

Abstract
Regulation of the profile and magnitude of toll-like receptor (TLR) responses is important for effective host defense against infections while minimizing inflammatory toxicity. The chemokine CXCL4 regulates the TLR8 response to amplify inflammatory gene and inflammasome activation while attenuating the interferon (IFN) response in primary monocytes. In this study, we describe an unexpected role for the kinase RIPK3 in suppressing the CXCL4 + TLR8–induced IFN response and providing signal 2 to activate the NLRP3 inflammasome and interleukin (IL)-1 production in primary human monocytes. RIPK3 also amplifies induction of inflammatory genes such as TNF, IL6, and IL1B while suppressing IL12B. Mechanistically, RIPK3 inhibits STAT1 activation and activates PI3K-Akt–dependent and XBP1- and NRF2-mediated stress responses to regulate downstream genes in a dichotomous manner. These findings identify new functions for RIPK3 in modulating TLR responses and provide potential mechanisms by which RIPK3 plays roles in inflammatory diseases and suggest targeting RIPK3 and XBP1- and NRF2-mediated stress responses as therapeutic strategies to suppress inflammation while preserving the IFN response for host defense.
Keywords: CXCL4, NLRP3 inflammasome, RIPK3, TLR8, gene regulation, inflammation
1. Introduction
Toll-like receptors (TLRs) are a family of pattern recognition receptors and play key roles in host defense against pathogen infection by inducing production of type I interferon (IFN-I) and inflammatory cytokines such as tumor necrosis factor (TNF) and interleukin (IL)-1β. However, imbalanced and excessive IFN-I and inflammatory cytokine expression could be detrimental to the host during defense against pathogen infection, such as influenza and COVID-191,2; uncontrolled TLR responses to molecules from host dying cells, such as HMGB1 and self-nucleic acids (NAs), also cause inflammation, which is related to inflammatory diseases and autoimmune disorders, such as rheumatoid arthritis and systemic lupus erythematosus.3–5 A moderate and balanced IFN-I response and nuclear factor κB (NF-κB)–induced inflammatory response is critical for host defense against pathogen infections and maintenance of homeostasis; however, the mechanisms that balance TLR responses are still not understood. One example is the balance between IFN and IL-1, as previous work has shown negative crossregulation between IFN-I and IL-1β through the regulation of NLRP3 inflammasome activation and IL-1 expression and the induction of eicosanoids by IL-1β, which is associated with Mycobacterium tuberculosis pathogenesis.6–9
Endosomal TLRs, including TLR3, TLR7, TLR8, and TLR9, sense NAs to induce IFN-I and cytokine production and are critical for antiviral and anti-intracellular bacterial responses. TLR8 recognizes single-stranded RNA (ssRNA) to induce IFN-I and cytokines. CXCL4 is a cationic chemokine and has been implicated in various inflammatory diseases including atherosclerosis and systemic sclerosis.10–14 In our recent study,10 we discovered that CXCL4 facilitates TLR8 ssRNA ligand ORN8L15 internalization through nanoparticle formation between CXCL4 and ORN8L; CXCL4 and TLR8 signaling crosstalk synergistically activates cytokine and chemokine genes involved in cytokine storm and inflammatory disease pathogenesis, and triggers a second signal-independent NLRP3 inflammasome activation and massive IL-1β production. In contrast to this superactivation, CXCL4 modestly attenuates TLR8-induced IFN responses, as determined by reduction of select IFN-stimulated gene (ISG) expression after CXCL4 + TLR8 costimulation relative to TLR8 alone. However, CXCL4 and TLR8 signaling crosstalk strongly activates TBK1/IKKϵ-IRF3 axis to induce a high amount of IFN-I expression compared with TLR8 alone. This suggests that CXCL4 and TLR8 signaling activates a feedback mechanism that negatively regulates IFN-I responses to set the balance between IFN responses and NLRP3 inflammasome activation and IL-1β production.
RIPK3 and RIPK1 are kinases that initiate apoptosis and necroptosis. RIPK1 is a death domain (DD)–containing protein, which can interact with DD of other proteins such as TNF receptor 1 and the death receptor Fas; RIPK1 also harbors an amino-terminal kinase domain and a bridging intermediate domain that contains an RIP homotypic interaction motif (RHIM). Unlike RIPK1, RIPK3 lacks a DD in the carboxy-terminal region but contains a RHIM protein: protein interaction motif.16 Although RIPK3 cannot directly interact with or be activated by DD-containing death receptors, it can bind proteins such as RIPK1, TRIF, and ZBP116 through RHIM domain–mediated interaction and become activated. RIPK3 can also activate itself through autophosphorylation at serine 227, which is generally achieved when RIPK3 interacts with other proteins, like its target pseudokinase mixed lineage kinase domain-like (MLKL).17,18 RIPK3 is the key kinase that activates MLKL to trigger a programed cell death termed necroptosis.16 Increasing evidence suggests that RIPK3 not only is a pleiotropic modulator of necroptosis, but also contributes to second signal-independent NLRP3 inflammasome activation and inflammatory responses and diseases.16,19–24 RIPK3 is tightly controlled at baseline by caspase-8; after activation of TNF receptor (TNFR) or TLR signaling, RIPK3 can be activated by its upstream kinase RIPK1 or other kinases when caspase-8 activity is inhibited, which leads to necroptosis and NLRP3 activation.22,25 RIPK3 contributes to lipopolysaccharide induction of inflammatory genes such as Tnf through activation of NF-κB in the absence of caspase-8 activity in mouse bone marrow–derived macrophages.26 RIPK3 also promotes kidney fibrosis through the AKT-dependent activation of adenosine triphosphate (ATP) citrate lyase, in which genetic or chemical inhibition of RIPK3 suppressed AKT activation and ATP citrate lyase in transforming growth factor β–experienced fibroblasts.27 RIPK3 plays a greater role in mouse models of inflammation and tissue injury than its necroptotic executor client MLKL.24
In this study, we observed that CXCL4 and TLR8 costimulation activated RIPK3 in human primary monocytes, and that RIPK3 promoted expression of inflammatory genes such as TNF and activated NLRP3 inflammasome and IL-1β production while suppressing IFN response and IL12B expression. Furthermore, we demonstrated that RIPK3 activated XBP1-mediated endoplasmic reticulum (ER) stress and NRF2-mediated oxidative stress responses in a PI3K-Akt–dependent manner in human monocytes upon CXCL4 and TLR8 costimulation. XBP1- and NRF2-mediated responses contributed to RIPK3-mediated regulation of inflammatory gene and ISG expression. In accord with a dampened IFN response after CXCL4 and TLR8 costimulation, RIPK3 suppressed STAT1 activation. In addition, RIPK3 played a role in the second signal-independent NLRP3 inflammasome activation and RIPK1 partnered with RIPK3 for NLRP3 inflammasome activation and IL-1β production when caspase-8 was inhibited. These findings support the important role of RIPK3 in inflammatory conditions by driving inflammatory gene expression and NLRP3 inflammasome activation, as has been suggested in nonalcoholic fatty liver disease21 and COVID-19.28 Dampening IFN responses and IL12B expression also suggests that RIPK3 may suppress T helper 1 and cytotoxic T lymphocyte immune responses, thereby dampening antipathogen29,30 or antitumor immunity.21,31,32
2. Materials and methods
2.1. Cell culture
Deidentified human buffy coats were purchased from the New York Blood Center following a protocol approved by the Hospital for Special Surgery Institutional Review Board. Peripheral blood mononuclear cells were isolated with Lymphoprep (Accurate Chemical) via density gradient centrifugation and monocytes were purified from peripheral blood mononuclear cells with anti-CD14 magnetic beads as recommended by the manufacturer (Miltenyi Biotec).10 Monocytes were cultured overnight at 37 °C, 5% CO2 in RPMI-1640 medium (Invitrogen) supplemented with 10% heat-inactivated defined fetal bovine serum (FBS) (HyClone; Fisher Scientific), penicillin-streptomycin (Invitrogen), L-glutamine (Invitrogen), and 20 ng/mL human macrophage colony-stimulating factor (M-CSF). After at least 12-h culture, the cells were treated as described in the figure legends and were harvested and prepared for total RNA extraction, protein extraction, and flow cytometry. Cells were stimulated with CXCL4 (Sigma) and/or TLR8 ligand ORN8L (ChemGenes) as previously described10; testing of various lots of CXCL4 using the Chromogenic LAL Endotoxin Assay Kit (Genscript) showed that CXCL4 preparations contributed 0.01 to 0.06 EU/mL final concentration of endotoxin in cultures, which does not contribute appreciably to synergy with ORN8L.10
2.2. RIPK3 overexpression
Monocytes (1 × 106 cells) were seeded in 24-well plates in complete RPMI-1640 medium supplemented with 10% heat-inactivated defined FBS, penicillin-streptomycin, L-glutamine, and 50 ng/mL human M-CSF for 5 d. The culture medium was replaced with fresh medium on day 3 during the culture. On day 5 of culture, the old medium was removed and replaced with 200 μL of RPMI-1640 medium without penicillin-streptomycin with a final concentration of 2% heat-inactivated defined FBS and 50 ng/mL human M-CSF. Enhanced green fluorescent protein (GFP) or GFP-h-RIPK3 adenoviral particles (multiplicity of infection = 100 plaque-forming units/cell; Vector Biolabs) were added to the cells accordingly. The cells were centrifuged at 1600 rpm for 30 min at room temperature (RT). After culturing for 12 h, 300 μL of complete RPMI-1640 medium supplemented with 10% heat-inactivated defined FBS, penicillin-streptomycin, L-glutamine, and 20 ng/mL human M-CSF was added to the cell culture. The cells were cultured for another day before harvesting for experiments.
2.3. RNA sequencing
After RNA extraction, libraries for sequencing were prepared using the NEBNext Ultra II RNA Library Prep Kit for Illumina following the manufacturer’s instructions (Illumina). Quality of all RNA and library preparations was evaluated with BioAnalyzer 2100 (Agilent). Libraries were sequenced by the Genomics Resources Core Facility at Weill Cornell Medicine using a NovaSeq, 50-bp paired-end reads at a depth of ~10 to 20 million reads per sample. Paired-end reads were preprocessed using fastp (0.19.10; https://github.com/OpenGene/fastp), which supports adapter trimming, low-quality base trimming, and calculation of additional quality control metrics. Trimmed reads were aligned to the human genome (hg38) genome using STAR (2.7.3a; https://github.com/alexdobin/STAR). Low quality reads and multimapping alignments were filtered using SAMtools (1.9; https://github.com/samtools/). Reads were counted within exons and summarized at the gene level using featureCounts (v2.0.1; https://subread.sourceforge.net) to produce a count matrix of reads per gene. Differential gene expression analysis was performed in R (4.1.0; R Foundation for Statistical Computing) using edgeR (3.34.1; https://bioconductor.org/packages/release/bioc/html/edgeR.html). Genes with low expression levels (<3 counts per million in at least 1 group) were filtered from all downstream analyses. The Benjamini-Hochberg false discovery rate procedure was used to correct for multiple testing.
2.4. Ingenuity Pathway Analysis
Ingenuity Pathway Analysis was used to analyze differentially expressed genes. The Ingenuity Canonical Pathways were used to predict activated or suppressed pathways based on the expression pattern of genes regulated by CXCL4 and ORN8L in human primary monocytes. The Upstream Regulator analytic was used to predict upstream regulators whose change in expression or function could explain the observed gene expression changes. The overall activation/inhibition states of canonical pathways and Upstream Regulators are predicted based on a z-score algorithm, for which a negative or positive value represents the predicted inhibition or activation of the pathway and upstream regulator, respectively.
2.5. Analysis of messenger RNA amounts
Total RNA was extracted from cultured human monocytes with an RNeasy Mini Kit (Qiagen) and was used for complementary DNA synthesis by reverse transcription with a RevertAid RT Reverse Transcription Kit (Thermo Fisher Scientific; catalog number: K1691). Real-time polymerase chain reaction (PCR) was performed with the Fast SYBR Green Master Mix and a 7500 Fast Real-Time PCR System (Applied Biosystems). The primer sequences for the quantitative PCR (qPCR) reactions are listed in Supplementary Table 1. Cycle threshold (CT) values of target gene were normalized to GAPDH expression and are shown as percentage of GAPDH (100/2^ΔCt).
2.6. Flow cytometric analysis of cell death
Single-cell suspensions were stained with Fixable Viability Dye eFluor 780 (eBioscience; cat. #65-0865-18) for 20 min at 4 °C. Then, cells were washed with FACS buffer (phosphate-buffered saline containing 2% calf serum and 1 mM EDTA) and were analyzed by flow cytometry. Dead cells were detected based on eFluor 780 fluorescence. Data were analyzed with FlowJo software version 10 (TreeStar).
2.7. Flow cytometric analysis of phospho-NRF2
After treatment with CXCL4 and ORN8L in the presence/absence of PI3K inhibitor Wortmanin for 3 h, cells were harvested and fixed with 4% paraformaldehyde for 15 min at RT. After washing with FACS buffer, the cells were incubated in cold 90% methanol for 10 min and then incubated with anti-NRF2 antibody for 2 h at RT. Cells were then incubated with PE-conjugated secondary antibody for 1 h at RT. Cells were analyzed using a BD FACSymphony A5 Cell Analyzer to measure PE fluorescence.
