

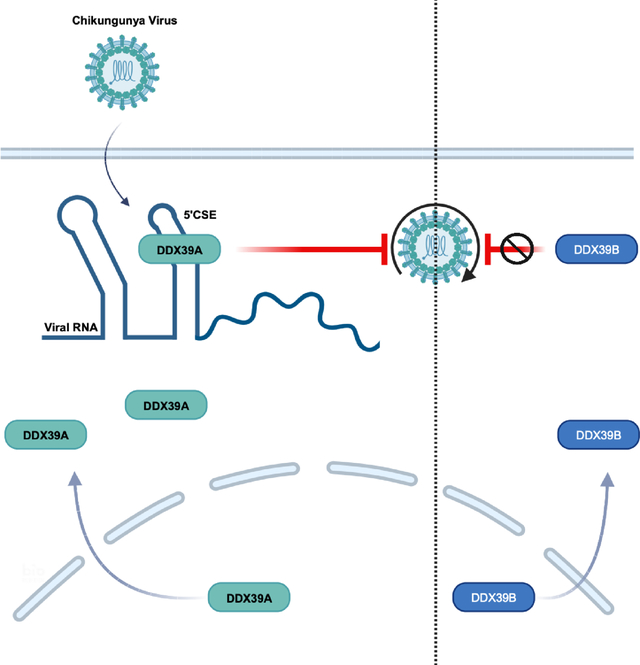
SUMMARY
Alphaviruses are a large group of re-emerging arthropod-borne RNA viruses. The compact viral RNA genomes harbor diverse structures that facilitate replication. These structures can be recognized by antiviral cellular RNA binding proteins, including DExD-box (DDX) helicases, that bind viral RNAs to control infection. The full spectrum of antiviral DDXs and the structures that are recognized remains unclear. Genetic screening identified DDX39A as antiviral against the alphavirus chikungunya virus (CHIKV) as well as other medically relevant alphaviruses. Upon infection, the predominantly nuclear DDX39A accumulates in the cytoplasm inhibiting alphavirus replication independent of the canonical interferon pathway. Biochemically, DDX39A binds to CHIKV genomic RNA, interacting with the 5ʹ conserved sequence element (5ʹCSE) which is essential for the antiviral activity of DDX39A. Altogether, DDX39A relocalization and binding to a conserved structural element in the alphavirus genomic RNA attenuates infection revealing a previously unknown layer to the cellular control of infection.
eTOC Blurb
The full spectrum of host RNA binding proteins that control viral infection remains unclear. Tapescu et al. show that DDX39A binds a conserved RNA structure encoded in the genome of alphaviruses inhibiting infection independent of the canonical interferon pathway.
Graphical Abstract
INTRODUCTION
Alphaviruses are medically relevant viruses that are a reemerging public health threat1. Based on their historic geographical distribution and clinical syndromes, alphaviruses are divided into Old World alphaviruses including chikungunya (CHIKV), Sindbis (SINV), O-nyong-nyong virus (ONNV), and into New World alphaviruses including Venezuelan equine encephalitis (VEEV)1. Infection with CHIKV leads to fever, headache, myalgia, and joint pain, in some cases leading to long-lasting and debilitating arthritis2. SINV, the prototypical alphavirus, transmitted between mosquitoes and migratory birds in Northern Europe causes self-limiting disease3. ONNV is endemic to sub-Saharan Africa and causes severe arthralgia and cervical lymphadenopathy4. VEEV predominantly circulates in the Americas and causes a spectrum of diseases. Around ~5–15% of infected individuals present with central nervous disease leading to fatal or long-lasting neurologic sequelae5. Despite being a major threat to human health, there are no specific treatments or vaccines approved for alphaviruses6,7.
Alphaviruses contain a compact positive-sense, single-stranded RNA genome of ~12 kb. Given this limited coding capacity, viral RNAs harbor structural elements to direct many activities required for the replication8–12. The replication cycle starts with alphavirus binding to specific receptors, internalization into an endocytic compartment, and release of the viral genome into the cytoplasm13. In the cytoplasm, the capped and polyadenylated viral genome with two open reading frames (ORFs) is immediately translated by host ribosomes14,15. ORF-1 is translated from the genomic RNA and encodes the nonstructural proteins nsP1–4, which are part of the viral replicase complex responsible for RNA synthesis. RNA synthesis by the viral replicase is facilitated by viral RNA structures, including the 5ʹ conserved sequence element (5ʹCSE) – the most conserved structural element across alphaviruses8,9,16. Moreover, during viral RNA replication, sequences and structures at the genome termini direct antigenomic (negative sense) RNA synthesis, which leads to the production of the double-stranded RNA (dsRNA) intermediate17. The antigenome serves as a template for the genome and the more abundant subgenomic RNA that is produced from an internal promoter in the antigenome RNA15,17,18. ORF-2, translated from the subgenomic RNA, encodes the alphaviral structural proteins required for the assembly and egress of virions. Additional RNA structures, including the packaging signal, direct efficient genome packaging within the virions for the ultimate release of infectious particles10,19.
To mount an antiviral response, cells must first sense the viral infection. Distinct features on viral nucleic acids, including dsRNA intermediates, are recognized by pathogen recognition receptors (PRRs) as foreign, which leads to the activation of antiviral pathways20. Two well-established PRRs are the retinoic acid-inducible gene I (RIG-I/DDX58) and melanoma differentiation-associated protein 5 (MDA-5/IFIH1), which recognize the virally encoded 5’ triphosphates and dsRNAs, respectively to induce interferon (IFN) signaling and the downstream interferon-stimulated genes (ISGs) that control infection20,21. However, alphaviruses have evolved diverse and potent strategies to evade and attenuate the canonical interferon pathway22–25. Given that alphaviruses suppress the canonical IFN program but are ultimately cleared, we hypothesized that host cells encode additional PRRs to control infection. Since RIG-I and MDA-5 are part of a larger family of DExH/D-box helicases that play diverse roles in RNA metabolism by binding to structural features rather than specific sequences, we hypothesized that this makes them well-suited to target structures on viral RNAs26–29. Indeed, emerging research suggests that in addition to RIG-I and MDA-5, other DDXs have critical roles in antiviral defense25,30–32. Furthermore, DDXs can bind RNA structural elements; however, the specificity of this large family is poorly understood33.
Altogether, given that we have an incomplete understanding of the host-encoded RNA helicases involved and the viral RNA structures they recognize, we set out to discover additional DDXs that control the alphavirus CHIKV. We performed a loss-of-function screen targeting 42 human DDXs and found that DDX39A is antiviral against CHIKV while its close paralog DDX39B is not. DDX39A is an RNA helicase with roles in mRNA export, innate immunity, and cancer, and has not been shown to have antiviral activity34–37. Furthermore, we found that DDX39A controls diverse alphaviruses, but not against other positive sense, such as coronaviruses and picornaviruses, or negative sense viruses, such as rhabdoviruses and bunyaviruses suggesting that DDX39A controls this group of viruses more broadly. Mechanistically, we found that DDX39A antiviral activity is independent of the canonical interferon pathway. During infection, DDX39A relocalizes from the nucleus to the cytoplasm where it binds CHIKV genomic RNA. We mapped the interaction using cross-linking immunoprecipitation (CLIP-Seq) and found an enrichment of DDX39A binding within the 5ʹCSE element of CHIKV RNA, which is the most highly conserved structure across diverse alphaviruses16. We showed the antiviral activity of DDX39A is dependent on the 5ʹCSE as loss of the structure renders CHIKV insensitive to the antiviral activity of DDX39A. Altogether, we found that DDX39A controls diverse alphaviruses independently of IFN through recognition of a conserved RNA structural element.
RESULTS
DDX39A is antiviral against alphaviruses
To identify DEAD-box helicases that impact CHIKV infection, we optimized an image-based RNAi screen in human osteosarcoma U2OS cells, which are permissive to alphavirus infections18,31. We included siRNAs targeting anti-apoptotic RNAs (siDeath) as a positive control for knockdown efficiency (~90% of cells die 3 days post-transfection) (Figure S1A). Furthermore, we used scrambled siRNA (siCon) as negative controls and siRNAs against the known antiviral factors IFIT1 and zinc finger antiviral protein (ZAP) as positive controls38,39. Infection with CHIKV (Ross strain, MOI 0.1, 24 h) led to an increase in CHIKV-infected cells upon loss of either IFIT1 or ZAP compared to non-targeting controls (Figure S1B). Using this validated assay, we performed a screen targeting 42 human DDXs (4 siRNAs per well, Table S1), and infected cells with CHIKV (Ross strain, MOI 0.1, 24h). We used automated imaging and image analysis to quantify cell number (Hoechst, nuclei +) and percent infection (Hoechst, nuclei+, and CHIKV+, using an anti-CHIKV antibody) (Figure 1A). We identified 3 helicases that when depleted led to an increase in infection (Z>1.5) in the absence of toxicity (Z<−2): DDX39A, DDX43, and DDX50 (Figure 1B, Table S1). DDX50 and DDX43 are known to stimulate the IFN signaling pathway40,41, and thus we did not pursue these candidates. DDX39A has a close paralog, DDX39B, with 90% homology, whose depletion did not impact infection (Figure 1B, Table S1), suggesting specificity. Therefore, we set out to explore the role of DDX39A in CHIKV infection.
Figure 1. DDX39A is an antiviral against alphaviruses.
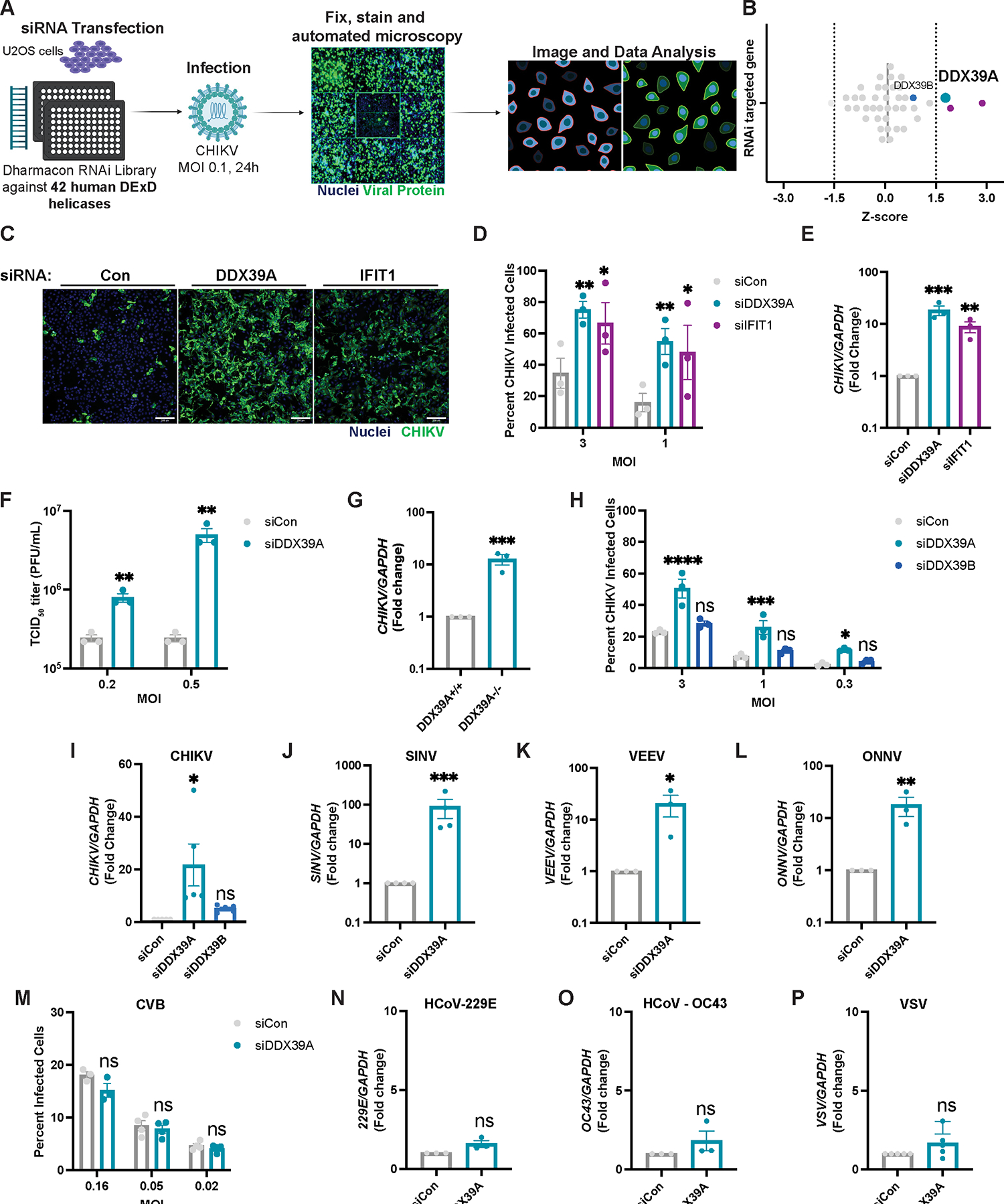
(A) Human U2OS cells were transfected with 42 siRNAs targeting human DDXs and infected with chikungunya (CHIKV, MOI 0.1, 24h). Cells were fixed and immunostained (infection, anti-CHIKV, and nuclei, Hoechst 33342). Automated imaging and image analysis were used to quantify cell number and percent infection. (B) Z-scores were graphed after removing cytotoxic candidates (Z<−2); 3 antiviral DDXs with a Z>1.5 are highlighted in purple, with DDX39A in turquoise. DDX39B is shown in blue. (C) Representative image of control, DDX39A, and IFIT1-depleted cells infected with CHIKV at the indicated MOIs for 24h and immunostained (nuclei, blue and infection, green). Magnification 10x; Scale bar 200 μm. Infection levels were quantified by (D) automated microscopy or by (E) reverse transcription-PCR (qPCR) for viral RNA. (F) Control or DDX39A-depleted U2OS cells were infected with CHIKV, and viral titers (median tissue culture infectious dose (TCID50)) were quantified 24hpi. (G) DDX39A−/− U2OS were generated using CRISPR/Cas9 and infected with CHIKV (MOI 0.5, 24h). CHIKV RNA was measured by qPCR. (H-I) Control, DDX39A, and DDX39B-depleted U2OS cells infected with CHIKV for 24h at (H) the indicated MOIs or at (I) MOI 0.5. Infection levels were quantified by (H) automated microscopy or by (I) qPCR for viral RNA. (J-M) Control and DDX39A-depleted U2OS cells were infected with (J) SINV (MOI 0.1, 24h), (K) VEEV (MOI 0.0005, 24h), (L) ONNV (MOI 0.005, 24h), (M) CBV (24h) at the indicated MOIs and infection was quantified by (J-L) RT-qPCR or (M) automated microscopy. (N-M) Control and DDX39A-depleted A549 cells were infected with (N) hCoV-229E (MOI 1.2, 24h), (O) hCoV-OC43 (MOI 1, 24h), and infection was assessed by qPCR. (P) Control and DDX39A-depleted cells were infected with VSV (MOI 1, 16h), and viral RNA was quantified by RT-qPCR. Data are presented as fold change vs. control (siCon) in (E, I-L). Dots represent individual knockdown experiments. GAPDH was used as a loading control gene for all qPCR experiments unless otherwise noted. Statistical analyses were performed using Student’s (unpaired, two-tailed) t-test (G, J, K, L, N, O, P), multiple unpaired t-tests (F), one-way ANOVA with Bonferroni corrections for multiple comparisons (E, I), two-way ANOVA with Bonferroni correction for multiple comparisons (D, F, H, M). *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001; error bars represent S.E.M.
