

Visual Abstract

Keywords: CKD, glomerular disease
Abstract
Background
Hypocomplementemia and complement co-deposition with monoclonal immunoglobulins in glomeruli are not rare in proliferative glomerulonephritis with monoclonal immunoglobulin deposits (PGNMID). Deposition of monoclonal immunoglobulins in glomeruli has been suggested to activate complement and cause kidney injury. However, the profiles of complement activation in PGNMID and their clinical and pathologic significance need to be clarified.
Methods
Forty-six patients with PGNMID were enrolled. Proteomic analysis of glomeruli using laser microdissection and mass spectrometry was performed for ten patients with PGNMID to determine the composition of glomerular deposits. Kidney deposition of complement components was detected by immunohistochemistry and immunofluorescence. Urinary and plasma levels of complement components were measured by an enzyme-linked immunosorbent assay. Group differences were assessed using t tests or Mann–Whitney U tests depending on the distribution. Correlation analysis was performed using Spearman rank correlation or Pearson correlation.
Results
Laser microdissection and mass spectrometry–based proteomic analysis showed that complement components were the most enriched proteins deposited in the glomeruli of patients with PGNMID. Glomerular deposition of C3c, C4d, and C5b-9 was detected in most patients. Levels of urinary and plasma C3a, C5a, soluble C5b-9, C4d, Bb, and C1q as well as urinary mannose-binding lectin were significantly higher in patients with PGNMID compared with healthy controls. The intensity of C3c and C4d deposition in glomeruli correlated with serum creatinine and the percentage of crescents, respectively. Furthermore, levels of urinary complement components correlated positively with serum creatinine, urinary protein excretion, percentage of crescents, and global glomerulosclerosis in kidney biopsies, whereas plasma levels of most complement components did not show a significant correlation with clinicopathologic parameters. In multivariable analysis, a higher level of urinary C4d was identified as an independent risk factor of kidney failure.
Conclusions
The complement system was found to be overactivated in PGNMID, and levels of urinary complements correlated with disease severity. A higher level of urinary C4d was identified as an independent risk factor of kidney failure.
Introduction
Monoclonal gammopathy of renal significance (MGRS) includes a group of hematologic disorders with monoclonal immunoglobulin-related kidney injury that do not cause tumor complications or meet any current hematologic criteria for specific therapy.1 Proliferative glomerulonephritis with monoclonal immunoglobulin deposits (PGNMID) is a particular form of MGRS that is limited to glomeruli.2 It is characterized by the histologic pattern of proliferative glomerulonephritis, predominantly membranoproliferative glomerulonephritis on light microscopy, a single immunoglobulin heavy chain subclass and a single light chain isotype on immunofluorescence (IF), and granular electron-dense deposits on electron microscopy.3,4 IgG3 is the most common subtype of monoclonal immunoglobulin, constituting 60%–68% of patients, followed by IgG1 (24%–29%).5,6
It is generally accepted that the mechanism of injury in PGNMID is deposition of monoclonal immunoglobulins in glomeruli.7 Apart from monoclonal immunoglobulins, glomerular co-deposition of C3 is observed in nearly 100% of patients, and co-deposition of C1q is observed in 54%–64% of patients. Hypocomplementemia is observed in 26%–36% of patients.5,6,8 Our previous study found that the intensity of glomerular C3 deposits in IgG3-subtype PGNMID was significantly higher than that in IgG1-subtype PGNMID.9 This finding is consistent with the fact that IgG3 has a higher complement-fixing ability than IgG110 and provides supporting evidence for the association between monoclonal immunoglobulins and complement activation. Moreover, Miller et al. reported a heterozygous missense mutation of complement factor H in a patient with PGNMID, along with higher levels of Bb and soluble C5b-9 (sC5b-9) and lower levels of C3 in the circulation, suggesting complement alternative pathway activation in the development of the disease.11 An intronic variant of complement factor H–related protein (CFHR) 3 and a heterozygous deletion of a large segment of CFHR1–CFHR3 were reported in another patient. Laboratory examination showed lower levels of complement C2, C3, C4, factor B, and factor P and higher Bb levels in the circulation. Combined with deposition of C1q in glomeruli, activation of the classical and alternative complement pathways was thus suggested.11 These findings suggest that complement activation is involved in the pathogenesis of PGNMID. Therefore, the profiles of complement overactivation in patients with PGNMID and their clinical and pathologic significance need to be clarified.
In this study, we investigated complement activation profiles in kidney specimens and plasma and urine from a large cohort of patients with PGNMID and analyzed correlations with clinical and histopathologic parameters.
Methods
Patients and Samples
This was a retrospective observational cohort study of 46 patients with kidney biopsy-proven PGNMID from January 2014 to September 2021 at Peking University First Hospital. The diagnostic criteria of PGNMID were as follows4: (1) glomerular monotypic immunoglobulin deposits on IF (a single heavy chain and a single light chain); (2) proliferative, membranoproliferative glomerulonephritis or atypical membranous nephropathy on light microscopy; (3) immune complex–like granular electron-dense deposits in glomeruli on electron microscopy; and (4) the absence of clinical manifestation of cryoglobulinemia. Patients with cryoglobulinemic glomerulonephritis were excluded. This series was reported in our previous study.9 Of the 46 patients, kidney biopsy specimens from 44 patients (frozen kidney biopsies from 12 patients were insufficient to detect all indicators) and plasma and urine samples from 43 patients collected on the day of kidney biopsies were available. Plasma and urine samples from 20 age- and sex-matched healthy individuals were collected as the healthy controls. The plasma and urine samples of patients and controls were centrifuged at 2500 rpm for 15 minutes at 4°C, and the supernatant was stored in aliquots at −80°C until use.
