

Abstract
Male contraceptive development has included use of testosterone (T) with or without a progestin or the use of a single molecule such as progestogenic androgens (PA) for suppression of testicular T production. Expanding upon the vast amount of data accumulated from nortestosterone (NT), NT analogs, and their prodrugs, a new series of PA, the C7 methyl, and ethyl α-substituted T analogs 7α-Methyltestosterone (7α-MT) and 7α-Ethyltestosterone (7α-ET), respectively, were hypothesized and designed to have superior androgenic and progestogenic activities when compared with parent T. Results from androgen receptor and progesterone receptor competitive binding and transcriptional activation assays showed favorable activities for these T analogs. Additionally, 7α-MT and 7α-ET were shown to be active substrates for aromatase in vitro, mitigating a potential negative impact on bone mineral density with long-term use. In conjunction with this observation, the diminished metabolism of these T analogs by 5α-reductase may reduce potential concerns for prostatic growth. In the Hershberger in vivo rat bioassay, 7α-MT and 7α-ET showed superior androgenic and anabolic activities as compared with T. These C7 α-substituted T analogs also showed clear progestogenic activity in the McPhail bioassay which evaluated endometrial glandular arborization in a rabbit model. The discovery of aromatizable molecules with reduced metabolism by 5α-reductase that have androgenic, anabolic, and progestogenic properties indicates that the core and/or prodrugs of 7α-MT and 7α-ET are promising molecules for further development as male contraceptive PAs.
Keywords: progestogenic androgen, male contraception, testosterone analogs, metabolism
Design, synthesis, in vitro, and in vivo studies of C7 α-substituted testosterone, a novel progestogenic androgen as a potential male contraceptive agent.
Graphical Abstract
Graphical Abstract.

Introduction
Exogenous testosterone (T) with or without a progestin or the use of a single progestogenic androgen (PA) acts via negative feedback on the hypothalamus and pituitary gland to suppress serum gonadotropins resulting in suppression of testicular T production [1–10]. Use of these entities results in T levels below the threshold needed to support spermatogenesis. If T suppression in the testes is maintained by a progestin, exogenous replacement of T is necessary to support normal androgen-dependent functions other than sperm production [11]. T-only dosing requires high physiologic doses to achieve contraceptive efficacy [12, 13], whereas a progestin–T combination requires T for the purpose of hormone replacement, while providing the added benefit of more rapid and reliable sperm suppression as compared with androgen-only administration [11].
Use of exogenous androgen for T suppression can also lead to impairment of estrogen production, and thus can affect bone mineral density. Given normal estrogen production is critical for maintaining bone mineral density (e.g. hypoestrogenemia can contribute to the development of osteopenia and osteoporosis among other related disorders), the conversion of T to estrogen through aromatase is a critical enzymatic reaction. Specifically, aromatase is responsible for converting androstenedione to estrone, and T to estradiol [14, 15]. Thus, it is desirable for candidate male contraceptive agents to maintain an adequate aromatization potential. Some of the PAs under development, in particular, nortestosterone (NT) analogs, undergo little to no aromatization which may seriously impact bone mineral density in the long-term. In general, NT and NT analogs such as 7α-Methylnortestosterone (MENT), 11β-Methylnortestosterone (11β-MNT), and Dimethandrolone (DMA) are not good substrates for aromatase because the critical C10 methyl group between rings A and B in the steroidal scaffold is absent. Indeed, while NT and MENT are aromatized to a lesser degree than T, 11β-MNT, and DMA show little to no aromatase activity as shown herein and by others [16]. Another key enzyme involved in T metabolism is 5α-reductase which is responsible for the irreversible conversion of T to dihydrotestosterone (DHT). As such, 5α-reductase can convert T analogs into their respective 5α-reduced form. Overabundance of DHT has been implicated in the development of prostate cancer in some studies; thus an ideal male contraceptive should be a poor substrate for 5α-reductase [17–19].
NT analogs with substitution at C7 and/or C11, such as MENT, 11β-MNT, and DMA, show improvements in both androgenic and progestogenic activities as compared with the parent molecule, NT. However, the implications of inadequate aromatization and increased metabolism by 5α-reductase are potential limitations to their use. To overcome these potential pitfalls, we synthesized and tested 7α-Methyltestosterone (7α-MT) and 7α-Ethyltestosterone (7α-ET) which are T analogs with a C7 α-substitution. In a manner similar to NT analogs, these T analogs could give rise to a more potent series of compounds with increases in both androgenic and progestogenic in vitro and in vivo activities. Herein we describe the design, synthesis, in vitro, and in vivo studies of C7 α-substituted T analogs, 7α-MT and 7α-ET, demonstrating them to be promising progestogenic-androgens for further development as potential male contraceptives.
Materials and methods
Synthesis of 7α-MT (4a) and 7α-ET (4b)
General Method: All reagents, chemicals, and solvents used for chemistry were obtained from commercial sources and used as supplied. Anhydrous solvents were obtained from commercial sources and used as supplied. Reactions were monitored by thin-layer chromatography (TLC) using silica gel plates (Analtech, Silica Gel GF). All final products were purified by flash column chromatography (EMD, Silica Gel 60, 230–400 Mesh ASTM). Chemical yields were not optimized. Purity of all compounds were determined using Agilent 1100/1200 HPLC system, and all assayed compounds were >95% pure. Melting points (m.p.) were determined using OptiMelt. NMR experiments were performed on Bruker or Agilent 400 MHz instruments. Chemical shifts are reported in parts per million (ppm) and coupling constants in hertz (Hz). For 1H NMR solvents, CDCl3 and DMSO-d6 were used with chemical shifts of 7.26 ppm and 2.5 ppm, respectively, as internal standards. For 1H spectra, the peak multiplicities are reported as: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet. Optical rotation measurements were performed on Autopol IV polarimeter. LC/MS experiments were performed on Agilent 6120/6130 mass spectrometer.
Abbreviations for Solvents: The following abbreviations were used for common solvents: DCM, dichloro-methane; DME, Dimethoxyethane; DMF, dimethylformamide; EtOAc, ethyl acetate; EtOH, ethanol; Et2O, diethyl ether; IPA, isopropanol; MeOH, methanol; MTBE, methyl tert-butyl ether; NBS, N-Bromosuccinimide; PE, petroleum ether; TBDMSCl, tert-Butyldimethylsilyl chloride; THF, tetrahydrofuran; CDCl3, deuterochloroform; DMSO-d6, deuterodimethyl sulfoxide; CD3OD, deuteromethanol.
(10R,13S,17S)-10,13-Dimethyl-3,5-diene-cyclopenta[a]phenanthrene-3,17-diyl diacetate (1). Under nitrogen, a mixture of T (5.0 g, 17.3 mmol), isopropenyl acetate (5.0 mL, 45.4 mmol) and p-Toluene sulfonic acid (0.17 g, 0.9 mmol) in DME (15 mL) was heated at reflux (heating mantle) for 4 h, TLC of a mini workup indicated completion. The reaction mixture was cooled slightly and diluted with IPA (10 mL). Phosphate buffer (pH 7.0, 45 mL) was added to the resulting slurry. The mixture was then stirred in an ice/water bath for 30 min, filtered and air-dried to constant weight (wt.) to obtain diacetate 1 as an off-white solid, 6.4 g (99% yield): m.p. 138–146°C; HPLC purity: 96.9%; 1H NMR (Partial, 400 MHz, CDCl3) δ 5.69 (d, 1H, J = 2.4 Hz, C4-H), 5.40 (t, 1H, J = 3.2 Hz, C6-H), 4.61 (dd, 1H, J = 9.2 & 8.0 Hz, C17-H), 2.14 (s, 3H, -CH3 of -OAc), 2.05 (s, 3H, -CH3 of -OAc), 1.01 (s, 3H, C10-CH3), 0.83 (s, 3H, C13-CH3); LC/MS: calculated for C21H31O3 [M + 1-Ac]: 331.5, found 331.3.
