

Abstract
Alzheimer’s disease cortical tau pathology initiates in the layer II cell clusters of entorhinal cortex, but it is not known why these specific neurons are so vulnerable. Aging macaques exhibit the same qualitative pattern of tau pathology as humans, including initial pathology in layer II entorhinal cortex clusters, and thus can inform etiological factors driving selective vulnerability. Macaque data have already shown that susceptible neurons in dorsolateral prefrontal cortex express a “signature of flexibility” near glutamate synapses on spines, where cAMP-PKA magnification of calcium signaling opens nearby potassium and hyperpolarization-activated cyclic nucleotide-gated channels to dynamically alter synapse strength. This process is regulated by PDE4A/D, mGluR3, and calbindin, to prevent toxic calcium actions; regulatory actions that are lost with age/inflammation, leading to tau phosphorylation. The current study examined whether a similar “signature of flexibility” expresses in layer II entorhinal cortex, investigating the localization of PDE4D, mGluR3, and HCN1 channels. Results showed a similar pattern to dorsolateral prefrontal cortex, with PDE4D and mGluR3 positioned to regulate internal calcium release near glutamate synapses, and HCN1 channels concentrated on spines. As layer II entorhinal cortex stellate cells do not express calbindin, even when young, they may be particularly vulnerable to magnified calcium actions and ensuing tau pathology.
Keywords: entorhinal cortex, calcium, cAMP, calbindin, immunoelectron microscopy, macaque, Alzheimer’s disease, stellate cells, PDE4D, HCN1, mGluR3
Introduction
Neurofibrillary tangle pathology in the cortex of patients with sporadic AD arises earliest in the rhinal cortices, especially distinct in stellate cells of the layer II entorhinal cortex (ERC) cell clusters (Hyman et al. 1986; Braak and Braak 1991, 1992; Braak and Del Trecidi 2015; Braak and Del Tredici 2015; Kaufman et al. 2018). Tau pathology can be seen initiating in these neurons even in young adults, with tangles appearing even in middle age (Braak and Del Tredici 2015; Liu and Li 2019). In contrast, tau pathology develops later in the association cortices, and only afflicts the primary visual cortex (V1) at end stage disease (Lewis et al. 1987). Studies in human and animal brains have provided evidence that tau pathology spreads from the ERC to interconnected glutamatergic neurons in the limbic and association cortices, as well as hippocampus, seeding pathology throughout higher brain circuits (Lewis et al. 1987; de Calignon et al. 2012; Ahmed et al. 2014; Calafate et al. 2015; Dujardin et al. 2018; Fu et al. 2018; Kaufman et al. 2018; Colin et al. 2020). Thus, understanding why layer II ERC cell clusters are so vulnerable to tau pathology is a major question for the etiology and potential prevention of sporadic AD.
Rhesus macaques naturally develop tau pathology with advancing age, with the same qualitative pattern and sequence as in humans, and thus can be used as an animal model to help learn why layer II ERC cell clusters are selectively vulnerable (Paspalas et al. 2018). As with humans, tau pathology appears in layer II cell clusters even in middle age, with AT8-labeled tangles appearing in the oldest macaques (Paspalas et al. 2018; Arnsten et al. 2019; Arnsten et al. 2021a; Arnsten et al. 2021b). These tangles are comprised of paired helical filaments with the same width and helical frequency as humans (Paspalas et al. 2018), providing an appropriate animal model to address this important question.
The origins of tau pathology have recently been examined in layer III of the rhesus macaque dorsolateral prefrontal cortex (dlPFC), in the pyramidal cell circuits necessary for working memory and higher cognition (Arnsten 2015; Arnsten et al. 2021c, 2022). For example, dlPFC “delay cells” generate and sustain flexible representations of visual space during a working memory task (Goldman-Rakic 1995), and these physiological representations rapidly change based on arousal state (Arnsten et al. 2021c). As with humans, layer III dlPFC neurons are especially vulnerable to tau pathology (Hof and Morrison 1991), but develop tau pathology at a later age than in ERC (Braak and Braak 1991), at a time when working memory deficits are prominent (Nelson et al. 2012). Findings to date point to dysregulated cAMP-calcium signaling as an early driver of tau pathology in dlPFC (Datta et al. 2021a), related to the special molecular needs of these cognitive circuits, where layer III dlPFC pyramidal cells express the molecular machinery for cAMP-PKA signaling to magnify internal calcium release near glutamate synapses in dendritic spines (Arnsten et al. 2021c). This is needed for dlPFC neurons to sustain NMDA receptor-dependent neuronal firing across the delay period without sensory stimulation, and to allow for dynamic changes in network connectivity through the opening of nearby potassium (K+) and HCN cation channels, needed for flexible representations in working memory (Arnsten et al. 2021c).
The molecular details of this “signature of flexibility” are schematically illustrated in Fig. 13(A), and consist of the following constellation of proteins near the glutamate synapse on dendritic spines: (1) evidence of cAMP-PKA signaling amplifying calcium release from the smooth endoplasmic reticulum (SER, known as the spine apparatus within dendritic spines), including expression of the phosphodiesterases PDE4A and PDE4D, which catabolize cAMP, next to the SER (Carlyle et al. 2014; Datta et al. 2021a); (2) K+ and HCN channels on the dendritic spine membrane that are opened by cAMP, PKA or calcium signaling (HCN, KCNQ2, SK channels, respectively) and (3) expression of neuromodulator receptors in the dendritic spine membrane that increase or regulate cAMP signaling (e.g. D1R vs. mGluR3, respectively). A number of cAMP-PKA signaling related proteins can be seen next to the spine apparatus in layer III dlPFC dendritic spines (Paspalas et al. 2013; Jin et al. 2018a; Datta et al. 2020; Datta et al. 2021a), including the phosphodiesterase PDE4D, which catabolizes cAMP (Houslay and Adams 2003; Baillie et al. 2019). PDE4D is of special interest, as it highly associated with the SER in dlPFC dendrites and dendritic spines (Datta et al. 2020) and is lost from dendrites with age (Lu et al. 2004; Datta et al. 2021a). Hyperpolarization-activated cyclic nucleotide-gated (HCN) channels on dendritic spines are also noteworthy (Wang et al. 2007; Paspalas et al. 2013), as their more typical localization is on distal dendrites where they regulate membrane voltage (Lorincz et al. 2002; Poolos et al. 2002; Notomi and Shigemoto 2004; Day et al. 2005; Fan et al. 2005; Thuault et al. 2013). In dendritic spines, HCN channels are localized near the synapse, and also in the spine neck, e.g. circling the thin “bottleneck” to maximize the role in gating (Wang et al. 2007; Paspalas et al. 2013). Recent evidence indicates that HCN cation channels in PFC dendritic spines form a complex with Slack K+ channels, and thus can directly regulate K+ conductance, gating out network inputs when they are opened by cAMP signaling (Wu et al. 2023). For example, dopamine D1R-cAMP signaling opens HCN channels in macaque dlPFC and can narrow the representation of visual space in working memory, reducing neuronal responses to nonpreferred stimuli, i.e. reducing “noise” (Vijayraghavan et al. 2007; Gamo et al. 2015; Wang et al. 2019). Thus, a dlPFC Delay cell under conditions of modest D1R stimulation might show persistent firing during the memory of multiple spatial locations, between a range of 150 ° and 220 °, but with higher levels of D1R stimulation would only fire if the stimulus had occurred more precisely between 170 ° and 180 ° (reviewed in Arnsten et al. 2015). This contrasts with primary visual cortex, where HCN channels are in their more typical locations on distal dendrites and presynaptic terminals, but not on dendritic spines, where they have more generalized, excitatory effects on neuronal firing (Yang et al. 2018). mGluR3 are traditionally considered astroglial and presynaptic receptors (Aronica et al. 2003) and they have prominent glial expression in dlPFC (Jin et al. 2018b). However, mGluR3 have an unusual, post-synaptic localization in dlPFC, concentrated on dendritic spines, where they enhance neuronal firing during working memory by regulating feedforward cAMP-calcium-K+ channel signaling, thus having therapeutic potential (Jin et al. 2017; Jin et al. 2018a).
Fig. 13.

Hypothetic model of “Signature of flexibility” in higher-order association cortices in primates. (A) Schematic diagram of the dlPFC layer III dendritic spines that partake in recurrent excitatory microcircuits subserving working memory. Our previous findings have revealed that glutamatergic synapses on dendritic spines in layer III of the dlPFC express feedforward Ca2+-cAMP-K+ channel signaling to dynamically gate network inputs, providing a “signature of flexibility” (Arnsten et al. 2021c). In these microcircuits, neurotransmission depends on NMDAR (with GluN2A and GluN2B subunits), and the molecular machinery for cAMP-PKA to magnify Ca2+ signaling needed to sustain persistent firing. This includes cAMP-PKA amplification of internal Ca2+ release from the SER spine apparatus, which in turn increases cAMP production, leading to feedforward cAMP-Ca2+ signaling. dlPFC layer III dendritic spines also express K+ channels (e.g. HCN1 cation channels, Slack K+ channels, KCNQ2) that are opened by cAMP-PKA signaling to provide dynamic changes in network connectivity. Under healthy conditions in the young adult dlPFC, these intracellular signaling pathways are tightly regulated by the calcium binding protein, calbindin, the phosphodiesterases PDE4A/D, which are anchored to the SER spine apparatus and catabolize cAMP, and receptors that inhibit cAMP production, e.g. mGluR3. In primate layer III dlPFC, mGluR3 are concentrated on dendritic spines, where they provide inhibitory regulation of cAMP-K+ channel signaling, strengthening connectivity and markedly enhancing delay cell firing. PDE4s are also found in dendrites near mitochondria, positioned to regulate cAMP drive on Ca2+ release from the SER into mitochondria (not shown). (B) The current study examined whether a similar “signature of flexibility” is expressed in ERC layer II circuits, examining the subcellular localization of PDE4D, HCN1 channels, and mGluR3. Our findings showed convergent anatomical patterns to dlPFC, with PDE4D and mGluR3 positioned to regulate internal calcium release near glutamate synapses, and nearby HCN1 channels to provide dynamic changes in synaptic strength. In contrast to dlPFC, this constellation was seen on dendrites as well as dendritic spines, consistent with the larger numbers of glutamate synapses on dendritic shafts in layer II ERC. As layer II ERC stellate cells do not express calbindin, even when young, they may be particularly vulnerable to magnified calcium actions and ensuing tau pathology with advanced age in AD.
