

Abstract
Background
Say‐Barber‐Biesecker‐Young‐Simpson (SBBYSS) variant of Ohdo syndrome is a rare, autosomal dominant and clinically heterogenous disorder, caused by pathogenic variants in the KAT6B gene located on chromosome 10q22.2. KAT6B encodes a highly conserved histone acetyltransferase belonging to the MYST family. Currently, diseases caused by pathogenic variants in KAT6B (KAT6B‐related disorders) comprise two allelic entities: SBBYSS variant of Ohdo syndrome and genitopatellar syndrome (GPS). Increase in the number of cases with overlapping GPS/SBBYSS phenotype which makes it necessary to redefine this group of phenotypes as KAT6B‐related disorders or KAT6B spectrum disorders.
Individuals with SBBYSS usually present with facial abnormalities, hypotonia, joint laxity, feeding problems, and long thumbs/great toes. This syndrome also typically involves skeletal problems including patellar hypoplasia/agenesis.
Methods
Here we report six SBBYS syndrome patients with the same dysmorphic features but a different course of the disease. One known and five novel KATB6 pathogenic variants were identified by molecular diagnostics using Next Generation Sequencing (NGS).
Results
We present a detailed phenotypic analysis of six individuals with KAT6B‐related disorders, in whom a heterozygous pathogenic variant in KAT6B gene was found. In all of our patients facial dysmorphism as well as developmental and speech delay were present. Additionally, all but one patients presented with hypotonia, ocular abnormalities and long thumbs. Most of our probands showed blepharophimosis and skeletal (mainly knee) defects. Contrary to previously reported severe patellar defects (hypoplasia/agenesis) anomalies presented by our patients were less severe (dysplasia, habitual dislocation, subluxation) referring to KAT6B‐related disorders.
Conclusion
While most of the anomalies found in our patients comply with SBBYSS criteria, phenotypic differences in our probands support a broader spectrum of the disease phenotype. To establish the range of this spectrum, a detailed analysis of clinical variability among patients with SBBYSS requires further investigation.
Keywords: dysmorphism, GPS, KAT6B gene, KAT6B–related disorder, SBBYSS
We present a detailed phenotypic analysis of six individuals with KAT6B‐related disorders, in whom a heterozygous pathogenic variant in KAT6B gene was found. We report six SBBYS syndrome patients with the same dysmorphic features but a different course of the disease. One known and five novel KATB6 pathogenic variants were identified by molecular diagnostics using Next Generation Sequencing (NGS). While most of the anomalies found in our patients comply with SBBYSS criteria, phenotypic differences in our probands support a broader spectrum of the disease phenotype. To establish the range of this spectrum, a detailed analysis of clinical variability among patients with SBBYSS requires further investigation.
1. INTRODUCTION
Say‐Barber‐Biesecker‐Young‐Simpson syndrome (SBBYSS), variant of Ohdo syndrome (OMIM: 603736, ORPHA: 3047) is an extremely rare autosomal genetic condition caused by heterozygous pathogenic variants in the KAT6B gene encoding a histone acetyltransferase enzyme. The SBBYS syndrome was first described in 1987 by Young and Simpson, who presented an individual with blepharophimosis, congenital heart defects, hypothyroidism and intellectual disability (Lonardo et al., 2019; Masuno et al., 1999) Underlying molecular effect was identified by Clayton‐Smith et al. (2011).
SBBYSS is a multiple congenital anomaly disorder with heterogenous presentation but typically characterized by specific facial features and skeletal defects (including long thumbs, great toes), as well as developmental delay/intellectual disability and feeding difficulties. Most patients with this syndrome have congenital heart defects, cleft palate, lacrimal duct anomalies, genital abnormalities, patellar anomalies (aplasia/hypoplasia), hearing loss, thyroid anomalies and dental anomalies (Lemire et al., 2012). The main dysmorphic features comprise: mask‐like facial appearance, blepharophimosis with downslanting palpebral fissures, flat broad nasal bridge, tubular/bulbous nose, long philtrum, thin upper lip, thin lip vermilion, prominent cheeks, low‐set and posteriorly rotated ears and micro−/retrognathia. Clinical symptoms suggesting an SBBYSS diagnosis have been divided into major and minor criteria. Affected patients with two major features or one major feature and two minor features are likely to have this KAT6B‐related disorder. Major features include long thumbs/great toes, immobile mask‐like face, blepharophimosis/ptosis, lacrimal duct anomalies and patellar hypoplasia/agenesis. Minor features include congenital heart defect, dental anomalies (hypoplastic teeth and/or delayed eruption of teeth), hearing loss, thyroid anomalies, cleft palate, genital anomalies (males: cryptorchidism), hypotonia, global developmental delay/intellectual disability (Lemire et al., 2012; Lonardo et al., 2019).
KAT6B mutations also can lead to a more severe presentation: genitopatellar syndrome (GPS, OMIM: 606170, ORPHA: 85201). Variable expressivity and broader phenotypic spectrum associated with KAT6B‐related GPS or SBBYSS is increasingly recognized. (Lonardo et al., 2019) Recently, several cases describing an intermediate phenotype between GPS and SBBYSS were reported (Gannon et al., 2015; Kim et al., 2017). Relative differences and similarities between the two conditions depend on the specific location of the causative variant in the KAT6B gene. When codons 1–1205 are affected, SBBYSS variant is predominant (group 1–2), for codons 1205–1350 it is GPS phenotype (group 3), while for 1350–1520 it may be GPS or SBBYSS or intermediate phenotype (group 4) and for 1520 to at least 1935 (the last affected codon closest to the C‐end, group 5) it is usually SBBYSS. The phenotype of predicted group 6 (codon 2073 at the C‐end) is yet unknown but probably mild (Radvanszky et al., 2017; Turkyilmaz & Ozden, 2021; Vlckova et al., 2015; Zhang et al., 2020). The differences in the clinical features are predicted to result from different molecular pathomechanism of the disturbed KAT6B gene. Defective transcripts escaping the process of nonsense‐mediated mRNA decay (NMD) and forming a truncated protein with reduced histone acetylase activity cause the more severe GPS phenotypic spectrum. Variants leading to NMD (located closest to C‐end but not affecting critical binding regions of the protein) more often correlate with SBBYSS and those causing haploinsufficiency of KAT6B (in the initiation region or whole‐gene deletions) result in a milder phenotype. Therefore, GPS is hypothesized to occur through a gain‐of‐function or dominant‐negative disease mechanism and SBBYSS through a loss‐of‐function (Brea‐Fernández et al., 2019; Campeau et al., 2012; Gannon et al., 2015).
