

Abstract
Cardiac amyloidosis (CA) is a condition caused by extracellular deposition of amyloid fibrils in the heart. It is an underdiagnosed disease entity which can present with a variety of cardiac and non-cardiac manifestations. Diagnosis usually follows an initial suspicion based on clinical evaluation or imaging findings before confirmation with subsequent imaging (echocardiography, cardiac magnetic resonance imaging, 3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy) in combination with biochemical screening for monoclonal dyscrasia (serum free light chains and serum and urine electrophoresis) and/or histology (bone marrow trephine, fat or endomyocardial biopsy). More than 95% of CA can be classified as either amyloid light-chain (AL) CA or amyloid transthyretin (ATTR) CA; these two conditions have very different management strategies. AL-CA, which may be associated with multiple myeloma, can be managed with chemotherapy agents, autologous stem cell transplantation, cardiac transplant and supportive therapies. For ATTR-CA, there is increasing importance in making an early diagnosis because of novel treatments in development, which have transformed this once incurable disease to a potentially treatable disease. Timely diagnosis is crucial as there may only be a small window of opportunity where patients can benefit from treatment beyond which therapies may be less effective. Reviewing the existing patient pathway provides a basis to better understand the complexities of real-world activities which may be important to help reduce missed opportunities related to diagnosis and treatment for patients with CA. With healthcare provider interest in improving the care of patients with CA, the development of an optimal care pathway for the condition may help reduce delays in diagnosis and treatment and thus enhance patient outcomes.
Keywords: cardiac amyloidosis, management, pathways, treatment
Introduction
Cardiac amyloidosis (CA) is a condition where there is extracellular deposition of amyloid fibrils in the myocardium. 1 The prevalence of the CA varies considerably depending on the population studied: rates of CA range from 0.2% of patients undergoing bone scans for non-cardiac reasons to 43% of people aged 75 years or older at autopsy. 2 It is an underdiagnosed disease entity that may be found incidentally or more often cause cardiac symptoms such as dyspnoea, palpitations, chest pain, presyncope, syncope and features of heart failure or systemic symptoms such as dyspepsia, nausea, constipation and symptoms related to neuropathy. 3 CA is a restrictive cardiomyopathy which is broadly classified according to the type of amyloid protein either as light-chain CA (AL-CA) or transthyretin CA (ATTR-CA), which can be further subdivided into its hereditary or wild-type forms. Both forms tend to lead to progressive heart failure and are associated with significantly reduced survival. 4 A clinical challenge is that many patients experience diagnostic delay and by the time diagnosis is made quality of life is often poor. 5
Diagnosis of CA
Once a patient is referred for clinical assessment there are many features which may prompt suspicion for a diagnosis of CA. AL amyloidosis may clinically manifest as renal dysfunction, proteinuria, cardiomyopathy, hepatic dysfunction, orthostatic hypotension, peripheral neuropathy, gastrointestinal abnormalities, hypothyroidism, macroglossia and periorbital purpura. 6 ATTR amyloidosis may present with cardiomyopathy, orthostatic hypotension, peripheral neuropathy, carpal tunnel syndrome, lumbar spinal stenosis, gastrointestinal abnormalities, spontaneous biceps tendon rupture and ocular floaters. 6 The diagnostic algorithm for the diagnosis of CA which calls for suspicion of the diagnosis if the left ventricular wall thickness is greater than 12 mm alongside one or more ‘red flags’ which could be a clinical symptom or sign such as peripheral polyneuropathy or skin bruising, a diagnosis such as bilateral carpal tunnel syndrome, abnormal elevations of plasma troponin levels, finding on imaging such as subendocardial or transmural late gadolinium enhancement or extracellular volume on cardiac magnetic resonance (CMR) imaging, electrocardiographic findings such as reduced QRS voltage to mass ratio, atrioventricular (AV) conduction disease and a possible family history.7,8 Confirmation of the diagnosis can then be made using the non-invasive algorithm for CA of ATTR type (established when there is grade 2 or 3 uptake at 3,3-diphosphono-1,2-propanodicarboxylic acid (DPD) scintigraphy in association with negative free light chains and negative serum and urine immunofixation and echocardiographic and/or CMR imaging supportive of CA) or via invasive methods for all amyloid types with cardiac biopsy that is positive for amyloid or extracardiac biopsy that is positive for amyloid and echocardiographic and/or CMR imaging supportive of CA.7,9 Other key findings from investigations that may point to a diagnosis of CA include low-voltage QRS (more typical for AL), poor R-wave progression, pathological ‘pseudo’ Q-waves, atrial fibrillation, AV conduction abnormalities, bundle branch block and QT prolongation on electrocardiogram; and echocardiographic findings of unexplained left ventricular hypertrophy, atrial dilatation, right ventricular free wall hypertrophy, thickened aortic valve and septum, diastolic left ventricular dysfunction, abnormal left ventricular global longitudinal strain with apical sparing and low-flow, low-gradient aortic stenosis. 