(10R,13S,17S)-17-Hydroxy-10,13-dimethyl-4,6-diene-cyclopenta[a]phenanthren-3-one (2). Under nitrogen, a thick slurry of diacetate 1 (6.3 g, 16.9 mmol) in DMF (13 mL) and water (0.3 mL, 16.7 mmol) was stirred in an ice/acetone bath. Solid NBS (3.2 g, 18.0 mmol) was then added portion-wise with temp <10°C during addition. After addition, the resulting slurry was stirred for 30 min in the ice bath, then quenched with 15% aq. Na2S2O3 (1.0 mL) to consume excess NBS. In the meantime, a mixture of LiBr (1.6 g, 18.4 mmol) and Li2CO3 (3.2 g, 43.3 mmol) in DMF (13 mL) was heated at 90°C under nitrogen. The slurry from above was added dropwise into the hot mixture with a temperature >85°C during addition. After addition, stirring was continued at 80–85°C for 1 h, then at room temperature (RT) overnight. The reaction mixture was diluted with EtOAc (10 mL) and filtered through a celite pad. The pad was rinsed with EtOAc (30 mL). The combined filtrate was poured into water (120 mL). The water layer was separated and re-extracted with EtOAc (2 × 30 mL). The combined organic extracts were washed with brine (1 × 20 mL), dried over MgSO4, filtered, and evaporated. The residual solid (6.4 g) was dissolved in warm MeOH (36 mL) and NaOMe/MeOH (2.0 g, 25 wt%, 9.3 mmol) was added, pH ~11. The resulting reaction mixture was stirred at RT for 3 h, then cooled in an ice/water bath before adjusted to pH ~6 with 10% aq. H3PO4 (3.0 mL). The resulting precipitate was collected by filtration and re-triturated in water (20 mL), filtered, and air-dried to give pure diene-one 2, 3.2 g (66% yield, m.p. 198–201°C) as 1st crop product. The filtrate was concentrated, the residue was triturated in water (40 mL), and the precipitate was collected by filtration and air-dried. The crude solid (1.5 g) was recrystallized from boiling IPA (8 mL) to afford 2nd crop product, 0.7 g (15% yield) with total yield of 81%, HPLC purity: 98.0%; 1H NMR (Partial, 400 MHz, CDCl3): δ 6.11 (m, 2H, C6-H, & C7-H), 5.68 (s, 1H, C4-H), 3.70 (m, 1H, C17-H), 1.13 (s, 3H, C10-CH3), 0.85 (s, 3H, C13-CH3); LC/MS: calculated for C19H27O2 [M + 1]: 287.4, found 287.3.
(10R,13S,17S)-17-((tert-Butyldimethylsilyl)oxy)-10,13-dimethyl-4,6-diene-cyclopenta[a]phenanthren-3-one. (3 ). A mixture of diene-one 2 (3.0 g, 10.5 mmol), imidazole (2.1 g, 31.4 mmol) and TBDMSCl (2.4 g, 15.7 mmol) in dry DMF (25 mL) was stirred at RT. After ~20 min, a clear solution was observed. The reaction mixture was stirred at RT overnight. The resulting slurry was poured into water (100 mL), stirred for ~10 min and filtered. The wet cake was triturated in cold MeOH (15 mL) cooled in an ice/water bath for 1 h, then filtered and air-dried to constant wt. to yield silyl ether 3 as an off-white solid, 3.8 g (91% yield): m.p. 112–115°C; HPLC purity: 99.3%; 1H NMR (Partial, 400 MHz, CDCl3): δ 6.10 (m, 2H, C6-H, & C7-H), 5.67 (s, 1H, C4-H), 3.59 (t, 1H, J = 8.0 Hz, C17-H), 1.12 (s, 3H, C10-CH3), 0.88 (s, 9H, t-butyl), 0.80 (s, 3H, C13-CH3), 0.12 (s, 6H, Si-(CH3)2); LC/MS: calculated for C25H41SiO2 [M + 1]: 401.7, found 401.3.
7α-MT (4a, CDB-4910): (7R,10R,13S,17S)-17-Hydroxy-7, 10, 13-trimethyl-1, 2, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17-tetradecahydro-3H-cyclopenta[a]phenanthren-3-one. Under nitrogen, a mixture of LiCl (0.3 g, 6.9 mmol) and CuI (0.6 g, 3.1 mmol) in dry THF (14 mL) was stirred at RT for 1 h. The resulting clear solution was added to a stirred, cold (−10°C) solution of silyl ether 3 (2.5 g, 6.2 mmol) in dry THF (14 mL), followed by the addition of TMSCl (4.3 mL, 34 mmol). The resulting mixture was cooled to −75°C (dry ice/acetone bath). The commercial 3.0 M MeMgCl in THF (5 mL, 15 mmol) was diluted with same amount of THF (5 mL) then added dropwise, over 1 h, into the reaction mixture with the temperature kept at approximately −75°C during addition. Stirring was continued for 3 h at −75°C, then water (10 mL) was added slowly and carefully with RT stirring overnight. The mixture was diluted with EtOAc (50 mL), and the organic layer was separated then washed successively with 50% conc. NH4OH (1 × 20 mL), 50% conc. NH4OH/conc. NH4Cl (1,1, 1 × 20 mL), water (1 × 20 mL) and brine (1 × 10 mL). All aqueous washings were re-extracted with EtOAc (1 × 30 mL). The organic extracts were combined, dried over MgSO4, and evaporated to dryness. The residual gum (~ 2.0 g, HPLC: 7α:7β ≈ 5:1) was dissolved in acetone (~4 mL) and stored in a freezer overnight. The crystallized solid was collected, rinsed with cold acetone, and air-dried to obtain 1st crop product (0.4 g). The filtrate was stripped, and the residue (1.6 g) was chromatographed over a flash silica gel column (50 g) using 0–5% acetone/CH2Cl2. Good TLC fractions were pooled and concentrated to obtain 2nd crop solid (0.2 g). These two solid crops were combined (~0.6 g), dissolved in boiling IPA (~6 mL), and cooled at RT overnight. The crystallized solid was collected, rinsed with cold IPA, and air-dried to constant wt. to give pure 7α-MT (4a, CDB-4910) as an off-white solid, 320 mg (17% yield): m.p. 211–214°C; HPLC purity: 99.0%; 1H NMR (Partial, 400 MHz, CDCl3): δ 5.73 (s, 1H, C4-H), 3.66 (m, 1H, C17-H), 1.20 (s, 3H, C10-CH3), 0.80 (s, 3H, C13-CH3), 0.76 (d, 3H, J = 6.8 Hz, C7-CH3); [α]26°C = + 105.6 (c = 1.0, CH3OH); LC/MS: calculated for C20H31O2 [M + 1]: 303.2, found 303.3.
7α-ET (4b, CDB-4918): (7R,10R,13S,17S)-7-ethyl-17-hydroxy-10,13-dimethyl-1, 2, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17-tetradecahydro-3H-cyclopenta[a]phenanthren-3-one. Under nitrogen, a mixture of LiCl (0.3 g, 6.9 mmol) and CuI (0.6 g, 3.1 mmol) in dry THF (14 mL) was stirred at RT for 1 h. The resulting clear solution was added to a stirred, cold (−10°C) solution of silyl ether 3 (2.5 g, 6.2 mmol) in dry THF (14 mL), followed by the addition of TMSCl (4.3 mL, 34 mmol). The resulting mixture was then cooled to −75°C (dry ice/acetone bath). The commercial 2.0 M EtMgCl in THF (8 mL, 16 mmol) was diluted with same amount of THF (8 mL) then added dropwise, over 1 h, into the reaction mixture and temperature was kept at approximately −75°C during addition. After addition, stirring was continued for 3 h at −75°C, then water (10 mL) was added slowly and carefully. The resulting acidic reaction mixture was stirred at RT overnight then the mixture was diluted with EtOAc (50 mL). The organic layer was separated then washed successively with 50% conc. NH4OH (1 x 20 mL), 50% conc. NH4OH/conc. NH4Cl (1,1, 1 × 20 mL), water (1 × 20 mL) and brine (1 × 10 mL). All the aq. washings were re-extracted with EtOAc (1 × 30 mL). The organic extracts were combined, dried over MgSO4, and evaporated to dryness. The residual solid (2.0 g, HPLC, 7α,7β ≈ 3,2) was dissolved in minimum boiling IPA (~16 mL) and stored at RT overnight. The crystallized solid was collected, rinsed with cold IPA, and air-dried to obtain 1st crop product [0.91 g, HPLC: 95.4% (7α) + 4.6% (7β)]. The filtrate was stripped, and the residue (1.0 g) was chromatographed over a flash silica gel column (40 g), using 5–15% EtOAc/CH2Cl2. Good TLC fractions were pooled and concentrated to obtain 2nd crop solid [0.29 g, HPLC: 98.2% (7α) + 1.0% (7β)]. These two solid crops were combined (1.2 g), dissolved in minimum boiling IPA (~13 mL), and cooled at RT overnight. The crystallized solid was collected, rinsed with cold IPA, air-dried to constant wt. to afford pure 7α-ET (4b, CDB-4918) as a white crystalline solid, 0.80 g (41% yield): m.p. 218–220°C; HPLC: 99.5% (7α) + 0.4% (7β); 1H NMR (Partial, 400 MHz, CDCl3): δ 5.73 (s, 1H, C4-H), 3.66 (m, 1H, C17-H), 1.20 (s, 3H, C10-CH3), 0.85 (t, 3H, J = 7.2 Hz, -CH3 of ethyl), 0.80 (s, 3H, C13-CH3); [α]26°C = + 87.5 (c = 1.0, CH3OH); LC/MS: calculated for C21H33O2 [M + 1]: 317.5, found 317.3.
