosomal pathway. To produce quantitative pH measurements/maps within cell lines, calibration curves must be obtained using each cell line. Cell type-dependent variation in nanosensor responses may be attributed to changes in lysosomal protein content and/or ionic strength39,55. Additionally, certain perturbations can result in modulation of the sensor response, such as drugs that cause large changes in the protein/nutrient content of the lysosome, as we found with strong mTORC1/2 inhibition. Similar challenges have been identified with pH-sensitive fluorescent dyes56, and such modulations must similarly be accounted for by acquiring calibration curves in the presence of the perturbation, potentially as part of a validation study in the case of drug screening investigations. Regarding the instrumentation required to use the nanosensor, conventional plate readers and microscopes that can measure fluorescence signals of conventional bioanalytical pH sensors cannot be used, although compatible instruments, incorporating a near-infrared detector, are now commercially available.33,44 Such detectors have been incorporated into plate readers,39 and additionally hyperspectral imaging devices (for spatial pH mapping) have been integrated into fluorescence microscopes44, and in vivo animal imaging systems33. For rapid measurements, a fiber-optic-based probe system can be used to measure the sensor fluorescence non-invasively in live mice in ≤ 5 seconds per mouse.55
Our findings suggest that it can be used generally for quantitative imaging and high-throughput measurements. The sensitivity is sufficient for measuring the lysosomal acidification/basification activities of autophagy-mediated hyper-acidification in multiple cell lines/in vivo. High-throughput measurements of sensor response in multiple experimental conditions were performed in a 96-well plate format. Regarding dynamic measurements, we found that the response of the sensor itself is immediate. For long-term measurements, the technology captured the dynamic pH response across 24 hours. In vivo studies demonstrated spatiotemporal imaging of drug action in live animals and longitudinal assessment of endolysosomal pH in the same mice for up to 72 hours. We anticipate that this technology will facilitate the understanding of the regulatory mechanisms of dysregulated biological systems in disease and accelerate the discovery of new therapeutic targets and interventions.
Methods
Purification of (6,5)-SWCNT.
Raw SWCNT material, CoMoCAT SG65i (Sigma-Aldrich) was dispersed in 1 wt/v% sodium deoxycholate (DOC, Sigma-Aldrich, 99.9%) aqueous solution at a nanotube concentration of 1 mg/mL using tip-sonication at 6W (Sonics & Materials, Inc) and 4 °C for 1h, followed by ultracentrifugation at 100,000 g for 30 min. The 85% supernatant was used to enrich (6,5)-SWCNT solution based on the previously reported protocol.57 The final purified (6,5)-SWCNTs were stabilized in 1.04% DOC solution to maintain long-term colloidal stability (> 6 months).
Covalent functionalization of purified (6,5)-SWCNT.
N,N-diethlyanimoaryl OCC were covalently functionalized to the purified (6,5)-SWCNTs via diazonium chemistry. N,N-diethyl-4-animobenzene tetrafluoroborate was freshly synthesized from N,N-diethyl-p-phenylenediamine (97%, Sigma-Aldrich) and nitrous acid following a modified literature method.58 The purified SWCNT solution was diluted with 1% sodium dodecyl sulfate (≥99.0%, Sigma-Aldrich) and mixed with the synthesized diazonium salts at the salt to carbon of (6,5)-SWCNT molar ratio of 3.17 to 1. The SWCNT-diazonium mixture was illuminated with a mercury arc lamp (X-Cite 120Q, Excelitas) at room temperature. After 20 minutes of illumination, the diazonium reaction was quenched by diluting the SWCNT solution with 1.04% DOC solution. The functionalized SWCNT solution was ultrafiltrated using Amicon® Ultra filters (100kDa MWKO).
DNA/DOC exchange for the OCC-functionalized SWCNTs.
To redisperse the OCC-SWCNTs to biocompatible polymers, we partially adapted the literature method by Streit et al.59 First, we sequentially added 25 μL of 25 w/v % polyacrylamide (10 kDa, Sigma-Aldrich), 30 μL of 10 mg/mL ssDNA (sequences = 5’- GTGTGTGTGTGTGTGTGTGTGTGTGTGTGT-3’ or 5’//Cy5/GTGTGTGTGTGTGTGTGTGTGTGTGTGTGT//3’, Integrated DNA Technologies), 270 μL of methanol (Anhydrous, 99.8%, Sigma-Aldrich), and 600 μL of 2-propanol (99.9%, Sigma-Aldrich) to the OCC-SWCNT solution. To precipitate DNA/polyacrylamide encapsulated OCC-SWCNTs, the solution was centrifuged at 17,000g for 2 seconds. The supernatant was further centrifuged for 2 minutes at the same speed and room temperature. The pellets from each centrifugation were combined and redispersed with 150 μL of water. The addition of 600 μL of 2-propanol and centrifugation were repeated one more time. The OCC-SWCNT pellets were then diluted in 1 mL of 4 mg/mL DNA in PBS and tip-sonicated for 1h at 6W and 4°C. The solutions were ultracentrifuged at 100,000 g and 4 °C for 30 min. 85% supernatant was collected and dialyzed against PBS to remove free DNA (Spectra-Por, Float-A-Lyzer, MWCO = 1MDa). The absorption spectra of the OCC-DNA complexes were obtained using a UV-Vis-NIR spectrophotometer (V-780, Jasco). The absorbance at (6,5) was used to estimate the relative OCC-DNA concentration.60
Near-Infrared Hyperspectral Fluorescence Microscopy.
A hyperspectral microscope (IMA™, Photon Etc.) was used to obtain spectrally and spatially resolved OCC-DNA emission in live cells. A continuous wave 808 nm laser (2W) injected into a multimode fiber to excite the nanosensors. The excitation beam passed through a beam-shaping module to produce a top-hat intensity profile with under 10% power variation on the imaged region of the sample. The power output at the sample stage was 425.8, 370.2, and 164.8 mW for ×20, ×50, and ×100 objectives, respectively. A long pass dichroic mirror with a cut-on wavelength of 875 nm (Semrock) was aligned to reflect the laser to the sample stage of an IX-71 inverted microscope equipped with LCPLN20XIR, LCPLN50XIR, and LCPLN100XIR IR objectives (Olympus). Hyperspectral microscopy was conducted by passing the emission through a volume Bragg grating placed immediately before a thermoelectrically cooled 2D InGaAs detector (ZephIR 1.7) in the optical path.
Analysis and Processing of Hyperspectral Data.
