

Abstract
Immune cells in the tumor microenvironment are not only powerful regulators of immunosuppression and tumorigenesis, but also represent a dominant cell type, with tumor-associated macrophages (TAMs) comprising up to 50% of total cell mass in solid tumors. Immunotherapies such as immune checkpoint inhibitors (ICIs) derive their efficacy from this cancer-immune cell interface, however, immune-related adverse events resulting from systemic blockade remain a significant challenge. To address this need for potent, yet highly tumor-specific immunotherapies, we developed Tumor-Immune Cell Targeting Chimeras (TICTACs), antibody conjugates that are capable of selectively depleting immune checkpoint receptors such as SIRPa from the surface of TAMs. These chimeric molecules consist of a synthetic glycan ligand that binds the C-type lectin CD206, a well-established TAM marker, conjugated to a non-blocking antibody that binds but does not inhibit the checkpoint receptor. By engaging CD206, which constitutively recycles between the plasma membrane and early endosomes, TICTACs facilitate robust removal of the checkpoint receptors from the surface of CD206high macrophages, while having no effect on CD206low macrophages. By decoupling antibody selectivity from its blocking function, we present a new paradigm for developing highly tumor-specific immunotherapies.
Introduction:
Over the past 10 years, cancer immune therapy has significantly improved the therapeutic landscape for multiple cancer subtypes.1 In particular, the PD-1 blocking immune checkpoint inhibitors (ICIs) pembrolizumab and nivolumab have become a mainstay in the first-line treatment of melanoma and non-small cell lung cancer (NSCLC).2 These antibody-based therapeutics act through blocking the inhibitory receptor PD-1 that is present on immune cells, thus potentiating the body’s immune response against the tumor. Nonetheless, significant challenges remain with respect to efficacy in diverse cancer subtypes as well as target-related side effects that can be dose limiting. FDA-approved ICI therapies against PD-1, CTLA-4,3 and LAG-34 target cell surface markers without further specificity for immune cell subtype, leading to pleiotropic effects including unwanted immune-related adverse events (irAEs).5–8 It has been proposed that ICIs with increased immune cell and tissue specificities would provide a path towards decreased irAEs without sacrificing efficacy. With this principle in mind, we sought to develop a platform for tumor-immune cell targeting with the eventual goal of interfacing with existing ICI technologies.
We focused on tumor-associated macrophages (TAMs), which represent a highly abundant immune cell type within the tumor microenvironment (TME).9–11 Unlike classically activated M1 macrophages that are akin to the inflammatory Type 1 T helper (Th1) immune response, TAMs predominantly adopt the M2-like, pro-tumor phenotype in response to cytokines such as IL-4 within the TME. These M2-like macrophages notably aid cancer cells in metastasis, angiogenesis, and proliferation via diverse anti-inflammatory mechanisms.12 CD206, also known as the macrophage mannose receptor (MMR), is one of the most prominent M2 markers, and its expression is correlated with a significantly worse overall survival of patients with solid malignancies (Fig. 1A).13–15
Figure 1. TICTACs target CD206 on M2-polarized macrophages.

A.) CD206 is upregulated on the surface of M2-polarized TAMs and binds multivalent mannose, fucose, and glucosamine via its C-type lectin-like domains (CTLDs) and sulfated galactose and galactosamine via its N-terminal R-type carbohydrate recognition domain (R-type CRD). B.) Two-step protocol for generating TICTACs, where X represents the linker, and the gray sphere represents the carbohydrate ligand. C.) Murine macrophages (RAW264.7) can be polarized to M2 upon treatment with IL-4, leading to increased expression of CD206. D.) Tris-dendron scaffold 1 enables CD206-targeting with mannose, fucose, and 3-SO4 galactose ligands 1a-c. E.) TICTAC-mediated internalization of fluorophore-conjugated rabbit IgG into M2-polarized macrophages. F.) Fold change in mean fluorescence intensity (MFI) relative to the control for M2-polarized RAW264.7 cells incubated at 37 °C for 3 h with 25 nM rabbit IgG-488 and 25 nM goat-anti-rabbit or various goat-anti-rabbit-TICTACs. MFI was determined by live cell flow cytometry. G.) MFI relative to control for M2 polarized murine macrophage cell line BMA3.1A7 and microglia cell line BV-2. H.) Knocking out CD206 ablates 1b and 1c-mediated uptake in RAW264.7 cells. For F, G, and H, error bars represent the SD from 3 independent experiments. For F and G, P values were determined by Welch’s two-tailed t-tests. Statistical significance was defined as P<0.05, and the asterisks * indicates P<0.1, ** indicates P<0.01, *** indicates P<0.001, and **** indicates P<0.0001.
Here we developed tumor-immune cell targeting chimeras (TICTACs), which are bifunctional small molecule-antibody conjugates that selectively bind CD206 on macrophages. Through a click-based two-step approach, we created a modular platform that enables facile optimization of the multivalent display of the small molecule ligand and show that these chimeras can be used to uptake cargo selectively into CD206+ cells (Fig. 1B, C). This antibody-based ligand screening platform led to the identification of a new low-molecular weight ligand for CD206. Moreover, this approach is modular with respect to the antibody employed, enabling virtually any antibody to be rapidly transformed into a TICTAC, including those targeting immune-checkpoint proteins. We demonstrate that TICTACs can facilitate robust depletion of immune checkpoint receptors from the surface of CD206high, but not CD206low macrophages. This approach may lead to a new class of immune therapies with reduced risk of systemic immune activation and associated toxicities.
Results
Prior work targeting CD206 largely employed mannose-functionalized glycopolymers that invoke avidity effects to compensate for the low monomeric affinity of mannose.16–22 However, the heterogeneous nature of these glycopolymers would render reproducible synthesis and characterization of the resulting antibody-conjugates challenging. Therefore, we sought alternative low molecular weight homogeneous ligands that bind CD206. A recent glycan array study identified L-fucose as a promising candidate: >60% of the top 20 binders were fucosylated, and fucose-containing glycans were structurally simpler than those containing mannose.23 We also explored sulfated glycans such as 3-SO4 galactose, which bind the N-terminal R-type carbohydrate recognition domain (R-type CRD) instead of the C-type lectin-like domains (CTLDs) commonly targeted by mannose (Fig. 1A).24 Therefore, we prepared a panel of propargyl-, azide-, or aminooxy-functionalized mannose, fucose, and 3-SO4 galactose mono, di, and trisaccharides (Supplementary Fig. 1). Antibody-glycan conjugates were generated in a two-step sequence, where goat-anti-rabbit secondary antibodies were first modified on lysine residues with linkers displaying alkynes, azides, or aldehydes in various valencies and orientations, then conjugated to the carbohydrate ligands via Cu-catalyzed or strain-promoted click reactions, or oxime ligations (Fig. 1B). Characterization of the antibody-glycan conjugates by MALDI-MS analysis revealed average ligand-to-antibody ratios between 10 and 16 (Supplementary Fig. 2–5).
