

Abstract
Proteins and enzymes generally achieve their function by creating well-defined 3D architectures that pre-organize reactive functionalities. Mimicking this approach to supramolecular preorganization is leading to the development of highly versatile artificial chemical environments, including new biomaterials, medicines, artificial enzymes, and enzyme-like catalysts. The use of beta-turn and alpha-helical motifs is one approach that enables the precise placement of reactive functional groups to enable selective substrate activation and reactivity/selectivity that approaches natural enzymes. Our recent work has demonstrated that helical peptides can serve as scaffolds for pre-organizing two reactive groups to achieve enzymelike catalysis. In this study, we used CYANA and AmberTools to develop a computational approach for determining how the structure of our peptide catalysts can lead to enhancements in reactivity. These results support our hypothesis that the bifunctional nature of the peptide enables catalysis by pre-organizing the two catalysts in reactive conformations that accelerate catalysis by proximity. We also present evidence that the low reactivity of monofunctional peptides can be attributed to interactions between the peptide-bound catalyst and the helical backbone, which are not observed in the bifunctional peptide.
Keywords: Peptide catalyst, CYANA, AmberTools, enzyme-like catalysts, alpha-helical peptides
Abstract Graphic:
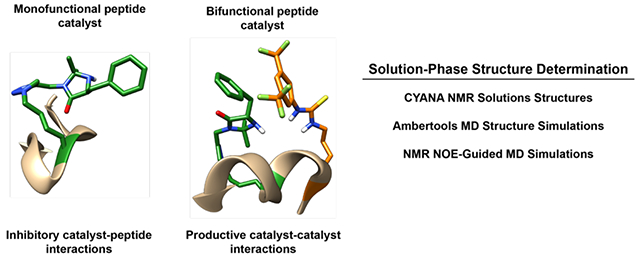
1. INTRODUCTION
The precisely tuned functions of proteins and enzymes are intimately connected to the exquisite control nature achieves over supramolecular 3-D structure. Significant interest has been invested in translating this strict control over the 3D structure from biological systems into artificial environments, including for new biomaterials and medicines,1–4 artificial enzymes,5–7 and enzyme-like catalysts.8–11 In the field of peptide catalysis, the use of beta-turn and alpha-helical motifs can enable the precise placement of reactive functional groups in close proximity to enable reactivity and selectivity that approaches that of natural enzymes.11 For example, work by Gellman has shown that having two different amine groups one turn apart on a helical alpha-beta-peptide can facilitate highly selective macrocyclization via condensations of aldehydes (Figure 1).12–13 Ball and coworkers have also used helical peptides to enable protein binding and selective protein modifications with dirhodium catalysts.14 Our group has utilized a helical peptide to pre-organize multiple catalysts to facilitate novel approaches to cooperative catalysis in Diels-Alder, indole alkylation, and nitroolefin addition reactions (Figure 1).15–16 We are particularly interested in using peptide secondary structures such as α-helices to develop highly efficient cooperative catalysts. Our efforts in this area,15–16 as well as the efforts of others,11 span the divide between biological and synthetic chemistry. Moreover, they require the development of new computational and mechanistic tools that investigate the impact of structure on cooperative reactivity and selectivity.4–7
Figure 1.
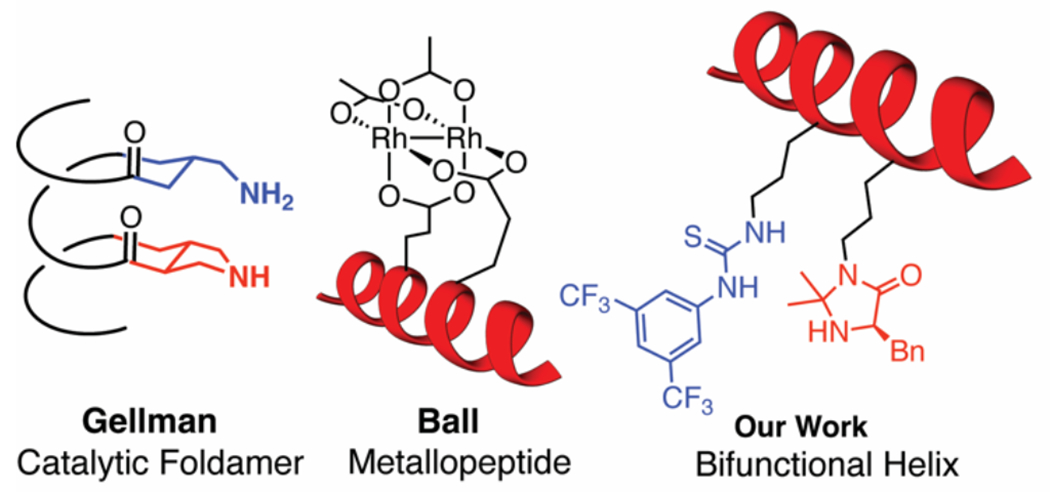
Examples of helical polypeptide-based catalysts.
