

Abstract
Sepsis-induced cardiac injury is associated with oxidative stress and mitochondrial dysfunction. Docosahexaenoic acid (DHA), an essential omega-3 fatty acid, protects the injured myocardium by modulating mitochondrial dysfunction. We aimed to confirm whether the cardioprotective effect of DHA is mediated via the alleviation of mitochondrial fragmentation in lipopolysaccharide (LPS)-induced cardiomyopathy in vitro. We found that DHA improved cell viability and alleviated cardiac cell apoptosis by reducing lactate dehydrogenase (LDH) release, expression levels of Cleaved caspase-3, and Caspase 3 activity. DHA attenuated oxidative stress as evidenced by decreased ROS production and increased superoxide dismutase activity. In addition, DHA ameliorated mitochondrial dysfunction by modulating mitochondrial respiratory chain injury and mitochondrial fragmentation, especially decreasing the mitochondrial fission-related protein p-Drp1(ser 616) but no effects on Drp1, p-Drp1(ser 637), and mitochondrial fusion-related protein. Our data suggest that DHA conferred cardioprotection by alleviating oxidative stress-induced apoptosis, which may be associated with alleviation of stress-induced mitochondrial fragmentation.
Keywords: Septic cardiomyopathy, DHA, Mitochondria dysfunction, Mitochondrial dynamics
Graphical abstract
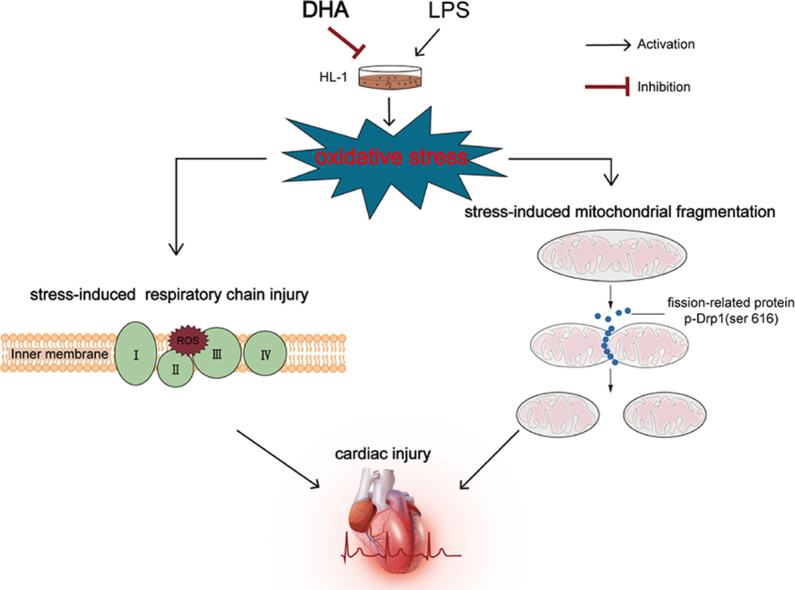
Highlights
-
•
DHA ameliorates LPS-induced cardiac injury by reducing oxidative stress.
-
•
DHA alleviates stress-induced mitochondrial fragmentation and respiratory chain injury.
-
•
Modulating mitochondrial fission-related protein p-Drp1(ser 616) plays a key role in the cardioprotection of DHA.
1. Introduction
Sepsis, a clinical condition characterized by critical organ dysfunction caused by an uncontrolled host response to infection, is associated with high mortality (28%–40 %), and sepsis-related organ dysfunction is still a leading cause of death in the intensive care unit (ICU) [1,2]. During the progression of sepsis-induced multi-organ dysfunction syndrome, the occurrence of septic cardiomyopathy is associated with increased morbidity and mortality. The fatality rate of sepsis combined with myocardial damage is much higher than that of sepsis without myocardial damage (70 % versus 20 %); hence, treatment of myocardial damage can improve the prognosis of sepsis [3].
Septic cardiomyopathy has a complex pathogenesis in which mitochondria dysfunction is believed to play a crucial role [[4], [5], [6], [7]]. Mitochondria are essential organelles contributing to several intracellular pathways. Mitochondrial dysfunction has been shown to contribute to oxidative stress and apoptosis in various heart diseases [8]. Furthermore, disequilibrium of mitochondrial dynamics, including mitochondrial fission and fusion, has been shown to play a key role in mitochondrial dysfunction [9,10]. Thus, a substance that can improve mitochondrial disequilibrium may be useful for the treatment of sepsis combined with myocardial damage.
Docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA) are omega-3 fatty acids (also called ω-3 FAs) [[11], [12], [13]]. Studies have shown that DHA has a variety of physiological functions, including anti-inflammatory, antioxidant, anti-atherosclerosis properties and mitochondria is a target of intracellular DHA [14,15]. However, whether DHA has a protective effect against cardiac impairment remains unclear [16,17]. Thus, in this study, we investigated whether DHA can alleviate oxidative stress-induced apoptosis in HL-1 cardiomyocytes by alleviating mitochondrial dysfunction, particularly mitochondrial fragmentation.
