

Abstract
Lymphocyte-specific protein tyrosine kinase (LCK) is an SRC-family kinase critical for initiation and propagation of T-cell antigen receptor (TCR) signaling through phosphorylation of TCR-associated CD3 chains and recruited downstream molecules. Until now, only one case of profound T-cell immune deficiency with complete LCK deficiency [1] caused by a biallelic missense mutation (c.1022T>C, p.L341P) and three cases of incomplete LCK deficiency [2] caused by a biallelic splice site mutation (c.188-2A>G) have been described. Additionally, deregulated LCK expression has been associated with genetically undefined immune deficiencies and hematological malignancies. Here, we describe the second case of complete LCK deficiency in a 6-month-old girl born to consanguineous parents presenting with profound T-cell immune deficiency. Whole exome sequencing (WES) revealed a novel pathogenic biallelic missense mutation in LCK (c.1393T>C, p.C465R), which led to the absence of LCK protein expression and phosphorylation, and a consecutive decrease in proximal TCR signaling. Loss of conventional CD4+ and CD8+ αβT-cells and homeostatic T-cell expansion was accompanied by increased γδT-cell and Treg percentages. Surface CD4 and CD8 co-receptor expression was reduced in the patient T-cells, while the heterozygous mother had impaired CD4 and CD8 surface expression to a lesser extent. We conclude that complete LCK deficiency is characterized by profound T-cell immune deficiency, reduced CD4 and CD8 surface expression, and a characteristic TCR signaling disorder. CD4 and CD8 surface expression may be of value for early detection of mono- and/or biallelic LCK deficiency.
Supplementary Information
The online version contains supplementary material available at 10.1007/s10875-023-01602-8.
Keywords: LCK deficiency, inborn errors of immunity, profound T-cell immune deficiency, TCR signaling, CD4 and CD8 co-receptor expression
Introduction
Lymphocyte-specific protein tyrosine kinase (LCK) is critical for T-cell development and activation. LCK is recruited to the T-cell antigen receptor (TCR) after ligation of the TCR by peptide:MHC complexes on the surface of antigen-presenting cells [3, 4]. LCK-dependent phosphorylation of immunoreceptor tyrosine-based activation motifs (ITAMs) in the cytoplasmic tails of CD3δ, -ε and -ζ chains create docking sites for ζ -chain associated protein kinase 70 (ZAP70) [5, 6]. LCK next phosphorylates ITAM-bound ZAP70, which transduces the signal to the linker of activated T-cells (LAT). LAT, together with SRC homology 2 (SH2) domain-containing leukocyte protein of 76kDa (SLP76), forms a signal amplification and diversification hub, ultimately leading to T-cell activation [7, 8]. Besides the CD3-chains and ZAP70, LCK phosphorylates additional members of the TCR signaling network, such as protein kinase Cθ (PKCθ) and interleukin-2 inducible T cell kinase (ITK), and is involved in signaling downstream of other receptors, most prominently the co-stimulatory molecule CD28 [9], all together contributing to the central role of LCK in T-cell biology.
LCK differs from other SRC-family kinases (SFKs), such as FYN and SRC, in its N-terminal SH4 domain targeting LCK to the plasma membrane and the unique domain (UD) mediating (weak) interaction with the co-receptors CD4 and CD8 [10, 11]. SH3 and SH2 domains, providing docking sites for intra- and intermolecular interactions, are connected by a linker region to the kinase domain (KD) and a C-terminal unstructured tail. Inactive LCK, phosphorylated on Y505 by the C-terminal SRC Kinase (CSK) [12], adopts a closed conformation with an intramolecular interaction of pY505 with the SH2 domain [13]. De-phosphorylation of pY505 by CD45 [14–16] opens LCK into a primed state, which allows trans-autophosphorylation of Y394 resulting in full LCK kinase activity [17].
LCK is found both free and co-receptor bound with varying distribution between CD4+ and CD8+ T-cells and their differentiation stages [18], likely serving different purposes [19–21]. Despite CD4 and CD8 co-receptors being structurally dissimilar, the non-covalent interaction of both coreceptors with LCK is mediated by two conserved cysteines forming a Zn2+-clasp structure [10]. Importantly, while CD4 is present as a monomer, CD8 forms CD8αβ hetero- or CD8αα homodimers, in conventional or unconventional T-cells, respectively, where LCK is only bound to CD8α [22].
Besides LCK, a T-cell-specific isoform of FYN (FYN-T) and SRC are expressed in T-cells, albeit the latter only at low levels. Despite a high similarity between FYN-T and LCK, their functions are non-redundant, partly owing to different subcellular localization determined by their different SH4 domains [3]. Nonetheless, FYN-T can compensate for some LCK functions. While lck-/- mice have an almost complete block in thymic positive selection, some peripheral mature T-cells develop [23]. In contrast, in lck-/- fyn-/- mice, no mature αβ T-cells are formed [24].
Owing to its critical role in T-cell biology, LCK loss of function (LOF) is expected to lead to a profound T-cell deficiency. Until now, only one case of complete LCK deficiency has been described [1]. A child suffering from recurrent, severe infections and immune dysregulation was found to have a biallelic missense mutation in LCK (c.1022T>C), resulting in an amino acid substitution in the kinase domain (p.L341P) with low protein expression and complete loss of LCK kinase activity. More recently, Li et al. described a homozygous splice site mutation in LCK (c.188-2A>G), predicted to affect the 3’ splice acceptor site of LCK exon 4, in a consanguineous family presenting with a partial CD4+ T-cell defect, and susceptibility to human papillomavirus (HPV) infections with atypical epidermodysplasia verruciformis (EV), as well as recurrent pneumonia [2]. Further, two reports of immune deficiency with reduced LCK protein expression and the presence of an aberrant isoform of LCK mRNA missing exon 7 have been published. However, pathogenic mutations in LCK were not identified in these reports, and signaling studies were not in line with defective LCK function [25, 26]. Lastly, beyond LCK, various inborn errors of immunity (IEIs) have been reported with deficiencies of proteins directly or indirectly activated by LCK, such as ZAP70 [27], LAT [28], SLP76 [29], ITK [30, 31] or components of the TCR-CD3 complex itself [32–35].
Here, we describe a novel complete biallelic LCK missense variant (c.1393T>C, p.C465R) in a patient with profound T-cell immune deficiency presenting with recurrent severe infections and characterize the molecular and cellular consequences of the variant for TCR signaling and T-cell function.
Methods
Human Samples
Blood samples were taken from the patient, relatives, and healthy volunteers who were treated at Mustafa Eraslan-Fevzi Mercan Children’s Hospital at Erciyes University and analyzed there and at the Dr. von Hauner Children’ Hospital at Ludwig-Maximilians-Universität München. Informed consent was obtained from both parents. This study was approved by Erciyes University local ethics committee (permit number: 2018/388) and conducted according to current ethical and legal guidelines and the Declaration of Helsinki.
Sequencing
Whole exome sequencing (WES) was performed at Intergen NGS facility, Ankara, Turkey. DNA was isolated using a magnetic bead capture method (MagPurix, Zinexts). Exome enrichment was done using the Twist capture kit (TwistBiosciences). Sequencing was performed on a MGIseq DNBSEQ-G400 (MGI Tech Co.). Data was analyzed and interpreted following the ACMG criteria [36]. PCR amplification was performed via in-house designed primers. Amplicons were checked by 2% agarose gel electrophoresis. Confirmation sequencing was performed by the next-generation sequencing method by Miseq-Illumina equipment (Illumina, San Diego, CA, USA) according to manufacturers’ instructions. Data were evaluated with IGV 2.3 (Broad Institute) software. Sanger sequencing for genotype confirmation was done using the following primers: 5′-ACCTCTAGTGTGACCTTACCA-3′ (forward), 5′-GCAGAGTCCACGCAACTACA-3′ (reverse) following standard protocols.
Lymphocyte Isolation and Cell Culture of T-Cell Blasts
Peripheral blood mononuclear cells (PBMCs) were isolated by density gradient centrifugation from peripheral blood samples using Ficoll-Paque Plus (Cytiva). PBMCs were frozen in human serum supplemented with 10% dimethyl sulfoxide (DMSO) in liquid nitrogen.
Primary T-cell blasts were generated from PBMCs of the patient and healthy controls (HCs). Frozen PBMCs stored in liquid nitrogen were quickly thawed and resuspended in prewarmed complete RMPI 1640 Glutamax (Invitrogen) supplemented with 10% FCS (Invitrogen) and Penicillin/Streptomycin 100U/ml (Invitrogen). PBMCs were stimulated with 5ng/ml phorbol-12 myristate-13-acetate (PMA) (Sigma), 1μM ionomycin (Sigma), and 200U/ml IL-2 (Novartis). After 2 days, cells were washed and cultured in complete RPMI with 100U/ml IL-2.