Importantly, layer III dlPFC pyramidal cells also express the calcium-binding protein, calbindin, which is typically found in GABAergic interneurons (Hof and Morrison 1991; Datta et al. 2021a). These exceptional, calbindin-expressing pyramidal cells in human dlPFC are the ones most vulnerable to tau pathology and autophagic neurodegeneration in AD when calbindin is lost with age (Hof and Morrison 1991; Datta et al. 2021a). PDE4D and mGluR3 are also lost from dendrites with age, leading to excessive cAMP-calcium-K+ signaling, correlating with the rise of tau pathology in dendritic spines and on microtubules in dendrites (Hernandez et al. 2018; Datta et al. 2021a; Datta et al. 2021b; Woo et al. 2022). Thus, the “signature of flexibility” becomes a “signature of vulnerability” when regulation of cAMP-calcium signaling is lost with age.
Whether layer II ERC neurons express a similar “signature of flexibility/vulnerability” is unknown, but recordings from rodent ERC suggests that this may indeed be the case. In rodents, layer II of the ERC contains grid cells, and time cells, neurons that dynamically represent points in space and time as they are occurring (Moser et al. 2014; Kraus et al. 2015; Moser et al. 2015; Sanders et al. 2015). Grid cells also have been detected in monkeys (Killian et al. 2012) and humans (Jacobs et al. 2013; Nadasdy et al. 2017), suggesting a cross-species function of these neurons with enhanced context-dependent scalability in primates compared to rodents. Studies in rodents show that ERC Grid cells, like layer III dlPFC Delay cells, are dependent on NMDAR neurotransmission, and show dynamic alterations in their grid representations involving HCN channel and D1R signaling, with slower HCN channel time constants correlating with larger grid scales (Giocomo et al. 2007; Burgess 2008; Giocomo et al. 2011), similar to dlPFC (Wang et al. 2007; Wang et al. 2013). Thus, the current study examined layer II of the macaque ERC to determine whether these circuits express evidence of feedforward cAMP-calcium signaling and a “signature of flexibility” in dendritic spines, similar to that in layer III dlPFC. We focused on PDE4D, mGluR3 and HCN channels, given their important expression patterns in dlPFC, and the therapeutic potential of agents that enhance mGluR3 signaling. Intriguingly, layer II ERC cell clusters do not express calbindin in macaques or humans even when young (Beall and Lewis 1992), suggesting that their extraordinary vulnerability may arise from having calcium magnification in the absence of protection from calbindin expression, as this would render these neurons particularly vulnerable to toxic calcium actions and tau pathology. This hypothesis was explored in the current study, which examined whether layer II ERC in macaque expresses patterns of PDE4D, HCN1, and mGluR3 expression consistent with the “signature of flexibility/vulnerability” seen in layer III dlPFC pyramidal cells.
Materials and methods
Animals and tissue processing
Three female young adult (8, 9, and 10 years) rhesus monkeys (Macaca mulatta) in this study were maintained and euthanized in accordance with the guidelines of Yale University Institutional Animal Care and Use Committee and National Institutes of Health “Guidelines for the Care and Use of Experimental Animals”. As described previously (Paspalas and Goldman-Rakic 2004; Paspalas et al. 2013), the primates were deeply anesthetized prior to transcardial perfusion of 100 mM phosphate-buffered saline, followed by 4% paraformaldehyde and 0.05% glutaraldehyde in 100 mM phosphate- buffered saline. All perfusates were administered ice-cold. Following perfusion, a craniotomy was performed, and the entire brain was removed. The brains were blocked coronally and blocks containing lateral and medial ERC were vibrosliced at 60 μm on a vibratome (Leica, Norcross, GA, USA) across the entire rostrocaudal extent. The sections were cryoprotected through increasing concentrations of sucrose solution (10, 20, and 30% each for overnight), cooled rapidly using liquid nitrogen and stored at −80 °C. Sections of ERC were processed for immunocytochemistry. To enable penetration of immunoreagents, all sections went through three freeze–thaw cycles in liquid nitrogen. Non-specific reactivity was suppressed with 10% normal goat serum (NGS) and 2% bovine serum albumin (BSA), and antibody penetration was enhanced with 0.3% Triton X-100 in 50 mM Tris-buffered saline (TBS).
Histology and immunoreagents
We used previously well-characterized primary antibodies raised in rabbit and mice. We used an affinity isolated polyclonal PDE4D protein (SAB4502128; Millipore Sigma Aldrich, Burlington, MA; RRID:AB_10744568) raised against amino acids 156–205 of PDE4D that recognizes human and rodent PDE4D based on sequence homology. The antibody is highly specific and detects endogenous levels of total PDE4D protein at a band migrating at ~ 91 kDa. The antibody is suited for a range of applications, including immunohistochemistry, immunoblotting and ELISA as per manufacturer’s recommendations. The specificity and selectivity of the PDE4D antibody has been previously characterized using immunohistochemistry in myocytes to identify a role of PDE4D-PRKAR1α in cardiac contractility (Bedada et al. 2016) and with immunohistochemistry and immunoEM in rhesus macaque dlPFC (Datta et al. 2020). For HCN1 channel immunolabeling we used a rabbit antibody against the human HCN1 channel subunit (HPA019195, Atlas, Bromma, Sweden; RRID:AB_2670094). We have previously shown that liquid-phase adsorption of the anti-HCN1 with the immunizing peptide (5–15 μg/ml; APrEST72946; Atlas) abolished all labeling (Yang et al. 2018). For mGluR3 immunoEM, a well-characterized rabbit polyclonal antibody raised against a synthetic peptide corresponding to the N-terminal (extracellular) domain of human mGluR3, and purified by peptide immunogen affinity column, was obtained commercially (G1545, Sigma-Aldrich; RRID:AB_476997) for use in this study. This antibody has been extensively tested and shown by other groups to specifically recognize mGluR3 in human brain using both immunohistochemical and immunocytochemical approaches. The specificity of this antibody has also been confirmed in in vitro assays and by Western blotting (WB) using small interfering RNA for genetic suppression of mGluR3 expression in rodent hypothalamic and cortical cultures (Park et al. 2011; Wang et al. 2012). This antibody has been validated by our own group in monkey dlPFC using immunohistochemistry and immunoEM (Jin et al. 2017; Jin et al. 2018a) and in rat prelimbic medial PFC (mPFC) using immunoEM and immunofluorescence (Woo et al. 2022).
The primary antibodies were used at appropriate dilutions (PDE4D, 1:200; HCN1, 1:500; mGluR3, 1:200) and were complexed with species-specific goat secondary antibodies. Normal sera and IgG-free BSA were purchased from Jackson Immunoresearch (West Grove, PA, USA). All chemicals and supplies for electron microscopy were purchased from Sigma Aldrich (St. Louis, MO, USA) and Electron Microscopy Sciences (Hatfield, PA, USA), respectively.
Single pre-embedding peroxidase immunocytochemistry
As described previously (Paspalas et al. 2009; Paspalas et al. 2013; Datta et al. 2020; Yang et al. 2022), the sections were incubated for 72 h at 4°C with primary antibodies in TBS, and transferred for 2 h at room temperature to species-specific biotinylated Fab’ or F(ab’)2 fragments in TBS. To reveal immunoperoxidase labeling, sections were incubated with the avidin-biotin peroxidase complex (1:300; Vector Laboratories, Burlingame, CA, USA) and then visualized in 0.025% 3,3-diaminobenzidine tetrahydrochloride (DAB; Sigma Aldrich, St. Louis, MO, USA) as a chromogen in 100 mM PB with the addition of 0.005% hydrogen peroxide for 8–12 min. After the DAB reaction, sections were exposed to osmification (concentration 1%), and dehydration through a series of increasing ethanol concentrations (70–100%), infiltrated with propylene oxide. Tissue blocks were counterstained with 1% uranyl acetate in 70% ethanol. Standard epoxy resin embedding followed typical immunoEM procedures followed by polymerization at 60 °C for 48 h. Omission of primary antibodies or substitution with non-immune serum resulted in complete lack of immunoperoxidase labeling. Similarly, labeling was nullified when blocking the biotinylated probes with avidin/biotin. Furthermore, we observed a lack of precipitate when control sections were treated with diaminobenzidine.
Electron microscopy and data analysis
All sections were processed as previously described (Paspalas et al. 2013). Briefly, blocks containing lateral and medial ERC layer II were sampled and mounted onto resin blocks. Our immunoEM analyses focused on ERC layer II focusing on stellate cell clusters where the neuropil includes proximal apical dendrites from ERC layer III pyramidal cells. The specimens were cut into 50 nm sections using an ultramicrotome (Leica, Norcross, GA, USA), mounted on individual slot grids and analyzed under a Talos L120C transmission electron microscope (Thermo Fisher Scientific). Individual grids were counterstained with 1% lead citrate. Several plastic blocks of each brain were examined using the 4th–12th surface-most sections of each block (i.e. 200–600 nm), to sample the superficial component of sections, avoiding penetration artifacts. Structures were digitally captured at x17,000–x90,000 magnification with a Ceta CMOS camera and individual panels were adjusted for brightness and contrast using Adobe Photoshop and Illustrator CC.2020.01 image editing software (Adobe Systems Inc.). Approximately, 1,800 micrographs of selected areas of neuropil with immunopositive profiles were used for analyses with well-defined criteria. For profile identification, we adopted the criteria summarized by Alan Peters (Peters et al. 1991).