2. Materials and methods
2.1. Cell culture
HL-1 cells are an immortalized mouse atrial cardiomyocyte tumor lineage with some characteristics of adult mouse cardiomyocytes, which were cultured at 37 °C in an atmosphere of air supplemented with 5 % CO2 in Claycomb medium (Sigma, 51800C) supplemented with 10 % fetal bovine serum (Gibco, Origin: Australia), 2 m ML-glutamine, 0.1 mM noradrenaline, 1 % penicillin/streptomycin [18,19]. HL-1 cells were purchased from Warner Bio Co., Ltd(Wuhan, China). The experiments were not performed until cells were cultured about 24 h to achieve 60–70 % confluence. The experimental groups were divided into as follows: (1) Control group: the HL-1 cells were incubated without DHA and LPS, (2) DHA group:the HL-1 cells were incubated with DHA (Sigma, D2534) diluted with absolute ethanol at a dose of 100 μM for 24h, (3) LPS group: the HL-1 cells were subjected to LPS (1 μg/ml) from E.coli0111:B4 (Sigma, L4391) treated for 24h, (4) DHA + LPS group: HL-1 cells were pretreated with DHA(100 μM) for 24h before being subjected to LPS (1 μg/ml) treatment for 24h.
2.2. Measurement of cell viability and lactate Dehydrogenase(LDH)
The methyl thiazolyl tetrazolium (MTT) cell proliferation assay kit (Nanjing jiancheng, China) was used to assess cell viability. Briefly, HL-1 cells were cultured at a density of 5000/well in 96-well plates. Media were replaced with 0.5 mg/mL MTT solution (20 μL/well) and incubated for 4 h at 37 °C. DMSO (150 μL/well) was then added before measuring the absorbance at 490 nm with an enzyme-labeled instrument. Optical density (OD) was used as a unit of cell viability. Cell viability was calculated according to the manufacturer's instructions: cell viability (%) = mean OD in test wells/mean OD in Control wells × 100 %. LDH Assay kit (Nanjing jiancheng, China) was used to quantify the LDH via the colorimetric method according to the manufacturer's instructions.
2.3. Measurement of oxidative stress
A reactive oxygen species assay kit (Beyotime Biotechnology, China) was used to detect dichlorofluorescein (DCF). Briefly, the cells were incubated in serum-free medium (SFM) diluted with DCF-diacetate (DCF-DA, 1:1000). After washing with SFM three times, reactive oxygen species (ROS) content was measured using a fluorescence microscope. The superoxide dismutase (SOD) level was measured by hydroxylamine method using a SOD kit (Nanjing jiancheng, China) according to the manufacturer's instructions.
2.4. Mitochondrial membrane potential
Mitochondrial membrane potential was assessed by JC-1 mitochondrial membrane potential assay kit (Beyotime Biotechnology, China). HL-1 cells were stained with JC-1 for 20 min at 37 °C. The fluorescence intensity of the JC-1 monomers/aggregates (green fluorescence for monomer, red fluorescence for aggregate) were taken by fluorescence microscopy, according to the instructions of JC-1 assay kit.
2.5. Caspase 3 activity assay
Caspase 3 activity was assessed by an assay kit (Nanjing jiancheng, China). According to the manufacturer's protocol, the cells were harvested and lysed with the lysis buffer for 30 min and then centrifuged for 5 min at 11,180 g, and 50 μL samples of supernatants were incubated with 50 μL of assay buffer including a specific caspase 3 substrate for 4 h at 37 °C. The activity of caspase 3 was measured via the absorbance at 405 nm in a microplate reader.
2.6. Electron microscopy
The HL-1 cells were harvested and fixed in 2 % paraformaldehyde and 0.1 % glutaraldehyde in 0.1 M phosphate buffer (pH 7.4) at room temperature for 1h. After dehydration, cells were subsequently embedded in 100 % Eponate resin and left to harden. Ultrathin sections were stained with uranyl acetate and lead citrate and then viewed with an FEI Tecnai G2 12 model transmission electron microscope equipped with a charge coupled device (CCD) camera. For quantification of the average optical density and area of mitochondria, image analysis was randomly performed on mitochondria obtained from at least three experiments using Image-Pro Plus 6.0.
2.7. Super resolution microscopy
Mitochondrial morphology was observed by immunofluorescence with Mitotracker Red (Yeasen Biotechnology, China). The cells in each group were incubated at a density of 4 × 104 cells/well using observation dishes. After 24-h incubation at 37 °C, the cell images were captured with super-resolution 3D-HISTECH.
2.8. Assessment of mitochondrial morphology in the cells
Mitochondrial volume and the number of mitochondria were analyzed and quantified. The Image J software was used to measure the roundness of mitochondria. The percentage of cells with fragmented mitochondria was determined.