Clinical and Laboratory Evaluation
Demographic, clinical, and laboratory data at kidney biopsy were obtained retrospectively from the electronic medical records as the baseline for analysis. Hematologic evaluation included serum/urine immunofixation, serum/urine protein electrophoresis, and serum free light chain and bone marrow examination. Bone marrow aspirate, biopsy, and flow cytometry were performed for 36 patients. Methods of kidney pathology are provided in the Supplemental Material. All missing data are summarized in Table 1, and patients with missing data were not included in the analysis of corresponding indicators.
Table 1.
Clinical and pathologic characteristics at presentation
| Characteristic | Value (N=46) |
|---|---|
| Age, yr | 52±13 |
| Male, n (%) | 25 (54) |
| 24-h urine protein, g/24 h | 5.3 (2.3–10.3) |
| Serum albumin, g/dl | 2.7±0.6 |
| Hematuria, n (%) | 41 (89) |
| Urine red blood cells, per µl | 61 (25–153) |
| Serum creatinine at biopsy, mg/dl | 1.7 (1.3–3.8) |
| eGFR at biopsy, ml/min per 1.73 m2 | 37 (16–62) |
| Low C3a, n (%) | 14/45 (31) |
| Low C4b, n (%) | 8/45 (18) |
| Hematologic studies, n (%) | |
| Serum or urine clone detection | 5/45 (11) |
| Positive serum immunofixation | 5/45 (11) |
| Positive urine immunofixation | 1/45 (2) |
| Positive serum protein electrophoresis | 2/19 (11) |
| Positive urine protein electrophoresis | 0/14 (0) |
| Abnormal serum free light chain ratioc | 0/37 (0) |
| Clone detection of bone marrow | 8/36 (22) |
| Positive serum cryoglobulin | 20/38 (53) |
| IF | |
| IgG1 κ/IgG3 κ/IgG3 λ/IgM λ | 5/31/9/1 |
| Deposition of C3, n (%) | 46 (100) |
| Deposition of C1q, n (%) | 38 (83) |
| Light microscopy | |
| No. of glomeruli | 30 (23–42) |
| % of globally sclerotic glomeruli | 6 (0–11) |
| Predominant histologic pattern | |
| Membranoproliferative glomerulonephritis, n (%) | 36 (78) |
| Mesangial proliferative glomerulonephritis, n (%) | 6 (13) |
| Endocapillary proliferative glomerulonephritis, n (%) | 3 (7) |
| Atypical membranous nephropathy, n (%) | 1 (2) |
| Crescents, n (%) | 18 (39) |
| Diffused, n (%) | 1 (2) |
| Electron microscopic, n (%) | |
| Location of granular electron-dense deposits | |
| Mesangial | 42/45 (93) |
| Subendothelial | 38/45 (84) |
| Subepithelial | 20/45 (44) |
| Intramembranous | 11/45 (24) |
IF, immunofluorescence.
The normal range of C3 is 60–150 mg/dl.
The normal range of C4 is 12–36 mg/dl.
The normal serum free light chain ratio is 0.26–1.65 for patients with eGFR ≥60 ml/min per 1.73 m2 and 0.37–3.10 for patients with eGFR >60 ml/min per 1.73 m2.
Diffuse, ≥50% of glomeruli.
Treatments and outcomes were collected through electronic medical records, outpatient records, and telephone follow-up. The end point of this study was kidney failure (defined as eGFR <15 ml/min per 1.73 m2 or requiring KRT). Kidney response was divided into complete remission, partial remission, persistent kidney dysfunction, and kidney failure, as described previously.5 The definitions are provided in the Supplemental Material.
The study was approved by the ethics committee of Peking University First Hospital. Informed written consent was obtained from all participants.
Proteomic Analysis of Glomeruli by Laser Microdissection and Mass Spectrometry
Proteomic analysis of kidney biopsies from ten patients with PGNMID was performed using laser microdissection and mass spectrometry (LMD/MS). Four specimens of nephrectomy tissue far from the kidney tumors were selected as normal controls. The method has previously been described.12 Peptide identifications were accepted if they could be established at a ≥95% probability as specified by the PeptideProphet algorithm. The differentially expressed protein screening conditions applied were fold change ≥2 or ≤1/2 to evaluate the fold change in protein expression levels between the samples by Student t tests with P < 0.05.13 Differentially expressed proteins were functionally enriched and analyzed according to the Gene Ontology database (http://geneontology.org) and the Kyoto Encyclopedia of Genes and Genomes database (https://www.kegg.jp). A more detailed description of the methods is given in the Supplemental Material.
Detection of Complement Components Deposited in Kidneys by Immunohistochemistry and IF
To study the complement activation products deposited in the kidneys of patients with PGNMID, C1q and C3c in 44 patients and Bb and mannose-binding lectin (MBL) in 32 patients were detected by IF using frozen kidney biopsies. C4d and C5b-9 were detected by immunohistochemistry on paraffin-embedded kidney tissues of 44 patients. IF and immunohistochemistry were performed as previously described.14 More details are provided in the Supplemental Material. Positive staining signals in glomeruli were quantified using Image-Pro Plus analysis software 6.0 (Media Cybernetics, Silver Spring, MD); positive signals were quantified as the mean optical density (integrated optical density/area).
Quantification of Complement Components in Plasma and Urine
Commercial enzyme-linked immunosorbent assay kits were used for the detection of the plasma and urinary complement components Bb, C3a, C4d, C5a, sC5b-9 (Quidel, San Diego, CA), MBL (Thermo Fisher Scientific, Waltham, MA), and C1q (Cusabio, Wuhan, China). Quantifications of complement components were performed according to the manufacturer's instructions.
