

Identification of resistance mechanisms to KRASG12C/SHP2 coinhibition highlights the need for additional combination therapies for lung cancer beyond on-pathway combinations and offers the basis for development of more effective combination approaches.
Abstract
Although KRASG12C inhibitors show clinical activity in patients with KRAS G12C mutated non–small cell lung cancer (NSCLC) and other solid tumor malignancies, response is limited by multiple mechanisms of resistance. The KRASG12C inhibitor JDQ443 shows enhanced preclinical antitumor activity combined with the SHP2 inhibitor TNO155, and the combination is currently under clinical evaluation. To identify rational combination strategies that could help overcome or prevent some types of resistance, we evaluated the duration of tumor responses to JDQ443 ± TNO155, alone or combined with the PI3Kα inhibitor alpelisib and/or the cyclin-dependent kinase 4/6 inhibitor ribociclib, in xenograft models derived from a KRASG12C-mutant NSCLC line and investigated the genetic mechanisms associated with loss of response to combined KRASG12C/SHP2 inhibition. Tumor regression by single-agent JDQ443 at clinically relevant doses lasted on average 2 weeks and was increasingly extended by the double, triple, or quadruple combinations. Growth resumption was accompanied by progressively increased KRAS G12C amplification. Functional genome-wide CRISPR screening in KRASG12C-dependent NSCLC lines with distinct mutational profiles to identify adaptive mechanisms of resistance revealed sensitizing and rescuing genetic interactions with KRASG12C/SHP2 coinhibition; FGFR1 loss was the strongest sensitizer, and PTEN loss the strongest rescuer. Consistently, the antiproliferative activity of KRASG12C/SHP2 inhibition was strongly enhanced by PI3K inhibitors. Overall, KRAS G12C amplification and alterations of the MAPK/PI3K pathway were predominant mechanisms of resistance to combined KRASG12C/SHP2 inhibitors in preclinical settings. The biological nodes identified by CRISPR screening might provide additional starting points for effective combination treatments.
Significance:
Identification of resistance mechanisms to KRASG12C/SHP2 coinhibition highlights the need for additional combination therapies for lung cancer beyond on-pathway combinations and offers the basis for development of more effective combination approaches.
Introduction
The RAS superfamily (KRAS, HRAS, and NRAS) of membrane-bound GTPases regulate a variety of processes linked to cell differentiation, proliferation and survival, by acting as molecular switches for signaling pathways including MAPK and PI3K–AKT. Mutations in the KRAS isoform, in particular codon-12 missense mutations, promote its constitutive activation and are common oncogenic drivers for human cancer (1–4). These mutations inhibit transition of active, GTP-bound KRAS to its inactive GDP-bound state by rendering it insensitive to the action of GTPase activating proteins that promote GTP hydrolysis (5–7). A common oncogenic KRAS mutation that bears a cysteine-for-glycine substitution at codon 12 (KRASG12C) is associated with residual GTP hydrolysis rates that are amenable to targeting the KRASG12C–nucleoside complex in its inactive (GDP-bound) form (8, 9). Covalent modification of the C12 thiol irreversibly traps KRASG12C in the inactive state by preventing GTP loading by nucleotide exchange factors such as Son of Sevenless (SOS), and several such covalent KRASG12C inhibitors (i)—with target specificity conferred by a dynamic binding pocket under the KRAS Switch II loop adjacent to the nucleoside binding site—have demonstrated clinical efficacy to date against KRASG12C-dependent solid tumors (10–12). Multiple mechanisms of KRASG12Ci treatment resistance and MAPK pathway reactivation have been identified, including KRAS mutations affecting drug binding or nucleoside cycling, KRAS amplification, mutations in upstream activators or downstream components of MAPK signaling, and alterations to orthogonal pathways such as PI3K and WNT (13–24).
Novel KRASG12Ci and rational combination strategies may help overcome or prevent some types of resistance. One such may be to combine a KRASG12Ci with an inhibitor of Src homology region 2-containing protein tyrosine phosphatase 2 (SHP2), which positively regulates RAS activation downstream of activated receptor tyrosine kinases (RTK) by forming a complex between SOS and SHP2-binding RTK signaling adaptors (25). Such a combination would be expected to reduce RTK-mediated GTP nucleoside exchange, elevating the pool of inactive GDP-bound KRASG12C and increasing KRASG12Ci target occupancy (13, 20). Furthermore, reduced nucleoside exchange might also impair KRASG12Ci treatment-associated reactivation of signaling by both mutant and wild-type RAS paralogues, irrespective of upstream RTK activity (13, 14, 18, 21). We have previously shown promising antitumor and pharmacokinetic/pharmacodynamic (PK/PD) activity in xenograft and PDX models for a combination of the novel KRASG12Ci JDQ443 (20, 26), and the SHP2i TNO155 (27), and such KRASG12Ci + SHP2i combinations are currently under clinical evaluation. Herein, we describe, in mouse models using a non–small cell lung cancer (NSCLC) xenograft, the durability and associated PK/PD of KRAS G12C-mutated tumor response to JDQ443, with and without concomitant TNO155 and/or inhibitors of the RAS-associated pathway components PI3K (PI3Ki) and cyclin-dependent kinase 4/6 (CDK4/6i). We also describe genes and pathways associated with sensitivity and adaptive resistance to KRASG12Ci ± SHP2i identified by genome-wide CRISPR knockout in a panel of KRAS G12C-mutated cell lines, and validate the combination benefit between KRASG12Ci and SHP2i with inhibitors of other pathways identified in the CRISPR screen.
Materials and Methods
Compounds and antibodies
All inhibitors were synthesized at the Novartis Institutes for BioMedical Research. Recombinant human FGF was purchased from R&D Systems (Catalog no. 3718-FB). All antibodies were obtained from Cell Signaling Technology: phospho-p44/42 MAPK (ERK1/2; Thr202/Tyr204; Catalog no. 4370); phospho-Akt (Ser473; #9271); phospho-S6 ribosomal protein (Ser235/236; #2211); phospho-S6 ribosomal protein (Ser240/244; #2215), cleaved PARP (Asp214; #9541); CAS9 (S. pyogenes; #14697); HRP-linked anti-rabbit IgG (#7074); HRP-linked anti-mouse IgG (#7076).
Xenograft generation and in vivo PD assessments
LU99 was provided by RIKEN BRC (SCR_003250) through the National BioResource Project of the MEXT, Japan. LU99 xenografting, JDQ443 target occupancy (LC/MS) and DUSP6 expression analyses (quantitative RT-PCR) were performed as previously described (20) under the same ethical and regulatory frameworks. Ras activation was assessed with pulldown assay kits (Cytoskeleton # BK008) following the manufacturer's protocol. All drugs were dosed orally as aqueous formulations: JDQ443 and TNO155 in 0.5% methylcellulose/0.1% Tween-80; alpelisib in 1% carboxymethylcellulose/0.5% Tween-80, and ribociclib in 0.5% methylcellulose. Target occupancy data were normalized using the pan-RAS (KRAS, HRAS, NRAS) wild-type peptide LVVGAGGVGK. Total RAS protein levels were determined by targeted mass spectrometry of the same pan-RAS peptide; KRASG12C by peptide LVVVGACGVGK; total KRAS by QGVDDAFYTLVR (KRAS1; KRAS-4B specific) or SFEDIHHYR (KRAS2; common to KRAS-4B and KRAS-4A); NRAS by SFADINLYR, and HRAS by SFEDIHQYR.