2.9. Western blot analysis
The HL-1 cells were plated into six-well plates and treated with reagents based on the indicated experimental designs. After the indicated treatments, cells were washed three times with PBS and lysed in lysis buffer. The supernatant was harvested after centrifugation at 12,000 g at 4 °C for 10 min. The protein concentrations of cell extracts were determined using the BCA Protein Assay Kit. Equal amounts of protein were loaded on SDS-polyacrylamide gels for SDS-PAGE, followed by standard immunoblot analysis. The antibodies we used in Western blot as follows: NDUFA9 (1:1000 dilution, ABclonal China, #A3196), SDHB(1:1000 dilution, ABclonal China, #A10821), COX IV (1:1000 dilution, Cell Signaling Technology USA,#3E11), UQCRC2(1:1000 dilution, ABclonal China,#A4181), VDAC (1:1000 dilution, Cell Signaling Technology USA,#4661), OPA1 (1:1000 dilution, ABclonal China, #A9833), Mfn2 (1:1000 dilution, ABclonal China, #A12771), Drp1 (1:1000 dilution, Cell Signaling Technology USA, #8570), p-Drp1 (Ser616) (1:1000 dilution, ABclonal China, #AP0849), p-Drp1 (Ser637) (1:1000 dilution, ABclonal China, #AP0812), cleaved caspase-3 (1:1000 dilution, Cell Signaling Technology USA, #9664), β-actin (1:1000 dilution, antgene China, #ant322), and HRP Goat anti Rabbit igG (h + l) (1:3000 dilution, antgene China, #ant022).The grey value of the target band was analyzed using Image-Pro Plus 6.0.
2.10. Statistical analysis
All data are presented as mean ± SD. GraphPad Prism software package (San Diego, CA) was used for data statistical analysis. One-way analysis of variance (ANOVA) was performed to detect significant differences between groups, followed by a t-test corrected for multiple comparisons (Student-Newman-Keuls). The p values < 0.05 were considered statistically significant.
3. Results
3.1. DHA protects HL-1 cardiac cells against LPS-induced injury
As shown in Fig. 1A, a marked decrease in cell viability was observed after treatment with LPS alone (p < 0.01), while DHA pretreatment increased the cell viability (p < 0.05). As shown in Fig. 1B, DHA pretreatment significantly reduced the level of LDH in comparison with the LPS group (p < 0.05). The results of the caspase 3 activity and the protein expression of Cleaved caspase 3 showed that DHA pretreatment decreased the apoptotic rate of HL-1 cells in comparison with the LPS group (p < 0.05, Fig. 1C and D). These results indicated that DHA pretreatment protected HL-1 cardiac cells against LPS-induced injury in the in vitro sepsis model.
Fig. 1.

Effects of DHA on cell death in the LPS-induced HL-1 cardiac cells. A) Cell viability was scored by methyl thiazolyl tetrazolium (MTT) assay. B) LDH content in the cell supernatant was utilized as a surrogate for cell death and quantified using Cytotoxicity Detection Kit. C) Caspase 3 activity was used to analyze apoptosis. D) The expression level of Cleaved caspase-3 was assayed by Western blot. n = 3 independent experiments. *P<0.05, **P<0.01, compared with the control group; #P<0.05, ##P<0.01compared with the LPS group.
3.2. DHA attenuates reactive oxygen species production in LPS-induced HL-1 cells
The results showed that LPS markedly increased the production of ROS (p < 0.01), while DHA pretreatment decreased these effects (p < 0.01) (Fig. 2A and B). Additionally, analysis of the level of SOD activity showed that DHA is an effective antioxidant in cardiomyocytes. As shown in Fig. 2C, LPS significantly decreased the level of SOD activity in HL-1 cells (p < 0.01), while DHA pretreatment reversed this effect (p < 0.05). These results indicated a protective effect of DHA against oxidative stress in LPS-induced HL-1 cardiac cells.
Fig. 2.

Effects of DHA on ROS production in the LPS-induced HL-1 cardiac cells. A) DCFH-DA was used to detect cytoplasmic H2O2 by fluorescence microscopy. B) DHA mitigated LPS-induced ROS production. n=6 independent experiments. C) DHA increased the level of SOD activity in LPS-induced HL-1 cells. n=3 independent experiments. **P<0.01, compared with the control group; #P<0.05, ##P<0.01compared with the LPS group.
