

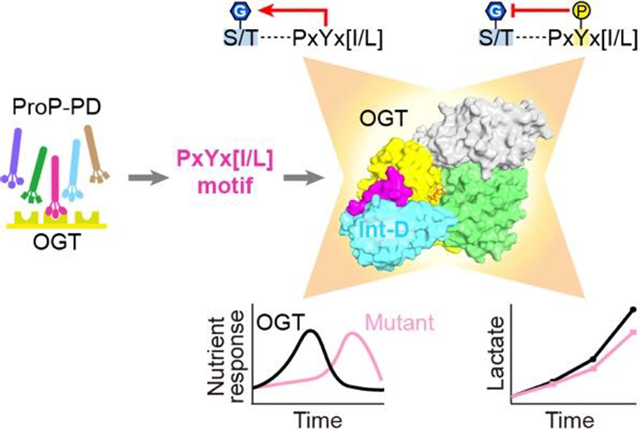
Abstract
The modification of intracellular proteins with O-linked β-N-acetylglucosamine (O-GlcNAc) moieties is a highly dynamic process that spatiotemporally regulates nearly every important cellular program. Despite its significance, little is known about the substrate recognition and regulation modes of O-GlcNAc transferase (OGT), the primary enzyme responsible for O-GlcNAc addition. In this study, we have identified the intervening domain (Int-D), a poorly understood protein fold found only in metazoan OGTs, as a specific regulator of OGT protein-protein interactions and substrate modification. Utilizing proteomic peptide phage display (ProP-PD) coupled with structural, biochemical, and cellular characterizations, we discovered a strongly enriched peptide motif, employed by the Int-D to facilitate specific O-GlcNAcylation. We further show that disruption of Int-D binding dysregulates important cellular programs including response to nutrient deprivation and glucose metabolism. These findings illustrate a mode of OGT substrate recognition and offer significant insights into the biological roles of this unique domain.
Graphical Abstract
Introduction
Regulation of intracellular primary metabolites is a delicate balance between excess and deficit, each of which can have deleterious effects on the health of a cell. One major signaling corridor for cellular nutrient sensing is the hexosamine biosynthetic pathway (HBP), which uses amino acids, nucleotides, carbohydrates, and fatty acids to produce uridine diphosphate N-acetylglucosamine (UDP-GlcNAc)1. This allows the level of each essential metabolite to influence HBP flux and cellular concentration of the nucleotide sugar. In turn, UDP-GlcNAc serves as the activated sugar donor of O-GlcNAc transferase (OGT), the single enzyme responsible for most mono-O-linked β-N-acetylglucosamine (O-GlcNAc) addition. O-GlcNAcylation dynamically and sensitively regulates numerous highly coordinated processes including the cell cycle2, gene expression3, and proteasomal degradation4, leading to its proposed role as a central nutrient sensing system5,6. Unlike many other enzymes, OGT does not recognize an apparent sequence motif at the modification site, and little is known about this enzyme’s substrate recognition and regulation modes.
Coordination of the spatiotemporally dynamic cellular environment requires enzymes to be in the right place at the right time to interact with their targets. For many enzymes that recognize a wide breadth of protein substrates, like OGT, this is achieved by association with larger protein machinery through specific protein-protein interactions (PPIs)7. Often these interactions utilize specific short linear motifs (SLiMs)8–10. However, these binding interactions can be difficult to map as they are typically low affinity and facilitated by poorly defined shallow grooves. It is therefore necessary to apply new technology to explore these complex regulatory networks.
OGT is a multidomain enzyme consisting of 13.5 tetratricopeptide repeats (TPRs) and a catalytic region (Fig. 1a)11–13. Tandem TPRs are common protein binding motifs, directing the focus of many studies toward identifying the TPR domain’s role in substrate recognition. Our group and others have identified key TPR residues, including parallel aspartate and asparagine ladders, that facilitate OGT substrate binding14–18. Despite these efforts, a full model of OGT substrate recognition and its connection to the O-GlcNAc regulatory network remains obscure. The OGT catalytic region is comprised of twin N- and C-catalytic lobes (N-Cat and C-Cat) separated by an intervening domain (Int-D) (Fig. 1a). Both catalytic lobes have Rossmann-like structures, typical of the GT-B superfamily of glycosyltransferases11. However, human OGT and its homologs in metazoans are the only known proteins to contain an Int-D. Since its identification over a decade ago, the function of the Int-D has remained mysterious.
Figure 1. ProP-PD screening identified a specific OGT binding motif.
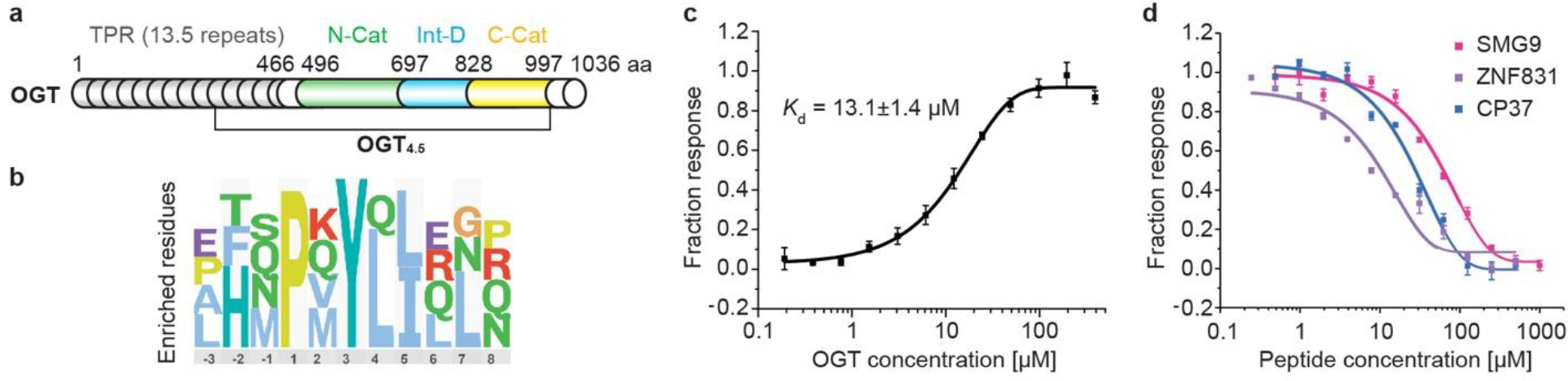
(a) Domain schematic of full-length OGT with tetratricopeptide repeat (TPR) domain in gray, N-terminal catalytic (N-Cat) domain in green, intervening domain (Int-D) in blue, and C-terminal catalytic (C-Cat) domain in yellow. The crystallization construct OGT4.5 with only 4.5 of 13.5 TPR repeats was also shown. (b) Sequence logo of highly enriched peptides from both OGT and OGT4.5 ProP-PD screens, aligned to the PxYx[I/L] motif. (c) Microscale thermophoresis (MST) binding assay of SMG9 peptide with OGT, n=3. (d) Competitive fluorescence polarization (FP) binding assay with fluorescently labeled 5-FAM-SMG9 peptide competing with unmodified SMG9, ZNF831, and consensus peptide 37 (CP37), for OGT binding, n=3. Data are presented as mean values +/− SD of three replicates.
To advance our understanding of this essential enzyme and its roles in the cell, we employ an innovative proteomic peptide phage display (ProP-PD)19 to profile PPI sites across the surface of OGT. Using X-ray crystallization and biochemical approaches, we identify a binding site in the OGT Int-D that is a motif dependent regulator of OGT binding and substrate specific O-GlcNAcylation. Moreover, we uncover a mode of indirect posttranslational modification (PTM) crosstalk through tyrosine phosphorylation of our motif. Revealing the specific binding site and mode of PTM crosstalk creates a structurally justifiable mechanism of cellular communication between phosphorylation and O-GlcNAcylation. We further demonstrate the surprising roles of this binding site in regulating the O-GlcNAcylation response to nutrient deprivation as well as lactate production. In addition to current hypotheses that propose HBP flux as a key driver of O-GlcNAcylation nutrient sensing20, we find that mutation of the Int-D can significantly delay the O-GlcNAcylation response to nutrient starvation. These findings reveal important roles of the OGT Int-D that has garnered interest for its unique character but to this point remained cryptic.
Results
ProP-PD identified a strongly enriched OGT binding motif
To directly identify biologically relevant OGT peptide binders, we employed an innovative phage display platform. The unique Proteomic Peptide Phage Display (ProP-PD) library of overlapping 16-mer peptides is designed around intrinsically disordered regions of the human proteome19,21. Compared to commonly used phage display platforms, ProP-PD features higher peptide copy number (~200 per virion) through display on the major coat protein P8 of the filamentous M13 phage, which enables enrichment of moderate to low affinity binders through avidity22. The targeted design allows us to only screen biologically relevant peptides that map to human proteins. This leads to identification of SLiMs that facilitate protein-protein interactions at amino acid resolution, which is difficult to obtain using traditional experimental approaches. We screened the ProP-PD library against two OGT constructs, full-length OGT (OGT) and the protein crystallization construct OGT4.5 (Fig. 1a, Supplemental Figure 1a). Enriched phage pools were analyzed with next-generation sequencing (NGS) and peptides were considered hits if they appeared in multiple replicates or if two overlapping sequences were identified. Remarkably, both OGT constructs yielded a similar peptide population which, using the SLiMFinder algorithm23,24, produced a strongly enriched motif PxYx[I/L] (Fig. 1b). Of the 24 most highly enriched peptides in the NGS dataset, 16 contain this exact motif or a single amino acid variant. Enrichment of a single motif, by OGT and OGT4.5, suggests this is the dominant peptide binding site localized to the OGT catalytic region. However, the absence of Ser/Thr in the motif implies that this is not a direct active site binder. Seven natural peptides and one consensus peptide (CP37) were selected for synthesis and further characterization (Extended data figure 1a).
We began assessing peptide interactions with OGT using the thermal shift assay (TSA), a rapid label-free binding assay that uses protein denaturation temperature (Tm) to evaluate interactions25. All but one peptide showed a significant Tm increase with both OGT and OGT4.5 (Extended data figure 1b). An advantage of ProP-PD is direct mapping of peptides to proteins with well-characterized biological functions. For instance, SMG9 protein is a critical component of cellular nonsense mediated mRNA decay (NMD), a process that targets transcripts with premature stop codons for degradation26. SMG9 also has a single major O-GlcNAcylation site at residue T114, located 30 amino acids upstream of the identified motif, making SMG9 an interesting target to investigate further27,28. Applying a fluorescently labeled 5-FAM-SMG9 peptide, we measured its binding affinity to OGT by microscale thermophoresis (MST)29. This binding assay showed a clear dose-response relationship and produced a Kd of 13.1 μM (Fig. 1c), which would be considered a moderately strong SLiM PPI. The remaining unlabeled, motif containing peptides were evaluated by competitive fluorescence polarization (FP) assay with the 5-FAM-SMG9 peptide (Extended data figure 1). Top binders (ZNF831 and CP37) demonstrated strong dose-response relationships with apparent binding affinity (EC50) similar to the SMG9 peptide (Fig. 1d, Extended data figure 1a).
Co-crystallization revealed a binding site in the OGT Int-D
Capitalizing on our expertise in protein crystallization, we sought to obtain structural insights into these interactions. We co-crystallized OGT4.5 with each of the three top binding peptides (SMG9: 140–155aa, ZNF831: 928–943aa, and CP37) in the presence of UDP-GlcNAc. We successfully determined the OGT4.5:UDP-GlcNAc:SMG9 and OGT4.5:UDP-GlcNAc:CP37 structures at 2.98 and 3.06 Å, respectively, while the OGT4.5:UDP-GlcNAc:ZNF831 structure was determined at 3.69 Å (Supplementary table 2 and Extended data figure 2a–d). Strikingly, unambiguous electron densities clearly show that all three peptides bind to the OGT Int-D in a highly conserved conformation (Fig. 2a, c and Extended data figure 2b, d). Peptide binding did not induce significant change to the OGT4.5:UDP-GlcNAc structure (Extended data figure 2e). Given the high similarity of these peptide binding conformations, we focus on the OGT4.5:UDP-GlcNAc:SMG9 structure in our following discussions.
Figure 2. OGT crystal structures reveal a binding site in the Int-D.
