

Significance
The pH-dependent response of CIL-LNPs (Cationic ionizable lipid-Lipid nanoparticles) is key to endosomal release and subsequently improved clinical outcome. We provide a systematic study of lyotropic order of three ionizable lipids and determine pH-dependent structural transitions in terms of crystal symmetry and spacings. The findings result in a refined perception of mode of action. Supported by molecular dynamics (MD) simulations, it stands to reason that the inverse micellar to inverse hexagonal transition of the excess CIL phase is the limiting step associated with release and less efficient in DD (DLin-DMA). This hypothesis is substantiated by delayed protein expression onset in DD-LNP-mediated transfection compared to MC3 and KC2. Our data suggest that not structure but structural transitions cause fusogenic activity.
Keywords: lipid nanoparticles, mRNA delivery, ionizable lipid, lyotropic mesophases, SAXS
Abstract
Lipid nanoparticles (LNPs) are advanced core-shell particles for messenger RNA (mRNA) based therapies that are made of polyethylene glycol (PEG) lipid, distearoylphosphatidylcholine (DSPC), cationic ionizable lipid (CIL), cholesterol (chol), and mRNA. Yet the mechanism of pH-dependent response that is believed to cause endosomal release of LNPs is not well understood. Here, we show that eGFP (enhanced green fluorescent protein) protein expression in the mouse liver mediated by the ionizable lipids DLin-MC3-DMA (MC3), DLin-KC2-DMA (KC2), and DLinDMA (DD) ranks MC3 ≥ KC2 > DD despite similar delivery of mRNA per cell in all cell fractions isolated. We hypothesize that the three CIL-LNPs react differently to pH changes and hence study the structure of CIL/chol bulk phases in water. Using synchrotron X-ray scattering a sequence of ordered CIL/chol mesophases with lowering pH values are observed. These phases show isotropic inverse micellar, cubic Fd3m inverse micellar, inverse hexagonal and bicontinuous cubic Pn3m symmetry. If polyadenylic acid, as mRNA surrogate, is added to CIL/chol, excess lipid coexists with a condensed nucleic acid lipid phase. The next-neighbor distance in the excess phase shows a discontinuity at the Fd3m inverse micellar to inverse hexagonal transition occurring at pH 6 with distinctly larger spacing and hydration for DD vs. MC3 and KC2. In mRNA LNPs, DD showed larger internal spacing, as well as retarded onset and reduced level of DD-LNP-mediated eGFP expression in vitro compared to MC3 and KC2. Our data suggest that the pH-driven Fd3m- transition in bulk phases is a hallmark of CIL-specific differences in mRNA LNP efficacy.
Decades of research, based on rational design and screening approaches, resulted in the achievement of lipid nanoparticles (LNPs), which have improved efficiency and safety of nucleic acid delivery over several orders of magnitude (1–3). First formulations based on LNP technology have been approved by the US Food and Drug Administration, including Onpattro, the first LNP-based drug delivering small interference RNA (siRNA) (4), as well as Moderna's and Pfizer/BioNTech’s mRNA LNP-based vaccines against SARS-CoV-2 (5, 6). It is expected that LNPs will play a vital role in future mRNA-based vaccines for cancer immunotherapy (6), chimeric antigen receptor T cell–based immunotherapies (7), and gene editing using CRISPR (8). LNPs exhibit favorable properties such as defined size, colloidal stability, and surface inertness. Efficient condensation and encapsulation of negatively charged nucleic acid cargo in LNPs is enabled via cationic lipid charge and rapid microfluidic mixing during the self-assembly process (9, 10). The choice of ionizable and biodegradable lipids reduces cytotoxicity and enables rapid elimination from plasma and tissue (11). Finally, LNPs efficiently mediate cellular uptake via plasma proteins and subsequent release of nucleic acid to the cytosol (12). It is understood that these properties come about by optimal choice of four lipid components including cholesterol (chol) that self-assemble into a well-defined core-shell architecture which distinguishes LNPs from other gene delivery particles. Typically, PEG-lipid and a structural helper lipid are employed to form a surface monolayer that stabilizes LNP size, while cationic ionizable lipid (CIL) and chol represent the components that condense and encapsulate nucleic acid in the LNP core. Cryo-TEM imaging provides some insight into LNP size and morphology (13–16). For LNPs formulated from ionizable lipid and siRNA, spherical morphology is observed with relatively homogeneous size and core textures indicating dense hexagonal or lamellar order (13, 15). The picture is in agreement with coarse-grained simulations of siRNA-LNPs reproducing core-shell particles with PEG-lipid segregated at the surface and a disordered, densely packed core phase (10, 17). The mRNA LNPs discussed here show spherical shapes with dense, homogenous cores in cryo-TEM images (14).
Despite clinical success, the mechanism of how LNPs mediate release of mRNA from endosomes into the cytosol is still not fully resolved. As a result of this, we are not yet in the position to design predictable nanomedicine from the bottom up. Small molecular variations in chemical structure have unexpectedly large effects on efficiency. While transport and biodistribution in the extracellular space as well as adhesion and uptake by target cells is controlled by surface properties, it is generally believed that the core structure and pH-dependent properties of the fusogenic ionizable lipid components enable endosomal release. Specifically, the claim was made that for optimal siRNA-mediated gene silencing, the effective pKa of ionizable lipids, as determined using the anionic fluorescent dye 2-(p-toluidino)-6-naphthalenesulfonic acid binding assay, should be in the range between 6.2 and 6.5 (18). The ionizable lipids DLin-DMA (DD), DLin-KC2-DMA (KC2) (19), and DLin-MC3-DMA (MC3) (18) have been a showcase of ionizable lipid optimization featuring dramatically improved in vivo activity (Fig. 1 A and B). However, despite the use of the same ionizable dimethylamino group and tail length, DD, KC2, and MC3 perform differently. It appears that the pH-dependent response of ionizable lipids is key to understand endosomal release, but the choice of an optimal ionizable lipid headgroup alone is not the full answer (13, 20).
Fig. 1.
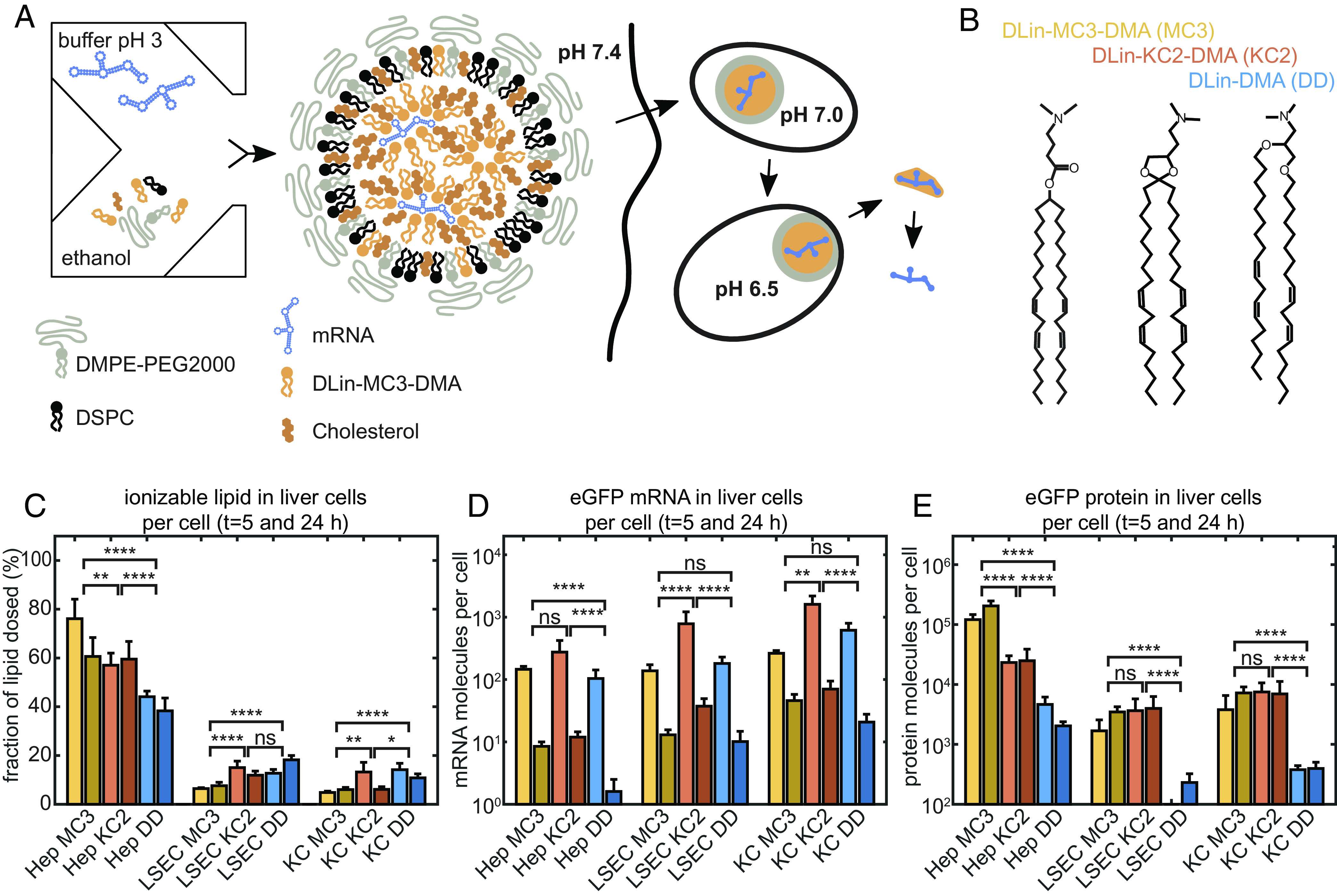
LNP-mediated mRNA delivery and protein expression in vivo. (A) Schematic drawing of production, internalization via endocytosis, and pH-dependent maturation of LNPs preceding endosomal release. mRNA LNP particles exhibit a core-shell structure consisting of a condensed core phase composed of mRNA, CIL, and chol, with an outer shell enriched in DSPC and a PEG corona. (B) The ionizable lipids MC3, KC2, and DD differ in head group area while sharing the same lipid tails and ionizable dimethylamino-group. (C) Mice study of ionizable lipid uptake in liver cells, (D) mRNA uptake in liver cells, and (E) in vivo protein expression per cell type showing same uptake yet unexplained order of protein expression efficacy MC ≥ KC2 > DD. Four mice in each group were used with an mRNA dose of 0.5 mg/kg. Error bars in figures (C–E) are the SEM. Significance levels are based on the Mann–Whitney method.