2.8. Western blotting
Cells were lysed with 50 μl of cold lysis buffer comprising 50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% (vol/vol) Triton X-100, 2 mM Na3VO4, 1 × phosSTOP EASYPACK, 1 mM Pefabloc, and 1× EDTA-free complete protease inhibitor cocktail (Roche), and incubated for at least 10 min on ice. Then, cell debris was pelleted using Eppendorf Centrifuge 5424 R at 21,130 g at 4 °C for 5 min. The protein fraction in the supernatant was mixed with 4× Laemmli Sample buffer (Bio-Rad; cat. #1610747) supplemented with 10% 2-mercroptoehanol (BME) (Sigma-Aldrich). Samples for Western blotting were subjected to electrophoresis on Bis-Tris gels (Invitrogen). Proteins were transferred to polyvinylidene difluoride membrane was as previously reported10 Membranes were blocked in 5% (w/v) bovine serum albumin in Tris-buffered saline (20 mM Tris, 50 mM NaCl, pH 8.0) with 0.1% (v/v) Tween 20 (TBST) at RT for at least 1 h with shaking. Membranes were then incubated with target primary antibodies at 4 °C overnight with shaking and then washed 3 times in TBST for 10 min, then incubated with anti-mouse or anti-rabbit immunoglobulin G secondary antibodies conjugated to horseradish peroxidase (GE Healthcare; cat: NA9310V and NA9340V) diluted in TBST at room temperature for 1 h with shaking. Next, membranes were washed 3 times in TBST at RT for with shaking. Antibody binding was detected using enhanced chemiluminescent substrates for horseradish peroxidase (ECL Western blotting reagents [PerkinElmer; cat: NEL105001EA]) or SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific; cat: 34,095) according to the manufacturer’s instructions, and visualized using premium autoradiography film (Thomas Scientific; cat: E3018). For membranes that required probing twice or more using different primary antibodies, Restore PLUS Western blotting stripping buffer (Thermo Fisher Scientific) was applied on the blots with shaking for 15 to 20 min at RT following first-time development. To detect multiple proteins on the same experimental filter, membranes were cut horizontally based on the molecular mass markers and molecular size of the target proteins. Antibodies used are identified in Supplementary Table 1.
2.9. Cytokine detection by enzyme-linked immunosorbent assay
Levels of IL-1β were measured in supernatants of cells using enzyme-linked immunosorbent assay kits (R&D Systems) according to the manufacturer’s instructions.
2.10. Statistical analysis
GraphPad Prism Version 8 for Windows (GraphPad Software) was used for all statistical analysis. Information about the specific tests used and the number of independent experiments are provided in figure legends. Two-way analysis of variance with Sidak correction for multiple comparisons was used for grouped data. Otherwise, 1-way analysis of variance with the Greenhouse–Geisser correction and Tukey’s post hoc test for multiple comparisons was performed. For paired data, when the data did not pass the normal distribution by F test the Wilcoxon signed rank test was performed; otherwise, a paired t test was used.
3. Results
3.1. RIPK3 suppresses ISG induction after CXCL4 + TLR8 costimulation
In our recent study, we found that CXCL4 and TLR8 costimulation synergistically activates inflammatory gene expression and mature IL-1 production at least in part via NF-κB, AP-1, and synergistic activation of TBK1-IRF5 signaling.10 As RIPK3 can promote NF-κB activity and inflammasome activation, we tested the role of RIPK3 in CXCL4 + TLR8–induced monocyte activation.23,26,33 Here, we found that treatment of human primary monocytes with the combination of CXCL4 and TLR8 ligand ORN8L activated RIPK3, as determined by increased phosphorylation at serine 227 compared with baseline (Fig. 1A and Supplementary Fig. 1B). Both CXCL4 and ORN8L contributed to this increase in a time-course experiment (Fig. 1A). CXCL4 + TLR8 stimulation induces production of TNF,10 which can potentially activate RIPK3 in an autocrine manner. We found that blockade of TNF signaling using anti-TNF neutralizing antibodies did not affect CXCL4 + TLR8–induced RIPK3 phosphorylation, suggesting that CXCL4 and TLR8 signaling–induced RIPK3 activation is TNF independent (Supplementary Fig. 1B). Strikingly, inhibition of RIPK3 using its specific inhibitor GSK87234,35 had a dichotomous effect on CXCL4 + TLR8–induced gene expression: induction of the NF-κB target gene TNF was nearly abolished, whereas induction of the ISG CXCL10 was increased (Fig. 1B); induction of IL12B, which is strongly potentiated by IFNs,36 was also increased, whereas IL6 and IL1B induction were less affected (Supplementary Fig. 1A).
Fig. 1.
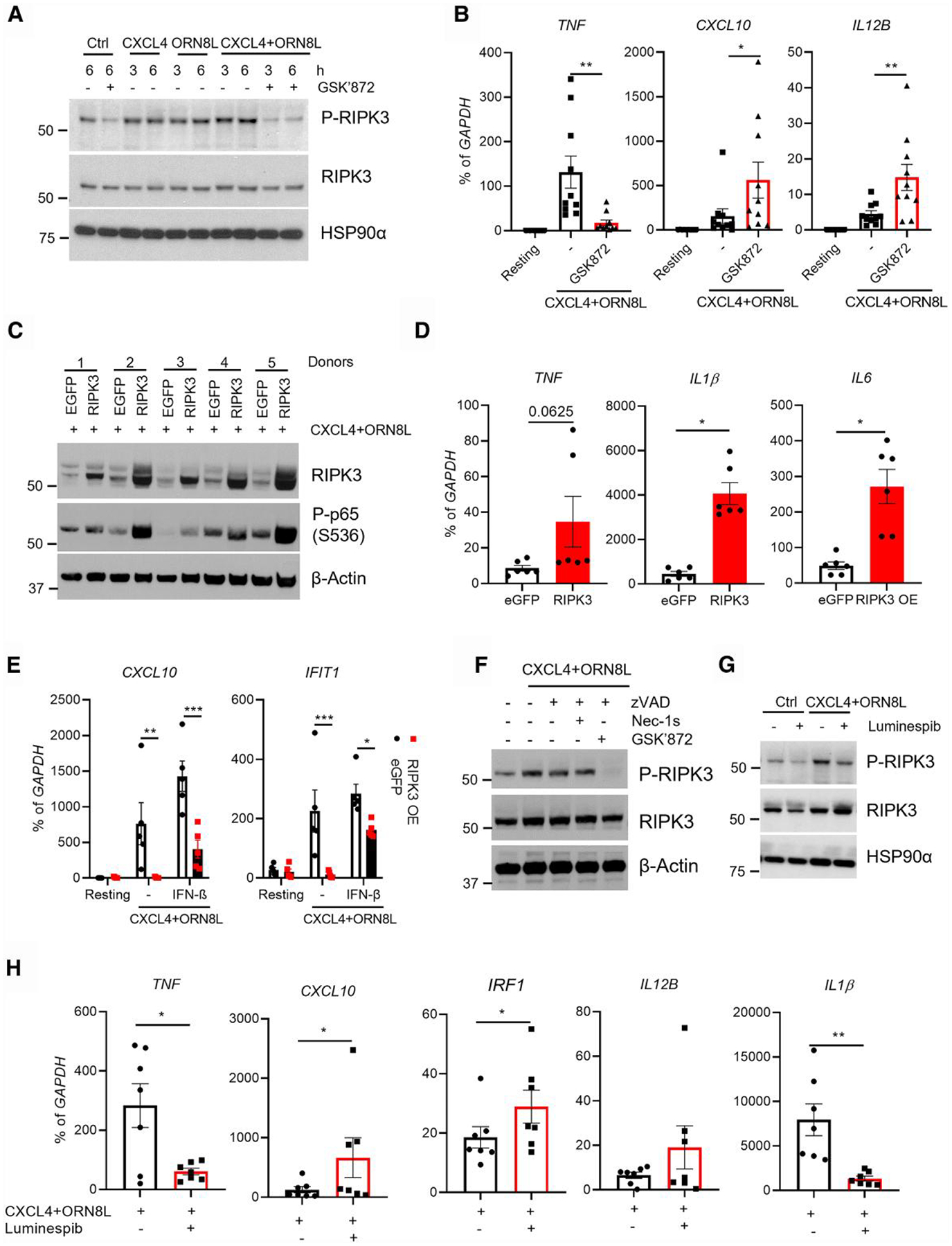
RIPK3 regulates CXCL4 and TLR8–induced inflammatory and IFN-stimulated genes. Human blood monocytes were isolated with CD14+ magnetic beads and rested overnight with M-CSF (20 ng/mL). (A) Immunoblot of phospho-RIPK3 and total RIPK3 using whole cell lysates of cells stimulated with CXCL4 (10 μg/mL) and TLR8 ligand ORN8L (20 μg/mL) in the presence/absence of GSK872 (10 μM) for the indicated times. (B) mRNA was measured by qPCR and normalized relative to GAPDH mRNA after 6 h of CXCL4 and TLR8 costimulation of human blood monocytes with/without RIPK3 inhibitor GSK872. (C) Immunoblot of RIPK3 and phospho-p65 with whole cell lysates of control enhanced GFP (eGFP)– or RIPK3-overexpressing cells stimulated with CXCL4 and ORN8L for 6 h. (D) mRNA of inflammatory genes was measured by qPCR and normalized relative to GAPDH mRNA in cells from panel C. (E) mRNA of CXCL10 and IFIT1 was measured by qPCR and normalized relative to GAPDH mRNA in cells from eGFP- or RIPK3-overexpressing cells stimulated with CXCL4 and ORN8L with/without IFN-β (100 ng/mL) for 3 h. (F, G) Immunoblot of RIPK3 and phospho-RIPK3 using whole cell lysates of cells stimulated with CXCL4 and ORN8L in the presence/absence of caspase-8 inhibitor zVAD (50 μM), RIPK1 inhibitor necrostatin-1 s (30 μM), and GSK872 (F) or of HSP90 inhibitor luminespib (1 μM) (G) for 6 h. (H) mRNA was measured by qPCR and normalized relative to GAPDH mRNA in cells stimulated with CXCL4 and ORN8L with/without luminespib for 6 h. Data are depicted as mean ± SEM of 5 to 11 healthy donors (B, D, E, and H) or 1 representative blot of at least 3 independent experiments (A, C, F, and G). ***P ≤ 0.001; **P ≤ 0.01; *P ≤ 0.05 by Wilcoxon matched signed rank test (B, D), paired t test (D, H), and 2-way analysis of variance with Sidak correction for multiple comparisons (E). OE = overexpressed.
Inhibition of RIPK3 did not affect NF-κB activation, as determined by p65 phosphorylation (Supplementary Fig. 1C). To further test the function of RIPK3 in regulation of the gene expression in CXCL4 + TLR8 costimulation, we took a complementary gain-of-function approach by overexpressing RIPK3 using adenoviral transduction of primary human macrophages and found that, in line with a previous report that RIPK3 contributes to lipopolysaccharide-induced NF-κB activation and inflammatory gene expression in mouse bone marrow–derived macrophages when caspase-8 is inhibited,26 forced expression of RIPK3 increased the amounts of NF-κB p65 phosphorylation under conditions of CXCL4 + TLR8 costimulation (Fig. 1C) and increased expression of NF-κB target inflammatory genes IL1B and IL6 with a trend toward increased TNF expression that was not significant but did not affect IL12B expression (Fig. 1D and Supplementary Fig. 1D). The results showing the effects of GSK872 and RIPK3 overexpression on p65 activation (Fig. 1C and Supplementary Fig. 1C) are consistent with a redundant role for endogenous RIPK3 in p65 activation in the context of primary human monocytes costimulated with CXCL4 + TLR8, but that the role of RIPK3 in this signaling pathway can be revealed by supraphysiological RIPK3 expression. The lack of IL12B superinduction after RIPK3 overexpression is consistent with the RIPK3-mediated suppressive pathway targeting IL12B but not TNF shown in Fig. 1B, which would counterbalance the positive effects of RIPK3-mediated NF-κB activation on IL12B expression. Strikingly, forced RIPK3 expression strongly suppressed induction of the ISGs CXCL10 and IFIT1 (Fig. 1E). Thus, both loss- and gain-of-function experiments concordantly show that RIPK3 attenuates induction of ISGs by CXCL4 + TLR8 costimulation, suggesting a feedback inhibition mechanism that may selectively target the IFN response.