KRAS mRNA expression
KRAS mRNA was assessed by quantitative RT-PCR in 384-well reaction plates (Applied Biosystems, #4309849) containing 20 ng/well of RNA. Each well also contained 10 μL of a reaction master mix comprising 5 μL of 2x concentrated One Step RT qPCR Mastermix (iTaq Universal Probes One-step kit, Bio-Rad, #172–5141), 0.5 μL primer, 20x concentrated KRAS probe (Applied Biosystems, #Hs00364282_m1) or beta actin probe (Integrated DNA Technologies, #Hs.PT.39a.22214847), 0.25 μL of the iScript RTase, and 1.75 μL RNase-free water. The plate was sealed with a MicroAmp clear adhesive film (Applied Biosystems, #4311971) and measured on a 7900HT Fast Real-Time PCR System (Thermo Fisher Scientific): 50°C for 10 minutes, 95°C for 3 minutes, then 40 cycles of 95°C for 15 seconds and 60°C for 1 minute. KRAS gene expression was normalized to beta actin using the delta Ct method of relative quantification (28) with induction fold change calculated relative to the vehicle (DMSO) control.
Cell culture
Human cancer cell lines were from the Cancer Cell Line Encyclopedia (SCR_013836; ref. 29), banked at Novartis Cell Bank, authenticated by SNP analysis and routinely tested as Mycoplasma-free. All cell lines were maintained at 37°C in 5% CO2. HEK293T, HEK293A and MIA PaCa-2 cells were cultured in DMEM medium (Gibco, #11995–040) supplemented with 10% FBS (Seradigm, #1500–500) and penicillin (100 units/mL)–streptomycin (100 mg/mL; Gibco, #15140–122). HCC44, NCI-H23, NCI-H1792, Calu1, NCI-H2030, NCI-H2122 and NCI-H1373 cells were cultured in RPMI1640 medium (Gibco, #22400–071) supplemented with 10% FBS and penicillin–streptomycin.
CRISPR screening, experimental procedure and data analysis
Genome-wide CRISPR knockout screening in the presence or absence of anchor drugs (KRASG12Ci1 ± SHP099) was undertaken as previously described (30) and data analyzed by the procedure of Dafflon and colleagues (31). The genome-wide single-guide RNA (sgRNA) library targeted 18,360 protein-coding genes (10 sgRNAs per gene) and was divided into 3 libraries and transduced into Cas9-expressing human KRAS G12C-mutated cancer cell lines at a multiplicity of infection of 0.5, aiming for an average coverage of 1,000 cells per sgRNA reagent. Selection was optimized by determining the puromycin dose required to achieve >95% cell killing in 72 hours. Cell viability was measured for a 6-point dose–response ranging from 0 to 20 mg/mL puromycin using the CellTiter-Glo assay (Promega, #G7570). Additional detail is given in the supplement.
Dose–matrix combination assays
Cells were seeded in 96-well plates at 5000 (NCI-H23), 4000 (NCI-H441), 3500 (Calu1, NCI-H1792, MIA PaCa-2, Calu1) or 2000 (NCI-H1373, HCC44) cells/well in 25 μL growth medium, and compounds added in a concentration matrix (triplicate assessments) the following day. Cell viability was determined at day 0 (untreated cells) and day 5 by CellTiter-Glo assay (Promega, #G7570). For a given (triplicate) assay signal S, vehicle signal V, and corresponding day 0 signal D0, growth inhibition was defined as
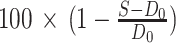 , where S < D0, or
, where S < D0, or
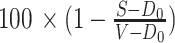 where S ≥ D0.
where S ≥ D0.
Colony formation assay
Cells (0.5 × 104 to 1.0 × 104/well) were seeded into 6-well tissue culture plates and left to adhere overnight in complete cell culture medium. The medium was replaced with fresh medium containing drugs or DMSO vehicle, and cells incubated for 7 to 10 days (DMSO) or 3 to 5 weeks (drugs) with medium/drug changed twice weekly. Remaining cells were washed with PBS, and fixed/stained with 0.2% crystal violet (Fisher Scientific, #C581–25) in 4% paraformaldehyde (room temp, 30 minutes) before being washed with water (×3) and dried in air. Cells were washed (×3) with water and allowed to air dry. Colony formation was quantified using an EPSON Perfection V600 scanner and the ColonyArea ImageJ plugin (32).
Protein extraction and immunoblotting
Cells were lysed in RIPA buffer (Thermo Fisher Scientific, #89901) supplemented with 100× Protease Inhibitor Cocktail (Sigma, #P8340), 100 × Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific, #78427) and 25 units/mL Benzonase Nuclease (Sigma, #E8263). Lysate was vortexed (30s × 2) and centrifuged for 10 minutes at 13,000 rpm (4°C). Protein concentration was determined using the DC Protein Assay Kit (Bio-Rad, #5000112) according to the manufacturer's instructions. Proteins were resolved by SDS-PAGE and transferred to nitrocellulose membranes (Bio-Rad, #1704159EDU) using a Trans-Blot Turbo Transfer System (Bio-Rad, #1704150EDU) according to the manufacturer's instructions. Membranes were blocked for 1 hour (room temperature) with 5% Blotting-Grade Blocker (Bio-Rad, #1706404) in Tris Buffered Saline with Tween 20 (TBST) (Sigma, #T9039), incubated overnight (4°C) with primary antibodies diluted in 5% BSA (Akron Biotechnology, #AK8917–0100), washed with TBST, and incubated with horseradish peroxidase–conjugated secondary antibody in 5% Blotting-Grade Blocker. Visualization was with Amersham ECL Western Blotting Detection Reagents (GE Healthcare, #RPN2106) or SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Fisher Scientific, #34580) and Amersham Hyperfilm ECL (GE Healthcare, #28906839).
Gene copy analysis
KRAS gene copy number was analyzed using a SYBR Green quantitative RT-PCR assay using the ER qPCR Supermix universal (#A10314, Invitrogen) on a 7900 HT RT PCR system (Applied Biosystems). KRAS primers (forward: TGGTCCTTTTCCGTGTGTAGG; reverse: AACTCGGAGGCAGATGCATTT) were derived from the EMBL database using the GCG Prime software package (Genetics Computer Group) and synthesized by MicrosynthAG. Long Interspersed Element-1 (LINE-1) was used as the reference gene amplimer (forward: AAAGCCGCTCAACTACATGG; reverse: TGCTTTGAATGCGTCCCAGAG). Genomic DNA (gDNA) samples were extracted from pulverized frozen tumor tissue using the Qiagen DNeasy Blood and Tissue kit (#69504) and 1.2 μg (2 μL) was amplified in triplicate in 384-well plates after addition of 8 μL/well of a mastermix containing 5 μL of 2× assay mix, 0.2 μL assay ROX (carboxyrhodamine) solution, 0.3 μL (300 nmol/L) Line-1 or 0.2 μL (200 nmol/L) KRAS primers, and water. Calibrator human gDNA (Promega, #G3041) was amplified in triplicate in a 1:4 serial dilution ranging from 0.024 ng/well to 100 ng/well. Amplification conditions were 95°C 10 minutes; 40 cycles of 95°C 10 s, 60°C 30 s; disassociation gradient 55°C to 5°C 30 seconds. KRAS gene copy number was calculated using the standard curve method previously described (33).
