

Metabolic reprogramming of bladder cancer cells driven by mutant FGFR3 increases serine synthesis that suppresses macrophage immunostimulatory functions to generate an immunosuppressive TME, which can be overcome by targeting PI3K.
Abstract
FGFR3 alterations are common in patients with bladder cancer. While the FGFR tyrosine kinase inhibitor erdafitinib has been approved as a targeted therapy for patients with FGFR3-altered (aFGFR3) bladder cancer, the response rate remains suboptimal, prompting development of strategies to improve treatment response. Here, we observed an immune-desert tumor microenvironment (TME) phenotype in human aFGFR3 bladder cancer and demonstrated that mutant FGFR3 indirectly induces a “cold” TME in mouse bladder cancer models. Single-cell RNA sequencing revealed the central role of macrophages in inducing the cold TME of aFGFR3 tumors. Macrophages in aFGFR3 tumors exhibited reduced T-cell recruitment and antigen presentation capabilities. Increased serine synthesis in bladder cancer cells that was induced by mutant FGFR3 activated the PI3K/Akt pathway in macrophages, shifting them to an immune-inert phenotype. Targeting PI3K in aFGFR3 tumors with duvelisib achieved promising efficacy by reversing the macrophage phenotype, and combination therapy with duvelisib and erdafitinib demonstrated increased antitumor activity. Overall, these findings reveal the critical role of enhanced serine synthesis efflux from cancer cells with mutant FGFR3 in shifting macrophages to an immune-inert phenotype. Reversing the macrophage phenotype holds promise for enhancing erdafitinib efficacy.
Significance:
Metabolic reprogramming of bladder cancer cells driven by mutant FGFR3 increases serine synthesis that suppresses macrophage immunostimulatory functions to generate an immunosuppressive TME, which can be overcome by targeting PI3K.
Graphical Abstract
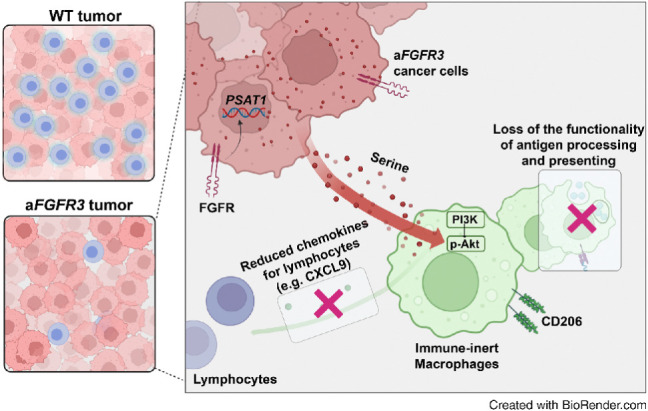
Introduction
Bladder cancer is one of the most common cancers worldwide (1). Platinum-based chemotherapy has traditionally been the standard of care for advanced bladder cancer, but the development of new therapies has led to the emergence of combination therapies that offer improved outcomes, including antibody–drug conjugate, immune checkpoint inhibitors (ICI), and erdafitinib for patients with alterations to FGFR (2). Previous research has shown that 17%–32% of bladder cancer harbors FGFR family alterations (including FGFR mutations, amplifications, and fusions), especially in early-stage disease (3, 4). FGFR3 alteration is the most common FGFR alteration in bladder cancer, accounting for approximately 47.5% of all FGFR alterations (5, 6). Nevertheless, the efficacy of erdafitinib, the pan-FGFR inhibitor and the only targeted drug approved by the FDA for bladder cancer, is only approximately 40% in patients with bladder cancer with FGFR alterations, indicating an urgent need for new therapies for erdafitinib nonresponders (7).
Traditionally, FGFR alterations were believed to solely promote cancer cell proliferation and survival (8). However, recent studies have suggested that FGFR alterations are also strongly associated with deleterious immune microenvironments. For example, FGFR alterations are associated with the luminal or luminal-papillary subtype, which is characterized by low CD8+ T-cell infiltration (9, 10). One study indicated that FGFR alterations facilitate bladder tumorigenesis by inhibiting acute inflammation (11). Others have shown that PD-L1 could be upregulated by FGFR activation in cancer cells through various pathways, leading to inactivation of T cells (12–14). Nonetheless, more efforts are needed to elucidate the association between FGFR activation and the deleterious tumor microenvironment (TME).
Metabolic reprogramming, one of the major hallmarks of cancers, has often been observed in cancer cells with oncogenic activation, such as FGFR family mutations (15). Studies have suggested that various oncogenes orchestrate metabolic reprogramming by inducing broad changes in gene expression (16). The Warburg effect is a well-known instance of metabolic rewiring in cancer, whereby cancer cells obtain surplus glycolytic intermediates utilized as inputs for a range of biosynthetic pathways (17). The significance of serine synthesis and metabolism in cancer cells has been emphasized in recent years as an auxiliary pathway of the Warburg effect, owing to its critical role in tumor progression. This metabolic pathway provides essential precursors for protein synthesis, influences the cellular antioxidative capacity, provides methyl groups for the one-carbon pools, and supports tumor homeostasis (18). Studies have suggested that cancer cell–derived serine promotes the development of a supportive tumor immune microenvironment. For instance, serine supplementation decreased the secretion of inflammatory cytokines from macrophages, including IL1β, IFNγ, and TNFα (19, 20). In addition, the enzyme phosphoglycerate dehydrogenase (PHGDH), which was essential for serine synthesis, was found to be a metabolic checkpoint of macrophage differentiation, and it was essential for the expression of key anti-inflammatory molecules (21). However, the underlying mechanism by which cancer cell–derived serine affects the surrounding immune and stromal cells has rarely been studied.
In this study, we discovered that increased serine synthesis derived from FGFR3-mutant (mFGFR3) cancer cells shifted macrophages to an immune-inert phenotype and elicited a cold TME. We revealed the mechanistic connections among FGFR3 alterations, serine metabolism, and antitumor immunity and further demonstrated that redirecting the macrophage phenotype with PI3K inhibitors is a potential therapeutic approach for mFGFR3 tumors.
Materials and Methods
The key resources and reagents used in this study are summarized in Supplementary Table S1. Methods regarding cancer cell supernatants preparation, mouse lymphocytes preparation, lymphocyte coculture assay, tumor-infiltrating lymphocyte classification, cell viability tests, Sun Yat-sen Memorial Hospital (SYSMH) cohort generation, and PSAT1 IHC evaluation were summarized in the Supplementary Materials and Methods.
Cells
The human bladder cancer cell line T24 and human monocyte cell line THP-1 were obtained from the ATCC. The murine breast cancer cell line MB49 was obtained from Millipore. The cells were grown according to standard protocols. Short tandem repeat authentication of the cells used in this study showed that there was no misidentification or contamination with other cells within 3 years. Cell lines were tested for Mycoplasma monthly using the Mycoplasma Detection Kit (catalog no. rep-mys-20, InvivoGen) and used within 10 passages.
Animals
All animal experiments were performed with the approval of the Institutional Animal Care and Use Committee (IACUC) of the South China University of Science and Technology (approval number 2022044) and according to the established guidelines. C57BL/6J mice and BALB/c nude mice used in this study were between 4 and 6 weeks old and purchased from Hunan Silaikejingda Experimental Animal Company Limited. huPBMC-NOG-dKO humanoid mice were purchased from Beijing Vital River Animal Company Limited. All mice were maintained in the South China University of Technology (Guangzhou, P.R. China) animal facility under specific pathogen-free conditions.
Lentivirus generation and gene transfer
The entire process of lentiviral construction and generation was entrusted to IGE Biotechnology. In brief, the cDNA sequence encoding human or mouse wild-type (WT) FGFR3b was cloned into the pCDH-CMV-MCS-EF1-copGFP-T2A-Puro vector and served as a template for site-directed mutagenesis. The activating mutation S249C (human) or S243C (mouse) was introduced into the FGFR3b cDNA by PCR using primers carrying the mutations, which was confirmed by sequencing. Lentiviral vectors encoding no insert or mutant human or mouse FGFR3b were transfected into HEK293T cells for packaging. Packaged lentivirus particles were collected from HEK293T cell supernatant and transduced into T24 or MB49 cells in the presence of polybrene (IGE Biotechnology). Successful infection was confirmed by the identification of green fluorescent cells. Infected cells were selected using increasing doses of puromycin (catalog no. 0219453925, MP). Successful infection was confirmed by the identification of green fluorescent cells, qRT-PCR, and Western blotting.
Preparation and culture of bone marrow–derived macrophages
Bone marrow cells were flushed from the femurs and tibias of 6 to 8 weeks old C57BL/6 mice. The erythrocytes were lysed with red blood cell lysis buffer (catalog no. CW0613S, CWBio). The remaining cells were seeded into 6-well or 12-well plates. Bone marrow–derived macrophages (BMDM) were cultured in DMEM supplemented with 10% FBS, 1% penicillin/streptomycin (Gibco), and 40 ng/mL murine MCSF (catalog no. 51112-MNAH, Sino Biological). Fresh culture medium was added to BMDMs every 3 days. On the 6th day of differentiation, the BMDMs were considered matured for in vitro stimulation.
Macrophages stimulation assay
Mature BMDMs were cocultured with tumor cell-conditioned medium for 48 hours and harvested for flow cytometry, qRT-PCR, or functional assays. Serine (2 mmol/L; catalog no. BS932, Biosharp), S-Adenosyl methionine (SAM, 1 mmol/L, Sigma-Aldrich, A7007), duvelisib (20 μmol/L; catalog no. A1720, APExBIO), or L-phenylglycine (50 μmol/L; catalog no. 2935-35-5, Sigma-Aldrich) was added. Lipopolysaccharide (100 ng/mL; catalog no. BS904, Biosharp) or murine IL4 (20 ng/mL; catalog no. 214-14-20, Peprotech) was added 24 hours before harvesting to induce proinflammatory or anti-inflammatory macrophages.