3.3. DHA inhibits LPS-induced mitochondrial damage in HL-1 cells
Transmission electron microscopy of HL-1 cells revealed no structural abnormality in the mitochondria in the control group and DHA pretreatment group. However, signs of mitochondrial damage such as mitochondrial swelling with loss of cristae and tubules were observed after treatment with LPS alone, while DHA pretreatment decreased the mitochondrial damage (Fig. 3A). DHA pretreatment significantly reduced the percentage of JC-1 monomeric cells induced by LPS (Fig. 3B). As shown in Fig. 4A and B, LPS treatment significantly increased the mitochondrial fragmentation (p < 0.01), which was reversed by DHA pretreatment (p < 0.01). We also assessed the effect of DHA on the expression of mitochondrial respiratory chain-associated proteins. Compared with the control group, LPS induced mitochondrial damage, down-regulated the membrane potential and the expression of NDUFA9, SDHB, and COX IV (p < 0.01, Fig. 5A, B and 5D), but did not affect the expression of UQCRC2 (p > 0.05, Fig. 5C). However, compared with the LPS group, the mitochondrial damage was attenuated and the expressions of NDUFA9, SDHB, and COX IV were increased in the DHA + LPS group (p < 0.01, p < 0.05), but no significant difference was detected in UQCRC2 (p > 0.05). These results indicate a protective effect of DHA on LPS-induced mitochondrial damage in HL-1 cardiac cells.
Fig. 3.

Effects of DHA on myocardial mitochondria in the LPS-induced HL-1 cardiac cells. A) Representative TEM images of HL-1 cells in control, DHA-treated, LPS-treated, and DHA + LPS-treated cells (scale bar = 1 μm). Black arrows indicate normal mitochondria, red arrows indicate abnormal mitochondria. B) Representative fluorescence images of JC-1 staining show the change in high mitochondrial membrane potential (red) and low mitochondrial membrane potential (green), which is used to evaluate mitochondrial damage. LPS treatment increased green fluorescence, whereas DHA decreased green fluorescence (scale bar = 20 μm).
Fig. 4.

Effects of DHA on mitochondrial fission in the LPS-induced HL-1 cardiac cells. A) Representative confocal microscopic images showing mitochondrial morphology stained by MitoTracker Red (scale bar = 10 μm). B) Percentage of HL-1 cells with fragmented mitochondria. n=6 independent experiments. ∗∗P<0.01, compared with the control group; ##P<0.01, compared with the LPS group.
Fig. 5.

Effects of DHA on the expression of mitochondrial respiratory chain-associated proteins in LPS-induced HL-1 cells. A-D) Representative Western blot images showing the effect of DHA on the expressions of NDUFA9, SDHB, UQCRC2, and COX IV. Quantitative analysis of above proteins among all groups n = 3 independent experiments. **P < 0.01, compared with the control group; #P<0.05, ##P<0.01, compared with the LPS group.
3.4. DHA improved mitochondrial dynamics-related dysfunction in LPS-induced HL-1 cells
The Western blots of mitochondrial fusion proteins showed no significant difference between any of the groups with respect to expression of Optic Atrophy 1 (Opa1) and Mitofusin 2 (Mfn2) proteins (p > 0.05, Fig. 6A, C and 6D). However, compared with the control group, the LPS group showed an increasing level of mitochondrial fission proteins p-Drp1(ser 616) (p < 0.01, Fig. 6B and E) but showed no changes in Drp1 and p-Drp1(ser 637) (p > 0.05, Fig. 6B and F). Compared with the LPS group, the expression of p-Drp1(ser 616) was reduced after pretreatment with DHA (p < 0.01, Fig. 6B and E). These results indicate that DHA attenuates mitochondrial dynamics-related dysfunction by inhibiting mitochondrial fission.
Fig. 6.

Effects of DHA on the expression of mitochondrial dynamics related proteins in LPS-induced HL-1 cells. A) Representative Western blot images showing the effect of DHA on the expressions of Mfn2 and Opa1. B) Representative Western blot images showing the effect of DHA on the expressions of Drp1, p-Drp1(Ser616), p-Drp1(Ser637) in LPS-induced HL-1 cells. C–F) Quantitative analysis of above proteins among all groups. n = 3 independent experiments. **P < 0.01, compared with the control group; ##P<0.01, compared with the LPS group.
4. Discussion
In the present study, we reported novel findings regarding the protective effect of DHA against LPS-induced dysfunction of mitochondrial dynamics in HL-1 cells. Our main findings were as follows: (1) DHA attenuated LPS-induced injury in HL-1 cardiac cells; (2) DHA markedly attenuated LPS-induced oxidative stress and apoptosis; (3) DHA inhibited LPS-induced mitochondrial damage in HL-1 cells; (4) DHA regulated the dynamic imbalance between mitochondrial fission and fusion, which possibly contributed to the antioxidant and antiapoptotic effect. To our knowledge, this is the first study to investigate the relative roles of DHA and stress-induced mitochondrial fragmentation in sepsis-induced cardiomyopathy.
