

Significance
More than two decades ago, a Gimap5 mutation was found responsible for autoimmune diabetes and lymphopenia in the BioBreeding (BB) rats. Later, studies of an ENU-induced Gimap5 mutation in mice, a Gimap5-null allele in mice, and loss of function mutations in GIMAP5 in humans uncovered an essential function of GIMAP5 in regulating liver homeostasis and opposing EMH. However, the mechanism(s) by which GIMAP5 controls these processes have not been described. Through forward genetic screening of ENU-mutagenized mice and proteomic analysis, we identified a tripartite protein complex, composed of MFSD1, GLMP, and GIMAP5, that supports lymphocyte development. We further determined that this protein complex operates in different cell types to assure normal hematopoiesis, liver homeostasis, and suppression of EMH.
Keywords: MFSD1, GLMP, GIMAP5, MTOR, extramedullary hematopoiesis
Abstract
We detected ENU-induced alleles of Mfsd1 (encoding the major facilitator superfamily domain containing 1 protein) that caused lymphopenia, splenomegaly, progressive liver pathology, and extramedullary hematopoiesis (EMH). MFSD1 is a lysosomal membrane-bound solute carrier protein with no previously described function in immunity. By proteomic analysis, we identified association between MFSD1 and both GLMP (glycosylated lysosomal membrane protein) and GIMAP5 (GTPase of immunity-associated protein 5). Germline knockout alleles of Mfsd1, Glmp, and Gimap5 each caused lymphopenia, liver pathology, EMH, and lipid deposition in the bone marrow and liver. We found that the interactions of MFSD1 and GLMP with GIMAP5 are essential to maintain normal GIMAP5 expression, which in turn is critical to support lymphocyte development and liver homeostasis that suppresses EMH. These findings identify the protein complex MFSD1-GLMP-GIMAP5 operating in hematopoietic and extrahematopoietic tissues to regulate immunity and liver homeostasis.
Membrane-bound solute carriers (SLCs) are essential to maintain physiological functions such as nutrient uptake, ion transport, and waste removal (1). Defects in SLC function are often associated with human diseases including metabolic disorders, immune deficiencies, and cancer (2). The SLC superfamily comprises about 400 transporters (1); however, many aspects of the cellular roles and mechanisms of SLC family members have not yet been addressed, leaving large areas of biology unexplored.
MFSD1 was first identified as a putative SLC protein belonging to the major facilitator superfamily (MFS), one of the largest families of transporters for a broad spectrum of solutes across cellular membranes (3–6). Recent studies have identified glycosylated lysosomal membrane protein (GLMP) as an accessory protein for MFSD1; together, they form a stable and functional lysosomal transporter complex necessary to maintain liver homeostasis in mice (7–11). However, the mechanisms through which MFSD1 or GLMP regulates lymphopoiesis have escaped understanding.
In this study, we identified three ENU-induced missense or null mutations in Mfsd1, each causing impairment of B lymphopoiesis in mice. Besides its effect on B cell development, we found that MFSD1 deficiency also significantly impaired survival, homeostasis, and function of T cells. Through mass spectrometry–based MFSD1 interactome analysis and biochemical characterization, we identified the existence of a lysosomal membrane resident protein complex composed of MFSD1, GLMP, and GIMAP5. Here, we describe how the complex functions to regulate lymphopoiesis, liver homeostasis, and EMH.
Results
Immunodeficiency Caused by a Mfsd1 Mutation.
In a forward genetic screen for defects in adaptive immunity, we identified several third-generation (G3) mice in three independent pedigrees showing low percentages of B220+ cells in the peripheral blood (Fig. 1A). The phenotypes, which we named white, schneeweiss, and edelweiss, were transmitted as recessive traits. By superpedigree mapping, a method that analyzes genotype vs. phenotype associations from multiple pedigrees sharing mutant alleles of a single gene (12), the three phenotypes were correlated with mutations in the gene Mfsd1 (Fig. 1B). Mfsd1 encodes MFS domain containing 1 (MFSD1), a lysosomal 12-transmembrane protein with unknown function in immunity. The white mutation was predicted to impair splicing of exon 6, resulting in a frameshift and premature stop at position 146 (glycine; G146*), in the fourth transmembrane helix of MFSD1 (Fig. 1C). The schneeweiss and edelweiss mutations affected the second and eighth transmembrane helices of MFSD1, respectively (Fig. 1C). Transient transfection of HEK293T cells with FLAG-tagged full-length or mutant MFSD1 (G146*) revealed decreased expression of the truncated MFSD1 compared to the wild-type protein (Fig. 1D), suggesting that the white mutation impairs protein stability. To date, the effects of MFSD1 deficiency in immunity have not been characterized. Mice expressing a CRISPR/Cas9-targeted germline null allele of Mfsd1 were generated, confirming that the mutations in Mfsd1 were responsible for the observed phenotype (Fig. 1 E–N). The Mfsd1−/− mice showed reduced numbers of white blood cells, lymphocytes, B220+ B cells, CD3+ T cells, NK cells, monocytes, red blood cells (RBC), and platelets in the peripheral blood compared to wild-type littermates (Fig. 1 E–L). Furthermore, T cell-dependent (TD) humoral immune responses to aluminum hydroxide precipitated ovalbumin (OVA) were diminished in Mfsd1−/− mice compared to wild-type littermates (Fig. 1M). However, the mice exhibited comparable T cell-independent (TI) antibody responses to 4-hydroxy-3-nitrophenylacetyl-Ficoll (NP-Ficoll; Fig. 1N). These results conclusively establish the hypomorphic nature of the Mfsd1 mutation and its causative relationship with the observed phenotype in Mfsd1−/− mice.
Fig. 1.
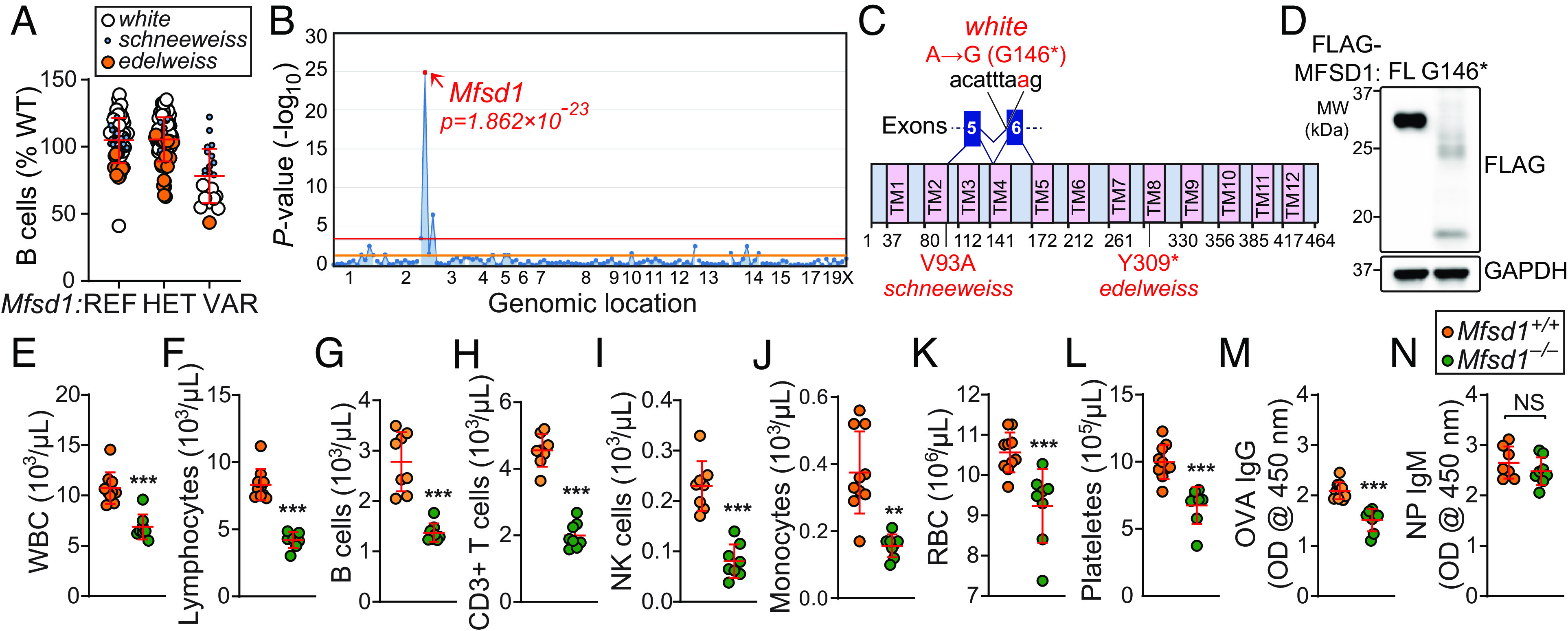
A heritable lymphopenia caused by MFSD1 deficiency in mice. (A) Frequency of B cells in the peripheral blood of third-generation (G3) mice from three pedigrees, each descended from a different ENU-mutagenized male mouse (G1), with REF (+/+), HET (+/mutant), or VAR (mutant/mutant) genotypes for Mfsd1 (n = 1 to 57 mice/genotype). Data were normalized to average B cell frequency of age-matched C57BL/6 mice at the time of experiment (n = 30 mice/experiment). Data point size is proportional to damage probability predicted by PolyPhen-2 of each Mfsd1 mutation identified in the three G1 founders of the pedigrees (13). (B) Manhattan plot showing P values for linkage of B cell frequency phenotype to mutations in the three affected pedigrees calculated using a recessive model of inheritance. The –log10 P-values (Y axis) are plotted vs. the chromosomal positions of mutations (X axis) identified in the three G1 founders of the affected pedigrees. Horizontal red or orange lines represent thresholds of P = 0.05 with or without Bonferroni correction, respectively. Causative mutation is in red. (C) Protein domains of mouse MFSD1 (464 aa in length). The location of the white, schneeweiss, and edelweiss mutations, which result in G146*, V93A, and Y309* in MFSD1, respectively, are indicated in red. * Premature stop codon. (D) HEK293T cells were transfected with either FLAG-tagged wild-type full-length (FL) or mutant (G146*) MFSD1. Total cell lysates were immunoblotted with antibodies against FLAG. GAPDH was used as a loading control. (E–L) Whole blood cell counts of white blood cells (E), lymphocytes (F), B cells (G), T cells (H), NK cells (I), monocytes (J), red blood cells (K), and platelets (L) in 10-wk-old Mfsd1−/− and wild-type littermates (n = 8 to 10 mice/genotype). (M and N) T cell–dependent (M) or T cell–independent (N) antibody responses after immunization with aluminum hydroxide precipitated ovalbumin (OVA/alum) or NP-Ficoll, respectively, in 8-wk-old Mfsd1−/− and wild-type littermates (n = 7 to 10 mice/genotype). Data are presented as absorbance at 450 nm. Each symbol represents an individual mouse (A, E–N). P values were determined by Student’s t test (E–N). Data are representative of one (A), two (D), or three (E–N) independent experiments. Error bars indicate SD. **P < 0.01; ***P < 0.001; and NS, not significant with P > 0.05.
Cell-Intrinsic MFSD1 Function in T Cell Survival, Homeostasis, and Expansion.