Hyperspectral data acquired were saved as a (512×640×76) 16-bit array, where the first two coordinates signify the spatial location of a pixel and the last coordinate is its position in wavelength space, ranging from 950 to 1250 nm with 4 nm interval. Custom codes, written using MATLAB software, were used to subtract background, correct for nonuniformities in excitation profile and wavelength-dependent quantum efficiency by each pixel, and compensate for dead pixels on the detector. A peak-finding algorithm was used to calculate the center wavelengths and intensities of and peaks for a given pixel. To reduce the spectral drift resulting from the movement of lysosomes during the hyperspectral cube acquisition, 8×8 pixels were combined into a single pixel and the spectral parameters were obtained from the averaged emission spectrum. Pixels that failed the peak-finding threshold, primarily due to low intensity above the background, were removed from the data sets. The remaining pixels were fit with a Lorentzian function.
Near-Infrared Fluorescence Spectroscopy of OCC-DNAs.
Fluorescence emission spectra of OCC-DNAs were acquired using a home-built near-infrared fluorescence spectroscopy. The SuperK EXTREME supercontinuum white-light laser source (NKT Photonics) was used with a VARIA variable bandpass filter accessory set to a bandwidth of 20 nm centered at 575 nm. The light passed through a 20× or 50× NIR objective equipped in an inverted IX-71 microscope (Olympus) and illuminated the samples in a 96-well clear flat bottom microplate (Corning). Emission from the OCC-DNAs was collected through the objective and passed through a dichroic mirror (875 nm cutoff, Semrock). A Shamrock 303i spectrograph (Andor, Oxford Instruments) with a slit width of 100 μm dispersed the emission using a 86 g/mm grating with 1.35 μm blaze wavelength. The spectral range was 723−1694 nm with a resolution of 1.89 nm. The light was collected by an iDus 1.7 μm InGaAs (Andor, Oxford Instruments) with an exposure time of 0.1–15 seconds. Background subtraction was conducted using a well in a 96-well plate filled with PBS or 10% FBS depending on the experiment. Following acquisition, the data was processed with custom codes written in MATLAB that applied the spectral corrections and background subtraction and fitted the emission peaks with Lorentzian functions.
OCC-DNA treatment in Live Cells for Near-Infrared Fluorescence Measurement.
The nanosensors were added at 4–12 ng/mL to cell culture media and incubated with cells at 30–60% confluency (8+ hours) at 37 °C. These sensor treatment conditions were chosen because they resulted in a strong sensor signal with an exposure time of less than 15 seconds from all the tested cell lines with high biocompatibility. Four hours after replacing to fresh media, cells were trypsinized (Gibco) and replated on a 96-well microplate (Corning) or 35 mm glass-bottom Petri dishes (MatTek). To compare uptake of OCC-DNAs in cells at different temperatures (4 and 37 °C), SKOV3 cells at 30% confluency in a 35mm glass-bottom dish were incubated in complete media containing 0.1 mg/L OCC-DNAs for 30 minutes. The cells were washed to remove cell surface-adsorbed OCC-DNA complexes and then incubated for 6h in the OCC-DNA-free media prior to fluorescence imaging. The fluorescence measurements were performed in a humidified chamber at 37 °C and 5% CO2.
In Vivo Hyperspectral Fluorescence Spectroscopy.
In vivo fluorescence imaging of the nanosensors was performed using a preclinical NIR hyperspectral mouse imaging system (IR VIVO™, Photon Etc.). We used 808 nm lasers to reduce tissue absorption and scattering of the excitation light in vivo. Excitation was provided by two continuous wave diode lasers each with an output power of 2 W. Excitation light was distributed over the entire mouse with a maximum power density of 340 mW/cm2. For hyperspectral animal imaging, the emission light was passed through a volume Bragg grating as described in the “Near-Infrared Hyperspectral Fluorescence Microscopy” section. The and hyperspectral cubes were scanned for 950–1050 nm with 10 nm step size and 1100–1200 nm with 4 nm step size, respectively.
pH Calibration Curves for Live Cells.
The buffers for generating the pH calibration curves were formulated with 125 mM KCl, 25 mM NaCl, 10 μM monensin (≥97%, Sigma-Aldrich), and 25 mM N-[2-hydroxyethyl]-piperazine-N-[2-ethanesulfonic acid] (HEPES, Sigma-Aldrich) for pH≥7 buffer or 25 mM 2-[N-morpholino] ethanesulfonic acid (MES, Fisher Scientific) for pH<7 buffers. Each buffer solution was adjusted to the appropriate final pH using 1 M NaOH or 1 M HCl. Cells treated with OCC-DNAs overnight were washed with fresh media. Four hours after the media exchange, the cells were washed with calibration buffer solutions 3 times. The cells in buffer solution were plated in a 96-well plate at the cell density of approximately 0.3–0.6*106 cells/well. The OCC-DNA fluorescence emission from the live cells were collected using a near-infrared fluorescence microscopy as described above.
Cell Culture Reagents and Conditions.
HEK293T, SKOV3, OVCAR3, HeLa, MEF cells were purchased from ATCC (Manassas, VA, USA).RM1 and Myc-CaP were gifts from the R. Blasberg lab (Memorial Sloan Kettering Cancer Center). All cells were grown at 37 °C and 5% CO2 incubator. HEK293T, SKOV3, HeLa, RM1, Myc-CaP, MEF cells were grown in Dulbecco’s modified Eagle’s medium (DMEM, Gibco) with 10% fetal bovine serum (FBS, Gibco); SKOV3 was supplemented with 100 μg/mL primocin (InvivoGen), and RM1 and Myc-CaP were supplemented with 1X penicillin/streptomycin. OVCAR3 cells were grown in RPMI-1640 medium with 20% FBS supplemented with 100 μg/mL primocin.
Cell Viability Assay.
The cell viability was performed using the CellTiter-Glo 2.0 Assay (Promega). Cells were plated (2,000 cells/well) in 96-well plates (Corning), and treated with media containing concentration gradient of the OCC-DNAs (0.08–0.313 ng/mL) or PBS as a solvent control in triplicate (a 100ul final volume of media). 72 h later, the viability was quantified using intracellular ATP levels. 15 uL of CellTiter-Glo substrate was added into the cell medium and incubated for 40 min at room temperature. Luminescence was measured using microplate reader (TECAN infinite M1000Pro) with 100 ms count time per well. For data processing, the luminescence value of PBS-treated cells was converted to 100% for each cell line, and pH sensor treated cells were normalized to the control as the relative viability.
NLRP3 Inflammasomes and LDH Cytotoxicity Assay.
Raw 264.7 cells stably expressing apoptosis-associated speck-like protein containing a C-terminal caspase recruitment domain (ASC) (RAW 264.7 cells do not endogenously express ASC) were treated with lipopolysaccharide (Invivogen, tlrl-3pelps) at 5 ug/mL for 16h. The cells were treated with imiquimod (100uM, Thermo Fisher Scientific, 107471G), MCC950 (10uM, Thermo Fisher Scientific, NC1363755), or OCC-DNA complexes (0.1 mg/mL) alone or in combination with an NLRP3 inhibitor, MCC950 (10uM) for 3h. Supernatants were analyzed for lactate dehydrogenase (LDH) activity using the Pierce LDH Cytotoxicity Assay Kit (Life Technologies, PI88953). LDH activity was quantified relative to a lysis control where cells were lysed by adding 8 μL of a 9% Triton X-100 solution. Cells were collected for western blotting. Cell death was analyzed by LDH assay and by immunoblot for PARP and GSDMD cleavage, an apoptosis and pyroptosis marker, respectively.