Statistical Analyses
Statistical analysis was performed using SPSS 21 (IBM Corp., Armonk, NY). Descriptive data are expressed as mean±SD for normally distributed data, median and interquartile range (IQR) for non-normally distributed data, and proportions for categorical variables. Differences in quantitative parameters between groups were analyzed using t tests or Mann‒Whitney U tests depending on the distribution. Categorical variables were tested by chi-squared tests or Fisher exact tests. Correlation analysis was performed using Spearman rank correlation and Pearson correlation for non-normally and normally distributed data, respectively. To adjust for the confounding effect of urinary protein excretion and higher levels of plasma complements on urinary complement concentration, the analysis of covariance model was used (details are described in the Supplemental Material). Data were censored at the time of loss to follow-up, death, or kidney failure or at the time of last examination of serum creatinine and urinary protein excretion during the follow-up. One patient who was lost to follow-up after 32 months was censored at the time of loss to follow-up in the Cox regression analysis. Survival analysis was performed by unadjusted survival analysis, multivariable Cox regression models, and Kaplan–Meier curves. Statistical significance was assumed at P < 0.05.
Results
Baseline Clinical and Pathologic Characteristics
Forty-six patients with PGNMID were included. The baseline clinical and pathologic data are presented in Table 1. Among the 36 patients undergoing bone marrow examination, clonal plasma cells were detected by flow cytometry in seven, with a median of 0.2% (IQR, 0.1%–0.4%); 17% clonal B cells with λ restriction were detected in one patient with lymphoma who had global deposition of IgM λ. Among these eight patients, clones were also detected in serum from three patients. Two patients with clones detected in serum and/or urine did not have detectable clones in bone marrow.
Proteomic Analysis
LMD/MS was performed for ten patients with PGNMID (P1–P10). Of the ten patients, the subtype of monotypic immunoglobulins was already determined by IF, showing monotypic IgG3 κ in four patients, IgG3 λ in two patients, and IgG1 κ in four patients. LMD/MS was used to detect overabundant Ig γ-1 or γ-3 chain constant regions in the ten patients, along with overabundant Ig κ or λ light chain constant and variable regions in six patients, which was consistent with the IF results. The dominant light chain subtype of the remaining four patients (P1, P7, P8, and P9) could not be identified by LMD/MS because of the small number of monoclonal immunoglobulins deposited in glomeruli.15 More details are presented in Supplemental Tables 1 and 2.
As shown in Figure 1A, complement proteins (C1q, C1r, C3, C4, C5, C6, C7, C8, and C9) and complement regulatory proteins (complement receptor type 1, complement factor H, CFHR1, CFHR3, CFHR5, C4-binding protein, and clusterin) were differentially expressed in patients with PGNMID compared with controls. Kyoto Encyclopedia of Genes and Genomes enrichment analysis and Gene Ontology analysis of the differentially expressed proteins between patients with PGNMID and controls showed that complement proteins, especially those of the classical and alternative pathways, were most enriched (Figure 1, B and C).
Figure 1.

Differentially expressed proteins between patients with PGNMID and healthy controls. (A) Heatmap of glomerular complement protein and complement regulatory protein abundance in patients with PGNMID and healthy controls. Each column represents an individual patient with PGNMID (P1–P10) or healthy control (C1–C4). Each row represents protein abundance. Red represents higher protein expression, and blue represents lower protein expression. Scale −2 to 2 reflects relative protein expression rescaled within each protein by centering at 0 and dividing by the SD. (B) Scatterplot for the top 20 pathways in KEGG enrichment of differentially expressed proteins between patients with PGNMID and healthy controls sorted by the −log10 P value. The enrichment score was calculated according to the number of annotated genes and that of all annotated genes in this pathway term (see Methods in detail). Lower P values indicate higher pathway enrichment. (C) Top ten gene ontology terms in the biological process of differentially expressed proteins between patients with PGNMID and healthy controls sorted by the −log10 P value. COVID-19, coronavirus disease 2019; KEGG, Kyoto Encyclopedia of Genes and Genomes; PGNMID, proliferative glomerulonephritis with monoclonal immunoglobulin deposits.
Deposition of Complements in Glomeruli
Immunohistochemical and IF examination revealed positive staining of C3c in 44 of 44 patients (100%), C5b-9 in 38 of 44 patients (86%), C4d in 42 of 44 patients (96%), C1q in 36 of 44 patients (82%), MBL in 10 of 32 patients (31%), and Bb in 14 of 32 patients (44%), along with the glomerular capillary walls and mesangium (Figure 2). The optical densities of C3c, C5b-9, C4d, C1q, MBL, and Bb in glomeruli were 6.22 (2.12–14.99), 0.01 (0.01–0.02), 0.03±0.01, 1.22 (0.55–3.82), 0.72 (0.38–1.04), and 2.26 (1.78–3.67), respectively. No obvious staining of complement components was detected in negative controls.
Figure 2.