To distinguish between hematopoietic and extrahematopoietic origins of the Mfsd1-associated immune cell defects, we reconstituted irradiated Rag2−/− (CD45.2) recipients with unmixed wild-type (CD45.1), Mfsd1−/− (CD45.2), or an equal mixture of wild-type (CD45.1) and Mfsd1−/− (CD45.2) bone marrow cells. The bone marrow cells from Mfsd1−/− mice were unable to repopulate cells of the lymphoid lineage, including B220+ B cells, CD4+ T, CD8+ T, and NK cells, in the spleens of irradiated recipients as efficiently as cells derived from wild-type donors (SI Appendix, Fig. S1A). In mixed chimeras, the proportions of lymphoid pools in the spleen showed that Mfsd1−/−-derived cells were at a competitive disadvantage in repopulating the lymphoid lineage compared with cells derived from wild-type donors (SI Appendix, Fig. S1B). These data indicate that the effects of MFSD1 deficiency on lymphoid development are intrinsic to the hematopoietic compartment and cell autonomous.
To test for a B cell– or T cell–intrinsic role of MFSD1, we generated mice in which Mfsd1 was conditionally deleted in B and T cells using Mb1- and Cd4-cre drivers, respectively. The frequencies of B220+ B cells and CD3+ T cells in peripheral blood were significantly diminished in Mb1-cre;Mfsd1fl/fl or Cd4-cre;Mfsd1fl/fl mice, respectively, compared to control littermates (Mfsd1fl/fl), suggesting there is a cell-intrinsic requirement for MFSD1 during B and T cell development or survival (Fig. 2 A and B). Furthermore, flow cytometry analysis of CD4+ and CD8+ T cells in the peripheral blood of Cd4-cre;Mfsd1fl/fl mice showed increased expression of the surface glycoprotein CD44, which encompasses recently activated, expanding, and memory phenotype cells (Fig. 2C). Immunoblot analysis of Mfsd1−/− CD8+ T cells in the spleen revealed constitutive phosphorylation of Akt and mitogen-activated protein kinase (p44/42 MAPK) relative to CD8+ T cells from wild-type littermates under basal conditions, a phenotype that can be seen in activated effector T cells (Fig. 2D). In addition, a higher percentage of peripheral CD3+ T cells from Mfsd1−/− mice were positive for annexin V alone (apoptotic) as well as annexin V and propidium iodide (PI; necrotic) under basal conditions compared to wild-type littermates (Fig. 2E). Increased proteolytic processing of caspases of the extrinsic (caspase-3) and intrinsic (caspase-9) apoptosis pathways were detected in splenic Mfsd1−/− CD8+ T cells relative to wild-type cells (Fig. 2F). These data suggest that Mfsd1−/− T cells in the periphery exist in an activated state that may be predisposed to apoptosis. The mixed bone marrow chimeras (SI Appendix, Fig. S1B) support the idea that T cell activation results from a cell-intrinsic dysfunctional state rather than being a consequence of lymphopenia.
Fig. 2.
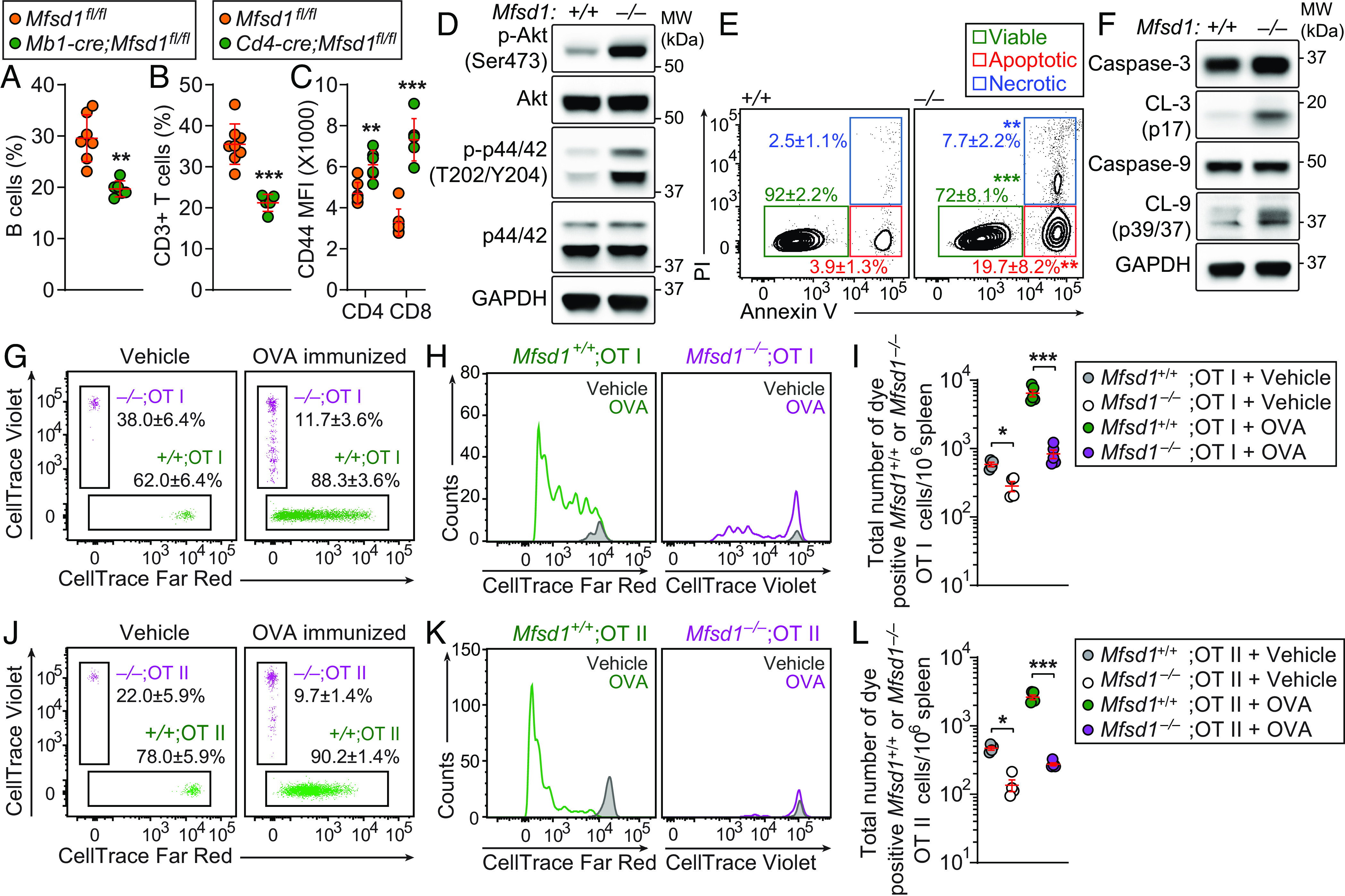
Cell-intrinsic MFSD1 function in T cell survival, homeostasis, and expansion. (A–C) Flow cytometry analysis of B220+ B cells (A), CD3+ T cells (B), and surface CD44 expression (mean fluorescence intensity, MFI; C) on CD3+ T cells in peripheral blood of 12-wk-old Mb1-cre;Mfsd1fl/fl or Cd4-cre;Mfsd1fl/fl mice (n = 5 to 8 mice/genotype). (D) Immunoblot analysis of phospho-Akt, Akt, phospho-p44/p42 MAPK, p44/p42 MAPK, and GAPDH in total cell lysates of splenic CD8+ T cells isolated from Mfsd1−/− or wild-type littermates. (E) Annexin V/PI staining of CD3+ T cells in peripheral blood obtained from 12-wk-old Mfsd1−/− or wild-type littermates (n = 6 to 7 mice/genotype). Numbers adjacent to outlined areas indicate percent cells (mean ± SD) in each. (F) Immunoblot analysis of caspase-3, cleaved caspase-3 (CL-3), caspase-9, cleaved caspase-9 (CL-9), and GAPDH in total cell lysates of splenic CD8+ T cells isolated from Mfsd1−/− or wild-type littermates. (G–L) Impaired antigen-specific expansion of MFSD1-deficient T cells. A 1:1 mixture of CellTrace Violet-labeled Mfsd1−/− (CD45.2) and Far Red-stained Mfsd1+/+ OT-I or OT-II T cells (CD45.2) was adoptively transferred into wild-type hosts (C57BL/6J; CD45.1). Representative flow cytometric scatter plots (G and J) and histograms (H and K), and quantification of total numbers (I and L) of CellTrace Violet- or Far Red-positive Mfsd1+/+ or Mfsd1−/− OT-I (G–I) or OT-II T cells (J–L) harvested from the spleens of wild-type (C57BL/6J; CD45.1) hosts, 96 h after immunization with soluble OVA or sterile PBS (vehicle control; n = 4 to 5 mice/group). Numbers adjacent to outlined areas indicate percent cells in each. Each symbol represents an individual mouse (A–C, I, and L). P values were determined by Student’s t test (A and B) or one-way ANOVA with Dunnett’s multiple comparisons (E, I, and L). Data are representative of two (D–F and G–L) or three (A–C) independent experiments. Error bars indicate SD. *P < 0.05; **P < 0.01; and ***P < 0.001.
It was unclear whether the survival defect of mature T cells was solely responsible for the peripheral T cell deficiency in Mfsd1−/− mice. This led us to test the proliferative response of peripheral T cells to expansion signals. To examine antigen-specific T cell expansion, an equal mixture of OVA-specific OT-I or OT-II T cells isolated from Mfsd1+/+ (CD45.2, Far-Red stained) or Mfsd1−/− (CD45.2, Violet stained) mice were transferred into wild-type CD45.1 recipients that were subsequently immunized with soluble OVA (Fig. 2 G–L). Wild-type OT-I or OT-II T cells proliferated as expected, but the Mfsd1−/− OT-I or OT-II T cells proliferated less as determined by CellTrace dye dilution (Fig. 2 G, H, J, and K) or by enumeration of total Mfsd1+/+ or Mfsd1−/− OT-I or OT-II T cells recovered from recipients in the spleen 4 d after immunization (Fig. 2 I and L). Although an equal mixture of wild-type and Mfsd1−/−OT-I or OT-II cells were adoptively transferred, significantly fewer Mfsd1−/− OT-I or OT-II T cells were recovered in the spleens of recipients given sterile PBS (vehicle control; Fig. 2 I and L). Together, these results indicate that cell-intrinsic MFSD1 function is critical for T cell survival, homeostasis, and expansion.
Identification of the MFSD1-GLMP-GIMAP5 Complex.
MFSD1 was first identified as a putative SLC protein belonging to the MFS, one of the largest transporter families, which transports a broad spectrum of solutes across cellular membranes (3–6). However, the mechanisms through which MFSD1 regulates lymphopoiesis have not been described. To gain insight into the immune function of MFSD1, we performed mass spectrometry–based interactome analysis. Immunoprecipitates prepared from EL4 T cells stably expressing FLAG-tagged MFSD1 contained GTPase IMAP family member 5 (GIMAP5), glycosylated lysosomal membrane protein (GLMP), and multiple subunits of the vacuolar H+-adenosine triphosphatase (v-ATPase) necessary for lysosomal amino acids to activate mTORC1 signaling (14) as putative interactors (Dataset S1).