We next sought to identify a suitable in vitro macrophage model system. Phorbol 12-myristate 13-acetate (PMA)-differentiated THP-1 or U-937 cells are frequently utilized as human macrophage surrogates, however, polarization with IL-4, IL-10, or IL-13 all failed to induce expression of CD206.25 We thus turned to the murine macrophage cell line RAW264.7, another extensively used model for studying macrophage biology.26 Gratifyingly, addition of 25 ng/mL IL-4 over 24 hours resulted in robust upregulation of CD206 (M2-polarized) in comparison to unpolarized cells (Fig. 1C).
We examined the ability of the antibody-glycan conjugates to internalize an extracellular target, rabbit IgG-488, via CD206, leading to the identification of tris-dendron TICTACs 1a–c (Fig. 1D, E). M2-polarized RAW264.7 cells were incubated with rabbit IgG-488 and goat-anti-rabbit (control) or goat-anti-rabbit TICTACs 1a–c for 3 hours, then analyzed by flow cytometry for intracellular 488 fluorescence (Fig. 1E). Because of the broad expression profile of CD206, cells were co-stained with an anti-CD206 antibody and gated for CD206high and CD206low populations. In the CD206high population, treatment with 3-SO4 Gal TICTAC 1c resulted in a 6-fold increase in intracellular fluorescence relative to the control, while Fuc TICTAC 1b and Man TICTAC 1a gave 2.6-fold and 2-fold increases, respectively (Fig. 1F). Direct triazole linkage at the anomeric position of mannose (1d) completely ablated internalizing activity. In the CD206low population, 1a–d did not result in significant increases in intracellular fluorescence.
We then evaluated whether TICTAC-mediated uptake was generalizable across other macrophage cell lines. Internalization of fluorophore-conjugated rabbit-IgG was observed for M2-polarized BMA3.1A727 and J774A.1 cell lines using TICTACs 1a-c (Fig. 1G left, Supplementary Fig. 6), with 1b and 1c showing superior uptake. The M2-polarized microglial cell line BV-2 was also treated with control, TICTACs 1b, 1c, or a tris-Gal analogue (negative control), resulting in internalization of 1b and 1c, but not tris-Gal (Fig. 1G right). Finally, to validate the mechanism of uptake, we generated a CD206-knockout RAW264.7 cell line and subjected it to M2-polarization conditions before treatment with fluorophore-conjugated rabbit IgG and control, 1b, or 1c (Fig. 1H). Uptake was abolished in the knockout cells, while preserved in cells that were treated with Cas9 without sgRNA (mock control).
We next asked whether the conjugated tris-fucose ligand in 1b could function independently as a small molecule CD206 ligand. We synthesized tris-Fuc 647 (2), wherein the tris-fucose ligand was conjugated to a 647-dye in a 1:1 stoichiometry (Fig. 2A). Polarized RAW264.7 cells were treated with various concentrations of the unconjugated dye or compound 2 for 2 hours, and intracellular fluorescence was analyzed by flow cytometry (Fig. 2B). Uptake was dependent on the concentration of 2, and significant differences were observed between 2 and the unconjugated control at concentrations as low as 0.1 nM. This result was confirmed by confocal microscopy, where treatment with 2 resulted in a high 647 signal that concentrated in certain cells over others, mirroring the broad expression profile of CD206 (Fig. 2C). Next, we tested whether 2 is internalized by primary human macrophages. Upregulation of CD206 was first confirmed upon polarizing the macrophages with IL-4/IL-10 over several days (Fig. 2D). M2-polarized and non-polarized macrophages were subjected to the unconjugated dye or 2 at various concentrations, and intracellular fluorescence was measured by flow cytometry (Fig. 2E). In addition to a significant increase in fluorescence observed by treatment with 2 compared to the unconjugated control, we observed a notable preference for M2-polarized over nonpolarized cells. CD206-knockout RAW264.7 cells were unable to internalize compound 2, confirming the selectivity of the ligand for CD206 (Fig. 2F). Binding of 2 to CD206 was also analyzed by surface plasmon resonance (SPR), which revealed an extremely slow off rate (< 2.4 × 10−4 min−1) that likely drives internalization even at very low nM concentrations (Supplementary Fig. 7).
Figure 2. Tris-fucose ligand functions independently as a small molecule CD206 binder.

A.) Tris-fucose ligand was independently synthesized and conjugated to a 647 dye to generate tris-Fuc-647 2. B.) Changes in 647 MFI in M2-polarized RAW264.7 cells (gated for CD206high population) upon treatment with unconjugated 647 dye or 2 at varying concentrations for 2 h at 37 °C. C.) Confocal microscopy imaging of M2-polarized RAW264.7 cells treated for 2 h with 2 or unconjugated 647 dye. D.) Monocytes were isolated from human PBMCs and differentiated into macrophages either in the absence of presence of IL-4 (20 ng/mL) and IL-10 (50 ng/mL). CD206 is upregulated in M2-polarized macrophages. E.) Changes in 647 MFI in M2-polarized human macrophages upon treatment with unconjugated 647 dye or 2 at varying concentrations for 2 h at 37 °C. F.) Knocking out CD206 ablates uptake of 2 in RAW264.7 cells.
For B, E, and F, error bars represent the SD from 3 independent experiments. For B and E, P values were determined by parametric two-tailed t-tests. Statistical significance was defined as P<0.05, and the asterisks ** indicates P<0.01, *** indicates P<0.001, and **** indicates P<0.0001.
Having demonstrated that TICTACs can efficiently internalize soluble extracellular cargo, we investigated whether they could mediate internalization of membrane proteins in M2-polarized macrophages. For proof-of-concept, we chose CD54 (ICAM-1) as a target due to its robust expression in several macrophage cell lines. TICTACs were constructed using a commercially available primary antibody against CD54 following a similar scheme to 1b and 1c to generate 3b and 3c (Fig. 3A). We treated RAW264.7 cells with 3b and 3c, then measured surface levels of CD54 by flow cytometry using an orthogonal detection antibody. TICTAC 3b resulted in >80% depletion of surface CD54 in CD206high RAW264.7 cells, whereas TICTAC 3c resulted in >60% depletion (Fig. 3B, left). In CD206low cells, this effect is greatly attenuated, with 3b and 3c treatment resulting in 30% and 15% depletion of surface CD54, respectively. We also evaluated the activity of 3b in BMA3.1A7 cells and J774A.1 cell lines. We observed >60% depletion of surface CD54 in CD206high BMA3.1A7 cells, while <30% depletion was observed in CD206low cells (Fig. 3B, right). J774A.1 cells also exhibited comparable reduction in cell surface CD54, which was dependent on expression levels of CD206 (Supplementary Fig. 13). Visualization of surface CD54 by confocal microscopy following TICTAC treatment showed significantly reduced membrane CD54, consistent with our flow cytometry observations (Fig. 3C). Reduction of cell-surface CD54 on CD206high RAW264.7 cells was concentration dependent, where 3b demonstrated a detectable decrease in surface CD54 at concentrations as low as 0.1 nM. Maximum activity of 3b was reached at 5 nM, where the same degree of depletion was maintained at higher concentrations up to 100 nM with no observable “hook effect” (Fig. 3D). Compounds 3b and 3c depleted cell-surface CD54 over 48 hours, where >50% reduction was observed for 3b after 3 hours, which further increased to >80% removal after 24 hours (Fig. 3E). Depletion of cell-surface CD54 mediated by 3c was markedly slower than by 3b. Finally, because CD206 expression is dependent on the cytokine IL-4, we reasoned that TICTAC activity must also be dependent on IL-4. We set up two parallel experiments where RAW264.7 or J774A.1 cells were either polarized with IL-4 or not prior to treatment with 3b. We observed 3b-mediated depletion of cell-surface CD54 that was dependent on the presence of IL-4, demonstrating that TICTACs can be “turned on” with IL-4 (Fig. 3F).