We validated the role of DDX39A in alphavirus infection using orthogonal assays. We transfected cells with either control siRNA (siCon), siIFIT1 as a positive control, or siRNAs against DDX39A and infected with CHIKV at various multiplicities of infection (MOI). Cells depleted of DDX39A or IFIT1 show a comparable increase in infection (Figure 1C–D) in the absence of cytotoxicity across MOIs (Figure S1C) as measured by automated microscopy. Immunoblot analysis also reveals similar increases in viral proteins upon depletion of either DDX39A or IFIT1 (Fig S1D). We also evaluated whether depletion of DDX39A impacted levels of CHIKV RNA by RT-qPCR and found that depletion of DDX39A leads to a ~10-fold increase in CHIKV viral RNA (Figure 1E). Furthermore, the depletion of DDX39A leads to increased viral titers (Figure 1F) and viral RNA (Figure S1E) at multiple MOIs. To confirm that the siRNAs were on-target, we tested the individual siRNAs and found that both siRNAs against DDX39A deplete DDX39A mRNA (Figure S1F) and protein (Figure S1G) and are active against CHIKV (Figure S1H). We also generated a U2OS DDX39A knockout cell line using CRISPR-Cas9, which we validated by immunoblot (Figure S1I). Consistent with our siRNA approach, loss of DDX39A leads to a ~10-fold increase in CHIKV RNA (Figure 1G) and increased viral protein (Figure S1I). Lastly, we tested whether DDX39A is active against CHIKV in other susceptible cell types42–44. We depleted DDX39A in human A549 (lung epithelial adenocarcinoma) and human HeLa (cervical epithelial adenocarcinoma) cells and found that loss of DDX39A leads to increased CHIKV replication as measured by microscopy (Figure S1J) and RT-qPCR (Figure S1K).
Our screen revealed an antiviral activity for DDX39A but not DDX39B, a ~90% homologous paralog45. While both helicases can rescue the lethality induced by deleting the yeast ortholog Sub2, DDX39A, and DDX39B have distinct activities in higher organisms34. We validated that DDX39B was depleted by the siRNAs at mRNA (Figure S2A) and protein level (Figure S2B) and that DDX39B-depletion had no impact on cell viability (Figure S2C) or CHIKV infection as measured by microscopy (Figure 1H) and by qPCR (Figure 1I). Together these data show that DDX39A, but not DDX39B, impacts CHIKV infection.
Since alphaviruses are a large group of medically relevant viruses, we evaluated the role of DDX39A in the infection of other globally important alphaviruses (Figure S2D). We found that both siRNA DDX39A-depleted and knockout cells are more susceptible to infection with SINV compared to controls as measured by RT-qPCR (Figure 1J, and Figure S1L), and by microscopy (Figure S2E) in the absence of cytotoxicity (Figure S2F). Moreover, DDX39A but not DDX39B is antiviral against SINV as knockdown of DDX39A but not of DDX39B results in an increased percentage of infected cells (Figure S2G). We also tested the activity of DDX39A against VEEV and ONNV and found that loss of DDX39A leads to an increase in VEEV (Figure 1K) and ONNV (Figure 1L) infection as measured by RT-qPCR. Alphaviruses are positive-sense RNA viruses and thus we were interested in the breadth of DDX39A against other positive and negative-sense viruses. We found that depletion of DDX39A does not impact infection with coronaviruses such as 229E (Figure 1M) and OC43 (Figure 1N) and picornaviruses such as the coxsackie B virus (CVB) (Figure 1N). In addition, knockdown of DDX39A does not impact infection with the negative sense viruses including the bunyavirus Rift Valley Fever virus (RVFV) (Figure S2H) and the rhabdovirus vesicular stomatitis virus (VSV) in either U2OS (Figure 1P and Figure S2I) or A549 cells (Figure S2J). Therefore, DDX39A is a broadly active anti-alphavirus factor.
DDX39A is antiviral independent of the interferon pathway
Upon binding of viral RNA ligands, the DExD/H-box helicases, RIG-I and MDA-5, upregulate interferons, which are secreted and bind to the interferon receptor. Binding activates JAK/STAT signaling to transcriptionally upregulate hundreds of ISGs that block infection20. However, alphaviruses antagonize the IFN axis23,46,47. Consistent with this, transfection of U2OS cells with poly (I:C), a dsRNA mimic, activates interferons and potently induces the ISG IFIT1 (Figure 2A). Similarly, infection with Sendai virus (SeV), another positive control for stimulation of the IFN pathway, strongly induces IFNB1 activation (Figure S3B), and ISGs such as IFIT1 (Figure S2C), IFIT2 (Figure S2D), IFIT3 (Figure S3E) and IFIT5 (Figure S3F). However, both CHIKV and SINV have a significantly more muted response; even at high MOIs, alphavirus infection results in attenuated induction of IFIT1 at both transcript (Figure 2A) and protein levels (Figure S3A) compared to poly (I:C) stimulation and SeV (Figures S3C–S3F). We first tested if DDX39A is itself an ISG. We found that, compared to the canonical ISG, IFIT1, DDX39A mRNA (Figure 2B) or protein (Figure 2C) is not induced following poly (I:C) treatment. Moreover, infection with either SINV, CHIKV, or SeV does not alter the levels of DDX39A (Figure 2D). Second, we determined if DDX39A promotes ISG induction. While treatment with poly (I:C) induces multiple ISGs (Figure S3G–K), loss of DDX39A does not decrease ISG production, suggesting that DDX39A is not antiviral by stimulating the IFN pathway.
Figure 2. DDX39A is antiviral independent of the interferon pathway.
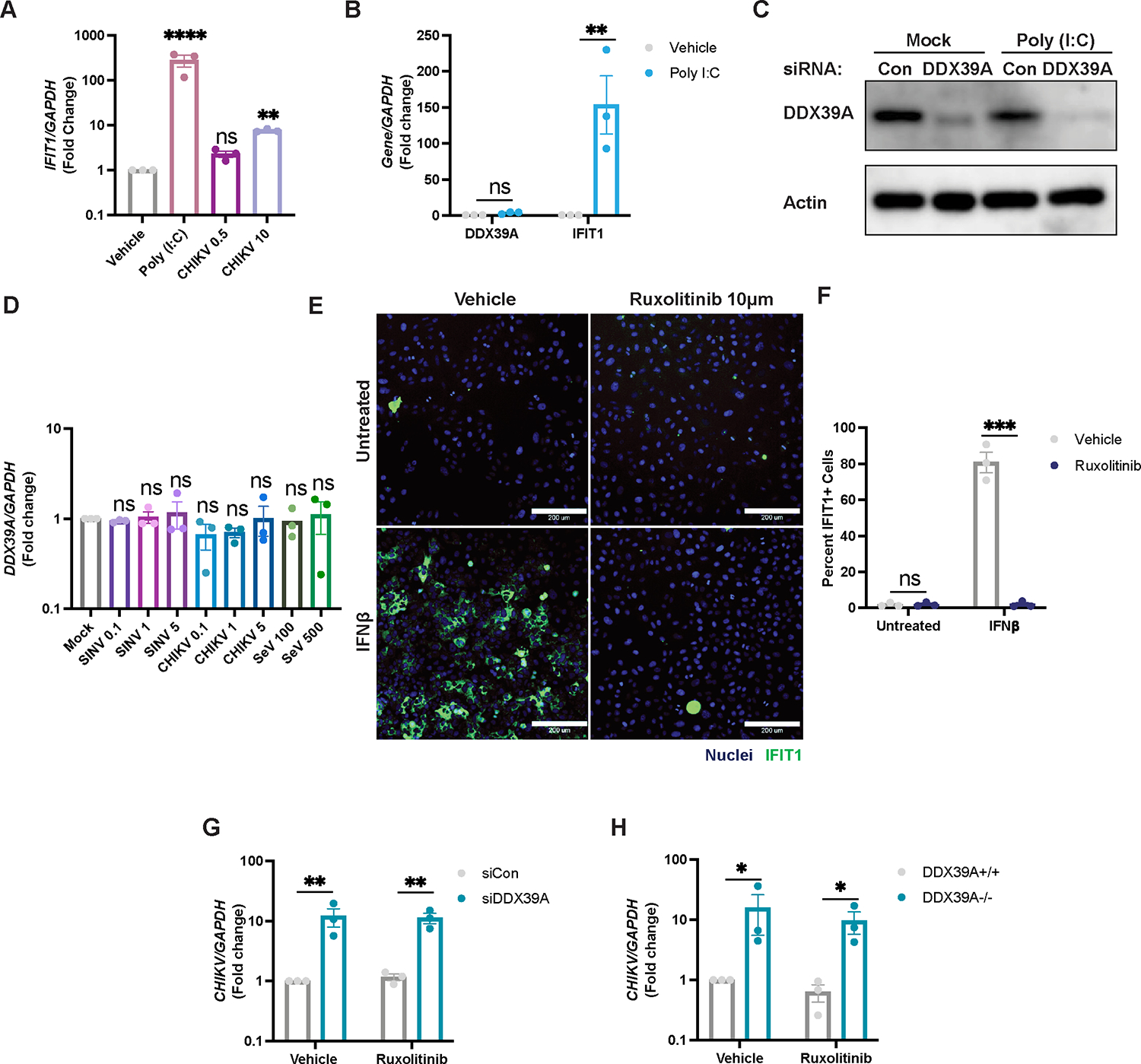
(A) U2OS cells were stimulated with poly (I:C) or infected with CHIKV at the indicated MOIs. IFIT1 levels were assessed by qPCR relative to uninfected vehicle control. (B-C) Levels of DDX39A or IFIT1 were measured by (B) qPCR for mRNA levels and by (C) immunoblot. A representative immunoblot image is shown. (D) U2OS cells were infected with SINV-mKate (MOIs 0.1,1,5), CHIKV (MOIs 0.1,1,3) or SeV (100 and 500 HAU/mL), and levels of DDX39A were assessed by qPCR compared to mock uninfected cells (E) Representative immunofluorescence image of U2OS cells pretreated with vehicle or Ruxolitinib (10 μM) for 2h and stimulated with IFNβ (10 ng/mL) for 6h, and immunostained (nuclei, Hoechst 33342 in blue and IFIT1+ cells in green). Magnification 10x; Scale bar 200 μm. (F) Percent IFIT1+ cells were calculated with automated microscopy. (G) Control or siRNA depleted-DDX39A, and (H) DDX39A+/+ or DDX39A−/− U2OS cells were pretreated with vehicle or Ruxolitinib (10 μM) and infected with CHIKV (MOI 0.5, 24h). Levels of viral RNA were measured by qPCR. Data are presented as fold change vs. vehicle-treated cells (A, B, F, G, H). Dots represent individual knockdown experiments. Immunoblots were performed in duplicate, n=3 for all other experiments as indicated. GAPDH was used as a loading control gene for all qPCR experiments. Beta-Actin was used as a loading control for all immunoblots as indicated. Statistical analyses were performed using one-way ANOVA with Bonferroni corrections (A, D), two-way ANOVA with Bonferroni corrections for multiple comparisons (B, E, G, H) *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001; error bars represent S.E.M.
We next evaluated if the antiviral effect of DDX39A is dependent on IFN signaling. For these studies, we pretreated cells with a JAK inhibitor, Ruxolitinib, and confirmed that this treatment prevents the IFN-dependent activation of ISGs (Figure 2E, F). Moreover, we found that the DDX39A-dependent increase in CHIKV infection is independent of JAK signaling as there is a comparable increase in infection in either the presence or absence of Ruxolitinib in both U2OS (Figure 2G and Figure 2H) and A549 cells (Figure S3L). Together, these data indicate that DDX39A is not regulated by the interferon pathway and that DDX39A has a mechanism of action independent of interferon signaling.
Infection with CHIKV leads to the accumulation of cytoplasmic DDX39A
Under basal conditions, we and others have found that DDX39A is predominately located in the nucleus as seen by confocal microscopy (Figure 3A)45. We next monitored the localization of DDX39A during CHIKV infection. We found that DDX39A accumulates in the cytoplasm concomitant with a decrease in nuclear DDX39A during CHIKV infection (Figure 3A). To further support our microscopy studies, we used biochemical fractionation to monitor DDX39A localization over time during infection. We assessed DDX39A protein expression using immunoblot, with GAPDH and Histone H3 as controls for our cytoplasmic and nuclear fractionations, respectively. We observed a small fraction of DDX39A in the cytoplasm basally and importantly a time-dependent increase in cytoplasmic DDX39A, concomitant with a decrease in nuclear DDX39A without a change in total protein levels during infection (Figure 3B). We next determined whether CHIKV replication was required for the cytoplasmic accumulation of DDX39A. For these studies, we infected cells with either wild-type or UV-inactivated CHIKV which cannot replicate. We validated that UV-inactivated CHIKV is unable to replicate by immunoblot (CHIKV E1) (Figure 3C). Notably, replication-competent CHIKV but not UV-inactivated CHIKV leads to cytoplasmic accumulation of DDX39A accompanied by a decrease in nuclear DDX39A (Figure 3C). These data together reveal a replication-dependent accumulation of DDX39A in the cytoplasm where RNA replication occurs. This cytoplasmic accumulation is in punctae and there are two well-studied cytoplasmic granules that appear as punctae: stress granules and P-bodies48. Moreover, helicases, such as DDX6 can drive the formation and accumulation of cytoplasmic P-bodies which are important for RNA decay49,50. Stress granules accumulate translationally arrested RNAs, are induced by alphavirus infection, and are thought to repress viral nSP synthesis51–54. We used confocal microscopy to monitor the localization of stress granules (TIAR) and P-bodies (RCK). We used arsenic as a positive control to induce these foci. We found that both arsenic and CHIKV infection induce these foci, but that DDX39A does not colocalize with either the stress granule marker TIAR or the P-body marker, RCK (Figure S4). Therefore, the DDX39A foci are not canonical stress granules or P-bodies.
Figure 3. DDX39A accumulates in the cytoplasm upon infection with CHIKV.