Transwell assay
For the transwell assay, 3 × 105 cancer cells or 5 × 105 stimulated macrophages were seeded into the bottom chamber (wells in a 24-well plate) with 500 μL DMEM containing 10% FBS. Next, 5 × 105 lymphocytes were added into the upper chamber (5-μm transwell inserts, catalog no. 3421-48EA, Corning) with 200 μL serum-free media. After 2.5 hours, the number of migrated lymphocytes was quantified by flow cytometry.
Antigen-presenting assay
A total of 5 × 105 BMDMs were cultured with cancer cell supernatants for 48 hours. Then, the cells were incubated with ovalbumin (OVA; 1 μg/mL, catalog no. 9006-59-1, Solarbio) for 24 hours. The cells were collected, washed with PBS, and stained with PE anti-mouse H-2Kb bound to SIINFEKL antibody (catalog no. 12-5743-81, Thermo Fisher Scientific, RRID: AB_925775) in PBS at 4°C for 1 hour.
RNA sequencing and in silico analysis
Public transcriptional databases and immune infiltration analysis
Transcriptome and clinical data in the IMvigor210 dataset obtained from patients with metastatic urothelial bladder cancer treated with atezolizumab were downloaded from http://research-pub.gene.com/IMvigor210CoreBiologies. The Cancer Genome Atlas (TCGA)-BLCA dataset was extracted from TCGA and used for further analyses. We integrated gene set enrichment analysis (GSEA) to estimate immune infiltration, and the signature used in this study is summarized in Supplementary Table S2.
Single-cell RNA sequencing of mouse MB49 tumor samples
A total of six samples (three per group) were used for single-cell RNA sequencing (scRNA-seq), and the samples of each group were mixed before sequencing. Droplet-based sequencing data from Singleron Matrix Single Cell Processing System was aligned and quantified against the mm10 mouse reference genome using CeleScope (https://github.com/singleron-RD/CeleScope) v1.9.0 pipeline. Seurat version 4.2.0 was used for cell filtering and subsequent analysis (22). The filtered data were normalized using a normalized data function with a scaling factor of 10,000. The top 2,000 highly variable genes were selected using the “find variable feature” function in the first method. Mutual nearest neighbors were used to remove the batch effects (23). FindNeighbors and FindClusters functions were employed with 10 dimensions and a resolution of 0.8. We used CellChat (1.1.0), a public repository of ligands, receptors, cofactors, and their interactions, for the inference and analysis of cell-to-cell communication. For the macrophage subcluster analysis, FindNeighbors and FindClusters functions were employed with five dimensions and a resolution of 0.3. GSEA (RRID: SCR_003199) was applied to identify significantly enriched pathways using the GSEA 4.1.0 desktop application.
qRT-PCR
Total RNA was extracted from macrophages and MB49 or T24 cancer cells using an RNA Quick Purification Kit (EZBioscience). qRT-PCR was performed using HieffTM qPCR SYBR Green Master mix (Yeasen Biotechnology) and analyzed using a LightCycler 480 instrument (Roche). The primer sequences are listed in Supplementary Table S3.
Serine quantification assay
We used a DL-Serine Assay Kit (catalog no. MAK352, Sigma-Aldrich) according to the manufacturer's instructions to analyze the relative serine concentration within cancer cells and BMDMs.
Western blotting
Conditioned cells were lysed using RIPA lysis buffer (catalog no. CW2333S, CWBio) in the presence of protease inhibitors (catalog no. CW2200S, CWBio) and phosphatase inhibitors (catalog no. CW2383S, CWBio). Proteins were quantified using a bicinchoninic acid assay (catalog no. CW0014S, CWBio) before denaturation. Protein samples were then subjected to SDS-PAGE and transferred to polyvinylidene difluoride membranes (catalog no. IPVH00010, Merck Millipore), which were blotted with anti-pERK1/2 (diluted 1:2,000, catalog no. 4370, Cell Signaling Technology, RRID: AB_2315112), anti-pERK5 (diluted 1:1,000; catalog no. 3371, Cell Signaling Technology, RRID: AB_2140424), anti-PSAT1 (diluted 1:5,000; catalog no. 10501-1-AP, Proteintech, RRID: AB_2172597), and anti-α-tubulin (diluted 1:20,000; catalog no. 66031-1-Ig, Proteintech, RRID: AB_11042766) antibodies, followed by incubation with horseradish peroxidase–conjugated anti-rabbit secondary antibodies (catalog no. GB23303, Servicebio) or anti-mouse secondary antibodies (catalog no. GB23303, Servicebio). Membrane proteins were visualized using SmartChemi 910 plus (Beijing Sage Creation Science Co, LTD) utilizing chemiluminescence (catalog no. WBKLS0500, Merck Millipore).
Flow cytometry of in vitro experiments
BMDMs were harvested for flow cytometry after the coculture assay as described above and blocked with mouse Fc Receptor Blocking Solution (catalog no. 101320, BioLegend, RRID: AB_1574975) before incubation with fluorochrome-labeled antibodies. The antibodies used for BMDM surface staining were BV510 anti-F4/80 (catalog no. 123135, BioLegend, RRID: AB_2562622), PE-Cy7 anti-CD11b (catalog no. 101215, BioLegend, RRID: AB_312798), and BV605 anti-IA/IE (catalog no. 107639, BioLegend, RRID: AB_2565894). A FIX & PERM Cell Permeabilization Kit (catalog no. GAS003, Thermo Fisher Scientific) was used to fix and permeabilize the cell membranes. The BMDMs were then incubated with Fixation Medium for 15 minutes, washed, and incubated Permeabilization Medium and PE anti-CXCL9 (catalog no. 515603, BioLegend, RRID: AB_2245490), PE anti-pAKT1 (catalog no. 12-9715-41, Thermo Fisher Scientific, RRID: AB_2637101), or AF647 anti-CD206 (catalog no. 141711, BioLegend, RRID: AB_10900240) antibodies for intracellular staining.
THP-1 cells were also harvested for flow cytometry after the coculture assay as described above and blocked with human Fc Receptor Blocking Solution (catalog no. 422302; BioLegend, RRID: AB_2818986). The PE anti-HLA-DR antibody (catalog no. 307605, BioLegend, RRID: AB_314683) was used for THP-1 surface staining. THP-1 cells were fixed and permeabilized as described above and incubated with BV421 anti-Human CD206 (catalog no. 566281, BD Biosciences, RRID: AB_2739655), or PE anti-CXCL9 (catalog no. 566013, BD Biosciences, RRID: AB_2739458) antibodies for intracellular staining.
Flow cytometry was performed using a CytoFLEX flow cytometer (Beckman Coulter) and analyzed using FlowJo (version 10, RRID: SCR_008520).
IHC
Formalin-fixed paraffin-embedded tumor tissues isolated were cut into 5-μm sections and used for IHC staining. For humanized tumor tissues, IHC staining of CD206 (diluted 1:1,500; catalog no. 24595, Cell Signaling Technology, RRID: AB_2892682) and CD8 (diluted 1:500; catalog no. MA5-14548, Thermo Fisher Scientific, RRID: AB_10984334) was performed. IHC staining for CXCL9 (diluted 1:1,000; catalog no. AF-492-NA, R&D Systems, RRID: AB_2086734) was performed for mouse tumor tissues. IHC staining for PSAT1 (diluted 1:200; catalog no. 10501-1-AP, Proteintech, RRID: AB_2172597) was performed in human bladder cancer tissues from the SYSMH cohort. Details of IHC were summarized in the Supplementary Materials and Methods.
Multicolor immunofluorescence
Multiplex immunofluorescence (IF) was performed using a PANO 5-plex IHC kit (catalog no. 10080100100, PANOVUE). The primary antibodies used for multiplex IF staining of mouse tumor tissues consisted of anti-F4/80 (diluted 1:3,000; catalog no. 70076, Cell Signaling Technology, RRID: AB_2799771); anti-CD206 (diluted 1:3,000; catalog no. 24595, Cell Signaling Technology, RRID: AB_2892682); anti-CD8 (diluted 1:1,200; catalog no. 98941, Cell Signaling Technology, RRID: AB_2756376); anti-granzyme B (diluted 1:2,000; catalog no. 46890, Cell Signaling Technology); and PE anti-IA/IE antibodies (diluted 1:200; catalog no. 107607, BioLegend, RRID: AB_313322). For human bladder cancer tissue staining, anti-CD68 (diluted 1:1,000; catalog no. ZM-0464, Zhongshan Golden Bridge); anti-HLA-DR (diluted 1:2,000; catalog no. ZM-0136, Zhongshan Golden Bridge); anti-CD206 (diluted 1:3,000; catalog no. 24595, Cell Signaling Technology, RRID: AB_2892682); anti-CXCL9 (diluted 1:1,000; catalog no. ab202961, Abcam); anti-CD8 (diluted 1:3,000; catalog no. MA5-14548, Thermo Fisher Scientific, RRID: AB_10984334); anti-granzyme B (diluted 1:1,000, catalog no. 46890, Cell Signaling Technology) was used. For primary antibodies without a fluorescent label, after incubation with the primary and secondary antibodies (catalog nos. MP-7451 and MP-7444, Vector), the sections were stained with Alexa Fluor Tyramides Opal 520, 570, 620, or 690 following the manufacturer's instructions. Finally, the slides were counterstained with DAPI, mounted with an anti-fade mountant, and scanned using the Vectra Polaris Automated Quantitative Pathology Imaging System (PerkinElmer). The evaluation was performed using Phenochart and inForm Tissue Analysis software (PerkinElmer). Alexa Fluor Tyramides, DAPI, and anti-fade mountant were all included in the kit.