Cloning and Plasmids
The 2nd generation lentiviral plasmid pCDH-CMV-insert-EF1a-LNGFR was provided by Dr. Thomas Magg, and the 3rd generation lentiviral plasmid pLJM1-EGFP[37] (Addgene #19319) was provided by Daniel Petersheim. The lentiviral helper plasmids psPAX2 (Addgene #10703) and pMD2.G (Addgene #12259) were provided by Dr. Oreste Acuto. Full-length LCK gene was amplified from the cDNA of healthy control PBMCs and subcloned into pCDH-CMV-insert-EF1a-LNGFR or pLJM1-EGFP. LCK-C465R was produced by site-directed mutagenesis using Q5 polymerase (New England Biolabs). A C-terminal HA-Tag was introduced into pCDH-LCK WT and C465R by PCR with a reverse primer specific for the C-terminus of LCK fused to a HA-Tag. All constructs were verified by Sanger sequencing.
| # | Primer | Sequence 5′-> 3′ |
| 1 | pCDH-LCK FW | ATATCTAGAGCCACCATGGGCTGTGGCTGCAGCTCACACC |
| 2 | pCDH-LCK RV | ATAGCGGCCGCTCAAGGCTGAGGCTGGTACTGGCCC |
| 3 | pCDH-LCK-HA RV |
ATAGCGGCCGCTCAAGCGTAGTCTGGGACGTCGTATGGGTA AGGCTGAGGCTGGTACTGGCCC |
| 4 | LCK C465R FW | GCATGGTGCGCCCTGACAACCGTCCAGAGGAGCTGTACCAA |
| 5 | LCK C465R RV | TTGGTACAGCTCCTCTGGACGGTTGTCAGGGCGCACCATGC |
| 6 | pLJM-LCK FW | ATAGCTAGCGCCACCATGGGCTGTGGCTGCAGCTCACACC |
| 7 | pLJM-LCK RV | ATAGAATTCTCAAGGCTGAGGCTGGTACTGGCCC |
Cells, Transfections, and Lentiviral Transductions
Cell lines were maintained at 37°C with 5% CO2 in a humidified incubator. Jurkat cells (clone E6.1, ATCC TIB-152), LCK-deficient Jurkat cells (J.CaM1.6, ATCC CRL-2063), and derived cell lines were maintained in RPMI 1640 Glutamax (Gibco) medium supplemented with 10% FCS (Invitrogen). The human embryonic kidney epithelial cells (HEK293) derivative Lenti-X 293T cells (Takara, Cat-No. 632180) were maintained in DMEM (Gibco) supplemented with 10% FCS and 4mM Glutamax (Gibco). Lentiviral particles were produced in Lenti-X 293T cells by co-transfection of the transfer plasmids pCDH or pLJM1 with the packaging plasmids psPAX2 and pMD2.G complexed with polyethylenimine (PEI, linear MW25K, Polyscienes, Cat-No. 23966-100). Forty-eight hours after transfection, viral supernatants were harvested, filtered, and used for the transduction of J.CaM1.6 cells in the presence of 5mg/ml polybrene. Twenty-four hours post-infection, cells were washed and re-suspended in RPMI 10% FBS. Forty-eight hours post-infection, puromycin selection was started on cells transduced with pLJM1 which contains a puromycin-resistance gene.
Stimulation for Immunoblotting
Cells were rested for 15 min on ice in RPMI 0% FCS. For anti-CD3/CD28 stimulation, cells were incubated for 15 min on ice with 1μg/ml soluble anti-CD3 (clone OKT3, BioLegend) with or without 5μg/ml anti-CD28 (clone CD28.2, BioLegend) and washed once with RPMI 0% FCS. In primary cells, antibodies were cross-linked with 10μg/ml goat anti-mouse IgG (BD) for 15 min on ice. In Jurkat cells, crosslinking was not performed. Stimulation was initiated by shifting cells to 37°C for the indicated times (2, 5, 15 min). Alternatively, cells were stimulated for 5 min at 37°C with 10ng/ml PMA and 1μM ionomycin or left untreated. The specific LCK inhibitor A770041 (Axxon Medchem) served as a negative control in healthy control cells. Cells were incubated with 10μM A770041 at 37°C for 10 min. After stimulation, cells were centrifuged at 4°C, and pellets were lysed immediately.
Cell Lysates and Immunoblotting
Stimulated or unstimulated cells were pelleted at 4 °C, and pellets were vigorously resuspended in ice-cold complete lysis buffer (50mM Tris-HCl (pH 7.6), 150mM NaCl, 10mM NaF, 1mM Na3VO4, 1% Triton X-100, proteinase inhibitor) for 15 min on ice. Lysates were cleared by centrifugation at 15,000×g, 4°C for 15 min. The leftover cleared lysates were boiled with Laemmli sample buffer (Sigma). For whole cell lysates of Jurkat E6.1, J.CaM1.6 and transduced cells for immunoblots for LCK expression, total protein was quantified by BCA Protein Assay Kit (Thermo Scientific). Lysates were separated by SDS-PAGE and transferred onto a nitrocellulose membrane. After a blocking step with 3% BSA TBS-T, immunoblotting was performed with the following antibodies: mouse anti-human FYN (clone E-3), mouse anti-human GAPDH (clone 6C5), mouse anti-human LCK (clone 3A5), mouse-IgGκ BP-HRP, rabbit-IgGκ BP-HRP, anti-mouse-IgG-HRP (all Santa Cruz Biotechnology) or rabbit anti-human pZAP70 pY319 (clone 65E4), rabbit anti-human polyclonal pSRC-family kinase (pSFK) pY416, rabbit anti-human pERK1/2 pT202/pY204 (clone 197G2), and rabbit anti-HA-tag (clone C29F4) (all Cell Signaling Technologies).
Surface and Intracellular Antigen Flow Cytometry Staining
0.5 Mio cells were harvested and washed twice with 1ml FACS buffer (PBS + 2% FCS). Cells were fixed with 150μl pre-warmed fixation solution (BD Cytofix®, BD Biosciences) for 10 min at 37°C. For staining of intracellular antigens, fixed samples were washed twice in 150μl permeabilization buffer (BD Perm/Wash I, BD Biosciences), re-suspended in 150μl permeabilization buffer, and incubated at RT for 30 min. Permeabilized cells were stained in 50μl permeabilization buffer containing the respective antibody dilution. For fluorescent-conjugated primary antibody staining, samples were incubated for 2h at 4°C. When fluorescent-conjugated secondary antibodies were used, they were diluted in 50μl permeabilization buffer and added to cells for 30 min at RT in the dark. Cells were washed 3 times with 1ml permeabilization buffer after each staining and twice with 1ml FACS buffer before surface staining. For staining of surface-expressed LNGFR, mouse anti-LNGFR-PE was added in 50μl of FACS buffer and stained for 30 min at RT in the dark. Cells were washed 3 times in FACS buffer, and samples were acquired at a LSRFortessa flow cytometer (BD Biosciences). The following antibodies were used:
| Antigen | Species | Fluorochrome | Clone | Supplier |
| HA | Rabbit | AF488 | C29F4 | CST |
| LCK | Mouse | n.a. | 3A5 | Santa Cruz Biotechnology |
| Mouse IgG (H+L) | Goat F(ab’)2 | AF647 | CST | |
| LNGFR (CD271) | Mouse | PE | ME20.4-1.H4 | Milteny Biotec |
Calcium-Flux Assay
Cells were harvested and rested for 2 h at 37°C, 5% CO2 in RPMI 1640 Glutamax (Gibco) 10mM Hepes without FCS. Cells were labeled with 5μM Indo-1-AM (Invitrogen) in RPMI 10mM Hepes for 30 min in the dark at 37°C, 5% CO2. After 30 min, RPMI 10mM Hepes 5% FCS was added to remove excess Indo-1-AM. After 30 min, cells were washed twice in RPMI 5% FCS and stained with anti-LNGFR-PE for 30 min in RPMI 5% FCS at RT in the dark. Cells were washed twice in 5ml RPMI 5% FCS and resuspended in RPMI 5% FCS before being acquired at a LSRFortessa flow cytometer (BD Biosciences). For calcium-flux measurement, cells were acquired for 1 min to record the baseline before addition of mouse anti-CD3 (BioLegend, clone OKT3) and anti-CD28 (clone CD28.2, BioLegend) both 2μg/ml followed by crosslinking with 4μg/ml goat anti-mouse IgG after 2 min. After 9 or 10 min, 1μM ionomycin was added to achieve maximum calcium-flux. Data were analyzed using FlowJo V9 (TreeStar).
Immunophenotyping of Peripheral Blood Mononuclear Cells
Patient’s and healthy control PBMCs were thawed and washed with PBS (Gibco). An antibody master mix was prepared in BD Brilliant stain buffer (BD Biosciences), and samples were stained for 15 min at RT. Sample acquisition was performed on a LSRFortessa flow cytometer (BD Biosciences), and data were analyzed using FlowJo V9 (TreeStar). The following antibodies were used:
| Antigen | Fluorochrome | Clone | Supplier |
| CD45 | BV480 | HI30 | BD |
| CD3 | BUV496 | UCHT-1 | BD |
| CD4 | BUV395 | RPAT4 | BD |
| CD8 | PE-Cy5 | RPA-T8 | BD |
| CD45RA | BUV737 | HI100 | BD |
| CD45RO | BB515 | UCHL1 | BD |
| CD25 | PE | M-A251 | BD |
| CD27 | APC-R700 | M-T271 | BD |
| CD28 | BB700 | L293 | BD |
| HLA-DR | BV711 | G46-6 | BD |
| CXCR3 | PE-CF594 | 1C6 | BD |
| CCR7 | BV421 | G043H7 | Biolegend |
| CCR6 | BV785 | G034E3 | Biolegend |
| CD127 | APC | A019D5 | Biolegend |
| CCR4 | PE-Cy7 | L291H4 | Biolegend |
| CXCR5 | BV605 | J252D4 | Biolegend |
| CD38 | BV650 | HB-7 | Biolegend |
Fixable viability stain 780 (BD Bioscience) was used for viability staining.