6 In addition, patients may have elevated serum cardiac biomarkers (NT-proBNP and troponin) which are helpful with staging and prognosis as well as to guide response to therapy. 10 Within ATTR-CA there is the further division of hereditary-type ATTR and wild-type ATTR, based on genetic testing which dependent on the TTR variant determines the phenotype and has implications for treatment and prognosis. 11 Once the diagnosis is made, there are further prognostic staging scores that then can be applied for AL amyloidosis12,13 and ATTR amyloidosis.14,15
Diagnostic delay in CA
Diagnostic delay in patients with CA has been highlighted previously.5,8,16 Historically, ATTR-CA was under-recognized with a typical patient attending hospital a median of 17 times in the 3 years prior to diagnosis. There is emerging evidence that the diagnosis is now being made earlier 16 helped by improved clinician awareness following the release of recent guidelines prompting consideration of the ATTR-CA in patients with aortic stenosis, heart failure with preserved ejection fraction and in those with a history of carpal tunnel syndrome, lumbar spinal stenosis, biceps tendon rupture and/or autonomic or sensory polyneuropathy. 17 Although in the United Kingdom, the median duration of symptoms before diagnosis has fallen from 36 months between 2002 and 2006 to 12 months between 2017 and 2021, there is still room for improvement. 16 An important factor permitting early diagnosis of ATTR-CA is the ability to access 99mtechnetium-labelled bone radiotracer scintigraphy which has a high sensitivity and specificity when this result is integrated with the non-biopsy diagnostic algorithm.9,18
Treatment of AL-CA
The management of AL-CA involves risk stratification and treatment with chemotherapy agents and/or autologous stem cell transplantation together with supportive therapies. 19 Agents such as bortezomib, melphalan, cyclophosphamide and dexamethasone may be used and supportive therapy includes salt restriction, diuretics, fitted stockings and midodrine for hypotension, pacemaker insertion and defibrillators for recurrent arrhythmias, gabapentin or pregabalin for neuropathic pain and nutritional support for adequate caloric intake. 14 While cardiac transplantation was once considered to be contraindicated due to high morbidity and mortality rates, the increased therapeutic options for amyloidosis and improved pre-transplantation screening practices has led to improvements in transplant outcomes over the past 10–15 years.20,21
Treatment of ATTR-CA
Over the last few years, there has been significant interest in the early diagnosis of CA because new treatments have transformed this once incurable disease to a potentially treatable disease. 4 For many years, liver or combined heart and liver transplantation were the only available treatments for patients with mutations causing ATTR and CA but now there are several pharmacological agents available or in the process of being tested. 22 There are three broad classes of TTR-ocused treatment, which can be classified as TTR stabilizers, TTR knockdown therapies and TTR removal therapies.
TTR stabilizers
Tafamidis is a drug which binds to transthyretin which prevents tetramer dissociation and amyloidogenesis. In the ATTR-ACT double-blind placebo controlled randomized controlled trial of 441 patients with transthyretin amyloid cardiomyopathy, tafamidis treatment was associated with lower all-cause mortality (29.5% versus 42.9%) compared to placebo at 30 months follow-up. 23 At longer-term follow-up (median 59 months), there were fewer deaths in the group on continuous tafamidis compared to placebo (44.9% versus 62.7%). 24 In real-world settings, an evaluation of 107 patients with ATTR with a median age of 84 years and 59% receiving tafamidis reported significantly higher median survival with treatment compared to no treatment (median survival 6.70 years versus 1.43 years, p < 0.001). 25 Another propensity match retrospective observational study using the TriNetX research network found that tafamidis therapy was associated with approximately 38% reduction in both heart failure exacerbation and all-cause mortality. 26 Changes in peak VO2 and baseline minute ventilation/carbon dioxide production slope in patients with CA after initiation of tafamidis has been reported and those patients with stable or improved peak VO2 with therapy showed more pronounced improvements in physical performance which highlights the importance of treatment with less advanced disease at baseline and the need for early diagnosis. 27 Despite the evidence from trials and real-world settings, the annual cost of tafamidis is approximately $250,000/year and a cost-effectiveness analysis shows that making tafamidis affordable will require a 92.6% reduction in its listed price. 28 As a result, in its initial review, the National Institute for Clinical Excellence (NICE) in the United Kingdom did not recommend the use of tafamidis for treating transthyretin amyloidosis with cardiomyopathy. 29 Acoramidis, is another oral TTR stabilizer which has been shown to be safe and effective for ATTR amyloid cardiomyopathy. The encouraging results of the phase III ATTRibute-CM trial were recently presented at a hotline European Society of Cardiology (ESC) session but are not yet published.30,31