Test and control articles
In addition to the synthesized 7α-MT and 7α-ET described above, the following molecules were evaluated as controls or test articles in the various assays described herein: T, Progesterone (P), and NT from Sigma-Aldrich (St. Louis, MO) and DMA, 11β-MNT, and MENT from Piramal Pharma Solutions (formerly Ash Stevens, LLC., Riverview, MI).
In vitro screening
Competitive radioligand binding
Eurofins Panlabs Discovery Services (New Taipei City, Taiwan) performed radioligand binding assays to the Androgen Receptor (AR) and Progesterone Receptor (PRβ). For AR binding, [3H] Methyltrienolone was used as the ligand and incubation was performed with human LNCaP clone FGC cells in 25 mM HEPES, pH 7.4, 10% Glycerol, 1 mM EDTA, and 10 mM Molybdate for 20 h at 4°C. For PRβ binding, [3H] Progesterone was used as the ligand and incubation was performed with human T-47D cells in 5 mM Na2HPO4/NaH2PO4, pH 7.4, 10% Glycerol, 10 mM Monothioglycerol, and 20 mM Sodium molybdate for 20 h at 4°C. The final vehicle concentration in both assays was 1% DMSO. Compounds were tested at concentrations ranging from 10−5 μM to 100 μM and IC50 values were determined by a non-linear, least squares regression analysis using MathIQ™ (ID Business Solutions Ltd., UK).
Transcriptional activation (transactivation)
Eurofins DiscoverX Corporation (Fremont, CA) performed transcriptional activation (transactivation) assays to AR and PRβ in agonist mode. Proprietary PathHunter® NHR cell lines were used for both AR and PRβ transactivation assays. For agonist determination, cells were incubated with compounds (10−8 μM to 10 μM) at 37°C or RT for 3–16 h to induce response. Final assay vehicle concentration was 1%. Assay signal was generated through a single addition of PathHunter Detection reagent cocktail, followed by a 1 h RT incubation. Microplates were read following signal generation with a PerkinElmer Envision™ instrument for chemiluminescent signal detection. Compound activity was analyzed using CBIS data analysis suite (ChemInnovation, CA). For agonist mode assays, percent activity was calculated using the following formula:
% Activity =100% × (mean RLU of test sample – mean RLU of vehicle control) / (mean MAX control ligand – mean RLU of vehicle control).
Metabolism by aromatase
For aromatase analysis, test articles (10 μM) were incubated at 37°C with human CYP19 + P450 reductase Supersomes™ (50 pmole/ml CYP450, 0.5 mg/ml protein; Corning Discovery Labware, Woburn, MA) and 3.3 mM MgCl2, with and without 2.5 mM NADPH in 0.1 M phosphate buffer, pH 7.4. Aliquots were removed at 0, 15, 30, and 60 min, mixed with acetonitrile containing 1000 ng/ml d3-T (Sigma-Aldrich); vortexed; centrifuged; and diluted with 0.1% formic acid in water for analysis by LC–MS/MS (see below LC–MS/MS Analysis).
Metabolism by 5α-reductase
For 5α-reductase analysis, test articles (1 μM) were incubated at 37°C with 0.5 mg/ml human liver cytosol (Xenotech, Kansas City, KS), 3.3 mM MgCl2, and 2.5 mM NADPH in 0.1 M phosphate buffer, pH 7.4, with and without 50 μM finasteride, a 5α-reductase type 2 specific inhibitor (Sigma-Aldrich). Aliquots were removed at 0, 15, 30, 60, 90, and 120 min; mixed with acetonitrile containing 100 ng/ml d3-T (Sigma-Aldrich); vortexed; centrifuged; and diluted with 0.1% formic acid in water for analysis by LC–MS/MS (see below LC–MS/MS Analysis).
LC–MS/MS analysis
Samples were analyzed on a Shimadzu LC-20 AD HPLC pump coupled with an API SCIEX 4000 Q TRAP system. Chromatographic separation was achieved on a Kinetex C8 or Luna C18(2) column [5 μm, 2 × 50 mm; Phenomenex (Torrance, CA)] maintained at 45°C with two mobile phases [0.2% formic acid in (A) water and (B) acetonitrile] and eluted using a gradient from 20% B to 90% B over 1.5 min at a flow rate of 0.65 ml/min. Detection was by multiple reaction monitoring after electrospray ionization in the positive ion mode.
Metabolism results were presented as % of parent test article remaining and calculated as follows: [peak area ratio (PAR) of parent at each time point / PAR of parent at T = 0 min] x 100. Area under the curve (AUC) was also calculated for each test article. The units for AUC were % remaining * min and converted to % of total AUC. In the aromatase assay, a low AUC indicated rapid metabolism, while a high AUC indicated slow metabolism. To distinguish the role of 5α-reductase, the difference between the AUC for incubations with and without finasteride were calculated. The larger the difference between the two AUC’s, the greater the potential that 5α-reductase was involved in test article metabolism.
In vivo studies
Animal care
All procedures occurred in an Association for Assessment and Accreditation of Laboratory Animal Care accredited facility under protocols approved by the Institutional Animal Care and Use Committee and in compliance with the National Research Council’s Guide for the Care and Use of Laboratory Animals [20]. Animals (Charles River Laboratory; Hollister, CA for rats and Saint-Constant, Quebec, Canada for rabbits) had ad libitum access to Envigo Tekland certified food (rodent diet #2018C or high fiber rabbit diet #2031; rabbits also received hay and fresh fruits/vegetables for dietary enrichment) and water and were housed ≤3 per cage (rats) and 1–2 per cage (rabbits) in separate rooms maintained on a 12-h light/dark cycle at controlled humidity and temperature (rats: 68–75°C, rabbits: 61–72°C). Animals were acclimated/quarantined ≥5 days prior to study start. Once dosing began, animals sharing a cage were always from the same treatment and dose level group. Dose administrations were by subcutaneous (SC) injection to 1 of 4 rotating dose sites on the dorsal surface. Animals were euthanized by injection with Euthasol® (Covetrus) ~24 h after the final dose. Prior to euthanasia, rabbits were sedated with a cocktail of acepromazine and torbugesic.
Hershberger bioassay
Hershberger study is a rat model using castrated weaning rats to evaluate relative androgenic and anabolic potencies [21]. Nursing Sprague Dawley intact male rat pups arrived with lactating foster mothers (8–10 male pups per dam) on Postnatal Day (PND) 13–16. At 21 days of age (PND 21), rats were weaned, castrated, and after surgery randomized by body weight into treatment groups. Starting on the day of castration (Day 1) and continuing for a total of seven consecutive days, rats were administered (0.2 mL SC injection/day irrespective of body weight) vehicle control, T reference control (6.9 μmol/rat; 0.99 μmol/injection), or test article (0.7, 2.8, and 11.2 μmol/rat; 0.1, 0.4, and 1.6 μmol/injection) all formulated in 10% EtOH/90% sesame oil from Sesame Indicum (Sigma-Aldrich). On Day 8, rats were weighed and euthanized, followed by excision, trimming, and weighing of ventral prostate glands, seminal vesicles, and levator ani muscles. The study was conducted as separate staggers. Each stagger contained its own set of vehicle and T reference control animals and a total of 30–32 male rats (n = 3–4 rats per control group; n = 8 rats per test article dose level; 3 dose levels per test article).