It has been argued for long that lipid polymorphism, i.e., the properties of lipids to aggregate into various liquid crystalline symmetries, plays a role in lipid-mediated delivery of nucleic acid (19, 21, 22). In the past, cationic lipid complexed with negatively charged DNA has been shown to form distinct mesophases with lyotropic lipid order and DNA superlattices (22–26). These condensed lipid phases are indexed with a superscribed “c” for complexed and constitute electrostatically neutral thermodynamic phases with defined DNA spacings. With increasing nitrogen–phosphate (N/P) ratio, complexed nucleic acid lipid phases coexist with excess cationic lipid. Phase behavior depends on composition, temperature, and salt conditions. Recently, lamellar CIL phases mixed with mRNA were studied and shown to be pH responsive (27–29). It has been shown that inverse hexagonal and cubic symmetries of complexed phases lead to increased nucleic acid delivery compared to lamellar order, a structure–activity relation that seems to hold both for lipoplexes (22–24) as well as LNPs (15, 16). In contrast to lipoplexes, that can be perceived as dispersed complexed mesophases without dedicated surface component, LNPs are core-shell particles with distinct surface and core composition. In fact, as shown in recent neutron scattering studies, the surface-active components PEG-lipid and DSPC are phase separated, for the most part, from ionizable lipid–chol that predominantly form the core phase (14). This finding is of fundamental value since it allows to study and improve surface and core properties separately. In this spirit, it is insightful to study phase behavior of macroscopic bulk samples that mimic the LNP core moiety. Yet, studies on CIL/chol bulk phases with and without nucleic acid are missing.
Here, we explore the in vivo delivery of eGFP (enhanced green fluorescent protein) mRNA LNPs based on MC3, KC2, and DD and quantify the cell-specific lipid and mRNA delivery as well as the consecutive protein expression. To rationalize these in vivo observations, we systematically study the mesophases of the core-forming LNP components, i.e., the CIL, MC3, KC2, and DD and chol. Using high-resolution synchrotron X-ray scattering, we identify lyotropic structures as a function of pH in pure CIL/chol/buffer phases and phases of CIL/chol/buffer and nucleic acid. We pursue the hypothesis that structures found in reconstituted bulk phases are meaningful for the clarification of LNP response inside endosomes. Throughout this manuscript, we will distinguish macroscopic “bulk phases” with defined lipid composition, from “LNP core phases”, which represent the inner lipid moiety of LNPs. We find for the three ionizable lipids, MC3, KC2, and DD, that first the pure lipid/chol phases exhibit ordered mesophases in the following order of appearance: , cubic micellar Fd3m, inverse hexagonal and bicontinuous cubic Pn3m, when lowering pH from pH7 to pH5. Second, if polyA (polyadenylic acid) as linear mRNA model is added to the bulk phases, coexistence of pure lipid phases and a complexed nucleic acid lipid phase is evidenced by separate sets of Bragg reflections. We show that the observed structures are consistent with SAXS (Small angle X-ray scattering) data from less ordered phases containing eGFP-mRNA as well as from LNPs. We find that the excess lipid phase for all three CIL studied undergoes an Fd3m- transition at about pH 6. However, DD exhibits considerably larger micellar nearest-neighbor distances in the transition region. Likewise, we observe that in LNPs, both spatial order and lipid mobility undergo a pronounced transition with pH. This finding goes hand in hand with a distinctly delayed onset and reduced level of eGFP expression in DD-meditated compared to MC3- and KC2-mediated mRNA LNP transfection. We discuss how the pH-sensitive transition from inverse spherical micellar to inverse hexagonal lipid structures in lipid excess phases explains the observed CIL-dependent difference in LNP endosomal release.
Results
In Vivo eGFP-mRNA LNP Dosing Experiment in Mice.
The LNPs studied here are prepared at pH 3 by rapid mixing of an aqueous mRNA citrate buffer phase and lipid/chol components dissolved in ethanol phase. The resulting particles are dialyzed against phosphate-buffered saline (PBS, pH 7.4) in order to adjust the pH and remove residual ethanol (Materials and Methods). LNPs are composed of ionizable lipid, DSPC, Chol and PEGylated lipid (DMPE-PEG2000) forming core-shell particles as schematically depicted in Fig. 1A. The N/P ratio of nitrogen atoms on the ionizable lipid and phosphor atoms on the mRNA chain was 3:1. In the following, we compare the three ionizable lipids DLin-MC3-DMA (MC3), DLin-KC2-DMA (KC2), and DD, which are outfitted with the same ionizable (dimethyl)-amino group and the identical (dilinoleyl) tail moiety as shown in Fig. 1B (Materials). However, the ionizable lipids differ in their linker group, and it has been noticed earlier that also the siRNA knockdown efficiency of the three lipids vastly differ both in vitro as well as in vivo (18). LNPs are internalized by cells via receptor-mediated endocytosis. Inside endosomes, pH is lowered over time and pH-induced structural changes are believed to induce LNP-endosomal membrane fusion and subsequent release of mRNA from LNPs (Fig. 1A). The different efficiency of the three ionizable lipids is believed to be routed in the endosomal release mechanism. In order to verify that this hypothesis is valid for mRNA delivery in vivo and furthermore that the mechanism is independent of cell type, we designed a comparative LNP dosing experiment in CD1 mice. Specifically, we measure uptake of the ionizable lipid component and the eGFP-mRNA via MC3-, KC2- and DD-based LNP formulations and compare relative uptake to protein expression. To this end, mice were injected with eGFP encoding mRNA LNPs, and the resulting mRNA and eGFP level in liver cells was analyzed. In order to quantify uptake into various types of liver cells in an unbiased way, cells from the liver were carefully isolated, thus removing extracellular components containing unsuccessful LNP material. Subsequently, cells were sorted to yield insight into possible cell type dependence. Specifically, hepatocytes, liver sinusoidal endothelial cells (LSECs), and Kupffer cells (KCs) were isolated from mouse liver. Hepatocytes make up 90% of the liver volume, LSECs account for most of the liver endothelial interface, and KCs comprise tissue-resident macrophages (30). There is a distinct dual blood clearance functionality between LSECs, which are specialized on uptake of macromolecules and nanoparticles <approx. 200 nm, and KCs, which are specialized on uptake of particles >approx. 200 nm (31). All three cell types are then subject to analysis of mRNA, eGFP, and ionizable lipid content. As shown in Fig. 1 C and D, the uptake of both ionizable lipid and mRNA in liver cells is similar between MC3-, KC2-, and DD-mediated delivery. In control mice given only PBS, analytical measurements of either the ionizable lipid, the mRNA or the protein was below the lower limit of quantification. A significant fraction of the lipid dose for all three LNPs is found in hepatocytes, likely because by both number and volume, these cells constitute the major part of the liver. Note that the study was designed with animals randomized in pairs as diagramed by two bars in the plot representing samples taken at 5 and 24h, respectively. Protein expression was most pronounced in the hepatocyte fraction vs LSECs and KCs. Furthermore, in the hepatocytes, eGFP expression varied in the order MC3 > KC2 > DD (Fig. 1E), despite similar lipid and mRNA uptake at the cellular lever (per cell). It should be noted that for LSEC and KC fractions, LNPs of MC3 and KC2 gave similar but higher expression vs DD LNPs. Furthermore, the order of expression level follows the same trend, albeit less pronounced, as for siRNA-LNP-mediated hepatic gene silencing (18, 19). Hence, the quest to search for difference in the pH-dependent response of the three different CIL-LNPs during endosomal release is of immediate importance for clinical applications.
Bulk CIL/Chol Mixtures Exhibit Sequence of Lyotropic Phases as a Function of pH.
In order to understand structural transitions that occur in the core of LNPs, we study macroscopic bulk phases of CIL/chol/water with and without nucleic acid as a function of pH. An overview of the high-resolution synchrotron SAXS data is given in Fig. 2. SI Appendix, Fig. S1 shows an overview of these SAXS measurements at a higher resolution. Adenoside monophosphate homopolymer, polyA, was selected as a model for mRNA because of similar biophysical properties. The experiments follow the investigations done in previous work (14) exploring bulk phase properties as proxies for LNP core phase behavior. In their study as well as in ours, the bulk phases were produced in a way that was as analogous to LNP production as possible. CILs, chol, and polyA were mixed and dialyzed against the same buffer used in the microfluidic mixer during the formation of LNPs (50 mM citrate pH 3 and ethanol, in a 3 to 1 volume ratio (SI Appendix, Fig. S2). Afterward, to simulate the process of LNP production, the bulk phases were dialyzed against PBS pH 7.4. Finally, a third dialysis step was used to bring the bulk phases to the required pH. The chosen pH range (pH 4.5 to pH 7.4) was adjusted to match the pH conditions experienced by LNPs within the cellular endosome (32) to capture the entire gamut of interactions of polyA with the lipids. Samples were equilibrated over 1 to 4 wk. The long dialysis and equilibration time are required to ensure the formation of equilibrated homogeneous phases as also seen in the evolution of SAXS data with time (SI Appendix). Note that according to the preparation protocol, water is added in excess and CIL/chol/buffer phases typically swim above supernatant buffer due to their lower density. Fig. 2A shows the scattering data of CIL/chol/buffer bulk phases dialyzed to a range of pH conditions for the three CILs, MC3, KC2, and DD. Sharp Bragg reflections emerge at pH values lower than about 6.5, indicating the formation of ordered phases. The crystal symmetries of the ordered phases are identified according to the relative peak positions (Fig. 2C and Methods). The overall progression of structures exhibits a general trend for all three CIL/chol/buffer phases: At neutral pH, the bulk phase is a disordered inverse micellar phase with one main structural peak (in the region of 0.1 Å−1) and a weak second-order peak at about 0.2 Å−1. As the conditions become more acidic, at a pH of around 6.0 to 6.5, an isotropic cubic inverse micellar phase (Fd3m) appears, which is followed by a transition to an inverse hexagonal phase. In the case of DD/chol/buffer, one additional phase, a bicontinuous cubic phase Pn3m, appears at very low pH values below pH 5.0. In rare cases, we observe an inverse hexagonal micellar phase with P63/mmc symmetry (33) preceding the Fd3m cubic phase at pH 7.0. All core phases, in all pH conditions, also showed two characteristic peaks at q values of 0.186 Å−1 and 0.368 Å−1 (Bragg spacings of 33.8 Å and 17.0 Å, respectively), which were noted previously (14) as chol monohydrate crystals (34). The chol crystals are already present during the preparation steps of the bulk phases. These crystals are not expected to be present in the LNPs due to the nano-sized precipitates rapidly formed by microfluidic mixing. The overall sequence of CIL mesophases observed in Fig. 2A follows well-known behavior in lyotropic liquid crystalline phase formation which are driven by the molecular shape factor, also known as critical packing parameter (CPP) (35). This interpretation suggests that at neutral pH the effectively charge neutral CIL monolayer possesses negative curvature favoring inverse micelles. Increasing protonation of the CIL headgroup will increase the headgroup area and hence shifts the spontaneous curvature of the CIL monolayer toward less negative values. In summary, the lipid bulk phases of the three CIL/chol/buffer mixtures show almost identical order of distinct mesophases following a trend from negative curvature toward zero curvature structure with decreasing pH.
Fig. 2.