The defining aspect of macromolecular catalysts such as our bifunctional peptides is that the well-defined, yet flexible nature of the peptide backbone can pre-organize catalytic groups to maximize reactivity. In addition, the chiral nature of a peptide scaffold can have a dramatic impact on optimizing the enantioselectivity of the desired transformation.14,16 We recently demonstrated that the placement of two reactive groups, an imidazolidinone catalyst, and a thiourea hydrogen bonding group, in proximity of each other on a peptide backbone leads to the recruitment and pre-organization of two substrates and enzyme-like acceleration of catalysis (Figure 2).15 The chiral nature of the peptide backbone also let to an increase in the enantioselectivity of the reactions. The rational design of such catalysts requires an in-depth understanding of the 3D structure of the peptide backbone and how it influences or interacts with the attached catalysts. To gain such an understanding, we set out to develop a modeling approach that could provide insights into how the peptide scaffold was interacting with the peptide-bound catalysts and influencing reactivity. To achieve this, we used NMR Nuclear Overhauser Effect spectroscopy (NOE) experiments, combined with CYANA17–21 and AmberTools22–23 calculations to model the solution-phase structures of our catalysts. Our studies compliment previous computational efforts to model peptide-based catalysts that focused on understanding the folded structure of short (3-4 residues) peptides and the importance of structure in catalysis.24–28 Our results support our hypothesis that the helical structure is present in solution and important for bifunctional catalysis. We also find that the low reactivity of our monofunctional imidazolidinone peptide likely results from non-productive interactions between the peptide and the catalyst that change the backbone structure and prevent catalyst interactions with the substrate. These studies are aiding in the design of new bifunctional catalysts capable of achieving improved cooperativity.
Figure 2.
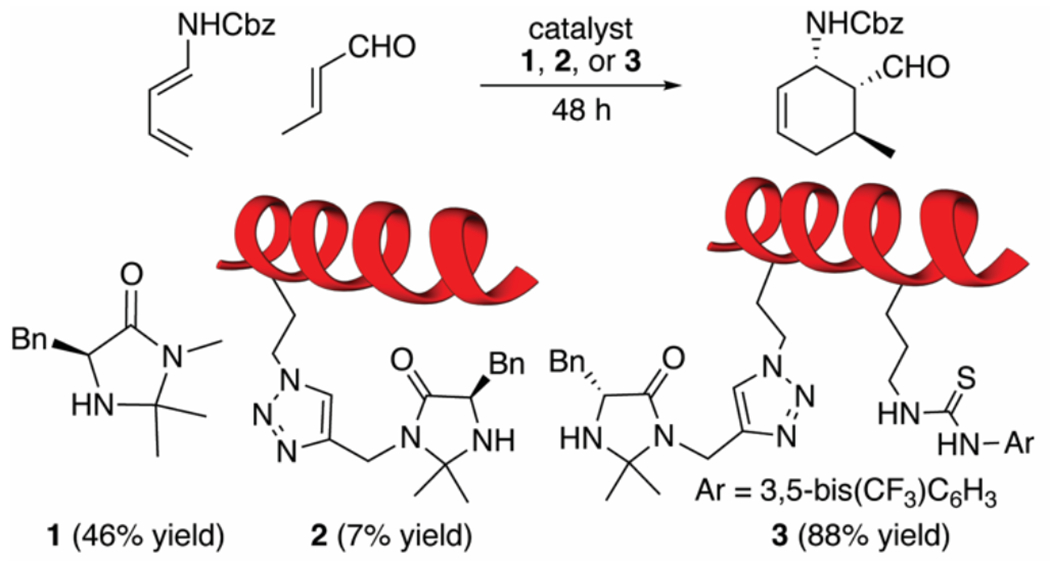
Catalyst structures and their reactivity in Diels-Alder reactions. Primary peptide sequence: AcNH-VXLBVXLBVAL-NH2; B=aminoisobutyric acid, X = Ala, or catalyst-modified reside.
2. RESULTS AND DISCUSSION
Our goal of modeling the structures of our peptide-based catalysts relies on a two-prong approach, where we used experimental NMR NOE data combined with both CYANA 2.1 calculations and AmberTools MD simulations. CYANA uses conformational constraints from nuclear magnetic resonance (NMR) experiments and an angle dynamics algorithm to determine the in-solution peptide structure.17–21 CYANA provides a torsional angle space for our structures but is limited because it does not account for bond length variability and electrostatic interactions. The benefit of CYANA-based computations is that they have low computational costs and provide rapid access to possible structures by simplifying structure determination to a few parameters. AmberTools, a molecular dynamic (MD) suite,22 provides a comprehensive model that complements CYANA’s deficiencies but is more computationally expensive. In addition, AmberTools-based structures can often become stuck in local minima,23 which can provide incorrect overall structures for our peptide catalysts. By comparing the results from CYANA with AmberTools models, we create a working model of the solution structures of our peptide catalysts, which provides an avenue to understand how peptide structure can impact catalysis in our system.
In our previous report on the development of bifunctional helical peptide catalysts,15 we found that the reactivity of imidazolidinone catalyst 1 was vastly different from that of peptide-based catalysts 2 and 3 (Figure 2). Mono-functional peptide 2 had much lower reactivity than 1 alone (7% vs 46% yield in 48 h), while bi-functional peptide 3 showed dramatically improved catalysis (88% yield). Non-functionalized peptides (containing no imidazolidinone catalyst) were unreactive. We also tested a non-helical bifunctional peptide catalyst (not shown) where the helical structure had been disrupted by introduction of helix-breaking proline residues. This non-helical peptide showed little to no catalysis, demonstrating that catalysis is supported by the helical backbone structure. From these studies, we hypothesized that the bifunctional peptide can bind both substrates in proximity, thus accelerating the reaction via cooperative effects. To obtain a better understanding of how peptide structure is influencing catalysis, we set out to develop a systematic approach to understanding the solution structure of our peptide catalysts.
2.1. CYANA Calculations.
Our first goal was to investigate the solution-phase structure of our peptide catalyst using the CYANA modeling software.17–21 Because our peptide catalysts are small (11 residues), we could use CYANA without the need to employ 14C and 15N isotopically labeled amino acids. We used a range of NMR experiments to fully assign the catalyst structures and the relevant NOE signals (see supporting information for details). We then used these assignments to obtain a series of probable calculated structures of our peptides using CYANA. We did not find the use of the CYANA software particularly intuitive and a tutorial of how we performed these calculations is included in the supporting information. For the non-natural, catalyst-containing side chains, we used Cylib 2.0 to generate the required force field parameters.29 Each of the computed structures for peptides 2 and 3 were evaluated using MolProbity30, a molecular structure evaluation and validation program for NMR structures. This software scores each structure by a series of evaluation tests based on a database of known structures.