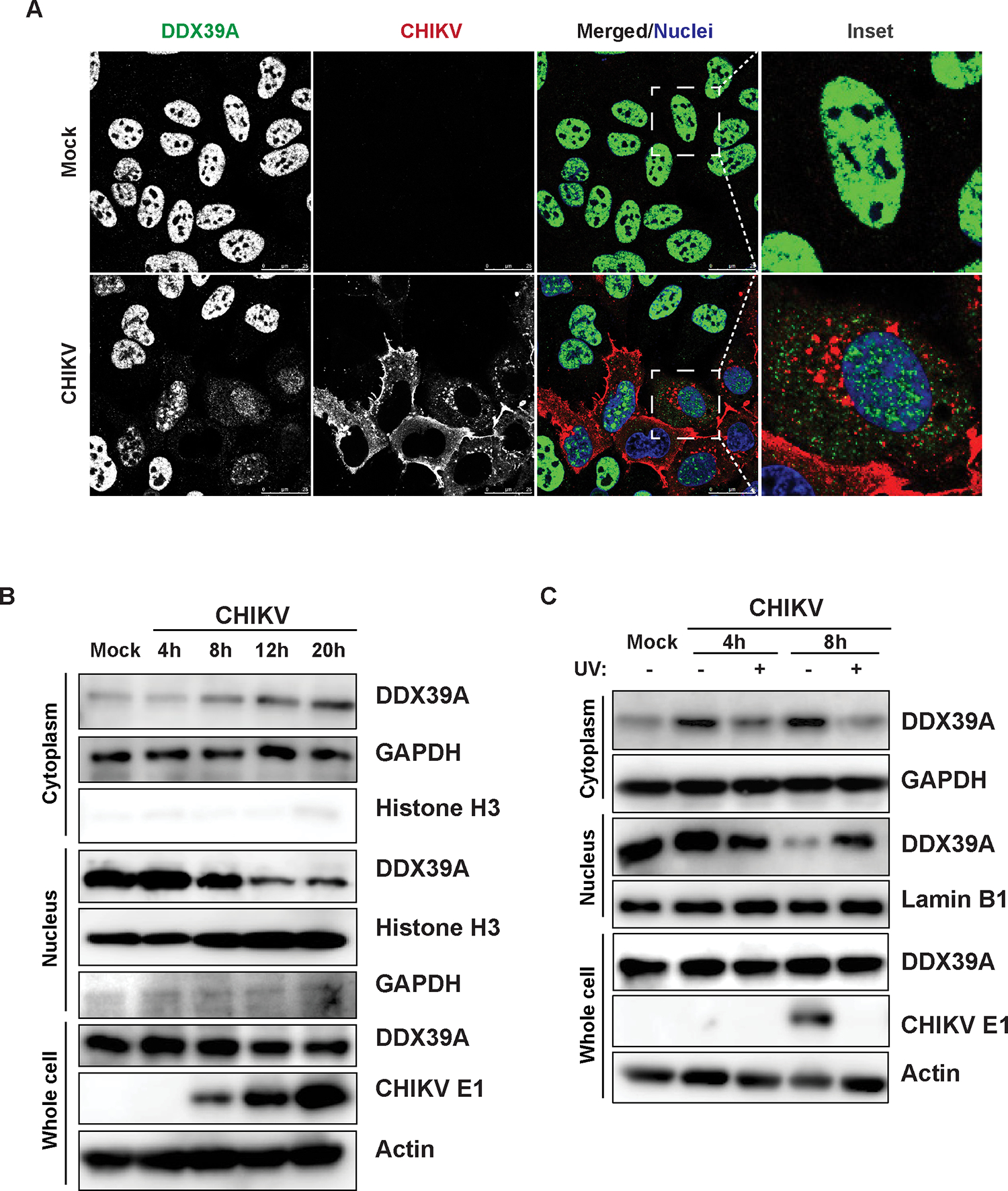
(A) Mock uninfected and CHIKV-infected U2OS cells (MOI 3, 20h) were fixed, stained (anti-DDX39A in green, anti-CHIKV in red, and nuclei, Hoechst 33342 in blue), and imaged with confocal microscopy, n=2; an inset is shown for an uninfected and a CHIKV-infected cell. Magnification 63x; Scale bar 25 μm. (B) Mock and CHIKV-infected U2OS cells MOI 5 were subjected to nuclear-cytoplasmic fractionation at the indicated time points. Representative blots are shown, n=4. (C) U2OS cells were infected with CHIKV MOI 5 or UV-inactivated CHIKV for 24h and subjected to nuclear-cytoplasmic fractionation at the indicated time points. Samples were immunoblotted with DDX39A or CHIKV E1 protein. Beta-Actin, histone H3/Lamin B1, and GAPDH were used as loading or fractionation purity controls, respectively. Representative blots are shown, n=3.
To further explore the specificity of this relocalization of DDX39A, we monitored DDX39B localization during infection. We observed that DDX39B also accumulates in the cytoplasm of infected cells (Figure S5A). These data suggest that infection is inducing a relocalization of multiple RNA binding proteins55,31, and since DDX39A and DDX39B are shuttling proteins56 perhaps a step in nuclear import is attenuated. Nevertheless, because DDX39A but not DDX39B impacts CHIKV infection, these data suggest that cytoplasmic localization is only one determinant of antiviral activity.
DDX39A affects early steps in viral RNA replication
Alphavirus replication begins with binding to specific alphavirus cellular receptors at the cell surface, and the receptors utilized by CHIKV, ONNV, VEEV, and SINV are distinct13,57. After binding, virions undergo endocytic internalization into a low pH compartment where fusion occurs and release of the viral genome into the cytoplasm13,14. To assess whether DDX39A impacts viral entry, we infected cells pretreated with cycloheximide to block new translation and quantified levels of CHIKV RNA that was internalized in either control or DDX39A-depleted cells. We observed no difference in viral RNA levels between control and DDX39A-depleted cells (Figure 4A), suggesting that DDX39A likely does not impact CHIKV entry.
Figure 4. DDX39A controls early steps in CHIKV RNA replication.
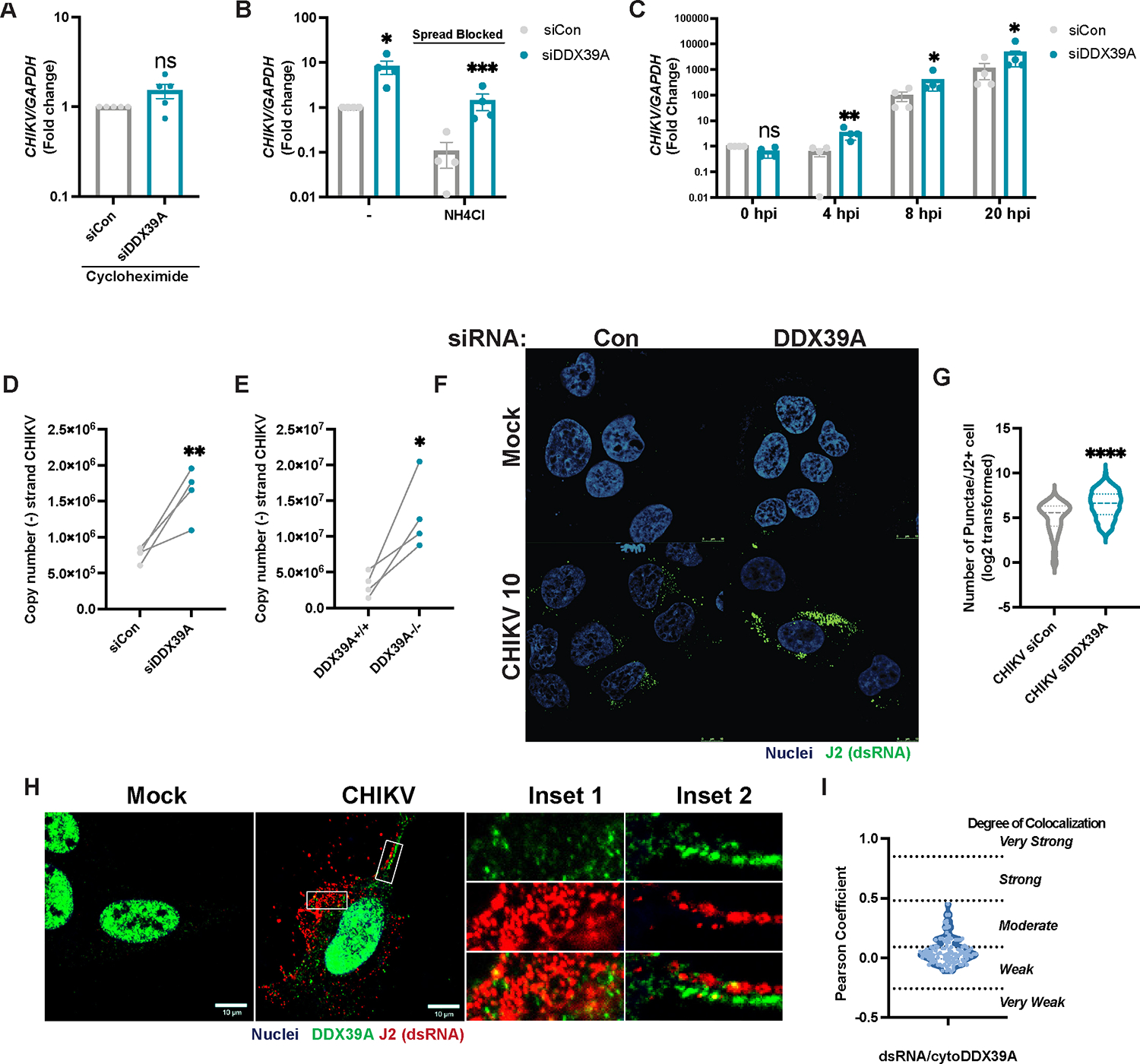
(A) Control and DDX39A-depleted U2OS cells were infected with CHIKV (MOI 20, 4h) in the presence of cycloheximide followed by removal of extracellular virions and quantification of intracellular viral RNA by qPCR. (B) Control and DDX39A-depleted U2OS cells were infected with CHIKV (MOI 0.5, 4h), followed by the addition of vehicle or ammonium chloride (NH4Cl) for 24h. CHIKV RNA was assessed by qPCR. (C) Control and DDX39A-depleted U2OS were infected with CHIKV (MOI 5, 1h at 4°C), washed, and incubated at 37°C. CHIKV RNA levels were quantified by qPCR at the indicated time points. Data are expressed as fold change relative to control at 0 hpi. (D) Control or DDX39A-depleted cells and (E) DDX39A+/+ or DDX39A−/− U2OS cells were infected with CHIKV (MOI 0.1, 8h) and (−) strand (antigenome) specific RT-qPCR was performed. (F) Control and DDX39-Adepleted cells were infected with CHIKV (MOI 10, 4h) and dsRNA/J2+ puncta were detected by immunofluorescence (nuclei, Hoechst 33342 in blue and dsRNA, anti-J2 in green). Magnification 63x; Scale bar 10 μm. (G) The number of puncta per infected cell was quantified by MetaXpress (granularity) in >67 cells per experiment in two independent experiments. (H) U2OS cells were infected with CHIKV (MOI 10, 8h) and stained with J2 (dsRNA, red), DDX39A (green), and Hoechst (nuclei, blue). Stars indicate colocalization. (I) Colocalization was assessed for J2 puncta and cytoplasmic DDX39A for 4 experiments (>24 cells/experiment) using the Coloc2 plugin in image J. The degree of colocalization was assessed based on Pearson cutoffs118. Dots represent individual experiments, n=3–4 for all experiments unless otherwise noted. GAPDH was used as a loading control gene for all qPCR experiments. Statistical analyses were performed using Student’s (unpaired, two-tailed) t-test (A, D, F), two-way ANOVA with Bonferroni corrections for multiple comparisons (B, C) *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001; error bars represent S.E.M.
Next, we tested whether DDX39A impacts viral spread. We infected cells for 4h to allow for entry and then blocked endosomal acidification with ammonium chloride which prevents subsequent entry of new virions, thus blocking a second round of infection. As expected, treatment with ammonium chloride decreases overall CHIKV infection levels; however, treatment does not abolish the phenotypic differences between control and DDX39A-depleted cells (Figure 4B). Altogether, our data suggest that DDX39A impacts a step in the replication cycle downstream of entry but upstream of spread.
These data implicate a role for DDX39A in an intracellular replication step in the viral lifecycle. We first determined how early the antiviral activity can be measured. We synchronized the infection by pre-binding virus for 1h at 4°C and quantified CHIKV RNA accumulation over time in DDX39A-depleted and control cells. While we observed no difference at 0hpi (1hpi at 4°C), prior to new replication, by 4h post-infection, when new viral RNA synthesis is occurring, DDX39A depletion leads to an increase in CHIKV RNA (Figure 4C). The earliest step in viral RNA synthesis is the generation of antigenome. Given the early timing of the antiviral phenotype, we hypothesized that DDX39A may impact the synthesis of antigenome copies. Thus, we quantified antigenome RNA accumulation using strand-specific and standard-curve RT-qPCR. We observed that loss of DDX39A leads to increased levels of antigenome copies at low (Figure 4D, Figure 4E) and high MOIs (Figure S3M). The synthesis of antigenome produces the dsRNA intermediate, which can be observed by microscopy. We infected control or DDX39A-depleted cells with CHIKV (MOI 10) and monitored infection 4hpi to capture the initial production of dsRNA foci. We found that loss of DDX39A leads to an increase in dsRNA foci compared to the control (Figure 4F–G).
To further explore the mechanism by which DDX39A impacts CHIKV replication, we used confocal microscopy to assess the localization of DDX39A relative to dsRNA intermediates. We observed that cytoplasmic DDX39A is found in the vicinity of dsRNA foci (Figure 4H) but displays weak to moderate colocalization (Figure 4I). Therefore, DDX39A inhibits the earliest steps in viral RNA synthesis and may prevent dsRNA formation by sequestering viral RNA away from replication complexes or by blocking their formation.
DDX39A binds to a conserved CHIKV RNA structure
We observed that DDX39A impacts early viral RNA synthesis and accumulates in the cytoplasm where the viral RNAs are localized. Therefore, we tested whether DDX39A binds to viral RNA to control infection. We infected cells with CHIKV and performed cross-linking RNA-immunoprecipitation followed by RT-qPCR (RIP-qPCR). Analysis by immunoblot showed that endogenous DDX39A was efficiently immunoprecipitated (Figure 5A). The associated RNAs precipitated with either rabbit IgG control or DDX39A were analyzed by RT-qPCR where we observed a striking enrichment of CHIKV RNA ~600-fold (Figure 5B) but not of other host mRNAs (Figure S5B). Since DDX39A is also antiviral against VEEV, but not VSV, we tested whether the ability of DDX39A to bind to these viral RNAs might account for the difference in phenotype. RIP-qPCR of DDX39A during VEEV (Figure S5C) or VSV infection (Figure S5D) demonstrates that DDX39A binds to VEEV RNA (Figure 5C), but not to VSV (Figure 5D), findings which paralleled the antiviral phenotype.
Figure 5. DDX39A binds to a conserved stem-loop in the CHIKV genome.
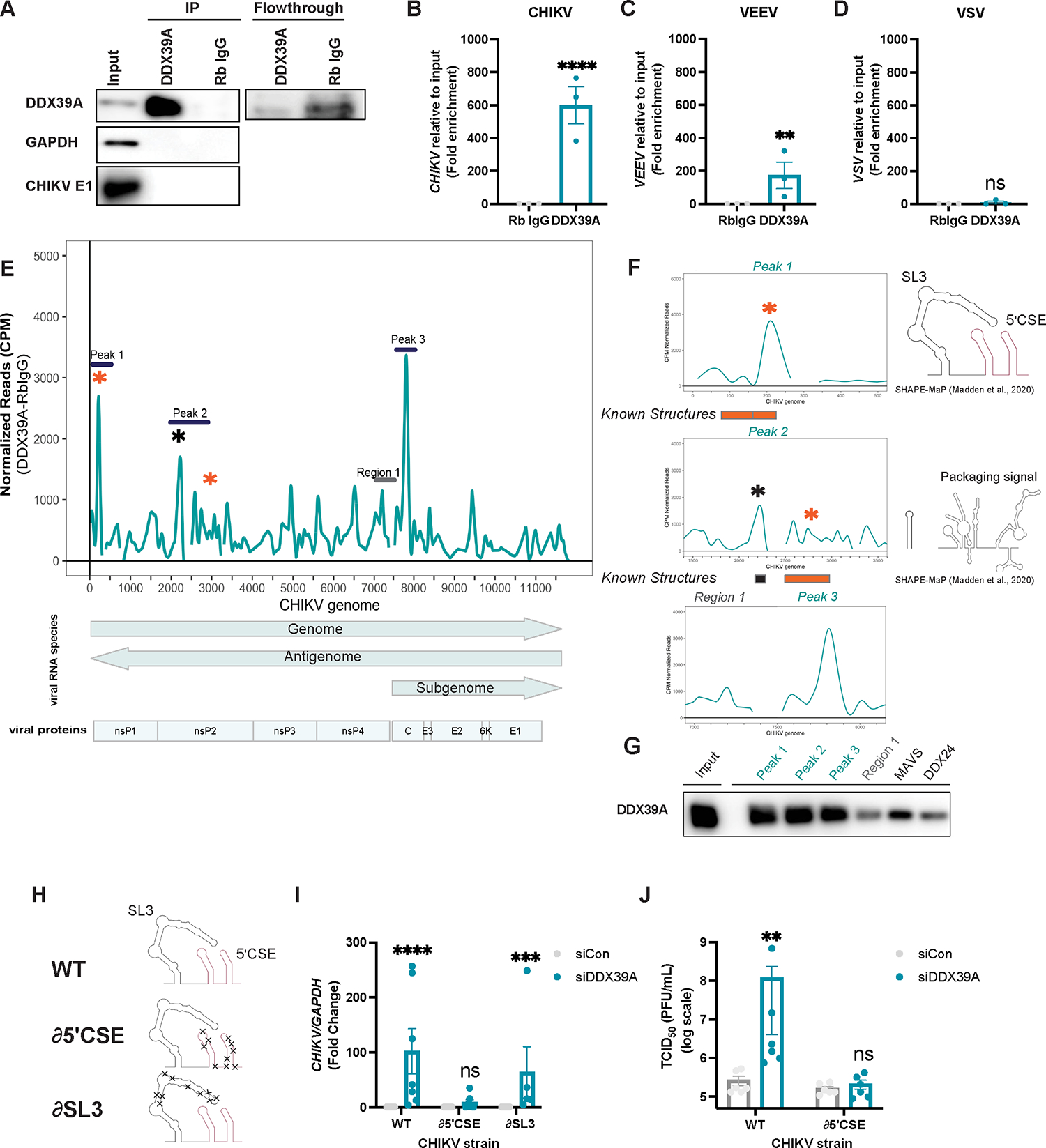
(A-D) U2OS cells were infected with (A-B) CHIKV (MOI 3, 20h), (C) VEEV (MOI 1, 20h), and (D) VSV (MOI 3, 16h), UV cross-linked and immunoprecipitated with DDX39A and rabbit IgG as shown in immunoblot (A). (B-D) Viral transcripts precipitated with DDX39A, and with the rabbit IgG control were analyzed by qPCR relative to input. (E) Graph shows coverage depth of viral reads after removal of RbIgG background reads, normalizing to counts per mapped reads (CPM) and transcript abundance. Viral proteins and RNAs are shown below the graph according to annotations in Table S3. The highly enriched 3 peaks are marked with lines; stars emphasize peaks overlapping known functional structures (orange) or SHAPE-characterized structures (black). (F) Zoomed in CLIP-seq coverage emphasizing the peaks bound by DDX39A with SHAPE-structures9. (G) In vitro transcribed RNAs mapping to Peaks 1–3 and Region 1 on the CHIKV genome and to DDX24 and MAVS (Table S5) were biotinylated, incubated with HEK293T cells transfected with DDX39A, and enriched using streptavidin-conjugated beads. The relative level of co-precipitated DDX39A onto each fragment was measured by immunoblot. (H) Schematics of wild-type (WT) parent, and mutated ∂5ʹCSE or ∂SL3 CHIKV strains with structure-disrupting mutations (Table S4) were generated with biorender based on SHAPE data9. (I) Control and DDX39A-depleted U2OS cells were infected with WT, ∂5ʹCSE, and ∂SL3 strains of CHIKV for 24h, and viral RNA was quantified by qPCR. (J) Control and DDX39A-depleted U2OS cells were infected with CHIKV WT and CHIKV ∂5ʹCSE strains, and viral titers (TCID50) were quantified at 24h. Dots represent individual experiments, n=3–7 for all experiments. Data represented as fold enrichment relative to input (B, C, D) and fold change relative to Rb IgG control. GAPDH was used as a loading control gene for all qPCR experiments unless otherwise noted. Statistical analyses were performed using Student’s (unpaired, two-tailed) t-test (B, C, D), two-way ANOVA with Bonferroni corrections with multiple comparisons (I) multiple unpaired t-tests (J). *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001; error bars represent S.E.M.