Results
Case Description of a Girl with Profound T-Cell Immune Deficiency
The female patient was born at term to consanguineous parents of Syrian descent (Fig. 1A). There were 4 healthy siblings and one sister, who had died of respiratory failure due to a fulminant respiratory infection at the age of 7 months. The patient’s postnatal presentation, including body weight, body height, and head circumference, was unremarkable. Newborn screening had not been performed, because the patient did not have access to a newborn screening program. She developed normally until the age of 6 months, when she was admitted to a community hospital with fever and coughing, a diffuse maculo-papular rash, and oral and perianal candidiasis. After 1 month, she was transferred with progressive respiratory failure to Erciyes University, Kayseri, for further evaluation and treatment. The chest X-ray showed diffuse bilateral infiltrates (Fig. 1B). At the time of transferal, inflammation markers and leukocyte count were normal, but she had lymphocytopenia [lymphocytes 1,550/mm3 (N. 4,000–13,500)] and thrombocytosis [platelets 439,000/mm3 (N. 310,000 ± 68,000)]. Suspicion of an IEI was raised, and flow cytometric analysis of lymphocyte subsets revealed T-cell lymphocytopenia with an absolute reduction in helper and cytotoxic T-cells, while B- and NK-cells numbers were normal [CD3+ T-cells, 667/mm3 (N. 2,400–8,100); CD4+ T-cells, 289/mm3 (N. 1,400–5,200); CD8+ T-cells, 332/mm3 (N. 600–3,000)] (Table 1, row 1). High viral loads of cytomegaly virus (CMV) (6.2 × 106 copies/ml), Epstein-Barr virus (EBV) (4.5 × 103 copies/ml), and adenovirus (ADV) (16.4 × 106 copies/ml) were detected in the blood and ganciclovir, and cidofovir treatment was initiated. In addition, because the patient had received BCG vaccination at the age of 2 months, treatment with rifampicin and isoniazid was started, but discontinued after 2 months because of hepatitis with elevated liver function tests (LFTs) (AST 1,166 U/l; ALT 632 U/l; GGT, 218 U/l; LDH 1,305 U/l). LFTs gradually decreased to baseline over the course of 4 weeks. While EBV and ADV became undetectable after 2 weeks of treatment, CMV viral load was still elevated after 40 days (3.6 × 106 copies/ml). After exclusion of ganciclovir resistance, foscarnet was added, and after 2 weeks of consecutive combined treatment, a decline of CMV viral load could be observed (427 copies/ml). Overall, her clinical condition stabilized such that after 3 months of anti-infective treatment, she could be discharged with trimethoprim-sulfamethoxazole, fluconazole, and acyclovir prophylaxis and was scheduled for control at the pediatric hematology-oncology outpatient clinic. Allogeneic hematopoietic cell transplantation was offered but declined by the caregivers. Unfortunately, the family was lost to follow-up, and the patient died at the age of 12 months, 4 months after discharge, most probably due to respiratory failure following severe pneumonia.
Fig. 1.

A novel biallelic missense variant in the LCK kinase domain. A Pedigree of the consanguineous family. The patient is indicated with an arrow. B Chest X-ray of the patient upon admission to Erciyes University Hospital in Kayseri. C Sanger sequencing of the index patient and indicated family members. Consensus amino acid and DNA sequence indicate on top, variant sequence below. D CADD vs. minor allele frequency (MAF)-plot of homozygous variants in LCK present in the gnomAD database and variants LCK C465R and LCK L341P [1]. Created with the PopViz webserver application [38]. The CADD cut-off was calculated with a 95% confidence interval. E Scheme of LCK protein: SH, Src homology region; UD, unique domain; KD, kinase domain. Numbers below indicate starting/ending aa residues of the individual domains. Arrows localize the variants L341P [1] and C465R. F (Right) Ribbon diagram of the X-ray structure of the kinase domain of human LCK in an open conformation (PDB ID: 3LCK) based on [17]. α-helices in purple, β-sheets in yellow, unstructured regions in grey. Residues Y394 and C465 are highlighted. (Left) Detailed view of the local structure surrounding C465. Carbon atoms depicted in green, nitrogen in blue, oxygen in red, and sulfur in yellow. Residues C465, P466, and Y469 are highlighted, and dashed lines indicate hydrogen bonds. Image created with Mol*Viewer [39] and RCSB PDB [40]. G Logo plot of LCK sequence conservation across the species indicated on the left. Created with Jalview [41]
Table 1.
| Reference | LCK mutation | Age (at onset) | Sex | Protein levels | Clinical phenotype | CD3+ T-cells | CD4+ T-cells | CD8+ T-cells | CD19+ B-cells | IgM | IgG | IgA | IgE (reference range) | LCK signaling analysis | Scientific evaluation | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Outcome | mm3 or % (reference range) | g/l (reference range) | |||||||||||||||
| 1 | Lanz et al. 2023 | c.1393T>C, p.C465R | 6 mos | F | Greatly reduced | Profound T-cell immune deficiency | Death due to respiratory failure | 667 (2,400–8,100) 32.7 % (60-85) | 289 (1,400–5,200) 14.2% (29-59) | 332 (600–3,000) 16.3% (19–48) | 828 (300–1,400) 40.6% (11-16) | 0.19 (0.36–0.77) | 4.86 (3.76–6.85) | 0.11 (0.09-0.38) | 0.37 U/ml (0-13) | Reduced Ca2+-flux, pZAP70 and pERK | Complete LCK deficiency |
| 2 | Li et al. 2016 [2] | Intronic c.188-2A>G (ΔExon3) | 23 ys | F | Not shown | Atypical epidermo-dysplasia verruciformis with T-cell defect | Death due to invasive squamous cell carcinoma (SSC) | Not shown | Not shown, but reduced | Not shown | Not shown | Not shown | Not shown | Not shown | Suggestive of hypomorphic LCK deficiency with residual LCK WT expression | ||
| 18 ys | M | SSC, death, reason unknown | |||||||||||||||
| 15 ys | F | Alive | |||||||||||||||
| 3 | Hauck et al. 2012 [1] | c.1022T>C, p.L341P | 18 mos | F | Greatly reduced | Profound T-cell immune deficiency | Death due to veno-occlusive disease d7 post-HSCT | 1,254 (2,100–6,200) | 154 (1,300–3,400) | 1,012 (620–2,000) | 506 (720–2,600) | 3.06 (0.5–1.53) | 8.7 (4.82–8.96) | 1.21 (0.33–1.22) | <2 (<2 kUI/L) | Reduced Ca2+-flux and pTyr incl. pERK and pZAP70 after anti-CD3 stimulation | Complete LCK deficiency |
| 4 | Sawabe et al. 2001 [26] | Not identified. 2 LCK isoforms: WT & ΔExon7 | 22 ys | M | 60% reduced | CVID | Alive, mild phenotype without treatment. | 200 (n/a) 23% (58–78) | 50 (n/a) 6% (33–52) | 150 (n/a) 18% (119–34) | 310 (n/a) 37% (3–13) | 0.031 (n/a) | 4.21 (n/a) | 0.72 (n/a) | <29 U/ml (n/a) | Not shown | Not LCK-deficient. Expression of LCK ΔExon7 may be consequence of an unknown CVID. |
| 5 | Goldman et al. 1998 [25] | Not identified. 2 LCK isoforms: WT & ΔExon7 | 2 mos | M | Greatly reduced | SCID | HSCT | 760 (2,000–7,035) | 330 (1,520–4,830) | 430 (640–2,730) | 710 (800–3,600) | 5 (33–155) 0.05 (3.3–15.5) a, b | 105 (252–708) 1.05 (2.52–7.08)a, b | 6 (22–129) 0.06 (0.22–1.29)a, b | Not shown | Unperturbed Ca2+-flux and pTyr incl. pERK after anti-CD3 stimulation | Not LCK-deficient. Expression of LCK ΔExon7 may be consequence of an unknown SCID. |
aValues at the age of 1 mo, before IVIG
bThe original publication gives no units for Igs. According to the reference range given, the values refer to mg/dl
A Novel LCK Variant Impairs LCK Protein Expression and Proximal TCR Signaling
The patient’s clinical presentation and immunological phenotype was suggesting a profound T-cell immune deficiency. Whole exome sequencing revealed a novel homozygous missense variant in exon 13 of LCK (HGNC:6524; c.1393T>C, p.C465R). Sanger sequencing confirmed a biallelic mutation in the patient, and both parents were found to be heterozygous supporting an autosomal-recessive inheritance pattern in this consanguineous family (Fig. 1C). Three of four healthy siblings (II.2, II.4, II.5) were heterozygous carriers, while one sister (II.3) was homozygous wildtype (Fig. 1C). The LCK variant was not present in the genome aggregation database (gnomAD) and predicted to be pathogenic by a combined annotation-dependent depletion (CADD) score [42] of 26.1 and a mutation significance cut-off (MSC) score [43] of 3.13. Only very few homozygous variants in LCK are found in gnomAD (Fig. 1D), none of them having similarly high CADD scores and low allele frequencies, and even heterozygous variants are rare (Fig. S1A). The gnomAD pLI score, which is a measurement of LOF-intolerance, is 0.99 for LCK, with a pLI ≥ 0.9 being LOF-intolerant [44], and a low observed/expected ratio (o/e) for pLOF variants (o/e = 0.067; 90% CI 0.027–0.21), suggestive of a haploinsufficient gene, is calculated [45]. For comparison, the pLI score for LOF variants in ZAP70 was 0.88 and the o/e ratio was 0.17 (90% CI 0.09–0.36)
The LCK variant resulted in an exchange of a small neutral cysteine to a large basic arginine (C465R) in a highly conserved region towards the end of the C-terminal lobe (C-lobe) of the KD [11] (Fig. 1E and F). C465 was not only conserved across species (Fig. 1G), but also in all members of the SRC kinases family (Fig. S1B), implying an important role for protein stability and/or function. In both open (PDB ID: 3LCK; Fig. 1F) and closed (PDB ID: 2pI0; Fig. S1C) forms of LCK, the sulfhydryl group of C465 forms a hydrogen bond with the neighboring residue P466, which in turn forms a hydrogen bond with Y469 in the α-helix H, and the introduction of an arginine was expected to disturb these interactions.