Dye Quenched-Bovine Serum Albumin (DQ-BSA) Assay.
Cells were plated 100,000 cells/well in clear, flat bottom CellBIND 48-well plates (Corning) and cultured until the cells reached 50–75% confluency. Cell media was replaced with 200 uL of 800 nM Calcein, AM (Invitrogen) in Live cell imaging solution (Molecular Probes) for each well. An hour later, Calcein, AM containing media was removed, and 200 uL of 25 μg/mL DQ-BSA, Red (Invitrogen) in complete media with 1 μg/mL Hoechst stain was added to each well. A negative control group was added with Hoechst stain in complete media only. After 1h incubation, the media was replaced to 200 uL of complete media for each well and T=0h imaging of Cytation 5 was performed using Cell Imaging Multi-Mode Reader (BioTek). 200 uL of complete media with 2X concentration of treatment of interest was added to appropriate wells, and performed imaging reads for every hour on Cytation 5. At least six fields of views between 100 to 1000 cells were acquired in each well, with each treatment performed in two technical replicate wells. At least three independent biological experiments were performed for each cell line.
Antibodies and Western Blotting.
Antibodies to LC3B (NB100–2220), ATP6V1A (NBP2–55148), and ATG7 (NBP2–67596) were from Novus Biologicals. Antibodies to PARP (9542), GAPDH (14C10), p70 S6 Kinase (2708), phospho-p70 S6 Kinase – Thr389 (9234), SQSTM1/p62 (39749) and beta Actin (8457) were from Cell Signaling Technology. Mouse GSDMD rabbit monoclonal antibody (ab209845) was from Abcam. IRDye 800CW Goat anti-Rabbit IgG was from LI-COR Biosciences. After treatment, cells were lysed in Pierce RIPA buffer (Thermo Scientific) containing Halt protease and phosphatase inhibitor cocktails (Thermo Scientific). Cell lysate was normalized using Quick Start Bradford Dye Reagent (Bio-Rad), prepared by adding 4X protein loading buffer (LI-COR Biosciences), heated at 98°C for 5 min, resolved by 4–20% Mini-PROTEAN TGX Precast Polyacrylamide Gels (Bio-Rad), immunoblotted, and visualized using the Odyssey Imaging System (LI-COR Biosciences).
Colocalization Study.
SKOV3 cells were incubated with nanosensors (20 ng/mL) overnight. The cells were then washed 3× with PBS and placed in fresh cell media. Four hours later, the cells were incubated with 5–50 nM LysoTracker Green DND-26 (Life Technologies) for 30 min in cell media, washed 3-times with PBS, and imaged immediately in fresh PBS. For confocal fluorescence imaging of live cells, the FITC and Cy5 channels were used for LysoTracker Green and pH sensor, respectively. Cell imaging was performed on a Zeiss LSM 880, AxioObserver microscope equipped with a Plan-Apochromat 63× oil 1.4 NA differential interference contrast M27 objective in a humidified chamber at 37 °C and 5% CO2. For colocalizing nanosensor emission and LysoTracker Green, after PBS wash, the cells were placed in fresh media with either DMSO, 100 μM of EN6, 250 nM of torin1, or 100 nM of Bafilomycin A1. The LysoTracker Green emission was collected using a EMCCD camera with a FITC channel and the broadband emission of pH sensor was collected using a 2D InGaAs camera of a near-infrared fluorescence microscopy as described above. The measurements were performed in a humidified chamber at 37 °C and 5% CO2. After the fluorescence microscopy measurements, the images were processed for colocalization analysis in ImageJ with split channels of Cy5 (or nanosensors) and LysoTracker Green. The JACoP plugin61 was used to obtain the Mander’s coefficients.
The colocalization analysis of the tumor tissue immunofluorescence images was performed on Cy5-ss(GT)15 wrapped pH sensor-injected SKOV3 tumors. Five-micron serial tissue sections were used for analysis. The fluorescent tissue images of Cy5-labelled CNTs were stained with DAPI, and the serial tissue sections were stained with DAPI and LAMP1. The slides were scanned with a 20x/0.8NA objective on a Pannoramic Confocal Scanner (3DHistech, Budapest, Hungary). DAPI images were used to superimpose two consecutive images using a customized MATLAB code. The superimposed images of DAPI, Cy5, and LAMP1 were processed for colocalization analysis using the JACoP plugin61 implemented in ImageJ.
Atomic Force Microscopy.
A stock solution of nanosensors at 7 mg/L in 1X PBS was diluted 20× in dH2O and plated on a freshly cleaved mica substrate (SPI) for 4 min before washing with 10 mL of dH2O and blowing dry with argon gas. An Olympus AC240TS AFM probe (Asylum Research) in an Asylum Research MFP-3D-Bio instrument was used to image in AC mode. Data was captured at 2.93 nm/pixel XY resolution and 15.63 pm Z resolution.
Animal Studies.
All animal studies were approved by and carried out in accordance with the Memorial Sloan Kettering Cancer Center Institutional Animal Care and Use Committee. Female Hsd:Athymic Nude-Foxn1nu mice were purchased from Envigo at 5 to 6 weeks of age. All control and experimental mice were age-matched and housed in air-filtered laminar flow cabinets with food and water ad libitum under a 12-hour light/dark cycle, 18–23 °C and 40–60% humidity. Approximately 1 million SKOV3 tumor cells were injected subcutaneously in a 1:1 mixture of serum-free media and Matrigel (BD Biosciences) into both flanks (200 μL per flank). When the tumor volume reached by 100 mm3, mice were randomized and 30 mL of the nanosensors (0.1 mg/L, diluted in PBS) or PBS (control) were intratumorally injected in one flank, After 24h, the mice were intraperitoneally injected with EN6 (50mg/kg), Baf A1 (0.125mg/kg), Torin 1 (20mg/kg) or vehicle (saline:DMSO:polyethylene glycol 400, v:v:v = 6:1:1). For in vivo imaging and spectroscopy, mice were anesthetized with 2% isoflurane before and during data collection. Animals were euthanized using CO2 inhalation. Tumors were harvested for histology analysis.
Immunohistochemistry and Immunofluorescence.
The immunohistochemical imaging was performed at the Weill Cornell Medicine, Department of Pathology and Laboratory Medicine, Center for Translational Pathology, and Memorial Sloan Kettering Cancer Center Molecular Cytology Core Facility. At the experimental endpoint, subcutaneously engrafted tumor tissue was retrieved and fixed in 10% formalin at 4°C for 24h and embedded in paraffin after dehydration. Five-micron sections were used for analysis.