McPhail bioassay
McPhail study is a rabbit model to assess progestogenic properties of test articles as determined by hematoxylin and eosin (H&E)-stained uteri cross sections grouped into five possible scores for evaluation of endometrial glandular arborization [22]. New Zealand White immature female rabbits (aged 6–7 weeks at first priming dose) were primed on Days 1–6 with 17β-estradiol SC injection (5 μg/day, Sigma-Aldrich) in 10% EtOH/90% castor oil (USP grade, Spectrum Chemical Manufacturing Corporation, New Brunswick, NJ) followed by five consecutive days (Days 7–11) of SC injection of vehicle, P reference control (0.15 mg/day, 0.48 μmol/day, 2.4 μmol/rabbit), or test article (0.16, 0.48, 1.44, and 4.32 μmol/day; 0.8, 2.4, 7.2, and 21.6 μmol/rabbit) all formulated in 10% EtOH/90% castor oil; T was tested at only one dose level (1.44 μmol/day, 2.4 μmol/rabbit). Each test article was tested in 6 rabbits per dose level with the exception that T was tested in 3 rabbits at a single 1.44 μmol/day dose level; vehicle and P were each tested in 9 rabbits. Animals were randomly assigned by body weight to treatment groups during the priming period on Day 5 or 6. On Day 12, rabbits were weighed and euthanized. Uteri were excised intact, and weights recorded. Formalin fixed, paraffin embedded, and H&E stained uteri sections (2 per uterine horn per rabbit) were microscopically evaluated for uterus mucosa ramification and proliferation (i.e. endometrial glandular arborization) based on the 0–4 scoring system of McPhail [22].
Statistical analysis
GraphPad Prism version 9.5 for Windows was used for graphing and AUC calculations, including calculation of the standard error of the AUC. AUC differences across groups were evaluated for statistical significance using a two-sample test procedure with 30 degrees of freedom for the pairwise comparisons and a P-value threshold of ≤0.01 to account for multiple testings. The statistical significance of the t-statistic was calculated in Excel (Microsoft 365).
Results & discussion
Chemical synthesis
The T analogs, 7α-MT (4a) and 7α-ET (4b) were synthesized from T as the starting material using standard chemistry (Scheme 1). The diacetate protected 3,4-diene 1 was synthesized from T using isopropenyl acetate and p-toluene sulfonic acid in DME under reflux. The bromination of diacetate 1 at C7 was achieved by using NBS in wet DMF at 0°C, followed by de-bromination in a hot mixture of LiBr/Li2CO3 in DMF at 90°C. The resulting crude product was hydrolyzed in NaOMe/MeOH at RT to afford diene-one 2. The C17-OH of diene-one 2 was then protected, using TBDMSCl and imidazole in DMF at RT, as silyl ether 3. Michael addition of RMgCl at C7 of silyl ether 3 was accomplished by slow addition of the Grignard reagent into a mixture containing silyl ether 3, LiCl, CuI, and TMSCl in THF at −75°C, followed by quenching with water at RT overnight. After aqueous workup, the crude products were purified via silica gel column chromatography followed by recrystallization, isolation of pure 7α-MT (4a) and 7α-ET (4b), and characterization. None of the steps in this synthesis were optimized.
Scheme 1.

Synthesis of C7-Substituted T Analogsa, aReagents and conditions: (a) Isopropenyl acetate, p-toluene sulfonic acid, DME, reflux, 4 h, 99%; (b) DMF, H2O, NBS, 0°C, 30 min;(c) LiBr, Li2CO3, DMF, 90°C, 1 h; (d) crude product, MeOH, NaOMe/MeOH, RT, 3 h, 81%; (e) TBDMSCl, imidazole, dry DMF, RT, overnight, 91%; (f) RMgCl, LiCl, CuI, dry THF, –75°C, 3 h, H2O, RT, overnight; (g) Column purification and crystallization.
In vitro studies
Binding & transactivation assays
The binding and transactivation abilities of 7α-MT, 7α-ET, T, NT, MENT, 11β-MNT, and DMA, plus P as a reference control, to the human AR and PRβ were evaluated as summarized in Table 1. The AR binding IC50 for the androgen analogs ranged from 1.5 to 3.0 nM. For the AR transactivation assay, all test articles showed androgenic activity with the most to least active ranked by EC50 as MENT ≈ DMA > 11β-MNT > 7α-MT > 7α-ET > T > NT (1.9 ≈ 2.2 > 4.8 > 7.1 > 10.4 > 29.2 > 38.9 nM, respectively). The rank order of the test articles was similar for competitive binding to PRβ based on IC50 values and transactivation of PRβ based on EC50. The most to least active progestins based on IC50 values were P > 11β-MNT ≈ MENT > DMA > NT > T > 7α-MT > 7α-ET (1.51 > 2.7 ≈ 3.0 > 5.5 > 10.8 > 150 > 210 > 240 nM, respectively). The most to least active progestins based on EC50 values were P > 11β-MNT > MENT > NT ≈ DMA > T > 7α-MT ≈ 7α-ET (9.4 > 41.8 > 95.4 > 129 ≈ 132 nM > 1.9 μM > 10 μM, respectively).
Table 1.
Binding and Transcriptional Activation Assays for AR and PRβ.
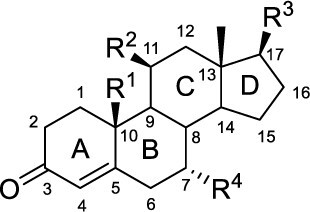
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| IC50 Binding (nM) | EC50 Transactivation (nM) | ||||||||
| Compound Name | Abbreviation | R1 | R2 | R3 | R4 | AR | PRβ | AR | PRβ |
| Progesterone | P | -CH3 | -H | -C(O)CH3 | -H | 3.6 | 1.5 | > 10 μM | 9.4 |
| Testosterone | T | -CH3 | -H | -OH | -H | 2.6 | 150 | 29.2 | 1.9 μM |
| 7α-Methyltestosterone | 7α-MT | -CH3 | -H | -OH | -CH3 | 2.6 | 210 | 7.1 | > 10 μM |
| 7α-Ethyltestosterone | 7α-ET | -CH3 | -H | -OH | -CH2CH3 | 1.8 | 240 | 10.4 | > 10 μM |
| Nortestosterone | NT | -H | -H | -OH | -H | 1.9 | 10.8 | 38.9 | 129 |
| 7α-Methylnortestosterone | MENT | -H | -H | -OH | -CH3 | 1.4 | 3.0 | 1.9 | 95.4 |
| 11β-Methylnortestosterone | 11β-MNT | -H | -CH3 | -OH | -H | 1.7 | 2.7 | 4.8 | 41.8 |
| Dimethandrolone | DMA | -H | -CH3 | -OH | -CH3 | 1.8 | 5.5 | 2.2 | 132 |
Given that the AR and PRβ binding results for T, 7α-MT, and 7α-ET were highly similar to one another, we conclude that addition of a methyl or ethyl group at the C7 position of T did not improve binding to AR or PRβ. Although all naturally occurring progestins including P have a C10 methyl, the absence of this methyl on exogenous androgens increased PRβ binding as shown by (a) improved IC50 values for T vs NT and 7α-MT vs MENT upon removal of the C10 methyl, (b) very weak binding to and transactivation of PRβ by 7α-MT and 7α-ET which both contain a C10 methyl, and (c) improved PRβ binding activities for NT and the NT analogs MENT, 11β-MNT, and DMA which all lack a C10 methyl.