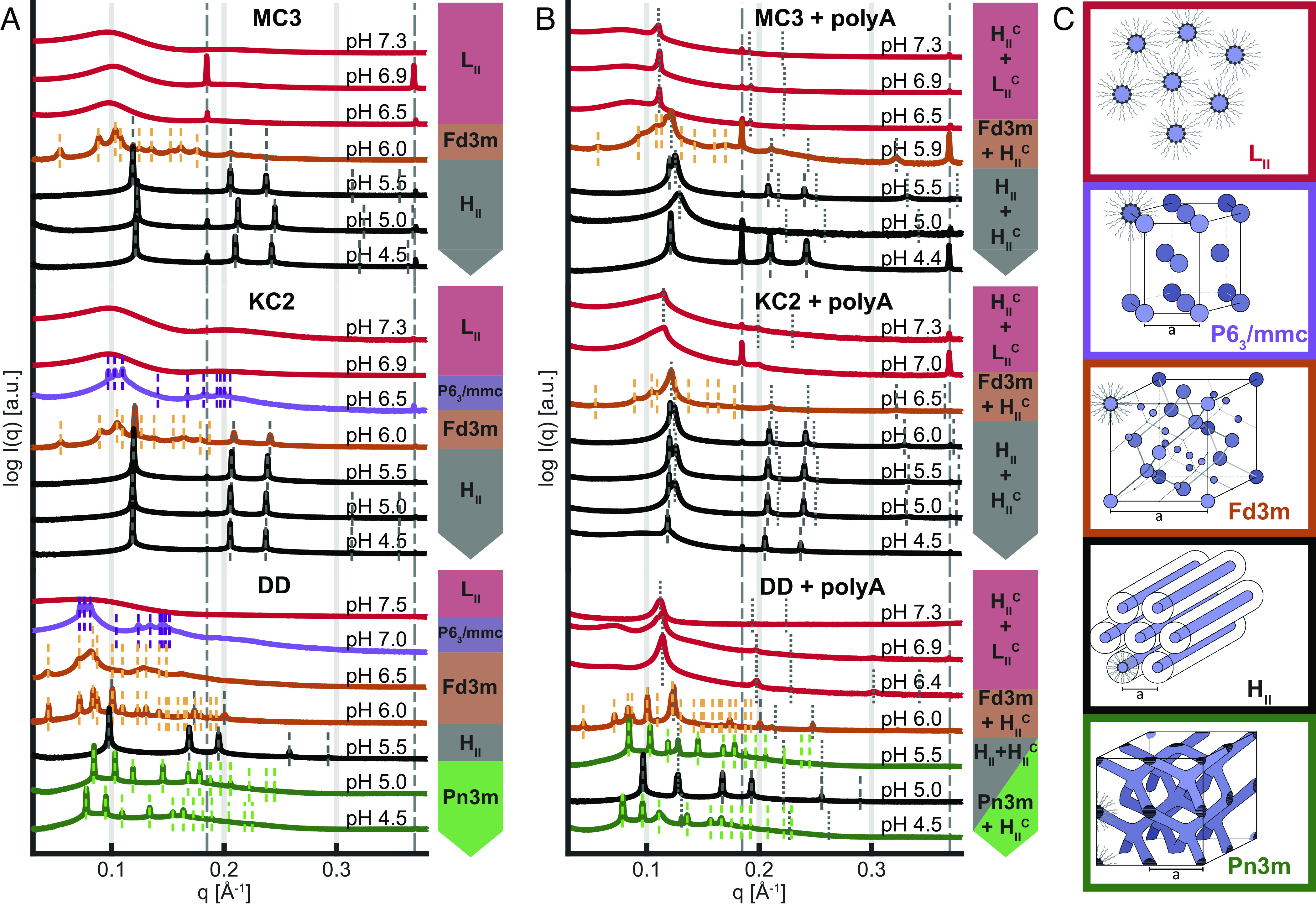
SAXS-based identification of pH-dependent mesophase transitions. (A) Ionizable lipid, chol, and buffer and (B) ionizable lipid, chol, buffer, and polyA. Data show the three ionizable lipids, MC3, KC2, and DD, for comparison. (C) Schematic representation of lipid phases with decreasing pH from top to bottom showing a trend from negative curvature toward zero curvature with increasing headgroup protonation: Inverse micellar fluid isotropic , inverse micellar cubic with P63/mmc symmetry, inverse micellar cubic with Fd3m symmetry, inverse hexagonal , and bicontinuous cubic Pn3m. In the presence of polyA (panel B), coexistence of lipid mesophases with a complexed, nucleic acid containing, phase is observed. In the pH range from 5.0 to 6.0 typically and coexist. See main text for further information.
polyA-Complexed CIL Phase Coexistence with Excess CIL.
If polyA is added to CIL/chol/buffer phases (Fig. 2B), we observe additional peaks in the SAXS data which correspond to nucleic acid complexed phases. The additional peaks appear on top of the phases without polyA, hence showing coexistence of an excess CIL phase with polyA-CIL complexed phases (SI Appendix). Most prominently, coexistence is observed in the pH range 5.5 to 6.5, where a complexed inverse hexagonal and the inverse lipid phases are seen by two closely spaced first-order reflections. Higher-order reflection with spacings 1:√3:√4 is found in some cases. At pH values above pH 6.5, when the ordered lipid phases turn into the disordered phase, the complexed phase seems to continue to exist as seen by the sharp peak on top of the phase signature. However, over periods of weeks, this peak broadens, shrinks, and eventually disappears (SI Appendix, Fig. S3). We investigated whether polyA was released from the bulk phase during this process but did not find increased nucleic acid content in the supernatant (SI Appendix, Fig. S4). Hence, we interpret the disappearance of the as unbundling of the hexagonal bundles into dispersed polyA filled inverse cylindrical micelles dissolved in the excess micellar lipid phase. As this equilibrated phase is stable over long time and SAXS pattern at long time scales do not show signs of phase coexistence, we term this isotropic complexed for a nucleic acid complexed micellar phase. Taken together, the addition of polyA to CIL/chol/buffer systems leads to complexation of polyA and CIL as indicated by the appearance of an additional inverse hexagonal condensed phase. At high pH values (pH 6 to pH 7), a less defined isotropic complexed phase forms.
Electron Density Profiles and Water Content of Inverse Phases for MC3.
Next, we study the electron density distribution and molecular conformations within the MC3/chol/buffer phases. We combine experiments and all-atom MD simulations by incorporating the experimental lattice symmetry and distances as constraints to the simulations and yield an accurate form factor from MD to model the SAXS intensities. Using this approach, the simulations provide detailed molecular insights into the lyotropic phases and and resolve the individual contributions to the scattering intensities. The measured Bragg peak intensities show that the lipid phases contain a water core surrounded by MC3 lipids with headgroups facing the water core and chol and MC3 lipid tails forming an outer oil-like moiety. We performed MD simulations of MC3/chol/water using our recently developed force fields for uncharged and charged MC3, which were validated based on neutron reflectometry data of MC3 lipid bilayers (36). Fig. 3A shows the inverse hexagonal structure with fully protonated MC3 obtained from the simulations. In the simulation, we assumed that MC3 at pH 5.5 is fully charged. Note that the exact degree of protonation of MC3 as a function of pH is difficult to determine and remains a matter of ongoing research. For the two extreme cases, we find that the hexagonal assembly with protonated headgroup is stable over the course of the simulation, while the corresponding simulation with uncharged MC3 headgroup is not. In the MD simulation, the water content is a free variable described by the molar ratio nW of water to CIL molecules. In order to determine the correct water content in the phase, we performed simulations with varying molar ratios nW. During the simulations, the box size and hence the lattice constant adapts to the external conditions. From these simulations, we selected the ratio that reproduced the experimentally measured lattice distance (SI Appendix, Fig. S5). Fig. 3B shows the electron density profile of an isolated inverse cylindrical micelle, i.e., a water channel surrounded by a monolayer of ionizable lipid and chol. The electron density profiles can be converted into effective volume fractions of the water and lipid components (see details in SI Appendix, Fig. S6). We obtain a water volume fraction for nW = 12 in the phase in good agreement with earlier experimental work for the core phase MC3 LNPs at neutral pH using neutron scattering. (14). Next, we calculated the scattering form factor from the radial MD electron density profile of an cylinder considering also neighboring cylinders (SI Appendix, Fig. S6C) and compared the result with the experimentally observed peak intensities. Fig. 3C shows a direct comparison of the SAXS data with a structure factor including Lorentzian line shapes and peak intensity given by the form factor from MD data with nW = 12 (orange line). The model agrees without any parameter adjustment, besides a global scaling factor, indicating that MD simulations reliably reproduce the structure. In the simulations, the electron density profiles of water, lipid headgroup, lipid tails, and chol can be calculated separately. In order to approach simulation of the disordered inverse micellar phase , we performed MD simulations of inverse spherical micelles with face-centered cubic symmetry providing periodic boundary conditions with twelve neighboring micelles. As before, the water content of the micelles was varied in multiple MD simulations (SI Appendix, Fig. S4 C and D). Note that the inverse spherical shape of the micelles is well reproduced by MD simulation even though the packing order of the simulation (fcc) and the more random packing in experiment differ. In particular, the simulations show that inverse micelles are stable if the MC3 head is uncharged, but unstable if the headgroup is charged. Moreover, we find a dramatic difference in the MC3 headgroup conformations in compared to phase. As the electron density profiles show, the charged MC3 headgroup sticks into the water column (Fig. 3B), while in case of spherical micelles, the uncharged MC3 headgroup hides within the hydrophobic lipid tail region (Fig. 3E). A cross-sectional view of the two phases clearly displays the different headgroup conformations (SI Appendix, Fig. S7). The experimental scattering data were fitted by the so-called Percus-Yevick structure factor with two free parameters for densely packed spheres and employing the form factor from MD simulations (Fig. 3F, red line). Moreover, the MD simulations are able to reproduce the to phase transition. As pH decreases, MC3 headgroups get charged. This is simulated by introducing charge to the MC3 headgroups at time t = 0 starting from an equilibrated conformation with uncharged MC3 headgroups (Fig. 3G). Within the first 120 ps, the headgroups swing out from an in-plane conformation toward a straight conformation that sticks into the water phase. Following this process on a time scale of 10 ns, we then see the transition of an inverse spherical to inverse cylindrical micelle, which is the basic local rearrangement in a transition from a disordered spherical water-in-oil microemulsion ( ) to an ordered anisotropic inverse hexagonal phase ( ) (Movie S1). To summarize, the MD simulations confirm the stability of the proposed liquid crystalline order for uncharged and fully charged MC3 headgroup. In addition, the combined approach allows us to assign the water content to the phase and to provide quantitative comparison with SAXS data by including the form factor from the all-atom simulations in the model fits.
Fig. 3.

MD simulations and electron density profiles of inverse phases for MC3. (A) Snapshot from an all-atom MD simulation of the phase for fully charged MC3 and chol. Water is shown in blue; cationic headgroup and tail of MC3 are shown in red and green, respectively. Chol is shown in yellow. (B) corresponding radial electron density profile for the individual components of a single cylindrical micelle from MD simulations. The black line indicates the total electron density; the vertical dash line is the half nearest neighbor distance (C) SAXS data are plotted as black dots, the fitted SAXS profile in blue and the form factor obtained from MD in orange. (D) Simulation of tightly packed inverse micelles for uncharged MC3 and (E) electron density profile. (F) Scattering of phase in red fitted in blue with the structure factor for densely packed spheres multiplied by the form factor obtained from MD. (G) Time series showing a cross-sectional view of an inverse-spherical to inverse-cylindrical micellar transition after charging the MC3 ionizable group at time t = 0. First, the MC3 headgroups swing into the water phase. Subsequently, a slower rearrangement from an inverse spherical to inverse cylindrical micelle takes place.
SAXS Signal of LNPs Is Broader but Retains Hallmarks of pH-Dependent Structural Changes.