The immunohistochemistry for p62/SQSTM1 (Enzo Life Science, Cat# BML-PW9860–0025, Antigen retrieval BOND Epitope Retrieval Solution 2 Catalog No: AR9640- ER2 (pH 9) for 20 minutes, Antibody dilution 1:100 with 15 minutes incubation time at room temperature) was developed on Leica Bond Rx with default protocol by Pathology Core at Weill Cornell Medical Center.
The immunohistochemical detection for pS6K and the immunofluorescence detection of LAMP1 were performed using Discovery XT processor or Ultra processor (Ventana Medical Systems-Roche). The tissue sections for pS6K were deparaffinized with EZPrep buffer (Ventana Medical Systems), antigen retrieval was performed with CC1 buffer (Ventana Medical Systems-Roche), and sections were blocked for 30 minutes with Background Buster solution (Innovex). A rabbit monoclonal anti-PS6R (Cell Signaling) was used in 0.36 mg/ml concentration. The incubation with the primary antibody was done for 6h, followed by 1h incubation with biotinylated goat anti-rabbit IgG (Vector labs) in 5.75 mg/ml. Blocker D, Streptavidin-HRP and DAB detection kit (Ventana Medical Systems) were used according to the manufacturer instructions. The slides were counterstained with hematoxylin and coverslipped with Permount (Fisher Scientific). Image quantitation of p62/SQSTM1 and pS6K staining, and nuclei count was taken by Qupath 0.2.62
For the LAMP1 staining, after 32 min of heat and standard retrieval via Cell Conditioning 1 (Ventana), the tissue sections were blocked first for 30 min in Background Blocking reagent (Innovex). A rabbit monoclonal LAMP1 (Cell Signaling) was used at 0.34 μg/ml. Primary antibody was incubated for 5h, followed by incubation with biotinylated goat anti-rabbit IgG (Vector labs) at 5.75 mg /ml for 1h. Blocker D, Streptavidin-HRP D (Ventana Medical Systems) and Tyramide-CF594 (Biotium) were prepared and applied according to manufacturer instruction in 1:2000 for 16 min. All slides were counterstained in 5ug/mL DAPI [dihydrochloride(2-(4-Amidinophenyl)-6-indolecarbamidine dihydrochloride], Sigma D9542, for 5 minutes at room temperature, mounted with anti-fade mounting medium Mowiol 4–88 (Millipore) and coverslipped.
Statistics and Reproducibility.
Statistical analysis was performed with GraphPad Prism version 8.4.3. All data met the assumptions of the statistical tests performed (i.e., normality, equal variances, etc.). Statistical parameters and significance are reported in figures or figure legends. Sample size decisions were based on the instrumental signal-to-noise ratios. AFM, hyperspectral and confocal microscopy images were repeated at least five times with comparable results. Immunohistochemistry and immunofluorescence staining were performed on all mice used in the studies (N=5 biological replicates per group). All cellular experiments and western blotting were performed at least three times with comparable results.
Extended Data
Extended Data Fig. 1 |. Characterization of OCC-DNA response in various buffer/media conditions.

Emission wavelengths of a-c, and d-f, of the OCC-DNA complexes at varying buffer pH and media conditions in phosphate buffered saline. The metal ion concentrations tested are physiologically relevant ranges. All data are presented as mean values and error bars denote standard deviation from N=3 technical replicates (a-f). g, Frequency distribution of standard deviations of wavelength shifts of triplicate measurements of a-f.
Extended Data Fig. 2 |. Protein concentration effects on OCC-DNA optical response.
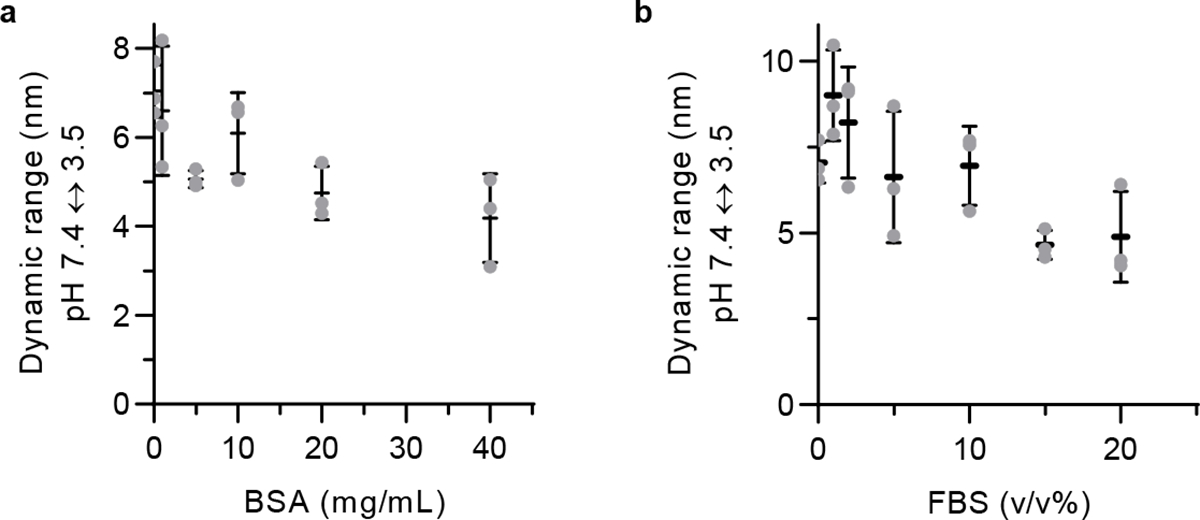
The dynamic range of the OCC-DNA response to pH at increasing concentrations of: a, bovine serum albumin and b, fetal bovine serum. All data are presented as mean values and error bars denote standard deviation from N=3 technical replicates (a,b).
Extended Data Fig. 3 |. Viscosity effects on OCC-DNA optical response.