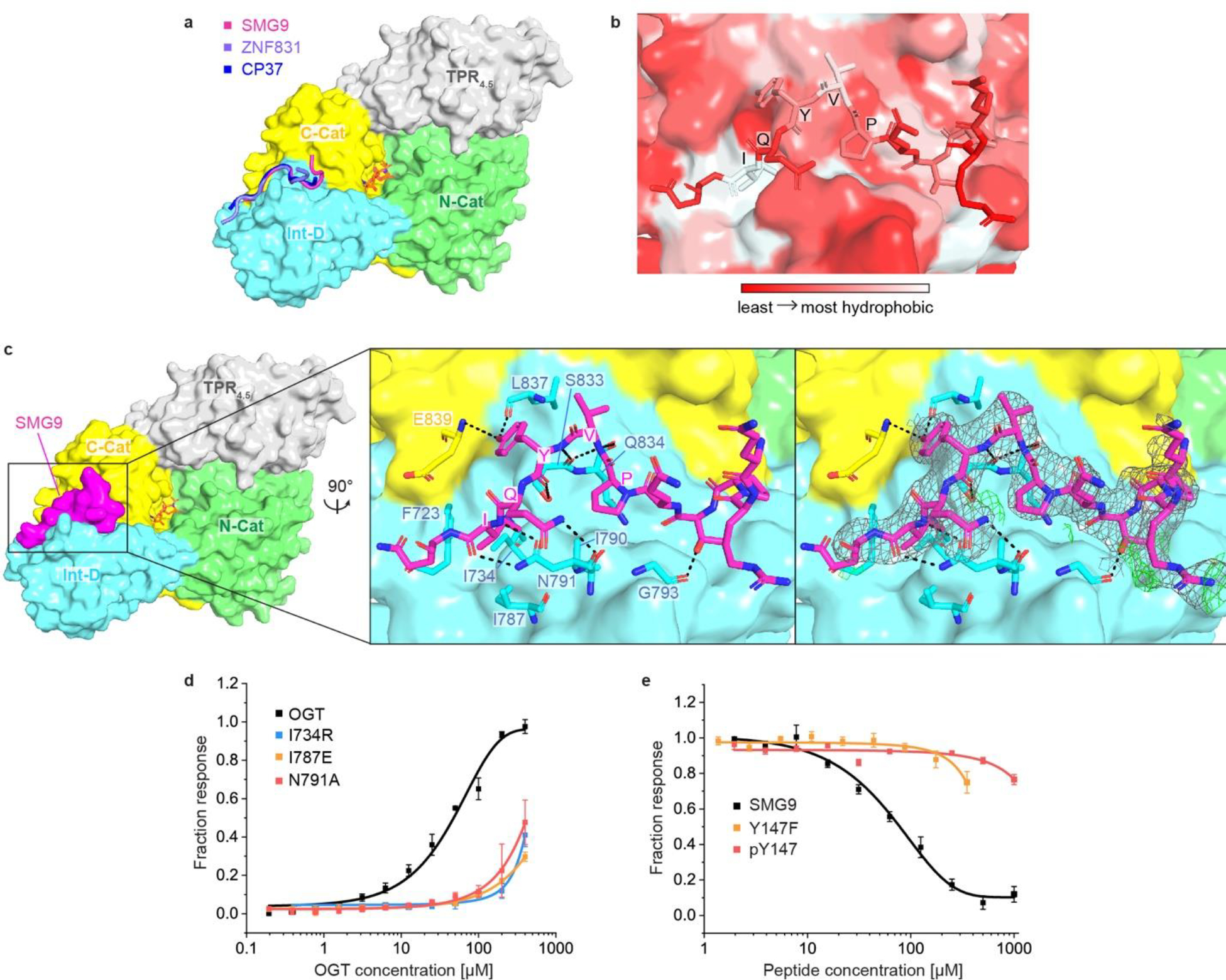
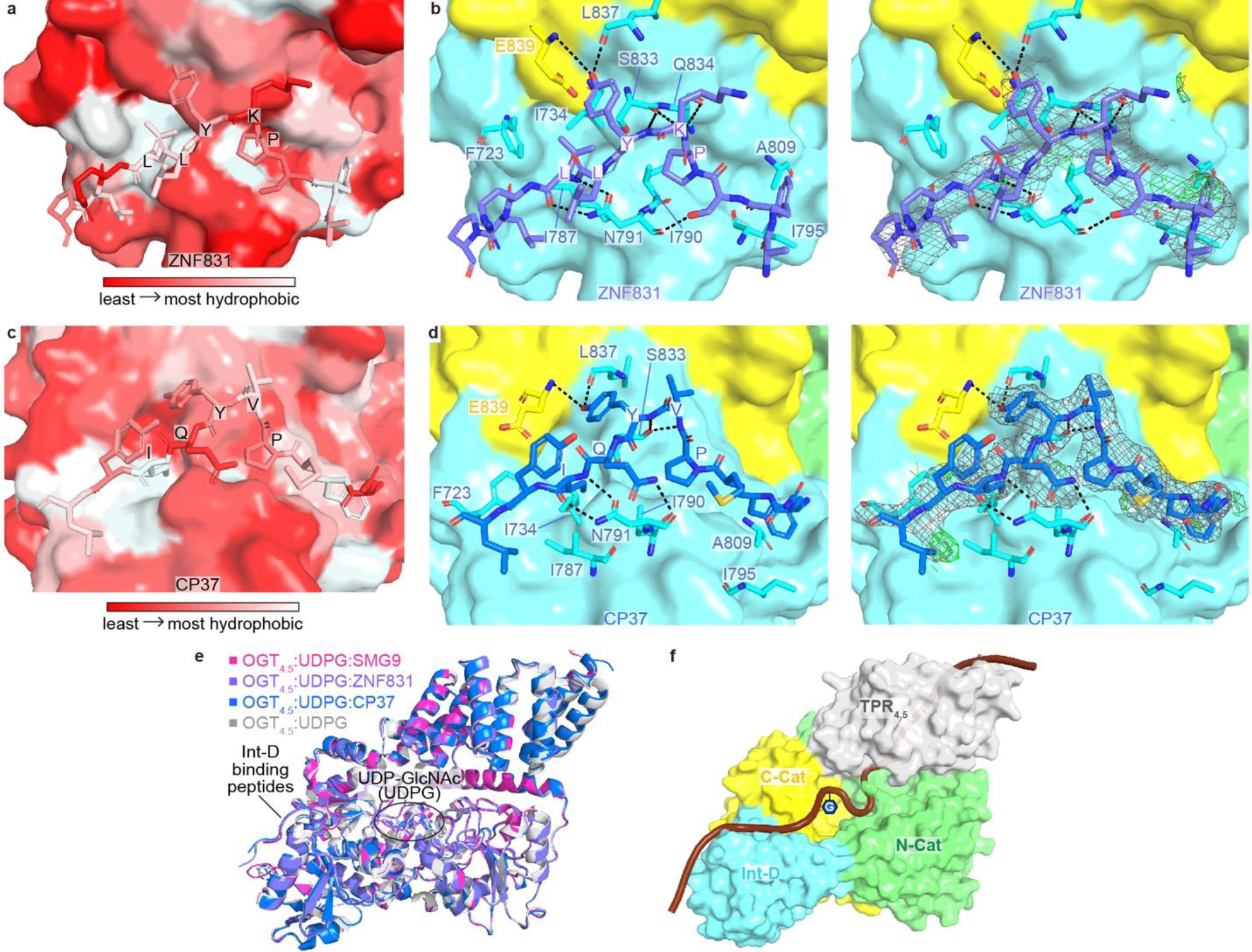
(a) OGT4.5 in complex with Int-D-binding peptides and UDP-GlcNAc. SMG9, ZNF831, and CP37 peptides are shown as cartoon in magenta, purple and blue, respectively. Protein domains in OGT4.5 are colored as in Figure 1a. UDP-GlcNAc is shown in orange sticks. (b) Zoom-in view at the Int-D binding site demonstrating hydrophobic interactions between SMG9 peptide (shown as sticks) and Int-D in OGT4.5 (shown as surface). Red to white color scale represent hydrophobicity. (c) Left: surface representation of SMG9 peptide bound to OGT4.5. Right: zoom-in view at the Int-D binding site demonstrating polar interactions between SMG9 peptide (shown as magenta sticks) and Int-D in OGT4.5 (shown as surface) with interacting OGT residues shown in sticks. 2Fo–Fc electron density map of SMG9 peptide is shown as grey mesh and contoured at 1.0 σ, Fo–Fc electron density map is shown as green mesh and contoured at 3.0 σ. (d) Saturation fluorescence polarization (FP) binding assay of fluorescently labeled 5-FAM-SMG9 peptide with wild-type (WT) OGT (black) and mutants I734R (blue), I787E (orange), and N791A (red), n=3. (e) Competitive FP binding assay of WT SMG9 (black), mutant SMG9 Y147F (orange) and phosphorylated SMG9 pY147 (red) peptides with fluorescently labeled 5-FAM-SMG9 peptide binding to OGT, n=3. Data are presented as mean values +/− SD of three replicates.
In the complex structure, SMG9 peptide binds in an elongated conformation in a shallow Int-D surface groove, displaying favorable shape complementarity (Fig. 2a, c). Peptide binding leads to 1,247 Å2 buried surface area, which is in the typical range for transient PPIs30,31. We observed strong electron density for the SMG9 peptide backbone and most of its side chains, allowing us to analyze this interaction with high confidence. The binding conformation of the SMG9 peptide is mainly supported by two types of interactions: hydrophobic (Fig. 2b) and hydrogen bonding (Fig. 2c). The motif isoleucine side chain of SMG9 peptide (corresponding to the I149 residue of SMG9 protein) reaches into a small hydrophobic pocket formed by OGT residues I734, I787 and F723. Additionally, the SMG9 motif proline (P145) residue interacts with hydrophobic I790, lining the Int-D surface groove, allowing appropriate positioning of the peptide motif. Besides these hydrophobic features, the carboxamide side chain of OGT Int-D N791 makes bidentate interactions with the backbone of SMG9 I149. Though not a motif residue, the carboxamide sidechain of Q148 in the SMG9 peptide donates a hydrogen bond to the N791 backbone carbonyl oxygen. The side-chain hydroxyl of SMG9 motif tyrosine Y147 forms double hydrogen bonds with OGT L837 and E839 backbones, while the Y147 backbone makes two additional hydrogen bonds to OGT S833. Moreover, the interactions between backbones of SMG9 V146 and OGT S833/Q834 further stabilize the peptide binding conformation. Collectively, these hydrophobic and hydrogen bonding interactions form a tangled network stabilizing the binding of SMG9 peptide in the OGT Int-D. Interestingly, while the OGT Int-D is unique to metazoans, many of the mentioned Int-D residues are only highly conserved in vertebrates (Extended data figure 3). This contrasts with the other OGT domains, which show an average of 86% sequence identity across metazoan homologs. This may suggest that the Int-D binding groove evolved specifically for vertebrate functions.
To confirm the SMG9 peptide binding mode in our crystal structure, we mutated each of the two primary binding features: the hydrophobic pocket (F723, I734, and I787) and the bidentate donor N791, and measured the changes in binding affinity. Each of the hydrophobic OGT residues were mutated to a glutamate or arginine, while the asparagine interaction was disrupted by mutation to alanine. Each mutated OGT construct was recombinantly expressed in E. coli, purified and brought to a high concentration for FP saturation binding assays with fluorescent SMG9 peptide (Supplemental Figure 1b). TSA of each OGT mutant showed no or slight changes in protein stability (Extended data figure 4a). Insertion of charged residues into the hydrophobic pocket (F723E, I734R, I787E, and I787R) as well as disruption of the bidentate backbone interaction (N791A) each shifted EC50 values by > 10-fold (Fig. 2d, Extended data figure 4b), showing that these mutations substantially disrupt SMG9 peptide binding. These results strongly support the peptide binding mode illustrated in our crystal structure and offer useful mutants for interrogation of this binding site’s role in cells.
SMG9 Y147 phosphorylation disrupts peptide binding to OGT
On the other side of this interaction, the SMG9 Y147 residue is highly conserved in our motif and makes substantial interactions with OGT Int-D (Fig. 2c), signaling its importance for binding. Using the same FP competition assay as before, mutant peptide SMG9 Y147F demonstrated > 10-fold shift in EC50 (Fig. 2e), further supporting the SMG9 binding modes observed in our crystal structure. Interestingly, Y147 is a major SMG9 phosphorylation site. Previous studies have linked phosphorylation at this site to transcriptional programs in epidermal growth factor signaling28. Using our FP competition assay, we tested the interaction of phosphorylated SMG9 peptide (pY147) with OGT. SMG9 pY147 disrupted peptide binding, to a similar degree as Y147F (Fig. 2e), consistent with our structural analysis. Tyrosine phosphorylation (pTyr) is a fundamental mechanism of cellular regulation32. Compared to the much more prevalent Ser/Thr phosphorylation, pTyr is relatively rare accounting for only 2% of the phosphoproteome. Interestingly, nearly 70% of O-GlcNAcylated proteins contain at least one pTyr site33. O-GlcNAcylation and pTyr share many commonalities with two primary functions of both modifications being signal transduction in response to extracellular stress and stimuli as well as regulation of protein localization and complex formation across various cellular processes. Dysregulation of pTyr can lead to a variety of diseases including cancer, which has made tyrosine kinases and phosphatases promising targets for therapeutic interventions34. More interestingly, tyrosine phosphorylation enzymes and the OGT Int-D appear to share similar evolutionary paths, as both primarily appear in metazoans and play more important roles as organismal complexity increases35. These parallels have long suggested cross communication between O-GlcNAcylation and tyrosine phosphorylation, but few studies have made progress in defining their interaction mechanisms.