TTR knockdown therapy
TTR knockdown therapy aims to reduce production of transthyretin in the liver and includes several drugs such as Patisiran, Inotersen, Vutrisiran, Eplontersen and NTLA-2001. Patisiran is a small interfering RNA (siRNA) drug which blocks TTR synthesis and in the APOLLO randomized control trial of 225 patients, was associated with significant improvements in quality of life, walking, nutritional status and activities of daily living. 32 Inotersen is an antisense oligonucleotide which has been shown to slow disease progression and help maintain quality of life scores in patients with hereditary ATTR polyneuropathy. 33 Eplontersen has also been shown in a phase III trial to improve neurological function in association with a reduction in level of TTR protein. 34 Vutrisiran, another RNA drug of the same class as Patisiran (second generation siRNA) has been shown to demonstrate similar benefits and has the advantage of being given subcutaneously only once every 3 months. 35 Finally, novel CRISPR-Cas9 in vivo gene technology has provided a modular platform to develop therapies such as NTLA-2001, designed to inactivate the TTR gene and thereby reduce production of the transthyretin protein. NTLA-2001 was tested in six patients with hereditary ATTR amyloidosis and achieved a greater than 90% sustained reduction in circulating TTR protein concentration through targeted knockout of TTR and a phase III trial is now planned. 36
TTR removal therapy
Amyloid removal therapy is the third class of drugs for ATTR-CA. A phase I, double-blind trial evaluated NI006 a recombinant human anti-ATTR antibody for the treatment of patients with ATTR cardiomyopathy and heart failure. 37 Treatment had an acceptable safety profile and appeared to be associated with a reduction in surrogate markers of TTR cardiac amyloid such as extracellular volume on CMR imaging.
There is significant interest in recent years in therapeutics for ATTR CA, but early diagnosis is important as there may only be a window of opportunity where patients are suitable for treatment after which therapies may be less effective.
Importance of pathways in CA
The concepts of patient pathways and missed opportunities can be applied to patients with CA. Patient pathways are the series of clinically relevant events that take place over time. 38 The key difference between the patient and care pathway is the patient decision making that is present in the patient pathway but not factored into the care pathway, and starts when the patient is seen by a healthcare professional. 39 As demonstrated previously in heart failure, the missed opportunities may relate to diagnosis and treatment.40,41 In general, as shown in Figure 1 the ideal scenario for patients with symptoms who are not incidentally found to have CA is that they present to healthcare professionals and the diagnosis is suspected and confirmed and appropriate treatment is started (Figure 1).
Figure 1.

Ideal pathway for patients with underlying cardiac amyloidosis.
In CA, it is important that the patients present to healthcare professionals for there to be a chance of the diagnosis being made and the initiation of appropriate treatment. Some of the real-world realities that arise are shown in Figure 2. Patients are in the community and they or may not be aware of their symptoms and their significance. For patients with early symptoms of cardiac dysfunction such as shortness of breath, orthopnoea or mild leg swelling, they may not present for evaluation and ignore these symptoms. This is notable as the missed opportunity relates to patient decision making rather than delays to diagnosis because of misdiagnosis related to clinical professional decision making. The patients with these early symptoms may also be seen by professionals for other reasons such as other co-existing medical or surgical problems and there could be an incidental opportunity at suspecting cardiac dysfunction and amyloidosis. It is also variable who the patient decides to present to, and it could be a general practitioner, emergency department or other healthcare professional in the community and they will have variable experience, expertise and understanding on how to manage a patient with underlying CA.
Figure 2.

Realities that arise in real-world settings which result in deviation from ideal pathway.