Immunoblotting for LCK in T-cell blasts from the patient and a healthy control revealed absent LCK protein expression in the patient (Fig. 2A, orange arrowheads), suggesting that C465R influenced either protein expression or stability, or both. Active LCK is phosphorylated on Y394, corresponding to Y416 of SRC. As expected, immunoblotting with an anti-SFK pY416 antibody showed almost complete loss of active SRC-family kinases corresponding to the absent active LCK doublet in the patient T-cells (Fig. 2B, red arrows). A minor band detectable most likely represented residual FYN-T expression in patient T-cells (Fig. 2B, black arrow). Consistent with the absence of LCK protein expression, phosphorylation of ZAP70 was absent after stimulation with either anti-CD3 or anti-CD3/CD28 as compared to a healthy control (Fig. 2B). Very low residual levels of pERK1/2 (loading corrected pERK1/2 band intensity: HC unstimulated = 1, patient unstimulated = 0.2) were detectable in unstimulated patient cells that did not increase after stimulation with either anti-CD3 or anti-CD3/CD28 (Fig. 2B). Importantly, although LCK and ZAP70 phosphorylation was not rescued by stimulation with PMA/ionomycin (P/I), pERK1/2 levels were normal after P/I stimulation in patient T-cells as compared to the healthy control (loading corrected pERK1/2 band intensity: HC unstimulated = 1, HC P/I = 5.8, patient unstimulated = 0.2, patient P/I = 5.8) and inhibition with the LCK-specific inhibitor A770041 abrogated ZAP70 and ERK1/2 phosphorylation in T-cells of the healthy control (Fig. 2B). Collectively, these results suggested that the LCK C465R variant most severely affected LCK protein expression and LCK-dependent proximal TCR signaling in patient T-cells.
Fig. 2.

Reduced protein expression and TCR signaling due to LCK C465R. A LCK and GAPDH immunoblots of whole cell lysates of patient or healthy control (HC) T-cell blasts unstimulated or stimulated with anti-CD3 (2 min), anti-CD3/CD28 (2 min), PMA/ionomycin (P/I, 5 min) or treated with the selective LCK-inhibitor A770041 (A77, 10 min). Orange arrowheads indicate LCK bands. B pZAP70, pSFK (pY416), pERK1/2 or GAPDH immunoblots, conditions as in A. Red arrowheads indicate pLCK bands, black arrowheads pFYN-T. Numbers below pERK1/2 blot indicate fold change pERK1/2 band intensity (HC unstimulated =1), intensity normalized to GAPDH loading (numbers below GAPDH blot; GAPDH signal intensity normalized to HC unstimulated) C LCK, HA-tag and GAPDH immunoblots of whole cell lysates of Jurkat E6.1, J.CaM1.6, and stable transduced cells line expressing either LCK WT or LCK C465R with or without on C-terminal HA-tag or transduced with pCDH containing only the expression cassette for the extracellular domain of the low-affinity nerve growth factor receptor (LNGFR) (J.CaM EV). D Overlay of Ca2+-flux measurement in the indicated cell lines. Experiment representative of 3 independent experiments. E pZAP70, pSFK (pY416), pERK1/2 immunoblots in J.CaM1.6 cells transduced with pLJM1 containing LCK WT or LCK C465R and stimulated with either anti-CD3 (2, 5 or 15 min), anti-CD3/CD28 (2 or 5 min), PMA/ionomycin (5 min) or left untreated
To verify the impact of the mutation on LCK expression and/or function in a model system, LCK wildtype (WT) or LCK C465R with or without a C-terminal HA-tag was expressed in LCK-deficient J.CaM1.6 Jurkat cells (J.CaM LCK WT or C465R) that were transduced with a lentiviral plasmid containing also the extracellular domain of the low-affinity nerve growth factor receptor (LNGFR, CD271) to identify transduced cells, or a puromycin-selectable plasmid (pLJM1). As in the patient cells, LCK C465R was poorly expressed in J.CaM1.6 Jurkat cells (Fig. 2C, orange arrowheads, and Figs. S2A and S2B). The defect in mounting a sufficient TCR signaling response was verified by Ca2+-flux measurements showing no response in J.CaM LCK C465R as opposed to J.CaM LCK WT (Fig. 2D and Figs. S2B and S2C). Immunoblotting for pY416 SFK, pZAP70, and pERK1/2 after stimulation with either anti-CD3 or anti-CD3/CD28 showed absent ZAP70 and LCK and ERK1/2 phosphorylation in J.CaM LCK C465R cells as compared to J.CaM LCK WT, while stimulation with P/I induced similar levels of pERK1/2 in J.CaM LCK C465R and J.CaM LCK WT (Fig. 2E and Fig. S2E), corroborating the defect seen in patient cells.
Aberrant Immune Phenotype of Bi- and Monoallelic LCK-Deficient T-Cells
To further characterize the impact of the LCK C465R variant, we performed immune phenotyping of cryopreserved PBMCs from the patient and her mother by flow cytometry. Two adult healthy controls were used alongside. We confirmed a severe loss of total CD3+ T-cells (patient 19.4% of CD45+ lymphocytes, mother 62%, HC1 58.6%, HC2 73.9%) and CD4+ T-cells (patient 1.81% of CD3+ lymphocytes, mother 23.0%, HC1 44.5%, HC2 55.2%) in the patient (Fig. 3A, upper row). CD8+ T-cells numbers were less affected, resulting in an inversed CD4/CD8 ratio of 0.02 (Fig. 3A, upper row). In addition, patient T-cells exhibited a reduction of CD4 and CD8 co-receptor surface expression, which has previously been described as pathognomonic for murine and human LCK deficiency [1, 23] (Fig. 3A and Figs. S3A and S3B). Unexpectedly, the heterozygous mother also displayed a reduction in CD4/CD8 ratio (0.36) and CD4 and CD8 co-receptor expression (Fig. 3A and Figs. S3A and S3B). Besides the decrease of CD4+ and CD8+ T-cells, the patient also showed a reduction of αβT-cell frequency with an increase of γδT-cells (Fig. 3A, lower row) expressing higher levels of TCRγδ on the surface as compared to healthy controls (Fig. S3B).
Fig. 3.

Immune phenotype of patient, mother, and two healthy controls (HC) by flow cytometry. Markers used for staining are indicated on the arrows next to the contour plots, gated populations above. For gating strategy, see Fig. S4. A (upper row) CD4 vs. CD8 expression on CD3+ lymphocytes, CD4/CD8 ratio in red; (lower row) αβ and γδT-cell frequency in CD3+ lymphocytes. B CD4+ T-cell differentiation (upper row) and CD57 expression (lower row). C CD8+ T-cell differentiation (upper row) and CD57 expression (lower row). D CD4+ CD45RO+ CCR6- T helper cell phenotype. F Treg cells
An almost complete loss of naïve CD4+ and CD8+ T-cells (1.67% and <1%, respectively) could be observed in the patient (Figs. 3B and C, upper rows). Most patient CD4+ T-cells were CD45RO+ CCR7- effector memory T-cells (EM: 70.3%), but we also noted an increase in the terminally differentiated CD45RO- CCR7- population (TEM: 3.2 % vs HC1: 0.52% and HC2: 0.072%) that has been described to expand in chronic virus infections [46] (Fig. 3B). Patient CD8+ T-cells were evenly distributed between the effector memory and terminally differentiated effector cell compartment (EM: 48.2%; TEM: 48.3%) (Fig. 3C). Further, both CD8+ and CD4+ CD45RO+ memory T-cells in the patient were displaying an unusual CD38hi HLA-DRhi double positive state, most likely reflecting exhaustion [47–50] (Fig. S3C). As with co-receptor expression, we observed that the T-cells of the heterozygous mother displayed an intermediate phenotype between the T-cells of the patient and the two healthy controls with partial loss of naivety of both CD4+ and CD8+ T-cells (19% and 6.7%, respectively) and a concurrent increase in CD4+ EM T-cells, while CD8+ T-cells were equally distributed between EM and TEM (Figs. 3B and C). Additionally, CD57, a marker of chronic immune activation and senescence [51], was increased on both CD4+ and CD8+ T-cells in the patient (36.3% and 82.8%, respectively), but also in the mother (14.9% and 70.7%, respectively) (Fig. 3B and C, lower rows). Additionally, CD8+ T-cells had almost no CD28 surface expression, another sign of terminal differentiation and chronic immune activation and senescence [52, 53] (Fig. S3D).
Further, analysis of CD4+ helper T-cell subsets revealed a decrease of CCR6- CXCR3+ CCR4- Th1 cells in the patient and the mother (4.31% and 6.51%) (Fig. 3E). The percentage of CCR6- CXCR3- CCR4+ Th2 cells in the patient was like the healthy controls (Fig. 3E). Within the CCR6+ T-cell population, percentages of CCR4+ Th17 cells were comparable between patient, mother, and healthy controls (37.5%, 42%, 47.8% and 44.6%, respectively). However, because of a 50% reduction in CD4+ CCR6+ T-cells (Fig. S3E, upper row), the absolute number of Th17 cells was decreased in the patient. Furthermore, both patient and mother had very low percentages of CCR6+ CXCR3+ Th17.1 cells (5.0 and 8.43%) (Fig. S3E, lower row).
CD4+ CD25hi CD127lo Treg cell frequency was elevated in the patient (20.2%), although we noticed that CD25 expression within the Treg gate was relatively dimmer as compared with the healthy controls, and thus activated conventional T-cells may have been included in this gate [54] (Fig. 3F). Patient Tregs showed either an activated (CD45RO+ HLA-DR+) or memory (CD45RO+ HLA-DR-) phenotype (Fig. S3F).
Taken together, the LCK-deficient patient showed a severe absolute loss of total CD4+ and to a lesser extent of CD8+ T-cells with a relative increase in γδT-cells and Treg cells and a decrease of Th1 cells. Remaining CD4+ and CD8+ T-cells, including Treg cells, showed an activated, memory phenotype with an increase in CD57 and loss of CD28, indicative of immune senescence. Importantly, surface expression levels of the CD4 and CD8 co-receptors were reduced. Of note, the heterozygous mother showed an intermediate T-cell immune phenotype.
Discussion
In the current study, we identified a novel biallelic missense LCK c.1393T>C, p.C465R variant in a patient from a consanguineous Syrian family with profound T-cell immune deficiency characterized by complete LCK protein expression deficiency and ensuing proximal TCR signaling- and CD4 and CD8-co-receptor-mediated functional and phenotypical defects. Both parents as well as three of four healthy siblings were monoallelic carriers of the LCK variant. Another child not amenable to analysis had passed away following a severe infection in the first year of life. Clinically, the patient presented in infancy with severe infections and passed away at the age of 12 months due to respiratory failure likely secondary to an infection, precluding more in-depth experimental analysis of primary T-cells.