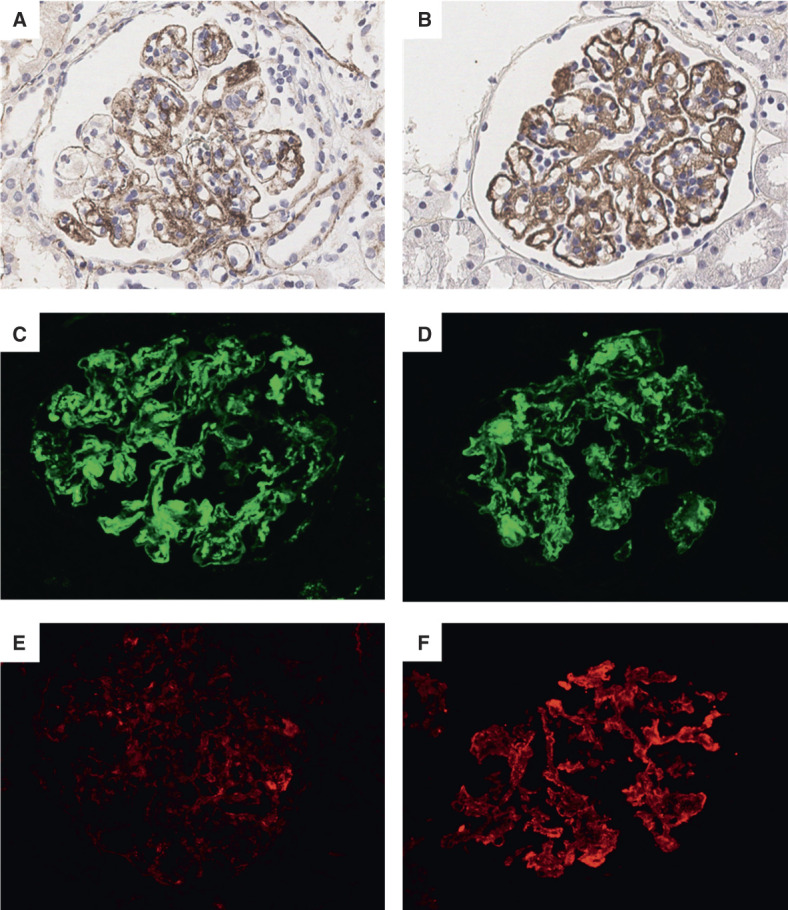
Staining for complement components in glomeruli detected by IHC and IF. Granular staining for C5b-9 (A) and C4d (B) along glomerular capillary walls and mesangial areas identified by IHC. Strong positive staining of C3c (C), C1q (D), and Bb (F) but weak positive staining of MBL (E) along glomerular capillary walls and mesangial areas were detected by IF. (A–F) Original magnification, ×400. IF, immunofluorescence; IHC, immunohistochemistry; MBL, mannose-binding lectin. Figure 2 can be viewed in color online at www.cjasn.org.
Levels of Complements in Urine and Plasma
Urinary levels of complements were normalized to urinary creatinine levels to correct for the influence of dilution. As shown in Figure 3, A–G, levels of urinary C3a/creatinine, C5a/creatinine, sC5b-9/creatinine, C4d/creatinine, C1q/creatinine, MBL/creatinine, and Bb/creatinine were significantly higher in patients with PGNMID than in healthy controls (24.49 [2.09–120.95] versus 0.00 [0.00–0.00] ng/mg, 34.25 [2.00–187.24] versus 0.04 [0.01–0.05] ng/mg, 214.85 [14.48–1018.92] versus 0.00 [0.00–5.17] ng/mg, 0.35 [0.03–1.44] versus 0.00 [0.00–0.00] µg/mg, 0.33 [0.11–1.39] versus 0.02 [0.00–0.16] ng/mg, 4.25 [0.72–27.57] versus 0.05 [0.03–0.08] ng/mg, 0.17 [0.05–2.25] versus 0.02 [0.02–0.04] µg/mg, respectively, all P values < 0.001). After adjusting for urinary protein excretion, all differences in the abovementioned comparisons remained significant, except for urinary Bb. As shown in Figure 3, H–N, levels of plasma C3a, C5a, sC5b-9, C4d, C1q, and Bb were significantly higher in patients with PGNMID than in healthy controls (5130.91 [3218.59–11779.67] versus 191.34 [178.25–211.16] ng/ml, 54.15 [12.17–397.23] versus 7.47 [4.34–11.83] ng/ml, 440.77 [292.54–520.05] versus 144.50 [104.12–186.73] ng/ml, 14.30 [8.51–17.46] versus 2.22 [1.16–3.68] µg/ml, 14,552.14±4706.18 versus 10,793.75±2077.91 ng/ml [t=4.40], 1.80±0.76 versus 0.95±0.32 µg/ml [t=6.31], respectively, all P values < 0.001). After adjusting for higher plasma levels of complements, all differences in the urinary complements between patients with PGNMID and healthy controls remained significant, except for urinary sC5b-9.
Figure 3.

Levels of urinary and plasma complement components in patients with PGNMID. (A) Urinary C3a. (B) Urinary C5a. (C) Urinary sC5b-9. (D) Urinary C4d. (E) Urinary C1q. (F) Urinary MBL. (G) Urinary Bb. (H) Plasma C3a. (I) Plasma C5a. (J) Plasma sC5b-9. (K) Plasma C4d. (L) Plasma C1q. (M) Plasma MBL. (N) Plasma Bb. Cr, creatinine; sC5b-9, soluble C5b-9. Figure 3 can be viewed in color online at www.cjasn.org.
Association between Complements and Clinicopathologic Parameters
The mean optical density of C3c and C4d deposited in glomeruli correlated positively with serum creatinine levels and the percentage of crescents, respectively (r=0.41, P = 0.01; r=0.55, P < 0.001). As summarized in Supplemental Table 3, levels of the urinary complement components C3a/creatinine, C5a/creatinine, sC5b-9/creatinine, C4d/creatinine, C1q/creatinine, MBL/creatinine, and Bb/creatinine correlated positively with serum creatinine at biopsy, urinary protein excretion, percentage of crescents, and percentage of global glomerulosclerosis, except for C1q/creatinine with serum creatinine. Urine red blood cells and levels of urinary complements did not correlate. There was no significant correlation between plasma complements and clinicopathologic features, except for the positive correlation between the plasma level of C4d and the percentage of global glomerulosclerosis (r=0.34, P = 0.03). No significant difference was observed in complements between patients with and without circulating monoclonal immunoglobulins and/or B/plasma cell clones in bone marrow.