We further assessed the specificity of the interaction between DDX39A and CHIKV RNA by testing whether the DDX39A paralog, DDX39B, which is not antiviral, binds to CHIKV RNA. We immunoprecipitated endogenous DDX39B (Figure S5E) and using RIP-qPCR, we found that DDX39B also binds to CHIKV RNA (Figure S5E), but potentially less efficiently compared to DDX39A (~60-fold vs. ~600-fold) (Figure 5B). Importantly, these differences in phenotype are not a consequence of differential infection levels between experiments (Figure S5G) or relative expression (Figure S5H), or cytoplasmic localization (Fig S5A). Altogether, these data suggest that viral RNA binding by DDX39A may be required for antiviral activity.
Thus, we set out to map where DDX39A binds to CHIKV RNA by performing cross-linking immunoprecipitation coupled with sequencing (CLIP-Seq)31,58. We immunoprecipitated endogenous DDX39A in CHIKV-infected cells (Figure S6A), radiolabeled and size-selected samples (Figure S6B), and prepared libraries (Figure S6C) for sequencing. We mapped the reads to the viral and human genome and found that 950 human RNAs had >10 reads across the 3 replicates (Table S6). Using CLIP-seq Analysis of Multi-mapped reads (CLAM)59 and the HOMER60 program, we identified two GC-rich motifs that were found in ~20% of DDX39A peaks and had a significant p-value (Figure S7). More strikingly, we found that approximately 15% of the total reads mapped to the viral genome while only ~ 0.02% of viral reads mapped to the antigenome (Figure S6D, Table S2), suggesting clear specificity for the genomic RNA. After removing background reads in the Rb IgG control and normalizing reads to counts per mapped (CPM) reads and relative viral species abundance18, we observed several enriched peaks (Figure 5E). We mined previous studies and found that two of the top peaks overlapped with previously characterized secondary structures (Figure 5F, orange asterisks, Table S3)9–11,61. This includes Peak 1 (Figure 5F, top panel), which encodes the 5ʹ conserved sequence element (5ʹCSE) and the upstream stem-loop 3 (SL3) that are important for viral replication (Madden et al., 2020). Peak 2 includes a stem-loop and the conserved alphavirus packaging sequence element (Figure 5F, middle panel). We also identified a third striking peak that does not encode any known structures (Figure 5F, bottom panel). To validate the CLIP-seq data, we performed a reverse RIP assay. First, we generated biotinylated in vitro transcribed RNAs (Figure S5E): Peak 1 (encompassing the known functional 5ʹCSE+SL3 structures) (Figure 5F top panel, Figure S5E), Peak 2 (which includes the packaging signal) (Figure S6E, Figure 5F, middle panel), Peak3 an enriched signal in ORF2 (Figure S6E, Figure 5F, bottom panel). We also included Region 1 with reduced reads as a negative control (Figure S6E. Figure 5F, bottom panel). Additionally, we included two cellular mRNA controls: MAVS as a positive control as it was shown by a previous study to be bound by DDX39A in uninfected cells35, and DDX24 as a cellular negative control as we observed no binding in our biochemical assay (Figure S5A). We added these biotinylated RNAs to lysates overexpressing DDX39A and used streptavidin beads to recover biotinylated RNAs. We observed that DDX39A preferentially binds to the enriched peaks observed by CLIP, and MAVS, but not the negative control fragment (Region 1) or DDX24 (Figure 5G).
DDX39A controls CHIKV infection through the conserved 5ʹ CSE element
The CHIKV genome has several structural elements, but few are conserved across alphaviruses16. While packaging signals exist across the family, the structures are not highly conserved16. The most conserved structural RNA element across the alphavirus genus is the 5ʹCSE, within our identified peak 1, which is important for the replication of CHIKV9, SINV62, and VEEV63. Since these alphaviruses are all sensitive to DDX39A depletion, and we observed a striking enrichment in CLIP Peak 1 (Figure 5E), and our biochemical binding assay (Figure 5G), we explored this interaction. We performed competition assays between Peak 1 and the negative control Region 1 (Figure S6F). We found that DDX39A preferentially binds to Peak1 RNA encompassing the SL3-+5ʹCSE elements since Region 1 was less able to compete for DDX39A binding (Figure S6G).
Peak 1 has two structures: the 5ʹ CSE or the SL3 stem-loop. We explored the role of each by taking advantage of recombinant CHIKV viruses with mutations in each of these regions as viruses with deletions in this region are not viable63. We utilized two distinct mutant strains: a strain with changes that disrupt the secondary structure of either the 5ʹ CSE (∂5ʹCSE’) or the SL3 (∂SL3) while maintaining the coding capacity and dinucleotide frequency (Figure 5H, Table S4)9. Both the 5ʹCSE and the SL3 structures are important but not essential for CHIKV RNA infectivity and CHIKV replication in some cell lines9 and we observed cell type-specific differences in titers where both mutants showed reduced titers in hamster BHK cells (Figure S6H). In contrast, in U2OS cells only the ∂SL3 showed reduced titers while the ∂5ʹCSE replicated similarly to WT (Figure S6I), these cell-type differences potentially reflecting a differential abundance of RNA-binding proteins that modulate the function of the CHIKV RNA structure. We infected control or DDX39A-depleted U2OS cells, with WT CHIKV, ∂5ʹCSE, or ∂SL3. We assessed CHIKV levels by qPCR (Figure 5I, Figure S6J), and immunofluorescence (Figure S6K). DDX39A knockdown resulted in a significant increase in WT CHIKV infection as well as ∂SL3; however, this antiviral effect was lost specifically against the ∂5ʹCSE mutant. We tested whether this was due to a loss of cytoplasmic localization of DDX39A upon infection with the ∂5ʹCSE virus. Using confocal microscopy, we observed that both the WT and the ∂5ʹCSE mutant lead to cytoplasmic accumulation of DDX39A (Figure S5L), suggesting phenotypic differences are not due to different localization of DDX39A. Furthermore, using our biochemical assay we compared DDX39A binding to either a ~500mer RNA fragment synthesized from either WT CHIKV or the ∂5ʹCSE strain that leaves SL3 intact (Figure S6M). We observed decreased binding of DDX39A to the RNA from ∂5ʹCSE compared to the WT virus (Figure S6N) further supporting a role for the 5ʹ CSE in DDX39A-dependent restriction. Importantly, we measured the viral titers of control or DDX39A-depleted cells infected with either WT CHIKV or the ∂5ʹCSE CHIKV strain. We found that loss of DDX39A leads to a ~ 200-fold increase in infection of the WT virus but that DDX39A has no impact on the titers of ∂5ʹCSE CHIKV (Figure 5J). Overall, these data demonstrate that binding of DDX39A to the CHIKV 5ʹ CSE is required for antiviral activity.
Some DEAD-box proteins interact with binding partners that increase the specificity for target RNAs64–68. DDX39A interacts with two RNA binding proteins, CIP29 and ALY/REF34,69. Therefore, we tested if depletion of either of these RNA binding proteins impacts alphavirus infection and observed that ALY/REF but not CIP29 has activity against CHIKV (Figure S8A) and SINV (Figure S8B).To determine whether ALY/REF works through interferon, we stimulated control and ALY/REF-depleted cells using poly(I:C) and we observed that ALY/REF has no effect on IFNB1 (Figure S8C) or IFIT1 levels (Figure S6D). Next, we tested if the antiviral activity of ALY/REF is dependent on IFN signaling. Thus, we pretreated cells with Ruxolitinib, and we observed that ALY/REF is antiviral in both vehicle and Ruxolitinib-treated cells (Figure S8E), confirming an IFN-independent phenotype as we observed with DDX39A. Next, we evaluated whether DDX39A and ALY/REF interact and potentially cooperate to control CHIKV infection. For these studies, we used a proximity ligation assay (PLA) to reveal interactions. As expected, we observed that under basal conditions, there is a strong nuclear PLA signal between DDX39A and ALY/REF (Fig S8F). As controls, we tested whether DDX39A interacts with an unrelated DEAD-box helicase DDX24 or GAPDH (Figure S8F) and found little interaction. Next, we challenged the cells with CHIKV and found that DDX39A and Aly/REF interact in the cytoplasm (Figure S8F, S8G). These results suggest that ALY/REF and DDX39A interact in the cytoplasm during CHIKV infection and likely cooperate to inhibit alphavirus replication.
DISCUSSION
Innate immune defense begins with the recognition of viral RNA by cellular sensors and the induction of antiviral effector pathways including the IFN pathway20. However, many viruses have evolved mechanisms to evade and dampen interferon-dependent responses22–25,70–72. Therefore, additional RNA binding proteins likely play roles in antiviral defenses outside of canonical interferon pathways. Key players at the host-pathogen interface, DEAD-box helicases are a conserved family of RNA binding proteins that bind RNA structures rather than sequences to regulate RNA metabolism26,30,33. While several DDXs inhibit viral replication by stimulating the IFN pathway, few RNA helicases have antiviral activities independent of IFN31,32,73,74. Using screening approaches, we identified DDX39A as antiviral against diverse Old and New World alphaviruses, but not against other positive or negative sense viruses, suggesting a direct effect. DDX39A controls infection independent of IFN signaling at the earliest stages of RNA replication. Biochemically we found that DDX39A binds a conserved RNA secondary structure and that binding the conserved 5ʹCSE RNA structure is required for DDX39A antiviral activity. Thus, DDX39A represents a class of antiviral effectors that recognize viral RNA structures to control infection outside of interferon pathways overcoming the virally encoded antagonism of this pathway.
Given their limited genetic space, RNA viruses have evolved complex RNA structures to regulate various aspects of their life cycles10,75–79. In the evolutionary arm-race between innate defense factors and viral evasion mechanisms, many restriction factors target evolutionarily conserved viral RNA features including the dsRNA intermediate produced during replication of all RNA viruses21,80. he DEAD-box helicase family is increasingly reMany alphavirus RNA structures have been characterized including packaging signals which direct encapsidation of the viral genome16. Interestingly, while every alphavirus contains a packaging signal, this signal is divergent between CHIKV and SINV or VEEV10,16,81. The most highly conserved structure across alphaviruses is the 5ʹCSE9,82. We demonstrate that DDX39A physically interacts with this structure on CHIKV RNA and disrupts its replication. The 5ʹCSE is important for replication events including the synthesis of both plus and minus strands by the viral replicase complex, and is essential, as deletion viruses are not viable, although the exact mechanism is unknown83. DDX39A attenuates early steps in viral replication including the production of dsRNA intermediates and negative strand antigenome copies. DDX39A and other RNA helicases have multiple activities including ATPase and helicase activity69, which enable them to unwind RNA structures and remodel RNA-protein complexes in the presence of target RNA84–86. Since mutations that disrupt the structure of the 5ʹCSE render CHIKV insensitive to DDX39A antiviral activity, this supports a model in which DDX39A binds to the CHIKV 5ʹCSE and either sequesters it away from the viral replication complex or alternatively, remodels the CSE to hinder its recognition by the viral replicase.
We found that DDX39A accumulates in the cytoplasm during infection with CHIKV. Subcellular localization of proteins clearly alters function, and viral infection can induce large-scale changes in protein localization31,32,46,55,87–89. Yet, the mechanisms underlying these changes remain unknown. As an mRNA export protein, DDX39A normally shuttles between the nucleus and cytoplasm to deliver cargo using a CRM1-independent pathway56. A cytoplasmic function for DDX39A has not been previously described. Here, we show that while DDX39A is predominantly nuclear basally, CHIKV infection leads to a redistribution of DDX39A to the cytoplasm and this is dependent on alphavirus replication. Like other DDX helicases72, DDX39A interacts with additional RNA binding proteins to facilitate function69 and we found that the known DDX39A binding partner ALY/REF controls alphavirus infection and interacts with DDX39A in the cytoplasm suggesting that DDX39A this complex inhibits CHIKV infection. Mechanistically, DDX39A impacts early steps in viral replication when a small amount of viral RNA is present, suggesting that either the basal cytoplasmic pool of DDX39A is sufficient to restrict the limited amount of viral RNA, or the initial translocated protein is sufficient to restrict replication but difficult to detect by low sensitivity immunoblot. We also found that the mutant virus that is insensitive to DDX39A also promotes the accumulation of DDX39A in the cytoplasm, suggesting that binding to the region alone does not drive cytoplasmic accumulation. Two well-established RNA foci include stress granules and P-bodies, which play roles in translational arrest and RNA decay 49–51. We found that while CHIKV infection induces these structures, DDX39A does not colocalize with these RNA granules suggesting distinct activity. Future studies will assess the composition of these foci along with which cellular signals or aspects of CHIKV replication are required for DDX39A relocalization. Moreover, defining the differential interactions of DDX39A with proteins, RNAs, or signaling pathways that alter the posttranslational modification of DDX39A may uncover additional regulation. Because the cytoplasmic accumulation of DDX39A during CHIKV infection is not colocalized with dsRNA replication foci or stress granules, we suggest that DDX39A binding of alphavirus genomic RNA sequesters this RNA away from replication machinery, thereby attenuating infection.