Animal experiments
For the humanized mouse experiment, T24 cells (5 × 106) were implanted subcutaneously into the right flank. In other animal experiments, MB49 cells (5 × 105) were inoculated subcutaneously for the subcutaneous tumorigenicity assay. The tumor diameter was monitored every 2–3 days using an electronic caliper. Tumor volumes were calculated as 0.5 × L × W2.
For the macrophage depletion study, neutral clodronate liposomes (catalog no. F70101C-N, FormuMax, 7 mg/mL, 200 μL, intraperitoneal) and control liposomes (catalog no. F70101-N, FormuMax, 200 μL, intraperitoneal) were given on the second, sixth, 10th, and 14th day after cell inoculation. Tumors were harvested on the 16th day.
For the duvelisib treatment study, from the 10th day after tumor cell inoculation, mice were injected with duvelisib (catalog no. A1720, APExBIO, 2 mg/kg, oral gavage) every day. For the erdafitinib treatment study, from the 10th day after tumor cell inoculation, the mice were injected with erdafitinib (catalog no. JNJ-42756493, MCE, 25 mg/kg, oral gavage) every day. Mice in the control group received vehicle treatment.
Flow cytometry for animal experiments
Antibodies were purchased for the animal experiment flow cytometric analyses, including Brilliant Violet 650 anti-CD8α (catalog no. 100741, BioLegend, RRID: AB_11124344); PE anti-CD3 (catalog no. 100206, BioLegend, RRID: AB_312663); PE/Cyanine7 anti-CD4 (catalog no. 100421, BioLegend, RRID: AB_312706); Alexa Fluor 647 anti-granzyme B (catalog no. 515406, BioLegend, RRID: AB_2566333); PE anti-CXCL9 (catalog no. 515603, BioLegend, RRID: AB_2245490); Brilliant Violet 605 anti-IA/IE (catalog no. 107639, BioLegend, RRID: AB_2565894); Brilliant Violet 510 anti-F4/80 (catalog no. 123135, BioLegend, RRID: AB_2562622); PE/Cyanine7 anti-CD11b (catalog no. 101215, BioLegend, RRID: AB_312798); and Brilliant Violet 421 anti-CD45 (catalog no. 103134, BioLegend, RRID: AB_2562559).
Tumor-bearing mice were euthanized at designated timepoints, and their tumors were isolated and stored in ice-cold 2% FBS. The collected tumors were then minced into 1–3 mm pieces using a scalpel before incubation with collagenase I (catalog no. GC305013, Servicebio), collagenase II (catalog no. GC305014, Servicebio), collagenase IV (catalog no. GC305015, Servicebio), and DNase (catalog no. 1121MG010, Servicebio) for 1 hour at 37°C on a shaker. The resulting tumor suspensions were then passed through a 40-μm cell strainer to remove large debris and generate single-cell suspensions.
Macrophage surface staining was performed by incubating 1 × 106 cells in 50 μL ice-cold PBS with fluorochrome-conjugated antibodies against CD45, CD11b, F4/80, and IA/IE. The cells were then washed again with PBS, fixed, permeabilized as described above, and stained using an intracellular anti-CXCL9 antibody.
Lymphocyte surface staining was performed by incubating 1 × 106 cells in 50 μL ice-cold PBS with fluorochrome-conjugated antibodies against CD3, CD4, CD45, and CD8. The cells were then washed again with PBS, fixed, permeabilized as described above, and stained using an intracellular anti-granzyme B antibody.
Statistical analysis and data presentation
All experiments in this study were independently performed three times. Quantitative data are presented as the mean ± SEM. The χ2 test was used for nonparametric variables, while Student t test and one-way ANOVA were used for parametric variables to identify statistically significant data. All data analyses were performed using GraphPad Prism version 9 (RRID: SCR_002798). A P value < 0.05 was considered to indicate significance. The proposed illustration of mechanism and experiments was created with BioRender.com.
Study approval
All human samples used in this study were obtained from patients with their written informed consent. The study was conducted in accordance with the Declaration of Helsinki and was approved by the Ethics Committee of the Sun Yat-sen Memorial Hospital, Sun Yat-sen University (approval number SYSKY-2023-422-01). The baseline information of patients included in the cohort was summarized in Supplementary Table S4. All animal experiments were performed with the approval of the IACUC of the South China University of Science and Technology (approval number 2022044).
Data availability statement
The IMvigor210 dataset analyzed in this study was downloaded from http://research-pub.gene.com/IMvigor210CoreBiologies. In addition, TCGA-BLCA cohort data analyzed in this study was extracted from TCGA at https://www.cancer.gov/ccg/research/genome-sequencing/tcga. The scRNA-seq data that support the findings of this study have been deposited into the Gene Expression Omnibus with accession number GSE243093 and the CNGB Sequence Archive (CNSA) of China National GeneBank DataBase (CNGBdb) with accession number CNP0004744. Other data supporting the findings of this study are available upon request from the corresponding author.
Results
FGFR3-altered bladder cancers indirectly shape a cold TME
FGFR3-altered (aFGFR3) bladder cancer has previously been reported to be associated with a non–T-cell–inflamed TME. We conducted a systematic review utilizing publicly available databases IMvigor210 and TCGA-BLCA to comprehensively assess the immune infiltration of aFGFR3 bladder cancer tumors (24). First, we utilized a previously established CD8+ effector T-cell signature to evaluate the infiltration level of CD8+ T cells (25). Our analysis revealed a significant decrease in the infiltration of CD8+ T cells in the aFGFR3 bladder cancer group compared with that in the WT group (Fig. 1A). In addition, we observed a negative correlation between the expression of CD8A and FGFR3 in both datasets (Fig. 1B). Tumor-infiltrating lymphocyte phenotypes were investigated, and a lower proportion of inflamed tumor tissues and a higher proportion of immune desert were observed in the aFGFR3 bladder cancer group in both datasets (Fig. 1C and D). We also conducted a cohort study at SYSMH to validate this finding. In this cohort, we observed a significant reduction in the infiltration of CD8+ granzyme B+ cells within the tumors of patients with aFGFR3 cancers (Fig. 1E).
Figure 1.

FGFR3-altered bladder cancers shape a cold TME indirectly. A, Dot plots showing the CD8+ T cells effector signature score in the WT and aFGFR3 groups in the IMvigor210 and TCGA-BLCA datasets. B, Spearman correlation analysis of the expression of CD8A with the expression of FGFR3. C, Stacked bar plots showing the proportion of immune-inflamed, immune-excluded, and immune-desert phenotypes in the WT and the aFGFR3 groups in the IMvigor 210 dataset. D, Representative images and stacked bar plots indicating the proportion of immune-inflamed, immune-excluded, and immune-desert phenotypes in the WT and the aFGFR3 groups in TCGA-BLCA dataset. Scale bar, black, 100 μm. E, Representative images and dot plot showing the CD8+ granzyme B+ cells infiltration in the WT and the aFGFR3 groups in the SYSMH cohort. Scale bar, white, 100 μm. F and G, Growth curves (F) and tumor weights (G) of the WT and mFgfr3 cancer cells in nude mice as well as C57BL/6 mice. H and I, Flow cytometry analysis and statistical analysis illustrating the proportion of CD3+ CD8+ cells within CD45+ cells (H) and the proportion of granzyme B+ cells within CD8+ T cells (I) in the indicated groups. J, Representative images and statistical analysis show the infiltration of CD8+ granzyme B+ cells in the indicated groups. K, Transwell assay of lymphocytes with cancer cell supernatant shows the cell count of migrated lymphocytes in the indicated groups. L, Flow cytometry analysis of in vitro coculture of cancer cells with lymphocytes illustrating the proportion of granzyme B+ cells within CD8+ T cells in the indicated groups. The data are shown as the mean ± SEM values (IMVigor 210 cohort: n = 190 in the WT group, n = 47 in the aFGFR3 group; TCGA-BLCA cohort: n = 350 in the WT group, n = 56 in the aFGFR3 group; SYSMH cohort: n = 24 in the WT group, n = 10 in the aFGFR3 group; n = 6 per group in F–L). P < 0.05 was considered a significant difference; ns, no significance (unpaired parametric Student t test and χ2 test). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
We generated an mFgfr3 MB49 cancer cell line to validate our findings in a mouse model. WT and mFgfr3 MB49 cells were inoculated into immunodeficient and immunocompetent mice. Surprisingly, there was no difference in tumor growth between WT and mFgfr3 cells in immunodeficient mice (Fig. 1F and G; Supplementary Fig. S1A and S1B). However, mFgfr3 tumors exhibited significantly accelerated growth in immunocompetent mice. These results suggested that the enhanced invasiveness of aFGFR3 cancers was not solely due to intrinsic proliferative changes but may involve immune evasion. We investigated the immune infiltration characteristics of mFgfr3 tumors and found a reduction in CD8+ granzyme B+ T cells through flow cytometry analysis and multiplex IF staining (Fig. 1H–J; Supplementary Fig. S1C and S1D).