OGT Int-D facilitates protein binding and O-GlcNAcylation
To begin investigating the biological roles of the OGT Int-D, we turned our attention to full-length protein interactions. First, we demonstrated that the OGT-SMG9 association occurs at the protein level by co-immunoprecipitation (co-IP) analysis of Flag-tagged OGT and cMyc-tagged SMG9 co-expressed in human embryonic kidney 293 cells (HEK293) (Fig. 3a and b). We then selected the SMG9-Y147F mutant as well as three OGT mutants (I734R, I787E, N791A) that most effectively disrupted SMG9 peptide binding for co-IP (Fig. 3a and b, Extended data figure 5a and b). Compared to wild-type (WT) SMG9, the SMG9-Y147F mutant substantially decreased association with OGT, further supporting the importance of Y147 side chain in their PPI. In addition, each of the OGT mutants significantly reduced the level of SMG9 association, though none of the individual mutants completely abolished SMG9 binding. A double mutant OGT (I787E-N791A) was used to co-IP SMG9 and produced similar results (Extended data figure 5c). This suggests that interaction via the Int-D site is the major facilitator of OGT-SMG9 association, but may not be the only interaction between the full-length proteins. Since all three of our tested OGT mutants disrupted SMG9 association similarly well, we decided to focus on N791A as its bidentate backbone interaction may be a more general binder. To evaluate the intrinsic activity of OGT and its Int-D mutant, we performed a radiolabeled activity assay with purified OGT or OGT-N791A, UDP-3H-GlcNAc and a CKII peptide substrate. We detected no significant difference in radio-ligand incorporation between OGT and OGT-N791A (Extended data figure 4c), demonstrating that OGT-N791A mutant retains similar intrinsic activity as WT OGT. In our previous studies, we have used O-GlcNAcase (OGA) as a model substrate and binding partner of OGT in vitro, as it contains a single major O-GlcNAcylation site at S405 that is easily detected by western blot17. OGA does not contain the PxYx[I/L] motif, or any closely related sequence variants. Indeed, co-IP of OGA with WT OGT and N791A mutant showed no difference in OGA association (Extended data figure 5d), supporting the Int-D site as a specific motif-dependent regulator of OGT protein binding.
Figure 3. Int-D binding site is a regulator of protein association and O-GlcNAcylation.
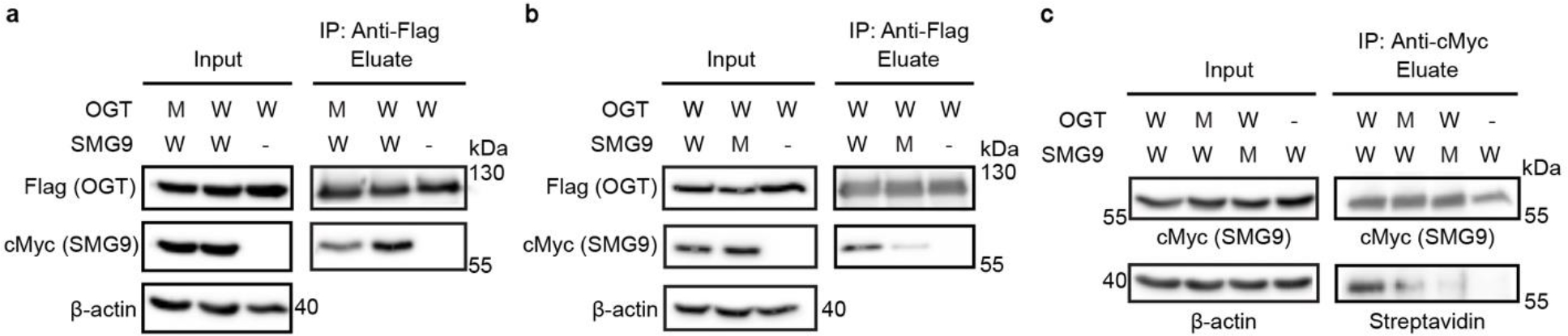
(a) Co-IP of cMyc-SMG9 with Flag-OGT (W) or Flag-OGT-N791A (M) from TRex-293 cells, followed by western blot detection. (b) Co-IP of cMyc-SMG9 (W) or cMyc-SMG9-Y147F (M) with Flag-OGT from TRex-293 cells, followed by western blot detection. (c) O-GlcNAcylation detection on SMG9 (W) or SMG9-Y147F (M) from cells co-expressed with Flag-OGT (W) or Flag-OGT-N791A (M). cMyc-tagged SMG9 (WT or Y147F mutant) was immunoprecipitated from TRex-293 cell lysate by cMyc-agarose, biotinylated by GalT assay, and detected by streptavidin-HRP far western blot. All whole cell lysates have endogenous OGT knocked down. Blots are representative of at least three biological replicates.
A recent study has reported a single O-GlcNAcylation site at T114 of SMG9, 30 amino acids upstream of our identified motif27. The proximity of this modification site to our Int-D motif implies a coordinated substrate binding model (Extended data figure 2f). To evaluate changes in SMG9 O-GlcNAcylation upon disruption of Int-D site interaction, we generated TRex-293 stable cell lines expressing Flag-tagged WT OGT or N791A mutant with endogenous OGT knockdown. Co-expression of cMyc-SMG9 or cMyc-SMG9-Y147F with each OGT construct, was followed by anti-cMyc immunoprecipitation and biotinylation of O-GlcNAc by GalT assay36. We observed dramatically reduced SMG9 O-GlcNAcylation in cells expressing mutants of the Int-D binding site as well as SMG9 motif (Fig. 3c). We similarly evaluated O-GlcNAcylation on recombinantly expressed Calmodulin-lysine N-methyltransferase (CaMKMT), another protein binder identified by the phage display screen. This protein has a single reported O-GlcNAcylation site 84 residues upstream of the Int-D binding motif and showed a similar decrease in O-GlcNAcylation when co-expressed with OGT N791A mutant (Extended data figure 5e). To determine if changes in O-GlcNAcylation are motif dependent, we also probed O-GlcNAcylation of Flag-tagged catalytically impaired OGA-D175N and OGT, two non-motif containing proteins, by western blot (Extended data figure 5f). Both proteins were comparably O-GlcNAcylated under WT OGT and N791A overexpression conditions. Collectively, this study provides evidence that the Int-D regulates OGT recognition and O-GlcNAcylation of specific substrates. Surprisingly, these results also implicate the OGT Int-D binding site as a direct facilitator of tyrosine phosphorylation crosstalk with O-GlcNAcylation. Further analysis of this binding site in silico and in cells will illuminate the extent of cross-regulation between these two important modifications.
Motif-containing proteins engage in diverse cellular roles
Leveraging the SLiMSearch algorithm37, we performed bioinformatic analysis of the PxYx[I/L] binding motif in the human proteome, excluding peptides with proline in positions 2 or 4 and those mapped to secreted or extracellular protein domains. We identified the motif in intrinsically disordered regions (IUPred score > 0.4) of 223 human proteins, 126 (56.5%) of which were found in the O-GlcNAc database with 89 proteins containing 374 assigned O-GlcNAcylation sites (Fig. 4a)38. Relative to the motif, 64 (17.1%) of reported O-GlcNAcylation sites are within 100 residues, showing modest enrichment (Fig. 4b). Gene ontology (GO) analysis of motif-containing proteins, using the Database for Annotation, Visualization, and Integrated Discovery (DAVID), showed significant enrichment (FDR < 0.05) across multiple cellular components and processes including transcription, transcription regulation, and chromatin regulation (Fig. 4c)39,40. These processes are all governed by complex systems of PPIs and PTMs that result in wide-reaching effects across the cell. Enrichment of protein binding molecular function along with other PTMs including phosphorylation, ubiquitination, and methylation, may lend further support for the Int-D binding motif as a regulator of cell signaling and protein complex formation.
Figure 4. Bioinformatic analysis of PxYx[I/L] motif containing proteins.
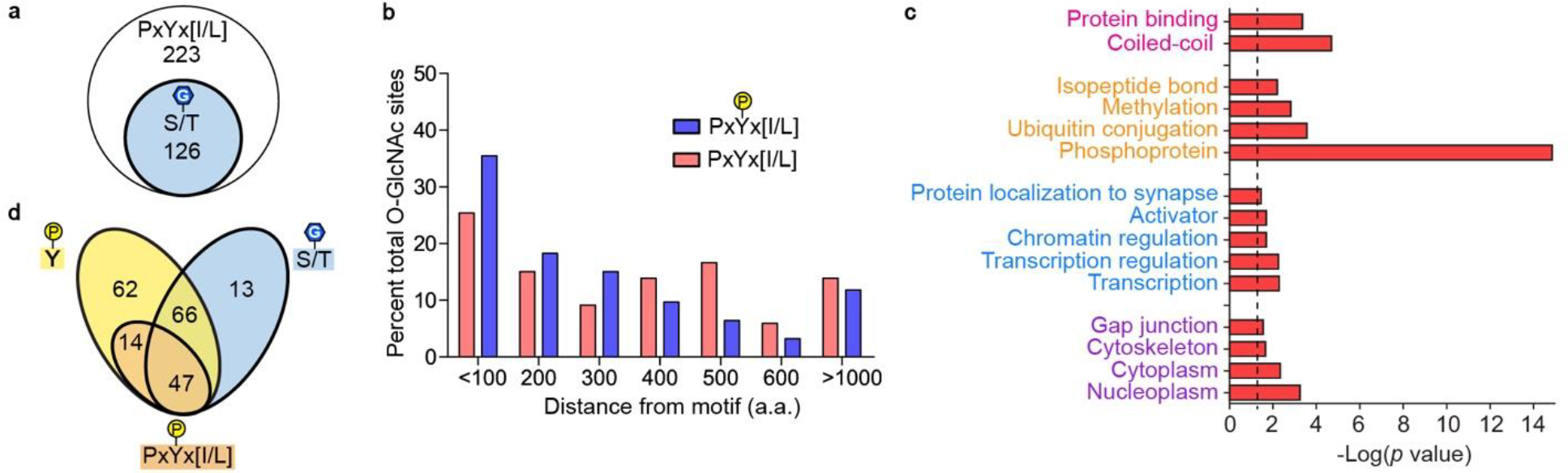
(a) O-GlcNAcylated proteins (blue circle), among the 223 PxYx[I/L] motif-containing proteins (white circle). (b) Distance of known O-GlcNAcylation sites from all PxYx[I/L] motifs (pink) and phosphorylated PxYx[I/L] motifs (blue). (c) Gene Ontology terms associated with motif-containing proteins. Molecular function and protein domain (pink), posttranslational modifications (orange), biological processes (blue), cellular components (purple). Dotted line represents p-value cutoff of 0.05. (d) Overlap analysis of motif-containing proteins with reported tyrosine phosphorylation anywhere on the protein (yellow oval), O-GlcNAcylation anywhere on the protein (blue oval), and tyrosine phosphorylation on the PxYx[I/L] (orange oval).
Comparing our list of motif-containing proteins to human tyrosine phosphorylation sites reported in PhosphositePlus, we identified 189 (84.8%) proteins with at least one pTyr site (Fig. 4d)41. Of these phosphorylated proteins, 61 (32.3%) are phosphorylated on our motif tyrosine, like SMG9, 47 (77.0%) of which have reported O-GlcNAcylation. Compared to all motif-containing proteins, these 47 motif phosphorylated proteins are enriched in proximal O-GlcNAcylation, with 33% of assigned O-GlcNAc modification sites falling within 100 residues of the motif (Fig. 4b blue columns). This further supports the Int-D site as a facilitator of O-GlcNAcylation cross regulation with pTyr. Interestingly, none of these motif phosphorylated proteins are receptor tyrosine kinases, pointing toward this mode of cross regulation occurring in downstream signal transduction on non-autocatalytic sites.
GO analysis of the 47 motif phosphorylated proteins demonstrated significant enrichment of only protein binding as a molecular function. Manual analysis of these proteins shows a variety of activities including kinase, phosphatase, and ubiquitin hydrolase, though the primary role of many proteins is non-catalytic binding to other proteins and protein complexes. BioGrid interaction analysis identified a set of 57 significantly enriched interactors of our motif phosphorylated protein set (FDR < 0.05)42. GO analysis of these 57 proteins showed strong enrichment of adherens and tight junction cellular components as well as other cell-cell adhesion functions. These results suggest roles for OGT Int-D and its direct crosstalk with tyrosine phosphorylation in regulation of cell-cell adhesion as well as signaling at the cell surface.
OGT Int-D regulates O-GlcNAc dynamics and lactate production
Our bioinformatic analysis showed motif-containing proteins, with and without reported O-GlcNAcylation sites, playing roles across a range of cellular processes. Previous studies have mutagenized and truncated the OGT TPR domain to disrupt substrate binding and detected significant changes in the global O-GlcNAc profile15–18,43,44. These results have established the TPR, especially those repeats closest to the catalytic domain, as broad facilitators of OGT substrate binding and O-GlcNAc modification. Unlike TPR mutations, we found that Int-D site mutants expressed in cells show no obvious changes in the global O-GlcNAc profile, compared to WT OGT (Fig. 5a and Extended data figure 6a). This supports that the motif-based substrate regulation is not a global mode of cellular O-GlcNAcylation and may play a more specific role.
Figure 5. OGT Int-D binding site is a regulator of O-GlcNAc dynamics during nutrient deprivation and lactate production.