Figure 3. Anti-CD54 TICTAC depletes cell-surface CD54.

A.) Proposed mechanism for the removal of cell-surface CD54 mediated by TICTACs. B.) Depletion of cell-surface CD54 in M2-polarized RAW264.7 and BMA3.1A7 cells determined by live cell flow cytometry following 24 h of treatment with 25 nM unconjugated anti-CD54 antibody or conjugates. C.) Visualization of cell-surface CD54 by confocal microscopy after 25 nM TICTAC treatments for 24 h. D.) Dose-response curve for cell-surface CD54 removal in M2-polarized RAW264.7 cells following treatment with unconjugated antibody or 3b at 0.1 nM, 1 nM, 5 nM, 10 nM, and 100 nM for 24 h. E.) Time-course of CD54 downmodulation in M2-polarized RAW264.7 cells incubated with 25 nM unconjugated antibody or conjugates at 3 h, 8 h, 24 h, and 48 h. F.) Depletion of cell-surface CD54 in RAW264.7 and J774A.1 cells that have been polarized with IL-4 (25 ng/mL) or not following treatment with 25 nM unconjugated antibody or 3b.
For B and F, error bars represent the SD from 3 independent experiments. P values were determined by Welch’s two-tailed t-tests. Statistical significance was defined as P<0.05, and the asterisks * indicates P<0.1, ** indicates P<0.01, *** indicates P<0.001, and **** indicates P<0.0001.
To determine whether internalized CD54 was subsequently degraded, we measured total CD54 levels in RAW264.7 cells following treatment with 3b or 3c by Western blot. No significant differences in total protein levels were observed between TICTAC treatment and the unconjugated control (Supplementary Fig. 8). The lack of degradation may be consistent with previous observations that CD206 primarily traffics cargo to a distinct class of early endosomes as opposed to the lysosome.28 Notably, these endosomes are stable and non-degradative, playing a critical role in storing and routing antigens for subsequent loading onto MHC-I for cross-presentation. Because internalized CD54 is not degraded, we wondered whether TICTAC-mediated depletion of surface CD54 was a reversible process. To test this, we conducted a series of washout experiments, wherein M2-polarized RAW264.7 cells were first exposed to media containing 25 nM 3b, 3c, or the unconjugated antibody for 1, 3, or 8 hours (treatment time), then allowed to grow in TICTAC- or antibody-free media for various time periods (washout period). For all treatment times, washout periods did not result in a rebound of surface CD54 levels. In particular, a 16-hour washout period following 8-hour treatment with 3b resulted in no significant change in cell-surface CD54, which remained >70% lower than that of the control (Supplementary Fig. 9). Taken together, these data suggest that TICTACs are non-degradative and downmodulate surface CD54 by altering its steady-state localization within the cell in a process that is not readily reversible.
We next sought to determine whether TICTACs could provide an alternative form of immune checkpoint blockade that was specific to M2-polarized macrophages, which are known to drive immune suppression in the tumor microenvironment. In a canonical checkpoint blockade, the antibody drug systemically blocks the immune checkpoint protein (ICP) and activates immune cells in healthy and cancerous tissue alike, leading to irAEs. We hypothesized that by generating TICTACs from non-blocking antibodies that bind but do not inhibit the ICP, we could selectively remove the ICP from TAMs while mitigating off-target effects (Fig. 4A). We targeted signal regulatory protein alpha (SIRPα), which is highly expressed on myeloid-derived immune cells and potently engages the “don’t eat me” signal CD47.29–33 Blocking antibodies against both SIRPα and CD47 are currently being evaluated as cancer immune therapies.31,34,35
Figure 4. Anti-SIRPα TICTACs downmodulate immune checkpoint protein SIPRα regardless of their ability to block CD47.
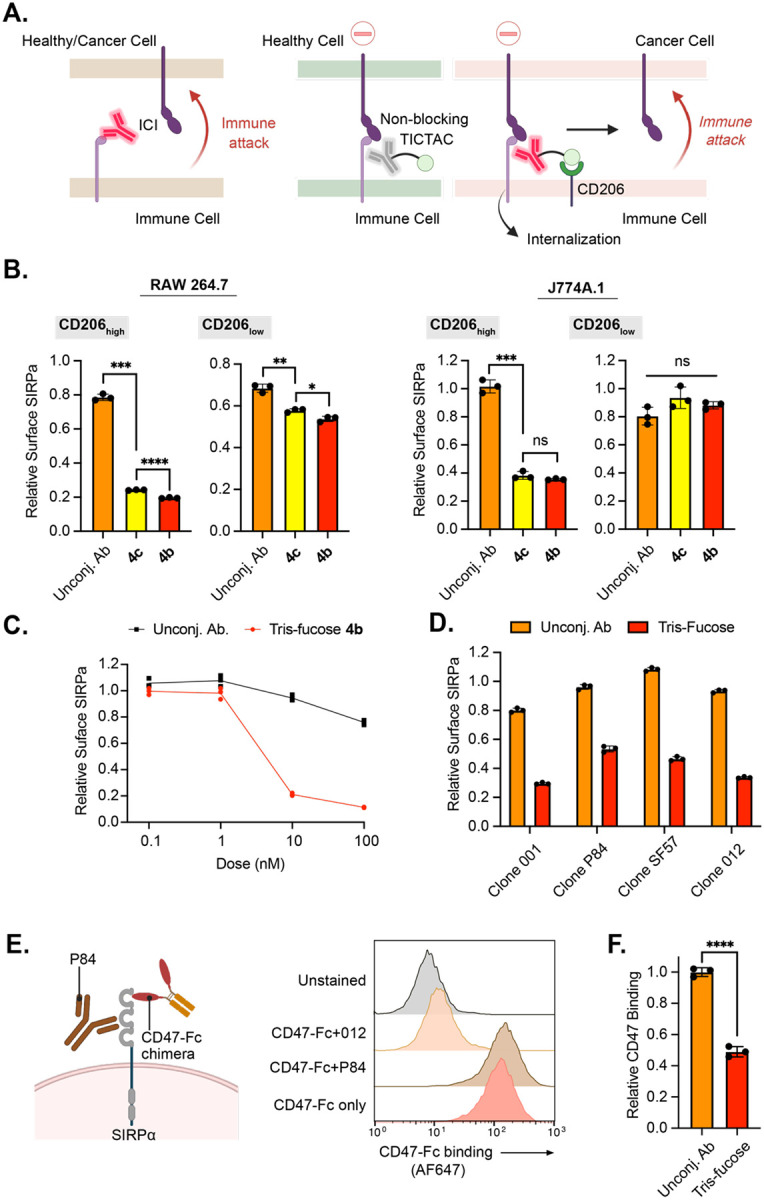
A.) Left: Canonical checkpoint inhibitor systemically blocks the ICP regardless of whether the interacting cell is healthy or cancerous; Right: Non-blocking TICTACs have no function-ablating activity in healthy tissue but can remove ICPs in CD206high TAMs. B.) Downmodulation of cell-surface SIRPα in M2-polarized RAW264.7 and J774A.1 cells determined by live cell flow cytometry following 24 h of treatment with 25 nM unconjugated anti-SIRPα antibody or conjugates (4b, 4c). C.) Dose-response curve for cell-surface SIRPα removal in M2-polarized RAW264.7 cells following treatment with unconjugated antibody or 4b at 0.1 nM, 1 nM, 10 nM, and 100 nM for 24 h. D.) TICTACs generated from multiple antibody clones targeting different epitopes on SIRPa downmodulate cell-surface SIRPα in M2-polarized RAW264.7 cells following treatment at 25 nM for 24 h. E.) Orthogonality of SIRPα antibody to CD47 was probed by live cell flow cytometry using an AF647-labeled CD47-Fc chimera. F.) CD47-binding to M2-polarized RAW264.7 cells was determined using an AF647-labeled CD47-Fc chimera following treatment with non-blocking clone P84 or its tris-fucose conjugate at 25 nM for 24 h. Data is normalized to unconjugated P84 antibody treatment.