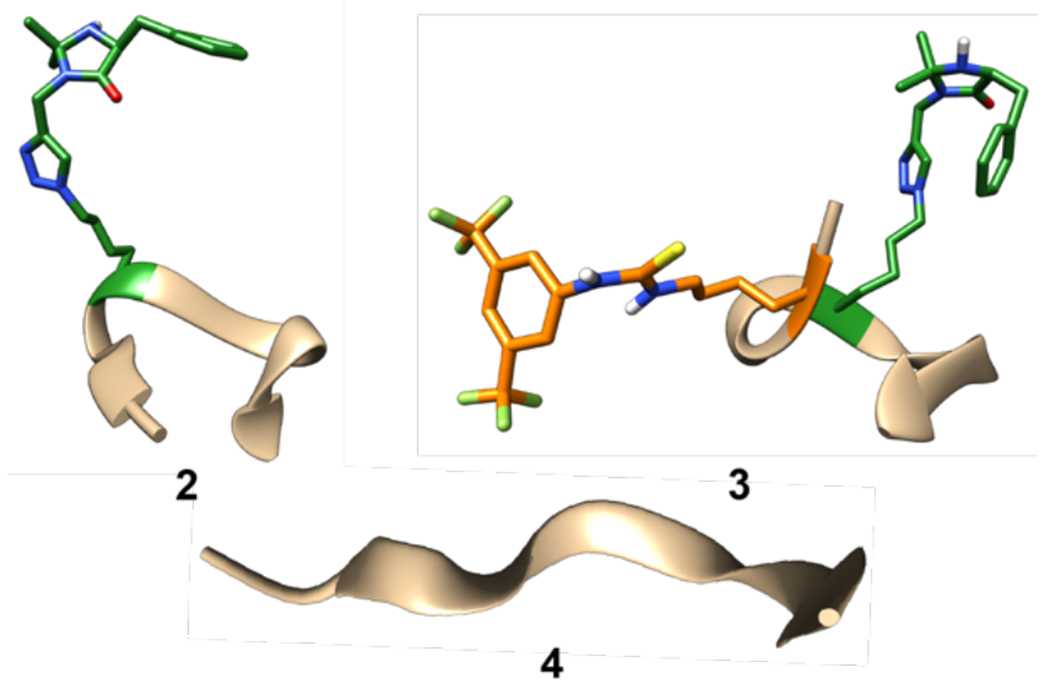
From our preliminary CYANA-computed structures (Figure 3), we already see indications of how structure can affect reactivity. For example, Figure 3 shows one of the CYANA structures of our mono-functional peptide catalyst 2 with the best MolProbity score. The computed structures show an overall loosely helical backbone with a reverse turn in the middle of the backbone. Moreover, from the Ramachandran plots31,32 of 2 (see supporting information), a wide range of dihedral angles (Phi and Psi) are in the helix and reverse helix regions. The reverse turn structure could be caused by attachment of the catalyst to peptide backbone, as a similar reverse turn is observed in the bifunctional catalyst 3 (Figure 3). The CYANA structure of 2 also shows the imidazolidinone interacting with the peptide backbone through H-bonding interactions. This interaction between the imidazolidinone and the peptide backbone could help explain the very low reactivity of monofunctional catalyst 2 when compared to catalyst 1. The imidazolidinone catalyst is tied up in stabilizing interactions with the peptide backbone, making it inaccessible to interact with substrates and undergo catalysis. Thus, minimizing these types of interactions with the backbone could be a productive method for improving catalysis (vide infra).
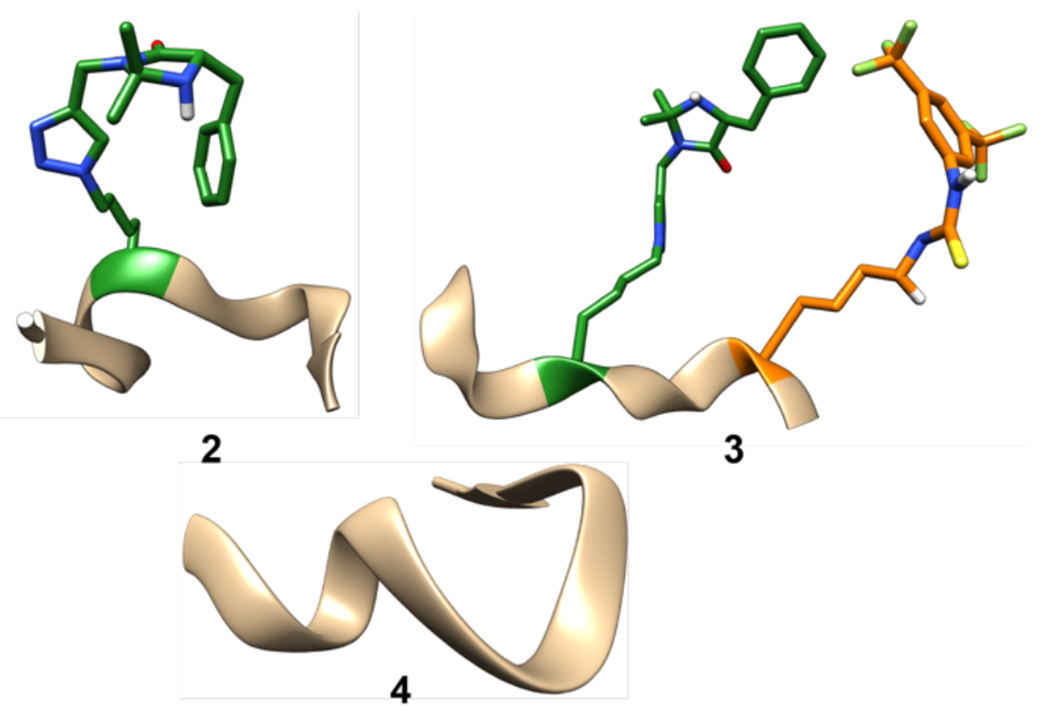
Figure 3.