Here we report six SBBYS syndrome patients with the same dysmorphic features but a different course of the disease. One known and five novel KATB6 pathogenic variants (standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology—ACMG/AMP) were identified by molecular diagnostics using Next Generation Sequencing (NGS). Four of those patients had severe symptoms, while in the remaining two milder symptoms were observed (Table 1).
TABLE 1.
Clinical symptoms in presented patients with KAT6B mutation.
| Proband 1 | Proband 2 | Proband 3 | Proband 4 | Proband 5 | Proband 6 | |
|---|---|---|---|---|---|---|
| Mutation (cDNA) Protein alteration | c.3852_3864del, p.Asp1284GlufsTer46 | c.4026dupA, p.Pro1343ThrfsTer4 | c.5819delA, p.Gln1940Argfs*11 | c.3147G > A, p.Pro1049= | c.4455dupT, p.Asn1486fs | c.5012del, p.Gly1671Alafs*44 |
| Inheritance | De novo | De novo | De novo | De novo | De novo | De novo |
| Gender | Male | Male | Female | Male | Female | Female |
| Age of diagnosis | 12 months | 5 years 9 months | 21 years | 24 months | 12 month | |
| Current age | Died at the age of 3 years | Died at the age of 6 years 8 months | 25 years | 3 years | 15 month | 20 years |
| Prenatal period | Bilateral hydronephrosis, fetal movements poorly | Impending preterm delivery from 32 week of pregnancy | Uncomplicated | Uncomplicated | Uncomplicated | Uncomplicated |
| Time of delivery (weeks) | 38 | 35 | 37 | 40 | 39 | 37 |
| Birth weight (g) | 3220 | 2700 | 3250 | 3490 | 3570 | 3150 |
| Points of Apgar | 8 | 5 | 10 | 9/10/10/10 | 4/6/6 | 9/10 |
| Neonatal period | Incision of the urethral valve posterior, left nephrostomy and ureteropelvisplasty | Asphyxia, congenital pneumonia, laryngomalacia | Uncomplicated | Retracted chin, decreased muscle tone, congenital pneumonia | Decreased muscle tone, dysmorphia, systolic murmur | Mixed muscle tone |
| Feeding difficulties | + | + | + | − | + | + |
| Agenesis of the corpus callosum | + | + | − | − | + | − |
| Congenital heart defects | PFO | VSD | − | − | ASD, VSD | ASD |
| Cleft palate | − | High palate | Cleft uvula | − | High palate | − |
| Skeletal defects | Shoulder exostoses | Valgus knees | Valgus knees and feet | Valgus knees | − | Club feet and hands |
| Patellar anomalies | − | Patellar dysplasia; habitual dislocation of the right patellae, subluxation of the left patellae | Subluxation | − | − | Subluxation of the patella |
| Genital anomalies | Bilateral cryptorchidism, scrotum excavatum/showl | Bilateral cryptorchidism, hypoplasia of the scrotum; subcoronal hypospadias | − | Bilateral cryptorchidism | − | Clitoral hypertrophy |
| Hearing loss | + | − | − | + | + | + |
| Ocular finding | Eye fixation disorders | Nystagmus, left eye strabismus, myopia, astigmatism | Hypermyopia | Hypermyopia | Hypermyopia, astigmatism | Astigmatism, nystagmus |
| Microcephaly | + | + | + | − | + | − |
| Mask‐like face | + | + | + | + | + | − |
| Downslanting palpebral fissures | − | − | + | + | + | − |
| Blepharophimosis | + | − | + | − | + | + |
| Lacrimal duct anomalies | + | − | − | − | + | − |
| Tubular/bulbous nose | + | + | + | − | + | + |
| Prominent cheeks | + | + | + | − | + | ‐ |
| Low‐set/posteriorly rotated ears | + | + | + | − | + | + |
| Thin upper lip/lip vermilion | + | − | + | + | + | − |
| Long philtrum | + | − | + | − | + | + |
| Micro−/retrognathia | + | + | + | + | + | + |
| Dental anomalies | + | Abnormal and delayed teeth eruption, first tooth at 13 months of age (upper incisors) | Malocclusion | − | − | Delayed teeth eruption |
| Thyroid anomalies | Hypothyroidism | Hypothyroidism | Hypothyroidism | − | Hypothyroidism | – |
| Hand and fingers | Long thumbs | Long fingerlike thumbs | Long thumbs | Long thumbs, overlapping fingers | + | Long fingers, poor dermatoglyphics, radioulnar synostosis |
| Feet and toes | Great toes, club feet | Long overlapping toes | Valgus feet | Flat feet | + | Club feet |
| Flexion contractures | + | − | − | − | − | |
| Hypotonia | + | + | + | + | + | – |
| Developmental delay | + | + | + | + | + | + |
| Speech delay | + | + | + | + | + | + |
| Intellectual disability | NA | + | IQ 40 | NA | NA | + |
| Others symptoms | − | Instability of the elbows and ankles; multicystic kidneys; barrel‐shaped chest; epilepsy | Cleft uvula | Concave nipples, red palms, drooling | Café au lait spots, recurrent infections | Ptosis |
| Death | Yes, in 3 y | Yes, in 7 y | No | No | No | No |
Abbreviations: NA, not available; PFO, patent foramen ovale; VSD, ventricular septal defect.