To confirm the interactions between MFSD1 and putative interactors, HEK293T cells were transiently transfected with either HA-tagged MFSD1, GIMAP5, or GLMP or with FLAG-tagged GIMAP5, ATP6V1B2, GLMP, LAMTOR1, mTOR, or glycoprotein 78 (GP78). LAMTOR1 and mTOR were included in this experiment because of their known interactions with the v-ATPase, subunits of which were identified as MFSD1 interactors. GP78 was also included, but soon excluded from further testing because we confirmed no functional relationship existed between GP78 and MFSD1 (SI Appendix, Fig. S2). Total cell lysates from transfected HEK293T cells were mixed in vitro and coimmunoprecipitation was performed using anti-FLAG M2 agarose beads. HA-tagged MFSD1 and GIMAP5 immunoprecipitated with each of the FLAG-tagged proteins (Fig. 3 A and B). However, we found that GLMP only bound to MFSD1, GIMAP5, and GP78, but not to ATP6V1B2, LAMTOR1, and MTOR (Fig. 3 A and C). We further confirmed specific recruitment of endogenous GIMAP5, LAMTOR1, and MTOR by stably expressed FLAG-tagged MFSD1, but not by the LAMP1-FLAG controls (Fig. 3D).
Fig. 3.
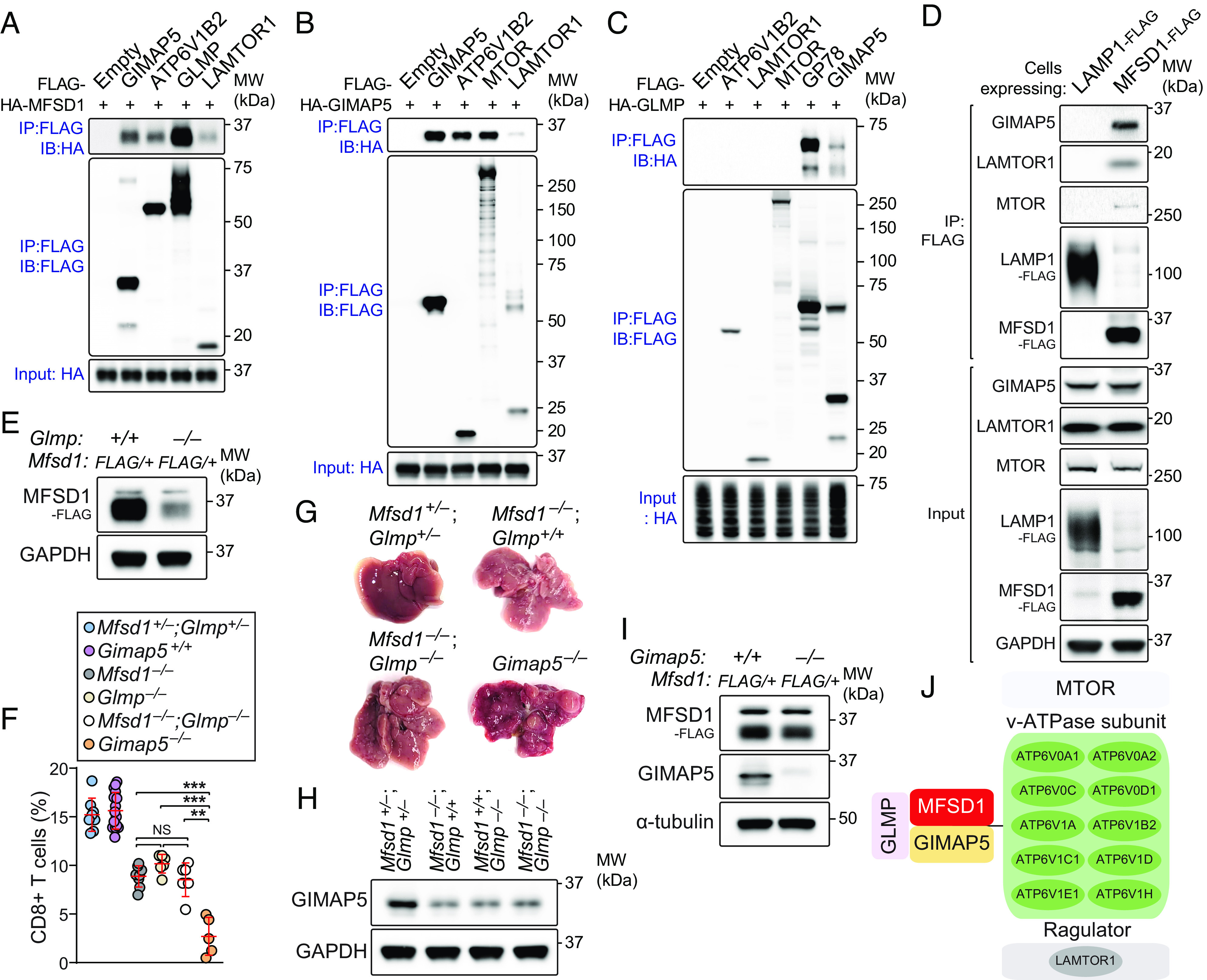
Identification of the MFSD1-GLMP-GIMAP5 complex. (A–C) HEK293T cells were transfected with FLAG-tagged GIMAP5, ATP6V1B2, GLMP, LAMTOR1, MTOR, GP78, or empty vector or with HA-tagged MFSD1, GIMAP5, or GLMP. Lysates of FLAG-expressing and HA-expressing cells were mixed as indicated and subsequently immunoprecipitated using anti-FLAG M2 agarose and immunoblotted with antibodies against HA or FLAG. (D) Recombinant FLAG-tagged MFSD1 coimmunoprecipitated endogenous GIMAP5 and the mTORC1 scaffolding complex proteins. Total cell lysates (TCLs) prepared from EL4 T cells stably expressing MFSD1-FLAG or LAMP1-FLAG (control) were subjected to immunoprecipitation using anti-FLAG M2 agarose beads. TCLs (Input) and immunoprecipitates (IP: FLAG) were analyzed by immunoblotting with the indicated antibodies. (E) Endogenous levels of FLAG-MFSD1 in splenic CD8+ T cells of the MFSD1-3X FLAG knock-in mouse (Mfsd1+/FLAG) on a GLMP-deficient (Glmp−/−) or wild-type background. (F) FACS analysis of CD8+ T cells in peripheral blood of mice with indicated genotype (n = 5 to 16 mice/genotype). (G) Representative photographs of livers isolated from mice with indicated genotype. (H) Immunoblot analysis of GIMAP5 expression in splenic CD8+ T cells isolated from mice with indicated genotype. (I) Endogenous levels of FLAG-MFSD1 in splenic CD8+ T cells of the MFSD1-3X FLAG knock-in mouse (Mfsd1+/FLAG) on a GIMAP5-deficient (Gimap5−/−) or wild-type background. (J) Summary of MFSD1 interactome identified by three independent mass spectrometry experiments. Data are representative of three independent experiments. Each symbol represents an individual mouse (F). P values were determined by one-way ANOVA with Dunnett’s multiple comparisons. Error bars indicate SD. **P < 0.01; ***P < 0.001; and NS, not significant with P > 0.05.
An essential role for GLMP in MFSD1 function has been described (9); GLMP is needed to maintain normal MFSD1 levels in lysosome membranes. Absent GLMP, MFSD1 expression is reduced in liver lysosomes, leading to splenomegaly and abnormal liver pathology. However, the effect of MFSD1 or GLMP deficiency in lymphocyte development was not recognized in the previous study. To test this, we generated mice expressing a CRISPR/Cas9-targeted germline null allele of Glmp and confirmed perfect phenocopy of MFSD1 deficiency in Glmp−/− mice (SI Appendix, Fig. S3 A–J). Since no commercial antibody to detect endogenous MFSD1 exists, we generated a CRISPR-based knock-in of a 3X FLAG-tag appended to the C terminus of endogenous MFSD1 in mice. A significant reduction in endogenous levels of FLAG-MFSD1 was found in splenic CD8+ T cells of the MFSD1-3X FLAG knock-in mouse on a GLMP-deficient background (Glmp−/−;Mfsd1+/FLAG) compared to those on a wild-type background (Glmp+/+;Mfsd1+/FLAG; Fig. 3E). This result further confirms the previous report that MFSD1-GLMP interaction is critical to protect both proteins from proteolytic degradation (9).
Gimap5 was first identified by positional cloning as a gene mutated in BioBreeding (BB) rats known to develop progressive lymphopenia and autoimmune diabetes (15). Later, we and others created an ENU-induced Gimap5 mutation, termed sphinx (16), as well as a targeted Gimap5-null mouse (17). Both strains recapitulated the severe lymphopenia observed in BB rats, although the mechanism(s) driving the phenotype remain unknown. We found that complete GIMAP5 deficiency resulted in more severe defects in lymphocyte development (Fig. 3F) and liver pathology (Fig. 3G) than those observed in Mfsd1−/−, Glmp−/−, or Mfsd1−/−;Glmp−/−mice. Importantly, a significant, yet incomplete, reduction in endogenous levels of GIMAP5 was found in splenic CD8+ T cells of the MFSD1 or GLMP single knockout, or double knockout mice compared to that seen in the Mfsd1+/−;Glmp+/− background (Fig. 3H). However, we found that GIMAP5 deficiency does not affect MFSD1 stability (Fig. 3I). Together, the above data provide strong evidence for an interdependent core MFSD1-GLMP-GIMAP5 (MGG) complex necessary for maintaining normal GIMAP5 levels to support lymphocyte development and liver homeostasis.
Compensatory mTOR Activation Caused by MGG Complex Deficiency.
It has been reported that GIMAP5 suppresses phosphoinositide 3-kinase (PI3K)/Akt/mTORC1 activation upon T cell receptor (TCR) cross-linking (18) to promote lymphocyte survival, proliferation and quiescence (16, 17). Considering the reduced GIMAP5 levels in MFSD1- or GLMP-deficient T cells and the overall similarity of immune phenotypes and liver pathology among Mfsd1−/−, Glmp−/−, and Gimap5−/− mice, we hypothesized that the interaction of MGG complex with mTOR, vATPase subunits, and LAMTOR1 (Fig. 3J) might regulate mTOR kinase activity, which in turn supports lymphocyte development and liver homeostasis.
To determine the functional relationship between the MGG complex and mTOR signaling, we examined steady-state mTOR kinase activity in Mfsd1−/−, Glmp−/−, and Gimap5−/− splenic CD8+ T cells. Immunoblotting revealed elevated S6K1 and 4E-BP1 phosphorylation under basal conditions in Mfsd1−/−, Glmp−/−, and Gimap5−/− CD8+ T cells compared to wild-type cells, indicative of mTOR activation (Fig. 4A). The GIMAP5 expression level was inversely correlated with the level of constitutive mTOR kinase activity when determined by phosphorylation status of its substrates S6K1 and 4E-BP1 (Fig. 4A). We analyzed splenic CD8+ T cells isolated from Cd4-cre;Mfsd1fl/fl mice and Mfsd1fl/fl littermates to test whether the constitutive mTOR kinase activity measured in Mfsd1-WT vs. -KO CD8+ T cells was cell intrinsic. In addition to constitutive phosphorylation of S6K1 and 4E-BP1 under basal conditions, sustained S6K1 and 4E-BP1 phosphorylation during amino acid withdrawal, as well as after amino acid or insulin stimulation, were observed in cells from Cd4-cre;Mfsd1fl/fl mice compared to those from Mfsd1fl/fl littermates (Fig. 4B). Treatment of cells with a potent mTOR inhibitor, Torin 1, completely suppressed amino acid- or insulin-induced phosphorylation of S6K1 and 4E-BP1 in MFSD1-deficient CD8+ T cells (Fig. 4B). This suggests that elevation of mTOR kinase activity caused by reduced GIMAP5 expression in CD8+ T cells is cell intrinsic. Furthermore, stimulation of cells with either glucose or cholesterol, alone or in combination with amino acids, also caused enhanced mTOR activation in Mfsd1−/− CD8+ T cells with respect to wild-type cells (SI Appendix, Fig. S4 A and B). These results also support our hypothesis that the MGG complex normally functions as a negative regulator of mTOR signaling in lymphocytes.