For B, D, and F, error bars represent the SD from 3 independent experiments. P values were determined by Welch’s two-tailed t-tests. Statistical significance was defined as P<0.05, and the asterisks * indicates P<0.1, ** indicates P<0.01, *** indicates P<0.001, and **** indicates P<0.0001.
We first generated TICTACs 4b and 4c from a commercially available primary antibody against SIRPα following a similar scheme to 1b and 1c. M2-polarized RAW264.7 and J774A.1 cells were treated with 4b and 4c, after which cell-surface SIRPα levels were quantified by flow cytometry using an orthogonal detection antibody. TICTAC treatment resulted in significant decreases in cell-surface SIRPa (>60%) for CD206high cells across both cell lines (Fig. 4B). 4b-mediated downmodulation of SIRPα was concentration dependent, reaching a maximum at 100 nM (Fig. 4C). Similar to CD54, measuring the total protein levels by Western blot revealed that 4b and 4c do not induce degradation of SIRPa (Supplementary Fig. 10).
To begin to test our hypothesis regarding non-blocking TICTACs, we first determined whether TICTACs could withstand changes in the binding epitope of the target protein. We generated tris-fucose TICTACs from four different monoclonal antibodies against SIRPα and tested their activity in M2-polarized RAW264.7 cells. TICTACs derived from all clones reduced surface SIRPα by >40% in CD206high cells (Fig. 4D). We next determined whether each of the antibodies inhibited CD47 binding by assessing orthogonality with a CD47-Fc chimera by flow cytometry. Only clone P84 was fully orthogonal to CD47, whereas other clones such as 012 resulted in near complete blockade of CD47 binding (Fig. 4E). Finally, we subjected RAW264.7 cells to P84-derived tris-Fuc TICTAC or the unconjugated P84 antibody and measured levels of CD47 binding after treatment. TICTAC treatment resulted in a 50% decrease in CD47 binding relative to the unconjugated control, demonstrating that TICTACs can selectively activate non-blocking ICP antibodies in the presence of CD206.
Having established the concept of non-blocking TICTACs for membrane targets, we wondered whether this technology could be applied to the selective clearance of soluble targets that are known to contribute to cancer immune modulation. Cytokines are ubiquitous signaling molecules that regulate a large number of processes, often having differing or even contrasting effects on different cell-types or tissues. We envisioned a strategy wherein TICTACs generated from non-blocking antibodies that do not disrupt binding of the cytokine to its receptor could result in tumor-specific clearance mediated by CD206, while having no inhibitory effect elsewhere (Fig. 5A). To test this strategy, we chose to target IL-4, a driver for M2 polarization in TAMs and immune suppression within the tumor microenvironment.36 We generated TICTACs 5b and 5c through conjugation of tris-Fuc and tris-3-SO4 Gal ligands with a known non-neutralizing anti-IL4 antibody (clone BVD6–24G2).37 Polarized RAW264.7 cells were subjected to 488-labeled murine IL4 and unconjugated anti-IL4 antibody, 5b, or 5c. In CD206high populations, 5b and 5c induced >10-fold increase in intracellular 488 fluorescence relative to the unconjugated antibody, whereas the change in CD206low populations was less than 2-fold. (Fig. 5B). These results suggest that non-blocking TICTACs can also be employed for tumor-specific removal of soluble targets such as cytokines.
Figure 5. CD206-dependent uptake of IL-4 using a non-neutralizing anti-IL-4 antibody.

A.) Non-blocking TICTAC against a soluble target would have no effect on tissue with low CD206 expression (top) but would internalize and eliminate soluble targets in the presence of TAMs (bottom). B.) Fold change in 488 MFI relative to the control for M2 polarized RAW264.7 cells incubated at 37 °C for 3 h with 25 nM 488-labeled IL-4 and 25 nM anti-IL4 antibody or various anti-IL-4 TICTACs 5b and 5c. MFI was determined by live cell flow cytometry. Error bars represent the SD from 3 independent experiments.
Discussion
While numerous strategies have been developed to target tumor cells and immune cells including antibody-drug conjugates and immune checkpoint inhibitors, few methods specifically target tumor-associated immune cells. Targeting the immune cells within the tumor microenvironment may be advantageous in mitigating irAEs and improving the efficacy and therapeutic window of existing therapies. Toward this objective, we developed tumor-immune cell targeting chimeras (TICTACs) that specifically target tumor-associated macrophages via the C-type lectin CD206. While previously reported synthetic ligands for CD206 mostly comprise mannose glycopolymers, we identified tris-Fuc and tris-3-SO4-Gal ligands that potently engage CD206 on their own and in the context of antibody conjugates. These conjugates drive the internalization of cell-surface CD54 and SIRPα from M2-polarized macrophages, and this activity is dependent on CD206 expression. We also established that TICTACs generated from non-blocking antibodies can still downmodulate SIRPα in the presence of CD206, which enables antibody selectivity to be decoupled from its blocking function. Finally, we demonstrated that non-blocking TICTACs can be utilized to clear soluble cytokines such as IL-4, which is known to contribute to immune suppression within the TME.
TICTACs alter the steady-state subcellular distribution of the target antigen without inducing degradation. This observation is consistent with the trafficking properties and biological role of CD206 in antigen cross-presentation pathways in APCs.38–40 Future work will explore the ability of TICTACs to enhance cross-presentation of relevant soluble and membrane-bound tumor antigens.41
Additionally, we demonstrated that a single tris-fucose ligand can function as a small molecule CD206 binder even when it is not multivalently displayed on an antibody. Small molecule binders to CD206 are scarce;42 previous approaches focused primarily on glycan-conjugated polymers, liposomes, nanoparticles, and other biomaterials. Further, this work presents a rare example of small molecule binders that are not based on mannose yet are highly selective for CD206 in M2-polarized macrophages.