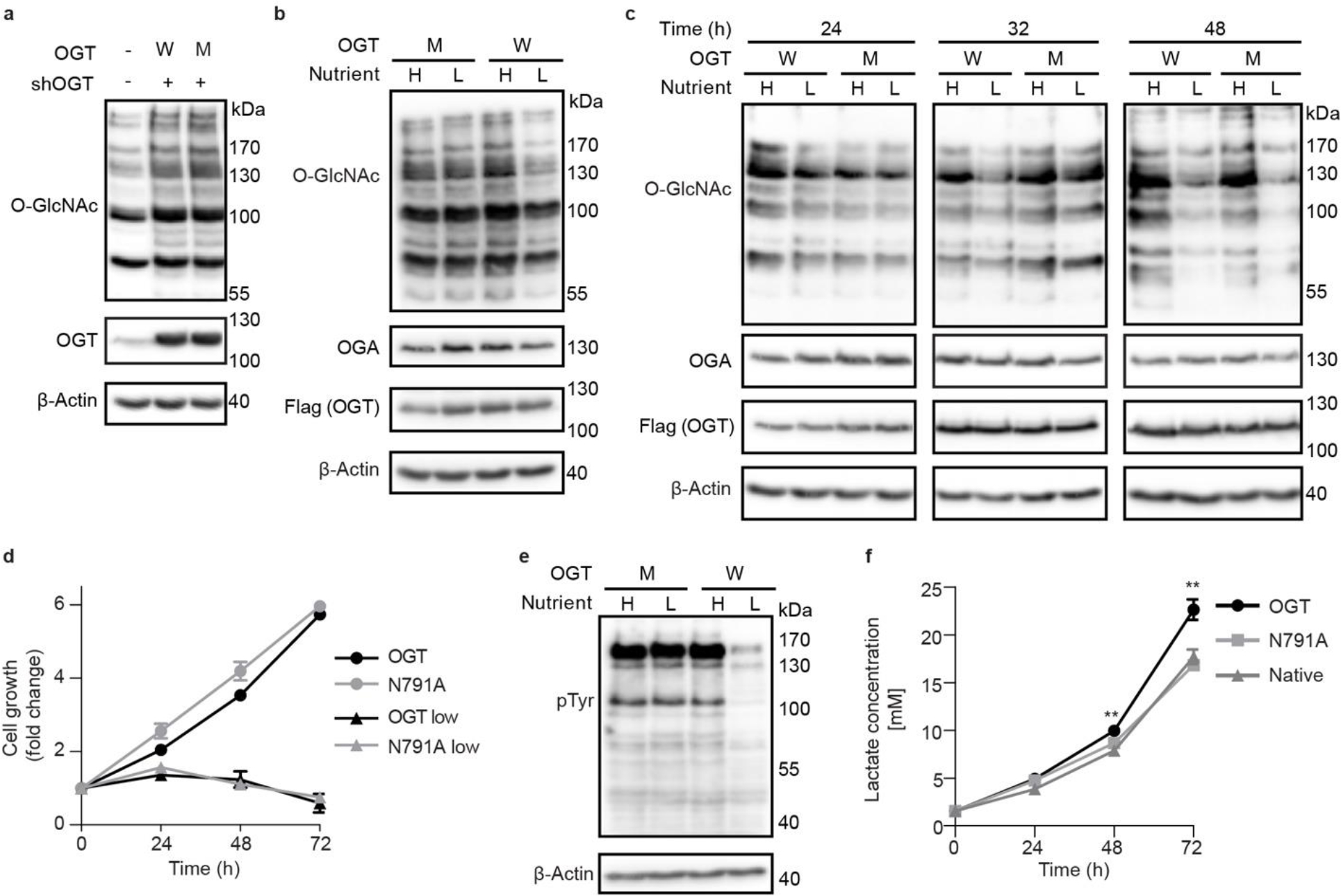
(a) Western blot detection of global O-GlcNAcylation in TRex-293 cells overexpressing OGT (W) or OGT-N791A (M) with endogenous OGT knockdown (shOGT). (b) Western blot of O-GlcNAcylation under high (H) and low (L) nutrient conditions with OGT (W) or OGT-N791A (M) overexpression. High nutrient conditions include DMEM with 4.5 g/L glucose and 10% FBS, low nutrient conditions include DMEM with 0.45 g/L glucose and 1% FBS. (c) Western blot showing time course study of O-GlcNAcylation response to a low nutrient condition with OGT (W) and OGT-N791A (M) overexpression. Cells were induced for 48 hours prior to high or low nutrient treatment, then samples were collected at 24, 32, and 48 hours of treatment. (d) Growth rate of cells overexpressing OGT (black lines) or OGT-N791A (gray lines) under high (circles) and low (triangles) nutrient conditions over 72 hours. Data are presented as mean values +/− SD of four biological replicates. No significant difference was detected between cell lines by two-tailed unpaired t-test in either nutrient condition. (e) Western blot of global phosphotyrosine signal from samples treated with the same conditions as panel b. (f) Media lactate concentration from native TRex-293 cells or those expressing WT OGT or N791A after 24, 48, and 72 hours. Lactate concentration, from LactateGlo assay, was normalized to cell number by CellTiter-Glo assay and quantified by lactate standard curve. Conditions were compared by two-tailed unpaired t-test, ** p=0.002. Data are presented as mean values +/− SD of four biological replicates. Western blots are representative of three biological replicates.
An important function of the O-GlcNAc system is sensing and responding to changes in environmental nutrients and growth factors. Some studies posit that the HBP, the pathway responsible for production of OGT’s sugar donor UDP-GlcNAc, is the principal integrator of primary metabolite signaling with minimal input from OGT and OGA themselves1. In standard cell culture conditions, the major sources of nutrients are glucose and serum. We tested changes in cellular O-GlcNAc profile under high (4.5 g/L glucose, 10% FBS) and low (0.45 g/L glucose, 1% FBS) nutrient conditions in our TRex-293 stable cell lines. Though these high and low glucose conditions would represent hyper- and hypoglycemia in vivo, cultured cells tolerate both conditions well. Individual glucose or serum stress from these relatively mild conditions resulted in minimal changes to O-GlcNAcylation for both WT OGT and N791A expressing cells (Extended data figure 6b and c). When depleted in combination, WT OGT expressing cells demonstrated a significant decrease in global O-GlcNAc signal while the N791A cells, surprisingly, did not (Fig. 5b). A follow-up time course study revealed that the O-GlcNAc response to nutrient withdrawal is delayed in N791A expressing cells (Fig. 5c). Cell growth assay showed no significant difference between the growth rate of WT OGT or N791A overexpressing cells in either nutrient condition (Fig. 5d). These results implicate the Int-D site as a temporal regulator of cellular O-GlcNAc dynamics.
To determine if this delayed response to nutrient deprivation can be generalized to other cell lines, we stably expressed WT OGT and N791A in the cervical cancer cell line HeLa. When generating the stable cell lines, we selected colonies that express WT OGT and N791A at moderate levels, unlike TRex-293 cells that express proteins at a very high level. These cell lines only required 8 hours of low nutrient treatment to show considerable O-GlcNAc downregulation, illustrating the variability of response speed across different cell types (Extended data figure 7 and Supplemental figure 2c). As in our TRex-293 cells, N791A expression delayed the decrease in global O-GlcNAcylation, relative to WT OGT expressing cells. We also observed a downregulation of OGT and OGA expression in both cell lines under the low nutrient condition. This is a typical response to nutrient deprivation that was not seen in TRex-293 cells, likely as a result of high OGT expression overwhelming natural regulatory systems. The striking alteration in global O-GlcNAcylation dynamics to nutrient withdrawal by a single OGT point mutation has not been observed in previous studies. Probing global tyrosine phosphorylation in the same HeLa and TRex-293 samples showed a similar pattern of decreased pTyr signal in only the WT OGT low nutrient samples, further connecting the two modifications to the Int-D (Figure 5e, Supplemental figure 2b and c). Cellular nutrient sensing is regulated by different systems at multiple levels across the cell, explaining the eventual reduction of O-GlcNAcylation level we observed in N791A cells. It is interesting that this effect is only seen when both free glucose and serum levels are reduced, suggesting that a combination of primary metabolites and growth factors are responsible. The complex composition of serum precludes us from specifically identifying the affected signaling pathways at this point, though these results already support the Int-D binding site as an important regulator of nutrient sensing. This also supports a model of cooperative regulation between OGT and the HBP.
Response to external cues and nutrient status is only one side of OGT’s nutrient sensing role. Regulation of downstream metabolic programs, including the balance of glycolysis and oxidative phosphorylation, is a critical function of OGT45. Under normal conditions, healthy cells will preferentially utilize the oxidative phosphorylation pathway to metabolize glucose. However, metabolically aberrant cancer cells drive glucose consumption through both the citric acid cycle and glycolysis pathways, resulting in increased lactate production46. OGT overexpression has been identified in a range of clinical cancers and associated with increased glycolysis45. Throughout culture of our stable cell lines, we noticed rapid media acidification of cell lines overexpressing WT OGT but not N791A. To evaluate differences in metabolic activity, we measured media lactate concentration in a time course using the LactateGlo assay and normalized the results to cell growth rate. WT OGT overexpressing cells show a significant increase in media lactate concentration after 72 hours of culture, compared to native TRex-293 cells (Fig. 5f). In line with the observed differences in media acidification, N791A mutation ablated this increased lactate production, implicating the Int-D as a regulator of cellular metabolism. These simple tests of lactate production and O-GlcNAcylation response to nutrient level have broad implications for the important roles of Int-D in health and disease, which requires further investigation.
Discussion
Previous studies of OGT substrate regulation and protein binding have focused on the TPRs of OGT, as they are well-known structural scaffolds found in many proteins across the cell. Our study has assumed a fundamentally different route, employing the unique ProP-PD library to profile OGT surface binding sites. Our agnostic approach led to the surprising discovery of a motif-based binding site in the OGT Int-D. Since its initial structural characterization over a decade ago, the role of the Int-D has remained mysterious. The unique fold of Int-D that intersects an otherwise typical GT-B enzyme catalytic domain is only found in metazoan OGTs. This is unlike the TPR domain whose structure and length are highly conserved across OGT homologs in prokaryotic and eukaryotic species. This may suggest that the Int-D arises with increased organismal structural and functional complexity. Our observations that proteins containing a PxYx[I/L] motif partake in processes typically associated with evolutionarily complex functions of multicellular organisms support this premise. As mentioned above, tyrosine phosphorylation is also typically associated with multicellular organisms and regulation of evolutionarily complex functions. Sequence alignment of OGT Int-D reveals a further line of genetic delineation as the Int-D binding site residues identified in this study are only highly conserved in vertebrates (Extended data figure 3)11. This is interesting as knockout of ogt in vertebrate systems (e.g., mouse, human) is embryonically lethal, however, ogt knockout is tolerated in the invertebrate C. elegans47,48. Several residues around the Int-D binding site, though not our identified interacting residues, have also been reported as sites of mutation in both cancer and X-linked intellectual disability samples. Our work leads to a hypothesis that the Int-D site is an important piece of OGT’s essential cellular functions and may be exploited for therapeutic interventions.
Our surprising finding that the Int-D binding site influences lactate production and global O-GlcNAc response to nutrient deprivation opens many questions. Disruption of an apparent substrate recognition site, leading to maintained O-GlcNAcylation in a low nutrient environment is not intuitive. We hypothesize that specific interactions, facilitated by Int-D, allow OGT to interface with and modify larger signaling networks. Thus, disruption of these signaling nodes desensitizes certain pathways. Another interesting possibility is motif containing proteins acting as OGT inhibitors that block substrate recognition under specific cellular contexts. Future multi-omic studies, across multiple timepoints of the low nutrient response, will illustrate the underlying dynamics of this process and provide valuable insight into the roles of Int-D binding in OGT’s larger cellular context.
Hallmark phenotypes of cancer and other deleterious diseases include dysregulation of nutrient sensing and metabolism. Aberrant OGT and O-GlcNAcylation have been intimately linked to the development of cancer, X-linked intellectual disability, and other metabolic disorders. The wide reach of O-GlcNAcylation across important cellular systems makes it a valuable target for therapeutic intervention, however, disruption of these processes can also present problems. Current OGT inhibitors primarily target the active site, triggering global disruption of O-GlcNAcylation, spurring concern over unanticipated side effects. Apparent maintenance of normal O-GlcNAcylation upon Int-D site mutation indicates that this binding site plays explicit roles in the cell. The structural uniqueness of the Int-D, coupled with its specific sequence recognition offer great opportunity to target OGT with exceptional specificity. Our findings for the Int-D’s roles in low nutrient response and lactate production warrant further investigation of this domain’s role in cancer and metabolic disorders.
Just prior to the submission of this manuscript, a study was published which utilized an mRNA display screening approach to identify macrocyclic peptide binders of OGT49. Though the paper focused on other hits, we noticed that a PxYxI motif was strongly enriched in their screening results. A second study by the same group, published concurrently with this one, screened a random library of 15-mer peptides using a phage display platform50. Followed by deep mutational analysis, this screen produced a [Y/F]xPxYx[I/M/F] motif that binds tightly to the Int-D binding site and may interfere with peptide substrate modification. Though this exact motif is rarely found in the proteome (9 proteins by SLiMSearch), we believe it can serve as a useful starting point for targeted OGT Int-D probe development. Independent identification of related binding motifs across multiple screening platforms supports the Int-D as an important binding domain of OGT.