Next, we investigated how RIPK3 was activated after CXCL4 and TLR8 costimulation. The pan caspase inhibitor zVAD, which inhibits caspase-8, or necrostatin-1s, which inhibits RIPK1, minimally affected RIPK3 activation; in contrast, inhibition of RIPK3 using its inhibitor GSK872 completely removed its phosphorylation (Fig. 1A, F). These results suggest that in our system RIPK3 is not predominantly activated by the RIPK1/Caspase-8 pathway and instead RIPK3 activation might depend on autophosphorylation. RIPK3 is a client of chaperone protein HSP90, and chemical inhibitors of HSP90 disrupt the HSP90-RIPK3 complex and prevent RIPK3 phosphorylation on serine 227 and activation.37–39 Strikingly, inhibition of RIPK3 using HSP90 inhibitor luminespib, which is under clinical development as a cancer therapy, suppressed RIPK3 phosphorylation (Fig. 1G) and phenocopied the effects of the RIPK3 inhibitor GSK872 on gene expression (Fig. 1H): superinduction of CXCL10, IRF1, and IL12B with concomitant suppression of TNF and IL1B expression. This result opens HSP90 inhibition as a novel approach to modulating the balance between TLR-induced inflammatory and IFN responses.
3.2. RIPK3 attenuates CXCL4 + TLR8–induced STAT1 activation
To investigate mechanisms by which RIPK3 attenuates ISG induction, we examined regulation of IRF1 and STAT1, 2 key components of the IFN-Jak-STAT-IRF pathway that activates ISG expression. As expected based upon a previously described IFN-β-mediated autocrine loop,40 CXCL4 + TLR8 costimulation activated STAT1 by inducing tyrosine 701 phosphorylation (Fig. 2A). STAT1 activation was increased when RIPK3 was inhibited (Fig. 2A), as was induction of its target gene IRF1 at both messenger RNA (mRNA) and protein levels (Fig. 2B, C). In a complementary gain-of-function experiment, RIPK3 strongly suppressed CXCL4 + TLR8–induced STAT1 activation and IRF1 induction (Fig. 2D, compare lane 4 with lane 3; densitometry showed >90% decreases in band intensity [Supplementary Fig. 1E]). Thus, RIPK3 suppresses CXCL4 + TLR8–induced STAT1 activation; however, RIPK3 did not significantly affect induction of IFNB (Fig. 2E). Moreover, adding exogenous IFN-β only partially rescued STAT1 phosphorylation and IRF1 protein expression (Fig. 2D, compare lane 6 with lane 5) and only partially rescued RIPK3-mediated suppression of ISGs in CXCL4 + TLR8 costimulation (Fig. 1E, columns 5 and 6). Additional experiments supported that RIPK3 does not suppress ISG induction by exogenous IFN-β (Fig. 2F). Thus, overall the data suggest that RIPK3 contributes to suppression of macrophage IFN responses. The suppression of IFN-induced gene expression under conditions of CXCL4 + TLR8 costimulation, but not when exogenous IFN was added alone, suggests induction of a cofactor(s) that cooperates with RIPK3 to regulate IFN-mediated STAT activation, which we next investigated using a transcriptomic approach.
Fig. 2.
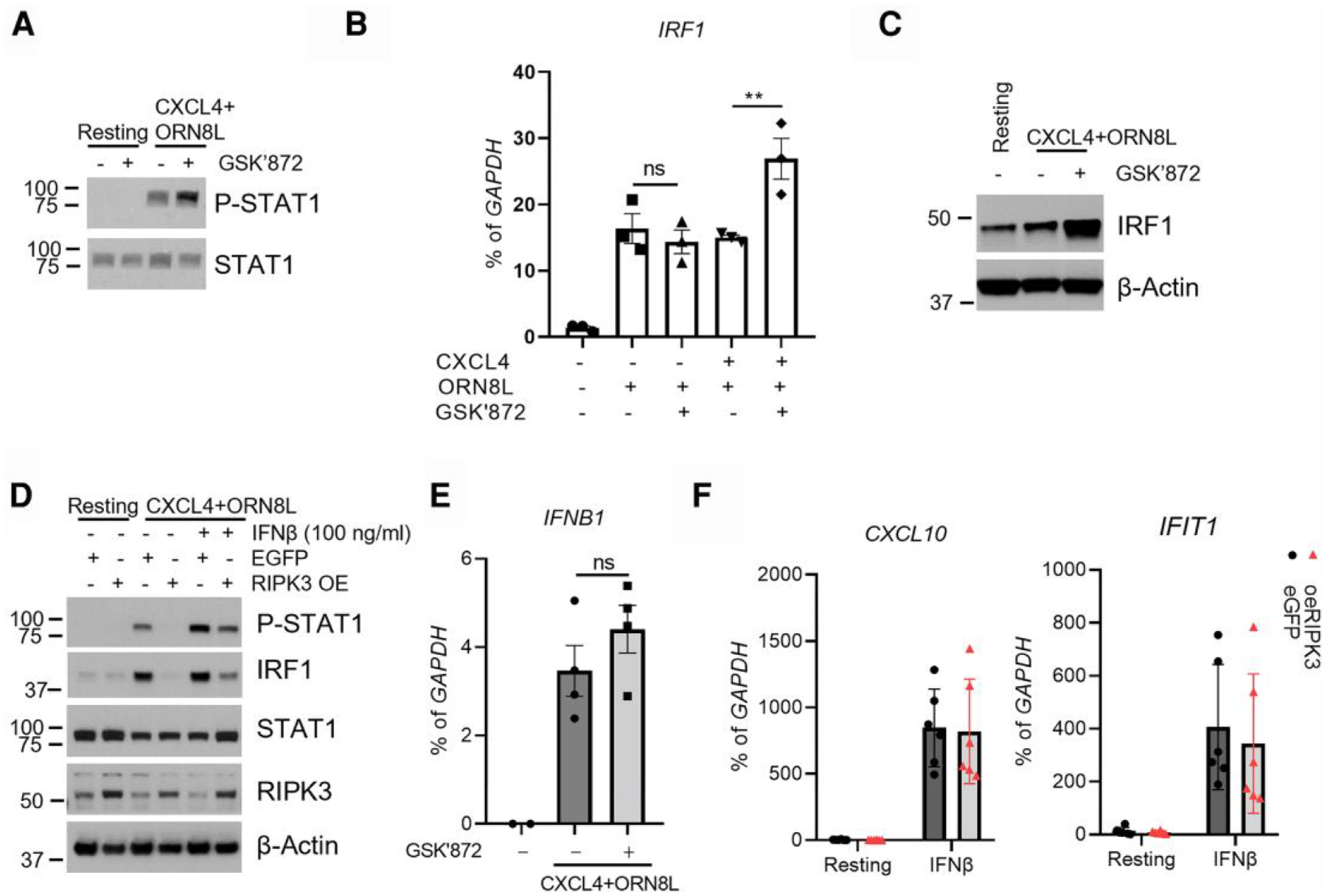
RIPK3 regulates the CXCL4 + TLR8–induced IFN response. (A) Immunoblot of STAT1 and phospho-STAT1 with whole cell lysates of cells stimulated for 3 h. (B, C) mRNA level (B) and protein level (C) of IRF1 measured in cells stimulated under the indicated conditions for 6 h. (D) Immunoblot of STAT1, phospho-STAT1, IRF1, and RIPK3 using whole cell lysates of eGFP- or RIPK3-overexpressing cells stimulated with CXCL4 and ORN8L with/without IFN-β (100 ng/mL) for 3 h. (E) mRNA of IFNB1 was measured by qPCR and normalized relative to GAPDH mRNA in cells stimulated with CXCL4 and ORN8L in the presence/absence of GSK872 for 3 h. (F) mRNA of CXCL10 and IFIT1 was measured by qPCR and normalized relative to GAPDH mRNA in cells from eGFP- or RIPK3-overexpressing cells stimulated with IFN-β (100 ng/mL) for 3 h. Data are depicted as mean ± SEM of 3 to 6 healthy donors (B, E, and F) or 1 representative blot from at least 3 independent experiments (A, C, and D). **P ≤ 0.01 by 1-way analysis of variance with Tukey’s post hoc test for multiple comparisons (B and E). ns = no significance; OE = overexpressed.
3.3. Genome-wide regulation of CXCL4 + TLR8–induced gene expression by RIPK3
We had previously observed gasdermin D (GSDMD) activation in CXCL4 + TLR8–stimulated monocytes,10 which can lead to cell death via pyroptosis (see the following) in addition to IL-1β release. RIPK3 can also induce necroptosis via activation of MLKL. To attenuate secondary effects of cell death on gene induction and to focus on the effects of upstream signaling pathways, we followed a standard approach in the field of suppressing cell death and inhibited MLKL and GSDMD activity using necrosulfonamide (NSA) (5μM),41 and used the selective RIPK3 inhibitor GSK872 to identify RIPK3 effects on gene expression. Inhibition of GSDMD had minimal effects on CXCL4 + TLR8–induced expression of inflammatory NF-κB targets with a trend toward increased expression of TNF (Supplementary Fig. 2A). In contrast, induction of the ISGs CXCL10 and IRF1, and of IL12B, was strongly suppressed (Supplementary Fig. 2A), and this suppression was reversed by RIPK3 inhibition (Supplementary Fig. 2B). These results suggest that inhibition of MLKL and GSDMD (and attendant cell death) more fully reveals the regulatory function of RIPK3 in gene expression, possibly related to prolonged signaling when cell viability is maintained, and supports using this system for studying RIPK3-mediated gene regulation. Thus, we performed transcriptomic analysis using RNA sequencing in primary human monocytes stimulated with CXCL4 + TLR8 in the presence of NSA and GSK872 (Supplementary Fig. 3A). Principal component analysis showed that GSK872 effectively reversed the effects of NSA (Supplementary Fig. 3A), further supporting the notion that inhibition of MLKL and GSDMD highlights the effects RIPK3 on gene expression. Significantly differentially expressed genes (n = 6,750; false discovery rate < 0.05, fold change >2) were clustered into 4 groups based on pattern of expression (Fig. 3A). In this study, we mainly focused on the genes that were upregulated by CXCL4 and TLR8 costimulation (clusters 1 and 4). Cluster 4 contained genes negatively regulated by RIPK3 and contained various ISGs (Fig. 3A, compare column 4 with column 3); pathway analysis showed most significant enrichment of interferon signaling pathways in cluster 4 (Fig. 3B). Accordingly, the most significantly enriched transcription factors (TFs) associated with these genes were IRFs and STAT1 (Fig. 3C). Increased expression of a representative panel of ISGs when RIPK3 was inhibited is displayed in a heat map in Fig. 3D. These results show that RIPK3 broadly suppresses the CXCL4 + TLR8–induced IFN response and suggest that RIPK3 activity may explain why ISG expression was suppressed in CXCL4 + TLR8 costimulation relative to TLR8 alone at late time points in our recent study.10
Fig. 3.
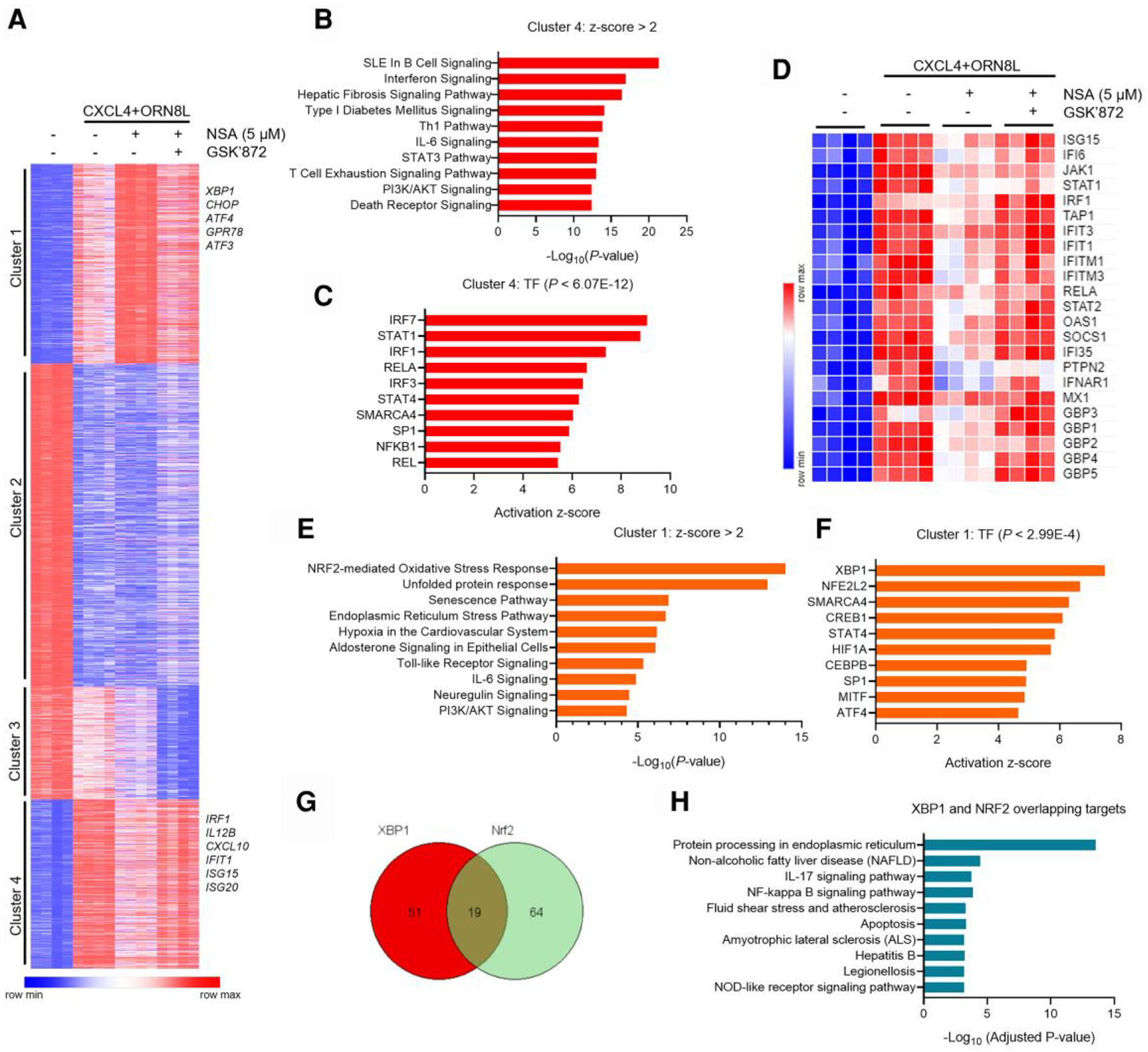
Transcriptomic analysis of RIPK3 function in the CXCL4 and TLR8 response. (A) K-means clustering (K = 4) of differentially expressed genes induced more than 2-fold with false discovery rate < 0.05 in indicated conditions (n = 4 healthy donors). (B, C, E, and F) Analysis of signaling pathways and upstream transcription factors by Ingenuity Pathway Analysis for the genes in clusters 1 and 4 defined in panel A. (D) Heatmap of representative ISGs regulated by RIPK3 in cluster 4. (G) Venn diagram showing the overlap of XBP1 and NRF2 target genes from panel F. (H) Top significant signaling pathways of the 19 overlapping genes in panel G analyzed with the Enrichr program. SLE = systemic lupus erythematosus.