The transactivation assay showed a similar trend in overall increases in potency for AR and PRβ for NT analogs as compared to T analogs. Unlike the binding study, the transactivation assay was more sensitive to the presence or absence of C7 substituents within each T or NT analog. For AR transactivation, 7α-MT with EC50 of 7.1 nM was ~4 times and 7 α-ET with EC50 of 10.4 nM was ~3 times more potent than T, indicating stronger androgenic and anabolic potential. This difference was more pronounced for NT and NT analogs. From this study, small substituents at C7 clearly increased AR transactivation potency for analogs of both T and NT. For PRβ transactivation, 7α-MT and 7α-ET were not active (EC50 > 10 μM) and trended in the opposite direction from T, indicating that this series may not have progestogenic activity. The presence of C7 α-substitution had a negative impact on PRβ transactivation for these T analogs. This was similar to the PRβ binding results for androgens with a methyl group at the C10 position. The in vitro PRβ transactivation results for C7 α-substituted T (i.e. 7α-MT and 7α-ET) showed a lack of progestogenic activity in a cell-based format. This contrasts with the original goal of increasing both androgenic and progestogenic binding and transactivation activities with a substitution at C7. The lack of definitive in vitro PRβ transactivation activity for 7α-MT and 7α-ET casts doubt on the original thoughts for the design and synthesis of this series; however, the in vivo study results presented below using the McPhail bioassay indicated functional progestogenic activity.
Aromatase
Aromatase is responsible for a key step in the biosynthesis of estrogens, specifically, the aromatization of androgens into estrogens [14, 15]. Aromatase is primarily expressed in the ovaries and testes. Leydig cells outside the seminiferous tubules are the major source of androgens for men. The conversion of testicular androgens to estrogen via aromatase occurs in Sertoli cells [23, 24]. Inhibition of T biosynthesis shuts down estradiol production due to impairment of the normal biosynthetic pathway for production of estrogens. The importance of aromatase in bone homeostasis in men is exemplified by aromatase deficient men displaying osteopenia and unfused epiphyses. In addition, elderly men with a gene polymorphism that alters aromatase activity, manifest a similar phenotype [24, 25]. Moreover, age-related decreases in estrogen production in men, similar to that seen in postmenopausal women, can lead to subsequent declines in bone mass due to enhanced bone resorption [24, 26]. As estrogens are critically important for bone homeostasis for both women and men, loss of gonadal steroids, largely T and estrogen, can result in a decline in bone mineral density. Thus, an optimized progestogenic-androgenic male contraceptive agent would ideally remain an active substrate for aromatase. To that end, the in vitro metabolism of T and the six test articles 7α-MT, 7α-ET, NT, MENT, 11β-MNT, and DMA were examined using human recombinant aromatase in the presence and absence of the co-factor NADPH.
As shown in Figure 1A, the rank order of test articles most to least metabolized by aromatase, based on % remaining after 30 min of incubation, were as follows: T ≈ 7α-ET > 7α-MT > MENT > NT > DMA > 11β-MNT (0.1, 0.2, 20.2, 63.2, 77.0, 87.3, and 98.5%, respectively). The same rank order of test articles from most to least aromatized was seen at the 15 and 60 min time points, as well as when AUC was used in the evaluation. AUC was calculated for % remaining of the parent molecule over time based on the graph of each test article, then converted to % of total AUC and displayed as a bar graph to present a quantifiable assessment of each test article (Figure 1B). A molecule that was completely inert to aromatase would have a calculated AUC of 6000% remaining*min or % of total AUC of 100%. Therefore, a low % of total AUC indicated that the test article was a very active substrate for aromatase.
Figure 1.

Human Aromatase Assay Results. (A) T and test articles % remaining depicted in solid lines when incubated with human CYP19 + P450 and co-factor NADPH at times 0, 15, 30, and 60 min. In dashed lines, T and test articles incubated in the absence of co-factor NADPH at times 0 and 60 min. (B) Calculated AUC (% remaining * min) of T and test articles (in the presence of NADPH, as presented in panel A) converted to % of total AUC. Low % of total AUC indicates rapid metabolism, while a high % of total AUC indicates slow to no metabolism by human aromatase CYP19.
These results clearly demonstrated the importance of a methyl group at the C10 position of the test articles for strong aromatase activity. Comparison of T to 7α-MT and 7α-ET, small sidechain analogs, showed little to no negative impact in aromatase activity with the addition of a C7 α methyl or ethyl group, implying C10 methyl is the primary driving force for the aromatase activity. Indeed, T and 7α-ET had nearly identical aromatase activities with 7α-MT following closely behind. Additionally, the NT and NT analogs which lack a C10 methyl had low to no aromatase activity as shown herein and reported previously [16]. Thus, the C10 methyl group appears important for maintaining the androgen’s aromatization potential and the C7 α-substitution does not hinder this activity. This assay clearly demonstrated that C7 α-substituted methyl and ethyl T analogs are substrates for human aromatase. The ability of 7α-MT and 7α-ET to aromatize to estradiol analogs could be beneficial to obviate potential bone density concerns when used long-term.
5α-reductase
The conversion of T to DHT occurs via the irreversible enzymatic pathway of 5α-reductase. The predominant tissues for this enzymatic action are localized to the prostate, skin, liver, and hair follicles. Additionally, there are other pathways in which DHT is converted from androgens other than T [27–30].
T and six test articles were evaluated as possible substrates of 5α-reductase using a male human liver cytosol assay. Incubations were performed in the absence and presence of the 5α-reductase type 2 specific inhibitor finasteride since recombinant 5α-reductase is not commercially available. The test article was considered a 5α-reductase substrate if a decrease of parent test article was observed in the absence of finasteride, while no decrease or significantly less decrease of parent (i.e. less metabolism) was observed in the presence of finasteride. If no difference was observed with or without finasteride, 5α-reductase was assumed to not be involved in metabolism of that test article. As shown in Figure 2A, 5α-reductase-mediated metabolism of.
Figure 2.

Human 5α-Reductase Assay Results. (A) In solid lines, T and test articles in the absence of 5α-reductase type 2 specific inhibitor finasteride at times 0, 15, 30, 60, 90, and 120 min. In dashed lines, T and test articles incubated in the presence of finasteride at times 0, 15, 30, 60, 90, and 120 min. (B) The difference between the AUC with finasteride and the AUC without finasteride was calculated for T and the test articles and then graphed as a % difference in AUC. The larger the % AUC difference the faster the metabolism, suggesting the test articles are substrates for 5α-reductase, while small to no % AUC difference suggests that the test articles are not substrates for 5α-reductase.
T, the natural substrate, was the most quickly metabolized of the test articles followed by 7α-MT which was the only other test article shown to be a substrate of 5α-reductase in this assay. The test articles 11β-MNT and MENT were very minimally metabolized while 7α-ET and DMA showed no difference in metabolism in the absence or presence of finasteride, indicating that these two compounds are unlikely to be substrates for 5α-reductase. Specifically, 7α-ET and DMA had 83.0 vs. 82.0% or 88.4 vs. 86.2% of parent remaining at 120 min in the presence or absence of finasteride, respectively. The results, calculated as % AUC difference in the absence and presence of finasteride, allowed for the ranking of potential 5α-reductase activity of the test articles as shown in Figure 2B: T > NT ≈ 7α-MT > 11β-MNT > MENT > DMA > 7α-ET (71.6% > 42.1% ≈ 40.1% > 10.1% > 4.0% > 0.6% > 0.1%, respectively). The % AUC difference of two negative controls, DHT and 7α-methyl dihydrotestosterone (7α-MDHT), each at 0.1% confirmed that they were not substrates of 5α-reductase. The finding that binding and transactivation of the DHT equivalent, 7α-methyl substituted DHT (7α-MDHT), approximately three and nine times less active as an androgen, respectively, than 7α-MT alleviates some concerns for prostate cancer risk as 7α-MDHT is not a potent androgen.
Because the addition of C7 α-methyl group to T (as occurred with 7α-MT) decreased 5α-reductase activity from 71.6 to 40.1% AUC, it appears that any substitution at this C7 center interferes with 5α-reductase enzymatic activity. Moreover, the NT analogs showed little to no 5α-reductase activity indicating that they are poor substrates for 5α-reductase.