2. CLINICAL REPORTS
2.1. Proband 1
Proband 1 belongs to group 3 according to the proposed genotype classification of KAT6B‐related disorder and presents classical phenotype of SBBYSS.
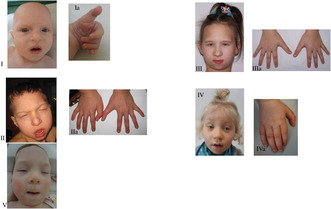
Proband 1 is a 2‐year‐old boy, second child of young, healthy, and unrelated parents. Pregnancy was complicated with bilateral hydronephrosis, shunted prenatally. During pregnancy his mother felt the fetal movements poorly. He was born at term with a weight of 3220 g (50‐90th percentile), length of 53 cm (90‐97th percentile), and occipitofrontal circumference (OFC) of 36 cm (90th percentile). In infancy, he was repeatedly admitted to hospitals where he underwent several procedures: incision of the posterior urethral valve, left nephrostomy and ureteropelvic junction plasty. As a newborn he presented with feeding difficulties. He showed features of generalized psychomotor delay, including gross/fine motor and social, feeding, social, speech and cognitive skills. He was first seen by a clinical geneticist at 7 months to establish the molecular cause of a congenital anomaly syndrome. At that time, physical examination showed dysmorphic features: blepharophimosis, bulbous nose, prominent cheeks, posteriorly rotated ears, long philtrum, thin upper lip (Figure 1). He had long thumbs (Figure 1), club feet, dysplastic nails, contractures of the knees, shoulder exostoses, bilateral cryptorchidism, bifid scrotum. Growth parameters at the time of examination were: weight 5.4 kg (<3rd percentile), OFC 40 cm (<3rd percentile). The boy was diagnosed with hypotonia, bilateral sensorineural hearing loss, hypothyroidism, and severe speech delay. He had frequent upper respiratory tract infections. Ophthalmologic examination revealed visual fixation problems and lacrimal duct anomalies. Transfontanelle ultrasonography showed agenesis of corpus callosum and ventricular dilatation. Echocardiogram result revealed a patent foramen ovale. The boy died at the age of 3 years (Table 1).
FIGURE 1.
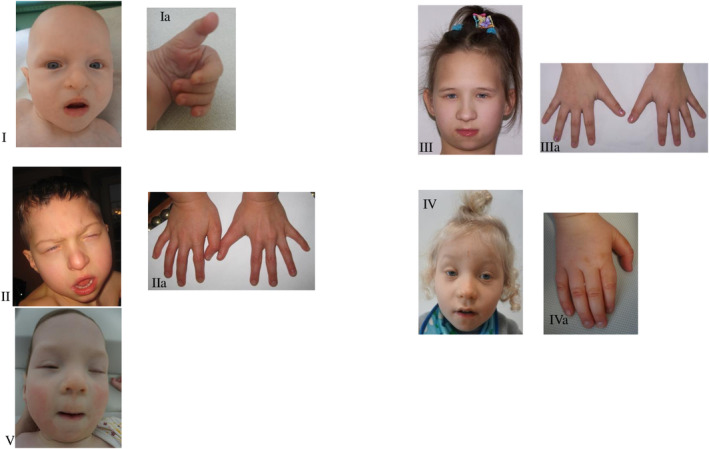
Phenotype of the patients. I—Proband 1, II—Proband 2, III—Proband 3, IV—Proband 4, V—Proband 5 Dysmorphia of the fingers‐long thumbs. Ia—Proband's 1 hand, IIa—Proband's 2 hands, IIIa—Proband's 3 hands, IVa—Proband's 4 hand.
2.2. Proband 2
Proband 2 belongs to group 3 according to the proposed genotype classification of KAT6B‐related disorder and presents a classical phenotype of GPS/SBBYSS.
Proband 2 was the first‐born child of young, healthy, and non‐consanguineous parents. Pregnancy was uncomplicated up to 32nd week, when hospital admission of the mother was necessary because of impending preterm delivery. The delivery was at 35 weeks; birth weight was 2700 g (50‐90th percentile), length 46 cm (10‐50th percentile), OFC 29 cm (3th percentile). Examination after birth revealed asphyxia, congenital pneumonia and laryngomalacia. He required mechanical ventilation and parenteral nutrition. At the age of 7 weeks he was referred to clinical geneticist for the evaluation of dysmorphic features and multiple malformations. Physical examination at the age of 14 months showed a weight of 7.5 kg (<3rd percentile), height of 80 cm (50th percentile), OFC of 43 cm (<3rd percentile), massive malnutrition and hypertonia. Dysmorphic features included: large fontanelle, small palpebral fissures, ptosis, big, bulbous nose, prominent cheeks, posteriorly rotated ears, small earlobes, small lips, high “gothic” palate and micrognathia (Figure 1). He presented with long overlapping toes, long thumbs, camptodactyly (Figure 1), genu valgums and patellar dysplasia in the form of habitual dislocation of the right patellae and subluxation of the left patellae. He had abnormal teeth with delayed eruption. Global developmental delay was recognized (he could not walk unassisted or speak a single word). At the age of 6 he was diagnosed with seizures treated by valproic acid. He had bilateral cryptorchidism, hypospadias, scrotal hypoplasia, hydronephrosis and hypothyroidism. MRI of the brain showed agenesis of the corpus callosum, while VSD type defect was noted on echocardiogram. Ophthalmological evaluation showed nystagmus, left eye strabismus, myopia and astigmatism. Electroencephalography revealed abnormalities. He died at the age of 6 years 8 months (Table 1).
2.3. Proband 3
Proband 3 belongs to group 6 according to the proposed genotype classification of KAT6B‐related disorder and presents mild phenotype of SBBYSS.