Association between Complements and Kidney Outcomes
The median duration of follow-up for the 46 patients was 21 (IQR, 8–36) months. One patient was lost to follow-up after 32 months. Treatments and outcomes are summarized in Table 2. Prognostic factors associated with kidney survival are presented in Table 3. Forty-three patients with complete clinical and laboratory data were included in multivariable Cox regression model analysis. Independent predictors for progression to kidney failure in multivariable analysis were a higher level of serum creatinine at biopsy, higher percentage of global glomerulosclerosis, and higher level of urinary C4d/creatinine. In receiver operating characteristic curve analysis based on progression to kidney failure, the best cutoffs were 1.4 mg/dl for serum creatinine (sensitivity 96%; specificity 54%), 6% for the percentage of global glomerulosclerosis (sensitivity 73%; specificity 77%), and 0.48 µg/mg for urinary C4d/creatinine (sensitivity 59%; specificity 86%). According to the Kaplan–Meier survival analysis, kidney survival was significantly poorer in the subgroup with serum creatinine ≥1.4 mg/dl (P = 0.001, Figure 4A), global glomerulosclerosis ≥6% (P < 0.001, Figure 4B), and urinary C4d/creatinine ≥0.48 µg/mg (P = 0.001, Figure 4C). The rate of kidney failure per 100 person-years was 16 (95% confidence interval [CI], 7 to 30) for patients with urinary C4d/creatinine ≥0.48 µg/mg, compared with 69 (95% CI, 37 to 119) for those with urinary C4d/creatinine <0.48 µg/mg. Both urinary levels of C1q/creatinine and MBL/creatinine correlated significantly with the urinary levels of C4d/creatinine (r=0.73; 95% CI, 0.51 to 0.86; P < 0.001, r=0.77; 95% CI, 0.64 to 0.85; P < 0.001, respectively).
Table 2.
Treatment and outcome
| Parameter | Value | Clone Detection |
|---|---|---|
| Treatment | ||
| Glucocorticoid and cyclophosphamide | 19 | Clonal plasma cells in bone marrow in three patients |
| Bortezomib-based chemotherapy | 15 | Clonal plasma cells in bone marrow and monoclonal IgG in serum in four patients; monoclonal IgG in serum and urine in one patient |
| Rituximab | 3 | Clonal B cells in bone marrow and monoclonal IgM in one patient; clonal plasma cells in bone marrow in one patient |
| Glucocorticoid alone | 1 | None |
| Renin‐angiotensin‐aldosterone system inhibitors alone | 3 | None |
| Other | ||
| Antiviral therapy to chronic viral hepatitis B | 3 | None |
| Lymphadenectomy | 1 | None |
| Termination of a pregnancy | 1 | None |
| Outcomea | ||
| Complete remission | 10 | — |
| Partial remission | 5 | — |
| Persistent kidney dysfunction | 9 | — |
| Kidney failure | 22 | — |
| Deathb | 2 |
Complete remission was defined as remission of proteinuria to <500 mg/d with normal kidney function; partial remission was defined as a reduction in proteinuria by at least 50% and to <2 g/d with stable kidney function (no more than a 20% increase in serum creatinine compared with the baseline); persistent kidney dysfunction was defined as failure to meet the criteria for either complete remission or partial remission but without kidney failure and included patients with unremitting proteinuria or progressive CKD; kidney failure was defined as eGFR <15 ml/min per 1.73 m2 or requiring KRT.
No patients died before receiving KRT.
Table 3.
Prognostic factors associated with kidney survival
| Parameter | Unadjusted Survival Analysis | Multivariable Analysis |
|---|---|---|
| Hazard Ratio (95% CI) | Hazard Ratio (95% CI) | |
| Age, yr | 1.02 (0.98 to 1.06) | |
| Male | 1.35 (0.58 to 3.12) | |
| 24-h urine protein, g/24 h | 1.02 (0.96 to 1.09) | |
| Urine red blood cells, per µl | 1.00 (1.00 to 1.00) | |
| Serum albumin, g/dl | 0.52 (0.24 to 1.12) | |
| Serum creatinine at biopsy, mg/dla,b | 1.55 (1.27 to 1.89) | 1.47 (1.15 to 1.87)c |
| Hemoglobin, g/La,b | 0.97 (0.95 to 0.99) | |
| Detection of clone | 0.62 (0.21 to 1.87) | |
| Percentage of global glomerulosclerosis, %a,b | 1.06 (1.02 to 1.10) | 1.05 (1.01 to 1.10)d |
| Percentage of crescents, %a,b | 1.04 (1.00 to 1.07) | |
| Bortezomib-based chemotherapyb | 0.52 (0.19 to 1.41) | |
| Urinary C3a/creatinine, ng/mg | 1.00 (1.00 to 1.00) | |
| Urinary C5a/creatinine, ng/mg | 1.00 (1.00 to 1.00) | |
| Urinary sC5b-9/creatinine, g/mga,b | 1.00 (1.00 to 1.00) | |
| Urinary C4d/creatinine, µg/mga,b | 1.39 (1.11 to 1.75) | 1.36 (1.00 to 1.83)e |
| Urinary C1q/creatinine, ng/mg | 1.04 (0.93 to 1.16) | |
| Urinary MBL/creatinine, ng/mg | 1.01 (1.00 to 1.02) | |
| Urinary Bb/creatinine, µg/mga,b | 1.06 (1.01 to 1.11) | |
| Plasma C3a, ng/ml | 1.00 (1.00 to 1.00) | |
| Plasma C5a, ng/ml | 1.00 (1.00 to 1.00) | |
| Plasma sC5b-9, ng/ml | 1.00 (1.00 to 1.00) | |
| Plasma C4d, µg/ml | 1.07 (0.98 to 1.16) | |
| Plasma C1q, ng/ml | 1.00 (1.00 to 1.00) | |
| Plasma MBL, ng/ml | 1.00 (1.00 to 1.00) | |
| Plasma Bb, µg/ml | 1.19 (0.69 to 2.06) | |
| Intensity of global C3ca,b | 1.07 (1.00 to 1.13) | |
| Intensity of global C5b-9×100 | 1.03 (0.73 to 1.46) | |
| Intensity of global C4d×100 | 1.15 (0.94 to 1.40) | |
| Intensity of global C1q | 1.06 (0.97 to 1.15) | |
| Intensity of global MBL | 0.63 (0.18 to 2.18) | |
| Intensity of global Bb | 1.09 (0.80 to 1.49) |
CI, confidence interval; sC5b-9, soluble C5b-9; MBL, mannose-binding lectin.