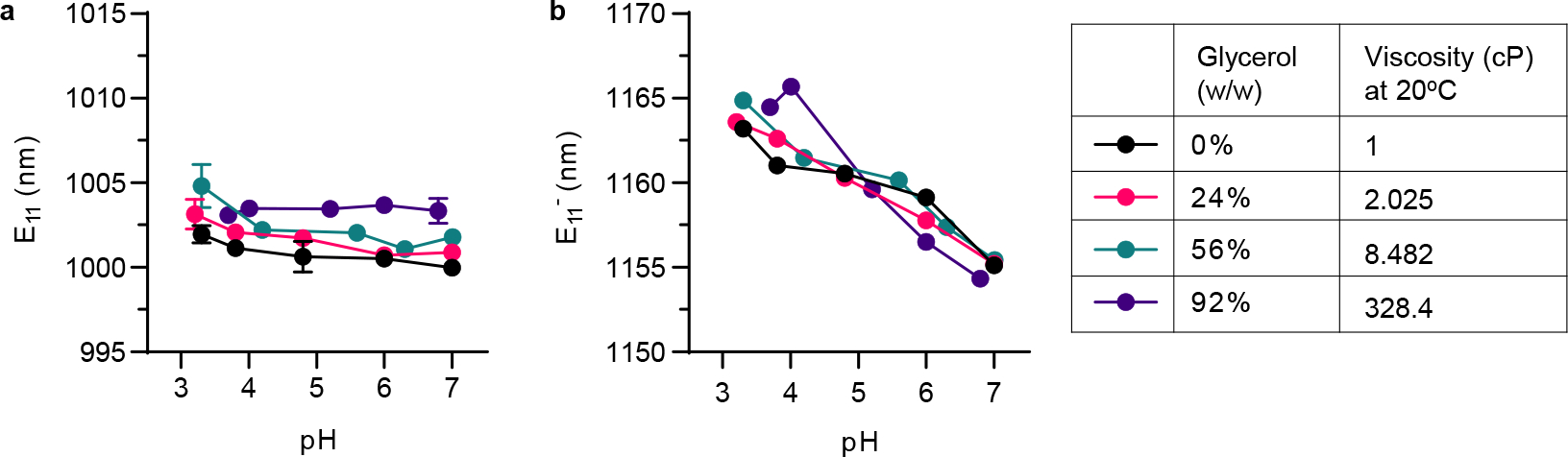
Emission wavelengths of a, and b, of the OCC-DNA complexes at varying buffer pH and glycerol. All data are presented as mean values and error bars denote standard deviation from N=3 technical replicates (a,b).
Extended Data Fig. 4 |. Cell viability in response to OCC-DNA complexes.
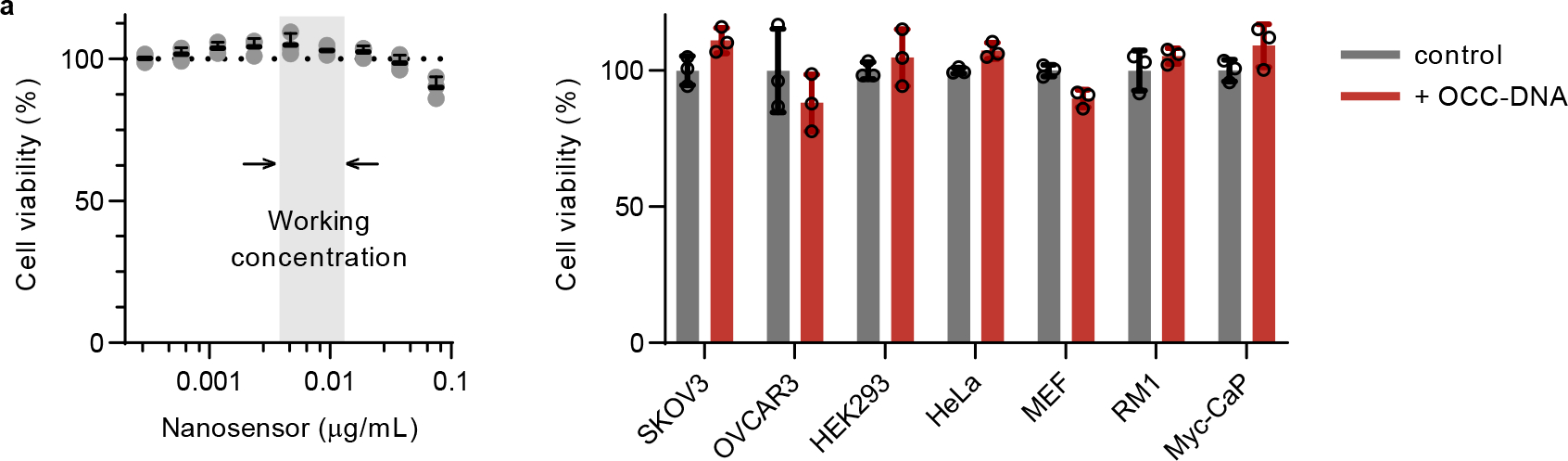
Single-dose (0.01 mg/L) OCC-DNA cell viability of SKOV3, OVCAR3, HEK293, HeLa, MEF, RM1, and Myc-CaP cell lines, measured via CellTiter-Glo 2.0, after 72 hours of incubation. No statistically significant differences were observed between the PBS control groups (gray) and the treatment groups (red) in all the tested cell lines. All data are presented as mean values and error bars denote standard deviation of triplicates for each condition.
Extended Data Fig. 5 |. OCC-DNA responses in 8 cell lines.

The emission response of OCC-DNAs in live cells upon exposure to HEPES or MES buffer solutions of varying pHs with monensin (see Methods). All data are presented as mean values and error bars denote standard deviation from N=3–25 biological replicates.
Extended Data Fig. 6 |. Inhibitor-mediated alterations of nanosensor response to pH.
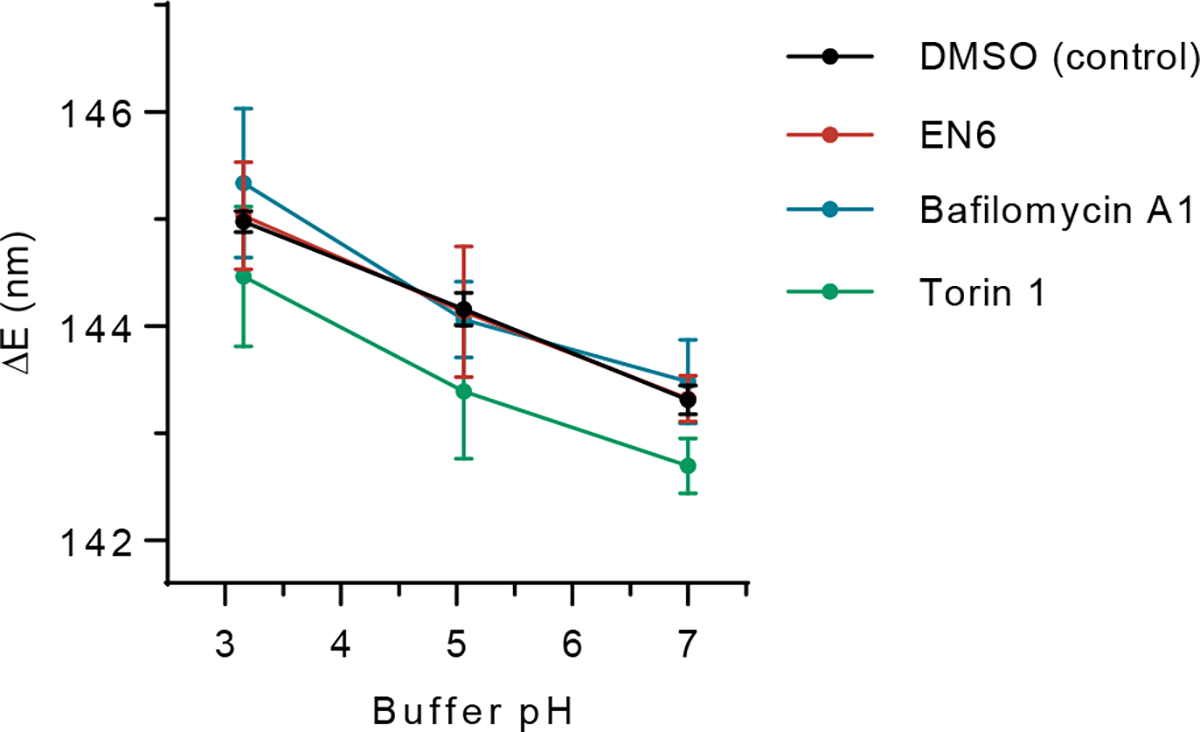
The emission response of nanosensors in live SKOV3 cells upon exposure to HEPES or MES buffer solutions of varying pHs with monensin (see Methods). Cells were treated with DMSO (black), 100 μM EN6 (red), 100 nM bafilomycin A1 (blue), 250 nM torin 1 (green) for 4 hours prior to pH measurements. Data are presented as mean values and error bars denote standard deviation from N=25 each DMSO, EN6, and Baf A1 point, and 25, 24, and 21 for pH 7, 5.06, and 3.16 for torin 1 biological replicates.