Sepsis-induced cardiomyopathy, first described by Parker in 1984, is a potentially fatal complication of severe sepsis. Many clinical and experimental studies have investigated potential therapeutic strategies for sepsis-induced cardiomyopathy [[20], [21], [22]]. Mitochondrial dysfunction is one of the main pathogenetic mechanisms of sepsis-induced cardiomyopathy, in which the imbalance of mitochondrial fission and fusion is one of the key reasons [21,23,24]. Therefore, mitochondrial dynamics is an important therapeutic target for sepsis, and exploration of strategies for improving mitochondrial dynamics-related dysfunction is a key research imperative.
DHA has been widely used in the study of inflammatory diseases due to its anti-inflammatory, antioxidant, and immunoregulatory properties [[25], [26], [27], [28], [29]]. It has been suggested that DHA supplementation can prevent cardiovascular disease. DHA was shown to increase the expression of uncoupling protein 3 (UCP 3) (which plays an important role in regulating mitochondrial division), and to reduce ROS production and myocardial cell apoptosis [30]. Consistent with these findings, in our study, DHA markedly attenuated LPS-induced cardiac apoptosis in HL-1 cells, which suggest that DHA may be a potential therapy to ameliorate sepsis-induced cardiomyopathy.
Mitochondrial dysfunction is believed to contribute significantly to the development and progression of sepsis-induced cardiomyopathy. Previous studies have demonstrated significant increase in ROS level during the pathogenesis of LPS-induced myocardial injury [[31], [32], [33]]. Elevated ROS levels consume and surpass the antioxidant capacity of the injured myocardium, leading to generation of oxidative stress resulting in mitochondrial lipid peroxidation and mitochondrial DNA (mt DNA) damage [34]. Overproduction of NO and ROS in the body can impair the function of mitochondrial respiratory chain complex, leading to the reduction of ATP production during oxidative phosphorylation, and the release of cytochrome c and apoptosis-related markers into the cytoplasm to promote apoptosis, which is closely related to mitochondrial dysfunction [35]. Additionally, the accumulation of damaged mitochondria promotes more ROS production, further accelerating mitochondrial abnormalities and mitochondrial membrane injury, leading to a vicious cycle [36,37]. It has been reported that DHA could alleviate oxidative stress injury and mitochondrial dysfunction induced by diabetes; the underlying mechanism was related to the activation of the expression of pyruvate dehydrogenase complex 4 (PDK-4) [29], promoting mitochondrial biogenesis and regulating myocardial metabolism. Thus, we hypothesized that DHA may participate in mitochondrial protection by regulating oxidative stress response to sepsis. In the current study, HL-1 cardiomyocytes treated with LPS exhibited oxidative stress and mitochondrial dysfunction, as evidenced by increased production of ROS production, decreased production of SOD, decreased mitochondrial membrane potential and decreased expression of mitochondrial respiratory chain-associated proteins. However, the LPS-induced mitochondrial dysfunction was reversed by DHA pretreatment in the present study, which is in line with previous experimental results [17,38]. These data demonstrated that DHA attenuated LPS-induced cardiac apoptosis by restoring mitochondrial function, regulating oxidative stress, and preserving cardiac function.
Mitochondrial dynamics, including imbalance between fission and fusion, have been recognized as a key component involved in oxidative stress and apoptosis in sepsis-induced cardiomyopathy [21]. Excessive mitochondrial fission induced by sepsis has been shown to trigger mitochondrial structural and function injury as well as cellular damage [21,22,24]. In our study, MitoTracker and transmission electron microscopy (TEM) revealed mitochondrial rupture or smaller mitochondria in LPS-treated cardiomyocytes, indicating that mitochondrial fission was increased and that DHA pretreatment alleviated this effect. In addition, mitochondrial dynamics are regulated by proteins including Drp1, Mff, and OPA1. Increasing evidence supports that the inhibition of Drp1 can reduce the death of cardiomyocytes [39]. Our data showed that LPS treatment upregulated the expression of p-Drp1(ser 616), while DHA pretreatment reversed this effect, which is consistent with the results observed by MitoTracker and TEM. These results suggested that DHA can inhibit mitochondrial fission and improved mitochondrial fragmentation. It has been reported that several antioxidative properties increased mitochondria fusion and/or improved stress-induced mitochondria fragmentation promoting cell survival. For example, metformin could ameliorate Pb-induced mitochondrial fragmentation via antioxidative effects originated from AMPK/Nrf2 pathway activation [40]. Another study found mitochondria-targeted antioxidant SkQ1 protected yeast cells against detrimental effects of benzalkonium chloride through improving mitochondrial fragmentation [41]. Thus, the improvement of mitochondrial dynamics and function may contribute to the inherent antioxidative capacity of DHA. In addition, mitochondrial dysfunction was also reported to directly induce apoptosis [[42], [43], [44], [45], [46]]. Taken together, we hypothesized that the mechanism of mitochondrial fragmentation, oxidative stress, and mitochondrial dysfunction may form an interactive cycle, eventually resulting in cardiomyocyte apoptosis. DHA may modulate the entire cycle and promote cardioprotection by improving mitochondrial fragmentation-targeted mitochondrial dysfunction.