On the basis of our observations thus far, we hypothesized that aFGFR3 cancer cells exert their influence on T-cell function and infiltration by excluding T cells from the proximity of cancer cells. We initially speculated that mFgfr3 cancer cells might decrease the expression of chemokines, thereby impairing lymphocyte chemotaxis. To test this hypothesis, we conducted a transwell assay, as described in Materials and Methods (Supplementary Fig. S1E). Surprisingly, we did not observe any significant difference in the number of lymphocytes crossing the transwell between the WT and mFgfr3 groups (Fig. 1K). In addition, we performed a coculture assay by incubating lymphocytes with either WT or mFgfr3 cancer cells (Supplementary Fig. S1F). Strikingly, no difference in lymphocyte cytotoxicity was observed between the two groups (Fig. 1L). These results collectively suggest that mFgfr3 cancer cells cannot modulate lymphocyte migration or directly suppress T-cell functionality.
scRNA-seq highlights the central role of macrophages in inducing the cold TME of the mFgfr3 tumor
To investigate the potential involvement of other cell types in mediating immunosuppression within the TME of mFgfr3 cancer cells, we conducted scRNA-seq analysis on WT and mFgfr3 MB49 tumor tissues. Following quality control and batch effect removal (refer to Materials and Methods: “scRNA-seq of mouse MB49 tumor samples”), we clustered a total of 28,458 single cells into major clusters. Specifically, 14,880 cells were analyzed in the WT group and 13,578 in the mFgfr3 group.
We utilized cluster-specific genes and classic markers described in previous studies to annotate the cell types: T cells (Cd3e+), CD8+ T cells (Cd8a+), CD4+ T conventional cells (Il7r+), proliferating T cells (Hmgb2+), B cells (Cd79b+), myeloid cells (Lyz2+), dendritic cells (Cst3+), polymorphonuclear leukocytes (Cxcr2+), and fibroblasts (Col1a1+; Fig. 2A; Supplementary Fig. S2A and S2B). Consistent with earlier observations, we identified a significant reduction in the infiltration of CD8+ T cells in the mFgfr3 tissues (Fig. 2B; Supplementary Fig. S2C).
Figure 2.

scRNA-seq highlights the central role of macrophages in inducing the cold TME of the mFgfr3 tumor. A, Uniform Manifold Approximation and Projection (UMAP) plot of scRNA-seq results of WT and mFgfr3 MB49 mice tumors colored by major cell types, 14,880 cells in the WT group and 13,578 cells in the mFgfr3 group. B, Pie chart shows the proportion of subtypes of T lymphocytes in the indicated groups. C, Circle plots and heatmap of CellChat analysis show the networks of the CXCL signaling pathway. D, UMAP plot of single cells of macrophages colored by subclusters. E, Volcano plot displays the differential genes of the subcluster-1 macrophages. F, GO analysis represents the enriched pathways in the subcluster-1 macrophages. G and H, Representative IF images showing the infiltration of CD206+ F4/80+ cells (G) and IA/IE+ F4/80+ cells (H) in the indicated groups. I, Representative IHC images showing the expression of CXCL9 in the indicated groups. J, GSEA showing the enrichment of antigen processing and presenting pathway and lymphocyte chemotaxis pathway in the IMvigor210 and TCGA-BLCA datasets. K, Representative multiplex IF images and statistical analysis showing the proportion of CD206+, HLA-DR+, and CXCL9+ cells within CD68+ macrophages in the SYSMH cohort; n = 24 in the WT group, n = 10 in the aFGFR3 group. L, Schematic illustration of macrophages depletion strategy with clodronate liposomes in MB49 tumor–bering mice. M, Flow cytometry analysis illustrating the proportion of macrophages in the splenocytes in the indicated groups. N and O, Flow cytometry analysis and IHC staining illustrating the proportion of macrophages in tumors in the indicated groups. P and Q, Flow cytometry analysis illustrating the proportion of CD3+CD8+ cells within CD45+ cells (P) and the proportion of granzyme B+ cells within CD8+ T cells (Q) in the indicated groups. R, Representative IF images and statistical analysis show CD8+ granzyme B+ cell infiltration in the indicated groups. The data are shown as the mean ± SEM values (n = 5 per group). P < 0.05 was considered a significant difference; ns, no significance (unpaired parametric Student t test). *, P < 0.05; **, P < 0.01; ***, P < 0.001. Scale bar, 100 μm. FMO, fluorescence minus one; Mφ, macrophages. SN, supernatants. (L, Created with BioRender.com.)
We next investigated changes in cell-to-cell communication in mFgfr3 tumors using the CellChat Package to understand the underlying immune suppression mechanism (26). Specifically, we aimed to identify the key signaling pathway responsible for mediating the immunosuppressive effects on CD8+ T cells. By comparing the incoming pathways between WT and mFgfr3 samples, we observed extensive downregulation of the CXCL pathway in the T cells of mFgfr3 tissues (Supplementary Fig. S2D). The CXCL signaling pathway is known to play a crucial role in chemotaxis and inflammation, suggesting that reduced CXCL signaling may contribute to the diminished infiltration of cytotoxic lymphocytes and the establishment of an immune-desert TME in mFgfr3 tumors. We traced the pathway to further investigate the sources of CXCL signaling and found that macrophages were the main contributors to CXCL signaling in CD8+ T cells in the WT samples (Fig. 2C). However, in the mFgfr3 tissues, we observed a significant reduction in the CXCL signaling pathway specifically in macrophages. These findings collectively indicate that macrophages play a pivotal role in driving immune suppression in the mFgfr3 TME.
We performed a reclustering analysis specifically focusing on macrophages to gain a comprehensive understanding of the characteristics of these cells in mFgfr3 tumors, identifying five distinct subclusters (Fig. 2D; Supplementary Fig. S2E). Despite the removal of batch effects, macrophages from the two tissue types still showed substantial differences in their expression patterns. Notably, subcluster 1 macrophages were exclusive to mFgfr3 tumors. We conducted a differential gene analysis to investigate this distinctive subcluster's molecular characteristics further and found a significant upregulation of Mrc1, which encodes CD206 (Fig. 2E; Supplementary Fig. S2F). Interestingly, the expression of chemokines (Cxcl9, Cxcl10, and Cxcl11), known for their potent ability to recruit lymphocytes, was notably downregulated in subcluster 1 macrophages. In addition, the expression levels of genes encoding MHC proteins, such as Cd74, H2-Ab1, and H2-Eb1, were also found to be decreased. Gene Ontology (GO) analysis revealed a negative enrichment of pathways associated with antigen processing and presentation and lymphocyte-mediated immunity in subcluster 1 macrophages (Fig. 2F). On the basis of these observations, subcluster 1 macrophages were named “immune-inert macrophages” considering their low expression of lymphocyte-attracting chemokines and a lack of effective T-cell immune response activation through MHC molecule antigen presentation. Collectively, these findings strongly suggest that the capacity of macrophages to process and present antigens and attract lymphocytes is substantially dampened in mFGFR3 tumor tissues.
We conducted multiplex IF and IHC staining to validate that macrophages adopt an immune-inert phenotype in response to mutant cancer cells. The results obtained were consistent with the scRNA-seq data: we observed an increase in CD206+ macrophages, a decrease in IA/IE+ macrophages, and reduced expression of CXCL9 in mFgfr3 tissues (Fig. 2G–I).
We further validated these findings in human bladder cancer cohorts. GSEA of TCGA-BLCA and IMvigor210 datasets revealed a reduction in the pathways related to antigen presenting and processing and lymphocyte chemotaxis in aFGFR3 tumors (Fig. 2J). IHC staining in bladder cancer tumor tissues of the SYSMH cohort confirmed elevated CD206 expression levels in macrophages from aFGFR3 tumors, while the expression levels of HLA-DR and CXCL9 were reduced (Fig. 2K).
We employed a macrophage depletion method using clodronate liposomes to investigate whether macrophages were the driving force behind immune suppression in aFGFR3 cancers (Fig. 2L; Supplementary Fig. S2G–S2I). We aimed to examine immune infiltration in the tissues following macrophage depletion. Both flow cytometry and IHC staining confirmed a significant reduction in macrophage expression in the spleen and tumor tissues (Fig. 2M–O; Supplementary Fig. S2J and S2K). In addition, in WT tumors, depleting macrophages led to a significant reduction in CD8+ granzyme B+ T-cell infiltration, suggesting that macrophages play a vital role in recruiting and activating T cells in the WT TME (Fig. 2P–R). In contrast, in the mFgfr3 TME, no difference was observed between the control group and the depletion group, validating the limited ability of macrophages in the mFgfr3 TME to recruit and activate CD8+ T cells. Taken together, these findings suggest that macrophages play a crucial role in inducing immunosuppression within the TME in tumors with activated FGFR3, leading to reduced infiltration of cytotoxic CD8+ T cells.
Next, we collected supernatants from these cancer cells and used them to stimulate BMDMs to investigate whether mFgfr3 cancer cells can directly influence macrophage phenotypes (Supplementary Fig. S3A). Flow cytometry and qRT-PCR analysis revealed a significant reduction in the expression of CXCL9 and MHC molecules, accompanied by an increase in CD206 expression in BMDMs stimulated with the mutant group's supernatant (Fig. 3A–C; Supplementary Fig. S3B).
Figure 3.

mFGFR3 cancer cells promote macrophage shift to an immune-inert phenotype. A–C, Flow cytometry analysis and statistical analysis illustrating the proportion of CD206+ cells (A), IA/IE+ cells (B), and CXCL9+ cells (C) within live BMDMs in the indicated groups. D, Antigen-presenting assays show the MHC-I SIINFEKL+ cells within the BMDMs. E, Transwell assay of lymphocytes with BMDMs previously stimulated by cancer cell supernatants shows the cell count of migrated lymphocytes in the indicated groups. F, Flow cytometry analysis of in vitro coculture of BMDMs previously stimulated with cancer cell supernatants with lymphocytes illustrating the proportion of granzyme B+ cells within CD8+ T cells in the indicated groups. G–I, Flow cytometry analysis and statistical analysis illustrating the proportion of CD206+ cells (G), HLA-DR+ cells (H), and CXCL9+ cells (I) within live THP-1 cells in the indicated groups. The data are shown as the mean ± SEM values (n = 6 per group). P < 0.05 was considered a significant difference (unpaired parametric Student t test). *, P < 0.05; **, P < 0.01; ***, P < 0.001. FMO, fluorescence minus one; SN, supernatants.