Our strategy for generating a library of antibody conjugates through combinatorial screening of various linker scaffolds and glycan ligands enables the design of a targeting chimera without a pre-optimized small molecule ligand in hand. Antibody conjugation greatly simplifies the purification of the small molecule ligands, where size-exclusion techniques such as molecular weight cut-off resins can be used to separate the conjugates from unreacted material. Importantly, we demonstrated that ligands developed in this way retain their function in the absence of the parent antibody. Thus, we anticipate that this workflow can be utilized to discover novel small molecule binders to other lectins and find applications in new drug delivery platforms.
Reprogramming the immune system represents a powerful strategy for cancer therapy. As more targeted approaches are being developed for tumor cell-specific therapies, we envision a parallel opportunity to develop of tumor immune cell-specific therapies that alleviate irAEs and expand the eligible patient population for cancer immune therapy. TICTACs that leverage CD206 present one such approach of selectively reprogramming tumor-associated macrophages. Further discovery and targeting of immune cell receptors that are upregulated within the TME will expand the scope of this strategy to other immune cell types and checkpoint molecules.
Methods
General Synthetic Chemistry Procedures
Unless otherwise noted, all reactions were carried out in flame-dried glassware sealed with rubber septa under a nitrogen atmosphere with Teflon-coated magnetic stir bars. Reaction progress was monitored using thin layer chromatography on Millipore Sigma glass-backed TLC plates (250 μm thickness, F-254 indicator) and visualized with 254 nm UV light or stained by submersion in a basic potassium permanganate solution or 5% H2SO4 in methanol solution. Flash column chromatography was performed on a Biotage instrument using pre-packed silica gel columns. Reagents were purchased in reagent grade from commercial suppliers and used as received, unless otherwise described. Anhydrous solvents (dichloromethane, tetrahydrofuran, methanol, and acetonitrile) were purchased as sure-sealed bottles and used as received, unless otherwise described. See Supplementary Information for detailed synthetic procedures and characterization of compounds.
TICTAC antibody conjugation
General procedure for antibody linker labeling
A 1 mg/mL solution of antibody was buffer exchanged into PBS using a 7KDa Zeba size-exclusion column. The antibody was reacted with 50 equiv. of NHS-linker (10 mM stock solution in DMSO), and the reaction was incubated overnight at room temperature. The resulting mixture was purified using a 7KDa Zeba size-exclusion column to yield the conjugate.
General procedure for Cu-catalyzed click reaction
To a 1 mg/mL solution of the antibody-linker conjugate was added 75 equiv. of the carbohydrate ligand (10 mM stock solution in H2O). BTTP-Cu(II) pre-catalyst was generated by mixing CuSO4 (aqueous solution) and BTTP (DMSO solution) in a 1:4 molar ratio. 150 μM BTTP-Cu and 3 mM sodium ascorbate (100 mM solution in H2O, prepared immediately prior to addition) were added, and the reaction was briefly vortexed. The reaction mixture was allowed to incubate at room temperature for 1 h and was filtered using a 7KDa or 40KDa Zeba size-exclusion column.
Cell Culture
All cell lines were purchased from the American Type Culture Collection (ATCC) unless otherwise noted. RAW26.7, J774A.1, BV-2, and U937 were cultured in DMEM supplemented with 10% heat-inactivated fetal bovine serum (HI FBS), 100U/mL Penicillin, and 100 μg/mL Streptomycin. THP-1 cells were cultured in RPMI supplemented with 10% HI FBS, 100U/mL Penicillin, and 100 μg/mL Streptomycin. The CB7BL/6-derived immortalized bone marrow macrophage cell line BMA3.1A7 was a generous gift from Dr. Kenneth Rock (University of Massachusetts Medical School), and the cells were maintained as previously described.43 Cells were cultured in T-75 flasks (Fisher Scientific) and incubated at 37 °C and 5% CO2 and tested negative for mycoplasma quarterly using a PCR-based assay.
Gene Knockout Pool
CRISPR-Cas9 mediated knockout cell pool of CD206 in RAW264.7 cells were generated by Synthego Corporation (Redwood City, CA). Cells were electroporated with Streptococcus pyogenes Cas9 (Sp-Cas9) and sgRNAs targeting CD206 using Synthego’s optimized protocol. 48 h post-electroporation, genomic DNA was extracted, PCR amplified, and sequenced using Sanger sequencing to verify editing efficacy. Resulting chromatograms were analyzed using Synthego Inference of CRISPR edits software (ice.synthego.com). Mock control cells were generated by electroporating Sp-Cas9 without sgRNA. Cells were cultured in DMEM supplemented with 10% heat-inactivated fetal bovine serum (FBS), 100U/mL Penicillin, and 100 μg/mL Streptomycin.
Surface plasmon resonance (SPR)
Sensor NTA chip, 350 mM EDTA, 0.5 mM NiCl2, N-Hydroxysuccinimide (NHS), 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC), and ethanolamine hydrochloride (EA) were obtained from GE Healthcare/Cytiva (Marlborough, MA).
SPR experiments were performed on a BIACORE T200 biosensor system (GE Healthcare/Cytiva) at 25 °C. The Sensor Chip NTA (GE Healthcare/Cytiva) was conditioned by injecting 350 mM EDTA regeneration solution over both reference and experimental surfaces for 60 s at a flow rate 30 μL/min. To capture the His-tagged mouse CD206 protein the experimental NTA flow cell surface was saturated with nickel by injecting nickel solution (0.5 mM NiCl2) for 60 s at 10 μL/min, followed by an Extra Wash with 3 mM EDTA to remove any remaining traces of nickel in the system. The His-tagged CD206 protein captured on the Sensor Chip NTA was stabilized by covalent coupling to the chip surface by amine coupling chemistry using N-hydroxysuccinimide (NHS) and N′-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) according to the manufacturer’s instructions. The reference surface was treated the same without the presence of His-tagged CD206 protein. Amino-PEG4-tris-α-L-fucose was diluted in PBS Assay Buffer and injected over the CD206-containing surface for 60 s at different concentrations at a flow rate of 30 μL/min. For each experiment at least 5 different concentrations of analyte molecules were injected over each experimental and control flow cell. Dissociation was allowed to occur at the same flow rate for 150 s followed by Regeneration Buffer (150 mM NaCl/NaAc pH 5) at a flow rate of 50 μl/min for 30 s and Assay Buffer alone at a flow rate of 30 μL/min to allow the baseline to stabilize. All data were corrected for unspecific binding by subtracting the signal measured in the reference flow cell lacking immobilized ligand.