Immunofluorescence with confocal microscopy
Calbindin immunofluorescence staining was carried out on free-floating sections as described previously (Yang et al. 2022). Antigen retrieval was performed with 10 mM Solution Citrate Buffer in a hot water bath for 30 min at high temperature. The free-floating sections were left to cool for 30 min at RT. After washing the sections in deionized water, they were transferred to 1X TBS for 10 min. Sections were then incubated in 0.5% Sodium Borohydride in 0.01 M TBS. Sections were blocked for 1 h at RT in 1X TBS containing 5% BSA, 2% Triton X-100, and 10% NGS. Sections were incubated for 48 h at 4 °C with calbindin antibody (1:500, Swant; Cat# 300; RRID:AB_10000347), followed by incubation overnight at 4°C with secondary antibodies (1:1,000, Alexa-Fluor conjugated, Invitrogen). Following incubation in secondary antibodies, they were incubated in 70% Ethanol with 0.3% Sudan Black B (MP Biomedicals, Cat# 4197-25-5), to decrease autofluorescence from lipofuscin, and counterstained with Hoechst (1:10,000, Thermo Fisher, Cat# H3570). The sections were mounted in ProLong Gold Antifade Mountant (Invitrogen, Cat# P36930).
Confocal images were acquired using a Zeiss LSM 880 Airyscan Confocal Microscope, with the Plan-Apochromat 20×/0.8 M27 objective (Zeiss) and C-Apochromat 40×/1.2 W Korr FCS M27 (Zeiss) water objective. Emission filter bandwidths and sequential scanning acquisition were set up to avoid possible spectral overlap between fluorophores. Images were merged employing Fiji software.
Results
Quantitative mapping of PDE4D, HCN1, and mGluR3 protein in ERC layer II neuropil
The ultrastructural location of PDE4D, HCN1, and mGluR3 in ERC layer II neuropil of the rhesus macaque was determined using quantitative analyses with immunoperoxidase labeling. For example, analysis of PDE4D immunolabeled profiles revealed that PDE4D was primarily expressed within dendritic shafts (47%) with prominent expression in dendritic spines (16%) and glia (17%) (Fig. 1A). Similarly, analysis of HCN1 immunolabeled profiles revealed that HCN1 was predominately expressed within dendritic spines (39%) in ERC layer II receiving a Type I asymmetric glutamatergic-like synapse, and within dendritic shafts (24%), with sparser immunolabeling in axons (9%) and glial (4%) subcompartments (Fig. 1B). Analysis of mGluR3 immunolabeled profiles revealed predominant expression within dendritic spines (20%) and dendritic shafts (16%), with the majority of immunolabeling in glial compartments (36%), the latter being consistent with the classic anatomical location of mGluR3 within cortical circuits (Fig. 1C).
Fig. 1.

Quantitative analysis of PDE4D, HCN1, and mGluR3 localization in macaque ERC layer II circuits. (A) The prevalence of PDE4D in various cellular subcompartments in layer II of the ERC neuropil, expressed as percentage of PDE4D profile (e.g. dendrite) per total PDE4D profiles. PDE4D is primarily expressed in postsynaptic subcompartments in ERC layer II microcircuits, with foremost expression in dendritic shafts, and significant expression within dendritic spines and astroglia. (B) The prevalence of HCN1 in various cellular subcompartments in layer II of the ERC neuropil, expressed as percentage of HCN1 profile (e.g. dendritic spine) per total HCN1 profiles. HCN1 is primarily expressed in postsynaptic subcompartments in ERC layer II microcircuits, with greatest expression in dendritic spines, and significant expression within dendritic shafts. (C) The prevalence of mGluR3 in various cellular subcompartments in layer II of the ERC neuropil, expressed as percentage of mGluR3 profile (e.g. glia) per total mGluR3 profiles. mGluR3 is predominantly expressed in glia, with significant expression in postsynaptic subcompartments in ERC layer II microcircuits, including in dendritic spines and dendritic shafts. For additional details regarding quantitative assessment and profile identification, see the “Materials and Methods” section. Nondetermined (N.D) are profiles that could not be unequivocally categorized.
General patterns of post-synaptic expression in ERC layer II
Ultrastructural analyses with immunoperoxidase labeling revealed that PDE4D, HCN1, and mGluR3 are concentrated in neurons in postsynaptic subcompartments within dendritic spines (Figs. 1 and 2), and in dendritic shafts (Figs. 1 and 3), near asymmetric, glutamate-like synapses in rhesus macaque ERC layer II. In dendritic spines, PDE4D is observed directly in association with the SER spine apparatus, while HCN1 and mGluR3 are localized on the dendritic spine plasma membrane in close proximity to the SER spine apparatus (Fig. 2). Within dendritic shafts, a similar spatial configuration is observed with HCN1 and mGluR3 localized on the plasma membrane at perisynaptic or extrasynaptic locations, and PDE4D and mGluR3 observed subjacent to the postsynaptic density (PSD), possibly in association with the SER (Fig. 3). HCN1 labeling was also seen on dendritic shafts, distant from synapses (see below), a common configuration for these channels. A detailed description of each labeling pattern is provided below.
Fig. 2.

Subcellular localization of PDE4D, HCN1, and mGluR3 in ERC layer II dendritic spines. Immunoperoxidase labeling revealed that PDE4D, HCN1, and mGluR3 were focused in dendritic spines near asymmetric, glutamate-like synapses in rhesus macaque ERC layer II. (A) PDE4D was observed directly in association with the SER spine apparatus (pseudocolored pink). (B) HCN1 and (C) mGluR3 were localized on the dendritic spine plasma membrane adjacent to the SER spine apparatus. Synapses are between black arrows. Color-coded arrowheads point to PDE4D (green), HCN1 (red), and mGluR3 (yellow) immunoreactivity. Profiles are pseudocolored for clarity. Ax, axon; Sp, dendritic spine; Mit, mitochondria. Scale bars, 200 nm.
Fig. 3.

Subcellular localization of PDE4D, HCN1, and mGluR3 in ERC layer II dendritic shafts. Immunoperoxidase labeling revealed PDE4D, HCN1, and mGluR3 are concentrated within dendritic shafts in young adult rhesus macaque ERC layer II. (A) PDE4D was observed in association with likely SER (pseudocolored pink) and on microtubules. (B) HCN1 and (C) mGluR3 localized on the plasma membrane in dendritic shafts at perisynaptic or extrasynaptic locations. As has been reported in human layer II ERC (Dominguez et al. 2021), dendritic shafts receive axodendritic asymmetric Type I glutamatergic-like synapses; synapses are between black arrows. Color-coded arrowheads point to PDE4D (green), HCN1 (red), and mGluR3 (yellow) immunoreactivity. Profiles are pseudocolored for clarity. Ax, axon; Den, dendritic shaft; Mit, mitochondria; Mvb, multivesicular body. Scale bars, 200 nm.
Subcellular localization of PDE4D in ERC layer II
PDE4D immunolabeling was localized within dendritic spines in macaque ERC layer II (Fig. 4). PDE4D was concentrated in dendritic spines receiving Type I glutamatergic-like asymmetric synapses in association with, or near the spine apparatus, the calcium-containing extension of the SER into the dendritic spine head, positioned to regulate cAMP-PKA-calcium signaling (Fig. 4A–F). PDE4D labeling was also observed subjacent to the PSD, exhibiting non-uniform expression along the length of the synaptic active zone (Fig. 4F), as well as near perisynaptic membranes flanking the glutamatergic-like synapse (Fig. 4G).
Fig. 4.

Postsynaptic expression of PDE4D within dendritic spines in ERC layer II. (A-E) PDE4D immunolabeling was expressed in dendritic spines, directly in association with or near the SER spine apparatus (pseudocolored pink), the calcium-containing extension of the SER into the dendritic spine head, positioned to regulate cAMP-PKA-calcium signaling. (F) PDE4D immunolabeling was observed subjacent to the PSD near the synaptic active zone. (G) PDE4D was also observed in perisynaptic compartments near the plasma membrane flanking the excitatory synapse. All dendritic spines receive axospinous Type I asymmetric glutamatergic-like synapses; synapses are between black arrows. Color-coded arrowheads point to PDE4D (green) immunoreactivity. Profiles are pseudocolored for clarity. Ax, axon; Sp, dendritic spine; Mit, mitochondria. Scale bars, 200 nm.
PDE4D labeling was also present within dendritic shafts in ERC layer II neuropil. PDE4D labeling was primarily associated with parallel bundles of microtubules within dendritic shafts, which could be seen in both horizontal (Fig. 5A–C) and perpendicular (Fig. 5D–E) planes, consistent with possible intracellular trafficking of PDE4D, as well as possible cAMP regulation of intracellular trafficking, including trafficking of mitochondria (Ogawa et al. 2016). Consistent with this, PDE4D immunoreactivity was also common in dendrites next to mitochondria (Fig. 5B, D, E). Although it is more difficult to recognize SER tubules in dendrites due to their nondescript morphology, they frequently concentrate near mitochondria to exchange calcium in mitochondrial-associated membranes (MAMs), a process regulated by cAMP-PKA signaling (Bravo-Sagua et al. 2019). Thus, it is possible that the PDE4D labeling concentrated in dendrites near mitochondria is also localized on MAMs.
Fig. 5.