This study unifies multiple lines of thinking that have been pervasive in the field but have lacked concrete evidence: 1) major regulatory mode(s) of OGT utilize its non-catalytic regions to coordinate interactions with specific substrates; and 2) due to its evolutionary interest, the Int-D plays one or more roles in OGT that make it distinct from other glycosyltransferases or even non-eukaryotic OGT homologs. Through multiple cellular assays we have identified the Int-D binding site as a motif-dependent regulator of protein association and O-GlcNAcylation. We further linked this site to tyrosine phosphorylation crosstalk, nutrient deprivation response, and metabolic regulation. Along with other important roles implicated by our bioinformatic analysis, these findings have radically changed our view of the Int-D and its importance to OGT. This opens exciting pathways to specifically dissect OGT’s regulation and functions in health and disease, creating numerous opportunities in the field.
Methods
ProP-PD and next generation sequencing (NGS) data analysis
Recombinantly purified OGT and OGT4.5 were used as bait proteins in selections against the ProP-PD library. Selections were performed following the published protocol19. Briefly, proteins were immobilized in 96 well Flat-bottom Immunosorp MaxiSorp plates for 4 °C overnight. Following blocking with 0.5% BSA in PBS, wells were washed four times with wash buffer (0.05% tween-20, PBS). The phage library, concentrated to 1011 phages in PBS per well, was first added to GST-coated wells for 1 hour at 4 °C then transferred to the bait protein-coated plates for 2 hours at 4 °C. After three washes with wash buffer, bound phages were eluted with log phase E. coli OmniMAX for 30 minutes at 37 °C. M13KO7 helper phages were added at 109 per well for 45 minutes at 37 °C then samples were transferred to 2xYT supplemented with carbenicillin, Kanamycin, and 0.3 mM isopropyl-β-D-1-thiogalactopyranoside (IPTG). Bacteria were grown at 37 °C for overnight, then spun down at 2,000 x g for 10 minutes. The phage supernatants were pH adjusted with 10x PBS and used for the next round of selection.
After at least three rounds of selection, enriched phage pools were collected, barcoded, and analyzed by next-generation sequencing on an Illumina MiSeq v3, as described previously1. Results were processed using in-house Python scripts19. Peptide sequences were annotated with PepTools and assigned confidence levels based on four criteria: occurrence in replicate selections, overlapping peptide sequence identification, high sequence counts, and sequences containing consensus motifs identified from the peptide pool. We only focused on the medium/high confidence peptides that fulfill at least three of these criteria and produced our identified motif.
Cell culture
Human HeLa (ATCC #CCL-2), HEK293T (ATCC #CRL-3216), HEK293 (ATCC #CRL-1573), and TRex-293 (ThermoFisher #R71007) cells were cultured in Dubelco’s modified Eagle’s medium (Corning) containing 10% fetal bovine serum (FBS) (Sigma), 10 U/mL penicillin, and 10 μg/mL streptomycin at 37 °C and 5% CO2. Inducible knockdown of endogenous OGT in TRex-293 and HeLa cells was generated using a standard second-generation lentiviral system. Briefly, HEK293T cells were co-transfected with pCMV-VSVG, psPAX2, and EZ-Tet-pLKO-shOGT-Puro by calcium phosphate transfection. Secreted virions were collected and used to infect TRex-293 and HeLa cells. Positive clones were selected by puromycin treatment and OGT knockdown upon doxycycline induction was evaluated. TRex-293 stable cell lines were generated following the supplier’s instructions. Briefly, pcDNA5/FRT/TO plasmids containing WT OGT or OGT-N791A were co-transfected with pOG44 into TRex-293 cells with endogenous OGT knockdown and positive colonies were selected by hygromycin treatment. Various doses of doxycycline were tested in each cell line to determine conditions for equal exogenous OGT expression. HeLa stable cell lines expressing WT OGT or OGT-N791A were similarly generated by lentiviral transduction using the pLenti transfer plasmid and zeocin selection.
Nutrient deprivation
For nutrient treatment experiments, cells were seeded at 4.5 × 105 cells/well in 6-well plates overnight then treated with normal DMEM (10% FBS, 4.5 g/L glucose) supplemented with doxycycline for 48 hours, to induce endogenous OGT knockdown and expression of our exogenous construct. Samples were then treated with the nutrient condition detailed in the results. Following low nutrient treatment, cells were washed with PBS and harvested by centrifugation. Cell pellets were lysed with RIPA total lysis buffer (150 mM NaCl, 1% nonidet-P40, 0.5% deoxycholate, 0.1% sodium dodecyl sulfate, 50 mM Tris-HCl, pH 7.4) supplemented with protease inhibitor cocktail (Millipore Sigma #P8340) and 10 μM Thiamet-G. Cell debris was removed by centrifugation at 16,000 g for 15 min at 4 °C. Samples were separated on an 8% SDS-PAGE gel and detected by western blot using antibodies: anti-O-GlcNAc (RL2, ThermoFisher scientific #MA1-072) 1:10000, anti-OGT (Santa Cruz #sc-74546) 1:5000, anti-OGA (Santa Cruz #sc-376429) 1:5000, anti-Flag (Millipore Sigma #F1804) 1:10000, anti-cMyc (Millipore Sigma #C3956) 1:5000, anti-phosphotyrosine (ThermoFisher scientific #14-5001-82) 1:2000, and anti-β-Actin (ThermoFisher scientific #MA1-140) 1:10000.
Protein expression and purification
Human OGT4.5, OGT, and mutants described above were expressed and purified as described previously11. Briefly, a pET24b plasmid with OGT4.5 gene inserted (a kind gift from S. Walker’s lab) was transformed into BL21(DE3) E. coli for protein expression. The bacteria were cultured in LB medium supplemented with 50 μg/mL kanamycin at 37 °C, 250 rpm. After reaching to an OD600 of 0.6–0.8, the 0.3 mM IPTG was added to induce protein expression at 16 °C, 220 rpm overnight. Bacterial cells were collected and resuspended in TBS buffer (150 mM NaCl, 20 mM Tris, pH 8.0) supplemented with 1 mM phenylmethylsulfonyl fluoride. The suspension was lysed by pressure cell homogenizer, clarified by centrifugation, and subjected to Ni-NTA affinity chromatography. The eluted OGT4.5 protein was incubated with HRV-3C protease to cleave the N-terminal His6-tag overnight. The protein sample was subsequently purified by a size exclusion chromatography column (Superdex 200 increase 10/300; Cytiva) on an AKTA FPLC system in TBS buffer with 0.5 mM tris(3-hydroxypropyl)phosphine. OGT4.5 protein was concentrated to 8 mg/mL for crystallization.
Crystallization
All peptides for crystallization were prepared by solid phase peptide synthesis (Ontores, ≥ 95% purity, HPLC). Hanging drop vapor diffusion method was used for co-crystallization. OGT4.5 was first incubated with 2 mM of peptide and 1 mM of UDP-GlcNAc on ice for two hours. After pre-incubation, 2 μL of protein complex was mixed with 1 μL of reservoir solution and equilibrated against 200 μL of reservoir solution at 20 °C. OGT4.5:UDP-GlcNAc:SMG9 and OGT4.5:UDP-GlcNAc:ZNF831 crystals were obtained in the condition consisting of 1.9 M ammonium sulfate, 1% xylitol, 0.1 M Tris, pH 8.5, while OGT4.5:UDP-GlcNAc:CP37 crystals were grown in the condition containing 1.45 M potassium phosphate, 1% xylitol, 10 mM EDTA, pH 8.0. Crystals were cryo-protected in mother liquor supplemented with 27% xylitol and then flash frozen in liquid nitrogen.
X-ray data collection and structure determination
All the X-ray data were collected on the Life Sciences Collaborative Access Team (LS-CAT) beam lines 21-ID-F (for OGT4.5:UDP-GlcNAc:SMG9) and 21-ID-D (for OGT4.5:UDP-GlcNAc:ZNF831 and OGT4.5:UDP-GlcNAc:CP37) in Argonne National Laboratory, IL. Data were processed using autoPROC51, which is a toolbox containing programs XDS and AIMLESS for data indexing and scaling52,53. Structures were solved by molecular replacement with Phaser using OGT4.5 (PDB 3PE3) as a search model11,54. Iterative model building was performed in COOT55, followed by refinement with PHENIX56. OGT4.5:UDP-GlcNAc:SMG9 and OGT4.5:UDP-GlcNAc:ZNF831 crystals belong to the space group P321, while OGT4.5:UDP-GlcNAc:CP37 crystal belongs to space group H3. All of these crystals contain four molecules in each asymmetrical unit, with each OGT4.5 in complex with one molecule of peptide and UDP-GlcNAc. All the structural figures were prepared using PyMOL (The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC).
Microscale thermophoresis (MST)
5-FAM labeled SMG9 peptide was prepared by solid phase peptide synthesis (Ontores, ≥ 95% purity, HPLC). Two-fold serial dilutions of OGT in TBS buffer supplemented with 0.05% tween-20 was prepared in strip-tubes, and then mixed with equal volume of 100 nM of 5-FAM SMG9 peptide in the same buffer. After 10 min pre-incubation, microscale thermophoresis upon OGT-peptide binding was measured in standard capillaries on Monolith NT.115 pico instrument (NanoTemper). MST was performed with three biological replicates. Dose-response curves were plotted, and the Kd values were calculated using Origin.
Thermal shift assay (TSA)
Thermal shift assay was carried out on a StepOnePlus Real-Time PCR System (Applied Biosystems). Reactions were set up in 96-well qPCR plates with optical seal at a total volume of 20 μL. Each reaction contained 2.5 μM protein, 3.2x SYPRO Orange dye (ThermoFisher, Cat# S6650) and 200 μM peptide in TBS buffer. At least three replicates were prepared for each reaction. Using the melt curve setting fluorescent readings were collected at 0.5 °C intervals from 25–95 °C using the ROX channel. Melting curves were plotted in StepOne software and the first derivatives of the fluorescence curves were used to determine Tm values.
Plasmids and cloning
The original OGT4.5, OGT, and OGA-D175N constructs, cloned into bacterial expression vectors, have been described previously. OGT cDNA, with codon optimization for mammalian expression, was provided by Harvard PlasmID (HsCD00045640). pDONR221-SMG9 was obtained from the DNASU plasmid repository (HsCD00533575). pHAGE-CaMKMT was a kind gift from Dr. Wade Harper as part of the BioPlex 3.0 library57. Mammalian OGT, OGA-D175N, SMG9, and CaMKMT were subcloned into pcDNA5/FRT/TO (Millipore Sigma) with an added N-terminal Flag tag on OGT and OGA, an N-terminal cMyc tag on SMG9, and N-terminal HA tag on CaMKMT. Site-directed mutagenesis was used to generate OGT mutants for bacterial and mammalian expression as well as the SMG9-Y147F mutant construct. Short hairpin RNA (shRNA) for endogenous human OGT knockdown was designed using the RNAi Consortium shRNA library and cloned into EZ-TET-pLKO-Puro (Addgene #85966). For viral transduction, Flag-tagged OGT constructs were cloned into the pLenti vector which was a kind gift from M. B. Jackson. Viral packaging and envelope plasmids pCDH and pVSVG were a kind gift from X. Zhao. All primers used in this study are listed in Supplemental Table 1.
Fluorescence polarization (FP) assays
All FP measurements were carried out in black small volume 384-well plates (Greiner Bio-One). For saturation binding assays, 2-fold serial dilutions of recombinantly purified OGT were prepared in TBS (150 mM NaCl, 20 mM Tris-HCl, pH 8.0). 5-FAM-SMG9 fluorescent peptide at 0.4 μM in TBS was added in equal volume to the OGT dilution series. For competition binding assays, 2-fold serial dilutions of unlabeled peptide were prepared in TBS. An equal volume of 20 μM OGT with 0.4 μM 5-FAM-SMG9 in TBS was added to the dilution series. After 20 min incubation in the dark at room temperature, FP was read on a BMG PheraStar multimode plate reader equipped with 488, 520, 520 FP filter cube (BMG Labtech).
Immunoprecipitation (IP)
For OGT-SMG9 co-immunoprecipitation experiments, HEK293 cells were co-transfected with the indicated protein constructs or an empty vector by calcium phosphate transfection. After 48 hours of transfection, cells were washed with PBS and collected by centrifugation. Pellets were gently lysed using IP lysis buffer (150 mM NaCl, 1% nonidet-P40, 20 mM Tris-HCl, pH 7.4) supplemented with protease inhibitor cocktail and 10 μM Thiamet-G. Total cell lysate was diluted to 1 mg/mL and incubated with Flag M2 agarose (Millipore sigma #A2220) for 16 hours at 4 °C. Samples were washed four times with ten bed volumes of IP lysis buffer and eluted by boiling with 2x SDS loading buffer. Samples were run on 8% SDS-PAGE gels and detected by western blots.