Data sharing
The data generated in this study are available within the article and its Supplementary Data files, and are also available upon request from the corresponding author. The lp-WGS and RNAseq data generated in this study are publicly available in the the DNA DataBank of Japan (DDBJ) at JGAS000643 (https://ddbj.nig.ac.jp/resource/jga-study/JGAS000643) and the CRISPR screening data in the SRA database at PRJNA1033544 (https://www.ncbi.nlm.nih.gov/sra/?term=PRJNA1033544).
Results
LU99 tumor progression after response to JDQ443 monotherapy is associated with upregulation of KRASG12C expression
Consistent with previous data (20, 26), oral JDQ443 treatment of mice bearing LU99 (KRASG12C NSCLC) tumor xenografts resulted in dose-dependent tumor growth inhibition over the range 10 to 100 mg/kg once daily, with regression occurring at the highest dose. However, growth resumed at 100 mg/kg after 14 days of treatment (Fig. 1A).
Figure 1.

A, Lu99 xenograft tumor growth curves under JDQ443 treatment. B, JDQ443 PK and PD at end of JDQ443 treatment (day 18/day 32). C, JDQ443 (100 mg/kg once daily) PK and PD during tumor regression (day 10). D, Tumor KRAS protein levels (G12C and wild type). E, Tumor wild-type pan-RAS protein levels at end of JDQ443 treatment. F, KRAS mRNA fold change versus vehicle at end of JDQ443 treatment. *, P < 0.05 versus vehicle; #, P < 0.05 versus each other; ns, nonsignificant, P ≥ 0.05. QD, once daily.
To investigate the PD and molecular mechanism of progression, tumors at all three dose levels were harvested and analyzed at end of treatment (10 mg/kg and 30 mg/kg at day 18, 100 mg/kg at day 32) when tumor volume had reached approximately 400 mm3. Dose-dependent reductions in DUSP6 mRNA expression (a PD measure of MAPK pathway suppression) and free KRASG12C (a PD measure of JDQ443 target occupancy) were observed at 6 hours post dose on day 18 for 10 mg/kg and 30 mg/kg treatment, but there was no additional reduction at day 32 in tumors progressing at 100 mg/kg. By 24 hours post dose, both these PD measures at the two lower doses had returned to approximately vehicle control levels, but in the 100 mg/kg group—where tumors were progressing after an initial period of regression—both PD measures had increased to well above the control (Fig. 1B). By contrast, both PD markers were inhibited at 24 hours, albeit to a lesser extent than at 6 hours, in this dosing group on day 10, where regression was still ongoing (Fig. 1C). The data therefore suggested that KRASG12C amplification may be linked to the resumption of tumor growth, which appears to be a common mechanism of resistance in preclinical and clinical settings (21, 34). Consistent with this, overall KRAS protein levels in progressing tumors were significantly (∼4-fold) elevated versus vehicle or the lower dosing levels that did not cause regression (Fig. 1D; Supplementary Fig. S1). Wild-type pan-RAS proteins were largely unaffected by dose or progression status (Fig. 1E), indicating a KRASG12C-specific upregulation leading to inadequate target occupancy and inhibition of the driver oncogene. KRAS mRNA levels reflected the protein data (Fig. 1F).
Combining JDQ443 with a SHP2 inhibitor delays JDQ443 resistance without altering the mechanism
To address the question of whether upregulation of KRASG12C expression in progressing tumors was affected by combination with a SHP2i, we studied the effects of JDQ443 + TNO155 treatment in LU99 xenografts.
SHP2 inhibition alone (TNO155 10 mg/kg BID) only partially inhibited tumor growth compared with strong regression (–58%) observed for JDQ443 100 mg/kg once daily monotherapy. By contrast, JDQ443 + TNO155 (5 or 10 mg/kg twice a day) in combination further improved tumor response (–66% to –76% regression) for both continuous and intermittent TNO155 dosing (Fig. 2A). A pronounced extension of regression durability to ∼24 to 27 days was also observed upon combination treatment, versus 16 days for JDQ443 monotherapy (Fig. 2A).
Figure 2.

A, LU99 xenograft tumor growth curves for JDQ443, TNO155, or both. B–D, PK and tumor PD of TNO155 10 mg/kg BID, JDQ443 100 mg/kg, or both, at end of treatment period. E, Tumor KRAS protein levels (G12C and wild type). F, Tumor wild-type pan-RAS protein levels. G, Tumor KRAS gene copy number at end of treatment; H, LU99 tumor growth curves for JDQ443 + TNO155 dosed together from day 1 or following early tumor progression on JDQ443 as a single agent. *, P < 0.05 versus vehicle; #, P < 0.05 versus each other; ns, nonsignifcant, P ≥ 0.05. BID, twice daily; QD, once daily.
As before, tumors were harvested and analyzed at end of treatment (day 21 for JDQ443 and TNO155 monotherapy, day 36 for combination treatment) at a volume of approximately 400 mm3. TNO155 alone only weakly inhibited DUSP6 mRNA (Fig. 2B). JDQ443 monotherapy of progressing tumors promoted stronger DUSP6 suppression at 3 hours post dose that declined to similar levels to TNO155 treatment by 24 hours, while free KRASG12C went from strong 3 hours suppression to slightly above vehicle levels at 24 hours (Fig. 2C). For combination treatment, in similarly-sized progressing tumors harvested later at day 36, there was no additional early suppression of DUSP6 while 24-hour DUSP6 and free KRASG12C were both substantially higher than vehicle controls (Fig. 2D). Overall KRAS protein levels were slightly (∼1.5-fold) but significantly elevated over controls at day 21 in progressing tumors treated with JDQ443 alone, but much more highly elevated (∼5-fold) at day 36 under combination treatment (Fig. 2E; Supplementary Fig. S2A). As before, these elevations were specific to KRASG12C, as pan-RAS wild-type protein levels (Fig. 2F) as well as individual HRAS and NRAS isoform levels (Supplementary Fig. S2B and S2C) were unaffected by time or treatment. Notably, this upregulation of KRAS protein was associated with a 3-fold increase in KRAS gene copy number (4 copies to 12 copies) in day 36 tumors progressing on combination therapy (Fig. 2G). We also evaluated whether delayed combination treatment with TNO155 could rescue early tumor progression on JDQ443 monotherapy. Intriguingly, the addition of TNO155 (10 mg/kg BID) on day 30 to progressing tumors receiving JDQ443 monotherapy (100 mg/kg once daily) stabilized tumor growth through the end of treatment at day 49 (Fig. 2H). These data therefore suggest that initiating SHP2i treatment in the context of early KRASG12Ci-induced KRAS amplification might provide clinical benefit, although clinical data will be needed to explore the duration and extent of this effect.