Proband 3 is a 24‐year‐old woman, who was born at 37 weeks of gestation with birth weight 3250 g (50‐90th percentile) and height 56 cm (>97th percentile) after uncomplicated pregnancy. As a baby she presented with feeding difficulties. She has typical facial dysmorphism including blepharophimosis, mask‐like face, downslanting palpebral fissures, microcephaly, long and bulbous nose, prominent cheeks, posteriorly rotated ears, small mouth, thin upper lip, long philtrum and micro−/retrognathia (Figure 1) The patient displayed skeletal deformities—long thumbs (Figure 1) genu/feet valgums, bilateral patellar subluxation. Her neurological and developmental examination demonstrated hypotonia, and global developmental, and speech delay. Ophthalmologic examination revealed hypermetropia. Additionally she has cleft uvula, teeth malocclusion and hypothyroidism (Table 1).
2.4. Proband 4
Proband 4 belongs to group 1–2 according to the proposed genotype classification of KAT6B‐related disorder and presents classical phenotype of SBBYSS.
Proband 4 is a 3‐year‐old boy, born at term to unrelated healthy parents with weight 3490 g (10‐50th percentile). Pregnancy was uncomplicated. After birth he had congenital pneumonia and decreased muscle tone. He displayed psychomotor and speech delay. Physical examination showed facial dysmorphism with downslanting palpebral fissures, flat broad nasal bridge, bulbous nose, long philtrum, thin upper lip and retracted chin (Figure 1). He presented with genu valgums, long thumbs (Figure 1), overlapping toes and flat feet. The patient had no thyroid problems. The patient's medical history revealed frequent drooling, hearing loss, hypermetropia, bilateral cryptorchidism, sunken nipples, and red palms of hands (Table 1).
2.5. Proband 5
Proband 5 belongs to group 4 according to the proposed genotype classification of KAT6B‐related disorder and presents classical phenotype of SBBYSS.
Proband 5 is a 15 month‐old girl, who was born to non‐consanguineous parents at 39 weeks of gestation after a normal pregnancy. The birth weight was 3570 g (50‐90th percentile). The Apgar scores at 1 and 5 min were 4 and 6 points, respectively. She presented with decreased muscle tone, systolic murmur and typical facial dysmorphism, including microcephaly (Figure 1) Clinical evaluation at 12 months revealed feeding problems, high palate and hearing loss. Ophthalmological evaluation showed ocular hypermetropia and astigmatism. ASD and VSD type defects were noted on echocardiogram. Transfontanelle ultrasonography revealed agenesis of the corpus callosum. Currently, she presents with features of developmental/speech delay. The child has hypothyroidism, café au lait spots and recurrent infections, but no skeletal or patellar problems (Table 1).
2.6. Proband 6
Proband 6 belongs to group 5 according to the proposed genotype classification of KAT6B‐related disorder and presents classical phenotype of GPS/SBBYSS.
Proband 6 is a 20‐year‐old woman, who was born at 37 weeks of gestation with birth weight 3150 g (50–90 percentile), length 52 cm (90 percentile and OFC 35 cm (90 percentile) after an uncomplicated pregnancy. The Apgar scores at 1 and 5 mins were 9 and 10 points, respectively. As a baby she presented feeding and swallowing difficulties. Her facial dysmorphism at birth was very prominent and included blepharophimosis, short and bulbous nose, low set and posteriorly rotated ears, long philtrum and micrognathia. The patient displayed skeletal deformities: long fingers, bilateral patellar subluxation, clubfeet and radioulnar synostosis. She also presented with delayed teeth eruption. Renal ultrasound revealed multicystic kidneys. He was diagnosed with an ASD type heart defect that closed on its own. Her neurological and developmental examination demonstrated variable muscle tension. She presented with developmental and speech delay. She was able to sit unsupported at 12 months, and walk unassisted at 24 months. At the age of 6 she could speak single sentences. Ophthalmologic examination revealed astigmatism and nystagmus. She was also diagnosed with bilateral hearing loss and additionally bilateral blepharophimosis was observed (Table 1).
3. GENETIC STUDIES
Genetic studies for all probands were performed using DNA samples extracted from the peripheral blood leukocytes. Target enrichment kits and basic sequencing statistics are summarized in Table 2. Enriched libraries were paired‐end sequenced (2 × 100 bp) on HiSeq 1500 (Illumina, San Diego, CA, USA). Raw sequencing data and variants prioritization were performed as previously described (Śmigiel et al., 2020), raw sequence data were aligned and mapped to the human genome reference sequence (GRCh38, hg38). Selected plausible disease‐causing variants were further subjected to family segregation study performed by amplicon deep sequencing (ADS) using Nextera XT Kit (Illumina) and sequenced as described for WES.
TABLE 2.
Detailed characteristics of identified KAT6B variants.