In unadjusted survival analysis, P < 0.05.
Variables were incorporated into multivariable Cox regression.
In multivariable analysis, P = 0.002.
In multivariable analysis, P = 0.01.
In multivariable analysis, P = 0.05.
Figure 4.

Kaplan–Meier curves demonstrating differences in kidney survival. (A) Kidney survival according to serum creatinine (P = 0.001). (B) Kidney survival according to the percentage of global glomerulosclerosis (P < 0.001). (C) Kidney survival according to the level of urinary C4d/creatinine (P = 0.001). Figure 4 can be viewed in color online at www.cjasn.org.
Discussion
Deposition of pathogenic monoclonal immunoglobulins in glomeruli has been suggested to activate complement and cause kidney injury in PGNMID.4,5 However, there is insufficient evidence of complement overactivation in the development of PGNMID. This study analyzed complements deposited in glomeruli as well as those in urine and plasma in a relatively large sample size to verify that overactivation of complement is involved in PGNMID and correlates with disease severity and outcome.
The classical, lectin, and alternative pathways, by which the complement system can become activated, converge at the process of C3 activation and the formation of the membrane attack complex (C5b-9), causing direct tissue damage. In this study, C1q, MBL, and Bb, markers of complement activation by the three pathways, can be detected in glomeruli, with co-deposition of C3c, C4d, and membrane attack complex. Coupled with higher levels of urinary and plasma complements, the complement system was overactivated in patients with PGNMID by all three pathways in the kidneys and the classical and alternative pathways in the circulation. Complements deposited in glomeruli and urinary levels of complement components correlated significantly with disease severity, whereas plasma levels of most complement components did not correlate significantly with clinicopathologic parameters. These results indicate the significance of local complement overactivation in the kidneys in the development of PGNMID.
Complement activation of the classical pathway may be initiated by binding of C1q and monoclonal immunoglobulins. This study found that the urinary C1q level was significantly higher in patients with IgG3-subtype PGNMID than in patients with IgG1-subtype PGNMID, which was consistent with the higher capability of IgG3 to bind C1q and activate the classical complement pathway.10 We speculate that the components in glomeruli or pathogenic monoclonal immunoglobulins that had glycosylation changes bind to MBL, resulting in complement activation of the lectin pathway.16 In addition, MBL can also bind to apoptotic or necrotic cells. Another possibility is that MBL bind to damaged glomerular tissue to initiate the lectin pathway.17 Complement overactivation of the alternative pathway further amplified the complement activation signals,18 which, in turn, activated downstream inflammatory mediators that promote infiltration of inflammatory cells, leading to injury and proliferation of endothelial and mesangial cells and, ultimately, glomerulonephritis. Several cases with genetic variation in complement factor H or CFHR have been reported, indicating that insufficient alternative pathway inhibition may be one of the etiologies of complement activation in PGNMID.11,19 The exact mechanisms of complement activation need further investigation in the future.
PGNMID is not exclusive to the relationship between MGRS and complement activation. The currently recognized pathogenesis of C3 glomerulopathy is associated with excessive activation of the alternative complement pathway. The disease is driven by multiple factors, including autoantibodies that target the C3 or C5 convertases and genetic variation in complement-related genes.20 Isolated cryoglobulins can activate both the classical and alternative complement pathways in vitro,21 and activation of the classical and lectin pathways has been proven in patients with cryoglobulinemic glomerulonephritis.22,23 By using mass spectrometry, a recent study showed high spectral counts of C3 and C4B in patients with immunotactoid glomerulopathy and monoclonal immunoglobulin deposition disease.24 Complement activation is not found in immunoglobulin-related amyloidosis.25,26
The kidney prognosis of patients with PGNMID is poor. In this study, approximately half of the patients experienced progression to kidney failure despite immunosuppressive therapy or bortezomib-based chemotherapy. In multivariable analysis, a higher level of urinary C4d was an independent risk factor of kidney failure. Differences between urinary C1q and MBL in correlation with urinary C4d were insignificant. These results suggest that complement overactivation by the classical and lectin pathways may be associated with progression of PGNMID. C4d, derived from C4b, persists in tissues through covalent binding to target cells and, therefore, serves as a marker for antibody-mediated tissue damage, such as antibody-mediated rejection in kidney allografts.27 In addition, C4d is considered a potential biomarker in other diseases in which antibodies can cause tissue damage, such as autoimmune diseases and pregnancy.28 In this study, we found that the level of urinary C4d, which is easy and inexpensive to detect, was associated with disease severity and progression in patients with PGNMID. Thus, urinary C4d might be used as a biomarker for disease surveillance of PGNMID. Treatment of complement inhibition may offer a promising therapeutic option for patients with PGNMID for whom chemotherapy is ineffective. However, further research is needed.