PI3K-mTOR-CDK4/6 signaling axis inhibition extends the duration of KRASG12Ci + SHP2i antitumor activity in vivo
Alpelisib (a PI3Kαi) and ribociclib (a CDK4/6i) augment JDQ443 antitumor activity in patient-derived mouse xenografts of KRAS G12C-mutant NSCLC and colorectal tumors (20). The impact of alpelisib and ribociclib on JDQ443 + TNO155 response was explored in LU99 xenografts, which in addition to KRAS G12C also harbor the PIK3CA T1025A mutation and a CDKN2A deletion. Neither alpelisib (50 mg/kg once daily) nor ribociclib (75 mg/kg once daily) had significant antitumor activity as single agents (Fig. 3A), potentially due to resistance conferred by the KRAS G12C mutation. In combination with JDQ443, however, both agents increased tumor regression over JDQ443 monotherapy to an extent comparable to that for seen for the TNO155 combination—with an approximately 70% tumor volume reduction at day 14 for both alpelisib or ribociclib combinations versus ∼50% for JDQ443 alone—and also comparably extended the duration of the response (Fig. 3B). Notably, triple combinations of JDQ443 + TNO155 with either alpelisib or ribociclib further extended the duration of response, while a combination of all four agents gave a further significant extension (Fig. 3B). The mechanism of tumor resistance remained the same for all combinations, with increased KRAS mRNA expression (Fig. 3C) and gene copy number (Fig. 3D) in progressing tumors relative to vehicle controls, with a trend towards increasing mRNA and gene copy with increasing number of combination drugs. Copy number analysis of the outgrowing tumors upon quadruple combination (vs. vehicle control) by lpWGS showed a heterogeneous pattern across tumors with a minimal, commonly amplified peak region containing the KRAS gene (Supplementary Fig. S3A). Sequencing counts revealed that the KRAS mutant but not the KRAS wt allele was amplified (Supplementary Fig. S3B). RNA-seq profiling and GSEA identified several gene signatures representing biological processes that might also contribute to the outgrowth of those tumors (Supplementary Fig. S3C).
Figure 3.

A, LU99 tumor growth curves for JDQ443, alpelisib, and ribociclib as single agents. B, Growth curves for JDQ443-based combination regimens. C and D, Fold changes versus vehicle in KRAS mRNA expression (C) and KRAS gene copy number (D) at end of combination treatment. *, P < 0.05. QD, once daily.
Genome wide CRISPR-CAS9 screening to identify genetic modifiers of response to KRASG12Ci ± SHP2i
An unbiased genome-wide loss of function genetic screen was performed to identify genes whose suppression affects the sensitivity of KRAS G12C-mutated NSCLC cell lines to KRASG12Ci ± SHP2i combinations. Screening was performed using CRISPR-Cas9 gene editing in a panel of five KRAS G12C-mutated NSCLC cell lines (NCI-H1373, NCI-H1792, HCC44, Calu1, NCI-H23) stably transfected to express Cas9, with selection by early tool inhibitors of SHP2 (SHP099; ref. 35) and KRASG12C (KRASG12Ci1 [Araxes WO2014/143659]; Supplementary Fig. S4) as anchor drugs (Fig. 4A). Target activity and appropriate inhibitor concentrations for screening were established by colony formation and PD analyses in 9 KRAS G12C-mutated cell lines comprising the CRISPR screening panel (non Cas9-expressing), the NSCLC lines H358, NCI-H2122, and NCI-H2030, and the pancreatic cancer line MIA PaCa-2. Combination treatment with KRASG12Ci1 + SHP099 showed greater dose-dependent colony inhibition (Supplementary Fig. S5) and PD inhibition of MEK and ERK1/2 phosphorylation than KRASG12Ci1 alone (Supplementary Fig. S6), as well as less GTP-bound active KRAS by GST-RBD pulldown assay (Supplementary Fig. S7A) and less free KRASG12C (Supplementary Fig. S7B). From these data, we estimated that adequate on-target response, particularly with respect to inhibition of KRAS activation and free KRASG12C, would be achieved by 0.75 μmol/L KRASG12Ci1 ± 10 μmol/L SHP099, and these concentrations were carried forward into CRISPR screening.
Figure 4.

A, Overview of the CRISPR screening approach. B and C, Individual sensitizing genes to the combination of KRASG12Ci1 and SHP099. D and E, Protein interaction networks for genes sensitizing genes identified by CRISPR screening. F, Individual rescuer genes to the combination of KRASG12Ci1 and SHP099. G, Protein interaction networks for rescuer genes.
Sensitizing and rescue genes to KRASG12C inhibition form distinct functional networks
Across the cell panel, strongly sensitizing genes to KRASG12Ci1 + SHP099 fell into two clusters: one involving heparan sulfate proteoglycan synthesis involved in RTK ligand presentation and the RTK FGFR1 itself (including FGFR1, B4GALT7, EXT1, EXT2 and EXTL3; Fig. 4B), and one involving adaptor and effector genes (including MAPK1, SHOC2, FRS2, MAPK14, PDCL and CRKL; Fig. 4C). The strongest individual sensitizer was FGFR1, followed by B4GALT7 and EXT2 (Table 1). Similar clustering occurred with KRASG12Ci1 as a single agent, although individual genes showed lower fold changes between wild-type and knockout cells, and the rank ordering was not the same as for the combination—particularly FGFR1 that had a substantially lesser significance in the absence of SHP099 (Supplementary Fig. S8). No significant sensitizers were observed for SHP099 alone.
Table 1.
Sensitizer and rescue genes to KRASG12Ci1 + SHP099 combination treatment.
| Sensitizers | Rescuers | |||||||
|---|---|---|---|---|---|---|---|---|
| Gene | LogFC | P value | Gene | LogFC | P value | Gene | LogFC | P value |
| FGFR1 | –1.7 | 0.0022 | PTEN | 3.1 | 0.0010 | TAOK1 | 1.2 | 0.0197 |
| B4GALT7 | –1.5 | <0.0001 | KCTD5 | 2.8 | 0.0013 | CIC | 1.2 | 0.0073 |
| EXT2 | –1.5 | <0.0001 | MED12 | 2.4 | 0.0153 | NF1 | 1.2 | 0.0004 |
| EXT1 | –1.4 | <0.0001 | TAF5L | 2.1 | 0.0156 | CTDSPL2 | 1.2 | 0.0117 |
| CRKL | –1.4 | 0.0038 | PPP2R4 | 2.0 | 0.0093 | PPP4C | 1.2 | 0.0338 |
| EXTL3 | –1.3 | <0.0001 | TIPRL | 2.0 | 0.0033 | MAU2 | 1.2 | 0.0268 |
| RER1 | –1.2 | 0.0027 | TSC2 | 1.9 | 0.0223 | SUPT20H | 1.1 | 0.0376 |
| MCL1 | –1.1 | 0.0221 | TADA1 | 1.7 | 0.0234 | DAPK3 | 1.1 | 0.0010 |
| TNPO1 | –1.1 | 0.0078 | SPRY2 | 1.6 | 0.0091 | C16orf72 | 1.1 | 0.0090 |
| MAPK1 | –1.0 | 0.0015 | TSC1 | 1.6 | 0.0226 | ARIH2 | 1.1 | 0.0074 |
| PDCL | –1.0 | 0.0014 | PDCD10 | 1.6 | 0.0003 | ROCK1 | 1.1 | 0.0079 |
| SHOC2 | –1.0 | 0.0127 | SOCS3 | 1.5 | 0.0207 | CCNC | 1.1 | 0.1064 |
| RAC1 | –1.0 | 0.0158 | CAB39 | 1.5 | 0.0060 | ROCK2 | 1.0 | 0.0015 |
| HS2ST1 | –0.9 | 0.0010 | NF2 | 1.4 | 0.0049 | KEAP1 | 1.0 | 0.0547 |
| RAF1 | –0.9 | 0.0271 | CYB5R4 | 1.4 | 0.0065 | UBE2F | 1.0 | 0.0025 |
| GRB2 | –0.8 | 0.0390 | CUL5 | 1.4 | 0.0007 | MED24 | 1.0 | 0.1189 |
| GLCE | –0.8 | <0.0001 | TADA2B | 1.3 | 0.0511 | |||
Abbreviation: LogFC, log-fold change.