Fig. 4.
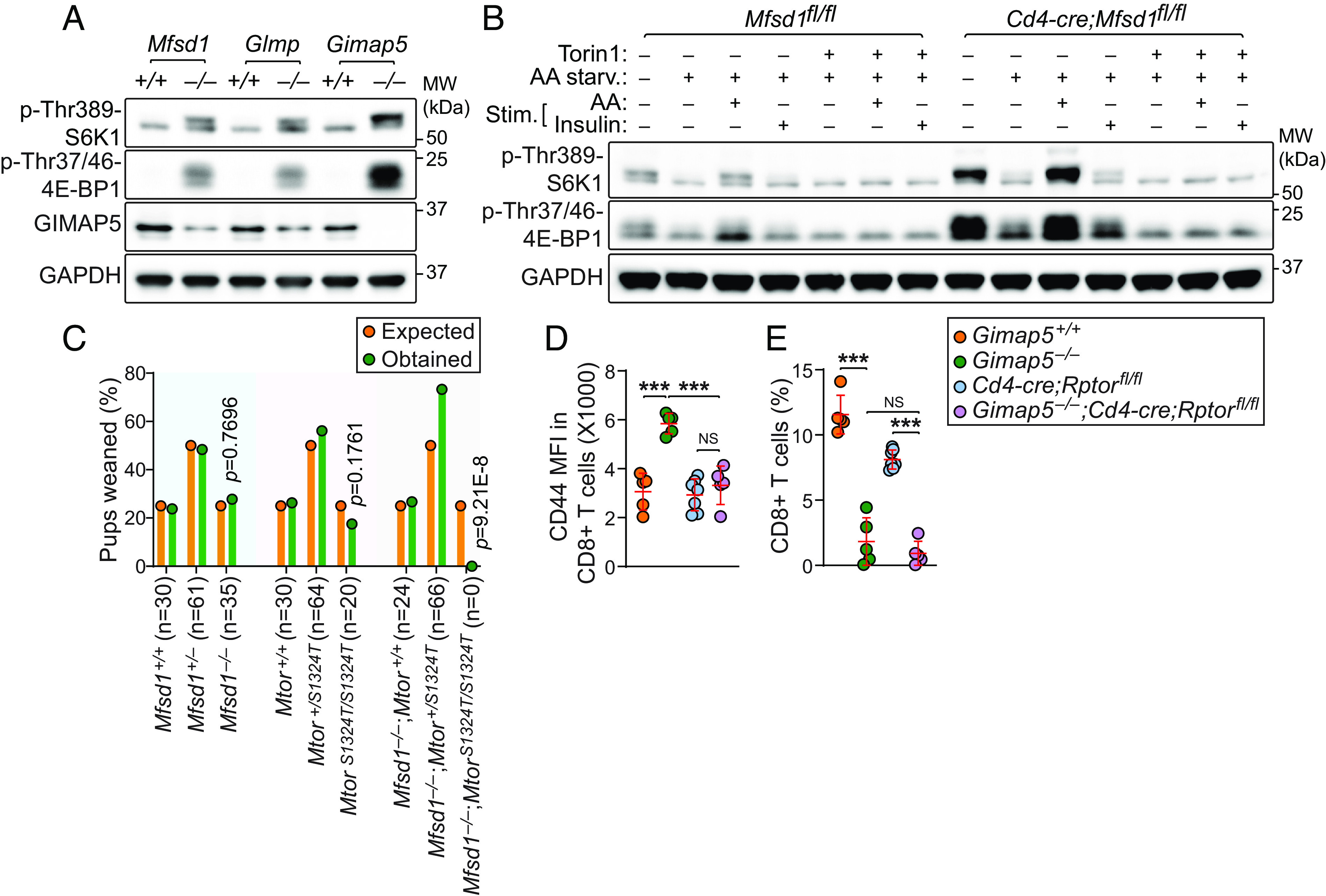
Cell-intrinsic mTOR activation is associated with reduced GIMAP5 expression in MFSD1- or GLMP-deficient T cells. (A) Steady-state mTORC1 kinase activity analyzed by the phosphorylation status of S6K1 at threonine 398 (p-Thr389-S6K1) and 4E-BP1 at threonine 37/46 (p-Thr37/46-4E-BP1) in splenic CD8+ T cells isolated from 10-wk-old Mfsd1−/−, Glmp−/−, Gimap5−/−, or wild-type littermates. Endogenous GIMAP5 levels were also measured. (B) Immunoblot analysis of p-Thr389-S6K1 and p-Thr37/46-4E-BP1 in splenic CD8+ T cells isolated from 10-wk-old Cd4-cre;Mfsd1fl/fl or Mfsd1fl/fl littermates. Cells were grown in amino acid (AA) free RPMI-1640 medium containing 10% dialyzed fetal bovine serum (FBS) for 2 h in the presence or absence of the mTOR inhibitor Torin 1 (250 nM) and then stimulated with AA (mTORC1) or insulin (mTORC2) for 30 min. (C) Offspring from crosses of Mtor mutant mice on a wild-type or MFSD1-deficient background. A χ2 test with the appropriate degrees of freedom was used to calculate P values. (D and E) Surface CD44 expression (D) and frequency (E) of peripheral blood CD8+ T cells from 10-wk-old mice with indicated genotype (n = 5 to 7 mice/genotype). Data are representative of two (D and E) or three independent experiments (A and B). P values were determined by Chi-square test (C) or one-way ANOVA with Dunnett’s multiple comparisons (D and E). Each symbol represents an individual mouse (D and E). Error bars indicate SD. ***P < 0.001; NS, not significant with P > 0.05.
To test the effect of exaggerated mTOR kinase activity on lymphopoiesis in Mfsd1−/− mice, we employed a mouse strain carrying a viable hypomorphic ENU-induced missense mutation resulting in a serine to threonine substitution at position 1324 (S1324T) in the mTOR protein (allele name motor); amino acid 1324 is within the last HEAT repeat (SI Appendix, Fig. S4C). Immunoblotting of total cell lysates from splenic CD8+ T cells revealed that the S1324T mutation impairs mTOR stability and kinase activity under basal conditions as well as following stimulation with amino acids (SI Appendix, Fig. S4D). Unlike the germline mTOR knockout that results in complete prenatal lethality (19), homozygous motor mice are viable, presumably due to residual protein function sufficient for normal development and postnatal survival. Since MFSD1-deficient mice harboring heterozygous MtorS1324T allele are fertile (Mfsd1−/−;MtorS1324T/+), we bred these animals to obtain MtorS1324T homozygotes on a MFSD1-deficient background for immune phenotyping, anticipating phenotype rescue. However, the cross yielded no MtorS1324T homozygous pups on a MFSD1-deficient background, leading us to conclude that the double mutation caused synthetic lethality (P = 9.21E−8; χ2 test; n = 90 mice: 24 Mfsd1−/−;Mtor+/+, 66 Mfsd1−/−;MtorS1324T/+; 0 Mfsd1−/−; MtorS1324T/S1324T; Fig. 4C).
To more directly test the hypothesis that reduced GIMAP5 expression caused by disruption of the MGG complex leads to mTOR activation that impairs T cell development, we used a conditional knockout system to delete RAPTOR, a component necessary for mTORC1 activity, in GIMAP5-deficient T cells. Gimap5 knockout mice with a Cd4-cre transgene and an allele of Rptor with exon 6 flanked by loxP sites (Gimap5−/−;Cd4-cre;Rptorflox/flox) were viable and able to suppress T cell activation (Fig. 4D); however, homozygous deletion of Rptor failed to rescue GIMAP5 associated T cell deficiency (Fig. 4E). This result rejected our hypothesis that the MGG complex serves as a negative regulator of mTOR signaling. Rather, it suggests that the exaggerated mTOR activation observed in MFSD1, GLMP, or GIMAP5-deficient T cells that have reduced GIMAP5 expression is a compensatory response supporting cell survival.
Extramedullary Hematopoiesis (EMH) and Dysregulated Lipid Metabolism Caused by Mfsd1 Mutations.
The identification of the MGG complex and the phenotypes shared in the three knockout strains prompted us to test whether MFSD1 or GLMP deficiency is associated with EMH as seen in Gimap5 mutant mice (16). To test this, we performed flow cytometry-based quantitation of hematopoietic stem and progenitor cell (HSPC) subsets in the bone marrow and liver of Mfsd1−/− or Glmp−/− mice. We found that both MFSD1 and GLMP deficiency caused severe bone marrow failure in mice (Fig. 5A and SI Appendix, Fig. S2K). Total numbers of bone marrow cells were reduced by ~60%, and HSPC subsets were similarly affected (Fig. 5A). Despite this, Mfsd1−/− or Glmp−/− mice had enlarged spleens and lymph nodes compared to those of wild-type littermates (Fig. 5B and SI Appendix, Fig. S2L). We found that a significant number of HSPCs, including LSK+, ST-HSC, CLP, CMP, MEP, and GMP, exist in adult Mfsd1−/− or Glmp−/− liver (Fig. 5C and SI Appendix, Fig. S2M). These results confirm that MFSD1 or GLMP deficiency each causes EMH just as GIMAP5 deficiency does.
Fig. 5.