Soluble target uptake assay via flow cytometry
For uptake experiments, cells were plated (60,000 cells/well in 24-well plates) two days before treatment in complete media supplemented with 25 ng/mL murine IL-4 (Peprotech) for M2-polarization or without any cytokines (M0). Cells were incubated with 250 μL of M2- or M0-media with fluorescent proteins and TICTACs/controls or conjugated/unconjugated dyes for the indicated times (<8 h). For overnight uptake experiments, cells were plated/polarized one day before treatment. After treatment, cells were washed with PBS, then treated with 200 μL of enzyme-free Cell Dissociation Buffer (Gibco) for 10 minutes to dissociate the cells. The resuspended cells were transferred to a pre-chilled 96-well v-bottom plate (Corning) and washed with cold blocking buffer (PBS supplemented with 0.5% BSA and 5 mM EDTA) two times, pelleting by centrifugation at 500g for 3 min at 4 °C between washes. For experiments requiring co-staining for CD206 levels, Fc-receptors were first neutralized by incubating cells on ice for 5–10 minutes with TruStain FcX (BioLegend) at 2 μg/mL in 100 μL of blocking buffer. The cells were then stained with the fluorescently labeled anti-CD206 antibody in 100 μL of blocking buffer for 30 minutes at 4 °C. Cells were washed two times with blocking buffer, then incubated with Sytox Blue according to the manufacturer’s specifications for 10 min at 4 °C. Flow cytometry was performed on either a MACSQuant Analyzer 10 (Miltenyi Biotec), Novocyte Quanteon (Agilent), or Novocyte Penteon (Agilent), and FlowJo software was used to gate on single cells, live cells, and CD206high/low cells for analysis.
Cell-surface protein internalization assay via flow cytometry
Cells were plated (60,000 cells/well in 24-well plates) one day before treatment in M2- or M0-media. Cells were incubated with 250 μL of M2- or M0-media with 25 nM TICTACs/controls for the indicated amount of time, washed with PBS, and dissociated with Cell Dissociation Buffer for 5–10 minutes. Resuspended cells were transferred to a 96-well v-bottom plate and washed with cold blocking buffer, Fc-neutralized, and incubated with primary antibodies for 30 min at 4 °C. Cells were washed two times with blocking buffer, then incubated with Sytox Blue for 10 min before flow cytometry analysis.
Isolation and differentiation of donor macrophages
LRS chambers were obtained from healthy anonymous blood bank donors. PBMCs were isolated using Ficoll-Paque (GE Healthcare Life Sciences) density gradient separation. Monocytes were isolated by plating ~1 × 108 PBMCs in T-25 flasks with serum-free RPMI and incubating for 1 h, followed by 3x rigorous washes with DPBS (PBS +Ca +Mg) to remove non-adherent cells and ensuring that the final wash results in clear supernatant. The media was replaced with IMDM supplemented with 10% Human AB Serum (Gemini), and the cells were allowed to differentiate into macrophages over 7–9 days. To generate M2 macrophages, the media was replaced on day 4 with IMDM supplemented with 10% Human AB Serum, 20 ng/mL IL-4 (Peprotech), 50 ng/mL IL-10, and 50 ng/mL TGF-β. No media changes were conducted for M0 macrophages throughout the differentiation period.
Western blot analysis
Cells were plated (60,000 cells/well in 24-well plates) one day before treatment in M2- or M0-media. Cells were incubated with 250 μL of M2- or M0-media with 25 nM TICTACs/controls for the indicated amount of time, washed with 3x with PBS, and lysed with RIPA buffer supplemented with protease inhibitor cocktail (Roche), 0.1% benzonase (Millipore-Sigma), and phosphatase inhibitor cocktail (Cell Signaling Technologies) on ice for 30 min. The cells were scraped and transferred to 1.5 mL Eppendorf tubes and centrifuged at 21,000g for 15 min at 4 °C. The supernatant was collected, and the concentration was determined using a BSA assay (Pierce). Equal amounts of protein were loaded onto a 4–12% Bis-Tris gel (BioRad) and separated by SDSPAGE. The gel was transferred to a nitrocellulose membrane, stained with REVERT Total Protein Stain (LI-COR) and blocked with Odyssey Blocking Buffer (PBS) (LI-COR) for 1 h at room temperature. The membrane was then incubated with primary antibody in Blocking Buffer overnight at 4 °C, then washed three times with PBS with 0.1% Tween-20 (PBS-T). The membrane was incubated with secondary antibody for 1 h at room temperature, washed three times with PBST, and visualized with an Odyssey CLx imager (LI-COR). Image Studio (LI-COR) was used to analyze the image and quantify band intensities.
Confocal microscopy
Membrane protein analysis
Cells were plated (20,000 cells per well in an 8-chamber Labtek plate) 3 days prior to treatment. Cells were incubated with 250 μL of complete growth medium with 25 nM TICTACs or control for the indicated amount of time. Cells were then washed 3x with DPBS (300 μL/wash) and fixed with 4% paraformaldehyde in PBS for 10 min at room temperature. Cells were washed 2x with PBS and incubated with primary antibody in 300 μL blocking buffer (PBS + 1% BSA) for 1 h at room temperature. After washing with PBS, cells were incubated with Hoechst stain in PBS for 10 min at room temperature. The wells were aspirated, and the chambers were removed according to the manufacturer’s specifications. 15 μL of mounting medium (Vector Laboratories) was added followed by a coverslip. The slides were cured for 30 min at room temperature and imaged with Nikon A1R confocal microscope using a Plan Fluor x60, 1.30-NA oil objective. The following laser settings were used: 405 nm violet laser, 488 nm blue laser, 561 nm green laser, and 639 nm red laser.
Matrix-assisted laser desorption/ionization-mass spectrometry
A 1 mg/mL solution of the antibody conjugate was buffer exchanged into ddH2O using a 7K Zeba size-exclusion column to remove excess salts. Samples were prepared by mixing 2 μL of sinapinic acid (SPA) matrix (10 mg/mL in 0.1% trifluoroacetic acid and 50% acetonitrile) and 2 μL of the antibody sample. The mixture was vortexed, and 1 μL was loaded onto a MALDI stainless steel plate. The sample was dried at room temperature for 15 min, and the MALDI-MS was acquired by AB SCIEX TOF/TOF and a 5800 CovalX High Mass Detector with a mass range of 10,000 – 250,000 Da and a fixed laser intensity of 5,900. Greater than 3 scans were taken per sample, and the spectra was analyzed and averaged using MALDIquant.44
Statistical analysis and software
Statistical analysis was performed in GraphPad Prism (version 9). For SPR data, association then dissociation models were used to calculate the dissociation constant. For EC50 data, one-site total and nonspecific binding models were used to determine the apparent dissociation constant for specific binding.
Flow cytometry data was analyzed using FlowJo software (version 10.8.1). NMR data was analyzed using MestreNova software (version 14.2.3). Figures were created using BioRender.com and Adobe Illustrator software (version 26.5).
Supplementary Material
Acknowledgements
We gratefully acknowledge Michael Eckert and Jessica Tran (PAN Facility at Stanford) for performing SPR experiments. We also acknowledge Abel Bermudez (Canary Center at Stanford) for assisting with MALDI-TOF-MS characterization. We thank Jonathan Yang, Kang Yong Loh, and Jessica Stark for helpful discussions.
Funding Sources
This work was supported by National Institutes of Health grant GM058867 (C.R.B.). N.T. was supported by F32 Postdoctoral Fellowship F32GM143843. We acknowledge the NIH High End Instrumentation grant (1 S10 OD028697-01) for the Bruker Neo-500 MHz instrument.
Funding Statement
This work was supported by National Institutes of Health grant GM058867 (C.R.B.). N.T. was supported by F32 Postdoctoral Fellowship F32GM143843. We acknowledge the NIH High End Instrumentation grant (1 S10 OD028697-01) for the Bruker Neo-500 MHz instrument.