Some limitations of our study should be considered. Firstly, the experiments were only conducted in an immortalized cell line. Although HL-1 cells are frequently utilized in cardiovascular research, there are still various discrepancies between cell lines and animal models. Secondly, further experiments specific to mitochondria should be performed to better characterize the interaction among mitochondrial fragmentation, oxidative stress, and mitochondrial dysfunction. Finally, DHA and EPA are the two representative ω-3 FAs; it is important to explore whether the combined use of these two fatty acids can enhance the therapeutic potential of DHA in preventing organ damage related to sepsis. Further research is still underway to fully understand the underlying mechanisms and effects of DHA on mitochondria and oxidative stress.
5. Conclusion
Our study shows that DHA interacts with mitochondrial fragmentation, oxidative damage, mitochondrial dysfunction, and cardiomyocyte apoptosis. We provide evidence that DHA pretreatment can confer cardioprotection against oxidative stress induced apoptosis by modulating mitochondrial fragmentation during sepsis-induced cardiomyopathy. Further studies are required to assess the potential clinical application of these findings.
Funding
This work was supported by National Natural Science Foundation of China (grant no.81772131).
Data availability statement
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
CRediT authorship contribution statement
Chenyang Wang: Writing – review & editing, Writing – original draft, Project administration, Investigation, Data curation, Conceptualization. Dong Han: Writing – review & editing, Writing – original draft, Data curation. Xiaojing Feng: Writing – original draft, Data curation. Li Hu: Writing – original draft, Data curation. Jing Wu: Writing – review & editing, Supervision, Project administration, Data curation.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Antonucci E., Fiaccadori E., Donadello K., Taccone F.S., Franchi F., Scolletta S. Myocardial depression in sepsis: from pathogenesis to clinical manifestations and treatment. J. Crit. Care. 2014;29(4):500–511. doi: 10.1016/j.jcrc.2014.03.028. [DOI] [PubMed] [Google Scholar]
- 2.Singer M., Deutschman C.S., Seymour C.W., et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3) JAMA. 2016;315(8):801–810. doi: 10.1001/jama.2016.0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zaky A., Deem S., Bendjelid K., Treggiari M.M. Characterization of cardiac dysfunction in sepsis: an ongoing challenge. Shock. 2014;41(1):12–24. doi: 10.1097/SHK.0000000000000065. [DOI] [PubMed] [Google Scholar]
- 4.Romero-Bermejo F.J., Ruiz-Bailen M., Gil-Cebrian J., Huertos-Ranchal M.J. Sepsis-induced cardiomyopathy. Curr. Cardiol. Rev. 2011;7(3):163–183. doi: 10.2174/157340311798220494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cimolai M.C., Alvarez S., Bode C., Bugger H. Mitochondrial mechanisms in septic cardiomyopathy. Int. J. Mol. Sci. 2015;16(8):17763–17778. doi: 10.3390/ijms160817763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lima M.R., Silva D. Septic cardiomyopathy: a narrative review. Rev. Port. Cardiol. 2023;42(5):471–481. doi: 10.1016/j.repc.2021.05.020. [DOI] [PubMed] [Google Scholar]
- 7.Wang R., Xu Y., Fang Y., et al. Pathogenetic mechanisms of septic cardiomyopathy. J. Cell. Physiol. 2022;237(1):49–58. doi: 10.1002/jcp.30527. [DOI] [PubMed] [Google Scholar]
- 8.Suliman H.B., Welty-Wolf K.E., Carraway M., Tatro L., Piantadosi C.A. Lipopolysaccharide induces oxidative cardiac mitochondrial damage and biogenesis. Cardiovasc. Res. 2004;64(2):279–288. doi: 10.1016/j.cardiores.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 9.Brown D.A., Perry J.B., Allen M.E., et al. Expert consensus document: mitochondrial function as a therapeutic target in heart failure. Nat. Rev. Cardiol. 2017;14(4):238–250. doi: 10.1038/nrcardio.2016.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu Y., Yao Y.M., Lu Z.Q. Mitochondrial quality control mechanisms as potential therapeutic targets in sepsis-induced multiple organ failure. J. Mol. Med. 2019;97(4):451–462. doi: 10.1007/s00109-019-01756-2. [DOI] [PubMed] [Google Scholar]