Protein interaction network analysis of sensitizers to combination treatment identified one distinct network comprising FGFR/MAPK/integrin signaling genes (Fig. 4D), and another involving genes for heparan sulfate proteoglycan synthesis/RTK ligand presentation (Fig. 4E).
Using a similar approach, strong rescuers from the activity of KRASG12Ci1 + SHP099 were also identified (Fig. 4F; Table 1), with the strongest being PTEN. As with the sensitizers, many of these rescue genes were also observed for KRASG12Ci1 as a single agent (Supplementary Fig. S8). Network analysis identified a number of interrelated nodes corresponding to both vertical and orthogonal pathways, including the MAPK, PI3K-mTOR-STK11/LKB1, Rho and YAP pathways, the latter through NF2 loss, plus genes involved in transcriptional initiation, and genes belonging to the E3 ubiquitin ligase family (Fig. 4G). A number of these genes, including NF1, YAP1, KEAP1, MED12, and CIC, have been previously identified as predictors of sensitivity to BRAF, MEK, and RTK inhibitors (36–40), and inactivating mutations in KEAP1 ± STK11 are known to reduce clinical response to KRASG12C inhibition (12).
KEAP1 and STK11 mutated KRAS G12C cells show distinct rescue networks to KRASG12Ci + SHP2i treatment
Given the association of KEAP1 and STK11 mutations in clinical KRASG12Ci response, a second analysis was undertaken to identify genetic modifiers of KRASG12Ci1 + SHP099 activity in KRAS G12C-mutated cells carrying mutations in KEAP1 (NCI-H1792) or KEAP1 and STK11 (NCI-H23, HCC44), as opposed to those wild-type for both genes (NCI-H1373, Calu1).
No specific sensitizing genes were identified in either KEAP1- or KEAP1/STK11-mutated cells that were not also present in the other two groups. By contrast, five sensitizing genes were identified in KEAP1 and STK11 wild-type cells that were not present in the mutated lines (ITGB1, ALG5, DLG5, TM2D1, and TM2D3).
For specific rescuers, 69 genes were identified in the KEAP1/STK11-mutated group (Supplementary Table S1), and S24 in the in the KEAP1-mutated group (Supplementary Table S2). In accordance with the role of STK11 and KEAP1 as regulators of metabolism (41), gene set enrichment analysis showed the majority of these genes to be involved in the mitochondrial respiration chain complex 1, the NADH dehydrogenase complex, the oxidative phosphorylation pathway, the oxidoreductase complex, or present in the inner mitochondrial matrix (Supplementary Fig. S9). Four rescue genes specific to KEAP1/STK11 wild-type cells were identified (KEAP1, RFWD2, ERF, and PSM6), mostly associated with NRF2 transcriptional activity.
FGF activity significantly attenuates KRASG12Ci + SHP2i treatment effects
The CRISPR data show FGF activity to be implicated in attenuating KRASG12Ci + SHP2i response, both directly via the sensitizing effect of FGFR1 knockout and indirectly through the biosynthesis of heparan sulfate proteoglycans, which act as coreceptors for FGFR1 and other signaling receptors (42). Consistent with this, and with previous reports that FGFR signaling is not fully mediated through SHP2 (43), supplementation of cell growth medium with exogenous FGF (50 ng/mL) partially rescued colony formation under KRASG12Ci1 + SHP099 treatment and pERK inhibition (Fig. 5A and B, respectively, for NCI-H23 cells; with similar results for other lines). Furthermore, supplementation of growth medium with the pan-FGFR inhibitor BGJ398 (44) augmented the antiproliferative activity of KRASG12Ci1 ± SHP099 in colony formation assays (Fig. 5C for NCI-H23), and further reduced levels of pERK and pAKT (Supplementary Fig. S10).
Figure 5.

A and B, Inhibition of NCI-H23 colony formation (A) and pERK (B) under KRASG12Ci1 + SHP099 with or without exogenous FGF (50 ng/mL). C, Effect of FGFR1 inhibition with BJG398 on NCI-H23 colony formation under treatment with KRASG12Ci1 ± SHP099.
Pan-PI3K inhibition shows combination benefit with KRASG12Ci ± SHP2i treatment in cell cultures
Matrix combination studies were undertaken in the CRISPR screening panel to explore the antiproliferative effects of combining KRASG12Ci1 ± SHP099 with inhibitors of upstream and downstream/orthogonal RAS pathway components – specifically erlotinib (EGFRi), BGJ398 (FGFRi), trametinib (MEKi), alpelisib (PI3Kαi), and GDC0941 (pan-PI3Ki; ref. 45).
Combination results were overall similar for both KRASG12Ci1 alone and KRASG12Ci1 + SHP099, although greater antiproliferative effects were seen with the KRASG12Ci1 + SHP099 treatment. Fig. 6A–E shows inhibitor matrices (NCI-H23) with KRASG12Ci1 + SHP099, and Supplementary Fig. S11 the corresponding NCI-H23 matrices with KRASG12Ci1 alone. Supplementary Figs. S12–S15 show inhibitor matrices for KRASG12Ci1 ± SHP099 in the other four cell lines in the testing panel. With the exception of NCI-H1792 (Supplementary Fig. S12), the strongest effect in all cells and combinations was observed with the pan-PI3Ki GDC0941, and, in general, the second strongest was with the PI3Kα-specific inhibitor alpelisib. Notably, NCI-H1792 showed an unusually high dependence on FGFR among the tested cells, with combination effects seen with BGJ398 + KRASG12Ci1 ± SHP099 that were directly comparable to pan-PI3K inhibition with GDC0941 + KRASG12Ci1 ± SHP099.
Figure 6.

NCI-H23 dose combination matrices for RAS-associated pathway inhibitors with KRASG12Ci1 + SHP009 (10 μmol/L) + BGJ398 (FGFRi; A), erlotinib (EGFRi; B), trametinib (MEKi; C), alpelisib (PI3Kαi; D), or GDC0941 (pan-PI3K; E). SS, synergy score (Loewe's index).
The combination benefit observed with pan-PI3K inhibition was also reflected in long-term (48-hour) cell signaling responses. Across cell lines, the triple combination of KRASG12Ci1 + SHP099 + GDC0941 showed the strongest inhibition of pERK, pAKT, and downstream MAPK/PI3K convergence nodes pS6-S235/236 and pS6-S240/244 (Supplementary Fig. S10).