Footnotes
Conflicts of Interest
C.R.B. is a cofounder and Scientific Advisory Board member of Lycia Therapeutics, Palleon Pharmaceuticals, Enable Bioscience, OliLux Bio, InterVenn Bio, Firefly Bio, Redwood Bioscience (a subsidiary of Catalent), Neuravid Therapeutics and GanNa Bio. C.R.B. is a member of the Board of Directors of Alnylam and OmniAb. M.M., N.A.T., and C.R.B. are coinventors on a patent application related to this work (U.S. Provisional Patent Application No. 63/546,720).
References
- (1).Waldman A. D.; Fritz J. M.; Lenardo M. J. A Guide to Cancer Immunotherapy: From T Cell Basic Science to Clinical Practice. Nat. Rev. Immunol. 2020, 20 (11), 651–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Kanabar S. S.; Tiwari A.; Soran V.; Balendran P.; Price M.; Turner A. M. Impact of PD1 and PDL1 Immunotherapy on Non-Small Cell Lung Cancer Outcomes: A Systematic Review. Thorax 2022, 77 (12), 1163–1174. [DOI] [PubMed] [Google Scholar]
- (3).Lisi L.; Lacal P. M.; Martire M.; Navarra P.; Graziani G. Clinical Experience with CTLA-4 Blockade for Cancer Immunotherapy: From the Monospecific Monoclonal Antibody Ipilimumab to Probodies and Bispecific Molecules Targeting the Tumor Microenvironment. Pharmacol. Res. 2022, 175, 105997. [DOI] [PubMed] [Google Scholar]
- (4).Goldberg M. V.; Drake C. G. LAG-3 in Cancer Immunotherapy. Curr. Top. Microbiol. Immunol. 2011, 344, 269–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Conroy M.; Naidoo J. Immune-Related Adverse Events and the Balancing Act of Immunotherapy. Nat. Commun. 2022, 13 (1), 392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Esfahani K.; Meti N.; Miller W. H.; Hudson M. Adverse Events Associated with Immune Checkpoint Inhibitor Treatment for Cancer. CMAJ Can. Med. Assoc. J. 2019, 191 (2), E40–E46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Yin Q.; Wu L.; Han L.; Zheng X.; Tong R.; Li L.; Bai L.; Bian Y. Immune-Related Adverse Events of Immune Checkpoint Inhibitors: A Review. Front. Immunol. 2023, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Wang S. J.; Dougan S. K.; Dougan M. Immune Mechanisms of Toxicity from Checkpoint Inhibitors. Trends Cancer 2023, 9 (7), 543–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Lin Y.; Xu J.; Lan H. Tumor-Associated Macrophages in Tumor Metastasis: Biological Roles and Clinical Therapeutic Applications. J. Hematol. Oncol.J Hematol Oncol 2019, 12 (1), 76. 10.1186/s13045-019-0760-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Pan Y.; Yu Y.; Wang X.; Zhang T. Tumor-Associated Macrophages in Tumor Immunity. Front. Immunol. 2020, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).kong P.; Yang H.; Tong Q.; Dong X.; Yi M.-A.; Yan D. Expression of Tumor-Associated Macrophages and PD-L1 in Patients with Hepatocellular Carcinoma and Construction of a Prognostic Model. J. Cancer Res. Clin. Oncol. 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Petty A. J.; Yang Y. Tumor-Associated Macrophages: Implications in Cancer Immunotherapy. Immunotherapy 2017, 9 (3), 289–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Heng Y.; Zhu X.; Lin H.; jingyu M.; Ding X.; Tao L.; Lu L. CD206+ Tumor-Associated Macrophages Interact with CD4+ Tumor-Infiltrating Lymphocytes and Predict Adverse Patient Outcome in Human Laryngeal Squamous Cell Carcinoma. J. Transl. Med. 2023, 21 (1), 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Haque A. S. M. R.; Moriyama M.; Kubota K.; Ishiguro N.; Sakamoto M.; Chinju A.; Mochizuki K.; Sakamoto T.; Kaneko N.; Munemura R.; Maehara T.; Tanaka A.; Hayashida J.-N.; Kawano S.; Kiyoshima T.; Nakamura S. CD206+ Tumor-Associated Macrophages Promote Proliferation and Invasion in Oral Squamous Cell Carcinoma via EGF Production. Sci. Rep. 2019, 9 (1), 14611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Debacker J. M.; Gondry O.; Lahoutte T.; Keyaerts M.; Huvenne W. The Prognostic Value of CD206 in Solid Malignancies: A Systematic Review and Meta-Analysis. Cancers 2021, 13 (14), 3422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Azad A. K.; Rajaram M. V. S.; Schlesinger L. S. Exploitation of the Macrophage Mannose Receptor (CD206) in Infectious Disease Diagnostics and Therapeutics. J. Cytol. Mol. Biol. 2014, 1 (1), 1000003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Hatami E.; Mu Y.; Shields D. N.; Chauhan S. C.; Kumar S.; Cory T. J.; Yallapu M. M. Mannose-Decorated Hybrid Nanoparticles for Enhanced Macrophage Targeting. Biochem. Biophys. Rep. 2019, 17, 197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Locke L. W.; Mayo M. W.; Yoo A. D.; Williams M. B.; Berr S. S. PET Imaging of Tumor Associated Macrophages Using Mannose Coated 64Cu Liposomes. Biomaterials 2012, 33 (31), 7785–7793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Li Y.; Wu H.; Ji B.; Qian W.; Xia S.; Wang L.; Xu Y.; Chen J.; Yang L.; Mao H. Targeted Imaging of CD206 Expressing Tumor-Associated M2-like Macrophages Using Mannose-Conjugated Antibiofouling Magnetic Iron Oxide Nanoparticles. ACS Appl. Bio Mater. 2020, 3 (7), 4335–4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Azad A. K.; Rajaram M. V. S.; Metz W. L.; Cope F. O.; Blue M. S.; Vera D. R.; Schlesinger L. S. γ-Tilmanocept, a New Radiopharmaceutical Tracer for Cancer Sentinel Lymph Nodes, Binds to the Mannose Receptor (CD206). J. Immunol. Author Choice 2015, 195 (5), 2019–2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Surasi D. S.; O’Malley J.; Bhambhvani P. 99mTc-Tilmanocept: A Novel Molecular Agent for Lymphatic Mapping and Sentinel Lymph Node Localization. J. Nucl. Med. Technol. 2015, 43 (2), 87–91. [DOI] [PubMed] [Google Scholar]