CYANA-calculated solution-phase structures of peptide 2 and peptide 3. NMR experiments were run in DMSO-d6 at room temperature. All amino acid side chains were omitted for clarity except the thiourea catalyst (orange) and the imidazolidinone catalyst (green). All images of our models were created using Chimera.33
In contrast to the monofunctional peptide 2, the structure of bi-functional peptide catalyst 3 suggests a tighter, more helical peptide backbone (Figure 3). The Ramachandran plots for 3 show a wide range of dihedral angles like 2, but with more groupings in the helical regions that suggests a tighter helical structure. As with peptide 2, a reverse turn is present in the middle, likely caused by the catalyst side chain. With peptide 3, we see two different converging structures from the different CYANA calculations. The first structure shows the two catalysts are interacting, causing the helix to fold slightly inward (3, Figure 3). The second structure has the catalyst not interacting with each other and the backbone forming a stronger helix instead (3’). These two structures show that the catalysts are spending most of their time interacting with each other or the solvent and not interacting with the peptide backbone. This means that both catalysts are more available to interact with their respective substrates, bring them into proximity, and facilitate catalysis. This observation is consistent with the observed reactivity of peptide 3, which displays higher catalysis than both monofunctional catalysts 2 and non-peptide catalyst 1. Importantly, introducing these types of interactions between catalysts and functional side chains, such as by introduction of H-bonding residues (Gln, Asn, Asp, Ser, etc), could be a strategy for helping enhance catalyst activity by minimizing interactions of the catalyst with the peptide backbone itself.
Overall, the CYANA solution-phase structures of our peptides provided preliminary insights into how the structure of our peptide catalyst can influence the observed reactivity. One remaining challenge from these calculations is that almost all the CYANA structures showed high Van der Waals collisions. This means that CYANA is not able to account for steric interactions that could change the computed structures of our peptides. To address this concern, we next turned to molecular dynamics simulations using AmberTools to correct and relax the solution-phase structures of our peptides.
2.2. AmberTools Calculations.
Our next goal was to compare our CYANA structures with molecular dynamics simulations obtained using AmberTools.22,23 To accomplish this, we first pursued two different approaches to generate MD simulations of our peptide catalysts without any physical data input. In the first approach, we made our peptide catalysts in a linear, non-folded confirmation, heated the structures in silico to eliminate any structural bias, then allowed the structures to relax into their most desired conformation. Second, we initiated the MD simulations with the peptides already folded into a helical or beta turn secondary structure. We then allowed the MD simulations to relax these starting structures into the most favored conformations. This allowed us to avoid local minima structures that we observed with the first method. Finally, we performed NOE-restricted MD simulations using the NOE data and CYANA structure to restrain the MD simulations. This final combination of NMR restraints and MD simulations provided a more reliable and detailed structure of our peptide catalysts and addressed some of the limitations of the CYANA modeling approach.
To perform our MD simulations on our non-natural peptide catalysts, we first had to develop force field parameters for our non-natural catalyst side chains. The Ambertools force fields34 are more descriptive than CYANA’s angle dynamics algorithm and can be more accurate if performed correctly. The accuracy of these force fields dramatically depends on the parameters or variables of the force field equations. However, more accurate force fields require more resources and time to develop highly accurate parameters. To achieve moderately accurate parameters without requiring excessively long calculations, our group used a combination of AmberTools ff14SB forcefields,35 and general force fields (GAFF2) with AM1-BCC charges.36 The later forcefields were used for our non-natural, catalyst-containing amino acid side chains. Because our peptide catalysis occurs in organic solvents, we used organic solvent models in our MD simulations that had been previously developed and tested 37–41.
2.2.1. AmberTools Approach One.
Our first MD approach uses a classic sampling method by starting with a linear conformation of the peptide backbone and allowing the system to evolve naturally over time. We used our previously reported CAN program to build coordinate files, including for catalyst-containing non-natural amino acids.37 We then heated the system above 500 K, allowed the system to cool, and then ran the simulation at the target temperature (273 K). Each calculation was allowed to run for 1200 ns duration. From these simulations, we sorted the top clusters using a bottom-up hierarchy algorithm22 and visualized some of the best MolProbity29 scored structures (Figure 4). This was the same method used to obtain our most representative CYANA structures. With monofunctional catalyst 2, we observed an overall loosely helical backbone with a reverse helix near the catalyst structure, like the CYANA structures. However, an essential difference between the CYANA structures and these MD structures is that they do not always anneal properly, even when given a generous time to fold (1200 ns). This occurs because the structures become stuck in a local minimum. Other methods that were developed to address this challenge, such as replica exchange,43–45 were attempted with our peptides catalysts, but similar results occurred despite the increase in computational cost. Figure 4 shows an MD structure of 2 with a similar backbone structure as obtained using CYANA, but the frequency of structures like this was low in our runs. Nevertheless, the Ramachandran plot of the MD structure for 2 does show the torsional angles of the amino acids in more favorable regions than the CYANA structures, which was the overall goal of our Ambertools MD simulations (see supporting information for details).