Extended Data Fig. 7 |. Nanosensor response in autophagy-defective cells.

a, ATG7 expression by western blotting confirmed the knockout of ATG7 in the HEK293T cell line used herein. The emission wavelength response of OCC-DNAs in live b, wild type and c, ATG7−/− HEK293T cells upon exposure to HEPES or MES buffer solutions of varying pHs in the presence of monensin (see Methods). Cells were treated with DMSO (black) or 250 nM torin 1 (blue) for 4 hours prior to pH measurements. All data are presented as mean values and error bars denote standard deviation from N=10 technical replicates (b,c). Original gel images are in Supplementary Fig. 24.
Extended Data Fig. 8 |. Time-dependent fluorescence intensity changes of intratumorally-injected nanosensors.
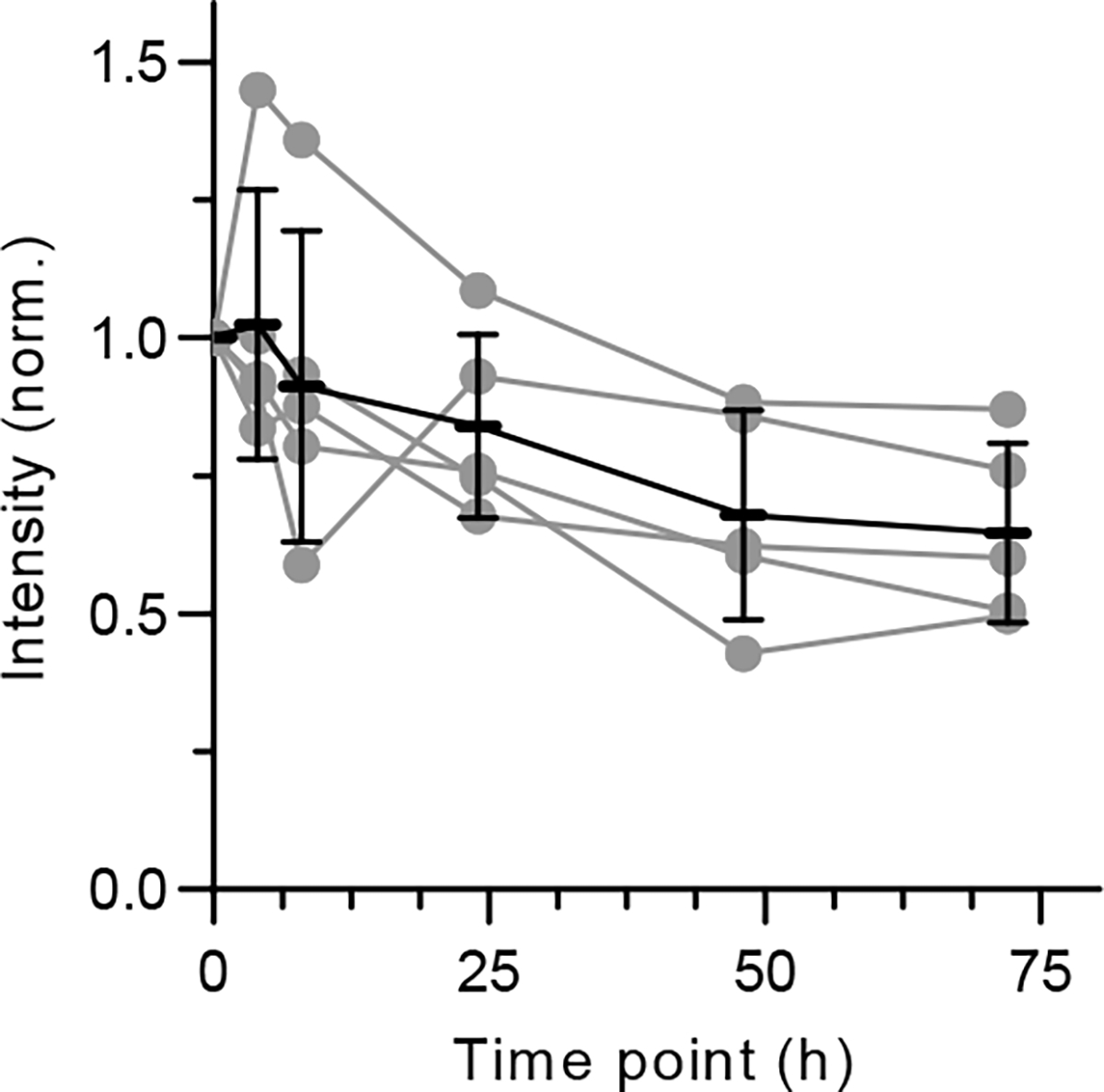
Quantification of total emission intensity of nanosensors in solid tumours of mice after injection. Fluorescence measurements were performed with a near-infrared preclinical hyperspectral imager with 730 nm excitation. Data are presented as mean values and error bars denote standard deviation from N=5 mice.