Genes in cluster 1 were positively regulated by RIPK3 and included TNF, in accordance with the reverse-transcription qPCR data (Fig. 1B). Notably, Ingenuity Pathway Analysis of cluster 1 genes showed most significant enrichment of NRF2-mediated oxidative stress response (OSR) and ER stress pathways and accordingly showed most significant enrichment of XBP1 and NFE2L2 (encoding NRF2) TFs as upstream regulators of these genes (Fig. 3E, F). The regulation of known XBP1 and NRF2 target genes is shown in Supplementary Fig. 3B. Out of the XBP1 and NRF2 target genes, 19 genes were regulated by both XBP1 and NRF2 (Fig. 3G); these genes mainly function in ER stress response (Fig. 3H), and a subset were dependent on RIPK3, such as ATF4 and MCFD2 (Supplementary Fig. 3C). In addition to stress pathways that regulate inflammatory gene expression,42 RIPK3 promoted expression of genes in the PI3K-Akt pathway (Fig. 3E), which has previously been implicated in context-dependent positive or negative regulation of TLR responses43 and also regulates stress responses.44–47 These findings led us to test whether RIPK3 regulates PI3K-Akt signaling and NRF2-mediated OSR and ER stress pathways even in the absence of MLKL and GSDMD inhibition, and whether these pathways contribute to downstream regulation of gene expression and suppression of ISGs.
3.4. PI3K-Akt pathway activity is dependent on RIPK3 and regulates ISGs and inflammatory genes
In accord with previously described activation of PI3K-Akt signaling by TLRs,48 treatment of monocytes with CXCL4 and the TLR8 ligand ORN8L increased Akt phosphorylation at serine 473 in most blood donors (Fig. 4A and Supplementary Fig. 4A). Inhibition of RIPK3 strongly diminished Akt phosphorylation in all 5 donors tested (Fig. 4A, Supplementary Fig. 4A, and data not shown). We next used inhibitors of PI3K, which is upstream of Akt, to test the role of PI3K-Akt signaling in CXCL4 + TLR8–induced gene activation. The PI3K inhibitor Wortmanin significantly increased CXCL10 and IRF1 induction and slightly suppressed TNF expression in CXCL4 and TLR8 costimulated human monocytes (Fig. 4B). This result was corroborated with inhibitors targeting individual PI3K catalytic subunits: α, β, δ, and γ or combined inhibition of the 4 subunits (Fig. 4C and Supplementary Fig. 4B, C). Strikingly, PI3K inhibition phenocopied RIPK3 inhibition: increased induction of ISGs and IL12B with suppression of TNF induction. Together, the results show that Akt activity is dependent on RIPK3 and support a role for PI3K-Akt signaling in mediating effects of RIPK3 on gene expression.
Fig. 4.
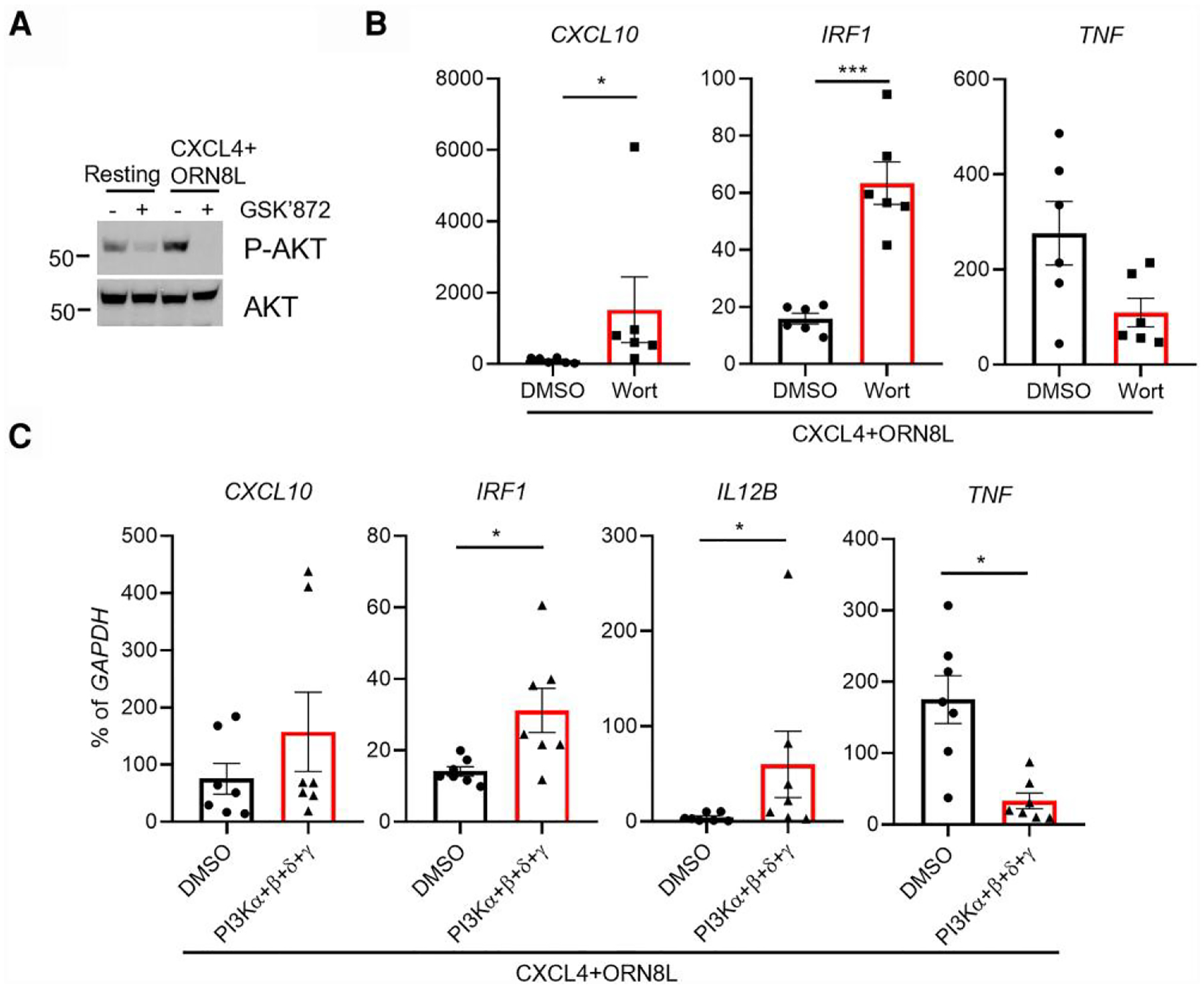
PI3K-Akt signaling is dependent on RIPK3 and regulates ISGs and inflammatory genes. (A) Immunoblot of phospho-AKT and AKT using whole cell lysates of cells stimulated (CXCL4 + ORN8L) in the presence/absence of GSK872 for 3 h. A representative blot for RIPK3 inhibition from 5 independent experiments; see also related data in Fig. S4A. (B, C) mRNA was measured by qPCR and normalized relative to GAPDH mRNA in CXCL4- and ORN8L-stimulated cells with/without PI3K inhibitor Wortmannin (Wort) (B) and combined inhibitors of PI3Kα/β/δ/γ (C) for 6 h. Data are depicted as mean ± SEM of 6 (B) and 7 (C) healthy donors. ***P ≤ 0.001; *P ≤ 0.05 by paired t test (C) or by Wilcoxon matched signed rank test (D). DMSO, dimethyl sulfoxide.
3.5. RIPK3 regulates XBP1 and NRF2 activation to regulate ISG and inflammatory gene expression
Emerging evidence shows that PI3K-Akt pathways activate both XBP1- and NRF2-mediated responses in regulating ER stress and the effector function and survival of human natural killer (NK) cells in regulating antioxidant function and growth factor gene expression and neuroinflammation and in maintaining memory CD4 T cells regulated by Akt-GSK3β–mediated NRF2 nuclear translocation and in preventing ferroptosis, respectively.44–47,49–51 Given the role of stress pathways in inflammatory responses42 and the RNA sequencing data that RIPK3 positively regulates ER stress and NRF2-mediated OSR (Fig. 3), we hypothesized that RIPK3 regulates ER stress and NRF2 pathways, which in turn affect ISG and inflammatory gene expression in human monocytes during CXCL4 and TLR8 costimulation. CXCL4 + TLR8 costimulation indeed activated ER stress, as determined by the upregulation of ER chaperone BIP and ER stress–related TF CHOP, and the increased ratio of spliced XBP1 (s-XBP1)/XBP1 (Fig. 5A, B). CXCL4 + TLR8 costimulation also significantly increased NRF2 activation, determined by phosphorylation at serine 40, relative to stimulation with either factor alone (Fig. 5C). Inhibition of RIPK3 using GSK872 significantly reduced BIP, CHOP, and the ratio of s-XBP1/XBP1 mRNA and NRF2 phosphorylation (Fig. 5A—C). Furthermore, inhibition of PI3K significantly reduced XBP1 activation, BIP expression, and NRF2 phosphorylation after CXCL4 and TLR8 costimulation of human monocytes (Fig. 5D, E). These findings indicate that RIPK3 activates a stress response mediated by XBP1 and NRF2 in a PI3K-Akt–dependent manner in CXCL4 + TLR8–costimulated monocytes.
Fig. 5.