O-GlcNAc detection by GalT assay
SMG9 immunoprecipitation for O-GlcNAc detection was carried out similarly to the above co-IP and GalT assisted biotinylation was performed as previously described36. Briefly, TRex-293 cells stably expressing the indicated OGT constructs were transfected with the indicated SMG9 construct by calcium phosphate transfection. 48 hours after transfection, cells were treated with 10 μM Thiamet-G for 16 hours to inhibit O-GlcNAc hydrolysis, allowing O-GlcNAc accumulation on SMG9. Cells were collected, lysed, and loaded on cMyc agarose as described above. Samples were washed four times with ten bed volumes of IP lysis buffer then eluted twice with soft elution buffer (0.2% SDS, 0.1% tween 20, 50 mM Tris-HCl, pH 8.0) for 10 min at 35 °C. Samples were precipitated with methanol at −20 °C overnight and spun down at 16,000 g for 30 min at 4 °C. Protein pellets were reconstituted in GalT buffer (0.6% nonidet-P40, 5 mM MnCl2, 150 mM NaCl, 20 mM HEPES, pH 7.5) with 25 μM uridine diphosphate N-azidoacetylgalactosamine (UDP-GalNAz) and 2.2 μM GalT-Y289L and incubated overnight at 4 °C. To biotinylate our modified SMG9, we used copper assisted click chemistry to add a Biotin-PEG4-Alkyne (Click Chemistry Tool #TA105-100) probe to the azide functionalized GalNAc residues. Samples were run on 8% SDS-PAGE gel and analyzed by far western blot with streptavidin-HRP (Cytiva #RPN1231) as well as western blot with cMyc and β-Actin antibodies mentioned above.
Radiolabeled activity assay of OGT
Purified OGT (500 nM of wild-type or N791A mutant) was incubated with 50 μM UDP-3H-GlcNAc (specific activity 0.3 Ci/mmol, PerkinElmer #NET434250UC) and 500 μM CKII peptide (YPGGSTPVSSANMM) in the reaction buffer (20 mM Tris pH 8.0, 150 mM NaCl, 0.5 mM THP) at 37 °C for 3 hours. Reactions were quenched by spotting the samples onto Amherst Protran 0.1 μm nitrocellulose membrane (Cytiva), air-dried, and washed for 5 min four times in PBS buffer. The radioactivity of each membrane was counted by a Tri-Carb 2900 TR Liquid Scintillation Analyzer (PerkinElmer). A reaction without OGT was set up as negative control. Another reaction without the washing steps was counted as a total of 50 μM of UDP-GlcNAc input to calculate the O-GlcNAcylation level of CKII peptide in each reaction. The experiment was performed in triplicate.
Cell growth assay
Cell viability was assessed using CellTiterGlo 2.0 reagent (Promega) according to the manufacturer’s instructions. Briefly, cells were seeded in 96-well tissue culture plates, with at least four biological replicates for each condition and time point. Cells were grown in the indicated nutrient condition for the specified amount of time before an equal volume of CellTiterGlo 2.0 reagent was added directly to the well and incubated at room temperature for 10 min in the dark. Luminescence was measured on a BioTek Synergy H1 plate reader.
Lactate production assay
Lactate production was measured using the LactateGlo assay kit (Promega) following manufacturer’s instructions. Briefly, cells were seeded in 96-well tissue culture plates with at least four biological replicates for each time point. At the specified time point, a sample of culture media was taken and diluted 1000x fold in PBS. A lactate standard curve from 0.2–200 μM in PBS was also prepared to quantify media lactate concentration. Samples were mixed with LactateGlo detection solution in low volume black 384-well plates and incubated at room temperature in the dark for 1 hour. Luminescence was measured on a BioTek Synergy H1 plate reader. Lactate concentration was normalized to cell number using the above cell growth assay protocol.
Statistical analysis
All of the data shown are mean or individual values with error bars representing standard deviation. Statistical significance was calculated in GraphPad Prism 5 and determined using two-tailed student’s t-test.
Extended Data
Extended Data Figure 1. OGT binding evaluation of motif-containing peptides.
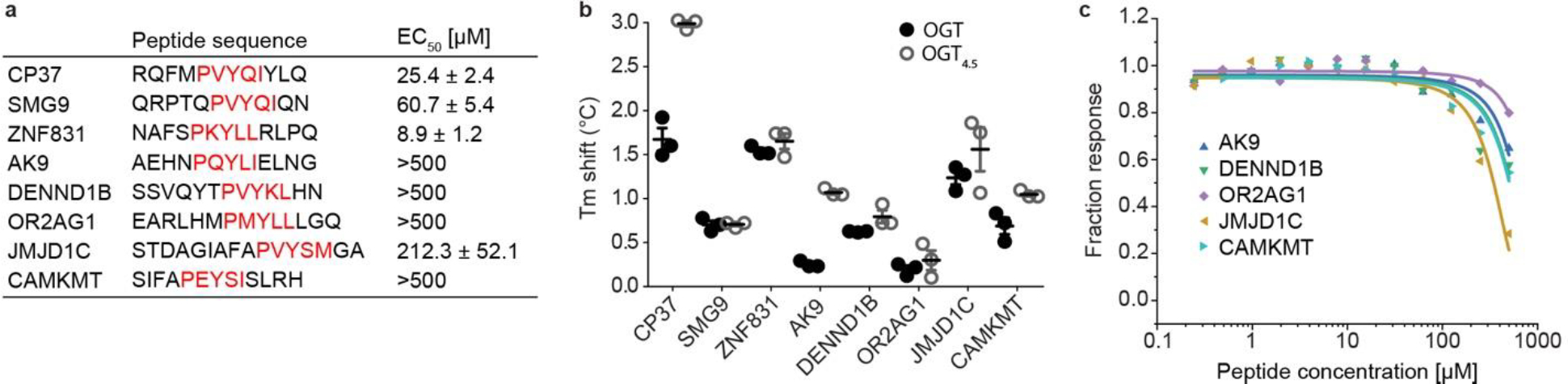
(a) Sequences of one consensus (CP37) and seven natural motif-containing peptides along with their apparent binding affinities (EC50) from competitive fluorescence polarization assay using 5-FAM-SMG9 peptide and OGT4.5. Standard deviations from three biological replicates are shown. PxYx[I/L] motif in each peptide is highlighted in red. (b) Thermal shift assay showing the shift in denaturation temperature (Tm) of OGT and OGT4.5 with peptide incubation, n=3. (c) Competitive fluorescence polarization assay between 5-FAM-SMG9 and motif-containing peptides AK9 (blue), DENND1B (green), OR2AG1 (purple), JMJD1C (brown), and CAMKMT (cyan) for OGT4.5 binding. Data are presented as individual measurements +/− SD of mean of three biological replicates.
Extended Data Figure 2.
(a) Zoom-in view at the Int-D binding site demonstrating hydrophobic interactions between ZNF831 peptide (shown as sticks) and Int-D in OGT4.5 (shown as surface). Red and white colors represent the least and most hydrophobic areas, respectively. (b) Zoom-in view at the Int-D binding site demonstrating polar interactions between ZNF831 peptide (shown as purple sticks) and Int-D in OGT4.5 (shown as surface) with interacting OGT residues shown in sticks. 2Fo–Fc electron density map of ZNF831 peptide is shown as grey mesh and contoured at 1.0 σ, Fo–Fc electron density map is shown as green mesh and contoured at 3.0 σ. (c) Zoom-in view at the Int-D binding site demonstrating hydrophobic interactions between CP37 peptide (shown as sticks) and Int-D in OGT4.5 (shown as surface). Red and white colors represent the least and most hydrophobic areas, respectively. (d) Zoom-in view at the Int-D binding site demonstrating polar interactions between CP37 peptide (shown as blue sticks) and Int-D in OGT4.5 (shown as surface) with interacting OGT residues shown in sticks. 2Fo–Fc electron density map of CP37 peptide is shown as grey mesh and contoured at 1.0 σ, Fo–Fc electron density map is shown as green mesh and contoured at 3.0 σ. (e) Superimposition of OGT4.5:UDP-GlcNAc (4GZ5, grey), OGT4.5:UDP-GlcNAc:SMG9 (magenta), OGT4.5:UDP-GlcNAc:ZNF831 (purple), and OGT4.5:UDP-GlcNAc:CP37 (blue) crystal structures showing peptide binding to Int-D does not change the overall structure of OGT4.5. (f) A proposed model of a substrate peptide (shown as brown cartoon) binding to OGT4.5 (shown as surface, domains colored as in Figure 1a). Int-D and TPR domain interactions facilitate substate glycosylation in the active site of OGT. G symbol represents GlcNAc moiety.
Extended Data Figure 3. Evolutionary conservation analyses of OGT intervening domain.
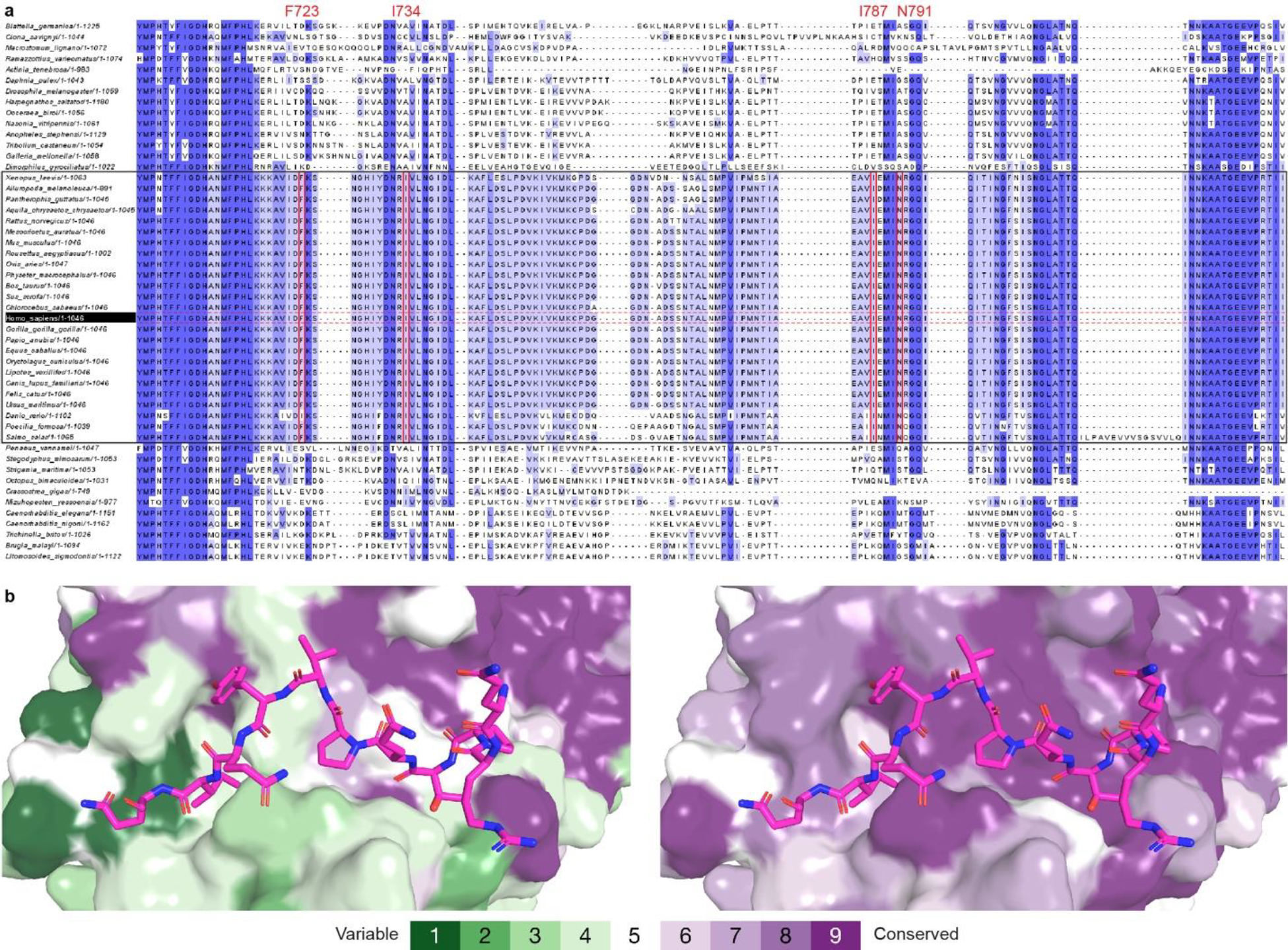
(a) Sequences of 50 metazoan OGT homologs, including 25 homologs from vertebrates (highlighted in black box), were aligned by Clustal Omega58. Jalview59 was used to review the alignment results and generate the figure. Residues are colored according to the percentage identity in each column (> 80 %, dark blue; > 60 %, blue; > 40%, light blue; < 40%, white). Int-D residues involved in motif interaction are highlighted in red boxes. (b) ConSurf60 analysis of metazoan (left) and vertebrate (right) OGT evolutionary conservation profile. Residues on the Int-D binding site are conserved among vertebrates but not invertebrates. Residues are colored from green to purple by variability across the aligned structures.
Extended Data Figure 4. OGT Int-D mutants reduce peptide binding but retain intrinsic activity.