The need to consider clinical pathways in the diagnosis of ATTR cardiomyopathy (ATTR-CM) has been described in several studies. Tini et al. 42 described the diagnostic pathways leading to the diagnosis of ATTRwt-CM in a retrospective multicentre Italian study which included patients who had initially been referred with heart failure or a suspicion of hypertrophic cardiomyopathy, as well as patients presenting with incidental findings. 42 These types of studies are important as they provide a framework for constructing clinical pathway decision tools that can be used to potentially aid cardiologists in making an early diagnosis of ATTR-CM. 43 It has been suggested that implementation of a clinical amyloidosis pathway can serve to increase physician awareness leading to decreases in diagnostic delay. 44 Pathways are important to diagnosis and need to take into account the complex interplay between racial and genetic differences, comorbidities and health disparities which can all influence the presentation of patients with ATTR-CM. 45 The recent expert consensus decision pathway produced by the American College of Cardiology suggests that the subtleties in the history of presentation of patients with CA are important to recognize, and that an early suspicion and referral to skilled subspecialists requires attention as misdiagnosis may have catastrophic consequences for patients. 46 Ideally, there should be adoption of an accurate diagnostic algorithm, 9 timely identification of multisystem involvement with appropriate multidisciplinary collaboration, and disease management strategies to optimize both survival and quality of life outcomes in patients with amyloidosis. 47
A treatment algorithm would be ideal for patients diagnosed with CA, but the reality is that the delivery of disease modifying treatments (i.e. TTR-directed therapy) will differ depending on the level of resource and services available specific to each country. As a result, although it is possible to refer to a diagnostic algorithm, 9 a treatment pathway needs to be tailored to the local setting. For example, in the United Kingdom at present tafamidis is not available for use in patients with ATTR-CM although the drug is in the process of undergoing a re-evaluation by the NICE. However, there are siRNA-directed therapies already approved for use in patients with hereditary ATTR amyloidosis and nerve involvement. 47 The National Amyloidosis Centre is currently commissioned by NHS National Specialised Services and funded by the Department of Health to provide a diagnostic and management advice service for the entire UK’s national caseload of patients with amyloidosis and related disorders. Patients with CA may or may not be referred from local clinicians for input to this service depending on the awareness of the individual clinician, their clinical judgement and the patient’s willingness or ability to travel. It is possible that patients with mild symptoms or those in more advanced stages of disease may not be referred. Patients with CA may or may not be referred from local clinicians for input to this service depending on the awareness of the individual clinician, their clinical judgement and the patient’s willingness or ability to travel. It is possible that patients with mild symptoms or those in more advanced stages of disease may not be referred. To address these barriers, there needs to be agreement between non-specialists and specialist centres on how they expect patients to be identified and where they are managed, together with agreement regarding patient monitoring and what types of treatments can be instigated. Due to increasing demand and a surge in the number of patients being referred, there is now an onus on the NHS to roll out the development of a UK network perhaps in the form of a hub-and-spoke model.
Missed opportunities in CA
We introduce the concept of missed opportunities as a mechanism to learn from real-world cases through evaluation of the management of patients so that clinicians may be able to learn how care can be improved. As the pathways may involve multiple clinicians there may be both upstream (non-specialist)-related delay or misdiagnosis contributing to missed opportunities, together with those further downstream related to the activities of the specialist (delay to diagnosis or delay to treatment). We believe the risk-benefit of treatments need to be individualized to the patient.
Some of the missed opportunities related to patients with underlying CA are shown in Figure 3. A key question related to the likelihood of identification of the underlying problems depends on the stage and pattern of disease but also which professional the patient presents to. Early symptoms and signs may be non-specific but later progression may result in more severe symptoms and more obvious signs of acute decompensated heart failure in CA, together with changes on cardiac imaging such as left ventricular hypertrophy with preserved ejection fraction and biatrial dilatation. 48 In the community, it may be challenging without multiple consultations for a general practitioner to suspect a diagnosis which merits confirmation by a secondary/tertiary care specialist with relevant expertise. Clinical encounters that take place in the emergency department may lead to diagnosis quicker because there is rapid access to tests such as bloods which may pick up underlying problems such as anaemia, hypercalcaemia, renal dysfunction or elevated NT-pro brain natriuretic peptide. There are also opportunities for incidental identification if patients with limited symptoms, but may have clinical signs of underlying CA such as incidental hydroxymethylene diphosphonate (HDP) myocardial tracer uptake in a patient with prostatic carcinoma undergoing a bone scan. For the patient to be identified incidentally, it is important that healthcare professionals are aware of the condition and the potential complications and treatments available that merit further referral for specialist review. Once, a patient presents to a healthcare professional for symptoms which may reflect underlying CA, they need to have access to the tests which may be helpful in suspecting the diagnosis. At an early stage, there should be access to specialists who can diagnose CA. For the screening and diagnosis of CA, basic investigations such as blood tests and echocardiography and more specialist investigations such as CMR imaging, DPD scans and cardiac biopsy need to be accessible locally. Moreover, to have access to specialist treatment, there may be the need for referral pathways so that patients in rural areas or those seen in centres where there is not the level of expertise required can refer patients for specialist input. Once patients have been reviewed by a clinician it is not uncommon for patients to be initially misdiagnosed with CA and there may be delay to eventual diagnosis. 49 In particular, it is vitally important for clinicians to determine the type of amyloid as there are very different treatment approaches and recognition of the potential of dual amyloid types is highly clinically relevant. 50 Diagnosis is important not only to attempt to educate the patient and modify the progression but also to prevent cardiac complications and manage potential comorbidities such as arrhythmias, heart failure, thromboembolism and aortic stenosis.