The muted activity of 5α-reductase for 7α-MT and lack of activity for 7α-ET is encouraging regarding potential concerns associated with prostate cancer and DHT. The role of DHT in prostate cancer is unclear as studies show mixed results. In a longitudinal, observational cohort study of 3638 men, the circulating DHT concentration was not associated with the incidence of prostate cancer. Likewise, other studies evaluating exogenous transdermal DHT administration showed that increasing serum DHT levels had no effect on prostate DHT concentrations, prostate size, and lower urinary tract symptoms. These clinical studies indicate that intracellular concentrations of DHT for androgen-sensitive tissues are not greatly impacted and virtually independent of circulating levels. In contrast, inhibition of 5α-reductase with 5α-reductase inhibitors (5ARIs) decreases intratissue DHT levels in tissue such as prostate, thus reducing prostate size and function. The 5ARIs have been used in benign prostate hyperplasia (BPH) to reduce prostate enlargement and the symptoms of lower urinary tract obstruction, also reducing the risk of developing prostate cancer. In a large population-based cohort study, men with lower urinary tract symptoms treated with a 5ARI for ˃2 years showed a decreased risk of death from prostate cancer as compared with men not treated with a 5ARI [18]. These findings are consistent with previous research showing treatment with a 5ARI is associated with a decrease in prostate cancer incidence. Thus, the importance or lack of serum DHT is not clear; however, a muted or lack of 5α-reductase response could be beneficial when used long-term.
In vivo studies
Hershberger bioassay
The Hershberger bioassay was conducted to evaluate the relative androgenic and anabolic potencies of T and six test articles using the castrated weanling rat model [21]. In this assay, after seven consecutive once daily SC administrations of test article, T reference control, or vehicle control, weanling castrated male rats were sacrificed; the ventral prostate glands, seminal vesicles, and levator ani muscle were excised; and these tissues were weighed. Increases in weights of ventral prostate glands and seminal vesicles in test article vs vehicle control treated rats are evidence of androgenicity while increases in weights of levator ani muscles demonstrate anabolic activity. Mean organ weights per test article per dose level are presented in Figure 3A–C. The calculated AUCs from the dose level versus organ weight graphs are presented in Figure 3D.
Figure 3.

Hershberger assay results evaluating androgenic and anabolic potencies in castrated weanling rats. Mean ventral prostate (A) and seminal vesicle (B) organ weights to evaluate androgenicity and levator ani muscle (C) organ weights to evaluate anabolic potency were determined on Day 8 after seven previous days of once daily SC administrations. (A–C) X-axis depicts total dose received per rat (μmol). Mean ± standard deviation. N = 8 rats/dose/test article. N = 20 rats for 6.9 μmol T condition and for vehicle (0 μmol). N = 8 rats for the other T dose levels. Test article symbols shown in panel A also apply to panels B and C. (D) Dose level versus organ weight AUC from panels A–C graphed ± standard error per test article per organ weight. Test articles with organ weight AUCs that differed with statistical significance from that of T are indicated with asterisks (P ≤ 0.01; MENT and DMA for ventral prostate and seminal vesicles and 7α-ET and DMA for levator ani).
Androgenic activity
All test articles showed dose dependent increases in weight for both ventral prostate and seminal vesicles, indicating an increase in androgenicity with increasing dose. MENT and DMA had similar and high androgenic potency, whereas 7α-MT, 7α-ET, and 11β-MNT had similar mid-range androgenic potency followed by T and then NT with the lowest androgenic activity (Figure 3A, B, D). In other words, the C7 substituted T analogs 7α-MT and 7α-ET were more potent androgens in vivo in rats than T but not as potent as DMA which has methyl groups at C7 and C11 but not at C10. Moreover, 7α-MT was a superior androgen than 11β-MNT. The AR transactivation results, and the in vivo Hershberger assay results taken together provided clear evidence of 7α-MT and 7α-ET being more potent androgens than T despite their muted in vitro AR binding data (Table 1).
Anabolic activity
As shown in Figure 3C, levator ani muscle weights increased in a dose dependent manner and relative to vehicle controls when rats were administered 7α-MT, 7α-ET, NT, 11β-MNT, MENT, and DMA. Very little to no dose response was observed for DMA suggesting that the mean levator ani muscle weight was high and similar irrespective of the dose level administered for this test article.
As shown in Figure 3D, conversion of the levator ani line graph to calculated AUC weight*dose bar graph for the six test articles and T allowed for relative anabolic potency ranking from highest to lowest as follows: MENT > DMA ≈ 7α-ET > 7α-MT > 11β-MNT > NT > T with 2.8 > 2.5 ≈ 2.5 > 2.3 > 2.1 > 1.8 > 1.7, respectively. However, DMA and 7α-ET were the only two test articles with AUCs for levator ani weights that differed with statistical significance from T (P ≤ 0.01) although the pairwise 7α-ET versus NT comparison also differed with statistical significance (P ≤ 0.01).
The anabolic potencies as measured by the weight of levator ani muscles (Figure 3C) did not show similar separation and clustering as observed for the weights of ventral prostate and seminal vesicles (Figure 3A and B). At the highest dose level of 11.2 μmol/rat, 7α-MT, 7α-ET, MENT, 11β-MNT, and DMA had similar anabolic potencies. At the low dose level of 0.7 μmol/rat, MENT and DMA were most potent with DMA showing very little to no increase in activity with increasing dose level. At this low dose, 7α-MT, 7α-ET, and 11β-MNT clustered as mid-range anabolic agents. Absorption of DMA with this sesame oil/ethanol formulation may have reached maximum because increased dose did not result in increased anabolic potency. In contrast, the rising slopes for T, 7α-MT, 7α -ET, 11β-MNT, and MENT indicated that these molecules may support further anabolic potential with increasing dose (Figure 3C).
McPhail study
A McPhail study was conducted to assess progestogenic activity in a rabbit model based on H&E-stained uteri cross sections evaluated for endometrial glandular arborization on a score of 0 to 4. As shown in Figures 4 and 5A, estrogen primed immature female rabbits administered vehicle control or 2.4 μmol T had McPhail Index scores of 0 while those administered 2.4 μmol P had scores of 4 with very pronounced proliferation of the uterus mucosa and pronounced ramification. Rabbits administered increasing dose levels of 7α-MT had increasing amounts of endometrial glandular arborization with mean scores of 0.17, 0.29, 1.25, and 2.21 when administered a total of 0.8, 2.4, 7.2, and 21.6 μmol 7α-MT, respectively. Administration of 7α-MT had clear progestogenic effects in this in vivo model, yet as shown in Figure 5, MENT, 11β-MNT, DMA, and NT were more potently progestogenic than 7α-MT.
Figure 4.
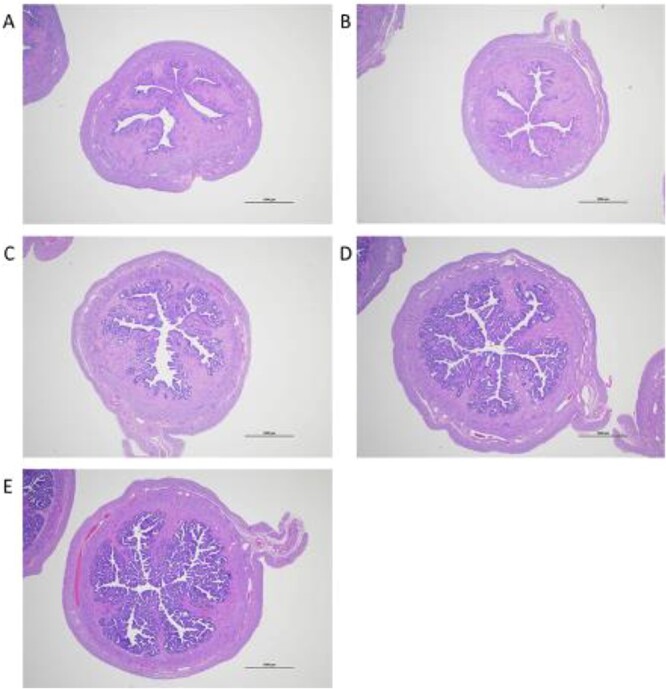
H&E-stained uteri cross sections representing each of the four possible McPhail Index scores for evaluation of endometrial glandular arborization. (A) Grade 0, vehicle control. (B) Grade 1, 7α-MT, 0.8 μmol total dose. (C) Grade 2, 7α-MT, 7.2 μmol total dose. (D) Grade 3, 7α-MT, 21.6 μmol total dose. (E) Grade 4, progesterone, 2.4 μmol total dose. Scale bar = 1000 μm.