We further investigated the functional changes in macrophages with a series of assays. An antigen-presenting assay using OVA was conducted, demonstrating that BMDMs stimulated with mFgfr3 supernatants displayed a reduced antigen-presenting ability (Fig. 3D). In addition, a transwell assay demonstrated that BMDMs stimulated with mFgfr3 supernatant had a significantly diminished capacity to attract lymphocytes compared with those stimulated with WT supernatant (Fig. 3E; Supplementary Fig. S3C). Furthermore, an in vitro coculture assay with supernatant-stimulated BMDMs and lymphocytes revealed a lower granzyme B level in CD8+ T cells cultured with mFgfr3 cell supernatant–treated BMDMs (Fig. 3F; Supplementary Fig. S3D).
We extended these experiments by applying supernatants from T24 WT and mFGFR3 cancer cells to stimulate THP-1 cells. The results were consistent with the data obtained from BMDMs stimulated with MB49 supernatants (Fig. 3G–I; Supplementary Fig. S3E).
mFGFR3 cancer cells upregulate serine synthesis, shifting macrophages to an immune-inert phenotype
We initially hypothesized that bioactive proteins, such as cytokines, might be involved in the mechanism by which mFgfr3 cancer cells promote macrophage polarization toward an immune-inert phenotype. To test this hypothesis, we boiled the cancer cell supernatants to denature any bioactive proteins and then used the boiled supernatants to stimulate BMDMs. The purpose was to determine whether the boiling process would abolish the immunosuppressive capacity of the supernatants. Surprisingly, the results showed no discernible difference between the groups treated with boiled and unboiled mutant supernatants. Macrophages stimulated with boiled mutant supernatants displayed an increase in CD206 expression, while the expression levels of MHC molecules and CXCL9 remained reduced (Fig. 4A–C). These findings suggested that heat-vulnerable proteins, such as cytokines, were likely not the bioactive factors involved in macrophage polarization. Thus, heat-resistant small-molecule factors are potential contributors.
Figure 4.

mFGFR3 cancer cells upregulate serine synthesis to shift macrophages to an immune-inert phenotype. A–C, Flow cytometry analysis illustrating the proportion of CD206+ cells (A), IA/IE+ cells (B), and CXCL9+ cells (C) within BMDMs stimulated with unboiled/ boiled WT/ mFgfr3 cancer cells supernatants. D, GSEA showing the metabolism of amino acid and derivatives pathway is enriched in the mFgfr3 cancer cells. E, Volcano plot displays the marker genes of the mFgfr3 cancer cells. F, FeaturePlot shows the expression of Psat1 in the WT and mFgfr3 cancer cells. G, Representative IHC pictures and statistic analysis show the expression of PSAT1 in the WT and aFGFR3 tumors; n = 24 in the WT group, n = 10 in the aFGFR3 group. H, Schematic illustration of the role of PSAT1 in the serine synthesis metabolism. I, Relative free serine concentration of the WT and mFgfr3 MB49 cells or WT and mFGFR3 T24 cancer cells. J, Relative free serine concentration of the BMDMs stimulated with WT and mFgfr3 cancer cell supernatants. K–M, Flow cytometry analysis and statistical analysis illustrating the proportion of CD206+ cells (K), IA/IE+ cells (L), and CXCL9+ cells (M) within BMDMs in the indicated groups. N, Antigen-presenting assay shows the MHC-I SIINFEKL+ cells within the BMDMs. O, Transwell assay of lymphocytes with BMDMs previously stimulated by cancer cell supernatants shows the cell count of migrated lymphocytes in the indicated groups. P, Relative free serine concentration of the BMDMs in the indicated groups. Q–S, Flow cytometry analysis and statistical analysis illustrating the proportion of CD206+ cells (Q), IA/IE+ cells (R), and CXCL9+ cells (S) within BMDMs in the indicated groups. T, Western blot analysis indicates the phosphorylation level of the MAPK pathway as well as the PSAT1 expression in the indicated groups. The data are shown as the mean ± SEM values (n = 6 per group in A–C, N–O, and Q–S; n = 4 per group in I; n = 5 per group in J–M and P) or are representative of three independent experiments (T). P < 0.05 was considered a significant difference; ns, no significance (one-way ANOVA with Holm-Sidak multiple comparison tests, unpaired parametric Student t test). *, P < 0.05; **, P < 0.01; ***, P < 0.001. Scale bar, white, 100 μm. FMO, fluorescence minus one; SN, supernatants. (H, Created with BioRender.com.)
We conducted a comprehensive analysis of scRNA-seq data to explore the biological characteristics of mFgfr3 cancer cells. GSEA revealed a significant upregulation of the pathway involved in amino acid metabolism in the mFgfr3 cancer cells compared with WT cells (Fig. 4D). Further examination of the differentially expressed genes identified Psat1 as a significantly upregulated gene in the mutant cells (Fig. 4E and F). This finding was validated by qRT-PCR, which confirmed the upregulation of Psat1 in mutant MB49 cells and PSAT1 in mutant T24 cells (Supplementary Fig. S4A and S4B). IHC staining was performed to validate these findings in the SYSMH bladder cancer cohort, revealing a significant upregulation of PSAT1 in aFGFR3 cancers (Fig. 4G; Supplementary Fig. S4C). PSAT1 encodes phosphoserine aminotransferase (PSAT1), an enzyme that plays a role in serine synthesis. Specifically, PSAT1 catalyzes the conversion of phosphohydroxypyruvate (pPYR), derived from glycolysis, to produce phosphoserine (pSER), which is then dephosphorylated to form serine (Fig. 4H). Serine quantification assays of cancer cells revealed that the relative serine concentration was elevated in mFGFR3 cells (Fig. 4I). We also examined the serine concentration in BMDMs following stimulation with mFgfr3 supernatants. The results showed a significantly higher serine concentration than that in BMDMs supplemented with WT supernatant (Fig. 4J). We supplemented the WT supernatant with serine for BMDM stimulation to further explore the role of serine in macrophage phenotype shifting. Flow cytometry analysis revealed that adding serine to the WT supernatant had similar effects to supernatant from mutant cells. Macrophages stimulated with serine-supplemented WT supernatant demonstrated increased expression of CD206 and reduced expression of CXCL9 and IA/IE (Fig. 4K–M). Consistent results were obtained when THP-1 cells were stimulated with serine-supplemented T24 WT supernatant (Supplementary Fig. 4D–F). Antigen-presenting assays and a transwell assay of serine-supplemented BMDMs were also performed, and their results also confirmed that adding serine to the WT supernatant showed effects comparable with those of the mutant cell supernatant (Fig. 4N and O).
Next, we aimed to investigate the functional consequences of blocking the serine influx in BMDMs using L-phenylglycine, a recognized inhibitor of serine transporters. A serine assay confirmed that the serine concentration in BMDMs stimulated with mFgfr3 supernatants and treated with L-phenylglycine was reduced and comparable with that of the WT group (Fig. 4P). Flow cytometry analysis was subsequently conducted, revealing that after blocking serine influx in BMDMs, the macrophage phenotype in the mFgfr3 group became similar to that of the WT group (Fig. 4Q–S). Based on these results, we can conclude that serine controls the macrophage phenotype in the mFgfr3 TME.
Because the MAPK pathway is regarded as the predominant downstream signaling pathway of FGFR, we hypothesized that activating FGFR3 might upregulate PSAT1 expression through MAPK signaling (Fig. 4T; ref. 27). Two ERK isoforms, ERK1/2 and ERK5, have been previously reported. In our study, increases in both pERK1/2 and pERK5 expression in mutant cancer cells were observed by Western blotting. Inhibition of ERK1/2 rather than ERK5 reduced the expression of PSAT1 in MB49 cancer cells, but in the T24 cells, inhibition of either ERK1/2 or ERK5 could prevent the upregulation of PSAT1 (Supplementary Fig. S4G).
Targeting the PI3K/Akt pathway in macrophages reverses macrophage phenotypes in mFGFR3 cancers
We investigated the underlying signaling pathway responsible for the induced phenotype shift to determine how to reverse the macrophage phenotype shifts in mFgfr3 cancers. Considering the various pathways associated with macrophage phenotype modulation, we performed GSEA on well-established signaling pathways (Supplementary Table S2; ref. 28). The analysis revealed the PI3K/Akt signaling pathway, known to play a crucial role in macrophage phenotype conversion, to be the most enriched pathway (Fig. 5A). This pathway is known to regulate the expression of CD206, CXCL9, and MHC in macrophages (29–32). We assessed the phosphorylation levels of Akt in macrophages upon stimulation with mFgfr3 or WT cell supernatant to validate the involvement of the PI3K/Akt pathway in mFgfr3 supernatant–induced macrophage polarization. Significantly elevated Akt phosphorylation was observed in BMDMs exposed to mFgfr3 cell supernatant compared with that in those stimulated with WT cell supernatant (Fig. 5B; Supplementary Fig. S5A). Furthermore, supplementation of serine in BMDMs cultured with WT cancer supernatant increased Akt phosphorylation (Fig. 5C; Supplementary Fig. S5B).
Figure 5.