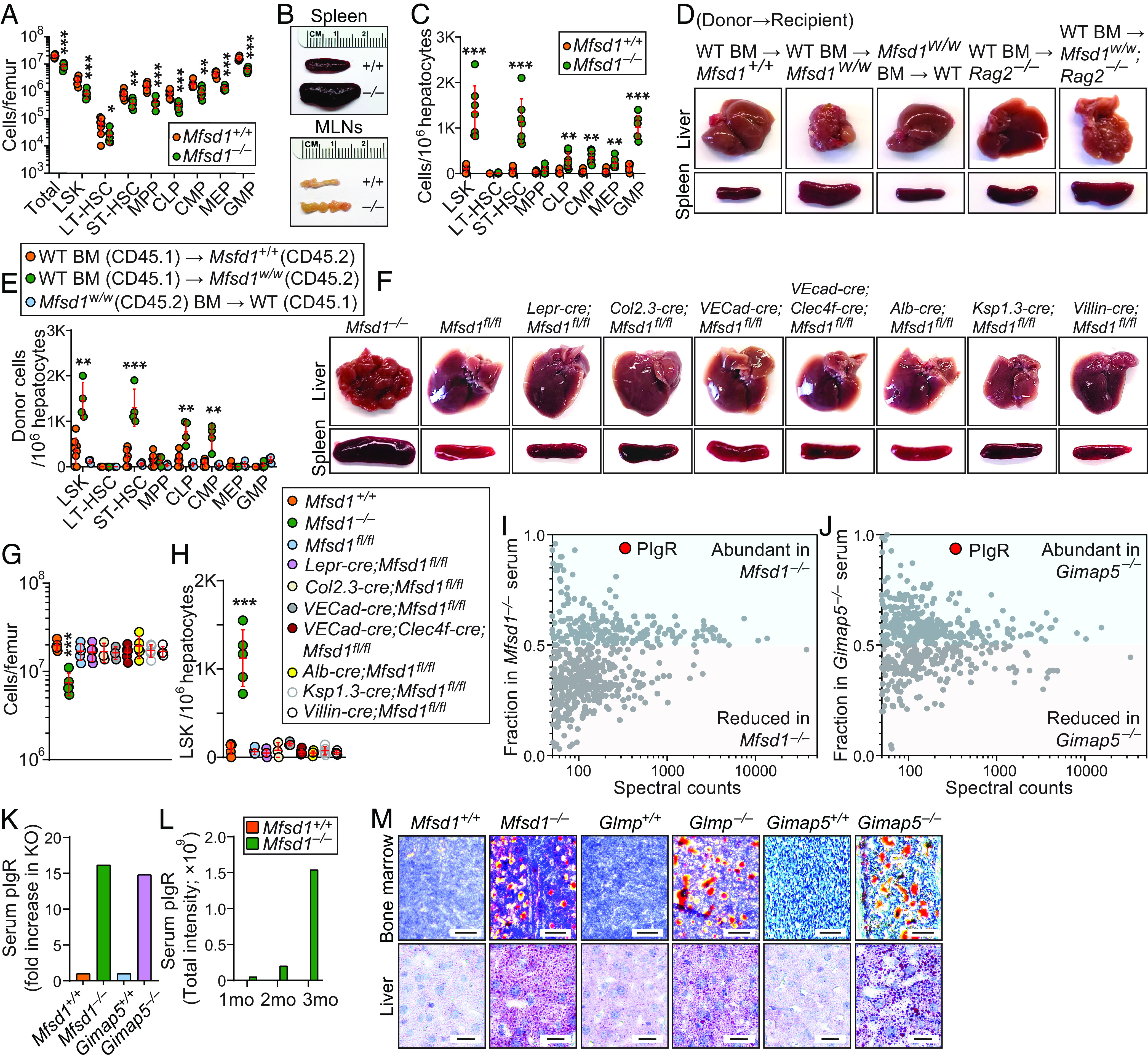
Extramedullary hematopoiesis and dysregulated lipid metabolism caused by MFSD1 deficiency in mice. (A) Numbers of HSPC subsets in the bone marrow of 14-wk-old Mfsd1−/− and wild-type littermates (n = 7 mice/genotype). (B) Representative photographs of spleens and mesenteric lymph nodes (MLNs) isolated from 14-wk-old Mfsd1−/− and wild-type littermates. (C) Numbers of HSPC subsets in the liver of 14-wk-old Mfsd1−/− and wild-type littermates (n = 7 mice/genotype). (D) Representative photographs of livers and spleens isolated from recipients rescued from lethal irradiation by engraftment of bone marrow with indicated genotype. (E) Numbers of donor-derived HSPC subsets in the liver 20 wk after reconstitution of recipients with bone marrow of the indicated genotype (n = 4 to 8 mice/genotype). (F) Representative photographs of livers and spleens isolated from 12- to 20-wk-old mice with indicated genotype. (G and H) Numbers of cells in the bone marrow (G) and LSK+ cells in the liver (H) of 12- to 20-wk-old mice with indicated genotype (n = 3 to 5 mice/genotype). (I and J) Mass spectrometry analysis of serum from Mfsd1−/− (I), Gimap5−/− (J), and wild-type littermates. Points above 0.5 or below 0.5 on Y axis denote proteins enriched or reduced, respectively, in either Mfsd1−/− or Gimap5−/− serum compared to wild-type serum. (K) Serum pIgR levels in 12-wk-old mice with indicated genotype. (L) Age-dependent increase in serum pIgR levels in Mfsd1−/−mice. (M) Representative Oil Red O staining of the bone marrow and liver from Mfsd1−/−, Glmp−/−, Gimap5−/− and wild-type littermates. Scale bars: 25 μm for bone marrow and 125 μm for liver. Each symbol represents an individual mouse (A, C, E, G, and H). P values were determined by Student’s t test (A and C) or one-way ANOVA with Dunnett’s multiple comparisons (E, G, and H). Error bars indicate SD. *P < 0.05; **P < 0.01; and ***P < 0.001. LSK, Lin−Sca1+c-Kit+ fraction of the bone marrow or liver; LT, long-term; ST, short-term; HSC, hematopoietic stem cells; MPP, multipotent progenitor; CLP, common lymphoid progenitor; CMP, common myeloid progenitor; MEP, megakaryocyte-erythrocyte progenitor; GMP, granulocyte–macrophage progenitor.
We investigated the cellular origins of Mfsd1-associated EMH. Using reciprocal bone marrow chimeras, we found that the liver and spleen of wild-type (CD45.1) recipients engrafted with Mfsd1 mutant (Mfsd1w/w; CD45.2) bone marrow appeared normal with no evidence of EMH up to 20 wk posttransplantation (Fig. 5D). However, reciprocal transplants of wild-type (CD45.1) bone marrow into Mfsd1w/w or Mfsd1w/w;Rag2−/− mutant mice (CD45.2) failed to rescue the liver pathology and splenomegaly (Fig. 5D). This is consistent with a previous report that EMH cannot be transferred with Gimap5 mutant bone marrow into wild-type recipients and that EMH in Gimap5 mutant mice does not require adaptive immune function (16). Flow cytometric analysis of liver cell suspensions showed a significant number of donor-derived wild-type (CD45.1) HSPCs in Mfsd1w/w liver compared to wild-type hosts (Fig. 5E). In contrast, very few donor-derived Mfsd1w/w (CD45.2) HSPCs were detected in wild-type (CD45.1) liver and spleen (Fig. 5E). This result led us to hypothesize that signals arising from radioresistant cells drive EMH in Mfsd1−/− mice.
It has been reported that endothelial-specific deletion of Mfsd1 using the Tie2-cre driver was sufficient to induce liver pathology [EMH was not recognized in this study; (9)]. Since our result indicates liver pathology caused by disruption of the MGG complex is associated with EMH, we hoped to test whether endothelial-specific Mfsd1 deletion causes EMH and validate the effect on liver pathology by breeding Mfsd1fl/fl mice to another endothelial-specific cre-expressing strain [VECad-cre; (20)] known to be more specific than the Tie2-cre driver, which shows cre recombinase activity in other nonendothelial cell types such as HSC (21), osteoblasts (22), and Kupffer cells [KCs; (9)]. To our surprise, the resulting endothelial-specific MFSD1-deficient mice (VECad-cre;Mfsd1fl/fl) had no aberrations of liver and spleen morphology (Fig. 5F) nor bone marrow cellularity (Fig. 5G), and did not develop EMH (Fig. 5H). To delineate the cell types necessary to trigger liver pathology and EMH in Mfsd1−/− mice, we further bred Mfsd1-floxed mice to several strains with cre expression in multiple extrahematopoietic cell types including leptin receptor+ mesenchymal stromal cells [Lepr-cre;Mfsd1fl/fl; (23)], osteoblasts [Col2.3-cre;Mfsd1fl/fl; (24)] which are the main sources of hematopoietic stem cell (HSC) niche factors responsible for HSC maintenance and retention in the bone marrow (25–28). To mimic the condition of Tie2-cre-mediated MFSD1 deletion that caused liver pathology [i.e., MFSD1 deletion in endothelial cells and KCs (9)], we knocked out Mfsd1 in both endothelial cells and KCs using a VECad-cre;Clec4f-cre double transgenic strain. Since MFSD1 is widely expressed in nonimmune tissues (SI Appendix, Fig. S5), we also knocked out MFSD1 specifically in hepatocytes [Alb-cre; (29)], renal cells [Ksp1.3-cre; (30)], and enterocytes [Villin-cre; (31)]. However, none of these tissue-specific MFSD1 deletions had an effect on liver pathology, bone marrow cellularity, or EMH (Fig. 5 F–H). Therefore, it is fair to conclude that the cells driving liver pathology and EMH in Mfsd1−/− mice remain uncharacterized.
Recent reports showed that MFSD1 deficiency in mice as well as loss of function mutations in GIMAP5 in both mice and humans results in capillarization of liver sinusoidal endothelial cells [LSECs; (9, 32)], a dedifferentiation of LSECs toward a common vascular endothelial cell type, which precedes liver fibrosis in various liver diseases including nonalcoholic steatohepatitis [NASH; (33)]. It is well established that lipid accumulation promotes LSEC capillarization in mouse models of nonalcoholic fatty liver disease (NAFLD) and in human NAFLD patients (34–37). However, it is unclear whether disruption of the MGG complex is also associated with NAFLD. If so, what are the factors that drive LSEC capillarization? To address this, we carried out mass spectrometry analysis of serum proteome using samples isolated from Mfsd1−/− or Gimap5−/− mice and wild-type littermates of each strain. We hypothesized that if Mfsd1−/− or Gimap5−/− mice have a deficiency or excess of the substrate molecule transported by the MGG complex, they might also display a deficiency or excess of a substrate carrier protein and proteomic analysis of serum might detect a difference between WT and mutant animals. We employed a protein fractionation strategy to facilitate detection of low-abundance serum proteins while minimizing interference by abundant proteins in serum such as albumin (38).
We obtained striking and unambiguous proteomic data in serum from both Mfsd1−/− and Gimap5−/− mice (Fig. 5 I and J and Dataset S2; highlighted in red). Polymeric immunoglobulin receptor (pIgR), a receptor that mediates transcytosis of dimeric IgA across mucosal epithelia, was significantly elevated in serum from Mfsd1−/− and Gimap5−/− mice compared to wild-type littermates (16.2-fold and 14.8-fold increase, respectively; Fig. 5K and Dataset S2). Previous studies found significant upregulation of pIgR in patients with NAFLD which was further validated in mouse models of NAFLD (39). Therefore, serum pIgR has been proposed as a new biomarker for NAFLD. Importantly, the onset of liver pathology and EMH in Mfsd1−/− and Glmp−/− mice is age-dependent. Young animals (<6 wk old) are relatively normal in liver morphology with no signs of EMH. The animals gain strikingly abnormal liver morphology characterized by irregular capsular surface and EMH around 10 to 15 wk of age (Fig. 5F). In agreement with these observations, Mfsd1−/− mice exhibited a progressive age-dependent increase in serum pIgR levels compared to wild-type littermates (Fig. 5L and Dataset S3).
It is known that a GLMP-deficient mouse strain (also known as Ncu-g1gt/gt mouse) develops lipid deposition in the liver due to increased de novo lipogenesis (7, 11). Since lipid accumulation in the liver promotes LSEC capillarization, we tested whether disruption of the MGG complex, especially in the setting of GIMAP5 deficiency, would result in lipid accumulation in the liver and other tissues. As mentioned earlier, GLMP stabilizes MFSD1 (Fig. 3E). Therefore, it was not surprising to see lipid deposition in livers from Mfsd1−/− mice, as in livers from GLMP-deficient mice (Fig. 5M). Interestingly, GIMAP5 deficiency also resulted in lipid accumulation in the liver, to a greater extent than seen in Mfsd1−/− or Glmp−/− mice. We also confirmed lipid accumulation in Mfsd1−/−, Glmp−/−, and Gimap5−/− bone marrow (Fig. 5M), a phenotype that is often associated with reduced bone marrow hematopoiesis (40), which explains the reduced bone marrow cellularity in Mfsd1−/− and Glmp−/− mice (Fig. 5A and SI Appendix, Fig. S2K). Together, these results indicate that dysregulated lipid metabolism caused by disruption of the MGG complex is a key pathogenic factor in liver pathology and possibly EMH.
Discussion
To maintain whole-organismal homeostasis, individual cells must meet physiological requirements by exchanging chemical matter across biological membranes via transporters. Cells of the immune systems are no exception, yet little is known of the regulation of these processes. SLC proteins are essential to maintain these biological processes by transporting a broad range of solutes across the cellular membranes. Despite the importance of SLCs to maintain physiological functions in cells, a significant portion (~30%) of the family has not yet been characterized, nor have the diseases that result from SLC dysregulation (1).