Figure 4.
Ambertools-generated structures with high MolProbity scores for peptides 2, 3, and non-functionalized peptide 4. Computations were run with a nitromethane solvent gradient at −10 °C for ~1200 ns. All natural amino acid side chains were omitted for clarity except the thiourea catalyst (orange) and the imidazolidinone catalyst (green).
As with monofunctional peptide 2, we were able to locate AmberTools MD structures for 3 with the same backbone shape as seen in our CYANA structure (helical ends and a reverse twist in the middle, Figure 4). This helical structure is one of the best MolProbity-scored structures from our runs. However, most of the structures did not converge and anneal properly, and an even lower frequency of helical structures was observed. This low frequency of helical structures could result from the high complexity of the two-catalyst system, which may need more time to evolve into the lower energy states. The Ramachandran plot of catalyst 3 does show many tortional angles in the helical regions, but also suggests significant linear character.
To benchmark our Ambertools structures, we next performed MD simulations on the non-functionalized peptide backbone (4) containing only natural amino acid side chains (catalyst residues were replaced with alanines). This peptide is a derivative of a known peptide (Boc-Val-Ala-Leu-Aib-Val-Ala-Leu-OMe) reported by Balaram and which is known to have significant helical character.46 This benchmark peptide would increase accuracy by using only ff14SB force fields when calculating the peptide structure. Figure 4 also shows one of the best MolProbity-scored structures of 4, which does display significant helical character. However, this structure is not as helical as peptides 2 and 3. Thus, it may be that our catalyst side chains have significant helix inducing character. The Ramachandran plot of 4 shows similar tortional angles to 2 and 3, but in less favorable regions.
When comparing the structures obtained from Ambertools vs CYANA, it is important to note that the computed structures for 2 and 3 from both methods have similar characteristics. However, CYANA structures show a greater definition in the helical structure of the backbone and AmberTools showes better energy profiles in its Ramachandran plots. Thus, we desired to improve upon these results by preorganizing the peptide backbones into helical structures prior to initiating the MD simulation to help minimize the number of simulations that got trapped in local minima.
2.2.2. AmberTools Approach Two.
For our second approach, we arranged each peptide in four different starting configurations (alpha-helix, beta-fold, 3-10 helix, and π-helix) to initiate each MD simulations. Our hypothesis was that preorganizing the peptides in this fashion would help avoid the structures getting stuck in local minima and provide more representative examples of low energy conformations. Each peptide simulation was heated to a target temperature (273 K) and run for ~400 ns. After the simulation was completed, we generated top clusters using the same bottom-up hierarchy algorithm and MolProbity scoring. The top MD structures for each of our peptides is shown in Figure 5. The structure of catalyst 2 shows a helical backbone with a reverse helix as observed previously, but these structures anneal better than with the approach one. The best starting structures for peptide 2 were the 3-10 helix or the alpha helix conformations. Thus, preorganizing the peptide into a proposed low energy conformation helps avoid long computational times (400 vs 1200 ns) and provides more representative low energy structures. The one drawback is that some notion of the low energy conformation of each peptide needs to be known to properly setup each run. In our case, we knew that our peptides existed in some form of a helical backbone due to the circular dichroism (CD) spectra obtained for peptides 2 and 3.15 However, our CD studies only provided information about the average structure of the peptides, and does not provide specific information about how helical the peptides are or what that helical structure looks like. The Ramachandran plot of 2 also shows better favorable regions than our CYANA structures. One additional feature of the new MD structure of peptide 2 is that we observe the imidazolidinone catalyst (green) interacting with the backbone of the catalyst in most structures. This result is consistent with the CYANA structure of peptide 2, which also indicates favorable interactions with the helix backbone and could be the cause of the low reactivity with peptide 2.
Figure 5.
Ambertools-generated structures with high MolProbity scores for peptides 2, 3, and non-functionalized peptide 4. Computations were run with a nitromethane solvent gradient at −10 °C for ~1200 ns. All natural amino acid side chains were omitted for clarity except the thiourea catalyst (orange) and the imidaz-olidinone catalyst (green).
The MD structure of peptide 3 was also somewhat improved when the starting conformation was either a 3-10 or an alpha helix (Figure 5). However, these structures were not as much improved as the monofunctional catalyst. The two-catalyst system (3) still didn’t fold properly most of the time. The frequency of correctly folded structures did increase, but not as much as with peptide 2. The structures obtained via method two also had a higher frequency of forming a full alpha helix without the reverse helix (not shown) as was observed in the CYANA structures. Thus, the fully alpha helical backbone may be a low energy conformation that the peptide accesses in solution that is similar in energy to the structure observed with CYANA and method one. Both helical structures obtained from this method have more favorable regions in their Ramachandran plots than the CYANA structure, indicating that they are likely lower in energy and more stable.
When we used approach two to calculate the structure of non-functionalized peptide 4, we found a lower frequency of helical structures than expected (Figure 5). This result confirms the results obtained with method one, which suggested that the backbone peptide wasn’t as structured as the monofunctional or bifunctional peptides 2 and 3. For peptide 4, the best structures came from the preorganized alpha helix starting point. However, the Ramachandran plots indicate few fully helical structures. This supports our hypothesis that the incorporation of catalysts, particularly in the two-catalyst peptide (3), helps improve the helical structure of our peptide.
2.2.3. AmberTools Approach Three.
Our final approach to MD simulations employed NMR-restraints to direct the backbone folding. These simulations use the information from NMR NOE experiments (Similar to CYANA) and force the structures into a predetermined configuration with some ability to relax. For all structures, we started the peptides in linear configurations, and used the restraints to help guide proper folding. These NMR-restrained simulations help the structure to start in and maintain a better configuration during the simulation. It also helped the simulation to avoid local minima. Each peptide was heated up to target temperature and ran for ~400 ns. After the simulation was completed, we generated the top clusters as described previously. The best scoring structures for peptides 2 and 3 are shown in Figure 6.