Discussion
In the LU99 mouse tumor model, loss of response to JDQ443 was associated with specific upregulation of KRASG12C protein, resulting from a gene amplification. This is reminiscent of the amplification seen in some clinical progressions on KRASG12C inhibitors (23), which in contrast to most low frequency mutations, seemed to mirror clinical progression (46). While dual, triple and quadruple on-pathway combinations of JDQ443 with TNO155, alpelisib and ribociclib significantly extended the durability of LU99 tumor response, especially for the four-drug combination, KRAS amplification and tumor progression ultimately occurred with every regimen. The stronger the treatment pressure the higher the KRAS G12C gene amplification that was observed to further outcompete drug binding and inhibition of the driver oncogene. Thus, in addition to the high risk of treatment-limiting toxicity with higher-order on-pathway combinations, such regimens also appear unlikely to prevent clinical resistance driven by KRAS amplification, underscoring the need for distinct approaches. It remains to be seen whether new inhibition modalities such as covalent ON-state inhibitors, pan-KRAS/pan-RAS inhibitors, or KRAS degraders (pan- or G12C-specific) will prevent or better control amplification-based resistance, as they might – respectively – fully block the oncogenic driver, prevent reactivation of the pathway via KRAS/RAS wild-type paralogues, or provide a further stoichiometric advantage through the processive nature of degradation. It also remains eminently important to assess whether alternative combination approaches with non-overlapping resistance mechanisms against KRAS-amplified subpopulations can overcome this mode of resistance, for example, via orthogonal combination approaches that shift the mechanism to cell killing.
While not preventing eventual resistance, the combination of a KRASG12Ci and a SHP2i provided a significant increase in LU99 tumor response over KRASG12C inhibition alone, and it is interesting that the benefit of JDQ443 + TNO155 was apparent whether the combination was administered ab initio (wherein tumor regression was extended and increased in magnitude) or following early loss of tumor response to JDQ443 as a single agent (wherein further tumor growth was curtailed for an extended period). Of note, recent clinical data for the KRASG12Ci sotorasib plus the SHP2i RMC-4630 in KRAS G12C-mutated NSCLC showed responses in three of six KRASG12Ci-naïve patients compared with none of six KRASG12Ci-experienced patients (47). However, mechanistic interpretation of these data is limited because neither time of initiation after initial KRASG12Ci failure nor the cause of KRASG12Ci resistance was fixed in this trial. Further in vivo studies to explore immediate versus delayed KRASG12Ci + SHP2i combinations in the context of KRAS amplification are merited.
Currently, about half of the clinical resistance landscape cannot be explained by mutational or copy number alterations and might be due to adaptive signaling resistance mechanisms. A complex and interconnected network of genes and pathways involved in modifying response to KRASG12Ci ± SHP2i activity was identified by genome-wide CRISPR knockout. The observation that FGFR1 inactivation was the strongest individual sensitizer to combination treatment across a panel of KRAS G12C-mutated cell lines was confirmed by the results on cell proliferation of both FGF supplementation and FGFR inhibition, and is consistent with the ability of FGFR signaling to bypass SHP2 in some settings (42, 48). Also of relevance, FGFRi + MEKi treatment is synergistic in KRAS-mutated cancers due to feedback engagement of FGFR1 under MEK inhibition (49). The identification of PTEN as the strongest rescuer from KRASG12Ci1 + SHP099 activity, along with TSC1 and TSC2, is consistent with the known reduction of MAPK-pathway inhibitor activity associated with hyperactivation of PI3K–AKT signaling (50). PI3K is a RAS effector; an association between RAS association and the catalytic domain p110α is essential for lung tumor maintenance (51), and its disruption by mutations in the p110α RAS-binding domain is protective against KRAS-mutant tumor formation in mouse models (52). However, as PIK3CA was not implicated in the CRISPR screen, it appears that orthogonal activation by RTKs was the primary mechanism of PI3K–AKT–mTOR pathway activity in this system, either alone or in combination with RAS-mediated activation.
Several other CRISPR-identified rescue genes and pathways are consistent with data in the literature. KEAP1 knock-out acting as rescuer to KRASG12C -inhibition is to be expected, given the poorer response to sotorasib observed in KEAP1-mutated KRAS G12C NSCLC (12) The association of multiple genes associated with mitochondrial respiration as rescuers in KEAP1- and KEAP1/STK11-mutated cells is of interest given that the KEAP1/NRF2 and STK11 pathways are known to promote RAS independence via metabolic reprogramming (53). These correlations are however based on a small number of models (2 vs. 3) and need to be validated in a larger cell panel.
The mTOR and YAP rescue pathways identified in our screen have also been discussed as promoting KRAS independence by metabolic reprogramming, and also by triggering the epithelial-to-mesenchymal transition (53); YAP pathway activation was recently identified as a potential adaptive mechanism in a patient with polymorphic resistance to sotorasib (54). Previous studies demonstrate Tead cooperation with AP1 transcription factors to coordinate target gene transcription in cancer (55) and show strong potential of combined targeting of the Hippo and MAPK pathways in KRAS-dependent tumors while YAP activation was shown as resistance mechanisms to MAPK inhibition (56, 57).
Finally, rescue genes identified from the CUL5 E3 ubiquitination complexes have been implicated as modifiers of RAS-associated pathways, with CUL5 and ARIH2 as resistance genes to thymidine kinase inhibitors targeting EGFR-mutant NSCLC (58).
In conclusion, we have identified a number of potential pathways of adaptive clinical resistance to KRASG12C inhibitor activity, with or without augmentation by inhibitors of SHP2, from polyclonal outgrowth of KRAS G12C-amplified cells to interconnected networks and pathways of highly variable function. CRISPR screening identified important roles for the FGFR1-MAPK and PI3K axes, but, notably, targeting these axes did not result in strong cell death in culture experiments, although it did augment KRASG12Ci + SHP2i antiproliferative activity. Taken together, this work identified the FGFR1 and PI3K nodes as potential combination partners in the resistance setting to KRASG12C + SHP2 inhibition that warrants further investigation to support potential clinical investigations. The more novel pathways identified, like the YAP, CUL5-SOCS3, SAGA and mediator complexes, and the mitochondrial respiration pathways identified as important rescuers in KRAS G12C/KEAP1 co-mutated cells, may also provide the basis for further exploring alternative combination approaches.
Supplementary Material
Supplementary methods, tables and figures
Acknowledgments
The work described herein was funded by Novartis Pharmaceuticals.
Editorial assistance with this manuscript was provided by Nick Fitch of ArticulateScience (London, United Kingdom) with funding from Novartis Pharmaceuticals. Due to citation limitations, we were unable to include all the relevant literature in this manuscript. We have included references that we believe are the most critical to support our findings and conclusions. We thank all authors who have contributed to the field, and their work has influenced our research.
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Authors' Disclosures
A. Weiss reports a patent for WO/2022/135346 pending and is employed and holds stocks at Novartis. H. Voshol reports other support from Novartis during the conduct of the study and outside the submitted work. K. Sprouffske reports other support from Novartis during the conduct of the study and outside the submitted work. M. Meistertzheim reports being employed by Novartis and owning shares in the company. M. Kazic-Legueux reports being a Novartis employee and having shares in the company. T. Kottarathil reports being employed at Novartis and owning shares at Novartis. L. Martinuzzi-Duboc reports being an employee of Novartis and having stocks at Novartis. A. Buhles reports being an employee of Novartis and having stocks at Novartis. F. Adler reports being an employee of Novartis and having stocks at Novartis. L. Tordella reports other support from Novartis Pharma outside the submitted work. L. Sansregret reports personal fees from Novartis during the conduct of the study. Sauveur-Michel Maira reports other support from Novartis Pharma AG during the conduct of the study. D. Graus Porta reports being an employee of Novartis during the conduct of the study and being a shareholder of Novartis shares. C. Fedele reports Novartis employment and being a shareholder. S.M. Brachmann reports a patent for WO/2022/135346 pending to Novartis and is an employee of Novartis and holds stocks of Novartis. No disclosures were reported by the other authors.