| Patient | Molecular variant on cDNA (NM_012330.4) | Genomic coordinates (GRCh38) | Protein prediction (NP_036462.2) | Variant effect | KAT6B exon | Known/new variant | Genetic databases | Variant frequency in gnomAD v3.1.2 | Patient's phenotype | Inheritance | Variant evaluation according to ACMG/AMP criteria |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | c.3852_3864del | chr10:g.75028676_75028688del | p.(Asp1284Glufs*46) | Frameshift | 18 | New | None | 0 | SBBYSS | de novo | Pathogenic (13 points): PVS1_very strong (8), PM2_moderate (2), PM6_moderate (2), PP4_supporting (1) |
| 2 | c.4026dup | chr10:g.75028850dup | p.(Pro1343Thrfs*4) | Frameshift | 18 | New | None | 0 | SBBYSS | de novo | Pathogenic (13 points): PVS1_very strong (8), PM2_moderate (2), PM6_moderate (2), PP4_supporting (1) |
| 3 | c.5819del | chr10:g.75030643del | p.(Gln1940Argfs*11) | Frameshift | 18 | New | None | 0 | SBBYSS | de novo | Pathogenic (13 points): PVS1_very strong (8), PM2_moderate (2), PM6_moderate (2), PP4_supporting (1) |
| 4 | c.3147G > A | chr10:g.75022006G > A | p.(Pro1049=) p.Ala1008Argfs*621 | Synonymous/splicing/frameshift | 16 | Known (6 unrelated patients)1,2 | HGMD (CM158570), Clinvar (ID 279815), dbSNP (rs886041207), ClinGen (CA10603048) | 0 | SBBYSS | de novo | Pathogenic (13 points): PS3_strong (4), PS4_strong (4), PM2_moderate (2), PM6_moderate (2), PP4_supporting (1) |
| 5 | c.4455dup | chr10:g.75029279dup | p.(Asn1486*) | Nonsense | 18 | New | None | 0 | SBBYSS | de novo | Pathogenic (13 points): PVS1_very strong (8), PM2_moderate (2), PM6_moderate (2), PP4_supporting (1) |
| 6 | c.5012del | chr10:g.75029836del | p.(Gly1671Alafs*44) | Frameshift | 18 | New | None | 0 | SBBYSS | de novo | Pathogenic (13 points): PVS1_very strong (8), PM2_moderate (2), PM6_moderate (2), PP4_supporting (1) |
In all probands a de novo variant in KAT6B (NM_012330.4) gene was detected, including one previously described synonymous substitution NM_012330.4:c.3147G > A, NP_036462.2:p.(Pro1049=) inducing aberrant splicing and partial loss of exon 16 at the RNA level and resulting in a p.Ala1008Argfs*62 frameshift and protein truncation (Gannon et al., 2015; Yilmaz et al., 2015), and four novel truncating variants in exon 18. Three changes caused a frameshift: NM_012330.4:c.3852_3864del, NP_036462.2:p.(Asp1284Glufs*46), NM_012330.4:c.4026dup, NP_036462.2:p.(Pro1343Thrfs*4), NM_012330.4:c.5819del, NP_036462.2:p.(Gln1940Argfs*11), NM_012330.4:c.5819del, NP_036462.2:p.(Gln1940Argfs*11) and one was nonsense variant NM_012330.4:c.4455dup NP_036462.2:p.(Asn1486*). Detailed characteristics of identified KAT6B variants are provided in Table 2.
4. DISCUSSION
To this date, 89 individuals with molecularly confirmed KAT6B‐related disorders have been reported in the literature, including 18 with GPS, 58 with SBBYSS, and 13 described as having an intermediate phenotype (Lemire et al., 2012). Respectively, Turkyilmaz and Ozden (2021), to this date, have reported a total of 89 variants in the KAT6B gene (36 missense/nonsense, 4 splicing, 29 small deletions, 14 small insertions, 2 small indels, 3 gross deletions, 1 complex rearrangement) and all of them have been characterized in the Human Gene Mutation Database (HGMD) (Radvanszky et al., 2017). Based on the current HGMD 2022.2 version, 111 of 156 total variants in the KAT6B gene are associated with SBBYSS/GPS phenotype, including missense/nonsense variants—36 (out of 65 reported); splicing variants—4 (out of 7 reported); small insertions—17 (out of 20 reported); small deletions—46 (out of 53 reported); small indels—3; large insertions (duplications)—2 (out of 3 reported) and large deletions—3 (out of 4 notified).
SBBYSS variant of Ohdo syndrome is inherited in an autosomal dominant pattern and the birth prevalence is estimated to be less than one in a million individuals (Kraft et al., 2011). KAT6B gene is located on chromosome 10 and encodes a highly conserved histone acetyltransferase belonging to the MYST family, functioning in a multi‐subunit complex and acetylating histone H3 in adult neural stem cells. Histone acetyltransferases epigenetically regulate genes that are important for early development, including development of the skeleton and nervous system (Kraft et al., 2011). Recently, more proximal pathogenic variants in KAT6B exons 3, 7, 11, and 14–17 (for GPS, only in exons 17 and 18) were identified in individuals with GPS, SBBYSS and the intermediate phenotype. The increasing identification of individuals with an intermediate phenotype having a variant previously identified in individuals with GPS or SBBYSS make phenotype predictions based on genotype alone imprecise (Zhang et al., 2020).
Kim et al. (2017) reviewed the 45 previously reported patients with Ohdo/SBBYS syndrome. In the study, developmental delay, feeding difficulties, hypotonia, long thumbs and dental anomalies were the most common symptoms. Ocular abnormalities and long thumbs are those of our findings that are in line with Kim et al. observations. Patients with Ohdo/SBBYSS generally have some degree of developmental delay and/or intellectual disability (Kim et al., 2017). Motor and speech delay occurred in all probands from our study. Three of our patients had agenesis of the corpus callosum, and four had congenital heart defects, hearing loss, hypothyroidism and feeding difficulties. All probands presented with hypotonia. None presented with cleft palate and one proband had cleft uvula. SBBYSS typically involves skeletal problems including patellar hypoplasia/agenesis. In our patients patellar anomalies were not as severe (dysplasia, habitual dislocation, subluxation). Interestingly, in one of our patients skeletal or patellar problems were not observed.
Facial appearance in SBBYSS is distinctive, with a mask‐like face, blepharophimosis, and ptosis as well as tubular/bulbous nose. Other features include prominent cheeks, low‐set and posteriorly rotated ears, downslanting palpebral fissures, flat broad nasal bridge, long philtrum, thin upper lip, thin lip vermilion, and micro−/retrognathia (Lemire et al., 2012). All of our patients presented with a typical SBBYSS face even though blepharophimosis, a major feature of SBBYSS, was observed in only 4/6 of our patients (Table 1).