DDX39A has a close paralog DDX39B. Indeed, though conserved in all three domains of life, DExD box helicases have expanded in part by gene duplication, resulting in many members with closely related paralogs90. While paralogs can functionally compensate for each other, the specific functions and the evolutionary advantage behind the diversification of paralogues remain poorly understood32,91–93. While either DDX39A or DDX39B can rescue the yeast Sub2 mutant, in human cells, DDX39A, and DDX39B play nonredundant roles in splicing, circRNA, mRNA export, and even cancer34,94–99. Similarly, other closely related paralogs such as DDX3X and DDX3Y (92% identical) have distinct biochemical properties100. Here, we found that while DDX39A controls alphavirus infection, DDX39B is dispensable even though both helicases have similar expression and localization56,95,101. Sequence divergence between DDX39A and DDX39B occurs outside the helicase core, which may explain the distinct binding affinities for RNA substrates56,102. How these two factors are divergently regulated is unclear, but we suggest that one driver of paralogs may include antiviral responses and future studies will explore this more directly.
The DEAD-box helicase family is increasingly recognized as an important pool of antiviral effectors that directly bind viral RNAs. These are well suited for this role as they can bind to conserved structural features. Indeed, viruses readily evolve different sequences while maintaining the structural motifs required for viral replication. Moreover, DDX proteins regulate diverse facets of RNA metabolism and thus can be highly regulated at the cell biological level103. We suggest that there may be a stress-induced program that regulates a subset of DDXs to expand their functions. Of note, these functions can be either proviral or antiviral depending on the virus or even cell type and both proviral and antiviral activities have been described in the same helicase31,35,104–114. Indeed, while we found that DDX39A restricts alphaviruses other studies found that DDX39A can promote VSV infection in HEK293 cells35. Therefore, a better understanding of the stimuli and mechanisms that drive these changes will further our understanding of how these proteins achieve functional specificity in the context of viral infections. There are many paralogs that have evolved in the DDX family, and we have shown differences in antiviral function. How and why this family has expanded may also provide additional insight into the new functions required by distinct organisms. While DDXs bind structural features, the determinants of these interactions are poorly understood. Our work defines an essential interaction between a host RNA binding protein and a conserved viral RNA structure highlighting the potential for structure-based antivirals. A better understanding of the DDXs that control infection and the structures they target will better characterize the interface between viruses and hosts and define the mechanisms by which DDXs interact with RNA. With many DEAD-box helicases dysregulated in cancers, autoimmune diseases, and other pathological conditions, continuing to define RNA features recognized and modified by DEAD-box helicases will be important for the development of therapeutics beyond viral infections115–117.
Limitations of the study
Our results demonstrate that DDX39A binds CHIKV RNA at the 5ʹCSE, and that viruses mutated in this structure are no longer sensitive to DDX39A restriction. However, we have not demonstrated how this interaction blocks viral replication. Furthermore, while we have shown that the 5’CSE structure is dispensable for CHIKV-induced relocalization, we have not identified the CHIKV-induced molecular signal that drives this cytoplasmic accumulation. Lastly, while we found that DDX39A binds cellular RNA with GC-enriched motifs, we could not correct for overall gene expression, thus future studies will define how this binding impacts RNA function.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Sara Cherry (cherrys@pennmedicine.upenn.edu).
Materials Availability
This study did not generate new unique reagents.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Cells and viruses:
U2OS, A549, BHK-21, Vero, HeLa, and HEK293T cells were obtained from ATCC and maintained at 37°C in Dulbecco’s modified Eagle’s medium (DMEM, Gibco) supplemented with 10% FBS, L-Glutamine (Gibco) and penicillin/streptomycin (Gibco). All cell lines were confirmed to be mycoplasma negative. CHIKV (strain Ross) was provided by Dr. David Weiner (University of Pennsylvania). VSV-enhanced GFP (VSV-eGFP) was a gift from J. Rose (Yale). CHIKV 181/25 WT, CHIKV 181/25 ∂CSE, CHIKV 181/25 ∂SL39, SINV-mKate (strain Girdwood) and VEEV (TC-83 strain) were obtained from M. Heise. Rift Valley Fever (RVFV, strain MP12) was a gift from Dr. Michael Diamond (Washington University in St. Louis). O’nyong’nyong (ONNV, strain SG650) was a gift from Dr. Thomas E. Morrison (University of Colorado). Viruses (CHIKV Ross, ONNV, RVFV, SINV-mKate, VEEV) were propagated in C636, CHIKV (181/25 WT, ∂CSE, and ∂SL3) in Vero CCL81 and (VSV-eGFP) in BHK cells as described119. Viral titers, represented as pfu/mL were determined by plaque assay or tissue culture infectious dose (TCID50) using Vero E6 cells or BHK cells.
METHODS DETAIL
Transfections:
U2OS cells were transfected with siRNAs using HiPerfect reagent (Qiagen) with a final siRNA concentration of 20nM for U2OS and 30nM for HeLa and for A549 cells following the manufacturer’s protocol for forward transfections. Unless specified, pooled siRNAs were used for all experiments. siRNAs for DDX39A, IFIT1, ALY/REF, CIP29, and DDX39B and controls were obtained from Ambion (SilencerSelect) or Sigma. AllStars Hs Cell Death siRNA (Qiagen #1027299) was used to verify knockdown in each experiment before infection. Transfection of DDX39A was performed with Viafect (Promega) according to the manufacturer’s protocol.
Cytokines and inhibitors:
Human interferon β (IFNβ) was purchased from Stem Cell Technologies, resuspended in phosphate-buffered saline (PBS), and used at 10 ng/mL. Ruxolitinib (JAK/STAT inhibitor) was purchased from Selleck Chemicals and added 2h prior to infection. Poly (I:C) (polyinosinic: polycytidylic acid) was used at 10μg/mL. Mock and untreated controls were “stimulated” with the appropriate vehicle at the volume/concentration used for the given experiment and reagent concentration.
High-throughput RNAi screening and analysis:
Pooled siRNAs (Dharmacon) targeting 42 human DEAD-box helicases were spotted in each well of a 96-well black tissue culture-treated plate. HiPerfect diluted in Opti-MEM was added to each siRNA-containing well (final siRNA concentration 20nM). U2OS cells (10,000/70μL) were then added and cells were incubated for 3 days at 37°C, 5% CO2. CHIKV (Ross strain 0.1) was then dispensed into each well, followed by 24h incubation. Cells were then fixed with 4% formaldehyde in PBS at room temperature, washed, and stained with anti-CHIKV (ATCC V-548–701- 438 562) for viral protein and Hoechst 33342 for nuclei. The cells were imaged at 10× (ImageXpress Micro, Molecular Devices) with nine sites per well in a 96-well plate. The number of infected cells and total nuclei per site were quantified using the MetaXpress software (Cell scoring). Percentage of Control [POC = (%Infectionsample siRNA/ Average %InfectioncontrolsiRNAs) 100] and Z-scores [Z = (%Infectionsample siRNA – Average %InfectioncontrolsiRNAs) / Standard Deviation %InfectioncontrolsiRNAs] were calculated with the Spotfire software (PerkinElmer).
Immunofluorescence, automated imaging and analysis:
Briefly, cells were transfected for 3 days with siRNA in a 96-well black tissue culture-treated plate using HiPerfect. Cells were then infected with SINV-mKate, VSV-GFP, VEEV, ONNV, or CHIKV (Ross or 181/25 strains) at the indicated MOIs, U2OS cells infected with SINV-mKate and VSV-GFP were fixed, washed with PBS and stained with Hoechst 33342. CHIKV and VEEV-infected U2OS cells were stained with anti-CHIKV (ATCC V-548–701-438 562) and anti-VEEV (ATCC V-532–711-562) primary antibodies, respectively, followed by incubation with anti-mouse Alexa488 and Hoechst 33342. The cells were imaged at 10× (ImageXpress Micro, Molecular Devices) with nine sites per well. The number of cells and the number of infected cells were measured using MetaXpress software (Cell Scoring Module) and the percentage of infected cells was calculated.
Generation of DDX39A U2OS knockout cell line by CRISPR/Cas9:
DDX39A-deficient U2OS cells were generated using CRISPR/Cas9 technology. A single guide RNA (sgRNA)–targeting DDX39A was cloned into a Cas9-expressing lentiviral transfer vector (lentiCRISPRv2)120. The oligonucleotides for sgRNAs were as previously described:5’-CACCGCAGCAGATTGAGCCTGTCAA-3; 5’AAACTTGACAGGCTCAATCTGCTGC-3’35. The plasmid was digested with BsmBI (Thermo #ER0451) and ligated and annealed oligonucleotide (DDX39A-sgRNA). The lentiCRISPRv2 plasmid was cotransfected into HEK293T cells with packaging vectors pCMV-VSV-G (Addgene) and psPAX2 (Addgene) using Lipofectamine 2000 (Invitrogen #11668027). One day after transfection media was changed, and two days after transfection, the viral supernatant was harvested and stored at −80°C. U2OS cells were transduced with virus supernatant containing 10 μg/mL polybrene for 48h and selected with 1.5 mg/ml puromycin. Single cell clonal populations were generated and DDX39A levels quantified via immunoblot.
RNA isolation, reverse transcription and quantitative reverse transcription PCR:
Cells were lysed in TRIzol, and RNA was extracted using on-column purification (Zymo Research #R1018) with DNase I treatment (Zymo Research #E1010). The concentration of the RNA was then determined, and cDNA was generated from random hexamers primers to prime reverse transcription reactions with Moloney murine leukemia virus (M-MLV) reverse transcriptase (Ambion). The qPCR was performed in triplicate with the diluted cDNA using a SYBR-based (ThermoScientific) qPCR assay on AppliedBiosystems equipment with Ta=60. Relative quantification was calculated according to the ΔΔCt method121 by normalization to the GAPDH loading control. Antigenome quantification was performed using strand-specific reverse transcription122. qPCR was performed with antigenome-specific primers. A standard curve was generated from a gBlock amplicon encoding 133 bp (IDT). A standard qPCR curve for antigenome (−) copy number was generated from a gBlock amplicon-based of 10-fold serial dilutions starting at 108 DNA copies and antigenome-specific qPCR primers.
Viral cycle assays:
Viral spread was assayed by infecting control or DDX39A-depleted cells for 4h with CHIKV, MOI 0.5 followed by the addition of 20 mM NH4Cl. Cells were lysed in TRIzol 20hpi and RNA was isolated for RT-qPCR as described above. The effect of DDX39A on entry was determined by infecting control and DDX39A-depleted cells with CHIKV, MOI 20 in the presence of 10 μg/mL cycloheximide (CHX) for 4h. Cells were then washed with PBS to remove unbound virus and then incubated in 0.25% trypsin-EDTA to remove bound but not internalized virus. Trypsin was neutralized with media and cells were collected and then lysed in 1 mL TRIzol for downstream RNA isolation.
Viral time course:
Control and DDX39A-depleted cells were synchronously infected with CHIKV, MOI 10 by allowing viruses to bind at 4°C for 1h. Cells were washed twice with PBS to remove unbound virus, complete culture media was added, and incubated at 37°C. At each time point, samples were washed with PBS and collected in TRIzol for downstream RNA extraction and qPCR analysis.
dsRNA accumulation assay:
Control and DDX39A-depleted cells were plated on a glass coverslip in a 24-well plate and infected with CHIKV, MOI 10 for 4h. Cells were then fixed with 4% formaldehyde, washed with PBS then processed for immunofluorescence staining with J2 antibody to measure dsRNA accumulation and Hoechst (#33342) to stain nuclei. Following PBS-T washes, secondary antibody treatment was done for 1h at room temperature. Following staining, coverslips were mounted onto glass slides with Vectashield (Vector Laboratories) and then imaged with Leica DM5500A confocal microscope. Images were imported in MetaXpress (Molecular Devices) software and quantified with the granularity module as previously described123.
Immunoblots:
For whole-cell analyses, cells were lysed in RIPA buffer, sonicated for 15 sec at 20% 3 times, and centrifuged for 10 minutes. Nuclear and cytoplasmic fractionation was performed following the manufacturer’s instruction (CST #9038). Briefly, cells were collected by trypsinization, and the cell pellet was resuspended in CIB buffer with protease inhibitors, vortexed, and incubated on ice for 5 minutes. After centrifugation, the supernatant was removed to obtain the cytoplasmic fraction. The pellet containing the nuclei was resuspended in CyNIB buffer supplemented with protease inhibitors and lysed after sonication for 5 sec at 20% power 3 times. Protein levels were quantified using the BCA Protein Assay (ThermoScientific) and equal amounts of protein were boiled in SDS buffer at 95°C. Samples were run on Tris-glycine SDS-PAGE gels and transferred to PVDF membranes (Immobilon) for western blot detection. Blots were exposed with ECL reagents (GE Healthcare) or SuperSignal West Femto (Thermo Fisher) and imaged with a CCD-based detection system (Amersham).
UV-inactivation of CHIKV:
Virus was spread on a 60 mm2 tissue culture dish and exposed to 1200 μJ × 100 of 254 nm UV light using a Stratalinker 2400 five times. The UV-inactivated virus was then placed in a new tube. Equivalent volumes of WT and UV-inactivated viruses were used for downstream studies.
Confocal microscopy:
Cells on glass coverslips were fixed with 4% formaldehyde and washed twice with phosphate-buffered saline (PBS) before blocking was performed for 30 min in PBS containing 1% Triton X-100 (PBS-T), 2% bovine serum albumin (BSA), and 0.02% sodium azide. Primary antibody in block buffer was added at room temperature for 1h. For the DDX39A localization experiment, the blocking buffer and antibody dilution buffers also had 0.1% saponin. Following three PBS-T washes, secondary antibody treatment was done for 1h at room temperature. Coverslips were then mounted with Vectashield (Vector Laboratories) and imaged using a 63x objective lens with a Leica DM5500A confocal microscope. Colocalization analysis of CHIKV-infected U2OS cells stained with DDX39A, J2(dsRNA), and Hoechst (nuclei) was performed using Coloc2 in Fiji. J2+ cells and nuclei were outlined using the polygon drawing tool and subtracted. The colocalization was determined on a single cell level between J2 and cytoplasmic DDX39A for 4 different experiments (>22 cells/experiment), the Costes randomizations were set to 10 and Pearson’s coefficient was calculated.
Viral titer quantification:
Control or DDX39A-depleted U2OS cells were infected with CHIKV at the indicated MOIs. At 4hpi, cells were washed with PBS, and media was replaced. Supernatant was collected at 24 hpi. Serial 10-fold dilutions of supernatants were used to infect either BHK or Vero E6 cells for tissue cultures infective doses (TCID50) analysis using the Reed and Muench method 124.
RNA-immunoprecipitation with qPCR (RIP-qPCR):
U2OS cells were infected with CHIKV, MOI 5, and 24hpi, cells were trypsinized, resuspended in Hank’s Balanced Salt Solution (HBSS) (GIBCO), and subjected to ultraviolet crosslinking. Cells were then lysed in Buffer A (30mM Hepes pH 7.4, 2mM MgOAc, 0.1% NP40) and 150 mM KOAc supplemented with fresh RNaseOUT Recombinant Ribonuclease Inhibitor (Invitrogen), PMSF and protease inhibitor cocktail (Sigma). Cell lysates were precleared by incubation with Protein A/G Agarose beads at 4 °C for 1 h with rotation. 10% of lysate was saved for input analyses of protein and RNA. The cell lysates were then incubated with Protein A/G Agarose beads and specific antibodies or rabbit IgG control overnight at 4 °C with rotation. Beads were then washed with Buffer A and 150mM KOAc, eluted with 0.2 M glycine pH 2.5, and neutralized with 1 M Tris pH 9. TRIzol was used to extract RNA. 5% of the washed beads were boiled in SDS loading buffer and analyzed by western blotting. Abundance of transcripts bound by DDX39A/DDX39B, or rabbit IgG control was analyzed by RT-qPCR relative to input.