Figure 3.

Common presentations and the sources of referrals to a specialist with an interest in cardiac amyloidosis.
AF, atrial fibrillation; CMR, cardiac magnetic resonance; HFpEF, heart failure with preserved ejection fraction; MGUS, monoclonal gammopathy of unknown significance; PPM, permanent pacemaker; TAVI, transcatheter aortic valve replacement.
Reducing missed opportunities in CA
A key consideration is whether the missed opportunity to make an early diagnosis could be avoided. The only way to influence patient decision-making is through education and public health measures to encourage the public to seek help and not ignore symptoms. Once patients seek an opinion from a healthcare professional, there is an expectation that the clinician should suspect a problem, but clinician awareness is determined by education of healthcare professionals. Screening tests that are readily available such as electrocardiogram, abnormal NT-proBNP or troponin and echocardiographic abnormalities should not be ignored. For example, a patients may have vague chest pains and found to have modestly elevated troponin but once their coronaries are examined and found to be normal, they may be discharged without investigation. Similarly, a patient with abnormal ECG such as high-grade AV block may present with syncope and receive a pacemaker but not checked for underlying amyloidosis. In addition, it is important for clinicians to be aware of the relevance of incidental DPD uptake on bone scintigraphy in prostate cancer. 51 To reduce diagnosis-related missed opportunities, education regarding CA may be necessary for echocardiographers who report scans and doctors to appropriately interpret bloods, ECG and imaging tests. Examples of common presentations and the sources of referrals to a specialist with an interest in CA are shown Figure 3. There may also be treatment-related missed opportunities in CA. The key consideration is whether the drug is available and how patients can be assessed if they are eligible for it. This is important as it may not be an avoidable missed opportunity if the health service does not provide the treatment, or the patient or healthcare service cannot afford to provide it as it is one of the most expensive drugs. Figure 4 depicts the sources of potential missed opportunities in CA.
Figure 4.
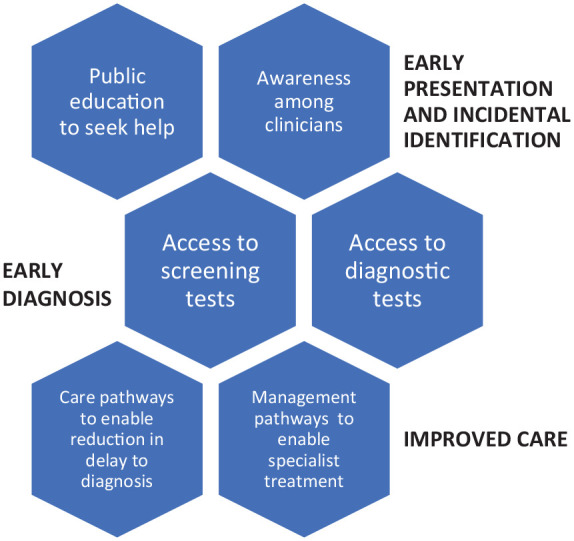
Missed opportunities in cardiac amyloidosis.
Care pathway development is key to providing high-quality local services. This may involve a specialist in the area taking an interest in CA so that referrals can be made to this specialist facilitating rapid access to investigations to confirm the diagnosis. Such services and pathways require investment of resources, so it is important that the burden of CA is significant in the location. In the United Kingdom, the NHS long-term plan highlights the importance of improving access for minority ethnicity, equality and diversity. 52 For many of the patients with ATTR, they may be old, frail and those with V122I mutation are often of Afro-Caribbean ethnicity. There may also be companies who develop novel drug therapies to help establish such pathways as there may be missed opportunities for their products to be utilized. 53 In addition, collecting data on the care of patients with CA and examining whether care could have been improved retrospectively has value on a local level to learn from past experiences. One means of doing this is the care pathway audit which compared real-world activities for patients to a defined standard that is expected. 36
Limitations
By the very nature of this article being a review, it does not provide new data. Given its authorship, it may also be biased towards a UK perspective. Nonetheless, this review has been conceived by researchers with experience in specialist amyloidosis care and patient and care pathways; its concept can be generalized and should prove helpful for planning further studies to better understand the real-world global activities related to the diagnosis and management of patients with CA. More studies are needed from around the world that explore the patient and care pathways for patients with CA.