Recent studies suggest a genotype–phenotype correlation for the specific KAT6B‐related disorder (GPS, SBBYSS or intermediate/overlapping), depending on the location of the variant in gene and on hypothetical patomechanism (not yet confirmed experimentally), which might be helpful in establishing the patient's diagnosis (Radvanszky et al., 2017; Turkyilmaz & Ozden, 2021; Vlckova et al., 2015). Nevertheless, the aminoacid boundaries of the phenotypic groups still need elucidation and may be imprecise due to a limited number of molecularly confirmed patients with detailed clinical description. Therefore, our work can contribute in further delineation of genotype–phenotype correlation (Table 3). According to the latest clinical suggestions from Radvanszky et al. (2017) and Zhang et al. (2020), one of our patient should be molecularly qualified for the intermediate variant (overlapping GPS/SBBYSS), but two others and different patients could be clinically qualified for this the GPS/SBBYSS phenotype.
TABLE 3.
Genotype–phenotype correlation.
| Group of KAT6B according to Radvanszky et al. (2017); Turkyilmaz and Ozden (2021); Vlckova et al. (2015) | Affected codons | Preducted effect | Phenotype | Polish genotype, number of patient | Polish phenotype, number of patient |
|---|---|---|---|---|---|
| 1–2 | 1–1205 | NMD | SBBYSS/mild | 4 | 4 (SBBYSS) |
| 3 | 1205–1350 | Truncated protein | GPS | 1 and 2 | 1 (SBBYSS) and 2 (GPS/SBBYSS) |
| 4 | 1350–1520 | Truncated protein | GPS/SBBYSS/intermediate | 5 | 5 (SBBYSS) |
| 5 | 1520–1935 | Truncated protein | SBBYSS | 6 | 6 (GPS/SBBYSS) |
| 6 (hypothesized) | Distal to p.1935 | Truncated protein | Unknown (SBBYSS/asymptomatic) | 3 | 3 (mild SBBYSS) |
Abbreviation: NMD, non‐mediated mRNA decay.
Patients 1, 2, 4 and 5 presented with a severe variant of KAT6B–related disorder; two of them (1 and 2) died of an infection and they did not have an autopsy exam. Patient 3 present milder symptoms. Genotypic analysis of all patients showed a de novo variant in KAT6B and in all patient identified variants were accessed as pathogenic (Table 2). Considering the most recent literature reports on the assignment of patients to phenotypic groups according to Radvanszky et al. (2017), Vlckova et al. (2015) and Turkyilmaz and Ozden (2021), proband 2 and 6 correspond to the GPS/SBBYSS phenotypic group while proband 1, 3, 4 and 5 present clinical symptoms of SBBYSS (Table 3).
Our genotype–phenotype analysis did not confirm genotype–phenotype correlation presented by Radvanszky et al. (2017), Vlckova et al. (2015) and Turkyilmaz and Ozden (2021). For example proband 1 and 5 presenting clinical symptoms of SBBYSS according to the assignment went to the GPS/SBBYSS mixed group. Proband 2 presenting severe clinical manifestations consistent with phenotypic assignment as GPS/SBBYSS belongs to GPS group according to genotype–phenotype correlation presented by above authors. Variant c.5819del p.Gln1940Argfs*11 identified in a proband 3 with mild type of SSBYSS phenotype, resulting in the most distal KAT6B truncating variant reported up‐to‐date which enables to stretch the group 6 upper limit of phenotypic correlation further down p.1935 codon (Table 3). Variants leading to GPS occur within the proximal portion of exon 18, always between amino acids 1150 and 1515 in this cohort and the literature variants within this region are associated with GPS in 60% of cases, SBBYSS in 19% of cases, and the intermediate phenotype in 19% of cases. Variants outside of this region cause SBBYSS and the intermediate phenotype in 84%, and 13% of cases, respectively, and never cause GPS (Radvanszky et al., 2017; Turkyilmaz & Ozden, 2021; Vlckova et al., 2015). Even within well‐described genotype–phenotype groups, the differences are observed. The pathomechanism is not yet experimentally verified so this gives room for discussion of deviations. Most of the anomalies found in our presented patients comply with SBBYSS clinical criteria. So observed differences in our probands could demonstrate a broader spectrum of the disease phenotype and more complicated difficulties in genotype–phenotype correlation in KAT6B–related disorder.
Recently, it was shown that molecular diagnosis of Mendelian disorders achieved by the standard DNA sequencing approaches may be significantly improved by identification of disease‐specific genomic DNA methylation pattern—episignature (Sadikovic et al., 2021). For KAT6B gene episignatures were previously reported (Aref‐Eshghi et al., 2020; Levy et al., 2021; Sadikovic et al., 2021), with two distinct DNA methylation signature for SBBYSS and GPS (PMID: 32109418). These results clearly demonstrate that detection of a specific DNA methylation patterns can help to establish and define a “molecular” phenotypes explaining the differences in clinical features seen between patients harboring pathogenic variants in the same gene. Moreover, episignatures influenced by different mutation types (missense, loss‐of‐function variants) or variant localizations (gene regions, codons, domains) may produce phenotypic variability within the same disease. Thus, our patients' cohort may be benefit from further study of DNA methylation signature which may improve our understanding of molecular etiology and phenotypic differences produced by KAT6B mutations.
In summary, our results suggest that additional molecular and clinical investigation as well as genotype–phenotype correlation study will be required to eventually understand the underlying pathological mechanism of KAT6B–related disorder.
5. AUTHOR CONTRIBUTIONS
Conceptualization: Klaniewska Magdalena, Bolanowska‐Tyszko Anna, Latos‐Bielenska Anna, Jezela‐Stanek Aleksandra, Szczaluba Krzysztof, Krajewska‐Walasek Malgorzata, and Zubkiewicz‐Kucharska Agnieszka. Investigation: Rydzanicz Małgorzata, Stawinski Piotr, Ciara Elzbieta, Pelc Magdalena, and Jurkiewicz Dorota. Data curation: Rydzanicz Małgorzata, Stawinski Piotr, Ciara Elzbieta, Pelc Magdalena, and Jurkiewicz Dorota. Writing–original draft preparation: Klaniewska Magdalena, Rydzanicz Małgorzata, Pelc Magdalena, and Smigiel Robert. Writing–review and editing: Klaniewska Magdalena, Rydzanicz Małgorzata, Smigiel Robert, and Ploski Rafal. Supervision: Ploski Rafal, and Smigiel Robert. Project administration: Smigiel Robert, Rydzanicz Małgorzata, and Klaniewska Magdalena; Funding acquisition: Klaniewska Magdalena, and Smigiel Robert. All authors have read and agreed to the published version of the manuscript.