There are three limitations of our study. First, this research is descriptive, and the mechanism of complement activation in PGNMID remains unclear. We hope to conduct further investigation to explore the pathogenesis and pathophysiologic link between the complement deposits and development of glomerulonephritis in the future. Second, sequential measurements of urinary C4d levels were unavailable during follow-up, and we could not determine whether lower levels of urinary C4d improved the kidney prognosis of patients, which needs further investigation. Third, the results are based on Chinese patients, and we cannot eliminate the existence of geographic geno-/phenotypic variations.
In summary, the complement system was overactivated in patients with PGNMID, and levels of urinary complement components correlated with disease severity. A higher level of urinary C4d was identified as an independent risk factor of kidney failure.
Supplementary Material
Acknowledgments
We would like to thank the patients for their participation in this study.
Disclosures
M.-h. Zhao reports consultancy for AstraZeneca, GSK, INFLARX, Kira, Novartis, and Roche. All remaining authors have nothing to disclose.
Funding
F.-d. Zhou: National Natural Science Foundation of China (82070747) and National High Level Hospital Clinical Research Funding (2022CR07).
Author Contributions
Conceptualization: Ming Chen, Meng-yao Liu, Xiao-juan Yu.
Data curation: Meng-yao Liu, Su-xia Wang, Xiao-juan Yu, Ming-hui Zhao, Fu-de Zhou.
Formal analysis: Yuan Li, Meng-yao Liu, Xiao-juan Yu.
Funding acquisition: Fu-de Zhou.
Investigation: Yuan Li, Meng-yao Liu, Guo-lan Xing, Xiao-juan Yu.
Methodology: Ming Chen, Yuan Li, Meng-yao Liu, Guo-lan Xing, Xiao-juan Yu.
Project administration: Su-xia Wang, Ming-hui Zhao.
Resources: Ming Chen, Yuan Li, Su-xia Wang, Guo-lan Xing, Xiao-juan Yu, Ming-hui Zhao, Fu-de Zhou.
Software: Yuan Li, Guo-lan Xing.
Supervision: Ming-hui Zhao, Fu-de Zhou
Validation: Meng-yao Liu, Xiao-juan Yu.
Visualization: Meng-yao Liu.
Writing – original draft: Meng-yao Liu.
Writing – review & editing: Ming Chen, Su-xia Wang, Fu-de Zhou.
Data Sharing Statement
All data are included in the manuscript and/or supporting information.
Supplemental Material
This article contains the following supplemental material online at http://links.lww.com/CJN/B811.
Supplemental Materials and Methods.
Supplemental Table 1. Identification of immunoglobulins deposited in glomeruli.
Supplemental Table 2. Proteomic analysis of glomeruli by laser microdissection and mass spectrometry associated with the IgG subtype and light chain isotype.
Supplemental Table 3. Correlation of urinary complements with clinicopathologic parameters.
References
- 1.Leung N Bridoux F Batuman V, et al. The evaluation of monoclonal gammopathy of renal significance: a consensus report of the International Kidney and Monoclonal Gammopathy Research Group. Nat Rev Nephrol. 2019;15(1):45–59. doi: 10.1038/s41581-018-0077-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sethi S, Rajkumar SV, D'Agati VD. The complexity and heterogeneity of monoclonal immunoglobulin-associated renal diseases. J Am Soc Nephrol. 2018;29(7):1810–1823. doi: 10.1681/ASN.2017121319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nasr SH Markowitz GS Stokes MB, et al. Proliferative glomerulonephritis with monoclonal IgG deposits: a distinct entity mimicking immune-complex glomerulonephritis. Kidney Int. 2004;65(1):85–96. doi: 10.1111/j.1523-1755.2004.00365.x [DOI] [PubMed] [Google Scholar]
- 4.Bridoux F, Javaugue V, Nasr SH, Leung N. Proliferative glomerulonephritis with monoclonal immunoglobulin deposits: a nephrologist perspective. Nephrol Dial Transplant. 2021;36(2):208–215. doi: 10.1093/ndt/gfz176 [DOI] [PubMed] [Google Scholar]
- 5.Nasr SH Satoskar A Markowitz GS, et al. Proliferative glomerulonephritis with monoclonal IgG deposits. J Am Soc Nephrol. 2009;20(9):2055–2064. doi: 10.1681/ASN.2009010110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhutani G Nasr SH Said SM, et al. Hematologic characteristics of proliferative glomerulonephritides with nonorganized monoclonal immunoglobulin deposits. Mayo Clinic Proc. 2015;90(5):587–596. doi: 10.1016/j.mayocp.2015.01.024 [DOI] [PubMed] [Google Scholar]
- 7.Leung N, Drosou ME, Nasr SH. Dysproteinemias and glomerular disease. Clin J Am Soc Nephrol. 2018;13(1):128–139. doi: 10.2215/CJN.00560117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guiard E Karras A Plaisier E, et al. Patterns of noncryoglobulinemic glomerulonephritis with monoclonal Ig deposits: correlation with IgG subclass and response to rituximab. Clin J Am Soc Nephrol. 2011;6(7):1609–1616. doi: 10.2215/CJN.10611110 [DOI] [PubMed] [Google Scholar]