Conclusion
ATTR-CA was once considered a condition with limited treatment options beyond supportive therapy. There has been increasing interest in CA because of the many recent novel therapies which have been or are currently being developed. For patients to benefit most from treatment, early diagnosis is vital, and this relies on patients seeking opinion from healthcare professionals and for those clinicians to suspect the diagnosis. There are still unacceptable delays to diagnosis, which results in progression of the disease and adverse patient outcomes. Patient pathways are useful ways to consider how clinically relevant events over time may have an influence on eventual outcomes and receipt of treatment. This approach enables consideration of missed opportunities for early diagnosis and timely initiation of appropriate therapy.
Acknowledgments
None.
Footnotes
ORCID iD: Chun Shing Kwok  https://orcid.org/0000-0001-7047-1586
https://orcid.org/0000-0001-7047-1586
Contributor Information
Chun Shing Kwok, Department of Cardiology, University Hospitals of North Midlands NHS Trust, Newcastle Rd, Stoke-on-Trent ST4 6QG, UK.
William E. Moody, Department of Cardiology, University Hospitals of Birmingham NHS Trust, Birmingham, UK Chun Shing Kwok is now affiliated to Department of Cardiology, University Hospitals of North Midlands NHS Trust, Stoke-on-Trent, UK; William E. Moody is also affiliated to University of Birmingham, Birmingham, UK.
Declarations
Ethical approval and consent to participant: Not applicable.
Consent for publication: Not applicable.
Author contributions: Chun Shing Kwok: Conceptualization; Writing – original draft; Writing – review & editing.
William Moody: Supervision; Writing – review & editing.
Funding: The authors received no financial support for the research, authorship, and/or publication of this article.
The authors declare that there is no conflict of interest.
Availability of data and materials: No original data was used for this review.
References
- 1. Bloom MW, Gorevic PD. Cardiac amyloidosis. Ann Intern Med 2023; 176: ITC33–ITC48. [DOI] [PubMed] [Google Scholar]
- 2. Rossi M, Varra GG, Porcari A, et al. Re-definition of the epidemiology of cardiac amyloidosis. Biomedicine 2022; 10: 1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sham P, Ahmed I. Cardiac amyloidosis. Treasure Island, FL: StatPearls; [Internet], 2023. [Google Scholar]
- 4. Kwok CS, Farzaneh-Far A, Mamas MA. Red flags in cardiac amyloidosis. Eur J Prev Cardiol 2019; 27: 1804–1805. [DOI] [PubMed] [Google Scholar]
- 5. Lane T, Fontana M, Martinez-Naharro A, et al. Natural history, quality of life, and outcome in cardiac transthyretin amyloidosis. Circulation 2019; 140: 16–26. [DOI] [PubMed] [Google Scholar]
- 6. Papingiotis G, Basmpana L, Farmakis D. Cardiac amyloidosis: epidemiology, diagnosis and therapy. E-J Cardiol Pract 2021; 19. https://www.escardio.org/Journals/E-Journal-of-Cardiology-Practice/Volume-19/cardiac-amyloidosis-epidemiology-diagnosis-and-therapy [Google Scholar]
- 7. Garcia-Pavia P, Rapezzi C, Adler Y, et al. Diagnosis and treatment of cardiac amyloidosis: a position statement of the ESC Working Group on Myocardial and Pericardial Disease. Eur Heart J 2021; 42: 1554–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vergaro G, Aimo A, Barison A, et al. Key to early diagnosis of cardiac amyloidosis: red flags from clinical, laboratory and imaging findings. Eur J Prev Cardiol 2020; 27: 1806–1815. [DOI] [PubMed] [Google Scholar]
- 9. Gillmore JD, Maurer MS, Falk RH, et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation 2016; 133: 2404–2412. [DOI] [PubMed] [Google Scholar]
- 10. Pregenzer-Wenzler A, Abraham J, Barrell K, et al. Utility of biomarkers in cardiac amyloidosis. JACC Heart Fail 2020; 8: 701–711. [DOI] [PubMed] [Google Scholar]