FUNDING INFORMATION
Contract grant sponsor: SUBZ.E250.23.020.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest. Corresponding author made all authors aware of the journal's conflict of interest policy.
ETHICS STATEMENT
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by Ethics Committee of Wroclaw Medical University, Poland (code: KB‐430/2018; date of approval: 23 July 2018).
PATIENT CONSENT STATEMENT
Written informed consent has been obtained from the patient's parents to publish this paper.
ACKNOWLEDGMENTS
Thanks to families and patients.
Magdalena, K. , Anna, B.‐T. , Anna, L.‐B. , Aleksandra, J.‐S. , Krzysztof, S. , Malgorzata, K.‐W. , Elzbieta, C. , Magdalena, P. , Dorota, J. , Piotr, S. , Agnieszka, Z.‐K. , Małgorzata, R. , Rafal, P. , & Robert, S. (2023). Clinical heterogeneity of polish patients with KAT6B–related disorder. Molecular Genetics & Genomic Medicine, 11, e2265. 10.1002/mgg3.2265
Ploski Rafal and Smigiel Robert contributed equally.
[Correction added on November 2, 2023 after first online publication. The fourth affiliation has been updated in this version.]
Contributor Information
Klaniewska Magdalena, Email: magdazdzie@gmail.com.
Smigiel Robert, Email: robert.smigiel@umw.edu.pl.
DATA AVAILABILITY STATEMENT
The data presented in this study are available on request from the corresponding author.
REFERENCES
- Aref‐Eshghi, E. , Kerkhof, J. , Pedro, V. P. , Groupe DI France , Barat‐Houari, M. , Ruiz‐Pallares, N. , Andrau, J. C. , Lacombe, D. , Van‐Gils, J. , Fergelot, P. , Dubourg, C. , Cormier‐Daire, V. , Rondeau, S. , Lecoquierre, F. , Saugier‐Veber, P. , Nicolas, G. , Lesca, G. , Chatron, N. , Sanlaville, D. , … Sadikovic, B. (2020, March 5). Evaluation of DNA methylation Episignatures for diagnosis and phenotype correlations in 42 Mendelian neurodevelopmental disorders. American Journal of Human Genetics, 106(3), 356–370. 10.1016/j.ajhg.2020.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brea‐Fernández, A. , Dacruz, D. , Eirís, J. , Barros, F. , & Carracedo, Á. (2019). Novel truncating variants expand the phenotypic spectrum of KAT6B‐related disorders. American Journal of Medical Genetics. Part A, 179A, 290–294. 10.1002/ajmg.a.60689 [DOI] [PubMed] [Google Scholar]
- Campeau, P. M. , Lu, J. T. , Dawson, B. C. , Fokkema, I. F. , Robertson, S. P. , Gibbs, R. A. , & Lee, B. H. (2012, November). The KAT6B‐related disorders genitopatellar syndrome and Ohdo/SBBYS syndrome have distinct clinical features reflecting distinct molecular mechanisms. Human Mutation, 33(11), 1520–1525. 10.1002/humu.22141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton‐Smith, J. , O'Sullivan, J. , Daly, S. , Bhaskar, S. , Day, R. , Anderson, B. , Voss, A. K. , Thomas, T. , Biesecker, L. G. , Smith, P. , Fryer, A. , Chandler, K. E. , Kerr, B. , Tassabehji, M. , Lynch, S. A. , Krajewska‐Walasek, M. , McKee, S. , Smith, J. , Sweeney, E. , … Black, G. (2011, November 11). Whole‐exome‐sequencing identifies mutations in histone acetyltransferase gene KAT6B in individuals with the say‐barber‐Biesecker variant of Ohdo syndrome. American Journal of Human Genetics, 89(5), 675–681. 10.1016/j.ajhg.2011.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gannon, T. , Perveen, R. , Schlecht, H. , Ramsden, S. , Anderson, B. , Kerr, B. , Day, R. , Banka, S. , Suri, M. , Berland, S. , Gabbett, M. , Ma, A. , Lyonnet, S. , Cormier‐Daire, V. , Yilmaz, R. , Borck, G. , Wieczorek, D. , Anderlid, B. M. , Smithson, S. , … Clayton‐Smith, J. (2015, September). Further delineation of the KAT6B molecular and phenotypic spectrum. European Journal of Human Genetics, 23(9), 1165–1170. 10.1038/ejhg.2014.248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, Y. R. , Park, J. B. , Lee, Y. J. , Hong, M. J. , Kim, H. T. , & Kim, H. J. (2017, June). Identifying the KAT6B mutation via diagnostic exome sequencing to diagnose say‐barber‐Biesecker‐young‐Simpson syndrome in three generations of a family. Annals of Rehabilitation Medicine, 41(3), 505–510. 10.5535/arm.2017.41.3.505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft, M. , Cirstea, I. C. , Voss, A. K. , Thomas, T. , Goehring, I. , Sheikh, B. N. , Gordon, L. , Scott, H. , Smyth, G. K. , Ahmadian, M. R. , Trautmann, U. , Zenker, M. , Tartaglia, M. , Ekici, A. , Reis, A. , Dörr, H. G. , Rauch, A. , & Thiel, C. T. (2011, September). Disruption of the histone acetyltransferase MYST4 leads to a Noonan syndrome‐like phenotype and hyperactivated MAPK signaling in humans and mice. The Journal of Clinical Investigation, 121(9), 3479–3491. 10.1172/JCI43428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemire, G. , Campeau, P. M. , & Lee, B. H. (2012, December 13). KAT6B disorders. In Adam M. P., Mirzaa G. M., Pagon R. A., Wallace S. E., Bean L. J. H., Gripp K. W., & Amemiya A. (Eds.), GeneReviews® [Internet]. University of Washington. [Google Scholar]