Figure 5.

Progestogenic effect of test articles on rabbit uteri after 17β-estradiol priming followed by 5 once daily SC administrations of test article. (A) Average McPhail Index of rabbit uteri per treatment. Uteri were collected on Day 12 (the day after the last test article dose), fixed, sectioned, stained, and scored based on the five-step scoring system (0–4) of McPhail to generate an average endometrial glandular arborization score for each rabbit. Data presented as average McPhail index ± standard deviation per treatment group (four sections scored per rabbit, N = 9 rabbits for vehicle and progesterone, N = 6 rabbits for each test article dose level, n = 3 rabbits for T that was tested only at 2.4 μmol/rabbit). (B) Mean uterus to body weight ratio (%) in the same rabbits depicted in panel A. (C) AUC for dose level versus McPhail score is plotted against the left y-axis in black bars ± standard error using the data graphed in panel A. AUC for dose level versus uterus body weight ratio is plotted against the right y-axis in white bars ± standard error using the data graphed in panel B. AUC values different with statistical significance (P ≤ 0.01) from that of 7α-MT are indicated by asterisks. None of the other pairwise AUC comparisons differed with statistical significance.
While NT analogs, 11β-MNT, MENT, and DMA reached their maximum and perhaps steady state McPhail Index of ~3 at ~2.4 μmole, 7α-MT and NT still showed signs of increasing progestogenic activity with increasing dose (Figure 5A and B). This suggests that the maximum McPhail indexes for both 7α-MT and NT may not have been achieved at the highest dose of 21.6 μmole tested in this study. Similar findings of steady state for higher concentrations for NT analogs and a dose dependent rise for 7α-MT and NT were also observed for the uterus to body weight ratio (Figure 5B and C).
As shown in Figure 5C, the AUC values derived from Figure 5A and B were calculated for the total dose versus the average McPhail index score and ratio of uterus to body weight per test article. The rank order of test articles based on the AUCs for McPhail index and uterus/body weight ratio both showed 11β-MNT > MENT > DMA ≈ NT > 7α-MT (60.2 > 55.1 > 51.9 ≈ 51.2 > 29, respectively, for McPhail index AUC and 5.6 > 4.7 ≈ 4.6 > 4.1 > 3.4, respectively, for uterus/body weight AUC). However, the only pairwise comparisons that met the level of statistical significance (P ≤ 0.01) for both McPhail index AUC and uterus ratio AUC were that of 7α-MT versus 11β-MNT and MENT. The relative progestogenic potency as typically determined through McPhail index mirrored the uterus/body weight ratio.
Among the five test articles, 7α-MT is the only molecule in this series that is not a NT analog. A direct comparison of 7α-MT and MENT (where the only difference is the presence or absence of methyl group at C10 position) showed more robust progestogenic activity without the methyl group at C10. This structural aspect is further observed when comparing T with NT. NT clearly showed high progestogenic activity while T at 2.4 μmole did not. Similar to the Hershberger results of androgenic and anabolic potencies, the absence of the methyl group at C10 appears to be an important factor in potency of progestogenic activity for this series of T and NT analogs. Although 7α-MT is a weaker progestin than MENT, 7α-MT had clear progestogenic activity despite the presence of the C10 methyl group. The progestogenic activity of 7α-MT is clearly derived from the presence of the C7 methyl group as compared to its parent scaffold, T. The structural feature of the C7 methyl group providing progestogenic activity is further supported when MENT is compared to NT. The only difference between the two molecules is the methyl group at the C7 position. A single substitution at C7 or C11 appears to effect progestogenic activity positively as observed in this in vivo McPhail study. However, having methyl groups at both C7 and C11 does not appear to be additive.
Conclusion
The C7 α-substituted T analogs 7α-MT and 7α-ET displayed androgenic and progestogenic activities. Despite somewhat muted in vitro binding and transactivation results, the in vivo studies for androgenic and progestogenic activities using rat and rabbit models, respectively, demonstrated a more promising outcome. The in vivo rat and rabbit models supported the rationale and design of the C7 α-substituted Ts, 7α-MT and 7α-ET. Results from the Hershberger study in rats showed superior in vivo androgenic and anabolic activities of the analogs as compared to T as determined by ventral prostate, seminal vesicle, and levator ani weights. Similarly, the McPhail study in rabbits showed 7α-MT and 7α-ET to have clear progestogenic activity as determined by evaluation of endometrial glandular arborization and the ratio of uterus to body weight. Additionally, both 7α-MT and 7α-ET readily undergo aromatization and are more resistant to 5α-reductase activity. These attributes are favorable in long-term for reducing prostate growth and maintaining bone mineral density. The encouraging in vitro outcome for these analogs could be potential improvements over NT analogs currently in the clinic.
The design and synthesis of 7α-MT and 7α-ET accomplished all the goals and objectives of achieving aromatizable molecules with reduced metabolism by 5α-reductase that have androgenic, anabolic, and progestogenic properties. Thus, the core and/or prodrugs of 7α-MT and 7α-ET are promising molecules for further development as male contraceptive PAs.
Acknowledgments
The authors would like to thank Alvin Chen (SRI) for project management support, SRI International’s team of researchers who provided technical support, Harold S. Javitz (SRI) for statistical analysis support, Lori Olson (SRI) for optical rotation measurements, Bharat Gurale (Piramal) for NMR and MS support, and Bashir Kaskar (Piramal) for discussions.
Footnotes
† Grant Support: This project has been funded with Federal funds from the National Institutes of Child Health and Human Development, National Institutes of Health, Department of Health and Human Services, under Contracts No. HHSN275201500002I and 75N94020D00003 held by SRI International and HHSN275201700003C held by Piramal Pharma Solutions (formerly Ash Stevens, LLC).
Contributor Information
Min S Lee, Contraceptive Development Program, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institute of Health, Bethesda, MD, USA.
Deborah I Bunin, Biosciences Division, SRI International, Menlo Park, CA, USA.
Anna M Furimsky, Biosciences Division, SRI International, Menlo Park, CA, USA.
Donna Nguyen, Biosciences Division, SRI International, Menlo Park, CA, USA.
Toufan Parman, Sangamo Therapeutics, Brisbane, CA, USA.
Kyuri Kim, Jazz Pharmaceuticals, Palo Alto, CA, USA.
Linda Rausch, Biosciences Division, SRI International, Menlo Park, CA, USA.
Ming-Teh Lin, Piramal Pharma Solutions, Riverview, MI, USA.
Pranab Gupta, Piramal Pharma Solutions, Riverview, MI, USA.
Jill E Brown, Department of Gynecologic Surgery & Obstetrics, Uniformed Services University, Bethesda, MD, USA.
Jeffrey M Kroopnick, Contraceptive Development Program, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institute of Health, Bethesda, MD, USA.
Diana L Blithe, Contraceptive Development Program, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institute of Health, Bethesda, MD, USA.
Conflict of interest
The authors report no conflict of interest.
Authors’ Contributions
K.K., T.P., A.M.F., D.N., L.R., D.I.B, and M.S.L. planned and oversaw the experiments, performed data analysis, and wrote the manuscript. M.-T.L. and P.G. synthesized the molecules and wrote chemistry methods. J.M.K., J.E.B., D.L.B, and M.S.L participated in in-house weekly meetings, data analysis, and discussions. M.S.L. participated in and directed all phases of the study.
Data availability
The authors declare that the data supporting the findings of this study are available within the paper.