Postsynaptic expression of PDE4D within dendritic shafts in ERC layer II. (A–E) in young adult macaque ERC layer II, PDE4D was predominantly concentrated in dendritic shafts and was associated with microtubules oriented in parallel bundles and likely SER tubules (pseudocolored pink), in both horizontal and perpendicular planes. In (A), a fortuitous section showing a dendritic spine emanating from a dendritic shaft, with PDE4D immunolabeling directly in association with microtubules. The dendritic spine receives an axospinous asymmetric glutamatergic-like Type I synapse. (B, D, and E) PDE4D immunolabeling within dendritic shafts was associated with mitochondrial profiles, potentially regulating calcium entry via MAMs, a process regulated by cAMP-PKA signaling. In (D) and (E), dendritic shafts receive axodendritic asymmetric Type I glutamatergic-like synapses; synapses are between black arrows. Color-coded arrowheads point to PDE4D (green) immunoreactivity. Profiles are pseudocolored for clarity. Ax, axon; Den, dendrite; Mit, mitochondria. Scale bars, 200 nm.
PDE4D labeling was also observed within axons, near the plasma membrane in both intervaricose axonal segments captured in continuity with glutamatergic-like axon terminals (Fig. 6A), and in presynaptic boutons (Fig. 6B–C) establishing axospinous synapses. However, the presynaptic PDE4D labeling was observed distant from the active release zone, and as in dendrites, was often near mitochondrial profiles (Fig. 6A–C).
Fig. 6.
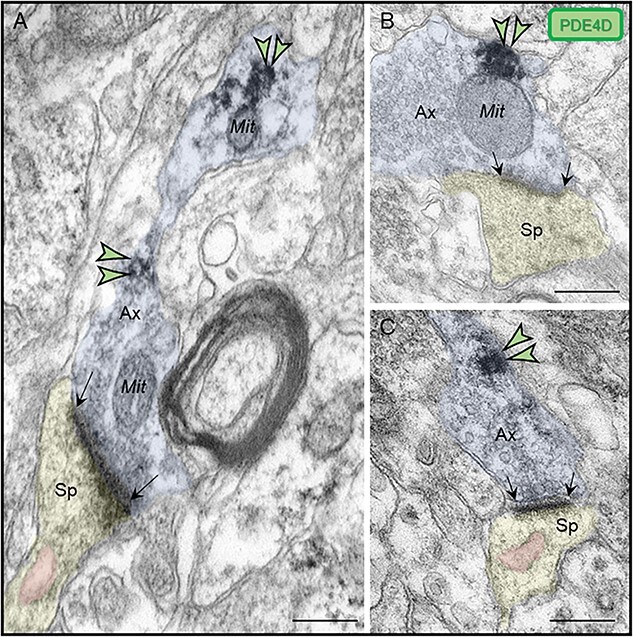
Presynaptic expression of PDE4D in ERC layer II. (A) PDE4D immunolabeling was observed in association with the plasma membrane in intervaricose axonal segments extending to form a glutamatergic-like axon terminal innervating a dendritic spine. (B-C) PDE4D is expressed in glutamatergic-like axonal terminals establishing axospinous Type I asymmetric synapses. The labeling is found in extrasynaptic subcompartments near the axon terminal plasmalemma, and not typically within the synaptic active zone. PDE4D immunolabeling in glutamatergic axon terminals was observed sometimes in close proximity to mitochondria (B). Synapses are between black arrows. Color-coded arrowheads point to PDE4D (green) immunoreactivity. Profiles are pseudocolored for clarity. Ax, axon; Sp, dendritic spine; Mit, mitochondria. Scale bars, 200 nm.
PDE4D was also expressed in astroglia as expected (Supplementary Fig. 1). Astroglial labeling was not uniformly distributed along the plasmalemma, but was instead concentrated near the synapse in perisynaptic astroglial processes (PAPs), near axospinous glutamatergic-like Type I asymmetric synapses (Supplementary Fig. 1A, B, D), or axodendritic glutamatergic-like Type I asymmetric synapses (Supplementary Fig. 1E), which are common on stellate cells in layer II ERC (Dominguez et al. 2021). The concentration of PDE4D in PAPS would be consistent with the known role of cAMP signaling in promoting calcium signaling in astrocytes (Sobolczyk and Boczek 2022).
Subcellular localization of HCN1 in ERC layer II
Similar to layer III dlPFC, HCN1 channels were localized within dendritic spines in ERC layer II where they were observed in perisynaptic or extrasynaptic locations on the plasma membrane near asymmetric, presumed glutamatergic-like synapses (Fig. 7A–F). HCN1 immunoperoxidase labeling on the dendritic spine membrane was often in close proximity to the SER spine apparatus (Fig. 7A, B, C, E, F). In favorable sections capturing the entire dendritic spine profile emanating from the dendritic shaft, a qualitative example of HCN1 immunoreactivity was observed occupying the dendritic spine neck along the plasma membrane (Fig. 7A).
Fig. 7.

Postsynaptic expression of HCN1 channels within dendritic spines in ERC layer II. (A–F) HCN1 channels were visualized with immunoperoxidase immunolabeling in perisynaptic and extrasynaptic portions of the plasmalemma adjacent to excitatory synapses within dendritic spines in ERC layer II. HCN1 channels were seen in close proximity to the SER spine apparatus (pseudocolored pink) (A, B, C, E, F). HCN1 channels were also observed in spine necks, especially in the narrow “bottleneck” (A, E). All dendritic spines receive axospinous Type I asymmetric glutamatergic-like synapses; synapses are between black arrows. Color-coded arrowheads point to HCN1 (red) immunoreactivity. Profiles are pseudocolored for clarity. Ax, axon; Sp, dendritic spine; Mit, mitochondria. Scale bars, 200 nm.
HCN1 channels were also concentrated in dendritic shafts in layer II ERC. HCN1 labeling could be seen on dendritic shafts near an asymmetric synapse (Fig. 3B), but was more commonly observed in discrete patches along the plasma membrane distant from any synapses (Fig. 8A–B). Clusters of HCN1 labeling distant from synaptic inputs are commonly seen on distal apical dendrites in both dlPFC and V1, likely the distal apical dendrites of layer V pyramidal cells traversing through layer III (Paspalas et al. 2013; Yang et al. 2018), as well as on the distal apical dendrites of pyramidal cells in rodent cortex (Lorincz et al. 2002). The labeling pattern in layer II ERC dendrites was qualitatively similar to these other structures.
Fig. 8.

Localization of HCN1 channels within dendritic shafts localized on the plasma membrane in ERC layer II. (A–B) Patches of HCN1 channel labeling were also observed along the plasma membrane in dendritic shafts, distant from any synapses, similar to patterns observed in apical dendrites in primates in both dlPFC and V1. Color-coded arrowheads point to HCN1 (red) immunoreactivity. Profiles are pseudocolored for clarity. Den, dendrite; Mit, mitochondria. Scale bars, 200 nm.
In addition to the postsynaptic location, immunoperoxidase labeling revealed HCN1 channels in axon terminals in ERC layer II in both perisynaptic compartments (Supplementary Fig. 2A) and in association with the active zone of asymmetric presumed glutamatergic-like synaptic terminals (Supplementary Fig. 2B).
Subcellular localization of mGluR3 in ERC layer II
Similar to dlPFC, mGluR3 labeling in layer II ERC was concentrated postsynaptically in or near asymmetric synapses on dendritic spines and shafts. In dendritic spines, mGluR3 labeling was seen at both perisynaptic and extrasynaptic locations (Fig. 9A, B, C, E), and within the synapse per se (Fig. 9D) and was often captured next to the SER spine apparatus. In dendrites, mGluR3 channels were observed on the plasma membrane near axodendritic glutamatergic-like excitatory synapses (Fig. 9F).
Fig. 9.

Localization of mGluR3 in postsynaptic compartments in dendritic spines and shafts in ERC layer II. (A–E) mGluR3 were expressed on dendritic spines, both in perisynaptic and extrasynaptic locations adjacent to the asymmetric synapse (A, B, C, E) and within the synapse per se (D1–D2), often near the SER spine apparatus (pseudocolored pink). Higher magnification image of mGluR3 in D2 revealed precise immunolabeling directly in association with the synaptic junction. The dendritic spine was visualized emanating from a dendritic shaft in D1. (F) Within dendritic shafts, mGluR3 channels were observed on the plasma membrane near axodendritic glutamatergic-like excitatory synapses; synapses are between black arrows. Color-coded arrowheads point to mGluR3 (yellow) immunoreactivity. Profiles are pseudocolored for clarity. Ax, axon; Sp, dendritic spine; Den, dendrite; Mit, mitochondria. Scale bars, 200 nm.
We also observed mGluR3 in presynaptic compartments, typically in discrete patches of the axonal plasma membrane distant from the active synaptic zone (Supplementary Fig. 3A–B), oftentimes associated with mitochondria (Supplementary Fig. 3B).
Finally, we observed mGluR3 immunolabeling in astroglial cells, as expected. As in dlPFC, mGluR3 labeling in astrocytes was focused in PAPs (Fig. 10A–G), consistent with the role of mGluR3 in modulating glutamate uptake into PAPs by increasing EAAT (excitatory amino acid transporter) expression via mitogen-activated protein kinase and phosphoinositide 3-kinase signaling (Aronica et al. 2003).
Fig. 10.

Astroglial expression of mGluR3 in ERC layer II. (A–G) Immunolabeling for mGluR3 was observed in PAPs with receptor expression being focused near the axospinous, excitatory synapse. A dendritic spine in (C) is also reactive against mGluR3 in extrasynaptic locations near the SER spine apparatus (pseudocolored in pink). Synapses are between black arrows. Color-coded arrowheads point to mGluR3 (yellow) immunoreactivity. Profiles are pseudocolored for clarity. Ax, axon; Sp, dendritic spine; Mit, mitochondria. Scale bars, 200 nm.