In vitro RNA synthesis:
Templates containing a T7 promoter were generated with the high-fidelity Phusion enzyme (NEB) from either uninfected U2OS cDNA (MAVS, DDX24) or cDNA generated from purified CHIKV RNA as follows: Peak 1 (nucleotides 1–517, overlaps the SL3 70–156 and the 5ʹCSE 165–216 structures), Peak 2 (nucleotides 1949–3131, overlaps a stem-loop 2276–2304, and the packaging signal 2501–3079), Region 1 (nucleotides 7049 −7525) and Peak 3 (nucleotides 7565–8050) (Table S6). After confirmation of the products by agarose gel electrophoresis and sequencing, in vitro RNA synthesis was performed using T7 MEGAScript Kit (AM1334). RNA products were biotinylated using ThermoScientific’s 3’ Biotinylation Kit (20160) according to the manufacturer’s protocol. After purification of the biotinylated RNA, biotinylation was confirmed by Streptavidin dot blot using Streptavidin-HRP antibody (CST #3999).
Biochemical RNA affinity pulldown:
Dynabeads Streptavidin C1 (Invitrogen #65001) were washed with Solution A (0.1 M NaOH, 0.05 NaCl) and Solution B (0.1 M NaCl) and then resuspended in Wash and Binding (W&B) buffer (10 mM Tris-HCl pH 7.5, 1mM EDTA, 2M NaCl) according to manufacturer’s instructions. Beads were then incubated with equal concentrations of biotinylated RNAs for 30 minutes at room temperature with rotation. Conjugated beads were then washed twice with W&B buffer and then equilibrated with Protein-RNA Binding Buffer (Thermo Scientific #20164). HEK293T cells transfected with DDX39A (PLX304-DDX39A from DNASU) for 24h and then cells were lysed in RIPA buffer. Protein levels were assessed via BCA assay and 150 μg of lysate was added to each condition. The protocol was adapted from the Pierce Magnetic RNA-Protein Pull-Down kit (Buffer (Thermo Scientific #20164). Briefly, beads were incubated with 150 ug protein from cell lysate, protein-RNA binding buffer, 50% glycerol, and RNaseOUT Recombinant Ribonuclease Inhibitor (Invitrogen) for 4h at room temperature with rotation. For competition assays, cell lysate was supplemented with excess non-biotinylated RNA. Beads were then washed three times washed with Wash Buffer (Thermo Scientific #20164) supplemented with NaCl (final concentration 500mM). Beads were then resuspended in RIPA buffer and boiled in SDS. The samples were then analyzed by immunoblot.
CLIP-Seq:
Cross-linking and immunoprecipitation (CLIP-seq) of DDX39A was performed using a modified protocol 31,58. Briefly, U2OS cells were plated in a 15-cm dish and infected with CHIKV (MOI, 5) for 20h. Cells were then trypsinized resuspended in HBSS and subjected to ultraviolet crosslinking. Cell lysates were then prepared in a DNase-free PMPG buffer and treated with Rnase T1. Samples were immunoprecipitated with anti-DDX39A (Proteintech #11723-1-AP) or rabbit IgG control (Bethyl # P120–201) antibodies bound to Dynabeads protein A beads (Thermo #10002D). A 32P-labeled RNA adapter was then ligated to the 3′end of RNA, and samples were then size-selected by running them on an SDS-PAGE gel and transferred to a nitrocellulose membrane. A 5′ RNA adapter with a degenerate barcode was ligated to the extracted RNA and samples were reversed transcribed and amplified by PCR using primers with specific indexes to allow for demultiplexing of samples following sequencing. Samples were pooled and sequenced on Illumina HiSeq at the University of Pennsylvania Next Generation Sequencing Core.
CLIP-Seq bioinformatics:
Sequencing results were generated for three independent sets of infections and immunoprecipitations. Raw FastQ reads were trimmed using Cutadapt125 and Trimmomatic126 and then collapsed exact duplicates using fastq2collapse127. Reads were aligned to a composite genome composed of the full human genome (hg38) and of the CHIKV genomic sequence (Ross strain; GenBank accession no. AF490259.3). To exclude cross-contaminating signals from our analysis of viral reads, reads aligned to the CHIKV genome were filtered to exclude any reads that matched in sequence, barcode, and mapping location between samples within each replicate. We used deeptools’ multiBamSummary and plotCorrelation (Spearman analysis) to confirm a correlation between replicates and merged samples to generate coverage plots128,129. Using deeptools’ bamcompare, we subtracted background reads from our Rb IgG control and normalized to counts per mapped reads (CPM) and relative viral genome abundance, as described in figure legends. We obtained host transcripts bound by DDX39A using the featureCounts function of the Rsubread package and used a cutoff of 10 reads in all replicates for further analysis into biotypes. We used samtools to assess read counts for various stages of data processing130. For motif analyses, RNA sequencing reads from three independent replicates of DDX39A and IgG (control) pulldowns were aligned to the hg38 genome using STAR 2.7.10a131. Preprocessing, realignment, and peak calling of BAM files was performed with CLAM 1.2.59 using the “—read-tagger-method start” arguments and the gencode version 109 annotation file. HOMER v4.1160 was used to identify motifs enriched in these peaks using the arguments “-rna -len 5,6,7 -p 20 -size 100 -S 10”. Analyses were run on the Penn Medicine Academic Computing Services High-Performance Computing System, and results were visualized in R (v 1.4.1106) and IGV (v 2.9.2). Figures were generated in R. Samtools130 was used to generate the CLIP-Seq read coverage on the CHIKV genome (Table S2).
Proximity ligation assay (PLA):
PLA was performed on formaldehyde-fixed U2OS cells either mock uninfected or infected with CHIKV Ross (MOI 1.5, 20h) using the Duolink PLA technology probes and reagents following the manufacturer’s instructions (Sigma-Aldrich). First, U2OS cells were permeabilized with DPBS + Triton X-100 0.2% for 10 min at room temperature followed by blocking in Duolink Blocking Buffer for 60 min at 37°C. Pairs of antibodies of different species were then added to each sample for 2 h at 37°C. Coverslips were then washed twice for 5 min with Wash Buffer A and then incubated with the anti-rabbit PLUS and anti-mouse MINUS PLA probes for 1h at 37°C. For technical controls, only one antibody was added to each coverslip. Coverslips were washed twice for 5 min with Wash Buffer A, and then the two oligonucleotides within the PLA probes are ligated by incubating with the Duolink ligase for 30 min at 37°C. After two additional washes, rolling circle amplification is performed by incubating with the polymerase in the kit for 100 min at 37°C. Coverslips were washed twice with Wash Buffer B for 10 minutes, then with 0.01X Wash Buffer B for 1 min followed by incubation with blocking buffer (PBS with 1% Triton X-100, 2% bovine serum albumin and 0.02% sodium azide) for 30 min. Coverslips were then incubated with anti-CHIKV (ATCC) antibodies overnight at 4°C, washed with PBS-T, and treated with secondary antibody for 1h. Coverslips were then mounted with Vectashield and imaged using a 63x objective lens using the Leica DM5500A confocal microscope.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical significance was assigned when p values were < 0.05 using Prism Version 8 (GraphPad). All statistics performed on fold change data were log2 transformed prior to analysis. Each dot represents an individual experiment. The specific tests and numbers of experiments are indicated in the Figure legends.
Supplementary Material
Table S1. DDX screen, Related to Figure 1.
Table S3. CHIKV genome annotations, Related to Figure 5.
Table S4. Recombinant virus sequences, Related to Figure 5 and S6.
Table S5. Oligonucleotide Sequences, Related to STAR Methods.
Table S2. CLIP reads on the CHIKV genome, Related to Figure S6.
Table S6. Host transcripts bound by DDX39A, Related to Figure S6.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| DDX39A | Proteintech | Cat#11723–1-AP |
| GAPDH | Sigma | Cat#CB1001 |
| Lamin B1 | Abcam | Cat#ab16048 |
| Tubulin | Sigma | Cat#T6199 |
| DDX39B | Santa Cruz | Cat#sc-271395 |
| Beta actin | Santa Cruz | Cat#sc-47778 |
| IFIT1 | Cell Signaling Technology | Cat#D2X9Z |
| CHIKV E1 | Novus | Cat# MAB97792 |
| CHIKV ascites | ATCC | Cat#V-548–701-562 |
| Histone H3 | Abcam | Cat#ab18521 |
| J2(dsRNA) | Absolute | Cat#Ab01299 |
| VEEV ascites | ATCC | Cat#V-532–711-562 |
| IAV NP | BEI Resources | Cat#NR-4282 |
| Rabbit IgG | Bethyl | Cat#P120–201 |
| VSV-M | Absolute | Cat#23H12 |
| DDX39A | Abnova | Cat# MAB6576 |
| DDX24 | Proteintech | Cat# 15769–1-AP |
| Streptavidin-HRP antibody | Cell Signaling Technology | Cat#3999 |
| ALY/REF | Novus | Cat#NB100670 |
| J2(dsRNA) | Cherry Lab | |
| DDX39B | Proteintech | Cat#14798–1-AP |
| RCK (p54) | MBL | Cat#PD009 |
| TIAR | BD Biosciences | Cat#610352 |
| Bacterial and virus strains | ||
| Coxsackie B Virus | Carolyn B. Coyne (University of Pittsburgh) | - |
| CHIKV, Ross strain | Dr. D. Weiner (Wistar Institute) | - |
| CHIKV 181/25 WT, δ5’CSE, δSL3 | M. Heise (University of North Carolina) | - |
| HCoV-OC43 | ATCC | Cat# VR-1558 |
| HCoV-229E | ATCC | Cat#VR-740 |
| O’nyong’ nyong (SG650) | Dr. Thomas E. Morrison (University of Colorado) | - |
| RVFV (MP12) | Dr. Michael Diamond (Washington University in St. Louis) | - |
| SeV | Charles River | Cat#10100774 |
| SINV-mKate | M. Heise (University of North Carolina) | - |
| VEEV (strain TC-83) | M. Heise (University of North Carolina) | - |
| VSV-EGFP | Jack Rose (Yale University) | - |
| NEB 5-alpha Competent E. coli | NEB | Cat#C2988J |
| Chemicals, peptides, and recombinant proteins | ||
| Random Primers | Invitrogen | Cat#48190011 |
| IFNβ | STEMCELL Technologies | Cat#78113 |
| M MLV Reverse Transcriptase | Invitrogen | Cat#28025013 |
| SYBR green master mix | Applied Biosystems | Cat#4368708 |
| Hoechst 33342 | Sigma Aldrich | Cat#B2261–25MG |
| HiPerFect reagent | Qiagen | Cat#301709 |
| TRIzol | Life Technologies | Cat#15596018 |
| Cycloheximide | Sigma Aldrich | Cat#01810 |
| Ruxolitinib | Invitrogen | Cat#tlrl-rux |
| Poly(I:C)-LMW | Invivogen | Cat# tlrl-picw |
| Critical commercial assays | ||
| Dynabeads Streptavidin C1 | Invitrogen | Cat#65001) |
| Zymo RNA clean and concentrator 25 | Zymo Research | Cat#R1018 |
| T7 MEGAScript Kit | Ambion | Cat#AM1334 |
| Cell Fractionation Kit | Cell Signaling Technology | Cat#9038 |
| 3’ Biotinylation Kit | ThermoScientific | Cat#20160 |
| Experimental models: Cell lines | ||
| Homo sapiens: U2OS | ATCC | HTB-96 |
| Homo sapiens: A549 | ATCC | CCL-2 |
| Homo sapiens: HeLa S3 | ATCC | CCL-185 |
| Homo sapiens: HEK 293T | ATCC | CRL-3216 |
| Mesocricetus auratus: BHK-21 | Mesocricetus auratus: BHK-21 | Mesocricetus auratus: BHK-21 |
| Cercopithecus aethiops: Vero E6 | ATCC | CRL-1586 |
| Cercopithecus aethiops: Vero CCL81 | ATCC | CCL81 |
| Oligonucleotides | ||
| Primers for qPCR analysis, see Table S5 | This paper | - |
| siRNA sequences, see Table S5 | This paper | - |
| Primers for RNA affinity experiments, see S5 | This paper | - |
| Deposited data | ||
| Microscope images and blots | Mendeley | Mendeley Data, V1, doi: 10.17632/fvzkrcb5n2.1 |
| Recombinant DNA | ||
| PLX304-DDX39A plasmid | DNASU | http://dnasu.org/DNASU/GetCloneDetail.do?cloneid=440517 |
| lentiCRISPR v2 | lentiCRISPR v2 was a gift from Feng Zhang | (Addgene plasmid # 52961; http://n2t.net/addgene:52961; RRID:Addgene_52961) |
| pCMV-VSV-G | pCMV-VSV-G was a gift from Bob Weinberg | Addgene plasmid # 8454; http://n2t.net/addgene:8454; RRID:Addgene_8454 |
| psPAX-2 | psPAX2 was a gift from Didier Trono | Addgene plasmid # 12260; http://n2t.net/addgene:12260; RRID:Addgene_12260) |
| Software and algorithms | ||
| Trimmomatic | (Bolger et al., 2014) | version 0.32 https://github.com/usadellab/Trimmomatic |
| Cutadapt | (Martin, 2011) | version 1.5 https://cutadapt.readthedocs.io/en/stable/index.html |
| STAR | (Dobin et al., 2013) | version 2.5.2a https://github.com/alexdobin/STAR |
| Samtools | (Li et al., 2009) | version 1.1 http://samtools.sourceforge.net/ |
| Picard | http://broadinstitute.github.io/picard | version 1.141 https://broadinstitute.github.io/picard/ |
| HOMER | (Heinz et al., 2010) | Version 4.11 http://homer.ucsd.edu/homer/ |
| CLIP-seq Analysis of Multi-mapped reads (CLAM) | (Zhang and Xing, 2017) | version 1.2.1 https://github.com/Xinglab/CLAM |
| Bedtools | (Quinlan and Hall, 2010) | version 2.30.0 https://bedtools.readthedocs.io/en/latest/ |
| RStudio | Team RS. 2016. RStudio: Integrated Development for R, RStudio, Inc., Boston, MA. | 1.4.1106 http://www.rstudio.com/. |
| MetaXpress | Molecular Devices Corporation | |
| Deeptools2 | (Ramırez et al., 2016) | version 2.0 https://deeptools.readthedocs.io/en/develop/content/installation.html |
| QuantStudio 6 1.3 | Applied Biosystems | N/A |
| PRISM 8.4.3 | GraphPad Software | N/A |
| Spotfire | PerkinElmer | N/A |
| Other | ||
| VECTASHIELD Antifade Mounting Medium containing DAPI | Vector Laboratories | H-1200 |
| 4 NUPAGE 4–12% Bis-Tris Gels | Invitrogen | Cat# NP0323 |
| Complete EDTA-free protease inhibitor cocktail | Sigma-Aldrich | Cat# 4693159001 |
| Dynabeads protein A | Invitrogen | Cat# 10002D |
| Nitrocellulose membrane | GE Healthcare Life Sciences | Cat#10600002 |
| SuperSignal™ West Femto Maximum Sensitivity | Thermo Fisher Scientific | Cat# 34095 |
| Lipofectamine 2000 | Thermo Fisher Scientific | Cat# L3000008 |
| RNase T1 | Thermo Fisher Scientific | Cat#AM2294 |
| RQ1 DNase | Promega | Cat# M6101 |
| SuperScript III Reverse Transcriptase Superscript RT buffer | Invitrogen | Cat#214351 |
| Accuprime Pfx supermix | Invitrogen | Cat# 12344040 |
| TURBO DNase | Thermo Fisher Scientific | Cat#AM2238 |
| ProteinaseK | Sigma-Aldrich | Cat# 3115828001 |
| T4 Polynucleotide Kinase | Thermo Fisher Scientific | Cat#EK003 |
| T4 RNA ligase | ThermoScientific | Cat#00864622 |
| T4 Polynucleotide kinase | Biolabs | Cat#M0201S |
Highlights.