MFSD1 is a poorly characterized SLC belonging to the MFS transporter clade. Through a forward genetic screen in ENU-mutagenized mice, we established that MFSD1 performs an essential function in lymphocyte development. Although a previous study described an essential role for MFSD1 and GLMP in maintaining liver homeostasis (9) the role of MFSD1 in immunity was not recognized in that study. Considering the physiological role of GLMP in MFSD1 stabilization, it was not surprising to find that GLMP deficiency also resulted in immune deficiency (as well as liver pathology) in mice.
By MFSD1 interactome analysis, we determined that MFSD1-GLMP directly interacts with GIMAP5, forming a complex that maintains normal GIMAP5 levels in lymphocytes. However, we demonstrated that the double knockout Mfsd1−/−Glmp−/− mice and each of the single knockout mice displayed similar CD8+ T cell phenotypes (Fig. 3 F and H). This indicates that MFSD1 and GLMP do not have independent roles in GIMAP5 stabilization. Since GLMP functions as an accessory subunit for MFSD1 and interacted exclusively with MFSD1 in our experiments, we suspect that reduced GIMAP5 expression in GLMP-deficient cells is a secondary effect caused by reduced MFSD1 expression.
Reduced GIMAP5 expression in Mfsd1 or Glmp knockout CD8+ T cells or complete GIMAP5 deficiency in Gimap5−/− mice leads to aberrant mTOR activation in lymphocytes under basal conditions and in response to several stimuli. It has been reported that aberrant mTORC1 activation upon deletion of its negative regulator TSC1 disrupts quiescence of T cells and predisposes them to apoptotic cell death (41–43). Therefore, we suspected that the MGG complex might act as a negative regulator of mTOR signaling in lymphocytes, like TSC1. However, further studies of the effect of mTORC1 loss of function in either Mfsd1−/− or Gimap5−/− mice suggested a different interpretation. Taking advantage of a mouse strain harboring a viable missense mutation of Mtor that significantly impairs the stability and kinase activity of mTOR, we were surprised to find that Mfsd1 and Mtor double mutation was lethal in mice. Moreover, conditional Rptor deletion in GIMAP5-deficient T cells also failed to rescue T cell deficiency, suggesting that aberrant mTOR activation caused by disruption of the MGG complex is a compensatory response to support lymphocyte survival. This conclusion is consistent with reports that insufficient mTOR activity also has deleterious consequences for lymphocyte development (44).
Besides immunity, MFSD1 and GIMAP5 support homeostasis of liver sinusoidal endothelial cells (LSECs). A recent paper reported that GIMAP5 deficiency promotes dedifferentiation of LSECs into capillarized endothelial cells, a process that causes portal hypertension (32). MFSD1 deficiency also causes LSEC capillarization in mice (9). Although the roles of MFSD1 and GIMAP5 in preventing LSEC degeneration appear clear, we question whether MFSD1 or GIMAP5 function in endothelial cells is essential to avert liver pathology and to suppress EMH for the following reasons: i) endothelial cell–specific Mfsd1 deletion using VECad-cre driver failed to recapitulate the liver pathology and EMH as seen in global Mfsd1 knockout mice, and ii) tamoxifen-induced endothelial cell–specific Gimap5 deletion using VECad-cre-ERT2 was sufficient to induce LSEC degeneration, but it was not stated that this was sufficient to cause liver pathology [it should be noted that EMH was not the focus of the study; (32)]. Based upon the data from mice with Tie2-cre driven Mfsd1 deletion that successfully recapitulated the liver pathology seen in Mfsd1−/− mice, we attempted to distinguish the cell type in which deletion of MFSD1 resulted in liver pathology and EMH. However, none of the endothelial- or non-endothelial-specific conditional knockouts we tested caused abnormal liver morphology and EMH. These results indicate that liver pathology and EMH may require MFSD1 mutation in multiple cell types simultaneously, which we have not yet tested (e.g., hepatic stellate cells, endothelial cells, and Küpffer cells). Disruption of the MGG complex results in lipid deposition in the liver, which is known to drive LSEC capillarization (34–37). Identifying cell types responsible for the increased de novo lipogenesis and neighboring cells that make close contact with them may help to pinpoint the necessary cell types that trigger liver pathology and EMH.
Without a functional lysosomal transmembrane MGG protein complex, normal hematopoiesis in the bone marrow fails and is driven to the spleen, liver, and perhaps other sites. Lack of the protein complex may affect lysosomal metabolism or solute contents. MSFD1 deficiency has been shown to diminish the size of lysosomes compared to those from wild-type cells (9). However, MFSD1 deficiency did not significantly alter lysosomal pH in cells. In the future, it will be crucial to identify the substrate cargo of the protein complex and identify upstream signals that the MGG complex responds to in support of lymphocyte development and liver homeostasis. This information may help to explain whether and how the static lymphocyte phenotypes are related to the progressive liver pathology in MGG mutant mice. Moreover, this substrate cargo or its deficiency might be important in other forms of pathology such as NASH and cirrhosis, and consequent portal hypertension. The present study provides insights into how MFSD1 works at the interface between lysosomal metabolism and immunity by uncovering essential proteins that cooperate with GIMAP5.
Materials and Methods
Mice.
As we have described previously (45–47), 8- to 10-wk-old C57BL/6J males purchased from The Jackson Laboratory were mutagenized with N-ethyl-N-nitrosourea (ENU). Mutagenized G0 males were bred to C57BL/6J females, and the resulting G1 males were crossed to C57BL/6J females to produce G2 mice. G2 females were backcrossed to their G1 sires to yield G3 mice, which were screened for phenotypes. Whole-exome sequencing and mapping were performed as described previously (12). To generate Mfsd1−/−, Glmp−/−, Gimap5−/−, Mfsd1+/FLAG, Mfsdfl/fl, and MtorS1324T/S1324T mice, female C57BL/6J mice were superovulated by injection of 6.5 U pregnant mare serum gonadotropin (PMSG; Millipore), followed by injection of 6.5 U human chorionic gonadotropin (hCG; Sigma-Aldrich) 48 h later. The superovulated mice were subsequently mated overnight with C57BL/6J male mice. The following day, fertilized eggs were collected from the oviducts and in vitro–transcribed Cas9 mRNA (50 ng/μL) and small base-pairing guide RNAs (50 ng/μL; Mfsd1: 5′-CCGCCTGGCGCACCGGCTCG-3′, Glmp: 5′-CTTGTGTGTTCAGCTCCATT-3′, Gimap5: 5′-TACAAGAATCTAGCTGCCTG-3′, Mfsd1+/FLAG: 5′-TGCAGGTAAAGCCGTGAGAA-3′, Mfsd1-floxed: 5′-floxed: 5′-CTTGTGGAAGTTCTTTGGAA-3′, 3′-floxed: 5′-GAAATCTAAGATGACCATCA-3′, MtorS1324T: 5′-TTCATTCAGCTCAGACCAGC-3′) were injected into the cytoplasm or pronucleus of the embryos. The injected embryos were cultured in M16 medium (Sigma-Aldrich) at 37 °C in 5% CO2. For the production of mutant mice, two-cell stage embryos were transferred into the ampulla of the oviduct (10 to 20 embryos per oviduct) of pseudo-pregnant Hsd:ICR (CD-1) female mice (Harlan Laboratories). Mice were housed in specific pathogen-free facilities at the University of Texas Southwestern Medical Center and all experimental procedures were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee.
C57BL/6.SJL (CD45.1; JAX:002014), Rag2−/− (JAX:008449), OT-I (JAX:003831), OT-II (JAX:004194), Mb1-cre (JAX:020505), Cd4-cre (JAX:022071), Lepr-cre (JAX:008320), VECad-cre (JAX:006137), Clec4f-cre (JAX:033296), Alb-cre (JAX:003574), and Rptorfl/fl (JAX:013138) mice were purchased from The Jackson Laboratory. [Col2.3-cre mice were obtained from Barbara Kream at University of Connecticut Health Center; (24). Gp78−/− mice were generated by us in our previous work (46). Mfsd1w/w;Rag2−/−, Mfsd1−/−;OT-I, Mfsd1−/−;OT-II, Mfsd1−/−; Glmp−/−, Cd4-cre;Mfsdfl/fl, Mb1-cre;Mfsdfl/fl, Lepr-cre;Mfsdfl/fl, Col2.3-cre;Mfsdfl/fl, VECad-cre;Mfsdfl/fl, VECad-cre;Clec4f-cre;Mfsdfl/fl, Alb-cre;Mfsdfl/fl, and Gimap5−/−;Cd4-cre;Rptorfl/fl mice were generated by intercrossing mouse strains.
Flow Cytometry.
As we have described previously (46, 48), bone marrow cells, thymocytes, splenocytes, or peripheral blood were isolated and red blood cell (RBC) lysis buffer was added to remove the RBCs. Cells were stained at a 1:100 to 1:200 dilution with mouse fluorochrome-conjugated monoclonal antibodies specific for the following murine cell surface markers encompassing the major immune lineages: B220 (clone RA3-6B2, BD), CD19 (clone 1D3, BD), IgM (clone R6-60.2, BD), IgD (clone 11-26c.2a, Biolegend), CD3ε (clone 145-2C11, BD), CD4 (clone RM4-5, BD), CD5 (clone 53-7.3, BD), CD11c (clone HL3, BD), CD44 (clone IM7, BD), CD43 (clone S7, BD), CD25 (clone PC61, Biolegend), CD21/CD35 (clone 7E9, Biolegend), CD23 (clone B3B4, BD), Ly-51 (clone BP-1, BD), CD8α (clone 53-6.7, Biolegend), CD11b (clone M1/70, Biolegend), NK1.1 (clone PK136, BD), F4/80 (clone BM8.1, Tonbo), CD62L (clone MEL-1, Tonbo), and in the presence of Fc shield (clone 2.4G2, Tonbo) for 1 h at 4 °C as described previously. After staining, cells were washed twice in PBS and analyzed by flow cytometry. To stain the hematopoietic stem cells, bone marrow and liver cell suspensions were isolated and stained with Alexa Fluor 700-conjugated lineage cocktail (B220, CD3, CD11b, Ly-6G/6C, and Ter-119; Biolegend), CD16/32 (93, eBioscience), CD34 (RAM34, eBioscience), CD135 (A2F10, Biolegend), c-Kit (2B8, Biolegend), IL-7Rα (SB/199, BD), and Sca-1 (D7, Biolegend) for 1 h at 4 °C. After staining, cells were washed twice in PBS and analyzed by flow cytometry. Data were acquired on an LSRFortessa (BD Bioscience) and analyzed with FlowJo software (Treestar).
Bone Marrow Transplantation.
Bone marrow chimeras were made using standard procedures as described in our previous paper. As we have described previously (46), 6- to 8-wk-old recipient mice were lethally irradiated with 15 Gy via gamma radiation (X-RAD 320, Precision X-ray Inc.). The mice were given an intravenous injection of 5 × 106 bone marrow cells derived from the tibia and femurs of the donors. Mice were maintained on antibiotics for 4 wk after irradiation. Twelve weeks after bone marrow engraftment, the chimeras were euthanized to assess lymphocyte and hematopoietic stem cell development in peripheral blood and liver by flow cytometry. Chimerism was assessed using congenic CD45 markers.
In Vivo T Cell Activation.