- 11.Martin J.M., Stapleton R.D. Omega-3 fatty acids in critical illness. Nutr. Rev. 2010;68(9):531–541. doi: 10.1111/j.1753-4887.2010.00313.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Calder P.C. Marine omega-3 fatty acids and inflammatory processes: effects, mechanisms and clinical relevance. Biochim. Biophys. Acta. 2015;1851(4):469–484. doi: 10.1016/j.bbalip.2014.08.010. [DOI] [PubMed] [Google Scholar]
- 13.Welty F.K. Omega-3 fatty acids and cognitive function. Curr. Opin. Lipidol. 2023;34(1):12–21. doi: 10.1097/MOL.0000000000000862. [DOI] [PubMed] [Google Scholar]
- 14.Li G., Li Y., Xiao B., et al. Antioxidant activity of docosahexaenoic acid (DHA) and its regulatory roles in mitochondria. J. Agric. Food Chem. 2021;69(5):1647–1655. doi: 10.1021/acs.jafc.0c07751. [DOI] [PubMed] [Google Scholar]
- 15.Chong S.Y., Wang X., van Bloois L., et al. Injectable liposomal docosahexaenoic acid alleviates atherosclerosis progression and enhances plaque stability. J. Contr. Release. 2023;360:344–364. doi: 10.1016/j.jconrel.2023.06.035. [DOI] [PubMed] [Google Scholar]
- 16.Afshordel S., Hagl S., Werner D., et al. Omega-3 polyunsaturated fatty acids improve mitochondrial dysfunction in brain aging--impact of Bcl-2 and NPD-1 like metabolites. Prostaglandins Leukot. Essent. Fatty Acids. 2015;92:23–31. doi: 10.1016/j.plefa.2014.05.008. [DOI] [PubMed] [Google Scholar]
- 17.Zhang T., Wu P., Zhang J.H., et al. Docosahexaenoic acid alleviates oxidative stress-based apoptosis via improving mitochondrial dynamics in early brain injury after subarachnoid hemorrhage. Cell. Mol. Neurobiol. 2018;38(7):1413–1423. doi: 10.1007/s10571-018-0608-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ruiz-Meana M., Minguet M., Bou-Teen D., et al. Ryanodine receptor glycation favors mitochondrial damage in the senescent heart. Circulation. 2019;139(7):949–964. doi: 10.1161/CIRCULATIONAHA.118.035869. [DOI] [PubMed] [Google Scholar]
- 19.Lee T.W., Lee T.I., Lin Y.K., Kao Y.H., Chen Y.J. Calcitriol downregulates fibroblast growth factor receptor 1 through histone deacetylase activation in HL-1 atrial myocytes. J. Biomed. Sci. 2018;25(1):42. doi: 10.1186/s12929-018-0443-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zou X., Xu J., Yao S., Li J., Yang Y., Yang L. Endoplasmic reticulum stress-mediated autophagy protects against lipopolysaccharide-induced apoptosis in HL-1 cardiomyocytes. Exp. Physiol. 2014;99(10):1348–1358. doi: 10.1113/expphysiol.2014.079012. [DOI] [PubMed] [Google Scholar]
- 21.Haileselassie B., Mukherjee R., Joshi A.U., et al. Drp1/Fis1 interaction mediates mitochondrial dysfunction in septic cardiomyopathy. J. Mol. Cell. Cardiol. 2019;130:160–169. doi: 10.1016/j.yjmcc.2019.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tan Y., Ouyang H., Xiao X., Zhong J., Dong M. Irisin ameliorates septic cardiomyopathy via inhibiting DRP1-related mitochondrial fission and normalizing the JNK-LATS2 signaling pathway. Cell Stress Chaperones. 2019;24(3):595–608. doi: 10.1007/s12192-019-00992-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shang X., Li J., Yu R., et al. Sepsis-related myocardial injury is associated with Mst1 upregulation, mitochondrial dysfunction and the Drp1/F-actin signaling pathway. J. Mol. Histol. 2019;50(2):91–103. doi: 10.1007/s10735-018-09809-5. [DOI] [PubMed] [Google Scholar]
- 24.Shang X., Zhang Y., Xu J., Li M., Wang X., Yu R. SRV2 promotes mitochondrial fission and Mst1-Drp1 signaling in LPS-induced septic cardiomyopathy. Aging (Albany NY) 2020;12(2):1417–1432. doi: 10.18632/aging.102691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ishikado A., Morino K., Nishio Y., et al. 4-Hydroxy hexenal derived from docosahexaenoic acid protects endothelial cells via Nrf2 activation. PLoS One. 2013;8(7) doi: 10.1371/journal.pone.0069415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saw C.L., Yang A.Y., Guo Y., Kong A.N. Astaxanthin and omega-3 fatty acids individually and in combination protect against oxidative stress via the Nrf2-ARE pathway. Food Chem. Toxicol. 2013;62:869–875. doi: 10.1016/j.fct.2013.10.023. [DOI] [PubMed] [Google Scholar]
- 27.Schwanke R.C., Marcon R., Bento A.F., Calixto J.B. EPA- and DHA-derived resolvins' actions in inflammatory bowel disease. Eur. J. Pharmacol. 2016;785:156–164. doi: 10.1016/j.ejphar.2015.08.050. [DOI] [PubMed] [Google Scholar]
- 28.Calder P.C. Omega-3 fatty acids and inflammatory processes: from molecules to man. Biochem. Soc. Trans. 2017;45(5):1105–1115. doi: 10.1042/BST20160474. [DOI] [PubMed] [Google Scholar]