- (22).Mastrotto F.; Pirazzini M.; Negro S.; Salama A.; Martinez-Pomares L.; Mantovani G. Sulfation at Glycopolymer Side Chains Switches Activity at the Macrophage Mannose Receptor (CD206) In Vitro and In Vivo. J. Am. Chem. Soc. 2022, 144 (50), 23134–23147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Feinberg H.; Jégouzo S. A. F.; Lasanajak Y.; Smith D. F.; Drickamer K.; Weis W. I.; Taylor M. E. Structural Analysis of Carbohydrate Binding by the Macrophage Mannose Receptor CD206. J. Biol. Chem. 2021, 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Fiete D. J.; Beranek M. C.; Baenziger J. U. A Cysteine-Rich Domain of the “Mannose” Receptor Mediates GalNAc-4-SO4 Binding. Proc. Natl. Acad. Sci. 1998, 95 (5), 2089–2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Tedesco S.; De Majo F.; Kim J.; Trenti A.; Trevisi L.; Fadini G. P.; Bolego C.; Zandstra P. W.; Cignarella A.; Vitiello L. Convenience versus Biological Significance: Are PMA-Differentiated THP-1 Cells a Reliable Substitute for Blood-Derived Macrophages When Studying in Vitro Polarization? Front. Pharmacol. 2018, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Taciak B.; Białasek M.; Braniewska A.; Sas Z.; Sawicka P.; Kiraga Ł.; Rygiel T.; Król M. Evaluation of Phenotypic and Functional Stability of RAW 264.7 Cell Line through Serial Passages. PLoS ONE 2018, 13 (6), e0198943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Kovacsovics-Bankowski Magdalena; Rock K. L. Presentation of Exogenous Antigens by Macrophages: Analysis of Major Histocompatibility Complex Class I and II Presentation and Regulation by Cytokines. Eur. J. Immunol. 1994, 24 (10), 2421–2428. [DOI] [PubMed] [Google Scholar]
- (28).van der Zande H. J. P.; Nitsche D.; Schlautmann L.; Guigas B.; Burgdorf S. The Mannose Receptor: From Endocytic Receptor and Biomarker to Regulator of (Meta)Inflammation. Front. Immunol. 2021, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).van Duijn A.; V. der Burg S. H.; Scheeren F. A. CD47/SIRPα Axis: Bridging Innate and Adaptive Immunity. J. Immunother. Cancer 2022, 10 (7), e004589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Logtenberg M. E. W.; Scheeren F. A.; Schumacher T. N. The CD47-SIRPα Immune Checkpoint. Immunity 2020, 52 (5), 742–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Qu T.; Li B.; Wang Y. Targeting CD47/SIRPα as a Therapeutic Strategy, Where We Are and Where We Are Headed. Biomark. Res. 2022, 10 (1), 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Willingham S. B.; Volkmer J.-P.; Gentles A. J.; Sahoo D.; Dalerba P.; Mitra S. S.; Wang J.; Contreras-Trujillo H.; Martin R.; Cohen J. D.; Lovelace P.; Scheeren F. A.; Chao M. P.; Weiskopf K.; Tang C.; Volkmer A. K.; Naik T. J.; Storm T. A.; Mosley A. R.; Edris B.; Schmid S. M.; Sun C. K.; Chua M.-S.; Murillo O.; Rajendran P.; Cha A. C.; Chin R. K.; Kim D.; Adorno M.; Raveh T.; Tseng D.; Jaiswal S.; Enger P. Ø.; Steinberg G. K.; Li G.; So S. K.; Majeti R.; Harsh G. R.; van de Rijn M.; Teng N. N. H.; Sunwoo J. B.; Alizadeh A. A.; Clarke M. F.; Weissman I. L. The CD47-Signal Regulatory Protein Alpha (SIRPa) Interaction Is a Therapeutic Target for Human Solid Tumors. Proc. Natl. Acad. Sci. 2012, 109 (17), 6662–6667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Zhang W.; Huang Q.; Xiao W.; Zhao Y.; Pi J.; Xu H.; Zhao H.; Xu J.; Evans C. E.; Jin H. Advances in Anti-Tumor Treatments Targeting the CD47/SIRPα Axis. Front. Immunol. 2020, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Son J.; Hsieh R. C.-E.; Lin H. Y.; Krause K. J.; Yuan Y.; Biter A. B.; Welsh J.; Curran M. A.; Hong D. S. Inhibition of the CD47-SIRPα Axis for Cancer Therapy: A Systematic Review and Meta-Analysis of Emerging Clinical Data. Front. Immunol. 2022, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Bouwstra R.; van Meerten T.; Bremer E. CD47-SIRPα Blocking-Based Immunotherapy: Current and Prospective Therapeutic Strategies. Clin. Transl. Med. 2022, 12 (8), e943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Mirlekar B. Tumor Promoting Roles of IL-10, TGF-β, IL-4, and IL-35: Its Implications in Cancer Immunotherapy. SAGE Open Med. 2022, 10, 20503121211069012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Finkelman F. D.; Madden K. B.; Morris S. C.; Holmes J. M.; Boiani N.; Katona I. M.; Maliszewski C. R. Anti-Cytokine Antibodies as Carrier Proteins. Prolongation of in Vivo Effects of Exogenous Cytokines by Injection of Cytokine-Anti-Cytokine Antibody Complexes. J. Immunol. 1993, 151 (3), 1235–1244. [PubMed] [Google Scholar]
- (38).Burgdorf S.; Kurts C. Endocytosis Mechanisms and the Cell Biology of Antigen Presentation. Curr. Opin. Immunol. 2008, 20 (1), 89–95. [DOI] [PubMed] [Google Scholar]
- (39).Muntjewerff E. M.; Meesters L. D.; van den Bogaart G. Antigen Cross-Presentation by Macrophages. Front. Immunol. 2020, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Burgdorf S.; Lukacs-Kornek V.; Kurts C. The Mannose Receptor Mediates Uptake of Soluble but Not of Cell-Associated Antigen for Cross-Presentation1. J. Immunol. 2006, 176 (11), 6770–6776. [DOI] [PubMed] [Google Scholar]
- (41).Modak M.; Mattes A.-K.; Reiss D.; Skronska-Wasek W.; Langlois R.; Sabarth N.; Konopitzky R.; Ramirez F.; Lehr K.; Mayr T.; Kind D.; Viollet C.; Swee L. K.; Petschenka J.; Kasmi K. C. E.; Noessner E.; Kitt K.; Pflanz S. CD206+ Tumor-Associated Macrophages Cross-Present Tumor Antigen and Drive Antitumor Immunity. JCI Insight 2022, 7 (11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Rare example of a small molecule glycomimetic scaffold to target CD206: Uehara K.; Harumoto T.; Makino A.; Koda Y.; Iwano J.; Suzuki Y.; Tanigawa M.; Iwai H.; Asano K.; Kurihara K.; Hamaguchi A.; Kodaira H.; Atsumi T.; Yamada Y.; Tomizuka K. Targeted Delivery to Macrophages and Dendritic Cells by Chemically Modified Mannose Ligand-Conjugated siRNA. Nucleic Acids Res. 2022, 50 (9), 4840–4859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Mazzaccaro R. J.; Gedde M.; Jensen E. R.; van Santen H. M.; Ploegh H. L.; Rock K. L.; Bloom B. R. Major Histocompatibility Class I Presentation of Soluble Antigen Facilitated by Mycobacterium Tuberculosis Infection. Proc. Natl. Acad. Sci. U. S. A. 1996, 93 (21), 11786–11791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Gibb S.; Strimmer K. MALDIquant: A Versatile R Package for the Analysis of Mass Spectrometry Data. Bioinformatics 2012, 28 (17), 2270–2271. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.