Splenic Mfsd1+/+;OT-I, Mfsd1−/−;OT-I, Mfsd1+/+;OT-II, and Mfsd1−/−;OT-II T cells (CD45.2) were purified using the EasySep™ Mouse CD8+ or CD4+ T Cell Isolation Kit (StemCell Technologies). As we have described previously (46), cells were labeled with 5 μM CellTrace Far Red (Mfsd1+/+ OT-I or OT-II) or 5 μM CellTrace Violet (Mfsd1w/w;OT-I or OT-II), and equal number of stained cells (2 × 106) cells were injected by retro-orbital injection into wild-type CD45.1 mice. The next day, recipients were injected with either 100 µg of soluble OVA in 200 μL of PBS or 200 μL of sterile PBS as a control. Splenocytes were prepared three days after OVA injection and stained with CD45.1, CD45.2, CD3, CD4, and CD8 antibodies. Antigen (OVA)-specific T cell expansion was analyzed by flow cytometry based on Far Red or Violet intensity of dividing OT-I or OT-II cells.
Detection of Apoptosis.
Annexin V/PI labeling and detection was performed with the FITC-Annexin V Apoptosis Detection Kit I (BD Bioscience) according to the manufacturer’s instructions as previously described (46).
Mass Spectrometry Analysis of MFSD1 Interactome.
A retrovirus packaging cell line, PT67 (ATCC), was transfected with a bicistronic IRES-GFP containing retroviral vector (49) encoding FLAG-tagged MFSD1 (pMCSV-IRES-GFP-MFSD1-FLAG) or empty vector (pMCSV-IRES-GFP-FLAG) using Lipofectamine 2000 reagent (Life Technologies). Forty-eight hours after transfection, cells were harvested, and GFP-expressing cells were sorted using a FACSAria II (BD). The sorted PT67 cells underwent three rounds of sorting to enrich GFP-expressing cells up to 90%. Retroviruses were harvested from culture media of GFP-expressing PT67 cells and concentrated using the Retro-X concentrator kit (Takara) according to the manufacturer’s instructions. Wild-type EL4 T cells were transduced with retroviruses and GFP expressing cells were FACS sorted as described above. The GFP-expressing EL4 T cells underwent three rounds of sorting to enrich GFP-expressing cells up to 95%. Immunoblot was performed to confirm expression of FLAG-tagged MFSD1.
Immunoprecipitation was performed using anti-FLAG M2 agarose beads (Sigma) for 4 h at 4 °C, and beads were washed six times in NP-40 lysis buffer. The proteins were eluted with 3X-FLAG peptides. Eluted proteins were precipitated overnight at 4 °C by addition of trichloroacetic acid (TCA) (Thermo) to 23% (v/v), followed by centrifugation (30 min at 16,100 × g). After acetone rinse, as written in ref. (50), “Proteins were solubilized in 8 M urea 100 mM tetraethylammonium bromide (TEAB) pH 8.5 and reduced with 5 mM Tris(2-carboxyethyl)phosphine hydrochloride (Sigma-Aldrich, product C4706) and alkylated with 55 mM 2-chloroacetamide (Sigma-Aldrich, product 22790). Proteins were digested for 18 h at 37 °C in 2 M urea 100 mM Tris pH 8.5, with 0.5 μg trypsin (Promega, product V5111). The digestion was stopped by addition of formic acid to 5%. Samples were analyzed using a QExactive HF (Thermo Scientific) or Orbitrap Fusion Lumos Tribrid mass spectrometer (Thermo Scientific). Protein and peptide identification were done with MSFragger [version 17.1; (51)] (https://fragpipe.nesvilab.org/) using a mouse protein database downloaded from UniProt (https://Uniprot.org) (one sequence per gene 9/13/2022, 21,984 entries), common contaminants and reversed sequences added. The search space included all fully tryptic peptide candidates with a fixed modification of 57.021464 on C, variable modification of 15.9979 on M and 42.0106 on the N terminus. MS1 quantification was done with total intensity and no match between runs. Protein intensity values were combined for replicates.”
Protein Fractionation of Serum.
Serum protein fractionation was performed by DiffPOP [Differential Precipitation of Proteins; (38)]. Briefly, 10 μL of serum was mixed with 100 μL of Phosphoprotein Buffer A (Takara) and 140 μL water. Precipitated proteins were isolated into separate fractions by centrifugation after addition of acidified methanol (90% MeOH, 1% acetic acid). Twelve protein fractions were isolated from each sample corresponding to acidified methanol of these percentages (0, 4, 9, 14, 20 ,26, 32, 38, 43, 52, 70, and 91). Protein pellets were washed with cold acetone, dissolved in 120 μL 8 M urea 100 μM triethylammonium bicarbonate (TEAB). One quarter of the pellet was reduced with 5 mM Tris(2-carboxyethyl)phosphine hydrochloride (Sigma-Aldrich, product C4706) and alkylated with 55 mM 2-chloroacetamide (Sigma-Aldrich, product 22790). Samples were diluted to 2 M urea and digested for 18 h with 0.5 μg trypsin (Promega). The digestion was stopped by addition of formic acid to 5%.
Samples were loaded onto Evosep tips and analyzed with timsTOF Pro (Bruker). Protein and peptide identification were performed with ProLucid using a mouse protein database downloaded from UniProt (uniprot.org) (one sequence per gene 9/13/2022, 21,984 entries), with common contaminants and reversed sequences added by IP2 Integrated Proteomics Pipeline (version 6.7.1) at The Scripps Research Institute [goldfish.scripps.edu; (52)]. The search space included all semi-tryptic peptide candidates with a fixed modification of 57.02146 on cysteine. Results were filtered with DTASelect with parameters: -p 2 -y 1 --trypstat --pfp 0.01 --extra --pI --DB --dm -in -t 1 (53).
Unfractionated Serum.
Three microliters of serum were added to 97 μL of water and proteins were precipitated overnight at 4 °C by addition of 30 μL of TCA. Proteins were pelleted by centrifugation (30 min at 16,100 × g). After acetone rinse, proteins were solubilized in 8 M urea 100 mM TEAB pH 8.5 and reduced with 5 mM Tris(2-carboxyethyl)phosphine hydrochloride (Sigma-Aldrich, product C4706) and alkylated with 55 mM 2-chloroacetamide (Sigma-Aldrich, product 22790). Proteins were digested for 18 h at 37 °C in 2 M urea 100 mM Tris pH 8.5, with 0.5 μg trypsin (Promega, product V5111). The digestion was stopped by addition of formic acid to 5%. Samples were analyzed using a QExactive HF (Thermo Scientific). Protein and peptide identification were done with MSFragger [version 17.1; (51)] (https://fragpipe.nesvilab.org/) using a mouse protein database downloaded from UniProt (https://uniprot.org) (one sequence per gene 9/13/2022, 21,984 entries), common contaminants and reversed sequences added. The search space included all fully tryptic peptide candidates with a fixed modification of 57.021464 on C, variable modification of 15.9979 on M and 42.0106 on the N terminus. MS1 quantification was done with Total Intensity and no match between runs. Protein intensity values were combined for duplicates.
Plasmids.
Full-length or mutant mouse Mfsd1, Gimap5, Atp6v1b2, Glmp, Mtor, Gp78, or Lamtor1 were tagged at the C terminus with FLAG or HA epitope. All plasmids were verified by capillary sequencing.
Cell Culture and Antibodies for Immunoblotting.
Standard tissue culture and immunoblot experiments were performed as described previously (46, 54, 55). The following primary antibodies were used: anti-FLAG (Sigma, #F1804), anti-HA-Tag (CST, #3274), anti-Akt (CST, #4691), anti-phospho-Akt (Ser473; CST, #4060), anti-phospho-p44/42 MAPK (Thr202/Tyr204; CST, #4370), anti-p44/42 MAPK (CST, #4695), anti-Caspase-3 (CST, #9662), anti-cleaved Caspase-3 (Asp175; CST, #9661), anti-Caspase-9 (CST, #9508), anti-cleaved Caspase-9 (Asp353; CST, #9509), anti-GAPDH (CST, #5174), anti-GIMAP5 (gift from Dr. Geoff Butcher, MAC421), anti-LAMTOR1 (CST, #8975), anti-mTOR (CST, #2983), anti-phospho-mTOR (Ser2448; CST, #5536), anti-phospho-p70 S6 Kinase (Thr389; CST, #9234), anti-p70 S6 Kinase (CST, #9202) anti-phospho-4E-BP1 (Thr37/46; CST, #2855), anti-4E-BP1 (CST, #9644), and anti-β-actin (CST, #4970).
Amino Acid, Glucose, and Cholesterol Starvation and Stimulation.
Splenic CD8+ T or B cells were isolated using the EasySep™ Mouse CD8+ T or pan B cell Isolation Kit (StemCell Technologies), respectively. Cells were washed twice with sterile PBS, and amino acid, glucose, or amino acid and glucose starvation was performed by incubating cells in amino acid–, glucose-, or amino acid– and glucose-free RPMI-1640 media (US Biological) containing 10% dialyzed FBS (Invitrogen) at 37 °C for 2 h. Amino acid–, glucose-, or amino acid– and glucose-free RPMI-1640 media were complemented with 2 g/L of sodium bicarbonate (Sigma), adjusted pH to 7.4, and filter sterilized. Cells were then stimulated by adding the standard 20 amino acids, glucose, or both to the final concentrations indicated in regular RPMI-1640 medium for 45 min. Where inhibitor treatment was performed, cells were incubated with 500 nM of Torin 1 (Invivogen) during the entire starvation and stimulation period as described previously (14).
Cholesterol starvation and stimulation were performed as described previously (56). Splenic CD8+ T cells were rinsed twice in sterile PBS and incubated in amino acid-free or regular RPMI-1640 media (US Biological) containing 0.5% methyl-β-cyclodextrin (MCD, Sigma) containing 0.5% lipid-depleted serum (LDS) for 2 h. LDS was prepared as described previously (57). Cells were then transferred to amino acid–free or regular RPMI-1640 containing 0.1% MCD and 0.5% LDS and stimulated with 50 µg/mL of water-soluble cholesterol (Sigma) for 2 h. mTOR activation was analyzed by immunoblot analysis as described above.
Histology.
Tissues for Oil Red O (ORO) staining were harvested from mice and fixed in methanol-free 4% (v/v) paraformaldehyde at 4 °C for overnight and equilibrated in 18% (w/v) sucrose for 48 h. Tissue sections were stained for ORO according to the manufacturer’s instructions (Sigma-Aldrich).
Statistical Analysis.
The statistical significance of differences between two or more groups was analyzed with GraphPad Prism using the indicated analyses in each figure. Differences in values were considered statistically significant when P < 0.05. P values are denoted by *P < 0.05; **P < 0.01; ***P < 0.001; and NS, not significant with P > 0.05.
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Acknowledgments
We would like to thank Drs. Geoff Butcher and Barbara Kream for generously providing the anti-GIMAP5 antibody (clone: MAC21) and Col2.3-cre mice, respectively. We also thank the University of Texas Southwestern Medical Center (UTSW) Proteomics and Histo Pathology Core facilities for assistance with proteomics and histology experiments, respectively. This work was supported by the NIH (AI125581 to B.B.).
Author contributions
X.Z., J.J.M., B.B., and J.H.C. designed research; X.Z., J.J.M., J.K.D., A.M.P., J.A.S., J.W., K.K., S.L., and J.H.C. performed research; X.Z., J.J.M., and J.H.C. analyzed data; and E.M.Y.M., B.B., and J.H.C. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
Reviewers: M.K., La Jolla Institute for Immunology; and B.S., University of California San Diego (UCSD).