DDX39A is antiviral against alphaviruses independent of the interferon pathway
Infection with Chikungunya leads to cytoplasmic accumulation of DDX39A
DDX39A binds to a conserved RNA structure on the alphavirus genomic RNA
Aided by ALY/REF, DDX39A binds this structure to control infection
ACKNOWLEDGEMENTS
We thank the Cherry lab members for the scientific discussion and assistance with data analysis and assays, especially Megha G. Basavappa. We thank the High Throughput Screening Core at the University of Pennsylvania for technical support with the screen. Funding: This work was supported by grants from the National Institutes of Health to S.C. (R01AI150246, RO1AI152362, 1R21AI151882, and R01AI140539) and M.T.H. (U19 AI 109680 and R21 AI138056), as well as funding from the Penn Center for Precision Medicine. E.A.M. was supported by T32AI007419. S.C. is a recipient of the Burroughs Wellcome Investigators in the Pathogenesis of Infectious Disease Award. Schematics in Figure 5 were generated using Biorender.com.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES:
- 1.Baxter VK, and Heise MT (2020). Immunopathogenesis of alphaviruses. Advances in virus research 107, 315–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burt FJ, Rolph MS, Rulli NE, Mahalingam S, and Heise MT (2012). Chikungunya: a re-emerging virus. The Lancet 379, 662–671. 10.1016/S0140-6736(11)60281-X. [DOI] [PubMed] [Google Scholar]
- 3.Reed KD (2018). Viral zoonoses. Reference Module in Biomedical Sciences. [Google Scholar]
- 4.Rezza G, Chen R, and Weaver SC (2017). O’nyong-nyong fever: a neglected mosquito-borne viral disease. Pathog Glob Health 111, 271–275. 10.1080/20477724.2017.1355431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guzmán-Terán C, Calderón-Rangel A, Rodriguez-Morales A, and Mattar S (2020). Venezuelan equine encephalitis virus: The problem is not over for tropical America. Annals of clinical microbiology and antimicrobials 19, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Griffin DE (2010). Emergence and re-emergence of viral diseases of the central nervous system. Progress in neurobiology 91, 95–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Erwin-Cohen RA, Porter AI, Pittman PR, Rossi CA, and DaSilva L (2012). Host responses to live-attenuated Venezuelan equine encephalitis virus (TC-83) Comparison of naive, vaccine responder and nonresponder to TC-83 challenge in human peripheral blood mononuclear cells. Human vaccines & immunotherapeutics 8, 1053–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kendall C, Khalid H, Müller M, Banda DH, Kohl A, Merits A, Stonehouse NJ, and Tuplin A (2019). Structural and phenotypic analysis of Chikungunya virus RNA replication elements. Nucleic Acids Research 47, 9296–9312. 10.1093/nar/gkz640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Madden EA, Plante KS, Morrison CR, Kutchko KM, Sanders W, Long KM, Taft-Benz S, Cisneros MCC, White AM, Sarkar S, et al. (2020). Using SHAPE-MaP To Model RNA Secondary Structure and Identify 3′UTR Variation in Chikungunya Virus. Journal of Virology 94, e00701–00720. 10.1128/JVI.00701-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim DY, Firth AE, Atasheva S, Frolova EI, and Frolov I (2011). Conservation of a packaging signal and the viral genome RNA packaging mechanism in alphavirus evolution. Journal of Virology 85, 8022–8036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Firth AE, Wills NM, Gesteland RF, and Atkins JF (2011). Stimulation of stop codon readthrough: frequent presence of an extended 3′ RNA structural element. Nucleic Acids Research 39, 6679–6691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chung BY-W, Firth AE, and Atkins JF (2010). Frameshifting in alphaviruses: a diversity of 3′ stimulatory structures. J. Mol. Biol. 397, 448–456. [DOI] [PubMed] [Google Scholar]
- 13.Holmes AC, Basore K, Fremont DH, and Diamond MS (2020). A molecular understanding of alphavirus entry. PLOS Pathogens 16, e1008876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuhn R (2007). Togaviridae: the viruses and their replication. Fields virology 1, 1001–1022. [Google Scholar]
- 15.Strauss JH, and Strauss EG (1994). The alphaviruses: gene expression, replication, and evolution. Microbiological reviews 58, 491–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kutchko KM, Madden EA, Morrison C, Plante KS, Sanders W, Vincent HA, Cruz Cisneros MC, Long KM, Moorman NJ, and Heise MT (2018). Structural divergence creates new functional features in alphavirus genomes. Nucleic Acids Research 46, 3657–3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rupp JC, Sokoloski KJ, Gebhart NN, and Hardy RW (2015). Alphavirus RNA synthesis and non-structural protein functions. The Journal of general virology 96, 2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pott F, Postmus D, Brown RJ, Wyler E, Neumann E, Landthaler M, and Goffinet C (2021). Single-cell analysis of arthritogenic alphavirus-infected human synovial fibroblasts links low abundance of viral RNA to induction of innate immunity and arthralgia-associated gene expression. Emerging microbes & infections 10, 2151–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brown RS, Kim L, and Kielian M (2021). Specific Recognition of a Stem-Loop RNA Structure by the Alphavirus Capsid Protein. Viruses 13, 1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schoggins JW, and Rice CM (2011). Interferon-stimulated genes and their antiviral effector functions. Current opinion in virology 1, 519–525. 10.1016/j.coviro.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brisse M, and Ly H (2019). Comparative structure and function analysis of the RIG-I-like receptors: RIG-I and MDA5. Frontiers in immunology 10, 1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang X, Li MM, Zhao J, Li S, MacDonald MR, Rice CM, Gao X, and Gao G (2016). Sindbis virus can exploit a host antiviral protein to evade immune surveillance. Journal of Virology 90, 10247–10258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fros JJ, Liu WJ, Prow NA, Geertsema C, Ligtenberg M, Vanlandingham DL, Schnettler E, Vlak JM, Suhrbier A, and Khromykh AA (2010). Chikungunya virus nonstructural protein 2 inhibits type I/II interferon-stimulated JAK-STAT signaling. Journal of Virology 84, 10877–10887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rogers K, Jones-Burrage S, Maury W, and Mukhopadhyay S (2020). TF protein of Sindbis virus antagonizes host type I interferon responses in a palmitoylation-dependent manner. Virology 542, 63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Boxel-Dezaire AH, Rani MS, and Stark GR (2006). Complex modulation of cell type-specific signaling in response to type I interferons. Immunity 25, 361–372. [DOI] [PubMed] [Google Scholar]
- 26.Linder P, and Jankowsky E (2011). From unwinding to clamping—the DEAD box RNA helicase family. Nature reviews Molecular cell biology 12, 505–516. [DOI] [PubMed] [Google Scholar]
- 27.Tanner NK, and Linder P (2001). DExD/H Box RNA Helicases: From Generic Motors to Specific Dissociation Functions. Molecular Cell 8, 251–262. 10.1016/S1097-2765(01)00329-X. [DOI] [PubMed] [Google Scholar]
- 28.Jankowsky E (2011). RNA helicases at work: binding and rearranging. Trends in biochemical sciences 36, 19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, and Ishii KJ (2006). Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441, 101–105. [DOI] [PubMed] [Google Scholar]
- 30.Taschuk F, and Cherry S (2020). DEAD-Box helicases: sensors, regulators, and effectors for antiviral defense. Viruses 12, 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taschuk F, Tapescu I, Moy RH, and Cherry S (2020). DDX56 Binds to Chikungunya Virus RNA To Control Infection. MBio 11, e02623–02620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moy Ryan H., Cole Brian S., Yasunaga A, Gold B, Shankarling G, Varble A, Molleston Jerome M., tenOever Benjamin R., Lynch Kristen W., and Cherry S (2014). Stem-Loop Recognition by DDX17 Facilitates miRNA Processing and Antiviral Defense. Cell 158, 764–777. 10.1016/j.cell.2014.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cordin O, Banroques J, Tanner NK, and Linder P (2006). The DEAD-box protein family of RNA helicases. Gene 367, 17–37. [DOI] [PubMed] [Google Scholar]
- 34.Yamazaki T, Fujiwara N, Yukinaga H, Ebisuya M, Shiki T, Kurihara T, Kioka N, Kambe T, Nagao M, and Nishida E (2010). The closely related RNA helicases, UAP56 and URH49, preferentially form distinct mRNA export machineries and coordinately regulate mitotic progression. Molecular biology of the cell 21, 2953–2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shi P, Guo Y, Su Y, Zhu M, Fu Y, Chi H, Wu J, and Huang J (2020). SUMOylation of DDX39A Alters Binding and Export of Antiviral Transcripts to Control Innate Immunity. The Journal of Immunology. [DOI] [PubMed] [Google Scholar]
- 36.Otake K, Uchida K, Ide S, Kobayashi Y, Kobayashi I, and Kusunoki M (2016). Identification of DDX39A as a potential biomarker for unfavorable neuroblastoma using a proteomic approach. Pediatric Blood & Cancer 63, 221–227. [DOI] [PubMed] [Google Scholar]
- 37.Sugiura T, Nagano Y, and Noguchi Y (2007). DDX39, upregulated in lung squamous cell cancer, displays RNA helicase activities and promotes cancer cell growth. Cancer biology & therapy 6, 957–964. [DOI] [PubMed] [Google Scholar]
- 38.Bick MJ, Carroll J-WN, Gao G, Goff SP, Rice CM, and MacDonald MR (2003). Expression of the zinc-finger antiviral protein inhibits alphavirus replication. Journal of Virology 77, 11555–11562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reynaud JM, Kim DY, Atasheva S, Rasalouskaya A, White JP, Diamond MS, Weaver SC, Frolova EI, and Frolov I (2015). IFIT1 differentially interferes with translation and replication of alphavirus genomes and promotes induction of type I interferon. PLOS Pathogens 11, e1004863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pallett MA, Lu Y, and Smith GL (2022). DDX50 Is a Viral Restriction Factor That Enhances IRF3 Activation. Viruses 14, 316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou X, Gao F, Lu M, Liu Z, Wang M, Cao J, Ke X, and Yi M (2022). DDX43 recruits TRIF or IPS-1 as an adaptor and activates the IFN-β pathway in Nile tilapia (Oreochromis niloticus). Molecular Immunology 143, 7–16. [DOI] [PubMed] [Google Scholar]
- 42.Zhang R, Kim AS, Fox JM, Nair S, Basore K, Klimstra WB, Rimkunas R, Fong RH, Lin H, and Poddar S (2018). Mxra8 is a receptor for multiple arthritogenic alphaviruses. Nature 557, 570–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sourisseau M, Schilte C, Casartelli N, Trouillet C, Guivel-Benhassine F, Rudnicka D, Sol-Foulon N, Le Roux K, Prevost M-C, Fsihi H, et al. (2007). Characterization of reemerging chikungunya virus. PLOS Pathogens 3, e89–e89. 10.1371/journal.ppat.0030089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hu D, Zhang J, Wang H, Liu S, Yu L, Sun L, and Qu Y (2014). Chikungunya virus glycoproteins pseudotype with lentiviral vectors and reveal a broad spectrum of cellular tropism. PLOS ONE 9, e110893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kapadia F, Pryor A, Chang T-H, and Johnson LF (2006). Nuclear localization of poly (A)+ mRNA following siRNA reduction of expression of the mammalian RNA helicases UAP56 and URH49. Gene 384, 37–44. [DOI] [PubMed] [Google Scholar]
- 46.Akhrymuk I, Kulemzin SV, and Frolova EI (2012). Evasion of the innate immune response: the Old World alphavirus nsP2 protein induces rapid degradation of Rpb1, a catalytic subunit of RNA polymerase II. Journal of Virology 86, 7180–7191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bae S, Lee JY, and Myoung J (2019). Chikungunya virus-encoded nsP2, E2 and E1 strongly antagonize the interferon-β signaling pathway. [DOI] [PubMed]
- 48.Beckham CJ, and Parker R (2008). P bodies, stress granules, and viral life cycles. Cell host & microbe 3, 206–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ayache J, Bénard M, Ernoult-Lange M, Minshall N, Standart N, Kress M, and Weil D (2015). P-body assembly requires DDX6 repression complexes rather than decay or Ataxin2/2L complexes. Molecular biology of the cell 26, 2579–2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Di Stefano B, Luo E-C, Haggerty C, Aigner S, Charlton J, Brumbaugh J, Ji F, Jiménez IR, Clowers KJ, and Huebner AJ (2019). The RNA helicase DDX6 controls cellular plasticity by modulating P-body homeostasis. Cell stem cell 25, 622–638. e613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tourrière H, Chebli K, Zekri L, Courselaud B, Blanchard JM, Bertrand E, and Tazi J (2003). The RasGAP-associated endoribonuclease G3BP assembles stress granules. The Journal of cell biology 160, 823–831. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 52.Liu L, Weiss E, Panas MD, Götte B, Sellberg S, Thaa B, and McInerney GM (2019). RNA processing bodies are disassembled during Old World alphavirus infection. Journal of General Virology 100, 1375–1389. [DOI] [PubMed] [Google Scholar]
- 53.Fros JJ, Domeradzka NE, Baggen J, Geertsema C, Flipse J, Vlak JM, and Pijlman GP (2012). Chikungunya virus nsP3 blocks stress granule assembly by recruitment of G3BP into cytoplasmic foci. J Virol 86, 10873–10879. 10.1128/jvi.01506-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jayabalan AK, Griffin DE, and Leung AK (2019). Alphavirus nsP3 ADP-ribosylhydrolase activity disrupts stress granule formation. bioRxiv, 629881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Garcia-Moreno M, Noerenberg M, Ni S, Järvelin AI, González-Almela E, Lenz CE, Bach-Pages M, Cox V, Avolio R, Davis T, et al. (2019). System-wide Profiling of RNA-Binding Proteins Uncovers Key Regulators of Virus Infection. Molecular Cell. 10.1016/j.molcel.2019.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Thomas M, Lischka P, Müller R, and Stamminger T (2011). The cellular DExD/H-box RNA-helicases UAP56 and URH49 exhibit a CRM1-independent nucleocytoplasmic shuttling activity. PLOS ONE 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Clark LE, Clark SA, Lin C, Liu J, Coscia A, Nabel KG, Yang P, Neel DV, Lee H, and Brusic V (2022). VLDLR and ApoER2 are receptors for multiple alphaviruses. Nature 602, 475–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vourekas A, and Mourelatos Z (2014). HITS-CLIP (CLIP-Seq) for mouse Piwi proteins. In PIWI-Interacting RNAs, (Springer; ), pp. 73–95. [DOI] [PubMed] [Google Scholar]
- 59.Zhang Z, and Xing Y (2017). CLIP-seq analysis of multi-mapped reads discovers novel functional RNA regulatory sites in the human transcriptome. Nucleic Acids Res 45, 9260–9271. 10.1093/nar/gkx646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589. 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hyde JL, Gardner CL, Kimura T, White JP, Liu G, Trobaugh DW, Huang C, Tonelli M, Paessler S, and Takeda K (2014). A viral RNA structural element alters host recognition of nonself RNA. Science 343, 783–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fayzulin R, and Frolov I (2004). Changes of the secondary structure of the 5′ end of the Sindbis virus genome inhibit virus growth in mosquito cells and lead to accumulation of adaptive mutations. Journal of Virology 78, 4953–4964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Michel G, Petrakova O, Atasheva S, and Frolov I (2007). Adaptation of Venezuelan equine encephalitis virus lacking 51-nt conserved sequence element to replication in mammalian and mosquito cells. Virology 362, 475–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Feng H, Bao S, Rahman MA, Weyn-Vanhentenryck SM, Khan A, Wong J, Shah A, Flynn ED, Krainer AR, and Zhang C (2019). Modeling RNA-binding protein specificity in vivo by precisely registering protein-RNA crosslink sites. Molecular Cell 74, 1189–1204.e1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jolma A, Zhang J, Mondragón E, Morgunova E, Kivioja T, Laverty KU, Yin Y, Zhu F, Bourenkov G, Morris Q, et al. (2020). Binding specificities of human RNA binding proteins towards structured and linear RNA sequences. bioRxiv, 317909. 10.1101/317909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rudolph MG, and Klostermeier D (2015). When core competence is not enough: functional interplay of the DEAD-box helicase core with ancillary domains and auxiliary factors in RNA binding and unwinding. Biological chemistry 396, 849–865. 10.1515/hsz-2014-0277. [DOI] [PubMed] [Google Scholar]