We detected very low levels, if any, of the variant LCK by immunoblotting in patient cells and LCK-deficient J.CaM1.6 cells transduced with LCK C465R, suggesting reduced protein expression or stability, or both. However, we do not know the exact mechanism that led to reduced LCK protein expression, i.e., whether protein translation or folding was disturbed, or if the protein was subject to faster degradation. It is likely that the replacement of a small neutral cysteine with a larger basic arginine disturbed the local configuration with possible consequences beyond the local structure. Interestingly, the adjacent α-helices H and I form part of the interface of c-SRC with the kinase CSK that phosphorylates the inhibitory tyrosine Y527 (Y505 in LCK) [55]. For a better understanding of the structural changes induced by C465R and potential functional consequences, further analysis with molecular dynamic simulation of the variant compared to wildtype LCK could be employed.
To our understanding, this is the third LCK variant leading to a LOF phenotype to be described in the biomedical literature [1, 2]. The first reported LCK deficiency was due to a pathogenic LCK c.1022T>C missense variant, leading to the residual expression of a signaling incompetent LCK p.L341P with abrogated protein tyrosine phosphorylation and Ca2+-flux (Table 1, row 3) [1]. The patient presented early in life with profound T-cell immune deficiency characterized by severe infections, autoinflammation, autoimmunity, and ensuing failure to thrive. Li et al. reported 3 siblings of a consanguineous family presenting with recurrent pneumonia and severe viral skin disease leading to malignant transformation [2]. The patients had an intronic LCK c.188-2A>G splice site variant resulting in skipping of exon 3 and mRNA decay. Although the impact on LCK protein level, TCR signaling, and T-cell immune phenotype was not reported, genetic and clinical data alongside with CD4+ T-cell lymphocytopenia suggested a hypomorphic LCK deficiency (Table 1, row 2).
Two additional studies have shown defective LCK protein expression in the context of severe combined immune deficiency (SCID) and common variable immune deficiency (CVID), respectively [25, 26]. Goldman et al. reported a boy presenting with SCID with severe infections and profuse diarrhea (Table 1, row 5) [25]. He was lymphocytopenic with a greater reduction in CD4+ than CD8+ T-cells and reduced B-cells with hypogammaglobulinemia. CD8+ T-cells had absent CD28 surface expression and reduced LCK expression; however, TCR proximal protein tyrosine phosphorylation, Ca2+-flux and ERK-phosphorylation were unperturbed. cDNA analysis from PBMCs revealed the presence of LCK wildtype cDNA and an additional LCK transcript lacking exon 7 (LCK ΔExon7) that genetically and mechanistically remained unexplained. The adult CVID patient reported by Sawabe et al. had almost asymptomatic bihilar lymphadenopathy, reduced CD4+ T-cells, class-switched memory B-cells, and IgG (Table 1, row 4) [26]. Two LCK transcripts, corresponding to LCK wildtype and to LCK ΔExon7, were detected in the patient’s PBMCs, and reduced LCK protein expression was noted. Exon 7 encodes a part of the kinase domain including the ATP binding site necessary for kinase activity (NP_005347.3, aa 212-262). Indeed, Germani et al. [56] and we (Hauck and Latour, unpublished data) showed that LCK ΔExon7 protein or a variant with a mutation in the active site (K273E) lost its catalytic kinase activity. LCK ΔExon7 is the sole transcript expressed in the J.CaM1.6 cell line that lacks LCK protein expression and has been generated under continuous mitogenic stimulation with PHA from Jurkat E6.1 cells [4, 57]. Low levels of the transcript coding for LCK ΔExon7 are detectable in the parental Jurkat E6.1 cell line, thus the chronic PHA stimulation might have given a selective survival advantage to the LCK protein-deficient cells. Furthermore, LCK ΔExon7 was shown to be expressed in PBMCs of healthy donors [58, 59]. Overall, we conclude that in both cases, the expression of LCK ΔExon7 was probably not the cause, but the consequence of the underlying genetically unresolved SCID and CVID, respectively.
The immune phenotype of the LCK-deficient patient described here (LCK p.C465R) was very similar to that reported by Hauck et al. (LCK p.L341P) as well as to that of the lck-/- mouse model [23, 60] with pronounced T-cell lymphocytopenia, inverted CD4/CD8 ratio, loss of T-cell naivety, exhausted memory phenotype in both CD4+ and CD8+ T-cells, and expansion of γδT-cells. However, we noted some differences between the two cases such as the percentage of Treg cells and the composition of immunoglobulin isotypes that may reflect either different functional consequences of the individual mutations or contact with different infectious agents and/or influences by additional genetic variants present in the patients. Immunologic workup of further LCK-deficient cases will help clarifying genotype-phenotype-correlations.
The immune phenotype of LCK deficiency reported here for the first time included T helper subsets showing a decrease of Th1, Th17, and Th17.1 cells. It is important to note, however, that due to the low frequency of CD4+ T-cells, the absolute numbers of events in the analysis of T helper subsets were low and analysis of further patients will give more clarity. The T helper subset phenotype is contrary to what has been reported from a mouse model with a post-thymic LCK gene deletion, which showed skewing towards Th1 responses in CD4+ T-cells [61], while peripheral T-cells in non-conditional lck-/- mice have not been analyzed in such detail. This may be due to the differences in the environment, in particular the chronic viral infections that the reported patient has suffered from.
A prominent feature of human and murine biallelic LCK deficiency is reduced surface expression of the co-receptors CD4 and CD8 on T-cells [1, 23], and here we corroborated this finding. Additionally, we noted reduced co-receptor expression in an individual with monoallelic LCK deficiency, which previously has been described for CD4 in lck+/- mice [23]. While CD4 internalization and degradation require phosphorylation of S408 by PKCθ and dissociation of LCK from CD4 to enable recognition of a dileucine motif by the clathrin adaptor AP2 [62–64], the mechanism for CD8 is less well understood and the shorter cytoplasmic tail of CD8 is devoid of both serin and dileucine motifs. This is in line with a recent study of a variant LCK unable to bind co-receptor, reporting differential regulation of CD4 and CD8 surface expression [21]. Thus, reduced LCK expression may increase CD4 endocytosis or by other mechanisms impede stabilization and reduce CD4 surface expression. Further experiments are needed to clarify the causes of reduced co-receptor expression in LCK deficiency, but we propose that it may be of value for early detection of mono- and/or biallelic LCK deficiency.
Besides reduced co-receptor expression, the mother with monoallelic LCK deficiency had further immune phenotypic alterations such as loss of CD4+ T-cells with an inverse CD4/CD8 ratio, CD4+ and CD8+ T-cell loss of naivety, and exhaustion. These changes could also be a sign of chronic viral infections, such as CMV [65]. Unfortunately, we were not able to acquire clinical information or blood samples for further analyses of the entire family. It is noteworthy that as opposed to ZAP70 or ITK deficiency, only one other case of complete LCK deficiency has been reported [1] and only two more are found in the ClinVar Database. The homozygosity in the case presented by Hauck et al. was due to a rare uniparental disomy; thus, only the mother was heterozygous for the LCK mutation [1]. Taken together, this raises the possibility that heterozygosity for a LOF variant in LCK may lead to clinical manifestations and purifying selection. Importantly, a recent study reported impaired proximal TCR signaling (pCD3 ζ, pZAP70, total-pY) in lck+/- mice relative to WT mice [18]. Thus, it will be of interest to carefully screen individuals with immune dysregulation for monoallelic LCK deficiency in the future.
In summary, we report the second case of complete biallelic LCK deficiency causing profound T-cell immune deficiency with CD4+ and CD8+ T-cell lymphocytopenia and reduced CD4 and CD8 cell surface co-receptor expression. In individuals with suspicion for mono- or biallelic LCK deficiency co-receptor expression should be analyzed and could streamline immediate genetic workup.
Supplementary Information
A CADD vs. minor allele frequency (MAF)-plot of heterozygous variants in LCK present in the gnomAD database and variants LCK C465R and LCK L341P[1]. Created with the PopViz webserver application[38]. The CADD cut-off was calculated with a 95% confidence interval. B Logo plot of sequence conservation across the human SRC-family kinases indicated on the left. Created with Jalview [41]. C (Right) Ribbon diagram of the X-ray structure of the kinase domain of human LCK in a closed conformation (PDB ID: 2PL0) based on [66] [17]. α-helices in purple, β-sheets in yellow, unstructured regions in grey. Residues Y394 and C465 are highlighted. (Left) Detailed view of the local structure surrounding C465. Carbon-atoms depicted in green, nitrogen in blue, oxygen in red and sulfur in yellow. Residues C465, P466 and Y469 are highlighted, and dashed lines indicate hydrogen bonds. Image created with Mol*Viewer [39] and RCSB PDB [40] (EPS 955 kb).