Additionally, we demonstrate that the structural hallmarks, which are clearly visible in reconstituted macroscopic bulk phases, persist in LNP core phases. To this end, we directly compare the SAXS scattering profiles of CIL+polyA against bulk CIL+mRNA and mRNA containing LNPs at both pH 5 and pH 7 (Fig. 4A). Here, we observe two effects. First, exchanging polyA with mRNA, in this case mRNA encoding for eGFP, the sharp peaks of the polyA containing complexed phase disappear. We attribute this finding to secondary mRNA structure, which is incompatible with long range inverse hexagonal order. We also see that the order of the lipid-only is disturbed, even though the 2nd- and 3rd-order peaks remain visible. In the course of this, the signatures of polyA-filled phase and the lipid-only phase become indistinguishable. Second, when we compare CIL-mRNA bulk phase with scattering from dispersed 100 nm-size mRNA LNPs we see an additional broadening of the remaining peak together with a small shift of the qmax position toward smaller q-values. We attribute this behavior to a finite size effect, which reduces long-range order of the inverse hexagonal phase even further. The fact that the real-space spacing increases is likely caused by the fact that the LNP core phase composition might slightly differ from the reconstituted CIL bulk phase, since small amounts of both CIL as well as chol are also present in the surface monolayer. Overall, we can state that the SAXS data from bulk phases are consistent with the observed LNP scattering. Most importantly CIL+mRNA bulk phases exhibit scattering that closely resembled the signal from LNPs suggesting that the main CIL transition from high pH to low pH phase also takes place in the LNP core phase. However, the highly ordered polyA condensed appears to be a special feature of the linear homopolymer polyA. At pH 6.5, the phase becomes metastable and dissolves into a disordered isotropic phase . We graphically depict the to transition in Fig. 4B showing an order hexagonal arrangement loosening into dispersed inverse wormlike micelles containing linear polyA molecules (37). If mRNA is mixed into CIL bulk phases, no is observed at any pH. We expect that mRNA retains secondary structure and that cylindrical CIL structures will cover the mRNA contours as shown as in Fig. 4B. Hence, the highly ordered CIL+polyA phases represent a rather artificial case. However, we show in the following that we can derive some general insights about nucleic acid-CIL complexes from the ordered phases.
Fig. 4.
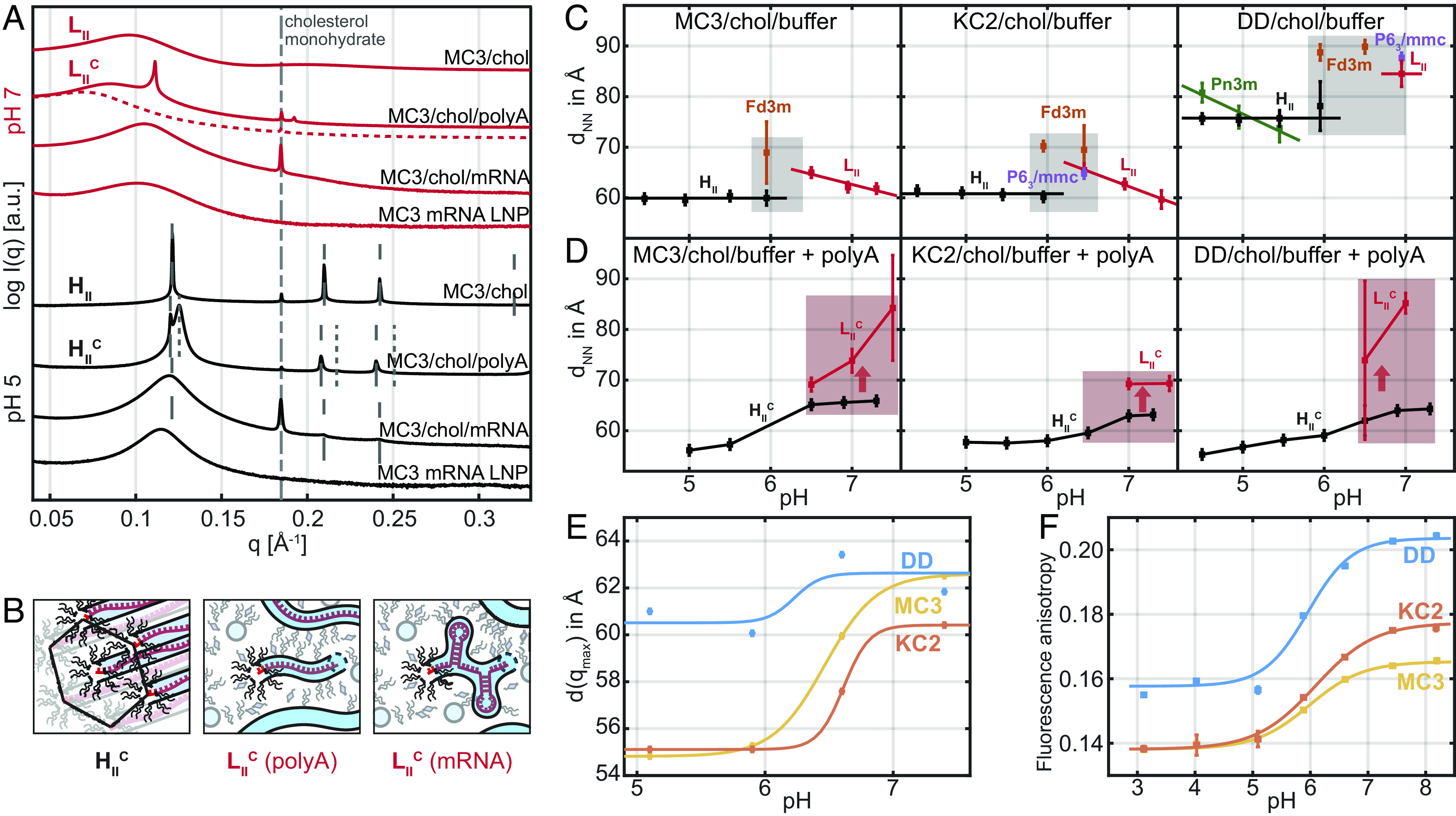
Scattering signature and pH-induced transitions in CIL bulk vs. LNP core phase. (A) SAXS scattering at pH 7 (red curves) of MC3/chol/water bulk phase, MC3/chol/water/polyA bulk phase, MC3/chol/water/mRNA bulk phase, and mRNA LNPs and corresponding SAXS data at pH 5 (black curves). The MC3/chol/water/polyA preparation at pH 7 showed time dependence with the peak dissolving over weeks. We denote the dashed line as micellar isotropic phase. The mRNA filled bulk phases and LNPs exhibit less long-range order due to mRNA secondary structure. (B) Schematic drawing of the polyA condensed phase at pH 5, the polyA condensed (polyA) phase at pH 7, and an mRNA condensed (mRNA) phase. mRNA is assumed to retain secondary structure in a disordered —micellar-like lipidic phase. (C) Next nearest neighbor distances as a function of pH showing pH-induced transitions in CIL bulk phases and (D) in polyA containing complexed phases. The gray area indicates the transition region with cubic phases. The reddish area indicates metastability of . (E) pH-dependent correlation distance in LNPs derived from SAXS data (F) Fluorescence anisotropy of a hydrophobic probe (DPH) as a function of pH for MC3-, KC2-, and DD-LNPs, respectively. Increasing anisotropy indicates decreasing mobility. The critical pH values of half-maximum are 6.0, 6.1, and 6.0 for MC3-, KC2-, and DD, respectively. Data shown in (C and D) are measured at room temperature, (E and F) at 37 °C.
pH-Induced Transitions in CIL Phases and LNPs.
Furthermore, we study the evolution of real-space distances as a function of pH. We define the nearest neighbor distance, dNN, as a figure of merit which can be deduced for all liquid crystalline phases shown in Fig. 2. In Fig. 4, the nearest neighbor distances are shown for all three CIL/chol/buffer mesophases Fig. 4C as well as for the condensed CIL/chol/polyA/buffer phases, Fig. 4D. At first glance, we note that MC3 and KC2 exhibit remarkably similar trends and spacings. The nearest neighbor distance in the inverse micellar phase increases with decreasing pH. This can be interpreted as protonation of the ionizable lipid headgroup concomitant with water uptake and swelling of the micelles. With increasing size, the degree of packing order increases, which ends up in cubic liquid crystalline phases at pH values around 6.5. In this pH regime, we almost always see coexistence with an inverse hexagonal phase , with considerably smaller nearest neighbor spacing of 60 Å. Interestingly, the nearest neighbor spacing does not change as pH values decrease further. In mesophases containing the ionizable lipid DD, the nearest neighbor distances are considerably larger, showing dNN, of about 85 to 89 Å for inverse micellar and inverse micellar cubic phase and dNN of 77 Å for the corresponding inverse hexagonal phase . Another peculiarity of DD is the coexistence of a bicontinuous cubic and an phase with the dNN spacing swelling with decreasing pH value. Hence, DD clearly shows features that are distinct from MC3 and KC2. At the same time, there is the common feature of all three ionizable lipid CIL/chol mesophases that the micellar-to-inverse hexagonal transition occurs at around the same pH value of 6.5 and in a regime, when the inverse micellar phase is most tightly packed with cubic order (highlighted as gray area in Fig. 4C). It is remarkable that the transition regime with coexistence of Fd3m and occurs at almost the same pH values for the three CILs. Fig. 4D shows the nearest neighbor spacing in polyA complexed phases to increase with increasing pH. At pH 6.5, the becomes metastable and dissolves into a disordered isotropic phase (metastable region highlighted in red). Before we discuss how these features possibly link to mechanistic action, we show that this feature and the distinction of DD are present in LNP SAXS data. Fig. 4E shows the spacing derived from LNP SAXS measurements that exhibit one maximum (SI Appendix, Fig. S8). We find that the characteristic internal spacing of MC3 and KC2 clearly exhibits the signature of a transition around the same pH value at which the transition occurs in the MC3 and KC2 mesophases. Strikingly, the spacing of DD-based LNPs does not exhibit a characteristic jump as dNN values in the bulk phases.
In order to further substantiate the existence of a structural pH-dependent transition in the LNP core phase, we carried out fluorescence anisotropy measurements using the hydrophobic tracer diphenylhexatriene (SI Appendix). Fluorescence anisotropy is inversely proportional to the tracer mobility in the lipid tail region (38). We find that fluorescence anisotropy, with increasing pH, exhibits a transition toward larger values around pH 6.0 for all three ionizable lipids. Again, MC3 and KC2 show closer agreement in absolute numbers, while DD exhibits the same trend but larger fluorescence anisotropy values indicating lower mobility in DD LNPs over the entire pH range. At the same time, the spherical to cylindrical transition does not significantly change the characteristic spacing in DD LNPs while it does in MC3 and KC2 LNPs.
DD-Mediated Transfection Exhibits Delayed Expression Kinetics vs. MC3 and KC2.