- 67.Silverman E, Edwalds-Gilbert G, and Lin R-J (2003). DExD/H-box proteins and their partners: helping RNA helicases unwind. Gene 312, 1–16. [DOI] [PubMed] [Google Scholar]
- 68.Mathys H, Basquin J, Ozgur S, Czarnocki-Cieciura M, Bonneau F, Aartse A, Dziembowski A, Nowotny M, Conti E, and Filipowicz W (2014). Structural and biochemical insights to the role of the CCR4-NOT complex and DDX6 ATPase in microRNA repression. Mol Cell 54, 751–765. 10.1016/j.molcel.2014.03.036. [DOI] [PubMed] [Google Scholar]
- 69.Sugiura T, Sakurai K, and Nagano Y (2007). Intracellular characterization of DDX39, a novel growth-associated RNA helicase. Experimental cell research 313, 782–790. [DOI] [PubMed] [Google Scholar]
- 70.Wu Y, Liu Q, Zhou J, Xie W, Chen C, Wang Z, Yang H, and Cui J (2017). Zika virus evades interferon-mediated antiviral response through the co-operation of multiple nonstructural proteins in vitro. Cell Discov 3, 17006. 10.1038/celldisc.2017.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nelemans T, and Kikkert M (2019). Viral innate immune evasion and the pathogenesis of emerging RNA virus infections. Viruses 11, 961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Thorne LG, Bouhaddou M, Reuschl A-K, Zuliani-Alvarez L, Polacco B, Pelin A, Batra J, Whelan MV, Hosmillo M, and Fossati A (2022). Evolution of enhanced innate immune evasion by SARS-CoV-2. Nature 602, 487–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bonaventure B, Rebendenne A, de Gracia FG, Mckellar J, Gracias S, Labaronne E, Tauziet M, Valadão ALC, Bernard E, and Briant L (2021). The DEAD box RNA helicase DDX42 is an intrinsic inhibitor of positive-strand RNA viruses. [DOI] [PMC free article] [PubMed]
- 74.Yamada T, Sato S, Sotoyama Y, Orba Y, Sawa H, Yamauchi H, Sasaki M, and Takaoka A (2021). RIG-I triggers a signaling-abortive anti-SARS-CoV-2 defense in human lung cells. Nature immunology 22, 820–828. [DOI] [PubMed] [Google Scholar]
- 75.Saberi A, Gulyaeva AA, Brubacher JL, Newmark PA, and Gorbalenya AE (2018). A planarian nidovirus expands the limits of RNA genome size. PLOS Pathogens 14, e1007314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Clyde K, and Harris E (2006). RNA secondary structure in the coding region of dengue virus type 2 directs translation start codon selection and is required for viral replication. Journal of Virology 80, 2170–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pirakitikulr N, Kohlway A, Lindenbach BD, and Pyle AM (2016). The coding region of the HCV genome contains a network of regulatory RNA structures. Molecular Cell 62, 111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yang D, and Leibowitz JL (2015). The structure and functions of coronavirus genomic 3′ and 5′ ends. Virus Research 206, 120–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liu Y, Zhang Y, Wang M, Cheng A, Yang Q, Wu Y, Jia R, Liu M, Zhu D, and Chen S (2020). Structures and functions of the 3′ untranslated regions of positive-sense single-stranded RNA viruses infecting humans and animals. Front. Cell. Infect. Microbiol. 10, 453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sauter D, and Kirchhoff F (2021). Evolutionary conflicts and adverse effects of antiviral factors. Elife 10, e65243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Brown RS, Anastasakis DG, Hafner M, and Kielian M (2020). Multiple capsid protein binding sites mediate selective packaging of the alphavirus genomic RNA. Nature communications 11, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hyde JL, Chen R, Trobaugh DW, Diamond MS, Weaver SC, Klimstra WB, and Wilusz J (2015). The 5′ and 3′ ends of alphavirus RNAs–Non-coding is not non-functional. Virus Research 206, 99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Frolov I, Hardy R, and Rice CM (2001). Cis-acting RNA elements at the 5′ end of Sindbis virus genome RNA regulate minus-and plus-strand RNA synthesis. RNA 7, 1638–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shen J, Zhang L, and Zhao R (2007). Biochemical characterization of the ATPase and helicase activity of UAP56, an essential pre-mRNA splicing and mRNA export factor. Journal of Biological Chemistry 282, 22544–22550. [DOI] [PubMed] [Google Scholar]
- 85.Wu G, Adachi H, Ge J, Stephenson D, Query CC, and Yu YT (2016). Pseudouridines in U2 snRNA stimulate the ATPase activity of Prp5 during spliceosome assembly. The EMBO journal 35, 654–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Choksupmanee O, Tangkijthavorn W, Hodge K, Trisakulwattana K, Phornsiricharoenphant W, Narkthong V, Tulakarnwong S, Ngamphiw C, Tongsima S, and Chimnaronk S (2021). Specific Interaction of DDX6 with an RNA Hairpin in the 3’ UTR of the Dengue Virus Genome Mediates G(1) Phase Arrest. J Virol 95, e0051021. 10.1128/jvi.00510-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cook KC, and Cristea IM (2019). Location is everything: protein translocations as a viral infection strategy. Current opinion in chemical biology 48, 34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Molleston JM, Sabin LR, Moy RH, Menghani SV, Rausch K, Gordesky-Gold B, Hopkins KC, Zhou R, Jensen TH, Wilusz JE, and Cherry S (2016). A conserved virus-induced cytoplasmic TRAMP-like complex recruits the exosome to target viral RNA for degradation. Genes & development 30, 1658–1670. 10.1101/gad.284604.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lai M-C, Lee Y-HW, and Tarn W-Y (2008). The DEAD-box RNA helicase DDX3 associates with export messenger ribonucleoproteins as well astip-associated protein and participates in translational control. Molecular biology of the cell 19, 3847–3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Paine I, Posey JE, Grochowski CM, Jhangiani SN, Rosenheck S, Kleyner R, Marmorale T, Yoon M, Wang K, and Robison R (2019). Paralog studies augment gene discovery: DDX and DHX genes. The American Journal of Human Genetics 105, 302–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Thompson NA, Ranzani M, van der Weyden L, Iyer V, Offord V, Droop A, Behan F, Gonçalves E, Speak A, and Iorio F (2021). Combinatorial CRISPR screen identifies fitness effects of gene paralogues. Nature communications 12, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Feng X, Tang M, Dede M, Su D, Pei G, Jiang D, Wang C, Chen Z, Li M, and Nie L (2022). Genome-wide CRISPR screens using isogenic cells reveal vulnerabilities conferred by loss of tumor suppressors. Science advances 8, eabm6638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Venkataramanan S, Gadek M, Calviello L, Wilkins K, and Floor SN (2021). DDX3X and DDX3Y are redundant in protein synthesis. RNA 27, 1577–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Huang C, Liang D, Tatomer DC, and Wilusz JE (2018). A length-dependent evolutionarily conserved pathway controls nuclear export of circular RNAs. Genes & development 32, 639–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pryor A, Tung L, Yang Z, Kapadia F, Chang TH, and Johnson LF (2004). Growth-regulated expression and G0-specific turnover of the mRNA that encodes URH49, a mammalian DExH/D box protein that is highly related to the mRNA export protein UAP56. Nucleic Acids Research 32, 1857–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sträßer K, and Hurt E (2001). Splicing factor Sub2p is required for nuclear mRNA export through its interaction with Yra1p. Nature 413, 648–652. [DOI] [PubMed] [Google Scholar]
- 97.Nakata D, Nakao S, Nakayama K, Araki S, Nakayama Y, Aparicio S, Hara T, and Nakanishi A (2017). The RNA helicase DDX39B and its paralog DDX39A regulate androgen receptor splice variant AR-V7 generation. Biochemical and Biophysical Research Communications 483, 271–276. [DOI] [PubMed] [Google Scholar]
- 98.Xu Z, Li X, Li H, Nie C, Liu W, Li S, Liu Z, Wang W, and Wang J (2020). Suppression of DDX39B sensitizes ovarian cancer cells to DNA-damaging chemotherapeutic agents via destabilizing BRCA1 mRNA. Oncogene 39, 7051–7062. [DOI] [PubMed] [Google Scholar]
- 99.He C, Li A, Lai Q, Ding J, Yan Q, Liu S, and Li Q (2021). The DDX39B/FUT3/TGFβR-I axis promotes tumor metastasis and EMT in colorectal cancer. Cell Death Dis 12, 74. 10.1038/s41419-020-03360-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Shen H, Yanas A, Owens MC, Zhang C, Fritsch C, Fare CM, Copley KE, Shorter J, Goldman YE, and Liu KF (2022). Sexually dimorphic RNA helicases DDX3X and DDX3Y differentially regulate RNA metabolism through phase separation. Molecular Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wisskirchen C, Ludersdorfer TH, Müller DA, Moritz E, and Pavlovic J (2011). Interferon-induced antiviral protein MxA interacts with the cellular RNA helicases UAP56 and URH49. Journal of Biological Chemistry 286, 34743–34751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hilbert M, Karow AR, and Klostermeier D (2009). The mechanism of ATP-dependent RNA unwinding by DEAD box proteins. Biological chemistry 390, 1237–1250. [DOI] [PubMed] [Google Scholar]
- 103.Rocak S, and Linder P (2004). DEAD-box proteins: the driving forces behind RNA metabolism. Nature reviews Molecular cell biology 5, 232–241. 10.1038/nrm1335. [DOI] [PubMed] [Google Scholar]
- 104.Pirinçal A, and Turan K (2021). Human DDX56 protein interacts with influenza A virus NS1 protein and stimulates the virus replication. Genetics and molecular biology 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Reid CR, and Hobman TC (2017). The nucleolar helicase DDX56 redistributes to West Nile virus assembly sites. Virology 500, 169–177. [DOI] [PubMed] [Google Scholar]
- 106.Rahman MM, Bagdassarian E, Ali MA, and McFadden G (2017). Identification of host DEAD-box RNA helicases that regulate cellular tropism of oncolytic Myxoma virus in human cancer cells. Scientific reports 7, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ullah R, Li J, Fang P, Xiao S, and Fang L (2022). DEAD/H-box helicases:Anti-viral and pro-viral roles during infections. Virus Research 309, 198658. 10.1016/j.virusres.2021.198658. [DOI] [PubMed] [Google Scholar]
- 108.Ma J, Rong L, Zhou Y, Roy BB, Lu J, Abrahamyan L, Mouland AJ, Pan Q, and Liang C (2008). The requirement of the DEAD-box protein DDX24 for the packaging of human immunodeficiency virus type 1 RNA. Virology 375, 253–264. 10.1016/j.virol.2008.01.025. [DOI] [PubMed] [Google Scholar]
- 109.Ma Z, Moore R, Xu X, and Barber GN (2013). DDX24 negatively regulates cytosolic RNA-mediated innate immune signaling. PLOS Pathogens 9, e1003721. 10.1371/journal.ppat.1003721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Serfecz JC, Hong Y, Gay LA, Shekhar R, Turner PC, and Renne R (2022). DExD/H Box Helicases DDX24 and DDX49 Inhibit Reactivation of Kaposi’s Sarcoma Associated Herpesvirus by Interacting with Viral mRNAs. Viruses 14, 2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Li G, Feng T, Pan W, Shi X, and Dai J (2015). DEAD-box RNA helicase DDX3X inhibits DENV replication via regulating type one interferon pathway. Biochemical and Biophysical Research Communications 456, 327–332. [DOI] [PubMed] [Google Scholar]
- 112.Loureiro ME, Zorzetto-Fernandes AL, Radoshitzky S, Chi X, Dallari S, Marooki N, Lèger P, Foscaldi S, Harjono V, and Sharma S (2018). DDX3 suppresses type I interferons and favors viral replication during Arenavirus infection. PLOS Pathogens 14, e1007125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Oshiumi H, Sakai K, Matsumoto M, and Seya T (2010). DEAD/H BOX 3 (DDX3) helicase binds the RIG-I adaptor IPS-1 to up-regulate IFN-β-inducing potential. European journal of immunology 40, 940–948. [DOI] [PubMed] [Google Scholar]
- 114.Soulat D, Bürckstümmer T, Westermayer S, Goncalves A, Bauch A, Stefanovic A, Hantschel O, Bennett KL, Decker T, and Superti-Furga G (2008). The DEAD-box helicase DDX3X is a critical component of the TANK-binding kinase 1-dependent innate immune response. The EMBO journal 27, 2135–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Cai W, Xiong Chen Z, Rane G, Satendra Singh S, Choo Z.e., Wang C, Yuan Y, Zea Tan T, Arfuso F, and Yap CT (2017). Wanted DEAD/H or alive: helicases winding up in cancers. JNCI: Journal of the National Cancer Institute 109, djw278. [DOI] [PubMed] [Google Scholar]
- 116.Hirano M, Galarza-Muñoz G, Schott G, Wang L, Nagasawa C, Antonia AL, Jain V, Yu X, Widen SG, and Briggs FB (2022). The RNA helicase DDX39B activates FOXP3 RNA splicing to control T regulatory cell fate. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Taroncher-Oldenburg G, Müller C, Obermann W, Ziebuhr J, Hartmann RK, and Grünweller A (2021). Targeting the DEAD-box RNA helicase eIF4A with rocaglates—a pan-antiviral strategy for minimizing the impact of future RNA virus pandemics. Microorganisms 9, 540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zinchuk V, Wu Y, and Grossenbacher-Zinchuk O (2013). Bridging the gap between qualitative and quantitative colocalization results in fluorescence microscopy studies. Scientific reports 3, 1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Moy RH, Gold B, Molleston JM, Schad V, Yanger K, Salzano M-V, Yagi Y, Fitzgerald KA, Stanger BZ, and Soldan SS (2014). Antiviral autophagy restricts Rift Valley fever virus infection and is conserved from flies to mammals. Immunity 40, 51–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nature methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Livak KJ, and Schmittgen TD (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2− ΔΔCT method. methods 25, 402–408. [DOI] [PubMed] [Google Scholar]
- 122.Meertens L, Hafirassou ML, Couderc T, Bonnet-Madin L, Kril V, Kummerer BM, Labeau A, Brugier A, Simon-Loriere E, Burlaud-Gaillard J, et al. (2019). FHL1 is a major host factor for chikungunya virus infection. Nature 574, 259–263. 10.1038/s41586-019-1578-4. [DOI] [PubMed] [Google Scholar]
- 123.Hackett BA, Dittmar M, Segrist E, Pittenger N, To J, Griesman T, Gordesky-Gold B, Schultz DC, and Cherry S (2019). Sirtuin inhibitors are broadly antiviral against arboviruses. MBio 10, e01446–01419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Reed LJ, and Muench H (1938). A simple method of estimating fifty per cent endpoints. American journal of epidemiology 27, 493–497. [Google Scholar]
- 125.Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. journal 17, 10–12. [Google Scholar]
- 126.Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Shah A, Qian Y, Weyn-Vanhentenryck SM, and Zhang C (2017). CLIP Tool Kit (CTK): a flexible and robust pipeline to analyze CLIP sequencing data. Bioinformatics 33, 566–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ramırez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Subgroup GPDP (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. DDX screen, Related to Figure 1.
Table S3. CHIKV genome annotations, Related to Figure 5.
Table S4. Recombinant virus sequences, Related to Figure 5 and S6.
Table S5. Oligonucleotide Sequences, Related to STAR Methods.
Table S2. CLIP reads on the CHIKV genome, Related to Figure S6.
Table S6. Host transcripts bound by DDX39A, Related to Figure S6.
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.