Figure 6.

NMR-restrained MD simulated structures with the highest MolProbity scores of peptides 2, and 3. Calculations were run in DMSO at room temperature for about ~400 ns. All natural amino acid side chains were omitted for clarity except the thiourea catalyst (orange) and the imidazolidinone catalyst (green).
From this NOE-guided approach, peptides 2 and 3 showed similar secondary structure as the structures obtained using CYANA. The main difference between these new structures and the CYANA structures for peptide 2 is that its Ramachandran plots show improvements in the number of angles that are in favorable regions for alpha helical structures. Likewise, the MolProbity scoring for the NMR-guided structure of 2 is much improved over the CYANA structure. These improvements in the structures of our peptide catalysts likely result from the improved forcefields used by the Ambertools MD simulations over the CYANA simulations. For peptide 3, however, the CYANA structure, based on the Ramachandran plot and the Molprobity scoring, had slightly better angles for a helical peptide and a higher scoring structure. Thus, both the CYANA and Ambertools calculations, guided by NMR NOE data, provide important tools for investigating the best solution structures of peptide-based catalysts.
Two important effects are confirmed for our peptide catalysts based on these most refined structures. First, the bifunctional peptide (3) has a well-defined helical structure that places the two catalysts into proximity. This facilitates interactions between the two catalysts that we believe are important for catalysis and prevents the catalysts from interacting with the peptide backbone. Second, favorable interactions between the imidazolidinone and the peptide backbone are observed with monofunctional peptide 2. These catalyst-peptide interaction are likely the cause of the low reactivity of peptide 2, when compared to the non-peptide bound imidazolidinone catalyst 1 (see Figure 2). To validate the importance of this interaction, we performed a time lapse study on the structure of 2 from approach three. What we observed is that over 600 ns, the imidazolidinone oxygen remains within two angstroms of the nearby valine alpha-hydrogen, which suggests that this folded structure persists over time (see Figure S1 in the supporting information).
3. CONCLUSIONS
The overarching goal of this work was to use MD simulations to investigate the solution-phase structure of short, non-natural peptide-based catalysts. Because this class of catalyst relies on well-defined 3-D arrangements of catalysts, understanding the structure of the peptide can help guide the design of new catalysts. We have demonstrated that the use of NMR-NOE restraints for either CYANA or Ambertools MD simulations provides the most reasonable solution-phase structures of our peptide catalysts. We employed Ramachandran plot analysis and MolProbity scoring to validate our structures to ensure that they were representative of the solution-phase structure. When comparing these two approaches, the Ambertools MD simulations provided higher scoring (MolProbity) structures that also showed better structural alignment in the Ramachandran plots. However, both methods provided similar overall structures, which confirms that the folded structures are likely those favored in solution. The structures obtained are also in agreement with previously obtained circular dichroism measurements, which suggested helical structures in solution.
We also used two different approaches to obtaining MD-simulated structures with Ambertools that did not rely on physical data such as NMR NOE interactions. Of the two methods reported, preorganizing the peptide into a folder structure (rather than a linear structure) before initiating the MD simulation provided better structures and avoided the peptide getting stuck in local minima. Thus, having some idea of the folded structure of our peptide catalysts was useful in obtaining potential structures without having to run excessively long computational runs.
By using MD simulations to investigate the solution phase structure of our peptide catalysts, we were also able to observe interactions that we believe are important for and influence catalysis. For example, our monofunctional peptide catalyst (2) experiences interactions between the catalyst and the peptide backbone that likely lower catalyst availability and reactivity. This result is consistent with the observed low reactivity with peptide 2. Thus, changes to the backbone structure that minimize this type of interaction could be important to enhance catalyst activity. We also showed that the bifunctional peptide (3) experiences interactions between that two catalysts that prevent interactions with the peptide backbone and may be essential for the observed cooperative catalysis with this peptide.
We believe that these results provide a useful framework for using MD simulations to understand the structure and reactivity of peptide-based catalysts that contain non-natural catalytic units. Our method can also be used to understand how interactions between the backbone peptide and the catalysts can influence reactivity. Our approach also provides several avenues for obtaining reasonable MD structures, depending on whether physical interactions data (NMR NOE data) is available for the peptide catalyst of interest. Our future work with these tools will involve the design of new catalytic systems that enhance cooperative effects by maximizing intercatalyst interactions and minimizing interactions between catalysts and the peptide. This application would allow a more direct and custom approach to discovering new and faster multifunctional catalysts.
Supplementary Material
ACKNOWLEDGMENT
We would like to thank the National Institutes of Health NIGMS for funding this work (1R15GM134476- 01). The authors also thank Brigham Young University and the Office of Research Computing, especially the Fulton Supercomputing Lab. The authors also acknowledge the Brigham Young University Simmons Research Endowment for partial funding of this work.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Computational methods (PDF)
Xyz coordinate files for key structures (.xyz).