Summary of immunoEM data
The current data show that the pattern of PDE4D, HCN1, and mGluR3 spine labeling in layer II of the macaque ERC is similar to that seen in macaque layer III dlPFC, with PDE4D and mGluR3 positioned to regulate cAMP enhancement of internal calcium release from the SER spine apparatus near glutamate-like synapses in postsynaptic compartments, and HCN channels on the dendritic spine membrane which may gate network inputs (Fig. 1). A parallel configuration was also seen near glutamate-like synapses on dendritic shafts, consistent with the high proportion of asymmetric synapses on ERC stellate cell dendritic shafts in layer II of human ERC (Dominguez et al. 2021). Some presynaptic labeling was seen, with mGluR3 and PDE4D particularly associated with mitochondria, and HCN1 labeling with the presynaptic zone. PDE4D, mGluR3, and HCN1 channel labeling was also seen in their typical locations, e.g. with PDE4D and mGluR3 concentrated in astrocytes, and HCN1 labeling on dendrites distant from synaptic inputs.
Calbindin-IR immunolabeling in ERC layer II
Pyramidal cells in dlPFC layer III express the calcium binding protein, calbindin, consistent with their reliance on high levels of calcium (Hof and Morrison 1991; Joyce et al. 2020; Datta et al. 2021a). A previous anatomical study of the ERC in humans and rhesus macaques has found prominent calbindin expression in layer III ERC pyramidal cells, but not in the layer II cell clusters that are so vulnerable to tau pathology (Beall and Lewis 1992). Thus, we examined whether calbindin was lacking from layer II ERC cell clusters in the current study. Multiple label immunofluorescence and immunoperoxidase immunohistochemistry showed that calbindin expression was most striking in interneurons across the cortical column, as expected (Supplementary Fig. 4A, Fig. 12). Calbindin expression could also be seen in layer III pyramidal cells, with much less expression in layer II and in deeper layers (Supplementary Figs. 4A–C and 12). A more extensive analysis of ERC layer II across horizontal tiles showed cell clusters devoid of calbindin labeling (Figs. 11 and 12). Thus, layer II excitatory neurons may be particularly vulnerable to elevations in calcium signaling.
Fig. 12.

Brightfield micrographs revealing calbindin immunoreactivity in ERC. MacBrain Resource Collection 6, Brain 79, female 9.80 years old. (A) Calbindin (CB) and (B) neuronal nuclear antigen (NeuN) staining in the entorhinal cortex. The CB section is 700 μm rostral to the NeuN one and they correspond to section number 38 in each series, publicly available at https://macbraingallery.yale.edu/collection6/#B79. The black arrows point to capillaries that were followed for spatial matching. White dashed ovals in (A) show layer II areas devoid of CB+ cells and in (B) the approximate corresponding areas. Scale bars in (A) apply to (B). (C) High magnification micrograph showing areas in ERC layer II cell clusters devoid of CB immunoreactivity (green arrow). Scale bar, 0.1 mm.
Fig. 11.

Calbindin immunoreactivity across a horizontal tile in young macaque ERC. (A) Calbindin 28 kDa immunoreactivity captures superficial layers I–III and highlights the presence of layer II stellate cell clusters (indicated with color-coded dashed circles) with low-level calbindin expression in young macaque. In contrast, calbindin-labeled interneurons are prominent in this layer. (B–D) magnified areas of cell clusters in ERC layer II at 40x, with color coding corresponding to (A). scale bars, (A) 50 μm; (B–D) 25 μm.
Discussion
The current study compared the ultrastructural expression of PDE4D, HCN1 channels, and mGluR3 in layer II of the macaque ERC to those in layer III of the recently evolved dlPFC (Arnsten et al. 2021c), and found many similarities, including PDE4D labeling on the SER spine apparatus, and mGluR3 and HCN1 channels on the dendritic spine plasma membrane. A similar configuration was seen on ERC dendrites next to glutamate-like synapses, consistent with the unusually high proportion of glutamate synapses on layer II stellate cell dendrites in primates (Dominguez et al. 2021). These data provide structural evidence supporting PDE4D and mGluR3 regulation of feedforward cAMP-calcium signaling in dendritic spines and dendrites, and a “signature of flexibility”, with HCN channels expressed on dendritic spines and shafts near glutamate-like synapses, positioned to gate synaptic inputs under conditions of higher cAMP signaling. Thus, layer II of ERC shares important features to that of layer III dlPFC (Fig. 13).
PDE4D association with the SER
PDE4 localization is often used as a proxy for cAMP, as cAMP expression is too fleeting to be captured using immunolabeling. Thus, the consistent localization of PDE4D on the SER and dendritic spine apparatus in the ERC (current study) and in layer III dlPFC (Datta et al. 2020) provides structural evidence for cAMP interactions with internal calcium release from the SER. cAMP-PKA signaling is known to increase internal calcium release from the SER through ryanodine and IP3 receptors (Soulsby and Wojcikiewicz 2005; Ozawa 2010), and cytosolic calcium can in turn increase the production of cAMP through activation of adenylyl cyclases (Halls and Cooper 2011), thus producing feedforward signaling. The current data show that PDE4D localizes on the SER spine apparatus, suggesting that feedforward cAMP-PKA-calcium signaling also occurs in layer II ERC dendritic spines where it is regulated by PDE4D. The localization of PDE4D near the mitochondria in dendrites suggests a similar relationship may occur on the SER in dendrites, especially in association with MAMs. PDE4D localization on dendritic microtubules may represent intracellular trafficking of PDE4D to other subcellular destinations but may also indicate cAMP-PKA signaling in these locations, e.g. where PKA is known to phosphorylate tau (Paspalas et al. 2018).
mGluR3 signaling
mGluR3 signaling is increasingly linked to higher cognitive and memory abilities in humans (Arnsten and Wang 2020; Zink et al. 2020). However, little is known about the functional role(s) of mGluR3 signaling in ERC, in part because antibodies and pharmacological agents that can dissociate mGluR3 from mGluR2 locations and actions are just recently becoming available. Like PDE4D, mGluR3 are known to regulate cAMP levels via Gi-mediated inhibition of adenylyl cyclase, reducing the production of cAMP (Tanabe et al. 1992; Li et al. 2022). The localization of mGluR3 on layer II ERC dendritic spine membranes suggests they are also positioned to regulate feedforward cAMP-calcium signaling, similar to what has been seen in layer III dlPFC (Jin et al. 2017, 2018a). Thus, agents that enhance mGluR3 regulation of cAMP-calcium signaling in dlPFC may also have therapeutic benefits in ERC (Jin et al. 2017; Datta et al. 2021b; Yang et al. 2022). As cAMP increases the open state of HCN channels, including in layer II of rodent ERC (Heys and Hasselmo 2012), the localization of HCN channels on layer II ERC dendritic spines with an SER spine apparatus suggests that feedforward calcium-cAMP-signaling may allow the rapid accumulation of cAMP to open HCN channels and gate out network inputs in response to an arousing event, similar to the dynamic gating by HCN channels found in dlPFC (Wang et al. 2007; Arnsten et al. 2012).
HCN channel signaling in rodent ERC
HCN channels have a broad spectrum of functions, based on their subcellular location and molecular interactions (Benarroch 2013). Evidence from several studies shows that they can play a gating role, e.g. gating out excitatory inputs onto distal hippocampal dendrites in rodents (Nolan et al. 2007), and onto dendritic spines in primate dlPFC, where they are concentrated on dendritic spine heads near glutamate-like synapses and in dendritic spine necks (Wang et al. 2007; Paspalas et al. 2013). HCN channels may play a similar gating role in layer II of the ERC, as they were localized in dendritic spine heads and necks in the same subcellular locations as in layer III dlPFC. Physiological recordings from “grid cells” in the rodent ERC show that HCN channel opening plays a major role in the organization of grid fields in the rodent ERC (Giocomo et al. 2007; Giocomo et al. 2011), where they dynamically alter the scale of the grid representation, leading to smaller spatial scales when HCN currents have a faster time constant (Giocomo et al. 2007; Giocomo and Hasselmo 2009; Giocomo et al. 2011) and can be modulated by dopamine signaling (Glovaci and Chapman 2019). Parallel findings are seen in rhesus macaque dlPFC, where dopamine D1R stimulation leads to increased cAMP-HCN channel signaling and a dynamic scaling of spatial representations in working memory, with smaller representations with increasing D1R-HCN actions (Arnsten 2015). These dynamic mechanisms would allow arousal state to rapidly alter neural representations, e.g. to have more precise information during an acute threat (Arnsten 2015).
It is of interest that HCN channels were not only seen near glutamate-like synapses on dendritic spines, but also near glutamate-like synapses on dendritic shafts. Excitatory synapses on dendritic shafts are normally a feature of inhibitory interneurons, not excitatory cells, but layer II ERC appears to be an exception. Thus, while excitatory inputs are predominately on dendritic spines in pyramidal cells in most circuits, layer II ERC stellate cells have an unusually high proportion (~25%) of glutamate synapses on dendrites in addition to the excitatory connections on dendritic spines (Dominguez et al. 2021). The current study found HCN channels, PDE4D, and mGluR3 near asymmetric synapses on dendrites, suggesting that cAMP opening of HCN channels may also dynamically gate network inputs onto dendritic shafts. Although physiological data from behaving macaques would be needed to test this hypothesis, the data from rodent ERC showing that knockout of the HCN1 subunit to cause slower time constants of HCN channels can produce increases in grid scales (Giocomo et al. 2011), suggest that HCN1 channels on dendritic spines and dendrites near glutamate-like synapses may contribute to this modulatory function.
HCN channels were also seen in more traditional locations in layer II primate ERC. For example, patches of HCN channel labeling were observed on dendrites that were distant from any synapses, similar to the distal dendritic HCN1 labeling on pyramidal cells in primate dlPFC and V1, as well as pyramidal cells in rodent hippocampus (Magee 2000; Poolos et al. 2002; Nolan et al. 2004) and sensory cortex (Strauss et al. 2004; Shah 2014). These HCN1 channels are thought to open in response to membrane hyperpolarization, depolarizing the distal dendrite (Shah 2014). In dlPFC, these dendritic HCN channels are not co-expressed with cAMP-related proteins as they are in dendritic spines, suggesting that they are regulated more by voltage than by cAMP signaling (Paspalas et al. 2013). Studies of layer II ERC stellate cells in wildtype vs. HCN1 knockout mice show that HCN1 channels shape resting membrane properties and control the pattern of action potential output by promoting recovery of the afterhyperpolarization (Nolan et al. 2007), and that adenosine can reduce layer II ERC cell firing via inhibition of HCN channel actions (Li et al. 2011).