Authors' Contributions
A. Prahallad: Conceptualization, resources, supervision, investigation, visualization, methodology, writing–original draft, writing–review and editing. A. Weiss: Conceptualization, formal analysis, supervision, validation, investigation, visualization, methodology, writing–original draft, writing–review and editing. H. Voshol: Resources, investigation, writing–review and editing. G. Kerr: Resources, data curation, software, formal analysis, investigation, visualization, writing–original draft, writing–review and editing. K. Sprouffske: Data curation, software, formal analysis, visualization, writing–review and editing. T. Yuan: Resources, formal analysis. D. Ruddy: Resources, data curation, software, validation, investigation, methodology, writing–review and editing. M. Meistertzheim: Formal analysis, investigation. M. Kazic-Legueux: Formal analysis, validation, investigation. T. Kottarathil: Formal analysis, validation, investigation. M. Piquet: Validation, investigation. Y. Cao: Investigation. L. Martinuzzi-Duboc: Formal analysis, investigation. A. Buhles: Formal analysis, investigation. F. Adler: Formal analysis, investigation. S. Mannino: Investigation. L. Tordella: Writing–review and editing. L. Sansregret: Writing–review and editing. S. Maira: Resources. D. Graus Porta: Validation, writing–review and editing. C. Fedele: Conceptualization, writing–original draft, writing–review and editing. S.M. Brachmann: Conceptualization, resources, supervision, validation, writing–original draft, project administration, writing–review and editing.
References
- 1. Cancer Genome Atlas Research N, Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, et al. The cancer genome atlas pan-cancer analysis project. Nat Genet 2013;45:1113–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, et al. COSMIC: mining complete cancer genomes in the catalogue of somatic mutations in cancer. Nucleic Acids Res 2011;39:D945–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Molina JR, Yang P, Cassivi SD, Schild SE, Adjei AA. Non–small cell lung cancer: epidemiology, risk factors, treatment, and survivorship. Mayo Clin Proc 2008;83:584–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer 2003;3:459–65. [DOI] [PubMed] [Google Scholar]
- 5. Donovan S, Shannon KM, Bollag G. GTPase activating proteins: critical regulators of intracellular signaling. Biochim Biophys Acta 2002;1602:23–45. [DOI] [PubMed] [Google Scholar]
- 6. Bos JL, Rehmann H, Wittinghofer A. GEFs and GAPs: critical elements in the control of small G proteins. Cell 2007;129:865–77. [DOI] [PubMed] [Google Scholar]
- 7. Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer 2007;7:295–308. [DOI] [PubMed] [Google Scholar]
- 8. Hunter JC, Manandhar A, Carrasco MA, Gurbani D, Gondi S, Westover KD. Biochemical and structural analysis of common cancer-associated KRAS mutations. Mol Cancer Res 2015;13:1325–35. [DOI] [PubMed] [Google Scholar]
- 9. Li C, Vides A, Kim D, Xue JY, Zhao Y, Lito P. The G protein signaling regulator RGS3 enhances the GTPase activity of KRAS. Science 2021;374:197–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. KRAS(G12C) inhibition with sotorasib in advanced solid tumors. N Engl J Med 2020;383:1207–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Janne PA, Riely GJ, Gadgeel SM, Heist RS, Ou SI, Pacheco JM, et al. Adagrasib in non–small cell lung cancer harboring a KRAS(G12C) Mutation. N Engl J Med 2022;387:120–31. [DOI] [PubMed] [Google Scholar]
- 12. Skoulidis F, Li BT, Dy GK, Price TJ, Falchook GS, Wolf J, et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N Engl J Med 2021;384:2371–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fedele C, Li S, Teng KW, Foster CJR, Peng D, Ran H, et al. SHP2 inhibition diminishes KRASG12C cycling and promotes tumor microenvironment remodeling. J Exp Med 2021;218:e20201414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hallin J, Engstrom LD, Hargis L, Calinisan A, Aranda R, Briere DM, et al. The KRAS(G12C) Inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discov 2020;10:54–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lou K, Steri V, Ge AY, Hwang YC, Yogodzinski CH, Shkedi AR, et al. KRAS(G12C) inhibition produces a driver-limited state revealing collateral dependencies. Sci Signal 2019;12:eaaw9450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Misale S, Fatherree JP, Cortez E, Li C, Bilton S, Timonina D, et al. KRAS G12C NSCLC models are sensitive to direct targeting of KRAS in combination with PI3K inhibition. Clin Cancer Res 2019;25:796–807. [DOI] [PubMed] [Google Scholar]
- 17. Molina-Arcas M, Moore C, Rana S, van Maldegem F, Mugarza E, Romero-Clavijo P, et al. Development of combination therapies to maximize the impact of KRAS-G12C inhibitors in lung cancer. Sci Transl Med 2019;11:eaaw7999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ryan MB, Coker O, Sorokin A, Fella K, Barnes H, Wong E, et al. KRAS(G12C)-independent feedback activation of wild-type RAS constrains KRAS(G12C) inhibitor efficacy. Cell Rep 2022;39:110993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ryan MB, Fece de la Cruz F, Phat S, Myers DT, Wong E, Shahzade HA, et al. Vertical pathway inhibition overcomes adaptive feedback resistance to KRAS(G12C) inhibition. Clin Cancer Res 2020;26:1633–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Weiss A, Lorthiois E, Barys L, Beyer KS, Bomio-Confaglia C, Burks H, et al. Discovery, preclinical characterization, and early clinical activity of JDQ443, a structurally novel, potent, and selective covalent oral inhibitor of KRASG12C. Cancer Discov 2022;12:1500–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xue JY, Zhao Y, Aronowitz J, Mai TT, Vides A, Qeriqi B, et al. Rapid non-uniform adaptation to conformation-specific KRAS(G12C) inhibition. Nature 2020;577:421–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives antitumor immunity. Nature 2019;575:217–23. [DOI] [PubMed] [Google Scholar]
- 23. Negrao MV, Araujo HA, Lamberti G, Cooper AJ, Akhave NS, Zhou T, et al. Comutations and KRASG12C inhibitor efficacy in advanced NSCLC. Cancer Discov 2023;13:1556–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sacher A, LoRusso P, Patel MR, Miller WH Jr., Garralda E, Forster MD, et al. Single-agent divarasib (GDC-6036) in solid tumors with a KRAS G12C mutation. N Engl J Med 2023;389:710–21. [DOI] [PubMed] [Google Scholar]
- 25. Bennett AM, Hausdorff SF, O'Reilly AM, Freeman RM, Neel BG. Multiple requirements for SHPTP2 in epidermal growth factor-mediated cell cycle progression. Mol Cell Biol 1996;16:1189–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lorthiois E, Gerspacher M, Beyer KS, Vaupel A, Leblanc C, Stringer R, et al. JDQ443, a structurally novel, pyrazole-based, covalent inhibitor of KRAS(G12C) for the treatment of solid tumors. J Med Chem 2022;65:16173–203. [DOI] [PubMed] [Google Scholar]
- 27. LaMarche MJ, Acker M, Argintaru A, Bauer D, Boisclair J, Chan H, et al. Identification of TNO155, an allosteric SHP2 inhibitor for the treatment of cancer. J Med Chem 2020;63:13578–94. [DOI] [PubMed] [Google Scholar]