REFERENCES
- 1.Hendrikse SIS; Contreras-Montoya R; Ellis AV; Thordarson P; Steed JW Biofunctionality with a twist: the importance of molecular organisation, handedness and configuration in synthetic biomaterial design. Chem. Soc. Rev 2022, 51, 28–42. [DOI] [PubMed] [Google Scholar]
- 2.Gray VP; Amelung CD; Jahan Duti I; Laudermilch EG; Letteri RA; Lampe KJ Biomaterials via peptide assembly: Design, characterization, and application in tissue engineering. Acta Biomater. 2022, 140, 43–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hamley IW Small Bioactive Peptides for Biomaterials Design and Therapeutics. Chem. Rev 2017, 117, 14015–14041. [DOI] [PubMed] [Google Scholar]
- 4.Tilocca A Molecular Dynamics Methods for Modeling Complex Interactions in Biomaterials. Methods Mol. Bio 2011, 285–301. [DOI] [PubMed] [Google Scholar]
- 5.de Pina Mariz B; Carvalho S; Batalha IL; Pina AS, Artificial enzymes bringing together computational design and directed evolution. Org. Biomol. Chem 2021, 19, 1915–1925. [DOI] [PubMed] [Google Scholar]
- 6.Damborsky J; Brezovsky J Computational Tools for Designing and Engineering Enzymes. Curr. Opin. Chem. Bio 2014, 19, 8–16. [DOI] [PubMed] [Google Scholar]
- 7.Nanda V; Koder RL Designing Artificial Enzymes by Intuition and Computation. Nature chemistry 2010, 2, 15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Akagawa K; Kudo K Development of Selective Peptide Catalysts with Secondary Structural Frameworks. Acc. Chem. Res 2017, 50, 2429–2439. [DOI] [PubMed] [Google Scholar]
- 9.Rink WM; Thomas F De Novo Designed a-Helical Coiled-Coil Peptides as Scaffolds for Chemical Reactions. Chem. Eur. J 2018, 25, 1665–1677. [DOI] [PubMed] [Google Scholar]
- 10.Zozulia O; Dolan† MA; Korendovych IV Catalytic Peptide Assemblies. Chem Soc Rev. 2018, 47, 3621–3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Girvin ZC; Gellman SH Foldamer Catalysis. J. Am. Chem. Soc 2020, 142, 17211–17223. [DOI] [PubMed] [Google Scholar]
- 12.Andrews MK; Liu X; Gellman SH Tailoring Reaction Selectivity by Modulating a Catalytic Diad on a Foldamer Scaffold. J. Am. Chem. Soc 2022, 144, 2225–2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Girvin ZC; Andrews MK; Liu X; Gellman SH Foldamer-templated catalysis of macrocycle formation. Science 2019, 366, 1528–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ball ZT Designing Enzyme-like Catalysts: A Rhodium(II) Metallopeptide Case Study. Acc. Chem. Res 2013, 46, 560–570. [DOI] [PubMed] [Google Scholar]
- 15.Kinghorn MJ; Valdivia-Berroeta GA; Chantry DR; Smith MS; Ence CC; Draper SRE; Duval JS; Masino BM; Cahoon SB; Flansburg RR; Conder CJ; Price JL; Michaelis DJ Proximity-Induced Reactivity and Product Selectivity with a Rationally Designed Bifunctional Peptide Catalyst. ACS Catal. 2017, 7, 7704–7708. [Google Scholar]
- 16.Wayment AX; Rodriguez Moreno M; Jones CJ; Smith GJ; Jarman P; Garcia Morin NJ; Coombs MJ; Parkman JA; Barlow CD; Allington Smith S; Burt SR; Michaelis DJ Optimizing the Local Chemical Environment on a Bifunctional Helical Peptide Scaffold Enables Enhanced Enantioselectivity and Cooperative Catalysis. Org. Lett 2022, 24, 2983–2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herrmann T; Güntert P; Wüthrich K. Protein NMR Structure Determination with Automated NOE Assignment Using the New Software Candid and the Torsion Angle Dynamics Algorithm DYANA. J. Mol. Bio 2002, 319, 209–227. [DOI] [PubMed] [Google Scholar]
- 18.Jee J; Güntert P Influence of the completeness of chemical shift assignments on the NMR structures obtained with automated NOE assignment. J. Struct. Funct. Genomics, 2003, 4, 179–189. [DOI] [PubMed] [Google Scholar]
- 19.Buchner Lena, and Güntert Peter. Systematic Evaluation of Combined Automated Noe Assignment and Structure Calculation with Cyana. J. Biomol. NMR 2015, 62, 81–95. [DOI] [PubMed] [Google Scholar]
- 20.Protein NMR Techniques, 2nd Ed.; Downing AK, Ed.; Humana Press, 2004. [Google Scholar]
- 21.Güntert P; Buchner L Combined Automated NOE Assignment and Structure Calculation with Cyana.” J. Biomol. NMR 2015, 62, 453–471. [DOI] [PubMed] [Google Scholar]
- 22.Case DA, Belfon K, Ben-Shalom IY, Brozell SR, Cerutti DS, Cheatham TE III., Cruzeiro VWD, Darden TA, Duke RE, Giam-basu G, Gilson MK, Gohlke H, Goetz AW, Harris R, Izadi S, Izmailov SA, Kasavajhala K, Kovalenko A, Krasny R, Kurtzman T, Lee TS, LeGrand S, Li P, Lin C, Liu J, Luchko T, Luo R, Man V, Merz KM, Miao Y, Mikhai-lovskii O, Monard G, Nguyen H, Onufriev A, Pan F, Pantano S, Qi R, Roe DR, Roitberg A, Sagui C, Schott-Verdugo S, Shen J, Simmerling CL, Skrynnikov NR, Smith J, Swails J, Walker RC, Wang J, Wilson L, Wolf RM, Wu X, Xiong Y, Xue Y, York DM, Kollman PA, AMBER 2020, University of California, San Francisco: 2020. [Google Scholar]