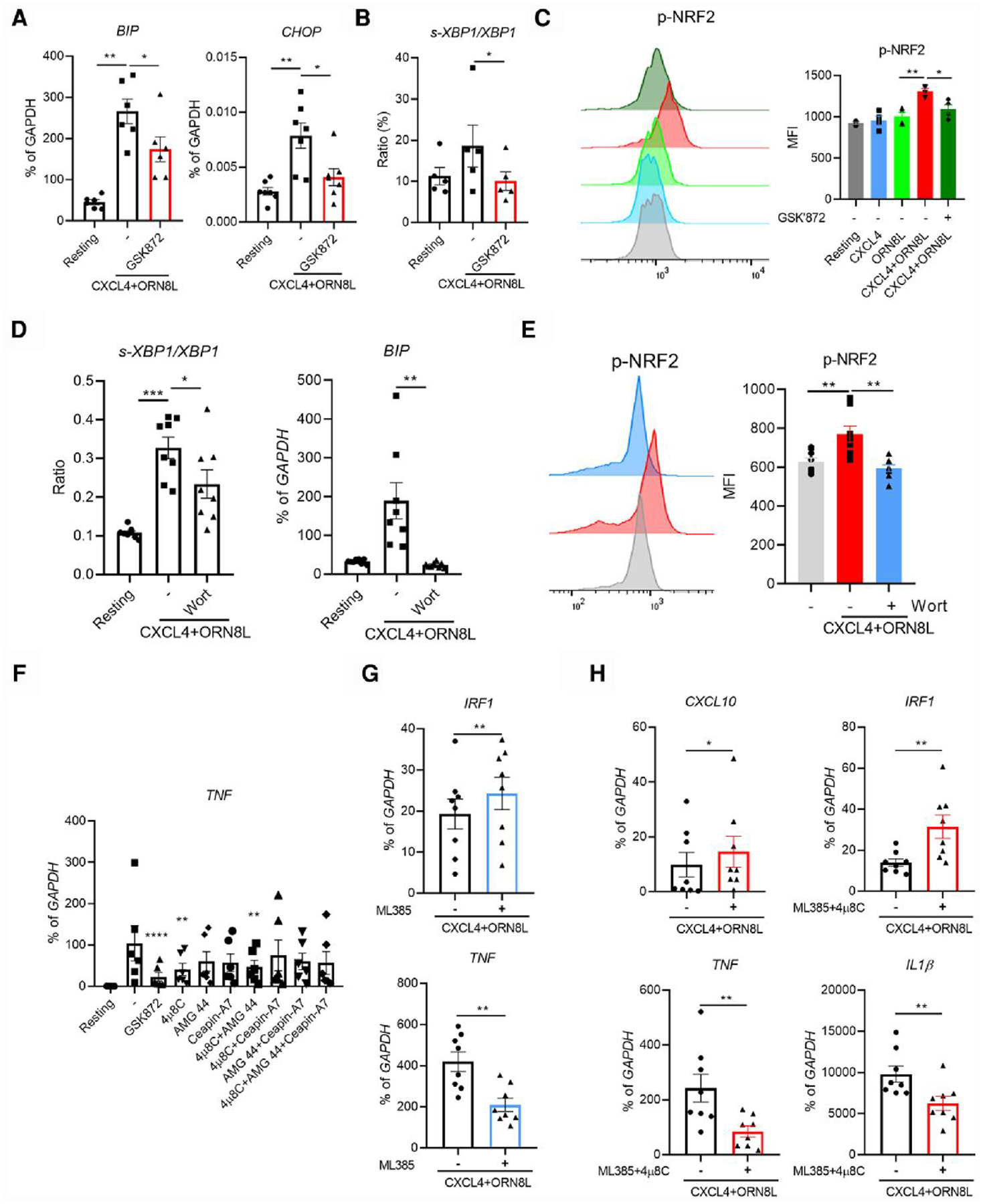
XBP1 and NRF2 activation contributes to RIPK3-AKT mediated regulation of gene expression. (A, B, and D) BIP, CHOP, and s-XBP1/XBP1 mRNA was measured by qPCR and normalized relative to GAPDH mRNA (n = 6 for panels A and B, n = 8 for panel D). (C, E) Representative flow plot and bar graph showing phospho-NRF2 in the indicated conditions. (F–H) Gene expression was evaluated by qPCR and normalized relative to GAPDH mRNA in the indicated conditions at 6 h (at least 6 healthy donors). Data are depicted as mean ± SEM. The representative flow plots are from at least 3 independent experiments (C and E). **P ≤ 0.01; *P ≤ 0.05 by 1-way analysis of variance with Tukey’s post hoc test for multiple comparisons (A, B, D, and F) or by Wilcoxon matched signed rank test (G and H).
Next, we tested the role of XBP1 and NRF2 in regulation of downstream ISG and inflammatory gene expression. Inhibition of XBP1 using its inhibitor 4μ8C, but not inhibition of other ER stress pathways, significantly reduced TNF expression without substantially affecting expression of other inflammatory genes or ISGs (Fig. 5F and Supplementary Fig. 5A). Inhibition of NRF2 using ML385 increased IRF1 and reduced TNF expression (Fig. 5G and Supplementary Fig. 5B). Similar to the effect of RIPK3 in regulation of the gene expression pattern, inhibition of both XBP1 and NRF2 together significantly increased ISG CXCL10 and IRF1 expression while reducing inflammatory TNF and IL1β expression (Fig. 5H). These data suggest that XBP1 and NRF2 contribute at least in part to RIPK3-mediated regulation of gene expression in CXCL4 and TLR8 costimulation of human monocytes.
3.6. RIPK3 and RIPK1 regulate NLRP3 inflammasome activation in CXCL4 and TLR8 costimulation
As RIPK3, together with RIPK1 and caspase-8, has been shown to contribute to noncanonical NLRP3 inflammasome activation (i.e. independent of second activation signals such as ATP and nigericin),19,25,52,53 we wished to test whether RIPK3 or RIPK1 contributed to CXCL4 + TLR8–induced NLRP3 inflammasome activation, which does not require a second activation signal such as ATP or nigericin.10 RIPK3 can be activated by recruitment to RHIM domain–containing signaling adaptors or in a cytoplasmic complex with RIPK1 and caspase-8, in which RIPK1 promotes RIPK3 activation and caspase-8 restrains this activation by proteolytic cleavage. Thus, inhibition of caspase-8 potentiates RIPK3 activation by RIPK1 in the RIPK1-RIPK3-caspase-8 complex.16
CXCL4 + TLR8–induced inflammasome activation, as assessed by cleavage of GSDMD, caspase-1, or IL-1β amounts in culture supernatants, was decreased upon inhibition of RIPK3 using GSK872 alone (in the absence of stabilization of the RIPK1-RIPK3-caspase-8 complex) (Fig. 6A, lane 6; Fig. 6B, column 7; Fig. 6C, lane 8). In contrast to RIPK3, inhibition of RIPK1 using necrostatin-1s alone had minimal effect on IL-1 production (Fig. 6B). However, when caspases were inhibited using pan caspase inhibitor zVAD to activate the RIPK1/RIPK3/caspase-8 axis, a significant inhibitory effect of necrostatin-1s on IL-1 production was observed (Fig. 6B, column 5). Overall, the results implicate RIPK3 in inflammasome activation in CXCL4 + TLR8–stimulated human monocytes; in this system, RIPK3 activity did not require inhibition of caspase-8, which is consistent with the results presented previously in Fig. 1. Our results also suggest that, in contrast to RIPK3, activation of RIPK1 by CXCL4 + TLR8 requires inhibition of caspase-8.
Fig. 6.
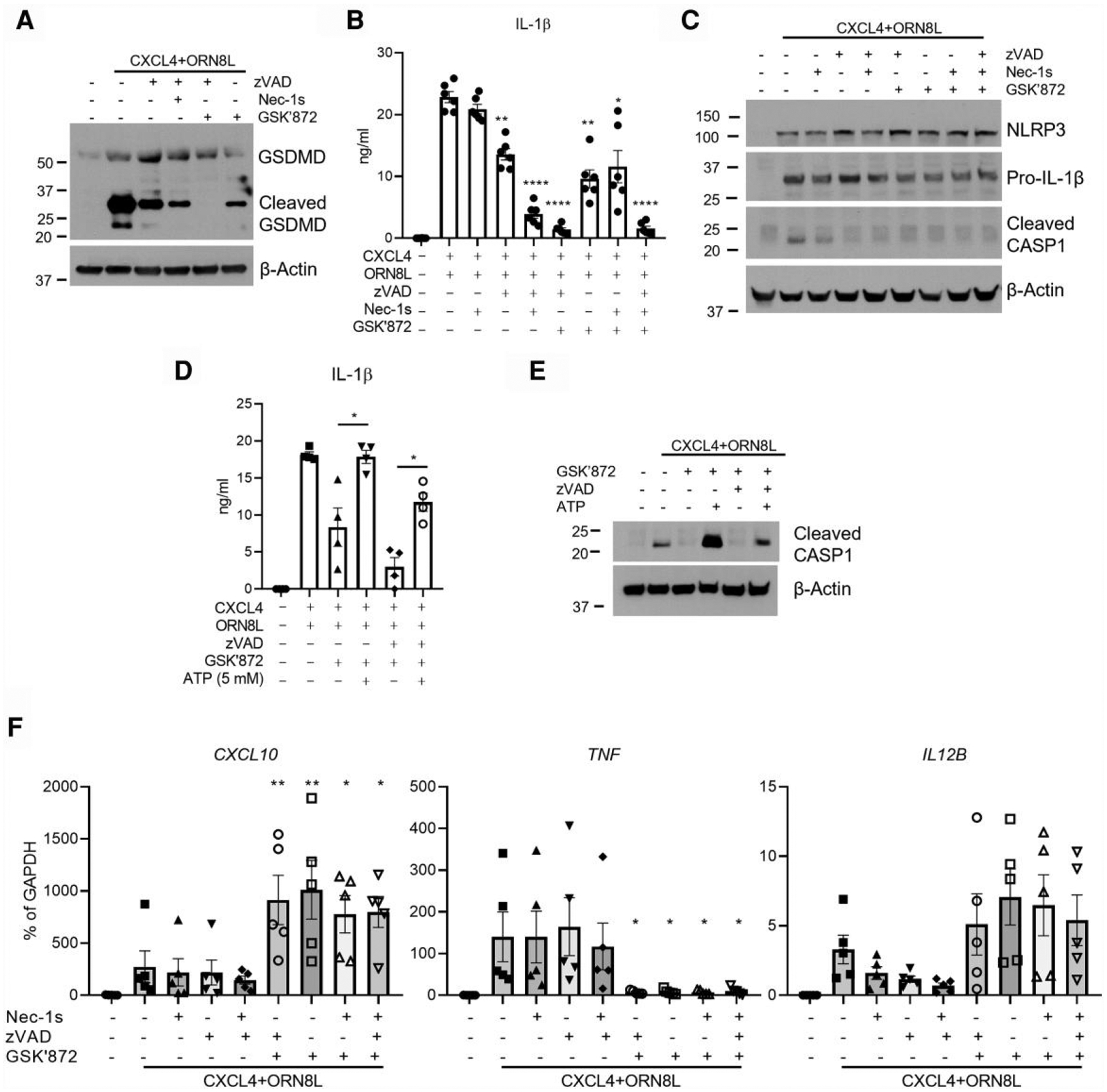
CASP8-RIPK1-RIPK3 axis regulates NLRP3 inflammasome. (A) Immunoblot of GSDMD with whole cell lysates in the indicated conditions at 6 h. (B, D) IL-1β protein concentrations in culture supernatants were measured by enzyme-linked immunosorbent assay after inhibition of CASP8, RIPK1, and/or RIPK3 (B) or after additionally adding ATP (5 mM) (D) (n = 4–6 healthy donors). (C) Immunoblot of NLRP3, IL-1β, and cleaved CASP1 using whole cell lysates in the indicated conditions at 6 h. The inhibitors targeting caspases, RIPK1, and RIPK3 are zVAD (50 μM), necrostatin-1 s (30 μM), and GSK872 (10 μM), respectively. (E) Immunoblot of cleaved CASP1 using whole cell lysates in the indicated conditions. (F) CXCL10, TNF, and IL12B mRNA was measured by qPCR and normalized relative to GAPDH mRNA (n = 5). Data are depicted as 1 of at least 3 representative blots (A, C, and E) and as mean ± SEM for the rest. ****P ≤ 0.0001; **P ≤ 0.01; *P ≤ 0.05 by 1-way analysis of variance with Tukey’s post hoc test for multiple comparisons (B, D, and F).
Interestingly, addition of exogenous ATP as a second signal increased IL-1β production and caspase-1 cleavage in CXCL4 + TLR8–stimulated monocytes under conditions of RIPK3 inhibition (Fig. 6D, E), which is consistent with the notion that RIPK3 signaling contributes to the second activation signal. In contrast to inflammasome activation, CXCL4 + TLR8–induced CXCL10 and TNF expression that was sensitive to RIPK3 inhibition was not affected by RIPK1 and/or caspase inhibition (Fig. 6F). Similar to CXCL10 and TNF expression, RIPK1 or caspase inhibition did not substantially or consistently affect the induction of NLRP3 and IL-1β precursor (Fig. 6C and data not shown), which occur at the level of gene activation; the role of RIPK1 in this system merits further study.
Activation of RIPK3 in a RIPK1/RIPK3/caspase-8 complex typically occurs downstream of TNF family receptors, requires inhibition of caspase-8 in in vitro cell culture systems, and activates MLKL, which not only induces necroptotic cell death, but also can contribute to inflammasome activation.16,54,55 We confirmed this model in our system using TNF stimulation of primary human monocytes and found a large increase in cell death after caspase-8 inhibition that was sensitive to MLKL inhibition using low-dose NSA (0.5 μM) (Supplementary Fig. 6A). In contrast to TNF, CXCL4 + TLR8–induced cell death occurred in the absence of caspase-8 inhibition and was not increased by caspase-8 inhibition (Supplementary Fig. 6B, C), which is consistent with an alternative activation pathway. However, under conditions of caspase-8 inhibition, cell death was decreased by inhibition of RIPK1 and RIPK3 (Supplementary Fig. 6B, C). Furthermore, when caspase-8 was inhibited, cell death was decreased in a dose-dependent way by NSA, which inhibits MLKL at a low concentration (0.5 μM) but begins to inhibit GSDMD, which induces pyroptotic cell death, at higher concentrations (5 μM) (Supplementary Fig. 6C). Overall, these findings are consistent with both pyroptosis and necroptosis contributing to CXCL4 + TLR8–induced cell death, at least under conditions of caspase inhibition, although this requires further investigation. Distinct types of cell death can occur concomitantly and can be difficult to distinguish, which is in accordance with emerging ideas about panoptosis.56
4. Discussion
CXCL4 and endosomal TLR signaling crosstalk augments TLR responses, which may play important roles in multiple inflammatory and autoimmune disorders.3,10,11,13,57–60 Recently, we identified signaling and epigenomic mechanisms by which CXCL4 boosts TLR8-mediated inflammatory gene induction in human monocytes.10 The major advances of the current study are to identify a role for RIPK3 in CXCL4 + TLR8–mediated responses, and to define activation of a RIPK3-Akt-stress pathway as a mechanism by which CXCL4 negatively regulates aspects of the TLR8 response, most notably ISG and IL12B induction. Moreover, RIPK3 plays a dichotomous role in regulating CXCL4 and TLR8 responses, as it also contributes to increased expression of inflammatory genes such as TNF and IL1B, and NLRP3 inflammasome activation. Negative regulation of IFN responses by RIPK3 targets STAT1 activation and involves PI3K-Akt activity that promotes XBP1-mediated ER stress and NRF2-mediated oxidative responses. The PI3K-Akt, XBP1, and NRF2 pathways also had a dichotomous role, as they promoted the expression of inflammatory genes while attenuating the IFN response. These results implicate a RIPK3-mediated signaling axis in regulating the balance between inflammatory gene, ISG, and inflammasome activation, which is important for mounting effective host defense while limiting concomitant inflammatory pathology.