Supplementary Material
Acknowledgments
We thank MSK Molecular Cytology Core Facility for assistance with AFM imaging, sample processing for immunofluorescence, and confocal microscopy, and image analysis. We also thank the Center for Translational Pathology at Weill Cornell Medicine for immunohistochemistry. We thank Claire O’Mara at Daniel Bachovchin lab (MSKCC) for the NLRP3 inflammasome experiment. We thank Prakrit Jena for assistance with image processing and analysis. Graphical abstract, Fig. 3a, Supplementary Fig. 2 and 11 were created with BioRender.com.
Funding:
This work was supported in part by the National Science Foundation CAREER Award (1752506, D.A.H.), the NCI (R01-CA215719, D.A.H. and Cancer Center Support Grant P30-CA008748, D.A.H. H.A., and Y.M.L.), the NIH Common Fund (DP2-HD075698, D.A.H.), the American Cancer Society Research Scholar Grant (GC230452, D.A.H.), the Ara Parseghian Medical Research Fund (D.A.H.), the Honorable Tina Brozman Foundation (D.A.H.), the Ovarian Cancer Research Alliance and the Edmée Firth Fund for Research in Ovarian Cancer (CRDGAI-2023-3-1003, D.A.H.), the Pershing Square Sohn Cancer Research Alliance (D.A.H.), the Expect Miracles Foundation–Financial Services Against Cancer (D.A.H.), the Louis and Rachel Rudin Foundation (D.A.H.), the Experimental Therapeutics Center of MSKCC (D.A.H. and Y.M.L.), Mr. William H. Goodwin and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research (D.A.H. and Y.M.L.), the JPB Foundation (Y.M.L.), and the William Randolph Hearst Fund in Experimental Therapeutics (Y.M.L.). M.K. was supported by the NIH (K99-EB033580) and the Marie-Josée Kravis Women in Science Endeavor Postdoctoral Fellowship. Z.Y. was supported by the Ann Schreiber Mentored Investigator Award (Ovarian Cancer Research Fund) and Young Investigator 2019 (Kaleidoscope of Hope). R.L. was supported by an NIH T32 training grant (T32-GM73546), J.W. was supported by an NIH T32 training grant (T32-GM136640-Tan). D.W. supported by NIH T32 training grants (T32-GM141949, T32-CA062948). R.F. was supported by the Alfred Benzon Foundation Fellowship, Y.H.W. acknowledges support from the National Science Foundation (CHE-1904488 and CHE-2204202).
Footnotes
Competing interests
D.A.H. is a co-founder and officer with equity interest in Lime Therapeutics, Inc., and co-founder with equity interest in Selectin Therapeutics Inc. and Resident Diagnostics, Inc., and a member of the scientific advisory board of Concarlo Therapeutics, Inc., Nanorobotics Inc., and Mediphage Bioceuticals, Inc. T.T. has research support from ONO Pharma USA., Inc. (unrelated to this work) and is a member of the scientific advisory board of Lime Therapeutics, Inc. with equity interest. The remaining authors declare no competing interests.
Ethics declarations
All animal procedures used in this study were performed in accordance with the protocol approved by the Memorial Sloan Kettering Cancer Center Institutional Animal Care and Use Committee.
Code availability
LABVIEW code for data acquisition and MATLAB codes for data analysis in this article are available in the public github repository (github.com/mijinee/HellerLab_MSKCC).
Data availability
All data presented in the manuscript or the supplementary materials, including the uncropped gels and the raw data are available in the Source Data files.
References
- 1.Lawrence RE & Zoncu R The lysosome as a cellular centre for signalling, metabolism and quality control. Nat. Cell Biol. 21, 133–142 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Mulcahy Levy JM & Thorburn A Autophagy in cancer: Moving from understanding mechanism to improving therapy responses in patients. Cell Death Differ. 27, 843–857 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Galluzzi L et al. Autophagy in malignant transformation and cancer progression. Embo j 34, 856–880 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhan L et al. Autophagy as an emerging therapy target for ovarian carcinoma. Oncotarget 7, 83476–83487 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jung S, Jeong H & Yu S-W Autophagy as a decisive process for cell death. Exp. Mol. Med. 52, 921–930 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lauzier A et al. Colorectal cancer cells respond differentially to autophagy inhibition in vivo. Sci. Rep. 9, 11316 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klionsky DJ et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 17, 1–382 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bellot G et al. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell Biol. 29, 2570–2581 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chavez-Dominguez R, Perez-Medina M, Lopez-Gonzalez JS, Galicia-Velasco M & Aguilar-Cazares D The double-edge sword of autophagy in cancer: From tumor suppression to pro-tumor activity. Front. Oncol. 10 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kimmelman AC & White E Autophagy and Tumor Metabolism. Cell Metab. 25, 1037–1043 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Janji B, Berchem G & Chouaib S Targeting autophagy in the tumor microenvironment: New challenges and opportunities for regulating tumor immunity. Front. Immunol. 9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mizushima N & Murphy LO Autophagy assays for biological discovery and therapeutic development. Trends Biochem. Sci. 45, 1080–1093 (2020). [DOI] [PubMed] [Google Scholar]
- 13.Poillet-Perez L et al. Autophagy promotes growth of tumors with high mutational burden by inhibiting a T-cell immune response. Nat. Cancer 1, 923–934 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuma A, Komatsu M & Mizushima N Autophagy-monitoring and autophagy-deficient mice. Autophagy 13, 1619–1628 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoshii Saori R. et al. Systemic analysis of Atg5-null mice rescued from neonatal lethality by transgenic ATG5 expression in neurons. Dev. Cell 39, 116–130 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Karsli-Uzunbas G et al. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 4, 914–927 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yim WW-Y & Mizushima N Lysosome biology in autophagy. Cell Discov. 6, 6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Williams RM et al. Harnessing nanotechnology to expand the toolbox of chemical biology. Nat. Chem. Biol. 17, 129–137 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aref M et al. Potentiometric pH nanosensor for intracellular measurements: Real-time and continuous assessment of local gradients. Anal. Chem. 93, 15744–15751 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma L, Ouyang Q, Werthmann GC, Thompson HM & Morrow EM Live-cell microscopy and fluorescence-based measurement of luminal pH in intracellular organelles. Front. Cell Dev. Biol. 5, 71 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Myochin T et al. Rational Design of Ratiometric Near-Infrared Fluorescent pH Probes with Various pKa Values, Based on Aminocyanine. Journal of the American Chemical Society 133, 3401–3409 (2011). [DOI] [PubMed] [Google Scholar]