- 29.Gui T., Li Y., Zhang S., et al. Docosahexaenoic acid protects against palmitate-induced mitochondrial dysfunction in diabetic cardiomyopathy. Biomed. Pharmacother. 2020;128 doi: 10.1016/j.biopha.2020.110306. [DOI] [PubMed] [Google Scholar]
- 30.Habicht I., Mohsen G., Eichhorn L., et al. DHA supplementation attenuates MI-induced LV matrix remodeling and dysfunction in mice. Oxid. Med. Cell. Longev. 2020;2020 doi: 10.1155/2020/7606938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crouser E.D. Mitochondrial dysfunction in septic shock and multiple organ dysfunction syndrome. Mitochondrion. 2004;4(5–6):729–741. doi: 10.1016/j.mito.2004.07.023. [DOI] [PubMed] [Google Scholar]
- 32.Celes M.R., Torres-Dueñas D., Prado C.M., et al. Increased sarcolemmal permeability as an early event in experimental septic cardiomyopathy: a potential role for oxidative damage to lipids and proteins. Shock. 2010;33(3):322–331. doi: 10.1097/SHK.0b013e3181b38ef6. [DOI] [PubMed] [Google Scholar]
- 33.Tsolaki V., Makris D., Mantzarlis K., Zakynthinos E. Sepsis-induced cardiomyopathy: oxidative implications in the initiation and resolution of the damage. Oxid. Med. Cell. Longev. 2017;2017 doi: 10.1155/2017/7393525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Williamson J., Davison G. Targeted antioxidants in exercise-induced mitochondrial oxidative stress: emphasis on DNA damage. Antioxidants. 2020;9(11):1142. doi: 10.3390/antiox9111142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bazán S., Mileykovskaya E., Mallampalli V.K., Heacock P., Sparagna G.C., Dowhan W. Cardiolipin-dependent reconstitution of respiratory supercomplexes from purified Saccharomyces cerevisiae complexes III and IV. J. Biol. Chem. 2013;288(1):401–411. doi: 10.1074/jbc.M112.425876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lancel S., Hassoun S.M., Favory R., Decoster B., Motterlini R., Neviere R. Carbon monoxide rescues mice from lethal sepsis by supporting mitochondrial energetic metabolism and activating mitochondrial biogenesis. J. Pharmacol. Exp. Therapeut. 2009;329(2):641–648. doi: 10.1124/jpet.108.148049. [DOI] [PubMed] [Google Scholar]
- 37.Huet O., Dupic L., Harrois A., Duranteau J. Oxidative stress and endothelial dysfunction during sepsis. Front. Biosci. 2011;16(5):1986–1995. doi: 10.2741/3835. [DOI] [PubMed] [Google Scholar]
- 38.Bou M., Torgersen J.S., Østbye T.K., et al. DHA modulates immune response and mitochondrial function of atlantic salmon adipocytes after LPS treatment. Int. J. Mol. Sci. 2020;21(11):4101. doi: 10.3390/ijms21114101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Papanicolaou K.N., Khairallah R.J., Ngoh G.A., et al. Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol. Cell Biol. 2011;31(6):1309–1328. doi: 10.1128/MCB.00911-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang L., Li X., Jiang A., et al. Metformin alleviates lead-induced mitochondrial fragmentation via AMPK/Nrf2 activation in SH-SY5Y cells. Redox Biol. 2020;36 doi: 10.1016/j.redox.2020.101626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rogov A.G., Goleva T.N., Sukhanova E.I., et al. Mitochondrial dysfunctions may Be one of the major causative factors underlying detrimental effects of benzalkonium chloride. Oxid. Med. Cell. Longev. 2020;2020 doi: 10.1155/2020/8956504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Olichon A., Baricault L., Gas N., et al. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J. Biol. Chem. 2003;278(10):7743–7746. doi: 10.1074/jbc.C200677200. [DOI] [PubMed] [Google Scholar]
- 43.Lin M.T., Beal M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443(7113):787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 44.Chistiakov D.A., Shkurat T.P., Melnichenko A.A., Grechko A.V., Orekhov A.N. The role of mitochondrial dysfunction in cardiovascular disease: a brief review. Ann. Med. 2018;50(2):121–127. doi: 10.1080/07853890.2017.1417631. [DOI] [PubMed] [Google Scholar]
- 45.Ding M., Feng N., Tang D., et al. Melatonin prevents Drp1-mediated mitochondrial fission in diabetic hearts through SIRT1-PGC1α pathway. J. Pineal Res. 2018;65(2) doi: 10.1111/jpi.12491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim S.H., Kim H. Inhibitory effect of astaxanthin on oxidative stress-induced mitochondrial dysfunction-A mini-review. Nutrients. 2018;10(9) doi: 10.3390/nu10091137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.