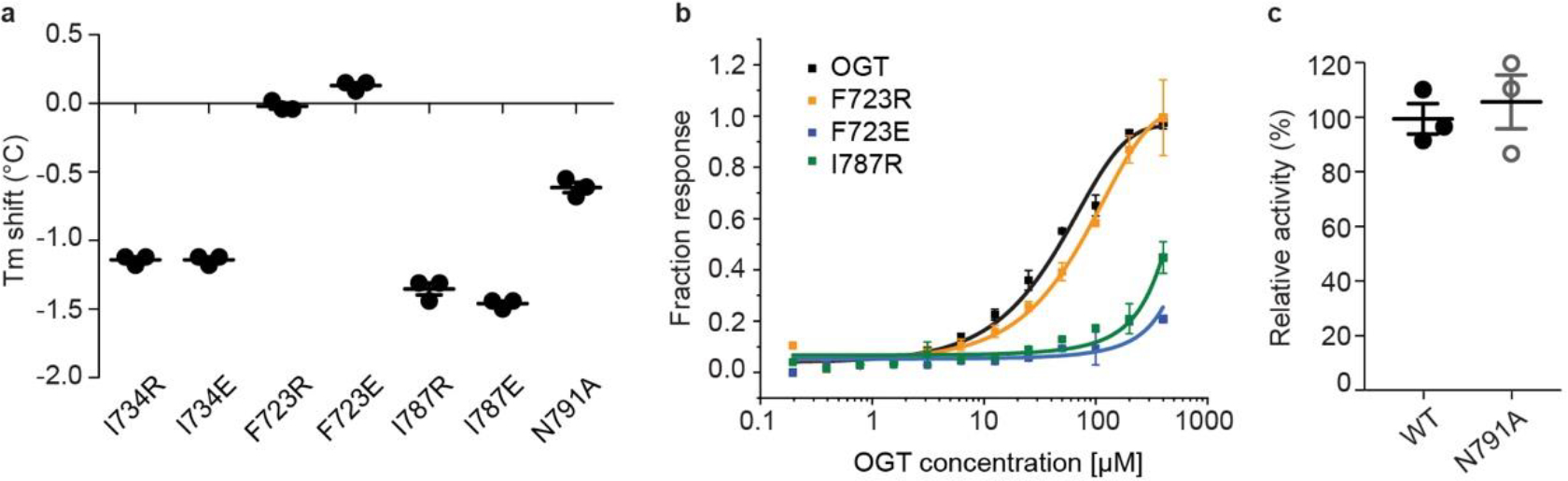
(a) Thermal shift assay showing the mutation induced change in OGT denaturation temperature (Tm). Data are presented as individual measurements +/− SD of mean of three biological replicates. (b) Saturation fluorescence polarization (FP) binding assay of fluorescently labeled 5-FAM-SMG9 peptide with WT OGT (black) and mutants F723R (orange), F723E (blue), and I787R (green). Data are presented as mean values +/− SD of three biological replicates. (c) Radiolabeled activity assay measuring integration of UDP-3H-GlcNAc onto CKII peptide by recombinantly purified WT OGT (WT) or OGT-N791A mutant (N791A). Data are presented as individual measurements +/− SD of mean of three biological replicates.
Extended Data Figure 5. The Int-D binding site is a motif-dependent regulator of protein association and O-GlcNAcylation.
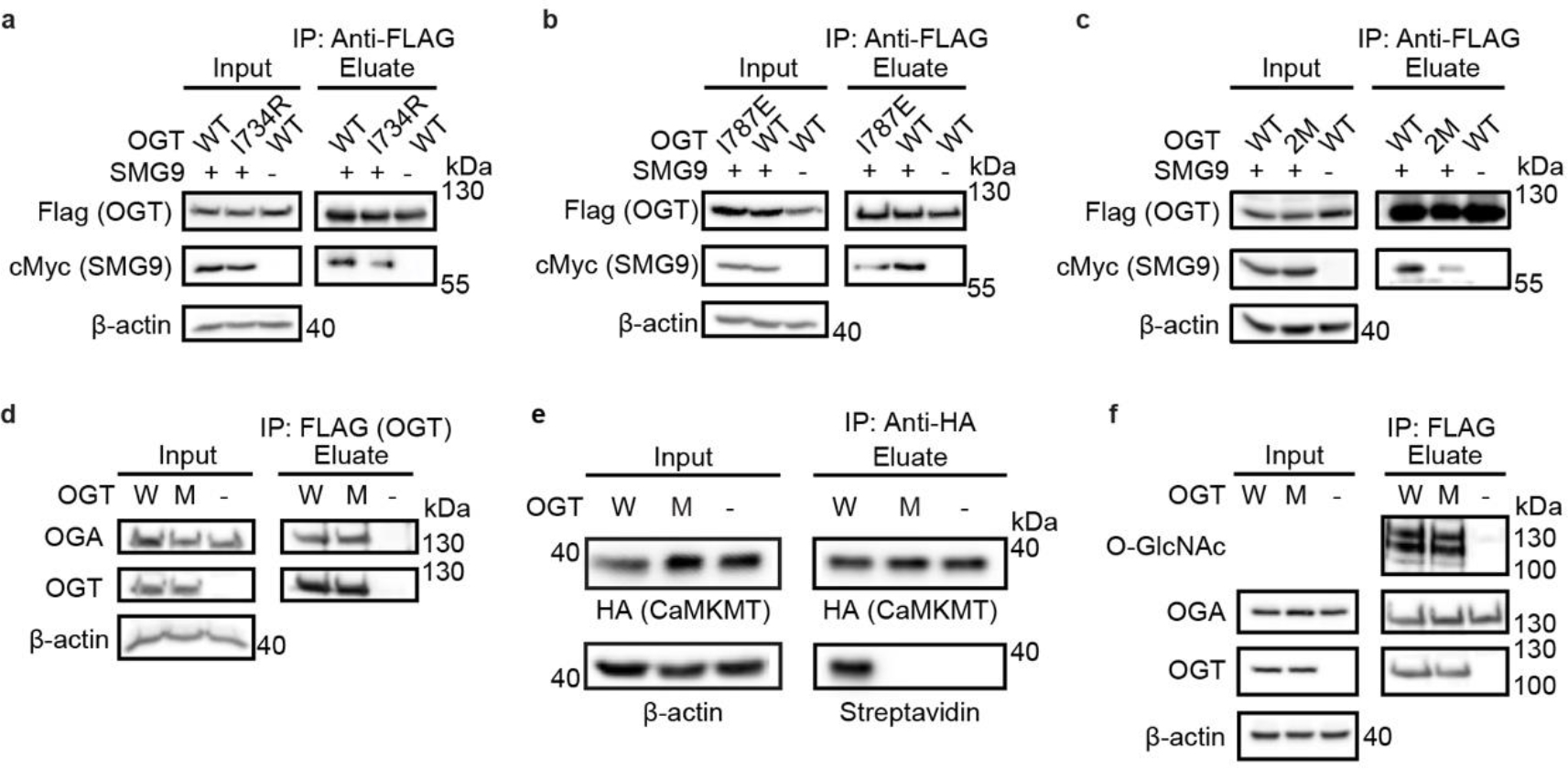
Co-immunoprecipitation of cMyc-SMG9 with Flag-OGT (WT) or (a) Flag-OGT-I734R (I734R), (b) Flag-OGT-I787E (I787E), (c) Flag-OGT-I787E-N791A (2M), from TRex-293 cells, followed by western blot. (d) Co-immunoprecipitation of endogenous OGA with Flag-OGT (W) or Flag-OGT-N791A (M) from TRex-293 cells, followed by western blot. (e) O-GlcNAcylation detection on CaMKMT from cells co-expressed with Flag-OGT (W) or Flag-OGT-N791A (M). HA-tagged CaMKMT was immunoprecipitated from TRex-293 cell lysate by anti-HA agarose and the O-GlcNAcylation was detected with the same GalT assay as SMG9. (f) O-GlcNAcylation detection on Flag-OGT (W), Flag-OGT-N791A (M), and Flag-HA-OGA-D175N. Flag-tagged OGT and OGA were immunoprecipitated from TRex-293 cell lysate by Flag-agarose, followed by western blot. All whole cell lysates have endogenous OGT knocked down. Blots are representative of at least three biological replicates.
Extended Data Figure 6. Global O-GlcNAcylation is unperturbed by Int-D site mutation and single nutrient deprivation.
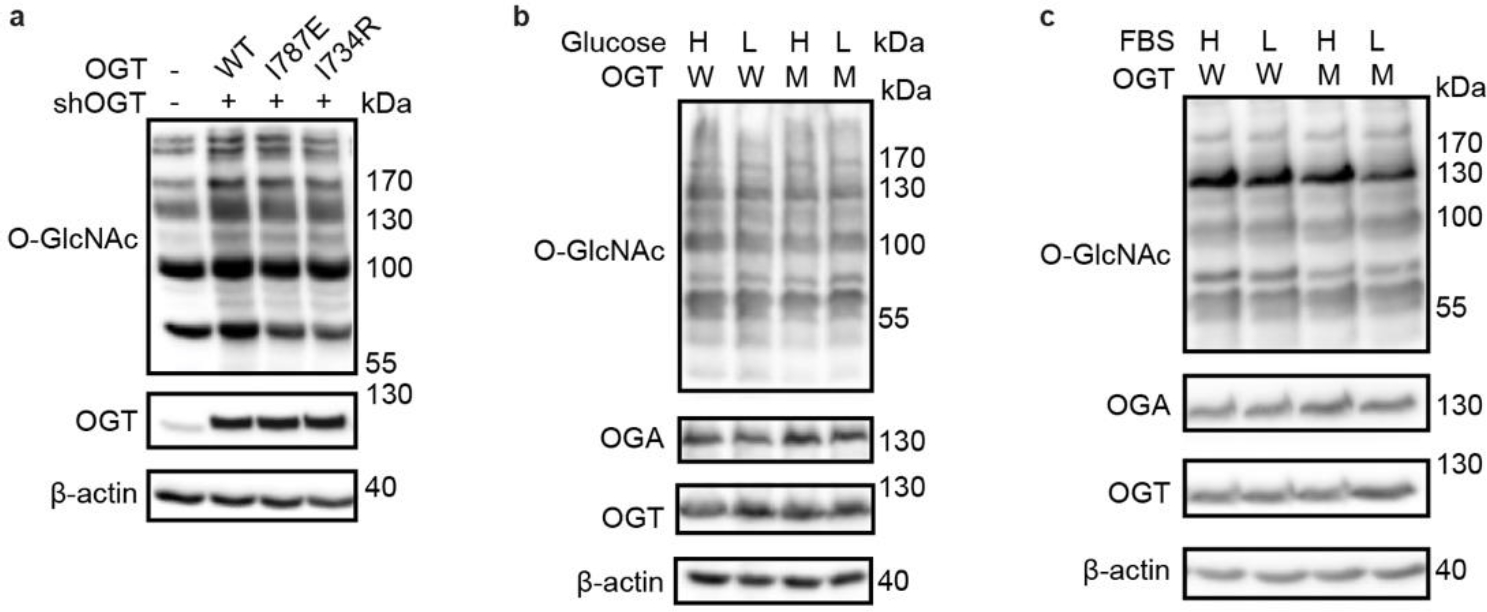
(a) Western blot detection of global O-GlcNAcylation in TRex-293 cells overexpressing OGT (WT), OGT-I787E (I787E), or OGT-I734R (I734R) with endogenous OGT knockdown (shOGT). Western blots of O-GlcNAcylation under high (H) and low (L) glucose (b) or serum (c) treatment with OGT (W) or OGT-N791A (M) overexpression. High nutrient conditions include DMEM with 4.5 g/L glucose and 10% FBS, low nutrient conditions include DMEM with 0.45 g/L glucose and 1% FBS. Blots are representative of at least three biological replicates.
Extended Data Figure 7. Delayed response to nutrient deprivation in HeLa cells.
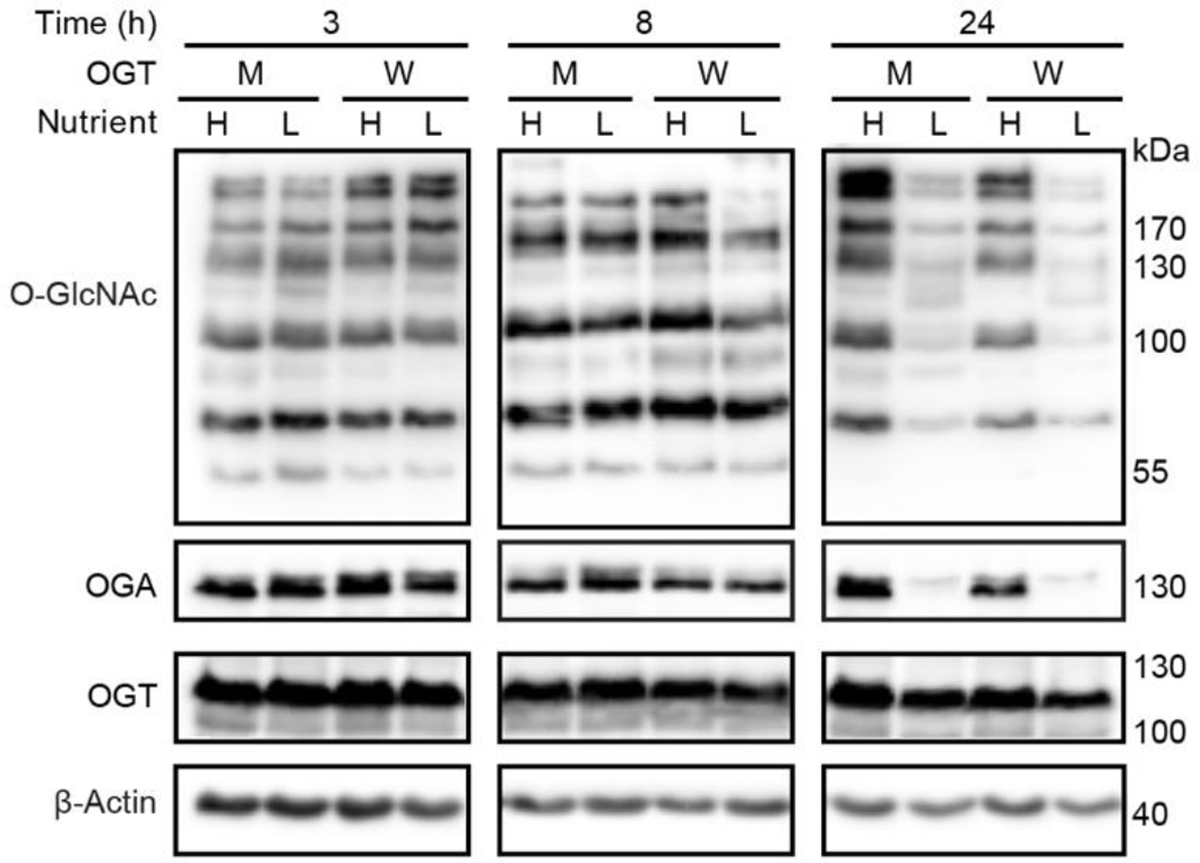
Western blot showing time course study of O-GlcNAcylation response to nutrient deprivation in HeLa cells stably expressing OGT (W) and OGT-N791A (M). Cells were induced for 48 hours prior to high or low nutrient treatment, then samples were collected at 3, 8, and 24 hours of treatment. Western blots are representative of three biological replicates.