In the current study, HCN channels were also seen on axon terminals in layer II ERC. This presynaptic localization is similar to what is seen in rhesus macaque V1 (Yang et al. 2018), but not in rhesus macaque dlPFC (Paspalas et al. 2013). In rodents, presynaptic HCN channels can increase transmitter release (Bender et al. 2007; Huang and Trussell 2014), although they have been shown to reduce transmitter release from layer III ERC pyramidal cells in mice (Huang et al. 2017). The identities of the labeled presynaptic terminals within layer II ERC in the current study are not known, but may arise from the perirhinal and parahippocampal cortices that project extensively into layer II of the primate ERC (Insausti and Amaral 2008). It is possible that the opening of these presynaptic HCN channels may combine with dendritic spine and dendritic HCN channel opening to dynamically gate network inputs and alter representations in the primate ERC.
Absence of calbindin protection may render layer II ERC neurons particularly vulnerable to tau pathology
In humans, tau pathology begins to appear in the lateral ERC layer II cell clusters as early as middle age, and becomes steadily more pronounced affecting medial ERC layer II with increasing age, with accompanying neurodegeneration across cortical layers (Braak and Braak 1991). A qualitatively similar pattern and sequence is seen in macaque ERC, where AT8-labeled dendrites are first apparent in layer II ERC cell clusters and become more numerous and widespread with increasing age (Paspalas et al. 2018). The ability to perform perfusion-fixation in macaques additionally allows detection of early stage, soluble phosphorylated tau (Paspalas et al. 2018), which is generally lost by post-mortem dephosphorylation in human brains. In these studies, PKA phosphorylation of tau at S214 was seen in neurons within the layer II cell clusters in young adult macaques, aggregating on microtubules, the SER, and near mitochondria in dendrites, and on the dendritic spine apparatus and within the PSD in dendritic spines (Paspalas et al. 2018). These are the same subcellular localizations where PDE4D was observed in the current study. As PDE4D levels are reduced with age in the macaque ERC (Bhatla, Datta, Nairn, and Arnsten, unpublished), dysregulation of cAMP-PKA signaling near microtubules and in dendritic spines may be an early driver of tau phosphorylation. The unusually large number of excitatory synapses on dendrites may further amplify this process, with dysregulated cAMP-calcium signaling within dendrites as well as dendritic spines, e.g. as documented by PKA phosphorylation of ryanodine receptors on the SER directly under glutamate-like synapses of layer II ERC dendrites (Paspalas et al. 2018).
It is likely that the absence of the calcium binding protein, calbindin, in neurons in the layer II ERC cell clusters in humans (Beall and Lewis 1992) and in rhesus macaques [(Beall and Lewis 1992), current study] may also play a role in their vulnerability to tau pathology. High levels of calcium are known to drive tau hyperphosphorylation, e.g. through calpain-2 cleavage of GSK3β, as well as autophagic degeneration (Arnsten et al. 2021a; Arnsten et al. 2021b). The current study provides structural evidence for feedforward cAMP-calcium signaling in layer II ERC dendrites and dendritic spines, and thus lower calbindin immunoreactivity in these neurons may render these cellular compartments particularly vulnerable to tau hyperphosphorylation. In contrast to layer II, pyramidal cells in layer III of ERC do express calbindin when young [(Beall and Lewis 1992), current study], and these neurons develop tau pathology later than the layer II cell clusters in both humans (Braak and Braak 1991) and rhesus macaques (Paspalas et al. 2018). Thus, the current data may help to explain the selective vulnerability of layer II cell clusters to tau pathology.
Supplementary Material
Acknowledgments
We thank Lisa Ciavarella, Sam Johnson, Tracy Sadlon, and Michelle Wilson for their invaluable technical assistance. The authors also would like to thank the Center for Cellular and Molecular Imaging, Electron Microscopy Facility at Yale Medical School for assistance with the work presented here.
Contributor Information
Dibyadeep Datta, Departments of Neuroscience, Yale University School of Medicine, New Haven, CT 06510, USA; Department of Psychiatry, Yale University School of Medicine, New Haven, CT 06510, USA.
Isabella Perone, Departments of Neuroscience, Yale University School of Medicine, New Haven, CT 06510, USA.
Yury M Morozov, Departments of Neuroscience, Yale University School of Medicine, New Haven, CT 06510, USA.
Jon Arellano, Departments of Neuroscience, Yale University School of Medicine, New Haven, CT 06510, USA.
Alvaro Duque, Departments of Neuroscience, Yale University School of Medicine, New Haven, CT 06510, USA.
Pasko Rakic, Departments of Neuroscience, Yale University School of Medicine, New Haven, CT 06510, USA.
Christopher H van Dyck, Department of Psychiatry, Yale University School of Medicine, New Haven, CT 06510, USA.
Amy F T Arnsten, Departments of Neuroscience, Yale University School of Medicine, New Haven, CT 06510, USA.
CRediT author statement
Dibyadeep Datta (Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Visualization, Writing—original draft, Writing—review and editing), Isabella Perone (Data curation, Investigation), Yury Morozov (Methodology), Jon Arellano (Methodology), Alvaro Duque (Funding acquisition, Investigation, Methodology, Writing—review and editing), Pasko Rakic (Funding acquisition, Project administration, Resources, Supervision, Writing—review & editing), Christopher Van Dyck (Funding acquisition, Project administration, Resources, Supervision, Writing—review and editing), Amy Arnsten (Conceptualization, Formal analysis, Funding acquisition, Project administration, Resources, Supervision, Visualization, Writing—original draft, Writing—review and editing).
Funding
This work was primarily supported by National Institute of Health (NIH) RO1 (grants AG061190) and NSF 2015276 to AFTA, NIH National Institute of Aging (grant 1R21AG079145-01 to D.D.), NIH NIDA (grant DA023999 to P.R.), and MacBrain Resource Center (NIH Grant MH113257 to A.D.). This work was also partially supported by the Alzheimer’s Disease Research Unit from CvD.
Conflict of interest statement: None declared.
Data availability
The raw data will be made available upon request. Please contact Dr. Dibyadeep Datta to request data.
References
- Ahmed Z, Cooper J, Murray TK, Garn K, McNaughton E, Clarke H, Parhizkar S, Ward MA, Cavallini A, Jackson S, et al. A novel in vivo model of tau propagation with rapid and progressive neurofibrillary tangle pathology: the pattern of spread is determined by connectivity, not proximity. Acta Neuropathol. 2014:127:667–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnsten AF. Stress weakens prefrontal networks: molecular insults to higher cognition. Nat Neurosci. 2015:18:1376–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnsten AFT, Wang M. The evolutionary expansion of mGluR3-NAAG-GCPII signaling: relevance to human intelligence and cognitive disorders. Am J Psychiatry. 2020:177:1103–1106. [DOI] [PubMed] [Google Scholar]
- Arnsten AF, Wang MJ, Paspalas CD. Neuromodulation of thought: flexibilities and vulnerabilities in prefrontal cortical network synapses. Neuron. 2012:76:223–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnsten AF, Wang M, Paspalas CD. Dopamine's actions in primate prefrontal cortex: challenges for treating cognitive disorders. Pharmacol Rev. 2015:67:681–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnsten AFT, Datta D, Leslie S, Yang ST, Wang M, Nairn AC. Alzheimer's-like pathology in aging rhesus macaques: unique opportunity to study the etiology and treatment of Alzheimer's disease. Proc Natl Acad Sci U S A. 2019:116:26230–26238. 10.1073/pnas.1903671116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnsten AFT, Datta D, Del Tredici K, Braak H. Hypothesis: tau pathology is an initiating factor in sporadic Alzheimer's disease. Alzheimers Dement. 2021a:17:115–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnsten AFT, Datta D, Preuss TM. Studies of aging nonhuman primates illuminate the etiology of early-stage Alzheimer's-like neuropathology: an evolutionary perspective. Am J Primatol. 2021b:83(11):e23254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnsten AFT, Datta D, Wang M. The genie in the bottle-magnified calcium signaling in dorsolateral prefrontal cortex. Mol Psychiatry. 2021c:26:3684–3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnsten AFT, Woo E, Yang S, Wang M, Datta D. Unusual molecular regulation of dorsolateral prefrontal cortex layer III synapses increases vulnerability to genetic and environmental insults in schizophrenia. Biol Psychiatry. 2022:92:480–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronica E, Gorter JA, Ijlst-Keizers H, Rozemuller AJ, Yankaya B, Leenstra S, Troost D. Expression and functional role of mGluR3 and mGluR5 in human astrocytes and glioma cells: opposite regulation of glutamate transporter proteins. Eur J Neurosci. 2003:17:2106–2118. [DOI] [PubMed] [Google Scholar]
- Baillie GS, Tejeda GS, Kelly MP. Therapeutic targeting of 3′,5′-cyclic nucleotide phosphodiesterases: inhibition and beyond. Nat Rev Drug Discov. 2019:18:770–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beall MJ, Lewis DA. Heterogeneity of layer II neurons in human entorhinal cortex. J Comp Neurol. 1992:321:241–266. [DOI] [PubMed] [Google Scholar]