Targeting the PI3K/Akt pathway in macrophages reverses macrophage phenotype in mFGFR3 cancers. A, Bubble chart of GSEA results involving the major pathways regulating macrophage phenotype in the macrophages cells from mFgfr3 cancers compared with the ones from WT cancers, along with enrichment results of the PI3K cascade pathway as well as PI3K/Akt/mTOR signaling pathway, Mφ, macrophages. B–D, Flow cytometry analysis illustrating the proportion of pAKT+ cells within BMDMs in the indicated groups. E–H, Flow cytometry analysis illustrating the proportion of pAKT+ cells (E), CD206+ cells (F), IA/IE+ cells (G), and CXCL9+ cells (H) within BMDMs in the indicated groups. I and J, Tumor growth curves (I) and tumor weights (J) of the indicated groups in the duvelisib monotherapy experiments. K, Representative images show the infiltration of CD206+ F4/80+ cells in the indicated groups. L and M, Flow cytometry analysis illustrating the proportion of IA/IE+ cells (L) and CXCL9+ cells (M) within macrophages in the indicated groups. N and O, Flow cytometry analysis illustrating the proportion of CD3+CD8+ cells within CD45+ cells (N) and the proportion of granzyme B+ cells within CD8+ T cells (O) in the indicated groups. P, Representative images show CD8+ granzyme B+ cell infiltration in the indicated groups. Q, Schematic illustration of duvelisib treatment strategy in huPBMC-NOG-dKO humanoid mice bearing mFGFR3 T24 tumors. R–T, Tumor growth curves (R), tumor weights (S), and the resected tumor picture (T) of the indicated groups in the duvelisib treatment experiment on the humanoid mice. U and V, IHC staining indicates the CD206 (U) and CD8 (V) infiltration in the indicated groups from the humanoid mice experiment. The data are shown as the mean ± SEM values (n = 6 per group in B, C, and E–J; n = 4 per group in R and S; n = 3 per group in U and V; n = 4–6 per group in D; n = 5–6 per group in L–O; n = 3–4 per group in K and P). P < 0.05 was considered a significant difference; ns, no significance (one-way ANOVA with Holm-Sidak multiple comparison tests). **, P < 0.01; ***, P < 0.001. Scale bar, 100 μm. FMO, fluorescence minus one; SN, supernatants. (Q, Created with BioRender.com.)
The mechanism by which serine activates the PI3K/Akt pathway in macrophages remains unclear. Serine is primarily involved in providing methyl donors for DNA and protein methylation. Thus, we hypothesized that elevated serine levels in mFgfr3 cancer cells promote PI3K/Akt pathway activation by enhancing intracellular methylation. Therefore, we supplemented the WT cell supernatant with SAM, a universal methyl donor. The results showed that SAM supplementation resulted in similar changes in Akt phosphorylation as those observed with elevated serine levels (Fig. 5D; Supplementary Fig. S5C). This finding suggests that exogenous serine may enhance intracellular methylation activities, thereby activating macrophage Akt.
Duvelisib is a potent inhibitor of PI3Kδ and PI3Kγ, constitutively expressed in leukocytes, and is FDA approved for certain hematologic malignancies (33). We treated BMDMs stimulated with cancer cell supernatant with duvelisib to investigate whether targeting the PI3K/Akt pathway could reverse the immunosuppressive effects of mFgfr3 cancer cell supernatant. Flow cytometry analysis showed that duvelisib successfully abolished the elevation of Akt expression in macrophages cultured with mFgfr3 supernatant (Fig. 5E; Supplementary Fig. S5D). Furthermore, duvelisib reduced the expression of CD206 and restored the expression of CXCL9 and MHC molecules in macrophages treated with mFgfr3 supernatant (Fig. 5F–H; Supplementary Fig. S5E–S5G). Meanwhile, cell viability assays indicated that duvelisib had no inhibitory effect on cancer cell proliferation (Supplementary Fig. S5H and S5I). We subsequently investigated the in vivo antitumor efficacy of duvelisib in C57BL/6 mice bearing WT or mFgfr3 MB49 tumors (Supplementary Fig. S5J). Consistent with previous findings, mFgfr3 tumors exhibited accelerated growth compared with that of WT tumors. However, duvelisib treatment effectively inhibited the growth of mFgfr3 tumors, resulting in reduced tumor weights (Fig. 5I and J; Supplementary Fig. S5K). In contrast, WT tumors did not show significant responsiveness to duvelisib treatment, with no considerable differences in tumor growth and weight between the treatment and control groups. Multiplex IF analysis confirmed the reduction in CD206+ macrophages within mFgfr3 tumor tissues following duvelisib treatment (Fig. 5K; Supplementary Fig. S5L). Flow cytometry analysis revealed decreased production of CXCL9 and expression of IA/IE by macrophages in the mFgfr3 group compared with that in the WT group (Fig. 5L and M; Supplementary Fig. S5M–S5O). Duvelisib treatment reversed the effects of mFgfr3 cancer cells and enhanced the secretion of CXCL9 and expression of IA/IE by macrophages. In addition, an increased frequency of CD8+ granzyme B+ T cells was detected in mFgfr3 tumors treated with duvelisib (Fig. 5N–P; Supplementary Fig. S5P–S5S).
The above results demonstrated that duvelisib significantly impacts mouse mFgfr3 MB49 tumors. Hence, we applied it in humanized mice bearing mFGFR3 T24 tumors (Fig. 5Q). The results demonstrated that duvelisib could also significantly delay the growth of mFGFR3 T24 tumors, and we noticed a reduction in tumor weights in this treatment group (Fig. 5R–T). Immune infiltration in the tumors was consistent with the above data: reduced CD206+ cells and increased CD8+ T cells were seen in the duvelisib treatment group (Fig. 5U and V; Supplementary Fig. S5T and S5U).
Duvelisib augments the antitumor efficacy of erdafitinib in combination therapy
Duvelisib effectively reprogrammed the TME of mutant tumors by restoring the immune-inert macrophages back to an active phenotype. Subsequently, we assessed the antitumor efficacy of duvelisib in combination with erdafitinib in mFgfr3 tumor-bearing C57BL/6 mice (Fig. 6A). In vitro cell viability assays indicated that erdafitinib has an inhibiting effect on cancer cell growth (Supplementary Fig. S6A). Compared with the vehicle treatment, duvelisib, and erdafitinib monotherapy significantly delayed tumor growth (Fig. 6B–D). Notably, compared with monotherapy, the combination of duvelisib and erdafitinib demonstrated enhanced suppression of tumor growth and reduced tumor weight. Multiplex IF analysis revealed a marked reduction in the number of CD206+ cells in the monotherapy groups, with even lower infiltration observed in the combination treatment group (Fig. 6E). Flow cytometry analysis demonstrated that both duvelisib and erdafitinib upregulated the expression of CXCL9 and IA/IE by macrophages (Fig. 6F and G). Consequently, CD8+ granzyme B+ T-cell infiltration increased (Fig. 6H–J). Importantly, the combination of erdafitinib with duvelisib further improved CXCL9 secretion, IA/IE expression, and CD8+ T-cell infiltration.
Figure 6.

Duvelisib augments the antitumor efficacy of erdafitinib in combination therapy. A, Schematic illustration of duvelisib and erdafitinib treatment strategy. B and C, Tumor growth curves (B) and tumor weights (C) of the groups receiving vehicle, duvelisib, erdafitinib, and combination therapy, respectively. D, Images of the dissected tumors from mice in the vehicle, duvelisib, erdafitinib, and combination treatment groups. E, Representative images and statistical analysis show CD206+ F4/80+ cell infiltration in the indicated groups. F and G, Flow cytometry analysis and statistical analysis illustrating the proportion of IA/IE+ cells (F) and CXCL9+ cells (G) within macrophages in the indicated groups. H and I, Flow cytometry analysis illustrating the proportion of CD3+CD8+ cells within CD45+ cells (H) and the proportion of granzyme B+ cells within CD8+ T cells (I) in the indicated groups. J, Representative images and statistical analysis show CD8+ granzyme B+ cell infiltration in the indicated groups. The data are shown as the mean ± SEM values (n = 5 per group in B–D; n = 4–5 per group in F–I; n = 3–4 per group in E and J). P < 0.05 was considered a significant difference (one-way ANOVA with Holm-Sidak multiple comparison tests). *, P < 0.05; **, P < 0.01; ***, P < 0.001. Scale bar, white, 100 μm. (A, Created with BioRender.com.)
We also investigated the efficacy of these combination therapies in nude mice with mFgfr3 MB49 tumors. Our findings demonstrated that treatment with erdafitinib as a monotherapy partially delayed tumor growth (Supplementary Fig. S6B–S6D). However, duvelisib showed no efficacy in immune-deficient mice when administered as a monotherapy. This observation further reinforces the notion that duvelisib can effectively slow tumor growth by modulating immune responses specifically in immune-competent mice. Notably, the combination therapy exhibited comparable results to erdafitinib monotherapy.
Discussion
This study provided evidence of aFGFR3 tumors being correlated with an immune-desert TME characterized by low numbers of inactive CD8+ T cells. We found that heightened serine synthesis in mFGFR3 cancer cells induces an immunologically cold state by shifting macrophages toward the immune-inert subtype, marked by impaired antigen presentation and T-cell chemotaxis. Moreover, we uncovered the mechanism through which increased serine efflux from mutant cancer cells activates Akt kinase activity, leading to the upregulation of CD206 expression and the downregulation of MHC molecules and CXCL9 expression in macrophages. We devised a novel therapeutic strategy using a PI3K inhibitor to inhibit macrophage reprogramming and enhance the treatment efficacy of erdafitinib in mFgfr3 bladder cancers.
Several studies have indicated the association of aFGFR3 tumors with an immune-desert TME. For example, it has been shown that aFGFR3 tumors are more prevalent in the luminal or luminal papillary subtypes, which are typically characterized by fewer T-cell inflammatory responses (9). In addition, according to the results of the BLC2001 study of erdafitinib, only 5% of aFGFR3 patients respond to prior ICIs treatment, suggesting that aFGFR3 cancers are associated with a more deleterious TME refractory to ICIs treatment (7). A similar trend was observed in a study of rogaratinib, which included 10 patients, with 90% displaying unresponsiveness to previous immunotherapy (34). Some people suggested that the PD-1/PD-L1 axis could be activated through FGFR activation in cancer cells (12, 13, 35). Nevertheless, this explanation does not account for the observed scarcity of cytotoxic T-cell infiltration or the limited response rate to ICIs treatment in these patients. In the current study, we demonstrate that cancer cells with FGFR3 alterations do not directly influence T-cell chemotaxis or functionality. Conversely, it was revealed that these cancer cells hijacked macrophages, inducing their phenotypic shift toward an immune-inert state, thus playing a pivotal role in establishing the immunosuppressive microenvironment.