Finally, we explore the kinetics of endosomal fusion and mRNA release in transfection experiments. As the endosome matures, its pH lowers and CILs become positively charged. The structural changes discussed above provide a reasonable basis to assume that LNP core phases become more fusogenic. However, little is known when endosomal fusion events occur and if MC3, KC2, and DD differ in expression kinetics. For this reason, we pursue single-cell eGFP transfection experiments using Cy5-labeled eGFP mRNA. Cells are seeded on micropatterned arrays with fibronectin coated squares in otherwise PEGylated, nonadhesive area (SI Appendix, Fig. S9). We monitor the Cy5 fluorescence of the Cy5-labeled mRNA and the onset of eGFP expression after LNP transfection (Fig. 5A). Typically, 12 view fields per channel are sequentially imaged in 10-min intervals. In total about 1,000 fluorescence time courses of single cells are recorded. LNPs were prepared as described in the Methods section. Microscopy channel slides are used to infuse cells with LNP transfection medium (dose 5 ng/µL mRNA) for a defined period of 1 h (gray bar in Fig. 5A). After the incubation period, the channels were washed with fresh medium. Setup and protocol has been described in the Methods section and previous work (39). We determine the time points of fluorescence onset, ton, after incubation, as well as the eGFP fluorescence intensity 5 h after ton for each single-cell trajectory. The fluorescence increase over the first 5 h is taken as a measure for the initial expression rate kCIL (32). Fig. 5B shows the single-cell scatter plot of eGFP onset time and expression rate, kCIL for MC3, KC2 and DD respectively. The mean eGFP onset times (see distribution functions SI Appendix, Fig. S10) show the order while the initial expression rates show opposite order (SI Appendix, Fig. S9). This inverse relation states that on average, early expression onset is likely to lead to larger expression levels. This finding agrees with the notion that endosomal release is associated with a window of opportunity (40). We also observe that the Cy5 fluorescence signal of the labeled Cy5-mRNA exhibits an abrupt increase over time (Fig. 5A). We attribute this signal to an dequenching of the Cy5 quantum efficiency as a result of an abrupt change in the environmental conditions. It stands to reason that this transition is associated with mRNA release from LNPs. Plotting Cy5 onset vs. onset of the eGFP signal exhibits similar timing for MC3 and KC2, while both Cy5 onset as well as eGFP onset are delayed for DD with respect to MC3 and KC2 (Fig. 5C). Hence, we conclude that endosomal fusion of DD-mediated transfection is delayed with respect to MC3 and KC2 and likely as a consequence of this, DD-mediated eGFP initial expression rate is reduced.
Fig. 5.
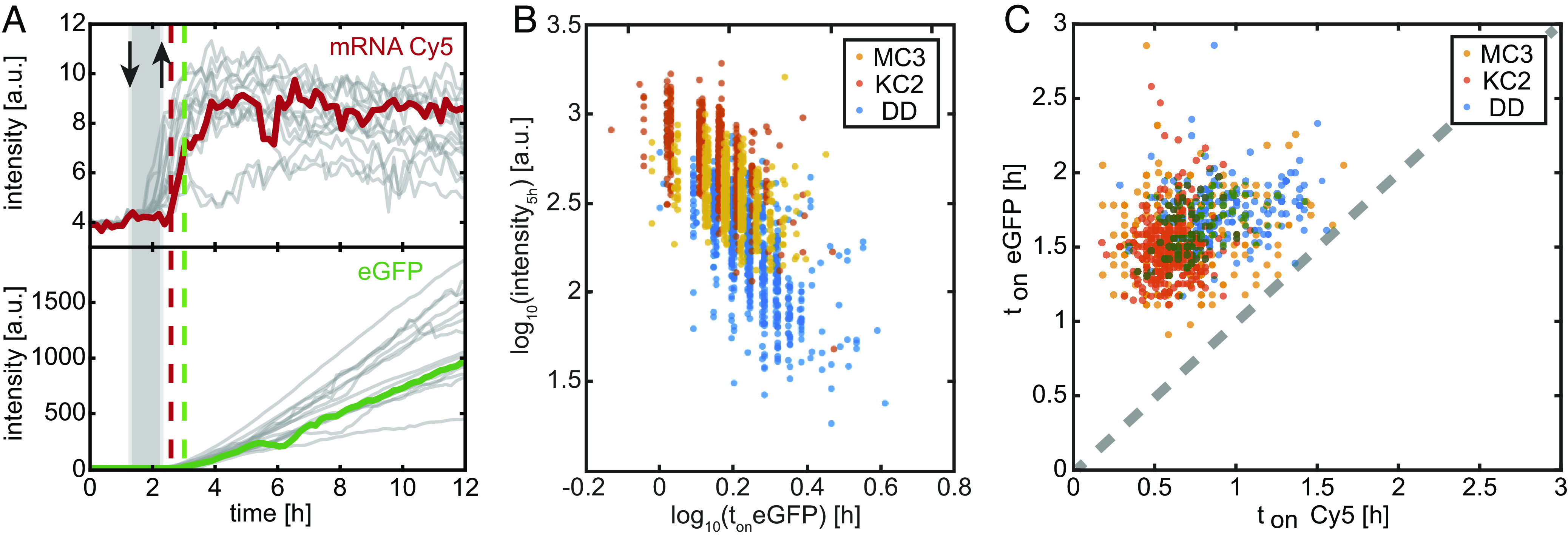
Expression kinetics after eGFP-mRNA LNP transfection in vitro. (A) Exemplary single-cell fluorescence time courses of mRNA-Cy5 and eGFP (B) Correlation of transfection efficiency and onset time of single cells reveals delayed and less efficient expression for DD vs. MC3 and KC2. (C) eGFP-onset typically occurs 0.9 h after the event of Cy5-signal increase.
Discussion
We showed that LNP-mediated delivery in mice experiments results in different protein expression of the three CILs DD, KC2 and MC3, while the lipid and mRNA uptake was similar for the three LNPs. From this finding, we concluded that there must be distinct differences in the behavior of LNPs inside endosomes that affects escape from endosomes and hence expression efficiency. To this end, we studied reconstituted mixtures of CIL and chol as proxy for the LNP core phase and explored the structural evolution of these model systems as a function of pH. We find ordered mesophases that appear in a distinct sequence. These observations are unique for the ionizable lipids MC3, KC2, and DD but fall in line with lyotropic phase behavior of binary or ternary lipid–water mixtures in general. Many amphiphilic liquid crystals display lyotropic order from inverse micellar ( ), isotropic cubic inverse micellar (I), inverse hexagonal ( ), and bicontinuous cubic (Q) toward lamellar ( ) lipid order depending on the preferred curvature of the amphiphiles (35, 41). In lyotropic liquid crystals, the CPP, which describes the ratio of headgroup to tail volume and hence the preferred curvature, predicts the observed sequence of mesophases. Applied to CILs the CPP will continuously increase with protonation of the ionizable lipid headgroups due to electrostatic headgroup repulsion explaining the sequence of all three ionizable lipids. In the specific case of MC3, all-atom MD simulations reveal that protonation of the lipid headgroup goes along with a remarkable relocation of the charged amine group from the hydrophobic part of the membrane into the aqueous phase. However, we notice CIL-specific differences in the SAXS data showing that first the nearest neighbor spacing is about 30% larger for DD vs. MC3 and KC2 phases and second that only DD exhibits a bicontinuous Pn3m phase coexisting with at low pH values. Both phenomena indicate a larger water content in DD compared to MC3 and KC2 for CIL/chol/water phases without polyA. When polyA is added to these phases at N/P ratio of three, we find coexistence of pure lipidic structures and polyA complexed structures. At pH above 6.5, the complexed inverse hexagonal structure dominates and decomposes at high pH into a disordered condensed phase over time periods of weeks. The phase agrees with previous work on DNA-lipoplexes (22) and general conjectures about a critical role of inverse hexagonal structures in LNP activity (19). However, the coexistence of two inverse hexagonal phases, a polyA filled one and an empty one, raises the question, which of the two phases relates to endosomal fusion. The data presented here support the notion that a structural transition in excess lipid is causal for fusion as key features of the lipid bulk phase transition seem to prevail in the LNP core phase. First, the curvature-driven transition from inverse spherical to inverse cylindrical structures is likely fusogenic as topological rearrangements include defects and fusogenic intermediates. In contrast, the polyA-condensed phase shows no abrupt transition at pH 6 rather coexistence of with a hypothetic isotropic wormlike phase at pH above 7. Second, the highest efficacy is observed at N/P ratio larger than one (42) demonstrating that excess lipid is adjuvant. Third, it is the Fd3m- transition that shows the biggest difference between DD and MC3 and KC2, namely increased spacing and degree of hydration. Larger spacing and hydration will lower the Gaussian curvature energies involved in the cubic-to-hexagonal transition possibly turning DD less fusogenic. It is striking that the pH-dependent shift of the one broad SAXS peak observed in nanoscopic mRNA LNPs occurs around pH 6 as the transition in excess lipid bulk phase. However, it remains an open question whether a phase-separated lipidic phase exists in the LNP core. While phase-separated structures have been recently observed in TEM images and SAXS data of oligonucleotide-loaded LNPs (15) and GMO-based LNPs (16), there is no direct evidence of phase separation in the mRNA LNPs presented here. This is most likely due to the fact that mRNA secondary structures obscure long ranged order. Our MD simulations show that excess lipid regions should at least locally favor inverse spherical micellar geometry and is likely to undergo a topological transition analogous to the inverse spherical to cylindrical transition shown in Fig. 3G. Such mechanism could possibly trigger LNP fusion with the endosomal membrane. This is in line with our finding that DD-LNP-mediated mRNA delivery showed delayed release compared to MC3 and KC2. Delayed release is closely connected to loss of LNPs and reduced delivery. Our work also suggests that “fusion” and “mRNA release” should be discerned as two distinct and deferred processes. If mRNA is embedded in an electrostatically neutral, salt-like nucleic acid lipid phase then endosomal membrane disruption will not immediately set free mRNA for translation. Little is known about the time it needs to dissolve the condensed ionizable lipid-mRNA state in the cytosol. In fact, our observation of an extremely persistent phase in macroscopic samples suggests that “unpacking” mRNA should follow endosomal fusion with some delay as evidenced in single-cell fluorescence time courses. A small percentage of internalized mRNA undergoes exocytosis which may impact the overall efficacy (43, 44). Little is known whether this externalization pathway depends on mRNA being associated with ionizable lipid.
In summary, we conclude from macroscopic phase transitions mimicking LNP core composition that pH-dependent response of excess lipid in LNPs is causing endosomal fusion. The observed bulk structures are consistent with SAXS scattering from LNP core phase and LNP cores exhibit hallmarks of pH-driven transitions. Nonetheless, we are aware that the constituents of the LNP core phase are in exchange with the LNP surface. Ionizable lipids, as well as chol molecules, partition in the surface monolayer to some degree. Recent SAXS data show that indeed changes induced by protein adsorption affects LNP core structure (45). Vice versa, a surface-core interrelation implies that pH-induced core phase transition will affect surface composition. In particular, chol partition plays a role, and there is evidence for lateral phase separation in the surface monolayer (46). While most literature strives to elicit structure–activity relations, we show that pronounced phase transitions with impetuous rearrangements might trigger activity. By comparing MC3, KC2, and DD, we conclude that structural changes in the lipidic spherical-to-cylinder transition make a difference. Similar transitions might exist in bulk phases of other ionizable lipids optimized for mRNA delivery, including ALC-0315 and SM-102, which deserve further studies. The objective of our work was to explain the distinct in vivo efficacy of mRNA LNPs made from structurally related ionizable lipids. Intravenous dosing in mice demonstrated the unequal in vivo efficiency of DD-LNPs vs. MC3- and KC2-LNPs which be ascribed to distinct behavior not in LNP uptake but in endosomal mRNA release efficiency. Elucidating further, the determinants of CIL structural transitions, their kinetics, and impact on fusion and release will contribute to rational design of improved mRNA LNP formulations for a broad spectrum of future medical applications.