- 22.Robinson KJ et al. Modified Organosilica Core–Shell Nanoparticles for Stable pH Sensing in Biological Solutions. ACS Sensors 3, 967–975 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Burgstaller S et al. pH-Lemon, a Fluorescent Protein-Based pH Reporter for Acidic Compartments. ACS Sensors 4, 883–891 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guha S et al. Approaches for detecting lysosomal alkalinization and impaired degradation in fresh and cultured RPE cells: Evidence for a role in retinal degenerations. Exp. Eye Res. 126, 68–76 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wäldchen S, Lehmann J, Klein T, van de Linde S & Sauer M Light-induced cell damage in live-cell super-resolution microscopy. Sci. Rep. 5, 15348 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Laissue PP, Alghamdi RA, Tomancak P, Reynaud EG & Shroff H Assessing phototoxicity in live fluorescence imaging. Nat. Methods 14, 657–661 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Heller DA et al. Peptide secondary structure modulates single-walled carbon nanotube fluorescence as a chaperone sensor for nitroaromatics. Proc. Natl. Acad. Sci. U.S.A. 108, 8544–8549 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim M et al. Detection of ovarian cancer via the spectral fingerprinting of quantum-defect-modified carbon nanotubes in serum by machine learning. Nat. Biomed. Eng. 6, 267–275 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bachilo SM et al. Structure-assigned optical spectra of single-walled carbon nanotubes. Science 298, 2361 (2002). [DOI] [PubMed] [Google Scholar]
- 30.Welsher K, Sherlock SP & Dai H Deep-tissue anatomical imaging of mice using carbon nanotube fluorophores in the second near-infrared window. Proc. Natl. Acad. Sci. U.S.A. 108, 8943 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mandal AK et al. Fluorescent sp3 Defect-tailored carbon nanotubes enable nir-ii single particle imaging in live brain slices at ultra-low excitation doses. Sci. Rep. 10, 5286 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jena PV et al. A carbon nanotube optical reporter maps endolysosomal lipid flux. ACS Nano 11, 10689–10703 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Galassi TV et al. An optical nanoreporter of endolysosomal lipid accumulation reveals enduring effects of diet on hepatic macrophages in vivo. Sci. Transl. Med. 10, eaar2680 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Galassi TV et al. Long-term in vivo biocompatibility of single-walled carbon nanotubes. PLOS ONE 15, e0226791 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brozena AH, Kim M, Powell LR & Wang Y Controlling the optical properties of carbon nanotubes with organic colour-centre quantum defects. Nat. Rev. Chem. 3, 375–392 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kwon H et al. Optical probing of local pH and temperature in complex fluids with covalently functionalized, semiconducting carbon nanotubes. J. Phys. Chem. C 119, 3733–3739 (2015). [Google Scholar]
- 37.Piao Y et al. Brightening of carbon nanotube photoluminescence through the incorporation of sp3 defects. Nat. Chem. 5, 840–845 (2013). [DOI] [PubMed] [Google Scholar]
- 38.Gravely M, Safaee MM & Roxbury D Biomolecular functionalization of a nanomaterial to control stability and retention within live cells. Nano Lett. 19, 6203–6212 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roxbury D, Jena PV, Shamay Y, Horoszko CP & Heller DA Cell membrane proteins modulate the carbon nanotube optical bandgap via surface charge accumulation. ACS Nano 10, 499–506 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hornung V et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol 9, 847–856 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Holtzmann E Lysosomes. (Plenum, New York, 1989). [Google Scholar]
- 42.Alberts B JA, Lewis J, et al. Molecular Biology of the Cell., Edn. 4. (Garland Science, 2002). [Google Scholar]
- 43.Jin H, Heller DA, Sharma R & Strano MS Size-dependent cellular uptake and expulsion of single-walled carbon nanotubes: Single particle tracking and a generic uptake model for nanoparticles. ACS Nano 3, 149–158 (2009). [DOI] [PubMed] [Google Scholar]
- 44.Roxbury D et al. Hyperspectral microscopy of near-infrared fluorescence enables 17-chirality carbon nanotube imaging. Sci. Rep. 5, 14167 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Han J & Burgess K Fluorescent indicators for intracellular pH. Chem. Rev. 110, 2709–2728 (2010). [DOI] [PubMed] [Google Scholar]
- 46.Yamamoto A et al. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell Line, H-4-II-E cells. Cell Struct. Funct. 23, 33–42 (1998). [DOI] [PubMed] [Google Scholar]
- 47.Mauvezin C & Neufeld TP Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome fusion. Autophagy 11, 1437–1438 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chung CY-S et al. Covalent targeting of the vacuolar H+-ATPase activates autophagy via mTORC1 inhibition. Nat. Chem. Biol. 15, 776–785 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Frost LS, Dhingra A, Reyes-Reveles J & Boesze-Battaglia K The use of DQ-BSA to monitor the turnover of autophagy-associated cargo. Methods Enzymol. 587, 43–54 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Corrotte M, Fernandes MC, Tam C & Andrews NW Toxin pores endocytosed during plasma membrane repair traffic into the lumen of MVBs for degradation. Traffic 13, 483–494 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Peña-Llopis S et al. Regulation of TFEB and V-ATPases by mTORC1. Embo J. 30, 3242–3258 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Settembre C et al. TFEB links autophagy to lysosomal biogenesis. Science 332, 1429 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thoreen CC et al. An ATP-competitive mammalian target of Rapamycin inhibitor reveals Rapamycin-resistant functions of mTORC1*. J. Biol. Chem. 284, 8023–8032 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sahani MH, Itakura E & Mizushima N Expression of the autophagy substrate SQSTM1/p62 is restored during prolonged starvation depending on transcriptional upregulation and autophagy-derived amino acids. Autophagy 10, 431–441 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Harvey JD et al. A carbon nanotube reporter of microRNA hybridization events in vivo. Nat. Biomed. Eng. 1, 0041 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang X-X et al. pH-sensitive fluorescent dyes: Are they really pH-sensitive in cells? Mol. Pharm. 10, 1910–1917 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods-only references
- 57.Subbaiyan NK et al. Role of surfactants and salt in aqueous two-phase separation of carbon nanotubes toward simple chirality isolation. ACS Nano 8, 1619–1628 (2014). [DOI] [PubMed] [Google Scholar]
- 58.Quintero B, Cabeza MC, Martínez MI, Gutiérrez P & Martínez PJ Dediazoniation of p-hydroxy and p-nitrobenzenediazonium ions in an aqueous medium: Interference by the chelating agent diethylenetriaminepentaacetic acid. Can. J. Chem. 81, 832–839 (2003). [Google Scholar]
- 59.Streit JK, Fagan JA & Zheng M A low energy route to DNA-wrapped carbon nanotubes via replacement of bile salt surfactants. Anal. Chem. 89, 10496–10503 (2017). [DOI] [PubMed] [Google Scholar]
- 60.Zheng M & Diner BA Solution redox chemistry of carbon nanotubes. J Am Chem Soc 126, 15490–15494 (2004). [DOI] [PubMed] [Google Scholar]
- 61.Bolte S & CordeliÈRes FP A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 224, 213–232 (2006). [DOI] [PubMed] [Google Scholar]
- 62.Bankhead P et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 7, 16878 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data presented in the manuscript or the supplementary materials, including the uncropped gels and the raw data are available in the Source Data files.