- Bedada FB, Martindale JJ, Arden E, Metzger JM. Molecular inotropy mediated by cardiac miR-based PDE4D/PRKAR1alpha/phosphoprotein signaling. Sci Rep. 2016:6:36803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benarroch EE. HCN channels: function and clinical implications. Neurology. 2013:80:304–310. [DOI] [PubMed] [Google Scholar]
- Bender RA, Kirschstein T, Kretz O, Brewster AL, Richichi C, Ruschenschmidt C, Shigemoto R, Beck H, Frotscher M, Baram TZ. Localization of HCN1 channels to presynaptic compartments: novel plasticity that may contribute to hippocampal maturation. J Neurosci. 2007:27:4697–4706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological staging of Alzheimer-related changes. Acta Neuropathol. 1991:82:239–259. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. The human entorhinal cortex: normal morphology and lamina-specific pathology in various diseases. Neurosci Res. 1992:15:6–31. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Trecidi K. Neuroanatomy and pathology of sporadic Alzheimer's disease. Adv Anat Embryol Cell Biol. 2015:215:1–162. [PubMed] [Google Scholar]
- Braak H, Del Tredici K. The preclinical phase of the pathological process underlying sporadic Alzheimer's disease. Brain. 2015:138:2814–2833. [DOI] [PubMed] [Google Scholar]
- Bravo-Sagua R, Parra V, Ortiz-Sandoval C, Navarro-Marquez M, Rodriguez AE, Diaz-Valdivia N, Sanhueza C, Lopez-Crisosto C, Tahbaz N, Rothermel BA, et al. Caveolin-1 impairs PKA-DRP1-mediated remodelling of ER-mitochondria communication during the early phase of ER stress. Cell Death Differ. 2019:26:1195–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess N. Grid cells and theta as oscillatory interference: theory and predictions. Hippocampus. 2008:18:1157–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calafate S, Buist A, Miskiewicz K, Vijayan V, Daneels G, Strooper B, Wit J, Verstreken P, Moechars D. Synaptic contacts enhance cell-to-cell tau pathology propagation. Cell Rep. 2015:11:1176–1183. [DOI] [PubMed] [Google Scholar]
- Carlyle BC, Nairn AC, Wang M, Yang Y, Jin LE, Simen AA, Ramos BP, Bordner KA, Craft GE, Davies P, et al. cAMP-PKA phosphorylation of tau confers risk for degeneration in aging association cortex. Proc Natl Acad Sci U S A. 2014:111:5036–5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colin M, Dujardin S, Schraen-Maschke S, Meno-Tetang G, Duyckaerts C, Courade JP, Buee L. From the prion-like propagation hypothesis to therapeutic strategies of anti-tau immunotherapy. Acta Neuropathol. 2020:139:3–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta D, Enwright JF, Arion D, Paspalas CD, Morozov YM, Lewis DA, Arnsten AFT. Mapping phosphodiesterase 4D (PDE4D) in macaque dorsolateral prefrontal cortex: postsynaptic compartmentalization in layer III pyramidal cell circuits. Front Neuroanat. 2020:14:578483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta D, Leslie SN, Wang M, Morozov YM, Yang S, Mentone S, Zeiss C, Duque A, Rakic P, Horvath TL, et al. Age-related calcium dysregulation linked with tau pathology and impaired cognition in non-human primates. Alzheimers Dement. 2021a:17:920–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta D, Leslie SN, Woo E, Amancharla N, Elmansy A, Lepe M, Mecca AP, Slusher BS, Nairn AC, Arnsten AFT. Glutamate carboxypeptidase II in aging rat prefrontal cortex impairs working memory performance. Front Aging Neurosci. 2021b:13:760270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day M, Carr DB, Ulrich S, Ilijic E, Tkatch T, Surmeier DJ. Dendritic excitability of mouse frontal cortex pyramidal neurons is shaped by the interaction among HCN, Kir2, and Kleak channels. J Neurosci. 2005:25:8776–8787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calignon A, Polydoro M, Suarez-Calvet M, William C, Adamowicz DH, Kopeikina KJ, Pitstick R, Sahara N, Ashe KH, Carlson GA, et al. Propagation of tau pathology in a model of early Alzheimer's disease. Neuron. 2012:73:685–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez-Alvaro M, Montero-Crespo M, Blazquez-Llorca L, Plaza-Alonso S, Cano-Astorga N, DeFelipe J, Alonso-Nanclares L. Analysis of the synaptic organization in the entorhinal cortex in Alzheimer's disease. eNeuro. 2021:8:ENEURO.0504-20.2021. 10.1523/ENEURO.0504-20.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dujardin S, Begard S, Caillierez R, Lachaud C, Carrier S, Lieger S, Gonzalez JA, Deramecourt V, Deglon N, Maurage CA, et al. Different tau species lead to heterogeneous tau pathology propagation and misfolding. Acta Neuropathol Commun. 2018:6:132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Fricker D, Brager DH, Chen X, Lu HC, Chitwood RA, Johnston D. Activity-dependent decrease of excitability in rat hippocampal neurons through increases in I(h). Nat Neurosci. 2005:8:1542–1551. [DOI] [PubMed] [Google Scholar]
- Fu H, Hardy J, Duff KE. Selective vulnerability in neurodegenerative diseases. Nat Neurosci. 2018:21:1350–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamo NJ, Lur G, Higley MJ, Wang M, Paspalas CD, Vijayraghavan S, Yang Y, Ramos BP, Peng K, Kata A, et al. Stress impairs prefrontal cortical function via D1 dopamine receptor interactions with hyperpolarization-activated cyclic nucleotide-gated channels. Biol Psychiatry. 2015:78:860–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giocomo LM, Hasselmo ME. Knock-out of HCN1 subunit flattens dorsal-ventral frequency gradient of medial entorhinal neurons in adult mice. J Neurosci. 2009:29:7625–7630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giocomo LM, Zilli EA, Fransen E, Hasselmo ME. Temporal frequency of subthreshold oscillations scales with entorhinal grid cell field spacing. Science. 2007:315:1719–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giocomo LM, Hussaini SA, Zheng F, Kandel ER, Moser MB, Moser EI. Grid cells use HCN1 channels for spatial scaling. Cell. 2011:147:1159–1170. [DOI] [PubMed] [Google Scholar]
- Glovaci I, Chapman CA. Dopamine induces release of calcium from internal stores in layer II lateral entorhinal cortex fan cells. Cell Calcium. 2019:80:103–111. [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS. Cellular basis of working memory. Neuron. 1995:14:477–485. [DOI] [PubMed] [Google Scholar]
- Halls ML, Cooper DM. Regulation by Ca2+−signaling pathways of adenylyl cyclases. Cold Spring Harb Perspect Biol. 2011:3:a004143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez CM, McQuail JA, Schwabe MR, Burke SN, Setlow B, Bizon JL. Age-related declines in prefrontal cortical expression of metabotropic glutamate receptors that support working memory. eNeuro. 2018:5:ENEURO.0164-18.2018. 10.1523/ENEURO.0164-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heys JG, Hasselmo ME. Neuromodulation of I(h) in layer II medial entorhinal cortex stellate cells: a voltage-clamp study. J Neurosci. 2012:32:9066–9072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hof PR, Morrison JH. Neocortical neuronal subpopulations labeled by a monoclonal antibody to calbindin exhibit differential vulnerability in Alzheimer's disease. Exp Neurol. 1991:111:293–301. [DOI] [PubMed] [Google Scholar]
- Houslay MD, Adams DR. PDE4 cAMP phosphodiesterases: modular enzymes that orchestrate signalling cross-talk, desensitization and compartmentalization. Biochem J. 2003:370:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Trussell LO. Presynaptic HCN channels regulate vesicular glutamate transport. Neuron. 2014:84:340–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Li G, Aguado C, Lujan R, Shah MM. HCN1 channels reduce the rate of exocytosis from a subset of cortical synaptic terminals. Sci Rep. 2017:7:40257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman BT, Van Hoesen GW, Kromer LJ, Damasio AR. Perforant pathway changes and the memory impairment of Alzheimer's disease. Ann Neurol. 1986:20:472–481. [DOI] [PubMed] [Google Scholar]
- Insausti R, Amaral DG. Entorhinal cortex of the monkey: IV. Topographical and laminar organization of cortical afferents. J Comp Neurol. 2008:509:608–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs J, Weidemann CT, Miller JF, Solway A, Burke JF, Wei XX, Suthana N, Sperling MR, Sharan AD, Fried I, et al. Direct recordings of grid-like neuronal activity in human spatial navigation. Nat Neurosci. 2013:16:1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin LE, Wang M, Yang ST, Yang Y, Galvin VC, Lightbourne TC, Ottenheimer D, Zhong Q, Stein J, Raja A, et al. mGluR2/3 mechanisms in primate dorsolateral prefrontal cortex: evidence for both presynaptic and postsynaptic actions. Mol Psychiatry. 2017:22:1615–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin LE, Wang M, Galvin VC, Lightbourne TC, Conn PJ, Arnsten AFT, Paspalas CD. mGluR2 versus mGluR3 metabotropic glutamate receptors in primate dorsolateral prefrontal cortex: postsynaptic mGluR3 strengthen working memory networks. Cereb Cortex. 2018a:28:974–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin LE, Wang M, Galvin VC, Lightbourne TC, Conn PJ, Arnsten AFT, Paspalas CD. mGluR2 vs. mGluR3 in primate prefrontal cortex: postsynaptic mGluR3 strengthen cognitive networks. Cereb Cortex. 2018b:28:974–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce MKP, Garcia-Cabezas MA, John YJ, Barbas H. Serial prefrontal pathways are positioned to balance cognition and emotion in primates. J Neurosci. 2020:40:8306–8328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman SK, Del Tredici K, Thomas TL, Braak H, Diamond MI. Tau seeding activity begins in the transentorhinal/entorhinal regions and anticipates phospho-tau pathology in Alzheimer's disease and PART. Acta Neuropathol. 2018:136:57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Killian NJ, Jutras MJ, Buffalo EA. A map of visual space in the primate entorhinal cortex. Nature. 2012:491:761–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus BJ, Brandon MP, Robinson RJ 2nd, Connerney MA, Hasselmo ME, Eichenbaum H. During running in place, grid cells integrate elapsed time and distance run. Neuron. 2015:88:578–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DA, Campbell MJ, Terry RD, Morrison JH. Laminar and regional distributions of neurofibrillary tangles and neuritic plaques in Alzheimer's disease: a quantitative study of visual and auditory cortices. J Neurosci. 1987:7:1799–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Fan S, Yan J, Li B, Chen F, Xia J, Yu Z, Hu Z. Adenosine modulates the excitability of layer II stellate neurons in entorhinal cortex through A1 receptors. Hippocampus. 2011:21:265–280. [DOI] [PubMed] [Google Scholar]