Potentiation of TLR8-induced inflammatory responses by CXCL4 has been attributed to a chaperone function that delivers increased amounts of NA ligands to endolysosomal locations to increase TLR8 activation, through synergistic activation of TBK1 signaling that drives inflammatory outcomes via IRF5-mediated gene induction and inflammasome-mediated IL-1β production, and by genome-wide regulation of chromatin accessibility,10 but signaling pathways in regulation of CXCL4 and TLR8–induced IFN response and other inflammatory gene induction such as TNF and IL1β as well as NLRP3 inflammasome activation have not been very well defined. RIPK3 plays critical role in a programed cell death, namely necroptosis, which contributes to effective host defense against intracellular pathogen infections while uncontrolled necroptosis also causes tissue damage and host injury. In addition to driving necroptosis, RIPK3 is upregulated in many conditions,27,61 and mounting evidence shows that RIPK3 plays roles in the activation signal–independent NLRP3 inflammasome activation, inflammatory diseases, and tumorigenesis.21,25,32 Instead of increasing RIPK3 expression, CXCL4 and TLR8 signaling induced RIPK3 activation in human primary monocytes, which can occur independently of RIPK1. In contrast to synergistic activation of TBK1 and IRF5 by CXCL4 + TLR8 costimulation,10 synergistic activation of P-RIPK3 by costimulation relative to activation by single agent treatments was not observed. This suggests that RIPK3 activity plays more of a regulatory role, being required for full activation of inflammatory genes and the inflammasome, while modulating and attenuating expression of other genes such as ISGs. Further, we show that the activation of RIPK3 by CXCL4 + TLR8 costimulation relies on HSP90 and autophosphorylation. It has been reported that HSP90 activates RIPK3 through an interaction that stabilizes RIPK338,39; however, at the 6-h time point where we conducted the experiments, blockade of HSP90 activity did not affect RIPK3 protein amounts. Thus, we speculate that HSP90 may act as a platform for RIPK3 conjugation and autophosphorylation. Additionally, the specific kinase inhibitor of RIPK3, GSK872, impeded CXCL4 and TLR8 signaling-induced RIPK3 phosphorylation, suggesting that the activation of RIPK3 we observed could be caused by RIPK3 autophosphorylation. We speculate that RIPK3 activation contributes to CXCL4 and TLR8–mediated synergistic effects. Inhibiting the kinase function of RIPK3 skewed the CXCL4 and TLR8–induced gene expression pattern: reducing the expression of inflammatory gene TNF while increasing ISG CXCL10 and T cell–activating cytokine IL-12 subunit IL12B expression. In line with a previous study that RIPK3 activates NF-κB and increases its target inflammatory gene expression,26 ectopic expression of RIPK3 increased CXCL4 + TLR8–induced NF-κB activation and its target inflammatory genes TNF, IL6, and IL1B but not IL12B. This suggests that there might exist a RIPK3-mediated suppressive pathway to balance NF-κB activation to attenuate IL12B expression in RIPK3 overexpressing cells. The role of RIPK3 in regulation of STAT1 activation and ISG expression in CXCL4 + TLR8 costimulation is also recapitulated in RIPK3-overexpressing cells. Moreover, administration of exogenous IFN-β only partially rescued STAT1 activation and ISG expression in CXCL4 + TLR8–stimulated cells. However, in the cells that did not experience CXCL4 + TLR8 costimulation, overexpression RIPK3 cannot suppress exogenous IFN-β response, suggesting that CXCL4 + TLR8 costimulation may induce a cofactor(s) that cooperates with RIPK3 to regulate IFN-mediated STAT activation, and that RIPK3 acquires the ability to regulate IFN response only in certain situations, which will be interesting to explore in future work.
Crosstalk between the RIPK and JAK-STAT pathways has been previously described, e.g. Yu et al.,62 which shows that a RIPK1-RIPK3 complex enhances IFN-γ-induced STAT1 activation and downstream gene expression in intestinal epithelial cells. This enhancement of IFN-γ signaling occurred via a proximal signaling mechanism, namely interaction of RIPK1-RIPK3 with JAK1, which increased STAT1 activation. In contrast, in our system using human monocytes, RIPK3 suppresses autocrine IFN-I responses by downstream activation of the PI3K-Akt and NRF2 pathways. Interaction between these 2 signaling pathways is likely to be complex and deserves future study.
One strength of our study is the use of freshly isolated primary human monocytes ex vivo, which closely reflects physiological regulation of primary cells. We previously found that as monocytes differentiate into macrophages during culture ex vivo, regulation of inflammasome activation changes as cells lose the ability to produce CXCL4 + TLR8–induced mature IL-1β without addition of a second signal such as ATP.10 Interestingly, the regulation of CXCL4 + TLR8 responses by RIPK3 also wanes during culture. This leads to a technical limitation of our system for studying RIPK3 function, as it precludes the ability to use genetic approaches such as short interfering RNA to study the RIPK3 functions described in this study.
The PI3K-Akt pathway regulates macrophage survival, migration, and proliferation and also participates in cell metabolism and regulation of inflammatory responses to TLR signals.63,64 The PI3K-Akt pathway restricts macrophage overreaction to TLR signals,64 but it can enhance IFN and ISG expression for host defense against intracellular pathogen infection.65,66 However, in the current study, instead of suppressing CXCL4 + TLR8–induced inflammatory gene expression, surprisingly, the PI3K-Akt pathway plays a more complex role in a dichotomous way, similar to RIPK3, to regulate TNF, IL12B, and ISG expression. Consistent with previous studies that RIPK3 can activate the Akt pathway in inflammatory conditions,27 RIPK3 is also important for PI3K-Akt pathway activity in CXCL4 and TLR8 costimulation in human monocytes. PI3K-Akt activation induced by TLR467 regulates inflammatory responses to overcome excessive inflammation and tissue damage.43,64 However, the unexpected activation of PI3K-Akt pathway by RIPK3 in the context of costimulation of TLR8 signaling with CXCL4 might execute different functions such as balancing IFN response while contributing to inflammatory gene expression and promoting tissue fibrosis.27
Our transcriptomic analysis and functional experiments indicate that RIPK3 activates ER stress responses and NRF2 oxidative response after CXCL4 and TLR8 costimulation of human monocytes. Stress pathway activation may reflect hyperactivation of monocytes during CXCL4 and TLR8 costimulation, as adequate ER stress responses and NRF2 oxidative response are protective mechanisms to maintain cellular homeostasis and cell survival.68,69 However, uncontrolled or excessive activation of ER stress and NRF2 oxidative responses also contribute to inflammation, contribute to cell death, and suppress antitumoral innate and adaptive immunity.42,68,70 In the current study, XBP1-mediated ER stress response coordinated with NRF2 oxidative response to contribute to RIPK3-mediated increases in inflammatory gene expression, while suppressing IFN response. IFN response is not only important for antipathogen response, but also critical for antitumor immunity through activating tumor microenvironment immune cells.71 Thus, we speculate that RIPK3-activated XBP1 and NRF2 might play pleiotropic roles in inflammatory diseases and host defense. How the activation of ER stress response and NRF2 is initiated in disease context (such as inflammatory diseases, tumorigenesis, and infections) is still not well defined. The PI3K-Akt pathway has been shown play critical roles in activating ER stress and NRF2 in multiple conditions, such as inflammation and cell survival,44–46,51 which is recapitulated in CXCL4 and TLR8 signaling crosstalk in human primary monocytes in this study. The study reveals that RIPK3 can initiate signaling by a PI3K-Akt-XBP1/NRF2 axis to modulate cellular responses.
In general, NLRP3 inflammasome activation requires 2 signals: a priming signal to upregulate the expression of inflammasome components NLRP3, caspase-1, and pro-IL-1β, and a second signal to promote the inflammasome complex assembly and activation of caspase-1, thereby cleaving pro-IL-1β to mature IL-1β and GSDMD to active N-terminal GSDMD, which induces plasma membrane pores that allow IL-1β secretion and can lead to pyroptotic cell death.25,52,72,73 In line with many studies,22,25,52,73 we demonstrate that RIPK3 and the caspase-8–RIPK1/3 axis contribute to an alternative NLRP3 inflammasome activation without affecting inflammasome component expression and the canonical second step of NLRP3 inflammasome activation by exogenous ATP. RIPK3 seems to play a key role in the NLRP3 inflammasome activation because inhibition of RIPK3 significantly affected NLRP3 inflammasome activation, while the involvement of RIPK1 only occurred when caspases were inhibited, which is in accordance with the established model that caspase-8 regulates RIPK1 kinase activation.16 Our study suggests that a distinct pathway from the RIPK1/RIPK3/Caspase-8 complex activates RIPK3, and RIPK3 plays a key role in CXCL4 + TLR8–induced NLRP3 inflammasome activation and balance of IFN response and inflammatory gene expression.23,27,74,75
Of the 3 RIPK3-regulated downstream outcomes of CXCL4 + TLR8 signaling assessed in the study—gene expression, inflammasome activation, and cell death—gene expression was entirely unaffected by RIPK1 inhibition, and inflammasome activation and cell death were modestly or minimally affected by RIPK1 inhibition under standard culture conditions, in the absence of a caspase-8 inhibitor. This argues for a predominant RIPK3 activation mechanism by CXCL4 + TLR8 that occurs independently of RIPK1 and the RIPK1/RIPK3/caspase-8 complex (i.e. activated by DD-containing receptors). Activation of RIPK3 independently of RIPK1 typically occurs via recruitment to RHIM domain–containing signaling molecules such as TRIF and ZBP116; this activation mechanism can also involve interaction with HSP90 and RIPK3 autophosphorylation.37–39 Our study supports a role for HSP90 in RIPK3 activation after CXCL4 + TLR8 costimulation, and future work will address interaction of RIPK3 with RHIM-containing or other signaling molecules activated by receptors engaged by CXCL4-ssRNA complexes.10 In cell culture systems, activation of RIPK3 by the RIPK1/RIPK3/caspase-8 complex typically occurs downstream of DD-containing receptors of the TNFR family and requires pharmacological inhibition of caspase-8, which prevents inactivating cleavage of these kinases. Interestingly, even when caspases were inhibited using the standard approach of adding zVAD in our system, RIPK1 played no role in regulation of gene expression; however, these conditions revealed a role for RIPK1 in inflammasome activation and cell death. It is possible that CXCL4 + TLR8 activates this caspase-8 and RIPK1–mediated pathway indirectly via induction of DD-containing receptors and or ligands.
In summary, RIPK3 modulates the CXCL4 and TLR8 response in human monocytes by suppressing STAT1 activation and activating NLRP3 and a PI3K-Akt-XBP1/NRF2 pathway that augments expression of select inflammatory genes while suppressing the IFN response (Fig. 7). These findings provide potential mechanisms by which RIPK3 calibrates TLR responses in inflammatory diseases and tumorigenesis, and suggest that RIPK3-PI3K-Akt-XBP1/NRF2 axis–mediated stress responses can be targeted for suppression of inflammation while preserving the IFN response for host defense against pathogen infections.
Fig. 7.