{kind=link}
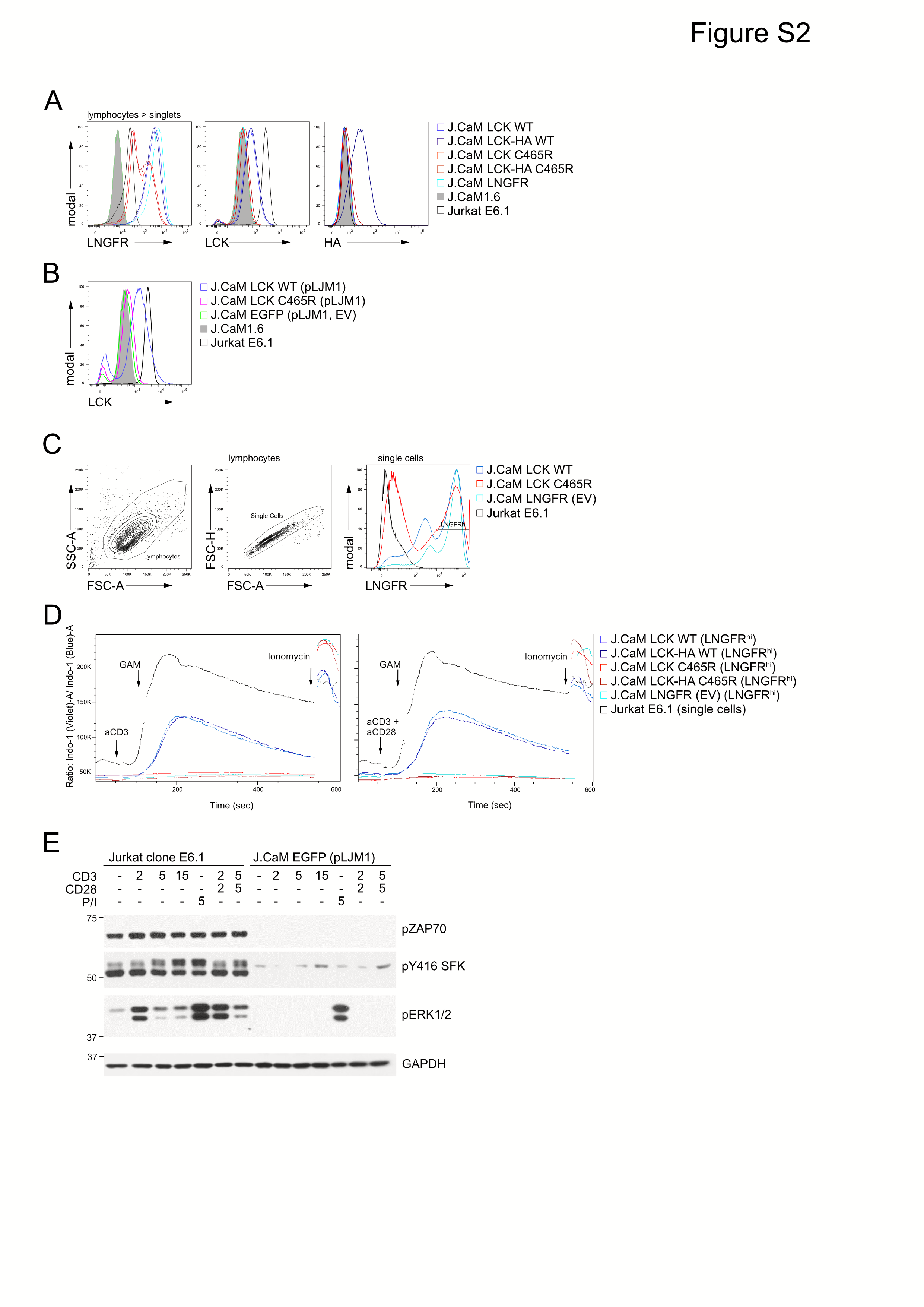
Supplementary Figure S2 A Histogram overlay of flow cytometry staining of Jurkat E6.1, J.CaM1.6 and stable transduced celllines with pCDH expressing either LCK WT or LCK C465R with or without an C-terminal HA-tag or transduced with pCDH empty vector containing only the expression cassette for the extracellular domain (ED) of the low-affinity nerve growth factor receptor (LNGFR) stained with anti-LNGFR-PE, anti-LCK + secondary anti-mouse AF647 and anti-HA-AF488. B Histogram overlay of flow cytometry staining of Jurkat E6.1, J.CaM1.6 and stable transduced celllines with pLJM1 expressing either LCK WT or LCK C465R transduced with empty vector pLJM1-EGFP anti-LCK + secondary anti-mouse AF647 and anti-HA-AF488. C Gating strategy used for Ca2+-flux measurements. J.CaM1.6 cells transduced with the lentiviral plasmid that co-expresses the extracellular domain (ED) of LNGFR were gated on LNGFRhi for Ca2+-flux measurements, while for untransduced Jurkat E6.1 that do not express LNGFR-ED single cells were acquired. D Ca2+-flux measurements in J.CaM1.6 expressing LCK WT (light blue), LCK-HA (dark blue), LCK C465R (light red), LCK-HA C465R (dark red), Jurkat E6.1 (black) and J.CaM1.6 transduced with empty vector (turquoise) stimulated with anti-CD3 (left) or anti-CD3/ anti-CD28 (right). E pZAP70, pSFK (pY416), pERK1/2 immunoblots in Jurkat E6.1 cells or J.CaM1.6 transduced with empty vector pLJM1-EGFP and stimulated with either anti-CD3 (2, 5 or 15 min), anti-CD3/CD28 (2 or 5 min), PMA/ionomycin (5 min) or left untreated. (PNG 704 kb)
Supplementary Figure S3 Immune phenotype of patient, mother and two healthy controls (HC) by flow cytometry. Markers used for staining as indicated on the arrows next to the contour plots, gated populations above. A Contour plot overlay of CD4 vs. CD8 staining of patient, mother, and healthy control (HC)1 (all in red) with HC2 (blue) B Histogram overlays of the patient, mother and 2 healthy controls for surface expression of CD4, CD8, TCRαβ and TCRγδ. C CD38 vs. HLA-DR staining on CD45RO+ memory CD4+ (upper row) and CD8+ (lower row) T-cells of the patient, mother and 2 healthy controls. D CD28 vs. CD27 staining on CD8+ T-cells, color-coded by CD45RA expression E (upper row) CCR6 expression on CD4+ CD45RO+ T-cells and (lower row) CD4+ CD45RO+ CCR6+ T helper cell phenotype F Treg phenotype characterized by CD45RO and HLA-DR expression. (EPS 2842 kb)
Supplementary Figure S4 Gating strategy used for flowcytometric immune phenotyping. (EPS 596 kb)
Acknowledgements
We thank the patient and her family for their personal contribution. We thank Gabriele Heilig for technical assistance with sequencing and the flow cytometry facility at the Dr. von Hauner Children’s Hospital for excellent technical support with immunophenotyping.
Author Contribution
Anna-Lisa Lanz, Serife Erdem, and Raffaele Conca performed experiments. Alper Ozcab, Gulay Ceylaner, Murat Cansever, Serdar Ceylaner, Turkan Patiroglu, and Ekrem Unal provided clinical information. Thomas Magg, Oreste Acuto, Sylvain Latour, Christoph Klein, Ahmet Eken, and Fabian Hauck conceptualized the study. Anna-Lisa Lanz and Fabian Hauck wrote the manuscript. Ahmet Eken and Fabian Hauck supervised experiments. All authors read and approved the final manuscript.
Funding
Open Access funding enabled and organized by Projekt DEAL. Fabian Hauck received funding from the Care-for-Rare Foundation (C4R, 160073), the Else Kröner-Fresenius Stiftung (EKFS, 2017_A110), and the German Federal Ministry of Education and Research (BMBF, 01GM2206D). Anna-Lisa Lanz received funding from the Care-for-Rare Foundation. This study was partly supported by grants from the Turkish Academy of Science GEBIP and Science Academy BAGEP awards to Ahmet Eken, and Erciyes University BAP grant TCD2021-10863 to Ekrem Unal.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Declarations
Ethics Approval
This study was approved by Erciyes University local ethics committee (Permit number: 2018/388) and conducted according to current ethical and legal guidelines and in line with the Declaration of Helsinki.
Consent to Participate
Written informed consent was obtained from the parents.
Consent for Publication
The authors affirm that human research participants or their legal guardians provided informed consent for publication of the images in Figures 1, 2, 3, and Supplementary Figures S1-4.
Conflict of Interest
The authors declare no competing interests.
Footnotes
The original online version of this article was revised due to revised Figure 2
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Anna-Lisa Lanz and Serife Erdem contributed equally to this work.
Ahmet Eken and Fabian Hauck contributed equally to this work.
Change history
1/16/2024
A Correction to this paper has been published: 10.1007/s10875-024-01654-4
Contributor Information
Ahmet Eken, Email: ahmet.eken@gmail.com.
Fabian Hauck, Email: fabian.hauck@med.uni-muenchen.de.