Materials and Methods
Materials.
The three ionizable lipids DLin-MC3-DMA [O-(Z,Z,Z,Z-heptatriaconta-6,9,26,29-tetraem-19-yl)-4-(N,N-dimethylamino)butanoate; C43H79NO2; mw 642.09 g mol–1, >99% purity], DLin-KC2-DMA [DLin-KC2-DMA; 2,2-dilinoleyl-4-(2-dimethylaminoethyl)-[1, 3]-dioxolane; C43H79NO2; mw 642.09 g mol–1, >99% purity], and DD (1,2-dilinoleyloxy-3-dimethylaminopropane; C41H77NO2; mw 616.06 g mol–1, >99% purity) were synthesized at AstraZeneca. 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC; C44H88NO8P; mw 790.15 g mol–1; 18:0 PC; >99% purity) was purchased from Avanti Polar Lipids; N-(Carbonyl-methoxypolyethyleneglycol 2000)-1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine (DMPE-PEG2000) from NOF Corporation (average molecular weight 2785 g mol−1); Chol (C27H45OH; 386.66 g mol−1; >98% purity) was purchased from Sigma-Aldrich. PolyA (600 to 4,000 bases according to the manufacturer) was purchased from Sigma-Aldrich. mRNA encoding for eGFP was purchased from Trilink, in both the unlabelled [Clean Cap eGFP (5 moU)] as well as the fluorescently labelled form (Cyanine 5 eGFP 5 Mec psi mRNA). PBS (1 mM KH2PO4, 155 mM NaCl, and 3 mM Na2HPO4.7H2O, pH 7.4) was purchased from Gibco (Life Technologies). Citrate buffer was purchased from Teknova. HyClone RNAse-free water was used for all LNP preparations with mRNA and was purchased from GE Healthcare Cell Culture.
Formulation and Characterization of LNPs.
DLin-MC3-DMA, DLin-KC2-DMA, and DLinDMA LNPs containing modified CleanCap eGFP mRNA (5-methoxyuridine) (TriLink Biotechnologies) were prepared by precipitating the mRNA with four different lipid components. These components consist of an ionizable lipid, DLin-MC3- DMA, DLin-KC2-DMA or DLinDMA, which are ionizable (cationic) at low pH, two helper lipids (DSPC and Chol) and a PEGylated lipid (DMPE-PEG2000). A solution of eGFP mRNA in water was prepared by mixing mRNA dissolved in HyClone-water, 100 mM citrate buffer pH 3 and HyClone-water to give a solution of 50 mM citrate. For cy5-labeled eGFP mRNA LNPs, the eGFP/cy5-eGFP molar ratio was 9:1. Lipid solutions in ethanol (99.5%) were prepared with a composition of four lipid components [Ionizable Lipid:Chol:DSPC:DMPE-PEG2000] = 50:38.5:10:1.5 mol% and a total lipid content of 12.5 mM. The mRNA and lipid solutions were mixed in a NanoAssemblr (Precision Nanosystems) microfluidic mixing system at a volume mixing ratio of Aq:EtOH = 3:1 and a constant total flow rate of 12 mL/min. At the time of mixing, the ratio between the nitrogen atoms on the ionizable lipid and phosphor atoms on the mRNA chain was 3:1. If “empty” LNPs were prepared, i.e., LNPs without any mRNA, the ethanol phase was mixed with only 50 mM citrate buffer pH 3. The initial 0.25 mL and the last 0.05 mL of the LNP solution prepared were discarded while the rest of the volume was transferred immediately to a Slide-a-lyzer G2 dialysis cassette (10,000 MWCO, Thermo Fisher Scientific Inc.) and dialyzed overnight at 4 °C against PBS (pH7.4). The volume of the PBS buffer was 650 to 800x the sample fraction volume. For some samples, the LNP suspension was concentrated using Amicon Ultra 30K centrifugation filters. For the characterization of formulated LNPs, following preparation, the intensity-averaged particle size (Z-average) was measured using a ZetaSizer Nano (Malvern Instruments Inc.). The final mRNA concentration and encapsulation efficiency were measured by the Quant-it RiboGreen Assay Kit (Thermo Fisher Scientific).
Animals.
Female CD1 mice, 10 to 11 wk of age, were purchased from Charles River Laboratory, Germany, and housed in the animal facility at AstraZeneca, Mölndal, Sweden. The mice were left to acclimatize for 5 d prior to study start. Mice were kept in groups of 2 mice per cage under standard conditions (21 °C RT, 12:12 h light–dark cycle, 45 to 55% air humidity) with access to a normal chow diet (R70, Lactamin AB) and water ad libitum. Environmental enrichment was provided (cartons, gnawing stick, and cotton nesting pads). Experimental procedures were approved (ethical application number 83‐2015) by the Regional Laboratory Animal Ethics Committee of Gothenburg, Sweden. The care and husbandry of animals was conducted in accordance with the European Directive (no.86/609) and the Swedish Animal Welfare Act and regulations (SJVFS 2012: 26.).
Inclusion and Exclusion Criteria.
All mice were in the expected weight range (23 to 26 g) and thereby met the inclusion criteria. Exclusion criteria were predetermined and set for unsuccessful performance of the liver perfusion or Intravenous injection. One mouse was excluded due to unsuccessful liver perfusion.
Study Design.
Animals were randomized in pairs, and cages with pairs allocated to dose groups based on body weight (ranged from 23 to 26 g), using an animal randomization tool at 3 d prior to study start. The dosing order was fixed and adapted to fit termination time points. The termination order was randomized and decided prior to the study start. The LNP formulations and Vehicle was coded and kept blinded for the operators during the cause of the study. At study start, eGFP mRNA LNPs (DLin-MC3-DMA, DLin-KC2-DMA, and DD) (0.5 mg/kg) (n = 4 per group) or PBS (n = 3) were intravenously injected in the tail vein of mice. At 5 and 24 h after iv injection, groups of mice were anesthetized using isoflurane (Baxter Medical AB, Kista, Sweden) and killed by cervical dislocation. The animal was placed under the microscope for performance of the liver perfusion.
Liver Cell Isolation.
Isolated hepatocytes, LSECs, and KCs were prepared by the following sequential steps (47): After perfusion with collagenase (LiberaseTM Research Grade bought from Roche Diagnostics, Oslo, Norway) through the portal vein, the liver was excised, and connective tissue and gallbladder removed before carefully transferring the liver to a Petri dish for weighing. The liver was then placed in cold albumin solution (1% bovine serum albumin in 10 mM phosphate buffer, pH 7.4) and Glisson’s capsule carefully removed with forceps before gently shaking of the liver tissue to generate a single-cell suspension. The dispersed cells were filtered through a 70 µm gauge filter, and the volume was measured. Glisson’s capsule and undigested liver residue that did not pass the filter were weighed. After a low-speed centrifugation (50 × g) of the liver cell suspension, the resulting supernatant depleted of hepatocytes and containing the majority of the LSECs and KCs was collected, whereas the pellet containing the majority of the hepatocytes was resuspended in buffer and subjected to four more low-speed centrifugation washing steps for further enrichment. The purity of the hepatocytes (>95%) was easily assessed by inspection in the light microscope, as these cells are readily distinguished from other types of liver cells due to their much larger size. The LSECs and KCs from the supernatants of the first and second low-speed centrifugations were pelleted and mixed into a 20 mL 20% Percoll solution (GE Healthcare, Nydalen, Norway). After centrifugation at 300 × g for 20 min, debris and dead cells moved to the top, whereas LSECs and KCs were pelleted. The pelleted cells were resuspended in MACS isolation buffer (Miltenyi Biotec Norden AB, Lund, Sweden) before being further purified by immunomagnetic cell isolation according to the instructions by the manufacturer. Briefly, 1/3 of the resuspended cells was incubated with magnetic beads (Miltenyi) conjugated with anti-CD146 antibodies for isolation of LSECs; in parallel, the remaining 2/3 of the resuspended cells were incubated with biotin anti-mouse F4/80 (Bio-Rad, Oslo, Norway) antibodies, and then with streptavidin-conjugated magnetic beads (Miltenyi) for isolation of KCs. Separation of the two cell types was achieved by passing the cell-beads complexes over two LS+ QuadroMACSTM separation columns, according to the manufacturer’s instructions. Liver cell fractions were stored in cryotubes at -80°C until assessment of the amounts of ionizable lipids, eGFP mRNA and eGFP protein.
Gradient UPLC for the analysis of ionizable lipids in liver cell fractions.
Each cell fraction was dissolved in 400 µL of a mixture of Triton X-100 in 0.5% formic acid (1% w/v). The samples were incubated at 37 °C for 30 min, centrifuged at 10,000 rpm for 10 min, and then injected on Acquity Ultra Performance LC coupled to a Single Quad Detector, SQD (Waters, Milford). The analytical column was a Waters Acquity UPLC® CSH C18, 1.7 μm, 2.1 × 100 mm, kept at 60 °C. The flow rate was 0.50 mL/min using a mobile phase of 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile (B). A gradient run was applied where 5% B at 0.0 min was increased to 83% B at 0.5 to 3.0 min and kept at 83% B to 6.5 min. A washing step of 99% B at 6.6 to 8.5 min was included in the gradient run. Then, 5% B was applied for conditioning from 8.6 min to 11.0 min. The separation between main peak of Triton X-100 and the cationic lipids was good under these conditions. Quantification was made using external standard solutions of DLin-MC3-DMA, DLin-KC2-DMA, and DD dissolved in Triton X-100 in 0.5% formic acid (1% w/v). The SQD was run using electrospray, positive mode and tuned using a solution of DLin-MC3-DMA. Recording of the cationic lipids was made using Single Ion Recording at M + 1 for each cationic lipid.
Branched DNA Analysis of Intact eGFP mRNA in Liver Cell Fractions.
Levels of eGFP mRNA were quantified using a commercial kit purchased from ThermoFisher (QuantiGene SinglePlex bDNA assay kit, QS0012). A bDNA probe set (eGFP SF-10526) specific for TriLink eGFP mRNA was designed and supplied by ThermoFisher. Prior to analysis, the cell pellet was lysed in bDNA working lysis buffer mix containing 33% Lysis Mix (QP0522) and 0.3% proteinase K (QS0510) to give a 400 cell/µL final concentration. The cell pellet was incubated for 30 min at 55 °C, after which time, the sample was vortexed for 1 min to ensure optimal lysate viscosity. Processed sample was then stored at −80 °C until analysis. To perform analysis, 60 µL of the working probe mix (33.3 µL Lysis mix, 25.4 µL water, 1.0 µL block reagent, and 0.3 µL eGFP probe set) was added to appropriate wells in the kit-supplied 96-well bDNA plate; following this, 40 µL of all samples and standards were added. The plate was sealed and incubated, without shaking, overnight at 55 °C. After washing the plate three times with washing buffer, 100 µL pre-amplification solution was added to each well, and the sealed plate was incubated, without shaking, for 90 min at 55 °C. The plate was then washed three times with washing buffer, and 100 µL amplification solution was added to each well and incubated without shaking for 90 min at 55 °C. The plate was then washed three times with washing buffer, and 100 µL label solution was added to each well and incubated without shaking for 90 min at 50 °C. The plate was then washed three times with washing buffer, and 100 µL substrate solution was added to each well and incubated in the absence of light and without shaking for 10 min at room temperature. Total luminescence was then measured on a SpectraMax iD3, and the analyte concentration in the samples was backcalculated from the standard curve (4pL fit, 1/y weighting).