- Li SH, Abd-Elrahman KS, Ferguson SSG. Targeting mGluR2/3 for treatment of neurodegenerative and neuropsychiatric diseases. Pharmacol Ther. 2022:239:108275. [DOI] [PubMed] [Google Scholar]
- Liu J, Li L. Targeting autophagy for the treatment of Alzheimer's disease: challenges and opportunities. Front Mol Neurosci. 2019:12:203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorincz A, Notomi T, Tamas G, Shigemoto R, Nusser Z. Polarized and compartment-dependent distribution of HCN1 in pyramidal cell dendrites. Nat Neurosci. 2002:5:1185–1193. [DOI] [PubMed] [Google Scholar]
- Lu T, Pan Y, Kao SY, Li C, Kohane I, Chan J, Yankner BA. Gene regulation and DNA damage in the ageing human brain. Nature. 2004:429:883–891. [DOI] [PubMed] [Google Scholar]
- Magee JC. Dendritic integration of excitatory synaptic input. Nat Rev Neurosci. 2000:1:181–190. [DOI] [PubMed] [Google Scholar]
- Moser EI, Roudi Y, Witter MP, Kentros C, Bonhoeffer T, Moser MB. Grid cells and cortical representation. Nat Rev Neurosci. 2014:15:466–481. [DOI] [PubMed] [Google Scholar]
- Moser MB, Rowland DC, Moser EI. Place cells, grid cells, and memory. Cold Spring Harb Perspect Biol. 2015:7:a021808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadasdy Z, Nguyen TP, Torok A, Shen JY, Briggs DE, Modur PN, Buchanan RJ. Context-dependent spatially periodic activity in the human entorhinal cortex. Proc Natl Acad Sci U S A. 2017:114:E3516–E3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, Alafuzoff I, Bigio EH, Bouras C, Braak H, Cairns NJ, Castellani RJ, Crain BJ, Davies P, Del Tredici K, et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol. 2012:71:362–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan MF, Malleret G, Dudman JT, Buhl DL, Santoro B, Gibbs E, Vronskaya S, Buzsaki G, Siegelbaum SA, Kandel ER, et al. A behavioral role for dendritic integration: HCN1 channels constrain spatial memory and plasticity at inputs to distal dendrites of CA1 pyramidal neurons. Cell. 2004:119:719–732. [DOI] [PubMed] [Google Scholar]
- Nolan MF, Dudman JT, Dodson PD, Santoro B. HCN1 channels control resting and active integrative properties of stellate cells from layer II of the entorhinal cortex. J Neurosci. 2007:27:12440–12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notomi T, Shigemoto R. Immunohistochemical localization of Ih channel subunits, HCN1-4, in the rat brain. J Comp Neurol. 2004:471:241–276. [DOI] [PubMed] [Google Scholar]
- Ogawa F, Murphy LC, Malavasi EL, O'Sullivan ST, Torrance HS, Porteous DJ, Millar JK. NDE1 and GSK3beta associate with TRAK1 and regulate axonal mitochondrial motility: identification of cyclic AMP as a novel modulator of axonal mitochondrial trafficking. ACS Chem Neurosci. 2016:7:553–564. [DOI] [PubMed] [Google Scholar]
- Ozawa T. Modulation of ryanodine receptor Ca2+ channels (review). Mol Med Rep. 2010:3:199–204. [DOI] [PubMed] [Google Scholar]
- Park WM, Wang Y, Park S, Denisova JV, Fontes JD, Belousov AB. Interplay of chemical neurotransmitters regulates developmental increase in electrical synapses. J Neurosci. 2011:31:5909–5920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paspalas CD, Goldman-Rakic PS. Microdomains for dopamine volume neurotransmission in primate prefrontal cortex. J Neurosci. 2004:24:5292–5300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paspalas CD, Selemon LD, Arnsten AF. Mapping the regulator of G protein signaling 4 (RGS4): presynaptic and postsynaptic substrates for neuroregulation in prefrontal cortex. Cereb Cortex. 2009:19:2145–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paspalas CD, Wang M, Arnsten AF. Constellation of HCN channels and cAMP regulating proteins in dendritic spines of the primate prefrontal cortex: potential substrate for working memory deficits in schizophrenia. Cereb Cortex. 2013:23:1643–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paspalas CD, Carlyle BC, Leslie S, Preuss TM, Crimins JL, Huttner AJ, Dyck CH, Rosene DL, Nairn AC, Arnsten AFT. The aged rhesus macaque manifests Braak stage III/IV Alzheimer's-like pathology. Alzheimers Dement. 2018:14:680–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A, Palay S, Webster H. The fine structure of the nervous system: neurons and their supporting cells. New York: Oxford University Press; 1991. [Google Scholar]
- Poolos NP, Migliore M, Johnston D. Pharmacological upregulation of h-channels reduces the excitability of pyramidal neuron dendrites. Nat Neurosci. 2002:5:767–774. [DOI] [PubMed] [Google Scholar]
- Sanders H, Renno-Costa C, Idiart M, Lisman J. Grid cells and place cells: an integrated view of their navigational and memory function. Trends Neurosci. 2015:38:763–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah MM. Cortical HCN channels: function, trafficking and plasticity. J Physiol. 2014:592:2711–2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobolczyk M, Boczek T. Astrocytic calcium and cAMP in neurodegenerative diseases. Front Cell Neurosci. 2022:16:889939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soulsby MD, Wojcikiewicz RJ. The type III inositol 1,4,5-trisphosphate receptor is phosphorylated by cAMP-dependent protein kinase at three sites. Biochem J. 2005:392:493–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss U, Kole MH, Brauer AU, Pahnke J, Bajorat R, Rolfs A, Nitsch R, Deisz RA. An impaired neocortical Ih is associated with enhanced excitability and absence epilepsy. Eur J Neurosci. 2004:19:3048–3058. [DOI] [PubMed] [Google Scholar]
- Tanabe Y, Masu M, Ishii T, Shigemoto R, Nakanishi S. A family of metabotropic glutamate receptors. Neuron. 1992:8:169–179. [DOI] [PubMed] [Google Scholar]
- Thuault SJ, Malleret G, Constantinople CM, Nicholls R, Chen I, Zhu J, Panteleyev A, Vronskaya S, Nolan MF, Bruno R, et al. Prefrontal cortex HCN1 channels enable intrinsic persistent neural firing and executive memory function. J Neurosci. 2013:33:13583–13599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayraghavan S, Wang M, Birnbaum SG, Williams GV, Arnsten AF. Inverted-U dopamine D1 receptor actions on prefrontal neurons engaged in working memory. Nat Neurosci. 2007:10:376–384. [DOI] [PubMed] [Google Scholar]
- Wang M, Ramos BP, Paspalas CD, Shu Y, Simen A, Duque A, Vijayraghavan S, Brennan A, Dudley A, Nou E, et al. Alpha2A-adrenoceptors strengthen working memory networks by inhibiting cAMP-HCN channel signaling in prefrontal cortex. Cell. 2007:129:397–410. [DOI] [PubMed] [Google Scholar]
- Wang Y, Song JH, Denisova JV, Park WM, Fontes JD, Belousov AB. Neuronal gap junction coupling is regulated by glutamate and plays critical role in cell death during neuronal injury. J Neurosci. 2012:32:713–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Yang Y, Wang CJ, Gamo NJ, Jin LE, Mazer JA, Morrison JH, Wang XJ, Arnsten AF. NMDA receptors subserve persistent neuronal firing during working memory in dorsolateral prefrontal cortex. Neuron. 2013:77:736–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Datta D, Enwright J, Galvin V, Yang ST, Paspalas C, Kozak R, Gray DL, Lewis DA, Arnsten AFT. A novel dopamine D1 receptor agonist excites delay-dependent working memory-related neuronal firing in primate dorsolateral prefrontal cortex. Neuropharmacology. 2019:150:46–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo E, Datta D, Arnsten AFT. Glutamate metabotropic receptor type 3 (mGlu3) localization in the rat prelimbic medial prefrontal cortex. Front Neuroanat. 2022:16:849937. 10.3389/fnana.2022.849937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, El-Hassar L, Datta D, Thomas M, Zhang Y, David PJ, DeLuca NJ, Chatterjee M, Gribkoff VK, Arnsten AFT, et al. Interaction between HCN and slack channels regulates mPFC pyramidal cell excitability and working memory; 2023bioRxiv:2023.2003.2004.529157. [DOI] [PubMed]
- Yang ST, Wang M, Paspalas CD, Crimins JL, Altman MT, Mazer JA, Arnsten AFT. Core differences in synaptic Signaling between primary visual and dorsolateral prefrontal cortex. Cereb Cortex. 2018:28:1458–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Datta D, Elizabeth W, Duque A, Morozov YM, Arellano J, Slusher BS, Wang M, Arnsten AFT. Inhibition of glutamate-carboxypeptidase-II in dorsolateral prefrontal cortex: potential therapeutic target for neuroinflammatory cognitive disorders. Mol Psychiatry. 2022:27:4252–4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zink CF, Barker PB, Sawa A, Weinberger DR, Wang M, Quillian H, Ulrich WS, Chen Q, Jaffe AE, Kleinman JE, et al. Association of Missense Mutation in FOLH1 with decreased NAAG levels and impaired working memory circuitry and cognition. Am J Psychiatry. 2020:177:1129–1139. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data will be made available upon request. Please contact Dr. Dibyadeep Datta to request data.