References
- 1.Hauck F, Randriamampita C, Martin E, Gerart S, Lambert N, Lim A, et al. Primary T-cell immunodeficiency with immunodysregulation caused by autosomal recessive LCK deficiency. J Allergy Clin Immunol. 2012;130(5):1144-52 e11. doi: 10.1016/j.jaci.2012.07.029. [DOI] [PubMed] [Google Scholar]
- 2.Li SL, Duo LN, Wang HJ, Dai W, Zhou EH, Xu YN, et al. Identification of LCK mutation in a family with atypical epidermodysplasia verruciformis with T-cell defects and virus-induced squamous cell carcinoma. Br J Dermatol. 2016;175(6):1204–1209. doi: 10.1111/bjd.14679. [DOI] [PubMed] [Google Scholar]
- 3.Palacios EH, Weiss A. Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation. Oncogene. 2004;23(48):7990–8000. doi: 10.1038/sj.onc.1208074. [DOI] [PubMed] [Google Scholar]
- 4.Straus DB, Weiss A. Genetic evidence for the involvement of the lck tyrosine kinase in signal transduction through the T cell antigen receptor. Cell. 1992;70(4):585–593. doi: 10.1016/0092-8674(92)90428-f. [DOI] [PubMed] [Google Scholar]
- 5.van Oers NS, Killeen N, Weiss A. Lck regulates the tyrosine phosphorylation of the T cell receptor subunits and ZAP-70 in murine thymocytes. J Exp Med. 1996;183(3):1053–1062. doi: 10.1084/jem.183.3.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Veillette A, Bookman MA, Horak EM, Samelson LE, Bolen JB. Signal transduction through the CD4 receptor involves the activation of the internal membrane tyrosine-protein kinase p56lck. Nature. 1989;338(6212):257–259. doi: 10.1038/338257a0. [DOI] [PubMed] [Google Scholar]
- 7.Acuto O, Di Bartolo V, Michel F. Tailoring T-cell receptor signals by proximal negative feedback mechanisms. Nat Rev Immunol. 2008;8(9):699–712. doi: 10.1038/nri2397. [DOI] [PubMed] [Google Scholar]
- 8.Brownlie RJ, Zamoyska R. T cell receptor signalling networks: branched, diversified and bounded. Nat Rev Immunol. 2013;13(4):257–269. doi: 10.1038/nri3403. [DOI] [PubMed] [Google Scholar]
- 9.Esensten JH, Helou YA, Chopra G, Weiss A, Bluestone JA. CD28 costimulation: from mechanism to therapy. Immunity. 2016;44(5):973–988. doi: 10.1016/j.immuni.2016.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim PW, Sun ZY, Blacklow SC, Wagner G, Eck MJ. A zinc clasp structure tethers Lck to T cell coreceptors CD4 and CD8. Science. 2003;301(5640):1725–1728. doi: 10.1126/science.1085643. [DOI] [PubMed] [Google Scholar]
- 11.Boggon TJ, Eck MJ. Structure and regulation of Src family kinases. Oncogene. 2004;23(48):7918–7927. doi: 10.1038/sj.onc.1208081. [DOI] [PubMed] [Google Scholar]
- 12.Bergman M, Mustelin T, Oetken C, Partanen J, Flint NA, Amrein KE, et al. The human p50csk tyrosine kinase phosphorylates p56lck at Tyr-505 and down regulates its catalytic activity. EMBO J. 1992;11(8):2919–2924. doi: 10.1002/j.1460-2075.1992.tb05361.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu W, Doshi A, Lei M, Eck MJ, Harrison SC. Crystal structures of c-Src reveal features of its autoinhibitory mechanism. Mol Cell. 1999;3(5):629–638. doi: 10.1016/s1097-2765(00)80356-1. [DOI] [PubMed] [Google Scholar]
- 14.Ostergaard HL, Shackelford DA, Hurley TR, Johnson P, Hyman R, Sefton BM, et al. Expression of CD45 alters phosphorylation of the lck-encoded tyrosine protein kinase in murine lymphoma T-cell lines. Proc Natl Acad Sci U S A. 1989;86(22):8959–8963. doi: 10.1073/pnas.86.22.8959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sieh M, Bolen JB, Weiss A. CD45 specifically modulates binding of Lck to a phosphopeptide encompassing the negative regulatory tyrosine of Lck. EMBO J. 1993;12(1):315–321. doi: 10.1002/j.1460-2075.1993.tb05659.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McNeill L, Salmond RJ, Cooper JC, Carret CK, Cassady-Cain RL, Roche-Molina M, et al. The differential regulation of Lck kinase phosphorylation sites by CD45 is critical for T cell receptor signaling responses. Immunity. 2007;27(3):425–437. doi: 10.1016/j.immuni.2007.07.015. [DOI] [PubMed] [Google Scholar]
- 17.Yamaguchi H, Hendrickson WA. Structural basis for activation of human lymphocyte kinase Lck upon tyrosine phosphorylation. Nature. 1996;384(6608):484–489. doi: 10.1038/384484a0. [DOI] [PubMed] [Google Scholar]
- 18.Horkova V, Drobek A, Mueller D, Gubser C, Niederlova V, Wyss L, et al. Dynamics of the coreceptor-LCK interactions during T cell development shape the self-reactivity of peripheral CD4 and CD8 T cells. Cell Rep. 2020;30(5):1504-14 e7. doi: 10.1016/j.celrep.2020.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu H, Littman DR. A kinase-independent function of Lck in potentiating antigen-specific T cell activation. Cell. 1993;74(4):633–643. doi: 10.1016/0092-8674(93)90511-n. [DOI] [PubMed] [Google Scholar]
- 20.Jiang N, Huang J, Edwards LJ, Liu B, Zhang Y, Beal CD, et al. Two-stage cooperative T cell receptor-peptide major histocompatibility complex-CD8 trimolecular interactions amplify antigen discrimination. Immunity. 2011;34(1):13–23. doi: 10.1016/j.immuni.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Horkova V, Drobek A, Paprckova D, Niederlova V, Prasai A, Uleri V, et al. Unique roles of co-receptor-bound LCK in helper and cytotoxic T cells. Nat Immunol. 2023;24(1):174–185. doi: 10.1038/s41590-022-01366-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rudd CE. How the discovery of the CD4/CD8-p56(lck) complexes changed immunology and immunotherapy. Front Cell. Dev Biol. 2021;9:626095. doi: 10.3389/fcell.2021.626095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Molina TJ, Kishihara K, Siderovski DP, van Ewijk W, Narendran A, Timms E, et al. Profound block in thymocyte development in mice lacking p56lck. Nature. 1992;357(6374):161–164. doi: 10.1038/357161a0. [DOI] [PubMed] [Google Scholar]
- 24.van Oers NS, Lowin-Kropf B, Finlay D, Connolly K, Weiss A. alpha beta T cell development is abolished in mice lacking both Lck and Fyn protein tyrosine kinases. Immunity. 1996;5(5):429–436. doi: 10.1016/s1074-7613(00)80499-9. [DOI] [PubMed] [Google Scholar]
- 25.Goldman FD, Ballas ZK, Schutte BC, Kemp J, Hollenback C, Noraz N, et al. Defective expression of p56lck in an infant with severe combined immunodeficiency. J Clin Invest. 1998;102(2):421–429. doi: 10.1172/JCI3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sawabe T, Horiuchi T, Nakamura M, Tsukamoto H, Nakahara K, Harashima SI, et al. Defect of lck in a patient with common variable immunodeficiency. Int J Mol Med. 2001;7(6):609–614. doi: 10.3892/ijmm.7.6.609. [DOI] [PubMed] [Google Scholar]
- 27.Arpaia E, Shahar M, Dadi H, Cohen A, Roifman CM. Defective T cell receptor signaling and CD8+ thymic selection in humans lacking zap-70 kinase. Cell. 1994;76(5):947–958. doi: 10.1016/0092-8674(94)90368-9. [DOI] [PubMed] [Google Scholar]
- 28.Keller B, Zaidman I, Yousefi OS, Hershkovitz D, Stein J, Unger S, et al. Early onset combined immunodeficiency and autoimmunity in patients with loss-of-function mutation in LAT. J Exp Med. 2016;213(7):1185–1199. doi: 10.1084/jem.20151110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lev A, Lee YN, Sun G, Hallumi E, Simon AJ, Zrihen KS, et al. Inherited SLP76 deficiency in humans causes severe combined immunodeficiency, neutrophil and platelet defects. J Exp Med. 2021;218(3) 10.1084/jem.20201062. [DOI] [PMC free article] [PubMed]
- 30.Huck K, Feyen O, Niehues T, Ruschendorf F, Hubner N, Laws HJ, et al. Girls homozygous for an IL-2-inducible T cell kinase mutation that leads to protein deficiency develop fatal EBV-associated lymphoproliferation. J Clin Invest. 2009;119(5):1350–1358. doi: 10.1172/jci37901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eken A, Cansever M, Somekh I, Mizoguchi Y, Zietara N, Okus FZ, et al. Genetic deficiency and biochemical inhibition of ITK affect human Th17, Treg, and innate lymphoid cells. J Clin Immunol. 2019;39(4):391–400. doi: 10.1007/s10875-019-00632-5. [DOI] [PubMed] [Google Scholar]
- 32.Arnaiz-Villena A, Timon M, Corell A, Perez-Aciego P, Martin-Villa JM, Regueiro JR. Brief report: primary immunodeficiency caused by mutations in the gene encoding the CD3-gamma subunit of the T-lymphocyte receptor. N Engl J Med. 1992;327(8):529–533. doi: 10.1056/NEJM199208203270805. [DOI] [PubMed] [Google Scholar]
- 33.Soudais C, de Villartay JP, Le Deist F, Fischer A, Lisowska-Grospierre B. Independent mutations of the human CD3-epsilon gene resulting in a T cell receptor/CD3 complex immunodeficiency. Nat Genet. 1993;3(1):77–81. doi: 10.1038/ng0193-77. [DOI] [PubMed] [Google Scholar]
- 34.Dadi HK, Simon AJ, Roifman CM. Effect of CD3delta deficiency on maturation of alpha/beta and gamma/delta T-cell lineages in severe combined immunodeficiency. N Engl J Med. 2003;349(19):1821–1828. doi: 10.1056/NEJMoa031178. [DOI] [PubMed] [Google Scholar]
- 35.Rieux-Laucat F, Hivroz C, Lim A, Mateo V, Pellier I, Selz F, et al. Inherited and somatic CD3zeta mutations in a patient with T-cell deficiency. N Engl J Med. 2006;354(18):1913–1921. doi: 10.1056/NEJMoa053750. [DOI] [PubMed] [Google Scholar]