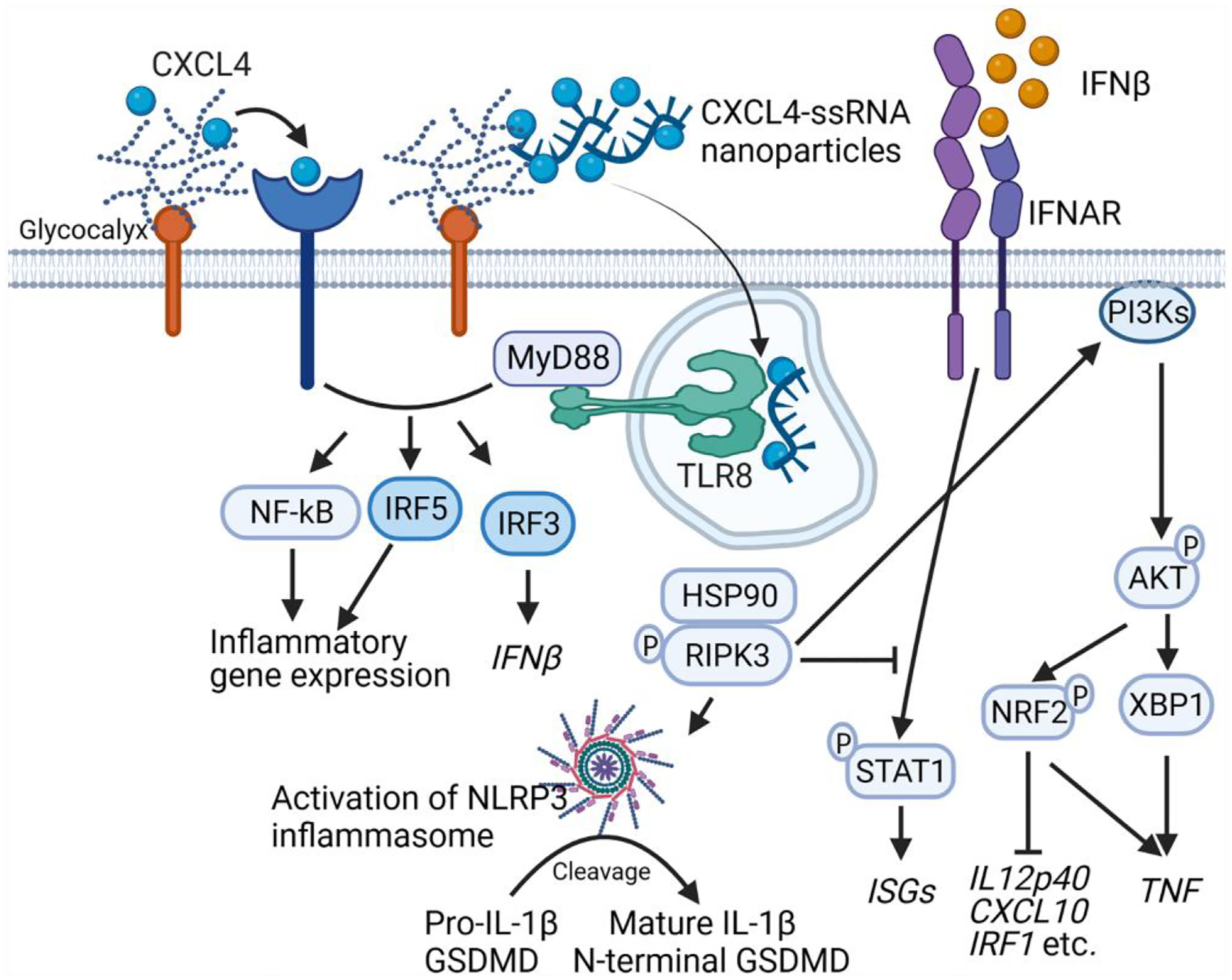
RIPK3 regulates the profile and balance of the CXCL4-costimulated TLR8 response. RIPK3 provides signal 2 for inflammasome activation and regulates stress pathways to amplify induction of inflammatory genes while suppressing STAT1 activation and IFN responses. The figure was created with BioRender.com.
Supplementary Material
Acknowledgments
The authors thank the Weill Cornell Medicine Genomic Resources Core Facility for next-generation sequencing, the Weill Cornell Medicine–HSS Flow Cytometry Core Facility for flow cytometry support, and David Oliver (HSS Genomics Center) for advice and discussions.
Funding
This work was supported by grants from the National Institutes of Health (L.B.I), from the National Institutes of Health 1R01AI132447 (F.J.B.), the Scleroderma Research Foundation (F.J.B.), and the Scleroderma Foundation (F.J.B.). The David Z. Rosensweig Genomics Center at HSS is supported by the Tow Foundation.
Footnotes
Supplementary material
Supplementary materials are available at Journal of Leukocyte Biology online.
Conflict of interest statement. F.J.B. is a founder of IpiNovyx, a startup biotechnology company. The other authors declare no relevant conflicts of interest.
Data availability
The datasets that support the findings of this study and were generated by the authors as part of this study have been deposited in the Gene Expression Omnibus database with the accession code GEO: GSE201909. Otherwise, the data are either contained within the manuscript or available from the authors on request.
Reference
- 1.Lind NA, Rael VE, Pestal K, Liu B, Borton GM. Regulation of the nucleic acid-sensing toll-like receptors. Nat Rev Immunol. 2021:22(4): 224–235. 10.1038/s41577-021-00577-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Laurent P, Yang C, Rendeiro AF, Nilsson-Payant BE, Carrau L, Chandar V, Bram Y, tenOever BR, Elemento O, Ivashkiv LB, et al. Sensing of SARS-CoV-2 by pDCs and their subsequent production of IFN-I contribute to macrophage-induced cytokine storm during COVID-19. Sci Immunol. 2022:7(75):eadd4906. 10.1126/sciimmunol.add4906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ah Kioon MD, Tripodo C, Fernandez D, Kirou KA, Spiera RF, Crow MK, Gordon JK, Barrat FJ. Plasmacytoid dendritic cells promote systemic sclerosis with a key role for TLR8. Sci Transl Med. 2018:10(423):eaam8458. 10.1126/scitranslmed.aam8458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barrat FJ, Elkon KB, A K. Fitzgerald: importance of nucleic acid recognition in inflammation and autoimmunity. Annu Rev Med. 2016:67(1):323–336. 10.1146/annurev-med-052814-023338 [DOI] [PubMed] [Google Scholar]
- 5.Andersson U, Erlandsson-Harris H. HMGB1 is a potent trigger of arthritis. J Intern Med. 2004:255(3):344–350. 10.1111/j.1365-2796.2003.01303.x [DOI] [PubMed] [Google Scholar]
- 6.Mayer-Barber KD, Andrade BB, Oland SD, Amaral EP, Barber DL, Gonzales J, Derrick SC, Shi R, Kumar NP, Wei W, et al. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature. 2014:511(7507):99–103. 10.1038/nature13489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mayer-Barber KD, Andrade BB, Barber DL, Hieny S, Feng CG, Caspar P, Oland S, Gordon S, Sher A. Innate and adaptive interferons suppress IL-1α and IL-1β production by distinct pulmonary myeloid subsets during Mycobacterium tuberculosis infection. Immunity. 2011:35(6):1023–1034. 10.1016/j.immuni.2011.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Novikov A, Cardone M, Thompson R, Shenderov K, Kirschman KD, Mayer-Barber KD, Myers TG, Rabin RL, Trinchieri G, Sher A, et al. Feng: mycobacterium tuberculosis triggers host type I IFN signaling to regulate IL-1β production in human macrophages. J Immunol. 2011:187(5):2540–2547. 10.4049/jimmunol.1100926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Förster I, Farlik M, Decker T, Du Pasquier RA, Romero P, et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity. 2011:34(2):213–223. 10.1016/j.immuni.2011.02.006 [DOI] [PubMed] [Google Scholar]
- 10.Yang C, Bachu M, Du Y, Brauner C, Yuan R, Ah Kioon MD, Chesi G, Barrat FJ, B L. Ivashkiv: CXCL4 synergizes with TLR8 for TBK1-IRF5 activation, epigenomic remodeling and inflammatory response in human monocytes. Nat Commun. 2022:13(1):3426. 10.1038/s41467-022-31132-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Du Y, Ah Kioon MD, Laurent P, Chaudhary V, Pierides M, Yang C, Oliver D, Ivashkiv LB, Barrat FJ. Chemokines form nanoparticles with DNA and can superinduce TLR-driven immune inflammation. J Exp Med. 2022:219(7):e20212142. 10.1084/jem.20212142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Silva-Cardoso SC, Tao W, Angiolilli C, Lopes AP, Bekker CPJ, Devaprasad A, Giovannone B, van Laar J, Cossu M, Marut W, et al. CXCL4 Links inflammation and fibrosis by reprogramming monocyte-derived dendritic cells in vitro. Front Immunol. 2020:11:2149. 10.3389/fimmu.2020.02149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gleitz HFE, Dugourd AJF, Leimkuhler NB, Snoeren IAM, Fuchs SNR, Menzel S, Ziegler S, Kroger N, Triviai I, Busche G, et al. Schneider: increased CXCL4 expression in hematopoietic cells links inflammation and progression of bone marrow fibrosis in MPN. Blood. 2020:136(18):2051–2064. 10.1182/blood.2019004095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Domschke G, A C. Gleissner: CXCL4-induced macrophages in human atherosclerosis. Cytokine. 2017:122:154141. 10.1016/j.cyto.2017.08.021 [DOI] [PubMed] [Google Scholar]
- 15.Lan T, Kandimalla ER, Yu D, Bhagat L, Li Y, Wang D, Zhu F, Tang JX, Putta MR, Cong Y, et al. Stabilized immune modulatory RNA compounds as agonists of toll-like receptors 7 and 8. Proc Natl Acad Sci U S A. 2007:104(34):13750–13755. 10.1073/pnas.0706059104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He S, Wang X. RIP Kinases as modulators of inflammation and immunity. Nat Immunol. 2018:19(9):912–922. 10.1038/s41590-018-0188-x [DOI] [PubMed] [Google Scholar]
- 17.Declercq W, Vanden Berghe T, Vandenabeele P. RIP Kinases at the crossroads of cell death and survival. Cell. 2009:138(2): 229–232. 10.1016/j.cell.2009.07.006 [DOI] [PubMed] [Google Scholar]
- 18.Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012:148(1): 213–227. 10.1016/j.cell.2011.11.031 [DOI] [PubMed] [Google Scholar]
- 19.Kang TB, Yang SH, Toth B, Kovalenko A, Wallach D. Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. Immunity. 2013:38(1):27–40. 10.1016/j.immuni.2012.09.015 [DOI] [PubMed] [Google Scholar]
- 20.Wen W, Chen J, Zhou Y, Li G, Zhang Y. Loss of Ripk3 attenuated neutrophil accumulation in a lipopolysaccharide-induced zebrafish inflammatory model. Cell Death Discov. 2022:8(1):88. 10.1038/s41420-022-00891-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Afonso MB, Rodrigues PM, Mateus-Pinheiro M, Simão AL, Gaspar MM, Majdi A, Arretxe E, Alonso C, Santos-Laso A, Jimenez-Agüero R, et al. Rodrigues: RIPK3 acts as a lipid metabolism regulator contributing to inflammation and carcinogenesis in non-alcoholic fatty liver disease. Gut. 2021:70(12):2359–2372. 10.1136/gutjnl-2020-321767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lawlor KE, Khan N, Mildenhall A, Gerlic M, Croker BA, D’Cruz AA, Hall C, Kaur Spall S, Anderton H, Masters SL, et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat Commun. 2015:6(1):6282. 10.1038/ncomms7282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moriwaki K, Balaji S, McQuade T, Malhotra N, Kang J, K F. Chan: the necroptosis adaptor RIPK3 promotes injury-induced cytokine expression and tissue repair. Immunity. 2014:41(4):567–578. 10.1016/j.immuni.2014.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Newton K, Dugger DL, Maltzman A, Greve JM, Hedehus M, Martin-McNulty B, Carano RA, Cao TC, van Bruggen N, Bernstein L, et al. RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ. 2016:23(9):1565–1576. 10.1038/cdd.2016.46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mangan MSJ, Olhava EJ, Roush WR, Seidel HM, Glick GD, Latz E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discov. 2018:17(8):588–606. 10.1038/nrd.2018.97 [DOI] [PubMed] [Google Scholar]
- 26.Najjar M, Saleh D, Zelic M, Nogusa S, Shah S, Tai A, Finger JN, Polykratis A, Gough PJ, Bertin J, et al. RIPK1 And RIPK3 kinases promote cell-death-independent inflammation by toll-like receptor 4. Immunity. 2016:45(1):46–59. 10.1016/j.immuni.2016.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Imamura M, Moon JS, Chung KP, Nakahira K, Muthukumar T, Shingarev R, Ryter SW, Choi AM, Choi ME. RIPK3 Promotes kidney fibrosis via AKT-dependent ATP citrate lyase. JCI Insight. 2018:3(3):e94979. 10.1172/jci.insight.94979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sefik E, Qu R, Junqueira C, Kaffe E, Mirza H, Zhao J, Brewer JR, Han A, Steach HR, Israelow B, et al. Inflammasome activation in infected macrophages drives COVID-19 pathology. Nature. 2022:606(7914):585–593. 10.1038/s41586-022-04802-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bian P, Ye C, Zheng X, Luo C, Yang J, Li M, Wang Y, Yang J, Zhou Y, Zhang F, et al. RIPK3 Promotes JEV replication in neurons via downregulation of IFI44L. Front Microbiol. 2020:11:368. 10.3389/fmicb.2020.00368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feng Y, Daley-Bauer LP, Mocarski ES. Caspase-8-dependent control of NK- and T cell responses during cytomegalovirus infection. Med Microbiol Immunol. 2019:208(3):555–571. 10.1007/s00430-019-00616-7 [DOI] [PubMed] [Google Scholar]