References
- 1. Brady BM, Amory JK, Perheentupa A, Zitzmann M, Hay CJ, Apter D, Anderson RA, Bremner WJ, Pollanen P, Nieschlag E, Wu FCW, Kersemaekers WM. A multicentre study investigating subcutaneous etonogestrel implants with injectable testosterone decanoate as a potential long-acting male contraceptive. Hum Reprod 2006; 21:285–294. [DOI] [PubMed] [Google Scholar]
- 2. Mommers E, Kersemaekers WM, Elliesen JR, Kepers M, Apter D, Behre HM, Beynon J, Bouloux PM, Costantino A, Gerbershagen H-P, Grønlund L, Heger-Mahn D, et al. Male hormonal contraception: a double-blind, placebo-controlled study. J Clin Endocrinol Metabol 2008; 93:2572–2580. [DOI] [PubMed] [Google Scholar]
- 3. Behre HM, Zitzmann M, Anderson RA, Handelsman DJ, Lestari SW, McLachlan RI, Meriggiola MC, Misro MM, Noe G, Wu FCW, Festin MPR, Habib NA, et al. Efficacy and safety of an injectable combination hormonal contraceptive for men. J Clin Endocrinol Metabol 2016; 101:4779–4788. [DOI] [PubMed] [Google Scholar]
- 4. Kamischke A, Heuermann T, Krüger K, Von Eckardstein S, Schellschmidt I, Rübig A, Nieschlag E. An effective hormonal male contraceptive using testosterone Undecanoate with oral or injectable Norethisterone preparations. J Clin Endocrinol Metabol 2002; 87:530–539. [DOI] [PubMed] [Google Scholar]
- 5. Meriggiola MC, Costantino A, Saad F, D’Emidio L, Morselli Labate AM, Bertaccini A, Bremner WJ, Rudolph I, Ernst M, Kirsch B, Martorana G, Pelusi G. Norethisterone Enanthate plus testosterone Undecanoate for male contraception: effects of various injection intervals on spermatogenesis, reproductive hormones, testis, and prostate. J Clin Endocrinol Metabol 2005; 90:2005–2014. [DOI] [PubMed] [Google Scholar]
- 6. Qoubaitary A, Meriggiola C, Ng CM, Lumbreras L, Cerpolini S, Pelusi G, Christensen PD, Hull L, Swerdloff RS, Wang C. Pharmacokinetics of testosterone Undecanoate injected alone or in combination with Norethisterone Enanthate in healthy men. J Androl 2006; 27:853–867. [DOI] [PubMed] [Google Scholar]
- 7. Thirumalai A, Ceponis J, Amory JK, Swerdloff R, Surampudi V, Liu PY, Bremner WJ, Harvey E, Blithe DL, Lee MS, Hull L, Wang C, et al. Effects of 28 days of oral Dimethandrolone Undecanoate in healthy men: a prototype male pill. J Clin Endocrinol Metabol 2019; 104:423–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wu S, Yuen F, Swerdloff RS, Pak Y, Thirumalai A, Liu PY, Amory JK, Bai F, Hull L, Blithe DL, Anawalt BD, Parman T, et al. Safety and pharmacokinetics of single-dose novel oral androgen 11β-Methyl-19-Nortestosterone-17β-Dodecylcarbonate in men. J Clin Endocrinol Metabol 2019; 104:629–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yuen F, Thirumalai A, Pham C, Swerdloff RS, Anawalt BD, Liu PY, Amory JK, Bremner WJ, Dart C, Wu H, Hull L, Blithe DL, et al. Daily oral Administration of the Novel Androgen 11β-MNTDC markedly suppresses serum gonadotropins in healthy men. J Clin Endocrinol Metabol 2020; 105:e835–e847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Anawalt BD, Roth MY, Ceponis J, Surampudi V, Amory JK, Swerdloff RS, Liu PY, Dart C, Bremner WJ, Sitruk-Ware R, Kumar N, Blithe DL, et al. Combined nestorone–testosterone gel suppresses serum gonadotropins to concentrations associated with effective hormonal contraception in men. Andrology 2019; 7:878–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu PY, Swerdloff RS, Anawalt BD, Anderson RA, Bremner WJ, Elliesen J, Gu Y-Q, Kersemaekers WM, McLachlan RI, Meriggiola MC, Nieschlag E, Sitruk-Ware R, et al. Determinants of the rate and extent of Spermatogenic suppression during hormonal male contraception: an integrated analysis. J Clin Endocrinol Metabol 2008; 93:1774–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Meriggiola MC, Farley TMM, Mbizvo MT. A review of androgen-progestin regimens for male contraception. J Androl 2003; 24:466–483. [DOI] [PubMed] [Google Scholar]
- 13. Wang C, Swerdloff RS. Male hormonal contraception. Am J Obstet Gynecol 2004; 190:S60–S68. [DOI] [PubMed] [Google Scholar]
- 14. Akhtar M, Wright JN. A review of 18O labelling studies to probe the mechanism of aromatase (CYP191A). J Steroid Biochem Mol Biol 2022; 216:106010. [DOI] [PubMed] [Google Scholar]
- 15. Di Nardo G, Gilardi G. Human aromatase: perspectives in biochemistry and biotechnology. Biotechnol Appl Biochem 2013; 60:92–101. [DOI] [PubMed] [Google Scholar]
- 16. Attardi BJ, Pham TC, Radler LM, Burgenson J, Hild SA, Reel JR. Dimethandrolone (7α,11β-dimethyl-19-nortestosterone) and 11β-methyl-19-nortestosterone are not converted to aromatic A-ring products in the presence of recombinant human aromatase. J Steroid Biochem Mol Biol 2008; 110:214–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Andriole GL, Bostwick DG, Brawley OW, Gomella LG, Marberger M, Montorsi F, Pettaway CA, Tammela TL, Teloken C, Tindall DJ, Somerville MC, Wilson TH, et al. Effect of Dutasteride on the risk of prostate cancer. New England J Med 2010; 362:1192–1202. [DOI] [PubMed] [Google Scholar]
- 18. Björnebo L, Nordström T, Discacciati A, Palsdottir T, Aly M, Grönberg H, Eklund M, Lantz A. Association of 5α-reductase inhibitors with prostate cancer mortality. JAMA Oncol 2022; 8:1019–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Thompson IM, Chi C, Ankerst DP, Goodman PJ, Tangen CM, Lippman SM, Lucia MS, Parnes HL, Coltman CA. Effect of finasteride on the sensitivity of PSA for detecting prostate cancer. JNCI: J National Cancer Instit 2006; 98:1128–1133. [DOI] [PubMed] [Google Scholar]
- 20. Council NR . Guide for the Care and Use of Laboratory Animals, Eighth ed. Washington, DC: The National Academies Press; 2011. [PubMed] [Google Scholar]
- 21. Hershberger LG, Shipley EG, Meyer RK. Myotrophic activity of 19-nortestosterone and other steroids determined by modified levator ani muscle method. Proc Soc Exp Biol Med 1953; 83:175–180. [DOI] [PubMed] [Google Scholar]
- 22. McPhail MK. The assay of progestin. J Physiol 1934; 83:145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dorrington JH, Armstrong DT. Follicle-stimulating hormone stimulates estradiol-17beta synthesis in cultured Sertoli cells. Proc Natl Acad Sci 1975; 72:2677–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Blakemore J, Naftolin F. Aromatase: contributions to physiology and disease in women and men. Phys Ther 2016; 31:258–269. [DOI] [PubMed] [Google Scholar]
- 25. Gennari L, Nuti R, Bilezikian JP. Aromatase activity and bone homeostasis in men. J Clin Endocrinol Metabol 2004; 89:5898–5907. [DOI] [PubMed] [Google Scholar]
- 26. Falahati-Nini A, Riggs BL, Atkinson EJ, O’Fallon WM, Eastell R, Khosla S. Relative contributions of testosterone and estrogen in regulating bone resorption and formation in normal elderly men. J Clin Investig 2000; 106:1553–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mostaghel EA. Steroid hormone synthetic pathways in prostate cancer. Transl Androl Urol 2013; 2:212–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Toorians AWFT, Kelleher S, Gooren LJ, Jimenez M, Handelsman DJ. Estimating the contribution of the prostate to blood Dihydrotestosterone. J Clin Endocrinol Metabol 2003; 88:5207–5211. [DOI] [PubMed] [Google Scholar]
- 29. Ishimaru T, Edmiston A, Pages L, Horton R. Direct conversion of testosterone to Dihydrotestosterone glucuronide in man. J Clin Endocrinol Metabol 1978; 47:1282–1286. [DOI] [PubMed] [Google Scholar]
- 30. Swerdloff RS, Dudley RE, Page ST, Wang C, Salameh WA. Dihydrotestosterone: biochemistry, physiology, and clinical implications of elevated blood levels. Endocr Rev 2017; 38:220–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors declare that the data supporting the findings of this study are available within the paper.