Metabolic alterations have often been observed in cancer cells with oncogenic activation, which was previously considered a cell-autonomous adaptation (36). Recent findings have suggested that oncogene activation plays a causative rather than a correlative role in reprogramming metabolic phenotypes in malignant cells (37). For example, in a recent study by Nan, activation of oncogenic tyrosine kinase receptors, such as FGFR activation in non–small cell lung carcinoma, enhanced aerobic glycolysis and recycled lactate (16). In melanoma, oncogenic BRAF activates the MAPK pathway, upregulates aerobic glycolysis, and enhances reliance on glutamine utilization (38). In this study, our results showed that FGFR3 activation in bladder cancer cells could upregulate PSAT1 expression and branch glycolysis to serine synthesis through the MAPK pathway. Serine is the fundamental building block essential for sustaining life. In addition to its role in protein synthesis, serine is involved in various metabolic pathways, including redox balance, nucleotide synthesis, and one-carbon metabolism (19, 39). One-carbon metabolism consists of the folate and methionine cycle and integrates carbon units from serine, generating various products, including substrates for methylation reactions (40). Intracellular nucleotide and protein methylation influence transcription, translation, and posttranslational protein function and stability. Cancer cells utilize these pathways to support uncontrolled proliferation and survival, with enhanced aerobic glycolysis resulting in serine overproduction.
The implications of serine metabolism in the TME extend beyond cancer cell proliferation. Serine metabolism also influences the functionality and phenotypes of immune cells. Previous studies have demonstrated that the metabolic status of immune cells is critical due to their dynamic activation states (20, 41). For example, Ma and colleagues (42) revealed that serine metabolism is required for effector T-cell expansion by fueling nucleotide biosynthesis and one-carbon metabolism. However, in the TME, most immune cells proliferate at a relatively slow rate, and serine synthesis in these cells is less active. In contrast, highly proliferating cancer cells that hyperactivate serine synthesis are the major source of serine in the TME (43). We believe this discrepancy in serine metabolism between cancer cells and immune cells has profound implications for immune cell function. In this study, our findings strongly suggest that the overproduction of serine from cancer cells, specifically mFGFR3 cells, plays a crucial role in macrophage phenotypic shifts toward an immune-inert state. This effect is evidenced by the downregulation of MHC molecules and the CXCL9 chemokines. The observed immune-inert phenotype in macrophages within the TME could potentially impair their ability to mount an effective immune response against cancer cells.
Some previous studies have suggested that in noncancer settings, macrophages are influenced by intracellular serine metabolism alteration. For example, a recent study demonstrated that serine metabolism deficiency in macrophages activated the p38-dependent JAK-STAT1 axis (44). In another study, intracellular serine metabolism was found to activate mTOR signaling and support IL1β production in macrophages (45). However, the precise mechanism by which macrophages are influenced by serine efflux remains controversial. In this study, we aimed to investigate the signaling pathways that regulate macrophage phenotype switching in response to serine efflux. Our screening process identified the PI3K/Akt pathway as the most upregulated pathway in macrophages from the mFgfr3 group. Subsequent experiments confirmed that serine supplementation increased Akt phosphorylation levels and induced substantial phenotype changes in macrophages. These findings provide novel insights into the mechanisms underlying the influence of serine efflux on macrophages. The upregulation of the PI3K/Akt pathway and its subsequent effects on macrophage phenotypes suggest a potential molecular mechanism involved in the serine-mediated modulation of immune responses.
The PI3K/Akt signaling has been best studied in tumorigenesis, where activating PI3K mutations or inactivating PTEN mutations represent frequent mutations in various cancer types (46). It is known that the PI3K/Akt pathway plays a vital role in macrophage phenotype modulation. For instance, Kaneda and colleagues indicated that PI3K inhibition enhanced the expression of MHC II and proinflammatory cytokines and inhibited the expression of immune suppressive factors in macrophages, indicating that PI3K controls the switch between immune suppression and immune stimulation (30). Other studies have validated this finding (31, 32). However, how this pathway is regulated in the macrophages remains largely unclear. According to studies on the PI3K/Akt signaling in cancer cells, in addition to the classic Akt-activating steps, several posttranslational modifications fine-tune Akt activity, including methylation, phosphorylation, and ubiquitination (47–49). Here, our studies showed that exogenous serine or SAM supplementation could enhance the phosphorylation level of Akt in macrophages, suggesting that enhancing intracellular methylation activities in macrophages promotes Akt activity. Several previous studies have demonstrated that increased Akt protein methylation promotes phosphorylation and enhances kinase activities (50, 51), which might be the trigger point of serine-induced Akt activation in macrophages.
PI3Ks consist of a regulatory subunit with a p110 catalytic subunit (p110α, β, γ, or δ; ref. 52). Herein, these heterodimeric complexes are referred to as PI3Kα, PI3Kβ, PI3Kγ, and PI3Kδ, with p110α, p110β, p110γ, and p110δ indicating the catalytic subunits themselves. p110α and p110β showed broad tissue distribution, whereas p110γ and p110δ were highly enriched in all leukocyte subtypes. Traditionally, the clinical application of pan-PI3K inhibitors in cancer settings has been limited because of their adverse cardiovascular and metabolic adverse effects (53). Several studies have indicated that compared with the pan-PI3K inhibitors, selective PI3K inhibitors in leukocytes have a better safety profile (54). Duvelisib, a selective PI3Kδ and PI3Kγ inhibitor, has been approved for the treatment of several types of lymphoma (33). In this study, we demonstrated that the selective inhibition of PI3K exhibits significant antitumor efficacy in aFGFR3 cancers due to the considerable upregulation of the PI3K/Akt pathway in macrophages.
Conclusion
In this study, we revealed that aFGFR3 cancers are associated with a non–T-cell–inflamed TME. We subsequently found that by activating the PI3K/Akt pathway, increased serine synthesis from mFGFR3 cancer cells shifted macrophages to an immune-inert phenotype characterized by reduced expressed chemokines for lymphocytes chemotaxis and MHC molecules, resulting in the promotion of a cold immune TME. We further suggest that redirecting the macrophage phenotype with PI3K inhibitors has the potential to enhance the antitumor efficacy of erdafitinib in aFGFR3 tumors.
Supplementary Material
Supplementary Figures 1-6
Supplementary Materials and Methods
Supplementary Table 1-4
Acknowledgments
This study was supported by the National Natural Science Foundation of China (grant no. 81825016 to Tianxin Lin; 81F961128027 to J. Huang; and 82373254 to Wenlong Zhong), Natural Science Foundation of Guangdong (grant no. 2023A1515010258 to W. Zhong), and Key Areas Research and Development Program of Guangdong (grant no. 2018B010109006 to T. Lin). The authors would like to thank Prof. Phei Er Saw for her advice on the experimental design.
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Authors' Disclosures
W. Zhong reports grants from National Natural Science Foundation of China and Natural Science Foundation of Guangdong during the conduct of the study. T. Lin reports grants from National Natural Science Foundation of China and Key Areas Research and Development Program of Guangdong during the conduct of the study. J. Huang reports grants from National Natural Science Foundation of China during the conduct of the study. No disclosures were reported by the other authors.
Authors' Contributions
Y. Ouyang: Conceptualization, formal analysis, validation, investigation, visualization, methodology, writing–original draft, writing–review and editing. Z. Ou: Conceptualization, investigation, methodology, writing–original draft. W. Zhong: Conceptualization, formal analysis, writing–original draft, writing–review and editing. J. Yang: Investigation. S. Fu: Resources, validation, investigation. N. Ouyang: Resources, validation, investigation. J. Chen: Methodology. L. Xu: Methodology. D. Wu: Investigation. J. Qian: Investigation. Y. Lin: Software. T. Lin: Supervision, writing–review and editing. J. Huang: Supervision, writing–review and editing.