- 31.Liu S, Joshi K, Denning MF, Zhang J. RIPK3 Signaling and its role in the pathogenesis of cancers. Cell Mol Life Sci. 2021:78(23): 7199–7217. 10.1007/s00018-021-03947-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jayakumar A, M AL. Bothwell: rIPK3-induced inflammation by I-MDSCs promotes intestinal tumors. Cancer Res. 2019:79(7): 1587–1599. 10.1158/0008-5472.Can-18-2153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wegner KW, Saleh D, Degterev A. Complex pathologic roles of RIPK1 and RIPK3: moving beyond necroptosis. Trends Pharmacol Sci. 2017:38(3):202–225. 10.1016/j.tips.2016.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li X, Zhang M, Huang X, Liang W, Li G, Lu X, Li Y, Pan H, Shi L, Zhu H, et al. Ubiquitination of RIPK1 regulates its activation mediated by TNFR1 and TLRs signaling in distinct manners. Nat Commun. 2020:11(1):6364. 10.1038/s41467-020-19935-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Najafov A, Chen H, Yuan J. Necroptosis and cancer. Trends Cancer. 2017:3(4):294–301. 10.1016/j.trecan.2017.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qiao Y, Giannopoulou EG, Chan CH, Park SH, Gong S, Chen J, Hu X, Elemento O, B L. Ivashkiv: synergistic activation of inflammatory cytokine genes by interferon-gamma-induced chromatin remodeling and toll-like receptor signaling. Immunity. 2013:39(3): 454–469. 10.1016/j.immuni.2013.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li D, Chen J, Guo J, Li L, Cai G, Chen S, Huang J, Yang H, Zhuang Y, Wang F, et al. A phosphorylation of RIPK3 kinase initiates an intracellular apoptotic pathway that promotes prostaglandin2α-induced corpus luteum regression. Elife. 2021:10:e67409. 10.7554/elife.67409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang CK, D S. He: heat shock protein 90 regulates necroptosis by modulating multiple signaling effectors. Cell Death Dis. 2016:7(3): e2126. 10.1038/cddis.2016.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li D, Xu T, Cao Y, Wang H, Li L, Chen S, Wang X, Shen Z. A cytosolic heat shock protein 90 and cochaperone CDC37 complex is required for RIP3 activation during necroptosis. Proc Natl Acad Sci U S A. 2015:112(16):5017–5022. 10.1073/pnas.1505244112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ivashkiv LB, T L. Donlin: regulation of type I interferon responses. Nat Rev Immunol. 2014:14(1):36–49. 10.1038/nri3581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rathkey JK, Zhao J, Liu Z, Chen Y, Yang J, Kondolf HC, Benson BL, Chirieleison SM, Huang AY, Dubyak GR, et al. Abbott: chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci Immunol. 2018:3(26):eaat2738. 10.1126/sciimmunol.aat2738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Song M, Cubillos-Ruiz JR. Endoplasmic Reticulum stress responses in intratumoral immune cells: implications for cancer immunotherapy. Trends Immunol. 2019:40(2):128–141. 10.1016/j.it.2018.12.001 [DOI] [PubMed] [Google Scholar]
- 43.Pourrajab F, Yazdi MB, Zarch MB, Zarch MB, Hekmatimoghaddam S. Cross talk of the first-line defense TLRs with PI3K/Akt pathway, in preconditioning therapeutic approach. Mol Cell Ther. 2015:3(1):4–4. 10.1186/s40591-015-0041-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Y, Zhang Y, Yi P, Dong W, Nalin AP, Zhang J, Zhu Z, Chen L, Benson DM, Mundy-Bosse BL, et al. The IL-15-AKT-XBP1s signaling pathway contributes to effector functions and survival in human NK cells. Nat Immunol. 2019:20(1):10–17. 10.1038/s41590-018-0265-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liao S, Wu J, Liu R, Wang S, Luo J, Yang Y, Qin Y, Li T, Zheng X, Song J, et al. A novel compound DBZ ameliorates neuroinflammation in LPS-stimulated microglia and ischemic stroke rats: role of Akt(Ser473)/GSK3beta(Ser9)-mediated Nrf2 activation. Redox Biol. 2020:36:101644. 10.1016/j.redox.2020.101644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang L, Chen Y, Sternberg P, Cai J. Essential roles of the PI3 kinase/Akt pathway in regulating Nrf2-dependent antioxidant functions in the RPE. Invest Ophthalmol Vis Sci. 2008:49(4):1671–1678. 10.1167/iovs.07-1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Song Q, Han CC, Xiong XP, He F, Gan W, Wei SH, Liu HH, Li L, Xu HY. PI3K-Akt-mTOR signal inhibition affects expression of genes related to endoplasmic reticulum stress. Genet Mol Res. 2016:15(3):gmr7868. 10.4238/gmr.15037868 [DOI] [PubMed] [Google Scholar]
- 48.Troutman TD, Bazan JF, Pasare C. Toll-like receptors, signaling adapters and regulation of the proinflammatory response by PI3K. Cell Cycle. 2012:11(19):3559–3567. 10.4161/cc.21572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Appenzeller-Herzog C, N M. Hall: bidirectional crosstalk between endoplasmic reticulum stress and mTOR signaling. Trends Cell Biol. 2012:22(5):274–282. 10.1016/j.tcb.2012.02.006 [DOI] [PubMed] [Google Scholar]
- 50.He F, Antonucci L, Yamachika S, Zhang Z, Taniguchi K, Umemura A, Hatzivassiliou G, Roose-Girma M, Reina-Campos M, Duran A, et al. NRF2 activates growth factor genes and downstream AKT signaling to induce mouse and human hepatomegaly. J Hepatol. 2020:72(6):1182–1195. 10.1016/j.jhep.2020.01.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang Y, Tian Q, Hao Y, Yao W, Lu J, Chen C, Chen X, Lin Y, Huang Q, Xu L, et al. The kinase complex mTORC2 promotes the longevity of virus-specific memory CD4(+) T cells by preventing ferroptosis. Nat Immunol. 2021:23(2):303–317. 10.1038/s41590-021-01090-1 [DOI] [PubMed] [Google Scholar]
- 52.Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. 2019;19(8):477–489. 10.1038/s41577-019-0165-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Speir M, Lawlor KE. RIP-roaring inflammation: RIPK1 and RIPK3 driven NLRP3 inflammasome activation and autoinflammatory disease. Semin Cell Dev Biol. 2021:109:114–124. 10.1016/j.semcdb.2020.07.011 [DOI] [PubMed] [Google Scholar]
- 54.Conos SA, Chen KW, De Nardo D, Hara H, Whitehead L, Nunez G, Masters SL, Murphy JM, Schroder K, Vaux DL, et al. Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc Natl Acad Sci U S A. 2017:114(6):E961–E969. 10.1073/pnas.1613305114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gutierrez KD, Davis MA, Daniels BP, Olsen TM, Ralli-Jain P, Tait SW, Gale M, Oberst A. MLKL activation triggers NLRP3-mediated processing and release of IL-1beta independently of gasdermin-D. J Immunol. 2017:198(5):2156–2164. 10.4049/jimmunol.1601757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Samir P, Malireddi RKS, Kanneganti T-D. The PANoptosome: a deadly protein Complex driving pyroptosis, apoptosis, and necroptosis (PANoptosis). Front Cell Infect Microbiol. 2020:10:238. 10.3389/fcimb.2020.00238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Aluri J, Bach A, Kaviany S, Chiquetto Paracatu L, Kitcharoensakkul M, Walkiewicz MA, Putnam CD, Shinawi M, Saucier N, Rizzi EM, et al. Cooper: immunodeficiency and bone marrow failure with mosaic and germline TLR8 gain of function. Blood. 2021:137(18):2450–2462. 10.1182/blood.2020009620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yeo L, Adlard N, Biehl M, Juarez M, Smallie T, Snow M, Buckley CD, Raza K, Filer A, Scheel-Toellner D. Expression of chemokines CXCL4 and CXCL7 by synovial macrophages defines an early stage of rheumatoid arthritis. Ann Rheum Dis. 2016:75(4): 763–771. 10.1136/annrheumdis-2014-206921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mok CC, Soliman S, Ho LY, Mohamed FA, Mohamed FI, Mohan C. Urinary angiostatin, CXCL4 and VCAM-1 as biomarkers of lupus nephritis. Arthritis Res Ther. 2018:20(1):6. 10.1186/s13075-017-1498-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lande R, Lee EY, Palazzo R, Marinari B, Pietraforte I, Santos GS, Mattenberger Y, Spadaro F, Stefanantoni K, Iannace N, et al. CXCL4 Assembles DNA into liquid crystalline complexes to amplify TLR9-mediated interferon-alpha production in systemic sclerosis. Nat Commun. 2019:10(1):1731. 10.1038/s41467-019-09683-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gautheron J, Vucur M, Schneider AT, Severi I, Roderburg C, Roy S, Bartneck M, Schrammen P, Diaz MB, Ehling J, et al. The necroptosis-inducing kinase RIPK3 dampens adipose tissue inflammation and glucose intolerance. Nat Commun. 2016:7(1): 11869. 10.1038/ncomms11869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yu X, Ma H, Li B, Ji Y, Du Y, Liu S, Li Z, Hao Y, Tian S, Zhao C, et al. A novel RIPK1 inhibitor reduces GVHD in mice via a nonimmunosuppressive mechanism that restores intestinal homeostasis. Blood. 2023:141(9):1070–1086. 10.1182/blood.2022017262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Song G, Ouyang G, Bao S. The activation of Akt/PKB signaling pathway and cell survival. J Cell Mol Med. 2005:9(1):59–71. 10.1111/j.1582-4934.2005.tb00337.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vergadi E, Ieronymaki E, Lyroni K, Vaporidi K, Tsatsanis C. Akt signaling pathway in macrophage activation and M1/M2 polarization. J Immunol. 2017:198(3):1006–1014. 10.4049/jimmunol.1601515 [DOI] [PubMed] [Google Scholar]
- 65.Tian J, Zhang X, Wu H, Liu C, Li Z, Hu X, Su S, Wang L-F, Qu L. Blocking the PI3K/AKT pathway enhances mammalian reovirus replication by repressing IFN-stimulated genes. Front Microbiol. 2015:6:886. 10.3389/fmicb.2015.00886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schabbauer G, Luyendyk J, Crozat K, Jiang Z, Mackman N, Bahram S, Georgel P : tLR4/CD14-mediated PI3K activation is an essential component of interferon-dependent VSV resistance in macrophages. Mol Immunol. 2008:45(10):2790–2796. 10.1016/j.molimm.2008.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.López-Peláez M, Soria-Castro I, Boscá L, Fernández M, Alemany S. Cot/tpl2 activity is required for TLR-induced activation of the Akt p70 S6k pathway in macrophages: implications for NO synthase 2 expression. Eur J Immunol. 2011:41(6):1733–1741. 10.1002/eji.201041101 [DOI] [PubMed] [Google Scholar]
- 68.Chen Y, Brandizzi F. IRE1: ER stress sensor and cell fate executor. Trends Cell Biol. 2013:23(11):547–555. 10.1016/j.tcb.2013.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang P, Geng J, Gao J, Zhao H, Li J, Shi Y, Yang B, Xiao C, Linghu Y, Sun X, et al. Macrophage achieves self-protection against oxidative stress-induced ageing through the Mst-Nrf2 axis. Nat Commun. 2019:10(1):755. 10.1038/s41467-019-08680-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Muri J, Wolleb H, Broz P, Carreira EM, Kopf M. Electrophilic nrf2 activators and itaconate inhibit inflammation at low dose and promote IL-1β production and inflammatory apoptosis at high dose. Redox Biol. 2020:36:101647. 10.1016/j.redox.2020.101647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G. Type I interferons in anticancer immunity. Nat Rev Immunol. 2015:15(7): 405–414. 10.1038/nri3845 [DOI] [PubMed] [Google Scholar]
- 72.Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016:16(7):407–420. 10.1038/nri.2016.58 [DOI] [PubMed] [Google Scholar]
- 73.He Y, Hara H, Nunez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci. 2016:41(12): 1012–1021. 10.1016/j.tibs.2016.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hildebrand JM, Murphy JM. The highway to hell: a RIP kinase-directed shortcut to inflammatory cytokine production. Immunity. 2016:45(1):1–3. 10.1016/j.immuni.2016.06.029 [DOI] [PubMed] [Google Scholar]
- 75.Saleh D, Najjar M, Zelic M, Shah S, Nogusa S, Polykratis A, Paczosa MK, Gough PJ, Bertin J, Whalen M, et al. Kinase activities of RIPK1 and RIPK3 can direct IFN-beta synthesis induced by lipopolysaccharide. J Immunol. 2017:198(11):4435–4447. 10.4049/jimmunol.1601717 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets that support the findings of this study and were generated by the authors as part of this study have been deposited in the Gene Expression Omnibus database with the accession code GEO: GSE201909. Otherwise, the data are either contained within the manuscript or available from the authors on request.