Supplementary Material
Acknowledgments
We would like to thank members of the Jiang lab for insightful discussions and suggestions. This research was funded by NIH R01 GM121718 (J.J.), University of Wisconsin-Madison Vilas Faculty Early Career Investigator Award (J.J.), National Science Foundation Graduate Research Fellowship under Grant No. DGE-1747503 (C.M.B.). We also thank Dr. Kenneth Satyshur for helpful suggestions on structure refinement and the staff members at LS-CAT for assisting with X-ray data collection, University of Wisconsin Carbone Cancer Center Small Molecule Screening Facility and Drug Development Core Medicinal Chemistry Center (supported by NIH P30 CA014520) for use of the facilities and services. The authors acknowledge support from the National Genomics Infrastructure in Stockholm funded byScience for Life Laboratory, the Knut and Alice Wallenberg Foundation and the Swedish ResearchCouncil, and SNIC/Uppsala Multidisciplinary Center for Advanced Computational Science forassistance with massively parallel sequencing and access to the UPPMAX computationalinfrastructure. We thank Norman E. Davey and Leandro Simonetti for support with the computational pipeline for NGS data analysis.
Footnotes
Competing Interests
The authors have no competing interests to declare.
Data availability
Atomic coordinates of the OGT4.5 co-crystal structures have been deposited in RCSB Protein Data Bank under accession codes 8FE6 (OGT4.5:UDP-GlcNAc:CP37), 8FE7 (OGT4.5:UDP-GlcNAc:SMG9), and 8FUF (OGT4.5:UDP-GlcNAc:ZNF831). The data that support the findings of this study are available within the main text and its Supplementary Information file. Source data are provided as Source Data files. Data are also available from the corresponding author upon request.
References
- 1.Chiaradonna F, Ricciardiello F & Palorini R The nutrient-sensing hexosamine biosynthetic pathway as the hub of cancer metabolic rewiring. Cells 7, 53 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu C & Li J O-GlcNAc: a sweetheart of the cell cycle and DNA damage response. Front. Endocrinol. 9, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parker MP, Peterson KR & Slawson C O-GlcNAcylation and O-GlcNAc cycling regulate gene transcription: emerging roles in cancer. Cancers 13, 1666 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ruan H-B, Nie Y & Yang X Regulation of protein degradation by O-GlcNAcylation: crosstalk with ubiquitination. Mol. Cell. Proteomics 12, 3489–3497 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ong Q, Han W & Yang X O-GlcNAc as an integrator of signaling pathways. Front. Endocrinol. 9, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hart GW Nutrient regulation of signaling and transcription. J. Biol. Chem. 294, 2211–2231 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kokot T & Köhn M Emerging insights into serine/threonine-specific phosphoprotein phosphatase function and selectivity. J. Cell Sci. 135, jcs259618 (2022). [DOI] [PubMed] [Google Scholar]
- 8.Van Roey K et al. Short linear motifs: ubiquitous and functionally diverse protein interaction modules directing cell regulation. Chem. Rev. 114, 6733–6778 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Tompa P, Davey NE, Gibson TJ & Babu MM A million peptide motifs for the molecular biologist. Mol. Cell 55, 161–169 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Kumar M et al. The eukaryotic linear motif resource: 2022 release. Nucleic Acids Res. 50, D497–D508 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lazarus MB, Nam Y, Jiang J, Sliz P & Walker S Structure of human O-GlcNAc transferase and its complex with a peptide substrate. Nature 469, 564–567 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jínek M et al. The superhelical TPR-repeat domain of O-linked GlcNAc transferase exhibits structural similarities to importin α. Nat. Struct. Mol. Biol. 11, 1001–1007 (2004). [DOI] [PubMed] [Google Scholar]
- 13.Meek RW et al. Cryo-EM structure provides insights into the dimer arrangement of the O-linked β-N-acetylglucosamine transferase OGT. Nat. Commun. 12, 6508 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iyer SPN & Hart GW Roles of the tetratricopeptide repeat domain in O-GlcNAc transferase targeting and protein substrate specificity. J. Biol. Chem. 278, 24608–16 (2003). [DOI] [PubMed] [Google Scholar]
- 15.Levine ZG et al. O-GlcNAc transferase recognizes protein substrates using an asparagine ladder in the tetratricopeptide repeat (TPR) superhelix. J. Am. Chem. Soc. 140, 3510–3513 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Joiner CM, Levine ZG, Aonbangkhen C, Woo CM & Walker S Aspartate residues far from the active site drive O-GlcNAc transferase substrate selection. J. Am. Chem. Soc. 141, 12974–12978 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kositzke A et al. Elucidating the protein substrate recognition of O-GlcNAc transferase (OGT) toward O-GlcNAcase (OGA) using a GlcNAc electrophilic probe. Int. J. Biol. Macromol. 169, 51–59 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Joiner CM, Hammel FA, Janetzko J & Walker S Protein substrates engage the lumen of O-GlcNAc transferase’s tetratricopeptide repeat domain in different ways. Biochemistry 60, 847–853 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davey NE et al. Discovery of short linear motif-mediated interactions through phage display of intrinsically disordered regions of the human proteome. FEBS J. 284, 485–498 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Hanover JA, Krause MW & Love DC The hexosamine signaling pathway: O-GlcNAc cycling in feast or famine. Biochim. Biophys. Acta 1800, 80–95 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Benz C et al. Proteome-scale mapping of binding sites in the unstructured regions of the human proteome. Mol. Syst. Biol. 18, e10584 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davey NE, Simonetti L & Ivarsson Y ProP-PD for proteome-wide motif-mediated interaction discovery. Trends Biochem. Sci. 47, 547–548 (2022). [DOI] [PubMed] [Google Scholar]
- 23.Davey NE, Haslam NJ, Shields DC & Edwards RJ SLiMFinder: a web server to find novel, significantly over-represented, short protein motifs. Nucleic Acids Res. 38, W534–W539 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Edwards RJ, Davey NE & Shields DC SLiMFinder: a probabilistic method for identifying over-represented, convergently evolved, short linear motifs in proteins. PLOS ONE 2, e967 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huynh K & Partch CL Analysis of protein stability and ligand interactions by thermal shift assay. Curr. Protoc. Protein Sci. 79, 28.9.1–28.9.14 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamashita A et al. SMG-8 and SMG-9, two novel subunits of the SMG-1 complex, regulate remodeling of the mRNA surveillance complex during nonsense-mediated mRNA decay. Genes Dev. 23, 1091–1105 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu J et al. Quantitative and site-specific chemoproteomic profiling of protein O-GlcNAcylation in the cell cycle. ACS Chem. Biol. 16, 1917–1923 (2021). [DOI] [PubMed] [Google Scholar]
- 28.Takeda S et al. Role of a tyrosine phosphorylation of SMG-9 in binding of SMG-9 to IQGAP and the NMD complex. Biochem. Biophys. Res. Commun. 410, 29–33 (2011). [DOI] [PubMed] [Google Scholar]
- 29.Jerabek-Willemsen M et al. MicroScale Thermophoresis: interaction analysis and beyond. J. Mol. Struct. 1077, 101–113 (2014). [Google Scholar]
- 30.Nooren IMA & Thornton JM Structural characterisation and functional significance of transient protein–protein interactions. J. Mol. Biol. 325, 991–1018 (2003). [DOI] [PubMed] [Google Scholar]
- 31.Janin J, Bahadur RP & Chakrabarti P Protein–protein interaction and quaternary structure. Q. Rev. Biophys. 41, 133–180 (2008). [DOI] [PubMed] [Google Scholar]
- 32.Ardito F, Giuliani M, Perrone D, Troiano G & Lo Muzio L The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review). Int. J. Mol. Med. 40, 271–280 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mishra S, Ande SR & Salter NW O-GlcNAc modification: why so intimately associated with phosphorylation? Cell Commun. Signal. 9, 1 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Taddei ML, Pardella E, Pranzini E, Raugei G & Paoli P Role of tyrosine phosphorylation in modulating cancer cell metabolism. Biochim. Biophys. Acta BBA - Rev. Cancer 1874, 188442 (2020). [DOI] [PubMed] [Google Scholar]
- 35.Hunter T The genesis of tyrosine phosphorylation. Cold Spring Harb. Perspect. Biol. 6, a020644 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thompson JW, Griffin ME & Hsieh-Wilson LC Methods for the detection, study, and dynamic profiling of O-GlcNAc glycosylation. Methods Enzymol. 598, 101–135 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krystkowiak I & Davey NE SLiMSearch: a framework for proteome-wide discovery and annotation of functional modules in intrinsically disordered regions. Nucleic Acids Res. 45, W464–W469 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wulff-Fuentes E et al. The human O-GlcNAcome database and meta-analysis. Sci. Data 8, 25 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang DW, Sherman BT & Lempicki RA Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009). [DOI] [PubMed] [Google Scholar]
- 40.Sherman BT et al. DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 50, W216–W221 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hornbeck PV et al. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 43, D512–D520 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oughtred R et al. The BioGRID database: A comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci. Publ. Protein Soc. 30, 187–200 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ramirez DH, Yang B, D’Souza AK, Shen D & Woo CM Truncation of the TPR domain of OGT alters substrate and glycosite selection. Anal. Bioanal. Chem. 413, 7385–7399 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hu C-W et al. Electrophilic probes for deciphering substrate recognition by O-GlcNAc transferase. Nat. Chem. Biol. 13, 1267–1273 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bacigalupa ZA, Bhadiadra CH & Reginato MJ O-GlcNAcylation: key regulator of glycolytic pathways. J. Bioenerg. Biomembr. 50, 189–198 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liberti MV & Locasale JW The Warburg effect: how does it benefit cancer cells? Trends Biochem. Sci. 41, 211–218 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shafi R et al. The O-GlcNAc transferase gene resides on the X chromosome and is essential for embryonic stem cell viability and mouse ontogeny. Proc. Natl. Acad. Sci. 97, 5735–5739 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Forsythe ME et al. Caenorhabditis elegans ortholog of a diabetes susceptibility locus: oga-1 (O-GlcNAcase) knockout impacts O-GlcNAc cycling, metabolism, and dauer. Proc. Natl. Acad. Sci. 103, 11952–11957 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alteen MG et al. Potent de novo macrocyclic peptides that inhibit O-GlcNAc transferase through an allosteric mechanism. Angew. Chem. Int. Ed. 62, e202215671 (2023). [DOI] [PubMed] [Google Scholar]
- 50.Alteen MG et al. Phage display uncovers a sequence motif that drives polypeptide binding to a conserved regulatory exosite of O-GlcNAc transferase. 2023.03.15.532872 Preprint at 10.1101/2023.03.15.532872 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods-only References
- 51.Vonrhein C et al. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. D Biol. Crystallogr. 67, 293–302 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Evans PR & Murshudov GN How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 69, 1204–1214 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Evans P Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 (2006). [DOI] [PubMed] [Google Scholar]
- 54.McCoy AJ et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Emsley P & Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 56.Adams PD et al. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 58, 1948–1954 (2002). [DOI] [PubMed] [Google Scholar]
- 57.Huttlin EL et al. Dual proteome-scale networks reveal cell-specific remodeling of the human interactome. Cell 184, 3022–3040.e28 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sievers F et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Waterhouse AM, Procter JB, Martin DMA, Clamp M & Barton GJ Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yariv B et al. Using evolutionary data to make sense of macromolecules with a “face-lifted” ConSurf. Protein Sci. 32, e4582 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic coordinates of the OGT4.5 co-crystal structures have been deposited in RCSB Protein Data Bank under accession codes 8FE6 (OGT4.5:UDP-GlcNAc:CP37), 8FE7 (OGT4.5:UDP-GlcNAc:SMG9), and 8FUF (OGT4.5:UDP-GlcNAc:ZNF831). The data that support the findings of this study are available within the main text and its Supplementary Information file. Source data are provided as Source Data files. Data are also available from the corresponding author upon request.