Determination of eGFP Protein in Liver Cell Fractions.
The eGFP protein levels were quantified using a commercial kit purchased from Abcam (ab171581). An extraction buffer (recommended by the kit vendor) was added to the cell samples, that were spun down and the supernatant stored at −80 °C until analysis. Fifty microliters of all samples and standards were added to appropriate wells in a 96-well plate. Then 50 µL of Antibody Cocktail, containing capture and detection antibodies were added to the wells. The plate was sealed and put on a shaker (400 rpm) for 1 h at room temperature. After washing the plate three times with washing buffer, 100 µL TMB substrate was added to each well, and the plate was put on a plate shaker for 10 min before 100 µL stop solution was added. The absorbance was then measured at 450 nm on a SpectraMax iD3, and the analyte concentration in the samples was backcalculated from the standard curve (4pL fit, 1/y weighting).
Preparation of Ionizable Lipid Bulk Phases.
CIL/chol/polyA bulk phases were produced at a range of pH conditions via three dialysis steps. First, the ionizable lipid and chol were dissolved in ethanol and mixed in a molar ratio of 3:1 (CIL:chol) to a total lipid concentration of 56.1 mg/mL. The ethanol mixture was introduced into a dialysis cassette with a molecular weight cutoff of 3.5 kDa (Slide-A-Lyzer™ G2, Thermo Scientific). An aqueous solution of polyA in buffer (20.1 mg/mL polyA in 50 mM citrate buffer at pH 3) was injected rapidly into the ethanol mixture to a final volume that resulted in a 3:1 CIL:nucleotide molar ratio. The dialysis cassette was placed into a beaker containing 500× the volume of dialysis buffer at 22 ± 2 °C. Samples were first dialyzed against 50 mM citrate buffer (pH 3) containing ethanol (at a volume ration of 3:1, buffer: ethanol) for 48 h. In the second dialysis step, the sample was dialyzed against PBS (1 mM KH2PO4, 155 mM NaCl, and 3 mM Na2HPO4 0.7H2O, pH 7.4) for 48 h. In the third step, the sample was dialyzed against a McIlvaine buffer with the required final pH (4.5, 5, 5.5, 6, 6.5, 7, and 7.5) for 48 h. During the dialysis process, large precipitates were formed from the polyA and lipids. The supernatant was removed from the cassette, and the solid precipitates were extracted for characterization by SAXS. CIL/chol bulk phases were prepared according to the same protocol, except without adding polyA.
SAXS.
Synchrotron small-angle X-ray scattering (SAXS) was carried out at the P12 BioSAXS EMBL beamline, PETRA III, DESY (Hamburg, Germany). The beamline instrumentation has been described elsewhere (48). The crystallographic space groups of the liquid crystalline phases were determined from relative peak positions. Corresponding Miller indices, calculation of the lattice constant and nearest neighbor distance are listed in SI Appendix, Tables S1 and S2. All measurements at P12 were performed in quartz capillaries (0.01 mm wall thickness, 1.5 mm outer diameter by Hilgenberg) at 10 keV. Scattering data background was subtracted by measuring empty capillaries. Fits of the phase (Fig. 4C) represent best fits of a rigid cylinder form factor P(q) and a structure factor S(q) as P(q)S(q)+bg(q), with bg(q) being a background term including the contribution of chol monohydrate in terms of two Lorentzian peaks at 0.185 Å−1 and 0.370 Å−1. The cylinder form factor P(q) is calculated from a radial electron density profile ρ(r) as retrieved from MD simulations. For data analysis, the numerical data were approximated by box model with ten partitions and fitted using the program SasView (49). The structure factor was approximated by Lorentzian profiles with peak spacings as discussed above. The fit of the phase (Fig. 4F) was obtained using P(q)S(q)+bg(q), with bg(q) being a constant, P(q) the form factor calculated from a radial electron density profile ρ(r) obtained from MD simulations (Fig. 4E) and the structure factor S(q). For S(q), the Percus–Yevick structure factor for densely packed spheres with hard core repulsive interaction was chosen (50). It describes the ordering of spheres with radius a and volume fraction . The nearest neighbor distance was approximated by assuming that micelles are in close contact as also suggested from MD simulations. Note that this approach agrees within 10% with the approximation , where qmax denotes the peak position with maximum intensity.
MD Simulations.
MD simulations are powerful tools to gain insights into the distribution of the various lipids (51). The Gromacs package (v-2019) (52) was used to perform the MD simulations. The AMBER Lipid 17 force field (53) was used to describe the chol. For the uncharged and cationic MC3, we used our recently developed force field parameters (36). These parameters have the advantage that they closely reproduce the structure of MC3 in lipid layers as judged by neutron reflectometry experiments. In addition, the parameters are compatible with the AMBER force field family. Ions were described using the Mamatkulov–Schwierz force field parameters (54), and the TIP3P water model (55) was used. After energy minimization using a gradient descent algorithm and a short equilibration run using the Berendsen thermostat and barostat, the production runs were carried out for 200 ns. The temperature was fixed at 293 K using the Nose–Hoover thermostat with a time constant of 1 ps. The pressure was fixed at 1 bar using semi-isotropic (for inverse hexagonal setup) or isotropic (for inverse micellar setup) Parrinello–Rahman barostat with a 5 ps time constant. Van der Waals interactions were truncated and shifted to zero at 1.2 nm. Short-range electrostatic interactions were cutoff at 1.2 nm, and long-range electrostatic interactions were evaluated using the Particle Mesh Ewald method with a Fourier grid spacing of 0.12 nm. All bonds involving hydrogens were constrained using the LINCS. A time step of 2 fs was used to integrate the equations of motion. A detailed description of the simulation’s setup for the inverse hexagonal and inverse micellar phase can be found in SI Appendix.
Single-Cell Time-Lapse Imaging.
Live-cell imaging on single-cell arrays was carried out as previously described (39, 56). In short, single-cell microarrays with adhesive squares were produced by microscale plasma-induced protein patterning. The adhesion sites are coated with fibronectin, and the interspace is passivated with PLL-g-PEG. Microstructured coverslips were attached to a six-channel slides (ibidi, GmbH Munich). Human hepatocyte-derived carcinoma, HuH7 cells (I.A.Z. Toni Lindl GmbH, Germany) were grown in modified RPMI containing GlutaMax supplemented with 5 mM HEPES, 1 mM sodium pyruvate, and 10% fetal bovine serum (FBS). Cells were seeded in the six-channel slide 4 h prior to the time-lapse measurement at a cell density of 10,000 cells per channel. The slide was connected to a tubing system allowing for transfection and subsequent rinsing during the time-lapse measurement. Scanning time-lapse imaging was performed on a motorized inverted microscope (Eclipse Ti-E; Nikon) with an objective lens (CFI PlanFluor DL-10x, Phase 1, N.A. 0.30; Nikon) equipped with a heating system (Okolab). Fluorescence image stacks with a time resolution of 10 min were acquired using a cooled CMOS camera (pco.edge 4.2; pco), a LED light source (SOLA-SE II, Lumencor), and a suitable filter cube for eGFP (BP450 to 490, FT510, LP 510 to 565; CHROMA Technology Corp.) or Cy5 (filter cube). First, all channels were rinsed with 37 °C warm PBS. Subsequently, LNP containing solution was added to each channel. After 1 h of incubation with the transfection complexes, all channels were washed with L15 medium supplemented with 10% FBS, which remains in the channels for the remaining 12 h of measurement. Analysis of single-cell fluorescence traces was done as described in earlier studies (56–58). The onset times of the Cy5 traces were determined manually, whereas the eGFP traces were determined using a hierarchical cluster analysis approach as described in ref. 39. Per LNP preparation, about 500 cells were evaluated (see scatterplot Fig. 5). At least two technical repeats were carried out.
Supplementary Material
Appendix 01 (PDF)
The video starts with a 90° round view of the inverse micellar phase (LII) with 12 nearest neighbors. All ionizable MC3 lipids are neutral. Subsequently, the cross section of the LII phase is shown. Mimicking the change in pH, the MC3 lipids are protonated and the molecular dynamics simulations start. The MC3 head groups (red) quickly enter the water phase. Subsequently, the topology changes from LII to HII and the water columns are formed. The dynamics stops and a 90° rotation of the inverse hexagonal phase (HII) is shown. In the video, head and tail groups of the MC3 lipids are shown in red and green, respectively. Water is shown in cyan and cholesterol in yellow
Acknowledgments
This work was supported by the German Federal Ministry of Education and Research through BMBF Project 05K18WMA and 05K18EZA within the framework of the Swedish–German research collaboration Röntgen-Ångström Cluster. A.R. and R.K acknowledge funding from the German Science Foundation through the Graduate School of Quantitative Biosciences (QBM), LMU. We would like to acknowledge Johan Broddefalk (AstraZeneca) for the synthesis of the CILs. This work benefited from the use of the SasView application, originally developed under NSF award DMR-0520547. SasView contains code developed with funding from the European Union’s Horizon 2020 research and innovation program under the SINE2020 project, grant agreement no. 654000. The authors gratefully acknowledge the scientific support and HPC resources provided by the Erlangen National High Performance Computing Center (NHR@FAU) of the Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU) under the NHR project b119ee.
Author contributions
L.L. and J.O.R. designed research; J.P., A.D., A.R., R.K., C.B., C.E.B., A.S., M.I., P.S., U.S., S.Ö., M.J., N.H., K.E., and B.S. performed research; J.P., A.D., A.R., K.F., B.N., and S.J. analyzed data; A.S., M.I., and N.S. performed simulations; S.Ö., M.J., N.H., K.E., and B.S. carried out animal study; and J.P., N.S., L.L., and J.O.R. wrote the paper.
Competing interests
A.D., P.S., U.S., S.Ö., M.J., N.H., K.E., B.S., and L.L. are employed by AstraZeneca R&D Gothenburg.
Footnotes
This article is a PNAS Direct Submission.
Data, Materials, and Software Availability
All study data are included in the article and/or supporting information.