Contributor Information
Bruce Beutler, Email: Bruce.Beutler@UTSouthwestern.edu.
Jin Huk Choi, Email: Jin.Choi@UTSouthwestern.edu.
Data, Materials, and Software Availability
Raw mass spectrometry data have been deposited in the MassIVE repository with accession no. MSV000092538 (https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?accession=MSV000092538; (58)) and MSV000092540 (https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?accession=MSV000092540; (59)).
Supporting Information
References
- 1.Cesar-Razquin A., et al. , A call for systematic research on solute carriers. Cell 162, 478–487 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Lin L., Yee S. W., Kim R. B., Giacomini K. M., SLC transporters as therapeutic targets: Emerging opportunities. Nat. Rev. Drug Discov. 14, 543–560 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chapel A., et al. , An extended proteome map of the lysosomal membrane reveals novel potential transporters. Mol. Cell Proteomics 12, 1572–1588 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perland E., et al. , The novel membrane-bound proteins MFSD1 and MFSD3 are putative SLC transporters affected by altered nutrient intake. J. Mol. Neurosci. 61, 199–214 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sreedharan S., Stephansson O., Schioth H. B., Fredriksson R., Long evolutionary conservation and considerable tissue specificity of several atypical solute carrier transporters. Gene 478, 11–18 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Yan N., Structural biology of the major facilitator superfamily transporters. Annu. Rev. Biophys. 44, 257–283 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Kong X. Y., et al. , Increased glucose utilization and decreased fatty acid metabolism in myotubes from Glmp(gt/gt) mice. Arch. Physiol. Biochem. 122, 36–45 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Kong X. Y., et al. , Loss of lysosomal membrane protein NCU-G1 in mice results in spontaneous liver fibrosis with accumulation of lipofuscin and iron in Kupffer cells. Dis. Model Mech. 7, 351–362 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Massa Lopez D., et al. , The lysosomal transporter MFSD1 is essential for liver homeostasis and critically depends on its accessory subunit GLMP. Elife 8, e50025 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lopez D. M., Kahlau L., Jungnickel K. E. J., Low C., Damme M., Characterization of the complex of the lysosomal membrane transporter MFSD1 and its accessory subunit GLMP. FASEB J. 34, 14695–14709 (2020). [DOI] [PubMed] [Google Scholar]
- 11.Kong X. Y., et al. , Lack of the lysosomal membrane protein, GLMP, in mice results in metabolic dysregulation in liver. PLoS One 10, e0129402 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang T., et al. , Real-time resolution of point mutations that cause phenovariance in mice. Proc. Natl. Acad. Sci. U.S.A. 112, E440–449 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adzhubei I. A., et al. , A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zoncu R., et al. , mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 334, 678–683 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.MacMurray A. J., et al. , Lymphopenia in the BB rat model of type 1 diabetes is due to a mutation in a novel immune-associated nucleotide (Ian)-related gene. Genome Res. 12, 1029–1039 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barnes M. J., et al. , Loss of T cell and B cell quiescence precedes the onset of microbial flora-dependent wasting disease and intestinal inflammation in Gimap5-deficient mice. J. Immunol. 184, 3743–3754 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schulteis R. D., et al. , Impaired survival of peripheral T cells, disrupted NK/NKT cell development, and liver failure in mice lacking Gimap5. Blood 112, 4905–4914 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen X. L., et al. , GIMAP5 deficiency is associated with increased AKT activity in T lymphocytes. PLoS One 10, e0139019 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hentges K. E., et al. , FRAP/mTOR is required for proliferation and patterning during embryonic development in the mouse. Proc. Natl. Acad. Sci. U.S.A. 98, 13796–13801 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alva J. A., et al. , VE-Cadherin-Cre-recombinase transgenic mouse: A tool for lineage analysis and gene deletion in endothelial cells. Dev. Dyn. 235, 759–767 (2006). [DOI] [PubMed] [Google Scholar]
- 21.Joseph C., et al. , Deciphering hematopoietic stem cells in their niches: A critical appraisal of genetic models, lineage tracing, and imaging strategies. Cell Stem. Cell 13, 520–533 (2013). [DOI] [PubMed] [Google Scholar]
- 22.Lin S. C., et al. , Endothelial-to-osteoblast conversion generates osteoblastic metastasis of prostate cancer. Dev Cell 41, 467–480 e463 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DeFalco J., et al. , Virus-assisted mapping of neural inputs to a feeding center in the hypothalamus. Science 291, 2608–2613 (2001). [DOI] [PubMed] [Google Scholar]
- 24.Liu F., et al. , Expression and activity of osteoblast-targeted Cre recombinase transgenes in murine skeletal tissues. Int. J. Dev. Biol. 48, 645–653 (2004). [DOI] [PubMed] [Google Scholar]
- 25.Christopher M. J., Liu F., Hilton M. J., Long F., Link D. C., Suppression of CXCL12 production by bone marrow osteoblasts is a common and critical pathway for cytokine-induced mobilization. Blood 114, 1331–1339 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Decker M., et al. , Leptin-receptor-expressing bone marrow stromal cells are myofibroblasts in primary myelofibrosis. Nat. Cell Biol. 19, 677–688 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ding L., Morrison S. J., Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature 495, 231–235 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ding L., Saunders T. L., Enikolopov G., Morrison S. J., Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 481, 457–462 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Postic C., et al. , Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J. Biol. Chem. 274, 305–315 (1999). [DOI] [PubMed] [Google Scholar]
- 30.Shao X., Somlo S., Igarashi P., Epithelial-specific Cre/lox recombination in the developing kidney and genitourinary tract. J. Am. Soc. Nephrol. 13, 1837–1846 (2002). [DOI] [PubMed] [Google Scholar]
- 31.Madison B. B., et al. , Cis elements of the villin gene control expression in restricted domains of the vertical (crypt) and horizontal (duodenum, cecum) axes of the intestine. J. Biol. Chem. 277, 33275–33283 (2002). [DOI] [PubMed] [Google Scholar]
- 32.Drzewiecki K., et al. , GIMAP5 maintains liver endothelial cell homeostasis and prevents portal hypertension. J. Exp. Med. 218 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang X. K., Peng Z. G., Targeting liver sinusoidal endothelial cells: An attractive therapeutic strategy to control inflammation in nonalcoholic fatty liver disease. Front. Pharmacol. 12, 655557 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Akyol G., Erdem O., Yilmaz G., Nonalcoholic fatty liver disease. Correlation with histology and viral hepatitis. Saudi. Med. J. 26, 1904–1910 (2005). [PubMed] [Google Scholar]
- 35.Bravo M., et al. , Restoration of liver sinusoidal cell phenotypes by statins improves portal hypertension and histology in rats with NASH. Sci. Rep. 9, 20183 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miyao M., et al. , Pivotal role of liver sinusoidal endothelial cells in NAFLD/NASH progression. Lab Invest. 95, 1130–1144 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Zhang Q., et al. , oxLDL induces injury and defenestration of human liver sinusoidal endothelial cells via LOX1. J. Mol. Endocrinol. 53, 281–293 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Pinto A. F. M., Diedrich J. K., Moresco J. J., Yates J. R. III, Differential precipitation of proteins: A simple protein fractionation strategy to gain biological insights with proteomics. J. Am. Soc. Mass. Spectrom. 34, 2025–2033 (2023), 10.1021/jasms.3c00182. [DOI] [PubMed] [Google Scholar]
- 39.Niu L., et al. , Plasma proteome profiling discovers novel proteins associated with non-alcoholic fatty liver disease. Mol. Syst. Biol. 15, e8793 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Naveiras O., et al. , Bone-marrow adipocytes as negative regulators of the haematopoietic microenvironment. Nature 460, 259–263 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.O’Brien T. F., et al. , Regulation of T-cell survival and mitochondrial homeostasis by TSC1. Eur. J. Immunol. 41, 3361–3370 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu Q., et al. , The tuberous sclerosis complex-mammalian target of rapamycin pathway maintains the quiescence and survival of naive T cells. J. Immunol. 187, 1106–1112 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang K., Neale G., Green D. R., He W., Chi H., The tumor suppressor Tsc1 enforces quiescence of naive T cells to promote immune homeostasis and function. Nat. Immunol. 12, 888–897 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lyszkiewicz M., et al. , LAMTOR2 (p14) controls B cell differentiation by orchestrating endosomal BCR trafficking. Front. Immunol. 10, 497 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Choi J. H., et al. , IgD class switching is initiated by microbiota and limited to mucosa-associated lymphoid tissue in mice. Proc. Natl. Acad. Sci. U.S.A. 114, E1196–E1204 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Choi J. H., et al. , LMBR1L regulates lymphopoiesis through Wnt/beta-catenin signaling. Science 364, eaau0812 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Georgel P., Du X., Hoebe K., Beutler B., ENU mutagenesis in mice. Methods Mol. Biol. 415, 1–16 (2008). [DOI] [PubMed] [Google Scholar]
- 48.Zhong X., et al. , Genetic and structural studies of RABL3 reveal an essential role in lymphoid development and function. Proc. Natl. Acad. Sci. U.S.A. 117, 8563–8572 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Holst J., et al. , Generation of T-cell receptor retrogenic mice. Nat. Protoc. 1, 406–417 (2006). [DOI] [PubMed] [Google Scholar]
- 50.Song R., et al. , Trans-Golgi protein TVP23B regulates host-microbe interactions via Paneth cell homeostasis and Goblet cell glycosylation. Nat. Commun. 14, 3652 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kong A. T., Leprevost F. V., Avtonomov D. M., Mellacheruvu D., Nesvizhskii A. I., MSFragger: Ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics. Nat. Methods 14, 513–520 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu T., et al. , ProLuCID: An improved SEQUEST-like algorithm with enhanced sensitivity and specificity. J Proteomics 129, 16–24 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tabb D. L., McDonald W. H., Yates J. R. III, DTASelect and Contrast: Tools for assembling and comparing protein identifications from shotgun proteomics. J. Proteome Res. 1, 21–26 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Choi J. H., et al. , Essential requirement for nicastrin in marginal zone and B-1 B cell development. Proc. Natl. Acad. Sci. U.S.A. 117, 4894–4901 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Choi J. H., et al. , Essential cell-extrinsic requirement for PDIA6 in lymphoid and myeloid development. J. Exp. Med. 217, e20190006 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Castellano B. M., et al. , Lysosomal cholesterol activates mTORC1 via an SLC38A9-Niemann-Pick C1 signaling complex. Science 355, 1306–1311 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Agnese S. T., Spierto F. W., Hannon W. H., Evaluation of four reagents for delipidation of serum. Clin. Biochem. 16, 98–100 (1983). [DOI] [PubMed] [Google Scholar]
- 58.Zhong X., et al. , Essential role of MFSD1-GLMP-GIMAP5 in lymphocyte survival and liver homeostasis. MassIVE. https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?accession=MSV000092538. Deposited 27 July 2023. [DOI] [PMC free article] [PubMed]
- 59.Zhong X., et al. , Essential role of MFSD1-GLMP-GIMAP5 in lymphocyte survival and liver homeostasis. MassIVE. https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?accession=MSV000092540. Deposited 27 July 2023. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Data Availability Statement
Raw mass spectrometry data have been deposited in the MassIVE repository with accession no. MSV000092538 (https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?accession=MSV000092538; (58)) and MSV000092540 (https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?accession=MSV000092540; (59)).