- 36.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008;320(5882):1496–1501. doi: 10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang P, Bigio B, Rapaport F, Zhang SY, Casanova JL, Abel L, et al. PopViz: a webserver for visualizing minor allele frequencies and damage prediction scores of human genetic variations. Bioinformatics. 2018;34(24):4307–4309. doi: 10.1093/bioinformatics/bty536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sehnal D, Bittrich S, Deshpande M, Svobodova R, Berka K, Bazgier V, et al. Mol* Viewer: modern web app for 3D visualization and analysis of large biomolecular structures. Nucleic Acids Res. 2021;49(W1):W431–W4W7. doi: 10.1093/nar/gkab314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The protein data bank. Nucleic Acids Res. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25(9):1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Itan Y, Shang L, Boisson B, Ciancanelli MJ, Markle JG, Martinez-Barricarte R, et al. The mutation significance cutoff: gene-level thresholds for variant predictions. Nat Methods. 2016;13(2):109–110. doi: 10.1038/nmeth.3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Harari A, Vallelian F, Pantaleo G. Phenotypic heterogeneity of antigen-specific CD4 T cells under different conditions of antigen persistence and antigen load. Eur J Immunol. 2004;34(12):3525–3533. doi: 10.1002/eji.200425324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Z, Zhu L, Nguyen THO, Wan Y, Sant S, Quinones-Parra SM, et al. Clonally diverse CD38(+)HLA-DR(+)CD8(+) T cells persist during fatal H7N9 disease. Nat Commun. 2018;9(1):824. doi: 10.1038/s41467-018-03243-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Du J, Wei L, Li G, Hua M, Sun Y, Wang D, et al. Persistent high percentage of HLA-DR(+)CD38(high) CD8(+) T cells associated with immune disorder and disease severity of COVID-19. Front Immunol. 2021;12:735125. doi: 10.3389/fimmu.2021.735125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ho HN, Hultin LE, Mitsuyasu RT, Matud JL, Hausner MA, Bockstoce D, et al. Circulating HIV-specific CD8+ cytotoxic T cells express CD38 and HLA-DR antigens. J Immunol. 1993;150(7):3070–3079. doi: 10.4049/jimmunol.150.7.3070. [DOI] [PubMed] [Google Scholar]
- 50.Chandele A, Sewatanon J, Gunisetty S, Singla M, Onlamoon N, Akondy RS, et al. Characterization of human CD8 T cell responses in dengue virus-infected patients from India. J Virol. 2016;90(24):11259–11278. doi: 10.1128/JVI.01424-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brenchley JM, Karandikar NJ, Betts MR, Ambrozak DR, Hill BJ, Crotty LE, et al. Expression of CD57 defines replicative senescence and antigen-induced apoptotic death of CD8+ T cells. Blood. 2003;101(7):2711–2720. doi: 10.1182/blood-2002-07-2103. [DOI] [PubMed] [Google Scholar]
- 52.Appay V, Dunbar PR, Callan M, Klenerman P, Gillespie GM, Papagno L, et al. Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat Med. 2002;8(4):379–385. doi: 10.1038/nm0402-379. [DOI] [PubMed] [Google Scholar]
- 53.Larbi A, Fulop T. From “truly naive” to “exhausted senescent” T cells: when markers predict functionality. Cytometry A. 2014;85(1):25–35. doi: 10.1002/cyto.a.22351. [DOI] [PubMed] [Google Scholar]
- 54.Cossarizza A, Chang HD, Radbruch A, Abrignani S, Addo R, Akdis M, et al. Guidelines for the use of flow cytometry and cell sorting in immunological studies (third edition) Eur J Immunol. 2021;51(12):2708–3145. doi: 10.1002/eji.202170126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Levinson NM, Seeliger MA, Cole PA, Kuriyan J. Structural basis for the recognition of c-Src by its inactivator Csk. Cell. 2008;134(1):124–134. doi: 10.1016/j.cell.2008.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Germani A, Malherbe S, Rouer E. The exon 7-spliced Lck isoform in T lymphocytes: a potential regulator of p56lck signaling pathways. Biochem Biophys Res Commun. 2003;301(3):680–685. doi: 10.1016/s0006-291x(02)03000-0. [DOI] [PubMed] [Google Scholar]
- 57.Goldsmith MA, Weiss A. Isolation and characterization of a T-lymphocyte somatic mutant with altered signal transduction by the antigen receptor. Proc Natl Acad Sci U S A. 1987;84(19):6879–6883. doi: 10.1073/pnas.84.19.6879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rouer E, Brule F, Benarous R. A single base mutation in the 5' splice site of intron 7 of the lck gene is responsible for the deletion of exon 7 in lck mRNA of the JCaM1 cell line. Oncogene. 1999;18(29):4262–4268. doi: 10.1038/sj.onc.1202720. [DOI] [PubMed] [Google Scholar]
- 59.Hubert P, Bergeron F, Ferreira V, Seligmann M, Oksenhendler E, Debre P, et al. Defective p56Lck activity in T cells from an adult patient with idiopathic CD4+ lymphocytopenia. Int Immunol. 2000;12(4):449–457. doi: 10.1093/intimm/12.4.449. [DOI] [PubMed] [Google Scholar]
- 60.Seddon B, Legname G, Tomlinson P, Zamoyska R. Long-term survival but impaired homeostatic proliferation of Naive T cells in the absence of p56lck. Science. 2000;290(5489):127–131. doi: 10.1126/science.290.5489.127. [DOI] [PubMed] [Google Scholar]
- 61.Kemp KL, Levin SD, Bryce PJ, Stein PL. Lck mediates Th2 differentiation through effects on T-bet and GATA-3. J Immunol. 2010;184(8):4178–4184. doi: 10.4049/jimmunol.0901282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Marsh M, Armes JE, Pelchen-Matthews A. Endocytosis and recycling of CD4. Biochem Soc Trans. 1990;18(2):139–143. doi: 10.1042/bst0180139. [DOI] [PubMed] [Google Scholar]
- 63.Shin J, Dunbrack RL, Jr, Lee S, Strominger JL. Phosphorylation-dependent down-modulation of CD4 requires a specific structure within the cytoplasmic domain of CD4. J Biol Chem. 1991;266(16):10658–10665. doi: 10.1016/S0021-9258(18)99274-4. [DOI] [PubMed] [Google Scholar]
- 64.Sleckman BP, Shin J, Igras VE, Collins TL, Strominger JL, Burakoff SJ. Disruption of the CD4-p56lck complex is required for rapid internalization of CD4. Proc Natl Acad Sci U S A. 1992;89(16):7566–7570. doi: 10.1073/pnas.89.16.7566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Almanzar G, Schwaiger S, Jenewein B, Keller M, Herndler-Brandstetter D, Wurzner R, et al. Long-term cytomegalovirus infection leads to significant changes in the composition of the CD8+ T-cell repertoire, which may be the basis for an imbalance in the cytokine production profile in elderly persons. J Virol. 2005;79(6):3675–3683. doi: 10.1128/JVI.79.6.3675-3683.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jacobs MD, Caron PR, Hare BJ. Classifying protein kinase structures guides use of ligand-selectivity profiles to predict inactive conformations: structure of lck/imatinib complex. Proteins. 2008;70(4):1451–1460. doi: 10.1002/prot.21633. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A CADD vs. minor allele frequency (MAF)-plot of heterozygous variants in LCK present in the gnomAD database and variants LCK C465R and LCK L341P[1]. Created with the PopViz webserver application[38]. The CADD cut-off was calculated with a 95% confidence interval. B Logo plot of sequence conservation across the human SRC-family kinases indicated on the left. Created with Jalview [41]. C (Right) Ribbon diagram of the X-ray structure of the kinase domain of human LCK in a closed conformation (PDB ID: 2PL0) based on [66] [17]. α-helices in purple, β-sheets in yellow, unstructured regions in grey. Residues Y394 and C465 are highlighted. (Left) Detailed view of the local structure surrounding C465. Carbon-atoms depicted in green, nitrogen in blue, oxygen in red and sulfur in yellow. Residues C465, P466 and Y469 are highlighted, and dashed lines indicate hydrogen bonds. Image created with Mol*Viewer [39] and RCSB PDB [40] (EPS 955 kb).
Supplementary Figure S2 A Histogram overlay of flow cytometry staining of Jurkat E6.1, J.CaM1.6 and stable transduced celllines with pCDH expressing either LCK WT or LCK C465R with or without an C-terminal HA-tag or transduced with pCDH empty vector containing only the expression cassette for the extracellular domain (ED) of the low-affinity nerve growth factor receptor (LNGFR) stained with anti-LNGFR-PE, anti-LCK + secondary anti-mouse AF647 and anti-HA-AF488. B Histogram overlay of flow cytometry staining of Jurkat E6.1, J.CaM1.6 and stable transduced celllines with pLJM1 expressing either LCK WT or LCK C465R transduced with empty vector pLJM1-EGFP anti-LCK + secondary anti-mouse AF647 and anti-HA-AF488. C Gating strategy used for Ca2+-flux measurements. J.CaM1.6 cells transduced with the lentiviral plasmid that co-expresses the extracellular domain (ED) of LNGFR were gated on LNGFRhi for Ca2+-flux measurements, while for untransduced Jurkat E6.1 that do not express LNGFR-ED single cells were acquired. D Ca2+-flux measurements in J.CaM1.6 expressing LCK WT (light blue), LCK-HA (dark blue), LCK C465R (light red), LCK-HA C465R (dark red), Jurkat E6.1 (black) and J.CaM1.6 transduced with empty vector (turquoise) stimulated with anti-CD3 (left) or anti-CD3/ anti-CD28 (right). E pZAP70, pSFK (pY416), pERK1/2 immunoblots in Jurkat E6.1 cells or J.CaM1.6 transduced with empty vector pLJM1-EGFP and stimulated with either anti-CD3 (2, 5 or 15 min), anti-CD3/CD28 (2 or 5 min), PMA/ionomycin (5 min) or left untreated. (PNG 704 kb)
Supplementary Figure S3 Immune phenotype of patient, mother and two healthy controls (HC) by flow cytometry. Markers used for staining as indicated on the arrows next to the contour plots, gated populations above. A Contour plot overlay of CD4 vs. CD8 staining of patient, mother, and healthy control (HC)1 (all in red) with HC2 (blue) B Histogram overlays of the patient, mother and 2 healthy controls for surface expression of CD4, CD8, TCRαβ and TCRγδ. C CD38 vs. HLA-DR staining on CD45RO+ memory CD4+ (upper row) and CD8+ (lower row) T-cells of the patient, mother and 2 healthy controls. D CD28 vs. CD27 staining on CD8+ T-cells, color-coded by CD45RA expression E (upper row) CCR6 expression on CD4+ CD45RO+ T-cells and (lower row) CD4+ CD45RO+ CCR6+ T helper cell phenotype F Treg phenotype characterized by CD45RO and HLA-DR expression. (EPS 2842 kb)
Supplementary Figure S4 Gating strategy used for flowcytometric immune phenotyping. (EPS 596 kb)
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.