Supporting Information
References
- 1.Wan C., Allen T. M., Cullis P. R., Lipid nanoparticle delivery systems for siRNA-based therapeutics. Drug Deliv. Transl. Res. 4, 74–83 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Yin H., et al. , Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 15, 541–555 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Cullis P. R., Hope M. J., Lipid nanoparticle systems for enabling gene therapies. Mol. Ther. 25, 1467–1475 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adams D., et al. , Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N. Engl. J. Med. 379, 11–21 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Hou X., Zaks T., Langer R., Dong Y., Lipid nanoparticles for mRNA delivery. Nat. Rev. Mater. 6, 1078–1094 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miao L., Zhang Y., Huang L., mRNA vaccine for cancer immunotherapy. Mol. Cancer 20, 41 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rurik J. G., et al. , CAR T cells produced in vivo to treat cardiac injury. Science 375, 91–96 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gillmore J. D., et al. , CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. N. Engl. J. Med. 385, 493–502 (2021). [DOI] [PubMed] [Google Scholar]
- 9.Belliveau N. M., et al. , Microfluidic synthesis of highly potent limit-size lipid nanoparticles for in vivo delivery of siRNA. Mol. Ther. Nucleic Acids 1, e37 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leung A. K. K., Tam Y. Y. C., Chen S., Hafez I. M., Cullis P. R., Microfluidic mixing: A general method for encapsulating macromolecules in lipid nanoparticle systems. J. Phys. Chem. B 119, 8698–8706 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Maier M. A., et al. , Biodegradable lipids enabling rapidly eliminated lipid nanoparticles for systemic delivery of RNAi therapeutics. Mol. Ther. 21, 1570–1578 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Francia V., Schiffelers R. M., Cullis P. R., Witzigmann D., The biomolecular corona of lipid nanoparticles for gene therapy. Bioconjug. Chem. 31, 2046–2059 (2020). [DOI] [PubMed] [Google Scholar]
- 13.Kulkarni J. A., et al. , On the formation and morphology of lipid nanoparticles containing ionizable cationic lipids and siRNA. ACS Nano 12, 4787–4795 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Yanez Arteta M., et al. , Successful reprogramming of cellular protein production through mRNA delivered by functionalized lipid nanoparticles. Proc. Natl. Acad. Sci. U.S.A. 115, E3351–E3360 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hammel M., et al. , Correlating the structure and gene silencing activity of oligonucleotide-loaded lipid nanoparticles using small-angle X-ray scattering. ACS Nano 17, 11454–11465 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zheng L., Bandara S. R., Tan Z., Leal C., Lipid nanoparticle topology regulates endosomal escape and delivery of RNA to the cytoplasm. Proc. Natl. Acad. Sci. U.S.A. 120, e2301067120 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rozmanov D., Baoukina S., Tieleman D. P., Density based visualization for molecular simulation. Faraday Discuss. 169, 225–243 (2014). [DOI] [PubMed] [Google Scholar]
- 18.Jayaraman M., et al. , Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo*. Angew. Chem. Int. Ed. 5, 8529–8533 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Semple S. C., et al. , Rational design of cationic lipids for siRNA delivery. Nat. Biotechnol. 28, 172–176 (2010). [DOI] [PubMed] [Google Scholar]
- 20.Carrasco M. J., et al. , Ionization and structural properties of mRNA lipid nanoparticles influence expression in intramuscular and intravascular administration. Commun. Biol. 4, 956 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hafez I., Maurer N., Cullis P., On the mechanism whereby cationic lipids promote intracellular delivery of polynucleic acids. Gene Ther. 8, 1188–1196 (2001). [DOI] [PubMed] [Google Scholar]
- 22.Koltover I., Salditt T., Rädler J. O., Safinya C. R., An inverted hexagonal phase of cationic liposome-DNA complexes related to DNA release and delivery. Science 281, 78–81 (1998). [DOI] [PubMed] [Google Scholar]
- 23.Leal C., Bouxsein N. F., Ewert K. K., Safinya C. R., Highly efficient gene silencing activity of siRNA embedded in a nanostructured gyroid cubic lipid matrix. J. Am. Chem. Soc. 132, 16841–16847 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Majzoub R. N., Ewert K. K., Safinya C. R., Cationic liposome–nucleic acid nanoparticle assemblies with applications in gene delivery and gene silencing. Philos. Trans. R. Soc. Math. Phys. Eng. Sci. 374, 20150129 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rädler J. O., Koltover I., Salditt T., Safinya C. R., Structure of DNA-cationic liposome complexes: DNA intercalation in multilamellar membranes in distinct interhelical packing regimes. Science 275, 810–814 (1997). [DOI] [PubMed] [Google Scholar]
- 26.Safinya C. R., Rädler J. O., Handbook of Lipid Membranes: Molecular, Functional, and Materials Aspects (CRC Press, ed. 1, 2021), 10.1201/9780429194078. (12 April 2023). [DOI] [Google Scholar]
- 27.Li Z., et al. , Acidification-induced structure evolution of lipid nanoparticles correlates with their in vitro gene transfections. ACS Nano 17, 979–990 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Uebbing L., et al. , Investigation of pH-responsiveness inside lipid nanoparticles for parenteral mRNA application using small-angle X-ray scattering. Langmuir 36, 13331–13341 (2020). [DOI] [PubMed] [Google Scholar]
- 29.Settanni G., Brill W., Haas H., Schmid F., pH-Dependent behavior of ionizable cationic lipids in mRNA-carrying lipoplexes investigated by molecular dynamics simulations. Macromol. Rapid Commun. 43, 2100683 (2022). [DOI] [PubMed] [Google Scholar]
- 30.Pertoft H., Smedsrød B., “Separation and characterization of liver cells” in Cell Separation: Methods and Selected Applications, Pretlow T. G., Pretlow T. P., Eds. (Academic Press, 1987), pp. 1–24. [Google Scholar]
- 31.Sørensen K. K., Smedsrød B., “The liver sinusoidal endothelial cell: Basic biology and pathobiology” in The Liver: Biology and Pathobiology, Arias I. M., et al., Eds. (John Wiley & Sons Ltd, ed. 34, 2020), pp. 422–434, chap. 6. [Google Scholar]
- 32.Paramasivam P., et al. , Endosomal escape of delivered mRNA from endosomal recycling tubules visualized at the nanoscale. J. Cell Biol. 221, e202110137 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shearman G. C., et al. , A 3-D hexagonal inverse micellar lyotropic phase. J. Am. Chem. Soc. 131, 1678–1679 (2009). [DOI] [PubMed] [Google Scholar]
- 34.Huang J., Buboltz J. T., Feigenson G. W., Maximum solubility of cholesterol in phosphatidylcholine and phosphatidylethanolamine bilayers. Biochim. Biophys. Acta 1417, 89–100 (1999). [DOI] [PubMed] [Google Scholar]
- 35.Yaghmur A., Rappolt M., “The micellar cubic Fd3m phase” in Advances in Planar Lipid Bilayers and Liposomes (Elsevier, 2013), pp. 111–145. [Google Scholar]
- 36.Ibrahim M., Gilbert J., Heinz M., Nylander T., Schwierz N., Structural insights on ionizable Dlin-MC3-DMA lipids in DOPC layers by combining accurate atomistic force fields, molecular dynamics simulations and neutron reflectivity. Nanoscale 15, 11647–11656 (2023), 10.1101/2023.02.28.529897. [DOI] [PubMed] [Google Scholar]
- 37.Hohner A., Bayer J., Rädler J. O., Wormlike lipid/DNA micelles in a non-polar solvent. Eur. Phys. J. E 21, 41–48 (2006). [DOI] [PubMed] [Google Scholar]
- 38.A. M. T. M. do Canto et al. , Diphenylhexatriene membrane probes DPH and TMA-DPH: A comparative molecular dynamics simulation study. Biochim. Biophys. Acta 1858, 2647–2661 (2016). [DOI] [PubMed] [Google Scholar]
- 39.Reiser A., et al. , Correlation of mRNA delivery timing and protein expression in lipid-based transfection. Integr. Biol. 11, 362–371 (2019). [DOI] [PubMed] [Google Scholar]
- 40.Wittrup A., et al. , Visualizing lipid-formulated siRNA release from endosomes and target gene knockdown. Nat. Biotechnol. 33, 870–876 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Salonen A., Guillot S., Glatter O., Determination of water content in internally self-assembled monoglyceride-based dispersions from the bulk phase. Langmuir 23, 9151–9154 (2007). [DOI] [PubMed] [Google Scholar]
- 42.Schoenmaker L., et al. , mRNA-lipid nanoparticle COVID-19 vaccines: Structure and stability. Int. J. Pharm. 601, 120586 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maugeri M., et al. , Linkage between endosomal escape of LNP-mRNA and loading into EVs for transport to other cells. Nat. Commun. 10, 4333 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nawaz M., et al. , Lipid nanoparticles deliver the therapeutic VEGFA mRNA in vitro and in vivo and transform extracellular vesicles for their functional extensions. Adv. Sci. 10, 2206187 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sebastiani F., et al. , Apolipoprotein E Binding drives structural and compositional rearrangement of mRNA-containing lipid nanoparticles. ACS Nano 15, 6709–6722 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eygeris Y., Patel S., Jozic A., Sahay G., Deconvoluting lipid nanoparticle structure for messenger RNA delivery. Nano Lett. 20, 4543–4549 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Elvevold K., Kyrrestad I., Smedsrød B., “Protocol for isolation and culture of mouse hepatocytes (HCs) in kupffer cells (KCs), and liver sinusoidal endothelial cells (LSECs) in analyses of hepatic drug distribution” in Antisense RNA Design, Delivery, and Analysis, Methods in Molecular Biology, Arechavala-Gomeza V., Garanto A., Eds. (Springer, US, 2022), pp. 385–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blanchet C. E., et al. , Versatile sample environments and automation for biological solution X-ray scattering experiments at the P12 beamline (PETRA III, DESY). J. Appl. Crystallogr. 48, 431–443 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.SasView, Version 5.0.5. https://github.com/SasView/sasview/releases. Github. Accessed 3 June 2022.
- 50.Botet R., Kwok S., Cabane B., Percus-Yevick structure factors made simple. J. Appl. Crystallogr. 53, 1570–1582 (2020). [Google Scholar]
- 51.Park S., Choi Y. K., Kim S., Lee J., Im W., CHARMM-GUI membrane builder for lipid nanoparticles with ionizable cationic lipids and PEGylated lipids. J. Chem. Inf. Model. 61, 5192–5202 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Abraham M. J., et al. , GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25 (2015). [Google Scholar]
- 53.Dickson C. J., et al. , Lipid14: The amber lipid force field. J. Chem. Theory Comput. 10, 865–879 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mamatkulov S., Schwierz N., Force fields for monovalent and divalent metal cations in TIP3P water based on thermodynamic and kinetic properties. J. Chem. Phys. 148, 074504 (2018). [DOI] [PubMed] [Google Scholar]
- 55.Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., Klein M. L., Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983). [Google Scholar]
- 56.Krzysztoń R., et al. , Single-cell kinetics of siRNA-mediated mRNA degradation. Nanomed. Nanotechnol. Biol. Med. 21, 102077 (2019). [DOI] [PubMed] [Google Scholar]
- 57.Leonhardt C., et al. , Single-cell mRNA transfection studies: Delivery, kinetics and statistics by numbers. Nanomed. Nanotechnol. Biol. Med. 10, 679–688 (2014). [DOI] [PubMed] [Google Scholar]
- 58.Ferizi M., et al. , Stability analysis of chemically modified mRNA using micropattern-based single-cell arrays. Lab. Chip 15, 3561–3571 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
The video starts with a 90° round view of the inverse micellar phase (LII) with 12 nearest neighbors. All ionizable MC3 lipids are neutral. Subsequently, the cross section of the LII phase is shown. Mimicking the change in pH, the MC3 lipids are protonated and the molecular dynamics simulations start. The MC3 head groups (red) quickly enter the water phase. Subsequently, the topology changes from LII to HII and the water columns are formed. The dynamics stops and a 90° rotation of the inverse hexagonal phase (HII) is shown. In the video, head and tail groups of the MC3 lipids are shown in red and green, respectively. Water is shown in cyan and cholesterol in yellow
Data Availability Statement
All study data are included in the article and/or supporting information.