References
- 1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin 2022;72:7–33. [DOI] [PubMed] [Google Scholar]
- 2. Cathomas R, Lorch A, Bruins HM, Comperat EM, Cowan NC, Efstathiou JA, et al. The 2021 updated European Association of Urology guidelines on metastatic urothelial carcinoma. Eur Urol 2022;81:95–103. [DOI] [PubMed] [Google Scholar]
- 3. Robertson AG, Kim J, Al-Ahmadie H, Bellmunt J, Guo G, Cherniack AD, et al. Comprehensive molecular characterization of muscle-invasive bladder cancer. Cell 2018;174:1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pietzak EJ, Bagrodia A, Cha EK, Drill EN, Iyer G, Isharwal S, et al. Next-generation sequencing of nonmuscle invasive bladder cancer reveals potential biomarkers and rational therapeutic targets. Eur Urol 2017;72:952–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Teo MY, Mota JM, Whiting KA, Li HA, Funt SA, Lee CH, et al. Fibroblast growth factor receptor 3 alteration status is associated with differential sensitivity to platinum-based chemotherapy in locally advanced and metastatic urothelial carcinoma. Eur Urol 2020;78:907–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Helsten T, Elkin S, Arthur E, Tomson BN, Carter J, Kurzrock R. The FGFR landscape in cancer: analysis of 4,853 tumors by next-generation sequencing. Clin Cancer Res 2016;22:259–67. [DOI] [PubMed] [Google Scholar]
- 7. Loriot Y, Necchi A, Park SH, Garcia-Donas J, Huddart R, Burgess E, et al. Erdafitinib in locally advanced or metastatic urothelial carcinoma. N Engl J Med 2019;381:338–48. [DOI] [PubMed] [Google Scholar]
- 8. Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer 2010;10:116–29. [DOI] [PubMed] [Google Scholar]
- 9. Robertson AG, Kim J, Al-Ahmadie H, Bellmunt J, Guo G, Cherniack AD, et al. Comprehensive molecular characterization of muscle-invasive bladder cancer. Cell 2017;171:540–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kamoun A, de Reynies A, Allory Y, Sjodahl G, Robertson AG, Seiler R, et al. A consensus molecular classification of muscle-invasive bladder cancer. Eur Urol 2020;77:420–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Foth M, Ismail NFB, Kung JSC, Tomlinson D, Knowles MA, Eriksson P, et al. FGFR3 mutation increases bladder tumourigenesis by suppressing acute inflammation. J Pathol 2018;246:331–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hu Y, Lu Y, Xing F, Hsu W. FGFR1/MAPK-directed brachyury activation drives PD-L1-mediated immune evasion to promote lung cancer progression. Cancer Lett 2022;547:215867. [DOI] [PubMed] [Google Scholar]
- 13. Li P, Huang T, Zou Q, Liu D, Wang Y, Tan X, et al. FGFR2 promotes expression of PD-L1 in colorectal cancer via the JAK/STAT3 signaling pathway. J Immunol 2019;202:3065–75. [DOI] [PubMed] [Google Scholar]
- 14. Lu M, Wang K, Ji W, Yu Y, Li Z, Xia W, et al. FGFR1 promotes tumor immune evasion via YAP-mediated PD-L1 expression upregulation in lung squamous cell carcinoma. Cell Immunol 2022;379:104577. [DOI] [PubMed] [Google Scholar]
- 15. Faubert B, Solmonson A, DeBerardinis RJ. Metabolic reprogramming and cancer progression. Science 2020;368:eaaw5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jin N, Bi A, Lan X, Xu J, Wang X, Liu Y, et al. Identification of metabolic vulnerabilities of receptor tyrosine kinases-driven cancer. Nat Commun 2019;10:2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liberti MV, Locasale JW. The warburg effect: how does it benefit cancer cells? Trends Biochem Sci 2016;41:211–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Amelio I, Cutruzzola F, Antonov A, Agostini M, Melino G. Serine and glycine metabolism in cancer. Trends Biochem Sci 2014;39:191–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Geeraerts SL, Heylen E, De Keersmaecker K, Kampen KR. The ins and outs of serine and glycine metabolism in cancer. Nat Metab 2021;3:131–41. [DOI] [PubMed] [Google Scholar]
- 20. He F, Yin Z, Wu C, Xia Y, Wu M, Li P, et al. l-Serine lowers the inflammatory responses during pasteurella multocida infection. Infect Immun 2019;87:e00677–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wilson JL, Nagele T, Linke M, Demel F, Fritsch SD, Mayr HK, et al. Inverse data-driven modeling and multiomics analysis reveals PHGDH as a metabolic checkpoint of macrophage polarization and proliferation. Cell Rep 2020;30:1542–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A, et al. Integrated analysis of multimodal single-cell data. Cell 2021;184:3573–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Haghverdi L, Lun ATL, Morgan MD, Marioni JC. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat Biotechnol 2018;36:421–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018;554:544–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rosenberg JE, Hoffman-Censits J, Powles T, van der Heijden MS, Balar AV, Necchi A, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet 2016;387:1909–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jin S, Guerrero-Juarez CF, Zhang L, Chang I, Ramos R, Kuan CH, et al. Inference and analysis of cell-cell communication using CellChat. Nat Commun 2021;12:1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yadav V, Zhang X, Liu J, Estrem S, Li S, Gong XQ, et al. Reactivation of mitogen-activated protein kinase (MAPK) pathway by FGF receptor 3 (FGFR3)/Ras mediates resistance to vemurafenib in human B-RAF V600E mutant melanoma. J Biol Chem 2012;287:28087–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol 2011;11:750–61. [DOI] [PubMed] [Google Scholar]
- 29. Kerneur C, Cano CE, Olive D. Major pathways involved in macrophage polarization in cancer. Front Immunol 2022;13:1026954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, et al. PI3Kgamma is a molecular switch that controls immune suppression. Nature 2016;539:437–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu X, Zhang W, Xu Y, Xu X, Jiang Q, Ruan J, et al. Targeting PI3Kgamma/AKT pathway remodels LC3-associated phagocytosis induced immunosuppression after radiofrequency ablation. Adv Sci 2022;9:e2102182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Goossens P, Rodriguez-Vita J, Etzerodt A, Masse M, Rastoin O, Gouirand V, et al. Membrane cholesterol efflux drives tumor-associated macrophage reprogramming and tumor progression. Cell Metab 2019;29:1376–89. [DOI] [PubMed] [Google Scholar]
- 33. Flinn IW, O'Brien S, Kahl B, Patel M, Oki Y, Foss FF, et al. Duvelisib, a novel oral dual inhibitor of PI3K-delta, gamma, is clinically active in advanced hematologic malignancies. Blood 2018;131:877–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schuler M, Cho BC, Sayehli CM, Navarro A, Soo RA, Richly H, et al. Rogaratinib in patients with advanced cancers selected by FGFR mRNA expression: a phase 1 dose-escalation and dose-expansion study. Lancet Oncol 2019;20:1454–66. [DOI] [PubMed] [Google Scholar]
- 35. Jing W, Wang G, Cui Z, Xiong G, Jiang X, Li Y, et al. FGFR3 destabilizes PD-L1 via NEDD4 to control T-cell-mediated bladder cancer immune surveillance. Cancer Res 2022;82:114–29. [DOI] [PubMed] [Google Scholar]
- 36. Vazquez A, Liu J, Zhou Y, Oltvai ZN. Catabolic efficiency of aerobic glycolysis: the warburg effect revisited. BMC Syst Biol 2010;4:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Levy P, Bartosch B. Metabolic reprogramming: a hallmark of viral oncogenesis. Oncogene 2016;35:4155–64. [DOI] [PubMed] [Google Scholar]
- 38. Ratnikov BI, Scott DA, Osterman AL, Smith JW, Ronai ZA. Metabolic rewiring in melanoma. Oncogene 2017;36:147–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yang M, Vousden KH. Serine and one-carbon metabolism in cancer. Nat Rev Cancer 2016;16:650–62. [DOI] [PubMed] [Google Scholar]
- 40. Locasale JW. Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat Rev Cancer 2013;13:572–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Leone RD, Powell JD. Metabolism of immune cells in cancer. Nat Rev Cancer 2020;20:516–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ma EH, Bantug G, Griss T, Condotta S, Johnson RM, Samborska B, et al. Serine is an essential metabolite for effector T cell expansion. Cell Metab 2017;25:345–57. [DOI] [PubMed] [Google Scholar]
- 43. Xia L, Oyang L, Lin J, Tan S, Han Y, Wu N, et al. The cancer metabolic reprogramming and immune response. Mol Cancer 2021;20:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shan X, Hu P, Ni L, Shen L, Zhang Y, Ji Z, et al. Serine metabolism orchestrates macrophage polarization by regulating the IGF1-p38 axis. Cell Mol Immunol 2022;19:1263–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rodriguez AE, Ducker GS, Billingham LK, Martinez CA, Mainolfi N, Suri V, et al. Serine metabolism supports macrophage IL-1beta production. Cell Metab 2019;29:1003–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hoxhaj G, Manning BD. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat Rev Cancer 2020;20:74–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Guo J, Wei W. Fine-tuning AKT kinase activity through direct lysine methylation. Cell Cycle 2019;18:917–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chan CH, Jo U, Kohrman A, Rezaeian AH, Chou PC, Logothetis C, et al. Posttranslational regulation of Akt in human cancer. Cell Biosci 2014;4:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Manning BD, Toker A. AKT/PKB signaling: navigating the network. Cell 2017;169:381–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Guo J, Dai X, Laurent B, Zheng N, Gan W, Zhang J, et al. AKT methylation by SETDB1 promotes AKT kinase activity and oncogenic functions. Nat Cell Biol 2019;21:226–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang G, Long J, Gao Y, Zhang W, Han F, Xu C, et al. SETDB1-mediated methylation of Akt promotes its K63-linked ubiquitination and activation leading to tumorigenesis. Nat Cell Biol 2019;21:214–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bilanges B, Posor Y, Vanhaesebroeck B. PI3K isoforms in cell signalling and vesicle trafficking. Nat Rev Mol Cell Biol 2019;20:515–34. [DOI] [PubMed] [Google Scholar]
- 53. Vanhaesebroeck B, Perry MWD, Brown JR, Andre F, Okkenhaug K. PI3K inhibitors are finally coming of age. Nat Rev Drug Discov 2021;20:741–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Curigliano G, Shah RR. Safety and tolerability of phosphatidylinositol-3-kinase (PI3K) inhibitors in oncology. Drug Saf 2019;42:247–62. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures 1-6
Supplementary Materials and Methods
Supplementary Table 1-4
Data Availability Statement
The IMvigor210 dataset analyzed in this study was downloaded from http://research-pub.gene.com/IMvigor210CoreBiologies. In addition, TCGA-BLCA cohort data analyzed in this study was extracted from TCGA at https://www.cancer.gov/ccg/research/genome-sequencing/tcga. The scRNA-seq data that support the findings of this study have been deposited into the Gene Expression Omnibus with accession number GSE243093 and the CNGB Sequence Archive (CNSA) of China National GeneBank DataBase (CNGBdb) with accession number CNP0004744. Other data supporting the findings of this study are available upon request from the corresponding author.