- 9.Liu M, Yu X, Wang S, Qin A, Zhou F, Zhao M. Proliferative glomerulonephritis with monoclonal immunoglobulin deposits: an entity associated with distinct diseases and comparison between IgG1 and IgG3 subtypes. J Nephrol. 2022;35(9):2363–2372. doi: 10.1007/s40620-022-01317-w [DOI] [PubMed] [Google Scholar]
- 10.Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol. 2014;5:520. doi: 10.3389/fimmu.2014.00520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller P Xiao AY Kung VL, et al. Progression of proliferative glomerulonephritis with monoclonal IgG deposits in pediatric patients. Pediatr Nephrol. 2021;36(4):927–937. doi: 10.1007/s00467-020-04763-5 [DOI] [PubMed] [Google Scholar]
- 12.Sethi S Theis JD Vrana JA, et al. Laser microdissection and proteomic analysis of amyloidosis, cryoglobulinemic GN, fibrillary GN, and immunotactoid glomerulopathy. Clin J Am Soc Nephrol. 2013;8(6):915–921. doi: 10.2215/CJN.07030712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen Z Zhou B Wang X, et al. Synergistic effects of mechanical stimulation and crimped topography to stimulate natural collagen development for tendon engineering. Acta Biomater. 2022;145:297–315. doi: 10.1016/j.actbio.2022.04.026 [DOI] [PubMed] [Google Scholar]
- 14.Xing G-q Chen M Liu G, et al. Complement activation is involved in renal damage in human antineutrophil cytoplasmic autoantibody associated pauci-immune vasculitis. J Clin Immunol. 2009;29(3):282–291. doi: 10.1007/s10875-008-9268-2 [DOI] [PubMed] [Google Scholar]
- 15.Said SM Sethi S Valeri AM, et al. Renal amyloidosis: origin and clinicopathologic correlations of 474 recent cases. Clin J Am Soc Nephrol. 2013;8(9):1515–1523. doi: 10.2215/CJN.10491012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roos A Rastaldi MP Calvaresi N, et al. Glomerular activation of the lectin pathway of complement in IgA nephropathy is associated with more severe renal disease. J Am Soc Nephrol. 2006;17(6):1724–1734. doi: 10.1681/ASN.2005090923 [DOI] [PubMed] [Google Scholar]
- 17.Nauta AJ Raaschou-Jensen N Roos A, et al. Mannose-binding lectin engagement with late apoptotic and necrotic cells. Eur J Immunol. 2003;33(10):2853–2863. doi: 10.1002/eji.200323888 [DOI] [PubMed] [Google Scholar]
- 18.Harboe M, Mollnes TE. The alternative complement pathway revisited. J Cell Mol Med. 2008;12(4):1074–1084. doi: 10.1111/j.1582-4934.2008.00350.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takehara E Mandai S Shikuma S, et al. Post-infectious proliferative glomerulonephritis with monoclonal immunoglobulin G deposits associated with complement factor H mutation. Intern Med. 2017;56(7):811–817. doi: 10.2169/internalmedicine.56.7778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith RJH Appel GB Blom AM, et al. C3 glomerulopathy - understanding a rare complement-driven renal disease. Nat Rev Nephrol. 2019;15(3):129–143. doi: 10.1038/s41581-018-0107-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wilson MR, Arroyave CM, Miles L, Tan EM. Immune reactants in cryoproteins. Relationship to complement activation. Ann Rheum Dis. 1977;36(6):540–548. doi: 10.1136/ard.36.6.540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haydey RP, de Rojas MP, Gigli I. A newly described control mechanism of complement activation in patients with mixed cryoglobulinemia (cryoglobulins and complement). J Invest Dermatol. 1980;74(5):328–332. doi: 10.1111/1523-1747.ep12543575 [DOI] [PubMed] [Google Scholar]
- 23.Ohsawa I Ohi H Tamano M, et al. Cryoprecipitate of patients with cryoglobulinemic glomerulonephritis contains molecules of the lectin complement pathway. Clin Immunol. 2001;101(1):59–66. doi: 10.1006/clim.2001.5098 [DOI] [PubMed] [Google Scholar]
- 24.Sethi S, Palma LMP, Theis JD, Fervenza FC. Proteomic analysis of complement proteins in glomerular diseases. Kidney Int Rep. 2023;8(4):827–836. doi: 10.1016/j.ekir.2023.01.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gupta N, Kaur H, Wajid S. Renal amyloidosis: an update on diagnosis and pathogenesis. Protoplasma. 2020;257(5):1259–1276. doi: 10.1007/s00709-020-01513-0 [DOI] [PubMed] [Google Scholar]
- 26.Ikura H, Endo J, Kitakata H, Moriyama H, Sano M, Fukuda K. Molecular mechanism of pathogenesis and treatment strategies for AL amyloidosis. Int J Mol Sci. 2022;23(11):6336. doi: 10.3390/ijms23116336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nickeleit V, Mihatsch MJ. Kidney transplants, antibodies and rejection: is C4d a magic marker? Nephrol Dial Transplant. 2003;18(11):2232–2239. doi: 10.1093/ndt/gfg304 [DOI] [PubMed] [Google Scholar]
- 28.Cohen D Colvin RB Daha MR, et al. Pros and cons for C4d as a biomarker. Kidney Int. 2012;81(7):628–639. doi: 10.1038/ki.2011.497 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data are included in the manuscript and/or supporting information.