- 11. Alreshq R, Ruberg F. Clinical approach to genetic testing in amyloid cardiomyopathy: from mechanism to effective therapies. Curr Opin Cardiol 2022; 36: 309–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kumar S, Dipenzieri A, Lacy MQ, et al. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J Clin Oncol 2012; 30: 989–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lilleness B, Ruberg FL, Mussinelli R, et al. Development and validation of a survival staging system incorporating BNP in patients with light chain amyloidosis. Blood 2019; 133: 215–223. [DOI] [PubMed] [Google Scholar]
- 14. Grogan M, Scott CG, Kyle RA, et al. Natural history of wild-type transthyretin cardiac amyloidosis and risk stratification using a novel staging system. J Am Coll Cardiol 2016; 68: 1014–1020. [DOI] [PubMed] [Google Scholar]
- 15. Gillmore JD, Damy T, Fontana M, et al. A new staging system for cardiac transthyretin amyloidosis. Eur Heart J 2018; 39: 2799–2806. [DOI] [PubMed] [Google Scholar]
- 16. Ioannou A, Patel RK, Razvi Y, et al. Impact of earlier diagnosis in cardiac ATTR amyloidosis over the course of 20 years. Circulation 2022;146:1657–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kittleson MM, Maurer MS, Ambardekar AV, et al. Cardiac amyloidosis: evolving diagnosis and management: a scientific statement from the American Heart Association. Circulation 2020; 142: e7–e22. [DOI] [PubMed] [Google Scholar]
- 18. Tahara N, Lairez O, Endo J, et al. 99m technetium-pyrophosphate scintigraphy: a practical guide for early diagnosis of transthyretin amyloid cardiomyopathy. ESC Heart Fail 2022; 9: 251–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Palladini G, Milani P, Merlini G. Management of AL amyloidosis in 2020. Blood 2020; 136: 2620–2627. [DOI] [PubMed] [Google Scholar]
- 20. Witteles RM. Cardiac transplantation and mechanical circulatory support in amyloidosis: JACC CardioOncol 2021; 3: 516–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Razvi Y, Porcari A, Di Nora C, et al. Cardiac transplantation in transthyretin amyloid cardiomyopathy: outcomes from three decades of tertiary center experience. Front Cardiovasc Med 2023; 9: 1075806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Emdin M, Aimo A, Rapezzi C, et al. Treatment of cardiac transthyretin amyloidosis: an update. Eur Heart J 2019; 40: 3699–3706. [DOI] [PubMed] [Google Scholar]
- 23. Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med 2018; 379: 1007–1016. [DOI] [PubMed] [Google Scholar]
- 24. Elliot P, Drachman BM, Gottlieb SS, et al. Long-term survival with tafamidis in patients with transthyretin amyloid cardiomyopathy. Circ Heart Fail 2022; 15: e008193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hussain K, Macrinici V, Wathen L, et al. Impact of tafamidis on survival in a real-world community-based cohort. Curr Probl Cardiol 2022; 47: 101358. [DOI] [PubMed] [Google Scholar]
- 26. Ghoneem A, Bhatti AW, Khadke S, et al. Real-world efficacy of tafamidis in patients with transthyretin amyloidosis and heart failure. Curr Prob Cardiol 2023; 48: 101667. [DOI] [PubMed] [Google Scholar]
- 27. Eslam RB, Ozturk B, Rettl R, et al. Impact of tafamidis and optimal background treatment on physical performance in patients with transthyretin cardiac amyloid cardiomyopathy. Circ Heart Fail 2022; 15: e008381. [DOI] [PubMed] [Google Scholar]
- 28. Mallus MT, Rizzello V. Treatment of amyloidosis: present and future. Eur Heart J Suppl 2023; 25: B99–B103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. National Institute of Clinical Excellence. Tafamidis for treating transthyretin amyloidosis with cardiomyopathy. Technology appraisal guidance [TA696], 2021. [Google Scholar]
- 30. Judge DP, Heitner SB, Falk RH, et al. Transthyretin stabilization by AG10 in symptomatic transthyretin amyloid cardiomyopathy. J Am Coll Cardiol 2019; 74: 285–295. [DOI] [PubMed] [Google Scholar]
- 31. Gillmore J. ATTRibute-CM: acoramidis (AG10) in patients with transthyretin amyloid cardiomyopathy. ESC Congress 2023, https://esc365.escardio.org/ESC-Congress/sessions/7125. (accessed 4 December 2023).