- 23.Purawat S; Ieong PU; Malmstrom RD; Chan GJ; Yeung AK; Walker RC; Altintas I; Amaro RE A Kepler Workflow Tool for Reproducible AMBER GPU Molecular Dynamics. Biophys. J 2017, 112, 2469–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Metrano AJ; Abascal NC; Mercado BQ; Paulson EK ; Hurtley AE; Miller SJ Diversity of Secondary Structure in Catalytic Peptides with beta-Turn-Biased Sequences. J. Am. Chem. Soc 2017, 139, 492–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rigling C; Kisunzu JK; Duschmalé J; Häussinger D; Wiesner M; Ebert M-O; Wennemers H Conformational Properties of a Peptidic Catalyst: Insights from NMR Spectroscopic Studies. J. Am. Chem. Soc 2018, 140, 10829–10838. [DOI] [PubMed] [Google Scholar]
- 26.Metrano AJ; Chinn AJ; Shugrue CR; Stone EA; Kim B; Miller SJ Asymmetric Catalysis Mediated by Synthetic Peptides, Version 2.0: Expansion of Scope and Mechanisms. Chem. Rev 2020, 120, 11479–11615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stone EA; Hosseinzadeh vP.; Craven TW; Robertson MJ; Han Y; Hsieh S-Y; Metrano AJ; Baker D; Miller SJ Isolating Conformers to Assess Dynamics of Peptidic Catalysts Using Computationally Designed Macrocyclic Peptides. ACS Catalysis 2021, 11, 4395–4400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yan XC; Metrano AJ; Robertson MJ; Abascal NC; Tirado-Rives J; Miller SJ; Jorgensen WL Molecular Dynamics Simulations of a Conformationally Mobile Peptide-Based Catalyst for Atroposelective Bromination. ACS Catalysis 2018, 8, 9968–9979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maden Yilmaz E; Güntert P NMR structure calculation for all small molecule ligands and non-standard residues from the PDB Chemical Component Dictionary. J. Biomol. NMR 2015, 63, 21–37. [DOI] [PubMed] [Google Scholar]
- 30.Davis IW; Leaver-Fay A; Chen VB; Block JN; Kapral GJ; Wang X; Murray LW; Arendall III WB; Snoeyink J; Richardson JS; Richardson DC MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007, 35, W375–W383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramachandran GN; Ramakrishnan C; Sasisekharan V Stereochemistry of polypeptide chain configurations, J. Mol. Biol 1963, 7, 95–99. [DOI] [PubMed] [Google Scholar]
- 32.Ramachandraw. PyPI, Ramachandran plot tool. https://pypi.org/project/RamachanDraw/.
- 33.Pettersen EF; Goddard TD; Huang CC; Couch GS; Greenblatt DM; Meng EC; Ferrin TE UCSF Chimera - a visualization system for exploratory research and analysis. J. Comput. Chem 2004, 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- 34.Hornak V; Abel R; Okur A; Strockbine B; Roitberg A; Simmerling C Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 2006, 65, 712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tian C; Kasavajhala K; Belfon KAA; Raguette L; Huang H; Migues AN; Bickel J; Wang Y; Pincay J; Wu Q; Simmerling C ff19SB: Amino-Acid-Specific protein backbone parameters trained against quantum mechanics energy surfaces in solution. J. Chem. Theory Comput 2020, 16, 528–552. [DOI] [PubMed] [Google Scholar]
- 36.Aronica PG; Fox SJ; Verma CS Comparison of Charge Derivation Methods Applied to Amino Acid Parameterization. ACS Omega 2018, 3, 4664–4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Caleman C; van Maaren PJ; Hong M; Hub JC; Costa LT; van der Spoel D Force Field Benchmark of Organic Liquids: Density, Enthalpy of Vaporization, Heat Capacities, Surface Tension, Isothermal Compressibility, Volumetric Expansion Coefficient, and Dielectric Constant, J. Chem. Theory Comput 8, 61–74 (2012). DOI [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fischer N; van Maaren PV; Ditz JC; Yildirim A; van der Spoel D Properties of organic liquids when simulated with long-range lennard-jones interactions, J. Chem. Theory Comput 2015, 11, 2938–2944. [DOI] [PubMed] [Google Scholar]
- 39.van der Spoel D; Ghahremanpour MM; Lemkul J Small Molecule Thermochemistry: A Tool for Empirical Force Field Development. J. Phys. Chem. A 2018, 122, 8982–8988. [DOI] [PubMed] [Google Scholar]
- 40.Zhang A Haiyang and Tan Tianwei and Feng Wei and van der Spoel David Molecular Recognition in Different Environments: β-Cyclodextrin Dimer Formation in Organic Solvents. J. Phys. Chem. B 2012, 116, 12684–12693. [DOI] [PubMed] [Google Scholar]
- 41.Ghahremanpour MM; van Maaren P; van der Spoel D The Alexandria Library: A Quantum-Chemical Database of Molecular Properties for Force Field Development. Sci. Data 2018, 5, 180062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Parkman JA; Barksdale CA; Michaelis DJ CAN: A new program to streamline preparation of molecular coordinate files for molecular dynamics simulations. J. Comput. Chem 2021, 42, 2031–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jaegil Kim J; Keyes T; Straub JE Generalized Replica Exchange Method. J. Chem. Phys 2010, 132, 224107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sugita Y; Okamoto Y Replica-exchange molecular dynamics method for protein folding. Chem. Phys Lett 1999, 314, 141–151. [Google Scholar]
- 45.Ostermeir K; Zacharias M Advanced replica-exchange sampling to study the flexibility and plasticity of peptides and proteins. Biochimica et Biophysica Acta 2013, 1834, 847–853. [DOI] [PubMed] [Google Scholar]
- 46.Karle IL; Flippen-Anderson JL; Uma K; Balaram P Unfolding of an α-helix in peptide crystals by solvation: Conformational fragility in a heptapeptide. Biopolymers 1993, 33, 827–837. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.