- 28. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001;25:402–8. [DOI] [PubMed] [Google Scholar]
- 29. Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012;483:603–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rago F, DiMare MT, Elliott G, Ruddy DA, Sovath S, Kerr G, et al. Degron mediated BRM/SMARCA2 depletion uncovers novel combination partners for treatment of BRG1/SMARCA4-mutant cancers. Biochem Biophys Res Commun 2019;508:109–16. [DOI] [PubMed] [Google Scholar]
- 31. Dafflon C, Gaulis S, Barys L, Kapur K, Cornacchione V, Schukur L, et al. DOT1L inhibition is lethal for multiple myeloma due to perturbation of the endoplasmic reticulum stress pathway. Oncotarget 2020;11:956–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Guzman C, Bagga M, Kaur A, Westermarck J, Abankwa D. ColonyArea: an ImageJ plugin to automatically quantify colony formation in clonogenic assays. PLoS One 2014;9:e92444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang TL, Maierhofer C, Speicher MR, Lengauer C, Vogelstein B, Kinzler KW, et al. Digital karyotyping. Proc Natl Acad Sci USA 2002;99:16156–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Awad MM, Liu S, Rybkin II, Arbour KC, Dilly J, Zhu VW, et al. Acquired resistance to KRAS(G12C) inhibition in cancer. N Engl J Med 2021;384:2382–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen YN, LaMarche MJ, Chan HM, Fekkes P, Garcia-Fortanet J, Acker MG, et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 2016;535:148–52. [DOI] [PubMed] [Google Scholar]
- 36. Whittaker SR, Theurillat JP, Van Allen E, Wagle N, Hsiao J, Cowley GS, et al. A genome-scale RNA interference screen implicates NF1 loss in resistance to RAF inhibition. Cancer Discov 2013;3:350–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang B, Krall EB, Aguirre AJ, Kim M, Widlund HR, Doshi MB, et al. ATXN1L, CIC, and ETS transcription factors modulate sensitivity to MAPK pathway inhibition. Cell Rep 2017;18:1543–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Krall EB, Wang B, Munoz DM, Ilic N, Raghavan S, Niederst MJ, et al. KEAP1 loss modulates sensitivity to kinase targeted therapy in lung cancer. eLife 2017;6:e18970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Huang S, Holzel M, Knijnenburg T, Schlicker A, Roepman P, McDermott U, et al. MED12 controls the response to multiple cancer drugs through regulation of TGF-beta receptor signaling. Cell 2012;151:937–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lin L, Bivona TG. The Hippo effector YAP regulates the response of cancer cells to MAPK pathway inhibitors. Mol Cell Oncol 2016;3:e1021441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Galan-Cobo A, Sitthideatphaiboon P, Qu X, Poteete A, Pisegna MA, Tong P, et al. LKB1 and KEAP1/NRF2 pathways cooperatively promote metabolic reprogramming with enhanced glutamine dependence in KRAS-mutant lung adenocarcinoma. Cancer Res 2019;79:3251–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ornitz DM, Itoh N. The fibroblast growth factor signaling pathway. Wiley Interdiscip Rev Dev Biol 2015;4:215–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lu H, Liu C, Huynh H, Le TBU, LaMarche MJ, Mohseni M, et al. Resistance to allosteric SHP2 inhibition in FGFR-driven cancers through rapid feedback activation of FGFR. Oncotarget 2020;11:265–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Guagnano V, Furet P, Spanka C, Bordas V, Le Douget M, Stamm C, et al. Discovery of 3-(2,6-dichloro-3,5-dimethoxy-phenyl)-1–6-[4-(4-ethyl-piperazin-1-yl)-phenylamino]-pyrimidin-4-yl-1-methyl-urea (NVP-BGJ398), a potent and selective inhibitor of the fibroblast growth factor receptor family of receptor tyrosine kinase. J Med Chem 2011;54:7066–83. [DOI] [PubMed] [Google Scholar]
- 45. Folkes AJ, Ahmadi K, Alderton WK, Alix S, Baker SJ, Box G, et al. The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-thieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J Med Chem 2008;51:5522–32. [DOI] [PubMed] [Google Scholar]
- 46. Yaeger R, Mezzadra R, Sinopoli J, Bian Y, Marasco M, Kaplun E, et al. Molecular characterization of acquired resistance to KRASG12C-EGFR inhibition in colorectal cancer. Cancer Discov 2023;13:41–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Falchook G, Li BT, Marrone KA, Bestvina CM, Langer CJ, Krauss JC, et al. OA03.03 Sotorasib in Combination with RMC-4630, a SHP2 Inhibitor, in KRAS p.GG12C-Mutated NSCLC and Other Solid Tumors. 2022; Vienna, Austria; August 6–9, 2022. p 17. Available from:https://wclc2022.iaslc.org/wp-content/uploads/2022/07/WCLC2022-Abstract-Book.pdf. [Google Scholar]
- 48. Ahmed TA, Adamopoulos C, Karoulia Z, Wu X, Sachidanandam R, Aaronson SA, et al. SHP2 drives adaptive resistance to ERK signaling inhibition in molecularly defined subsets of ERK-dependent tumors. Cell Rep 2019;26:65–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Manchado E, Weissmueller S, Morris JP, Chen CC, Wullenkord R, Lujambio A, et al. A combinatorial strategy for treating KRAS-mutant lung cancer. Nature 2016;534:647–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wee S, Jagani Z, Xiang KX, Loo A, Dorsch M, Yao YM, et al. PI3K pathway activation mediates resistance to MEK inhibitors in KRAS mutant cancers. Cancer Res 2009;69:4286–93. [DOI] [PubMed] [Google Scholar]
- 51. Castellano E, Sheridan C, Thin MZ, Nye E, Spencer-Dene B, Diefenbacher ME, et al. Requirement for interaction of PI3-kinase p110alpha with RAS in lung tumor maintenance. Cancer Cell 2013;24:617–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gupta S, Ramjaun AR, Haiko P, Wang Y, Warne PH, Nicke B, et al. Binding of ras to phosphoinositide 3-kinase p110alpha is required for ras-driven tumorigenesis in mice. Cell 2007;129:957–68. [DOI] [PubMed] [Google Scholar]
- 53. Adachi Y, Kimura R, Hirade K, Ebi H. Escaping KRAS: gaining autonomy and resistance to KRAS inhibition in KRAS mutant cancers. Cancers 2021;13: 5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tsai YS, Woodcock MG, Azam SH, Thorne LB, Kanchi KL, Parker JS, et al. Rapid idiosyncratic mechanisms of clinical resistance to KRAS G12C inhibition. J Clin Invest 2022;132:e155523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zanconato F, Forcato M, Battilana G, Azzolin L, Quaranta E, Bodega B, et al. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat Cell Biol 2015;17:1218–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kapoor A, Yao W, Ying H, Hua S, Liewen A, Wang Q, et al. Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell 2014;158:185–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lin L, Sabnis AJ, Chan E, Olivas V, Cade L, Pazarentzos E, et al. The Hippo effector YAP promotes resistance to RAF- and MEK-targeted cancer therapies. Nat Genet 2015;47:250–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zeng H, Castillo-Cabrera J, Manser M, Lu B, Yang Z, Strande V, et al. Genome-wide CRISPR screening reveals genetic modifiers of mutant EGFR dependence in human NSCLC. eLife 2019;8:e50223. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary methods, tables and figures