- 32. Adams D, Gonzalez-Duarte A, O’Riordan WD, et al. Patrisian, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med 2018; 379: 11–21. [DOI] [PubMed] [Google Scholar]
- 33. Brannagan TH, Wang AK, Coelho T, et al. Early data on long-term efficacy and safety of Inotersen in patients with hereditary transthyretin amyloidosis: a 2-year update from the open-label extension of the NEURO-TTR trial. Eur J Neurol 2020; 27: 1374–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Astrazeneca. Eplontersen met co-primary and secondary endpoints in interim analysis of the NEURO-TTRansform Phase III trial for hereditary transthyretin-mediated amyloid polyneuropathy (ATTRv-PN, astrazeneca.com (2022, accessed 4 December 2023). [Google Scholar]
- 35. Adams D, Tournev IL, Taylor MS, et al. Efficacy and safety of vutrisiran for patients with hereditary transthyretin-mediated amyloidosis with polyneuropathy: a randomized clinical trial. Amyloid 2023; 30: 1–9. [DOI] [PubMed] [Google Scholar]
- 36. Gillmore JD, Gane E, Taubel J, et al. CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. N Engl J Med 2021; 385: 493–502. [DOI] [PubMed] [Google Scholar]
- 37. Garcia-Pavia P, Aus dem Siepen F, Donal E, et al. Phase 1 trial of antibody NI006 for depletion of cardiac transthyretin amyloid. N Engl J Med 2023; 389: 239–250. [DOI] [PubMed] [Google Scholar]
- 38. Kwok CS, Muntean EA, Mallen CD. The patient pathway review: a new method of evaluating clinical practices to understand the complexities of real-world care. Crit Pathw Cardiol 2021; 20: 213–219. [DOI] [PubMed] [Google Scholar]
- 39. Kwok CS, Water D, Phan T, et al. Should audits consider the care pathway model? A new approach to benchmarking real-world activities. Healthcare 2022; 10: 1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kwok CS, Burke H, McDermott S, et al. Missed opportunities in the diagnosis of heart failure: evaluation of pathways to determine sources of delay to specialist evaluation. Curr Heart Fail Rep 2022; 19: 247–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kwok CS, Satchithananda D, Ahmed FZ, et al. A critical evaluation of patient pathways and missed opportunities in treatment for heart failure. J Cardiovasc Dev Dis 2022; 9: 455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tini G, Milani P, Zampieri M, et al. Diagnostic pathways to wild-type transthyretin amyloid cardiomyopathy: a multicentre network study. Eur J Heart Fail 2023; 25: 845–853. [DOI] [PubMed] [Google Scholar]
- 43. Muller SA, Oerlemans MIFJ. Implementing clinical pathways to enable early diagnosis and treatment of wild-type transthyretin amyloid cardiomyopathy. Eur J Heart Fail 2023; 25; 854–856. [DOI] [PubMed] [Google Scholar]
- 44. Muller SA, van der Meer MG, Oerlemans MIFJ. Racial and genetic differences: possible health disparities in transthyretin amyloid cardiomyopathy patients? JACC Heart Fail 2023; 11: 130. [DOI] [PubMed] [Google Scholar]
- 45. Martyn T, Saef J, Dey AR, et al. Racial and genetic differences in presentation of transthyretin amyloid cardiomyopathy with impaired left ventricular function. JACC Heart Fail 2022;10:689–691. [DOI] [PubMed] [Google Scholar]
- 46. Kittleson MM, Ruberg FL, Ambardekar AV, et al. 2023 ACC expert consensus decision pathway on comprehensive multidisciplinary care for the patient with cardiac amyloidosis: a report of the American College of Cardiology Solution Set Oversight Committee. J Am Coll Cardiol 2023; 81: 1076–1126. [DOI] [PubMed] [Google Scholar]
- 47. Porcari A, Fontana M, Gillmore JD. Transthyretin cardiac amyloidosis. Cardiovasc Res 2022; 118: 3517–3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Moody WE, Turvey-Haigh L, Knight D, et al. British Society of Echocardiography guideline for the transthoracic echocardiographic assessment of cardiac amyloidosis. Echo Res Pract 2023; 10: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lousada I, Maurer MS, Warner MT, et al. Amyloidosis research consortium cardiac amyloidosis survey: results from the patients with AL and ATTR amyloidosis and their caregivers. J Cardiac Fail 2019; 25: S69. [Google Scholar]
- 50. Hasib Sidiqi M, McPhail ED, Theis JD, et al. Two types of amyloidosis presenting in a single patient: a case series. Blood Cancer J 2019; 9: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Delaney FT, Dempsey P, Welaratne I, et al. Incidental cardiac uptake in bone scintigraphy: increased importance and association with cardiac amyloidosis. BJR Case Rep 2021; 7: 20200161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. The NHS Long Term Plan. Equality and health inequalities impact assessment – Annex 1, ehia-ltp-annex.pdf (england.nhs.uk) (2019, accessed 18 July 2023). [Google Scholar]
- 53. Kwok CS, Muntean EA, Foster W, et al. Patient pathways in cardiology: should pharmaceutical and medical device companies care? Crit Pathw Cardiol 2022; 21: 57–60. [DOI] [PubMed] [Google Scholar]