- Levy, M. A. , McConkey, H. , Kerkhof, J. , Barat‐Houari, M. , Bargiacchi, S. , Biamino, E. , Bralo, M. P. , Cappuccio, G. , Ciolfi, A. , Clarke, A. , DuPont, B. R. , Elting, M. W. , Faivre, L. , Fee, T. , Fletcher, R. S. , Cherik, F. , Foroutan, A. , Friez, M. J. , Gervasini, C. , … Sadikovic, B. (2021, December 3). Novel diagnostic DNA methylation episignatures expand and refine the epigenetic landscapes of Mendelian disorders. HGG Advances, 3(1), 100075. 10.1016/j.xhgg.2021.100075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonardo, F. , Lonardo, M. S. , Acquaviva, F. , Della Monica, M. , Scarano, F. , & Scarano, G. (2019, February). Say‐barber‐Biesecker‐young‐Simpson syndrome and Genitopatellar syndrome: Lumping or splitting? Clinical Genetics, 95(2), 253–261. 10.1111/cge.13127 [DOI] [PubMed] [Google Scholar]
- Masuno, M. , Imaizumi, K. , Okada, T. , Adachi, M. , Nishimura, G. , Ishii, T. , Tachibana, K. , & Kuroki, Y. (1999, May 7). Young‐Simpson syndrome: Further delineation of a distinct syndrome with congenital hypothyroidism, congenital heart defects, facial dysmorphism, and mental retardation. American Journal of Medical Genetics, 84(1), 8–11. [DOI] [PubMed] [Google Scholar]
- Radvanszky, J. , Hyblova, M. , Durovcikova, D. , Hikkelova, M. , Fiedler, E. , Kadasi, L. , Turna, J. , Minarik, G. , & Szemes, T. (2017, February). Complex phenotypes blur conventional borders between say‐barber‐Biesecker‐young‐Simpson syndrome and genitopatellar syndrome. Clinical Genetics, 91(2), 339–343. 10.1111/cge.12840 [DOI] [PubMed] [Google Scholar]
- Sadikovic, B. , Levy, M. A. , Kerkhof, J. , Aref‐Eshghi, E. , Schenkel, L. , Stuart, A. , McConkey, H. , Henneman, P. , Venema, A. , Schwartz, C. E. , Stevenson, R. E. , Skinner, S. A. , DuPont, B. R. , Fletcher, R. S. , Balci, T. B. , Siu, V. M. , Granadillo, J. L. , Masters, J. , Kadour, M. , … Alders, M. (2021, June). Clinical epigenomics: Genome‐wide DNA methylation analysis for the diagnosis of Mendelian disorders. Genetics in Medicine, 23(6), 1065–1074. 10.1038/s41436-020-01096-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Śmigiel, R. , Biela, M. , Szmyd, K. , Błoch, M. , Szmida, E. , Skiba, P. , Walczak, A. , Gasperowicz, P. , Kosińska, J. , Rydzanicz, M. , Stawiński, P. , Biernacka, A. , Zielińska, M. , Gołębiowski, W. , Jalowska, A. , Ohia, G. , Głowska, B. , Walas, W. , Królak‐Olejnik, B. , … Płoski, R. (2020, July 13). Rapid whole‐exome sequencing as a diagnostic tool in a neonatal/pediatric intensive care unit. Journal of Clinical Medicine, 9(7), 2220. 10.3390/jcm9072220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turkyilmaz, A. , & Ozden, A. (2021, October 1). A novel frameshift variant in proximal exon 18 of KAT6B gene associated with an overlapping genitopatellar/say barber Biesecker‐young‐Simpson syndrome phenotype. Clinical Dysmorphology, 30(4), 197–200. 10.1097/MCD.0000000000000376 [DOI] [PubMed] [Google Scholar]
- Vlckova, M. , Simandlova, M. , Zimmermann, P. , Stranecky, V. , Hartmannova, H. , Hodanova, K. , Havlovicova, M. , Hancarova, M. , Kmoch, S. , & Sedlacek, Z. (2015, October). A patient showing features of both SBBYSS and GPS supports the concept of a KAT6B‐related disease spectrum, with mutations in mid‐exon 18 possibly leading to combined phenotypes. European Journal of Medical Genetics, 58(10), 550–555. 10.1016/j.ejmg.2015.09.004 [DOI] [PubMed] [Google Scholar]
- Yilmaz, R. , Beleza‐Meireles, A. , Price, S. , Oliveira, R. , Kubisch, C. , Clayton‐Smith, J. , Szakszon, K. , & Borck, G. (2015, December). A recurrent synonymous KAT6B mutation causes say‐barber‐Biesecker/young‐Simpson syndrome by inducing aberrant splicing. American Journal of Medical Genetics. Part A, 167A(12), 3006–3010. 10.1002/ajmg.a.37343 [DOI] [PubMed] [Google Scholar]
- Zhang, L. X. , Lemire, G. , Gonzaga‐Jauregui, C. , Molidperee, S. , Galaz‐Montoya, C. , Liu, D. S. , Verloes, A. , Shillington, A. G. , Izumi, K. , Ritter, A. L. , Keena, B. , Zackai, E. , Li, D. , Bhoj, E. , Tarpinian, J. M. , Bedoukian, E. , Kukolich, M. K. , Innes, A. M. , Ediae, G. U. , … Campeau, P. M. (2020, August). Further delineation of the clinical spectrum of KAT6B disorders and allelic series of pathogenic variants. Genetics in Medicine, 22(8), 1338–1347. 10.1038/s41436-020-0811-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.