

Abstract
Applying rational design, we developed 17 kDa near-infrared (NIR-I) fluorescent protein, miRFP718nano, with emission peak at 718 nm. miRFP718nano structure revealed molecular basis of its red-shift. miRFP718nano has emission tail in short-wavelength infrared (SWIR) region (1000–1700 nm), allowing deep-penetrating off-peak fluorescence imaging in vivo. We demonstrated superiority of miRFP718nano-enabled SWIR imaging over NIR-I imaging of microbes in digestion system, mammalian cells in mammary gland, and NF-kB activity in inflammatory liver.
Keywords: NIR FP, iRFP, cyanobacterichrome, short-wavelength infrared, deep tissue imaging
Biological tissues have strong optical attenuation in the visible wavelength range (350–700 nm), due to the absorption of hemoglobin and melanin, as well as the tissue scattering, which fundamentally limits the imaging depth of high-resolution optical technologies. By contrast, shifting the optical wavelengths to the near-infrared (NIR) range can significantly improve optical imaging at larger depths1, 2. Recently, the second NIR window (NIR-II, 1000–1700 nm), so-called the short-wavelength infrared (SWIR) window, has gained tremendous interest by providing superior penetration depth, spatial resolution and image contrast in deep tissues, over the NIR-I window (700–1000 nm)3–5. Red-shifted NIR fluorescent proteins (FPs) are much needed for imaging applications6.
The state-of-the-art NIR FPs were engineered from bacterial phytochrome photoreceptors (BphPs)7. BphP-based FPs consist of PAS and GAF domains and have a relatively high molecular weight. We recently reported miRFP670nano protein, the first single-domain NIR FP developed from a GAF domain of cyanobacteriochrome (CBCR), and its improved variant miRFP670nano38, 9. Compared to the BphP-derived NIR FPs, miRFP670nanos are 2-fold smaller, naturally monomeric, and have higher stability in vitro and mammalian cells8, 9.
To develop a small red-shifted NIR FP miRFP718nano, we applied a structure-based rational design to miRFP670nano, followed by directed molecular evolution (Supplementary Note 1 and Supplementary Fig. 1, 2). We introduced point Cys mutation to bind BV chromophore via its C32 atom. In such chromophore, the double bond between C2 and C3 atoms is preserved and is engaged in conjugation resulting in the red-shifted emission.
With excitation and emission maxima at 690 nm and 718 nm, miRFP718nano exhibited substantially red-shifted fluorescence and absorbance spectra compared to miRFP670nano (Fig. 1a–c and Supplementary Table 1). Importantly, miRFP718nano has a long off-peak fluorescence emission tail that extends into the SWIR window. miRFP718nano fluorescence intensity above 1000 nm was 3-fold higher compared to miRFP670nano and about 2- and 1.5-fold higher than that of miRFP703 and miRFP709, respectively (Fig. 1d and Supplementary Table 1). Thus, the spectral properties of miRFP718nano make it a highly promising probe for SWIR imaging.
Figure 1. Properties of red-shifted miRFP718nano in comparison with parental miRFP670nano.
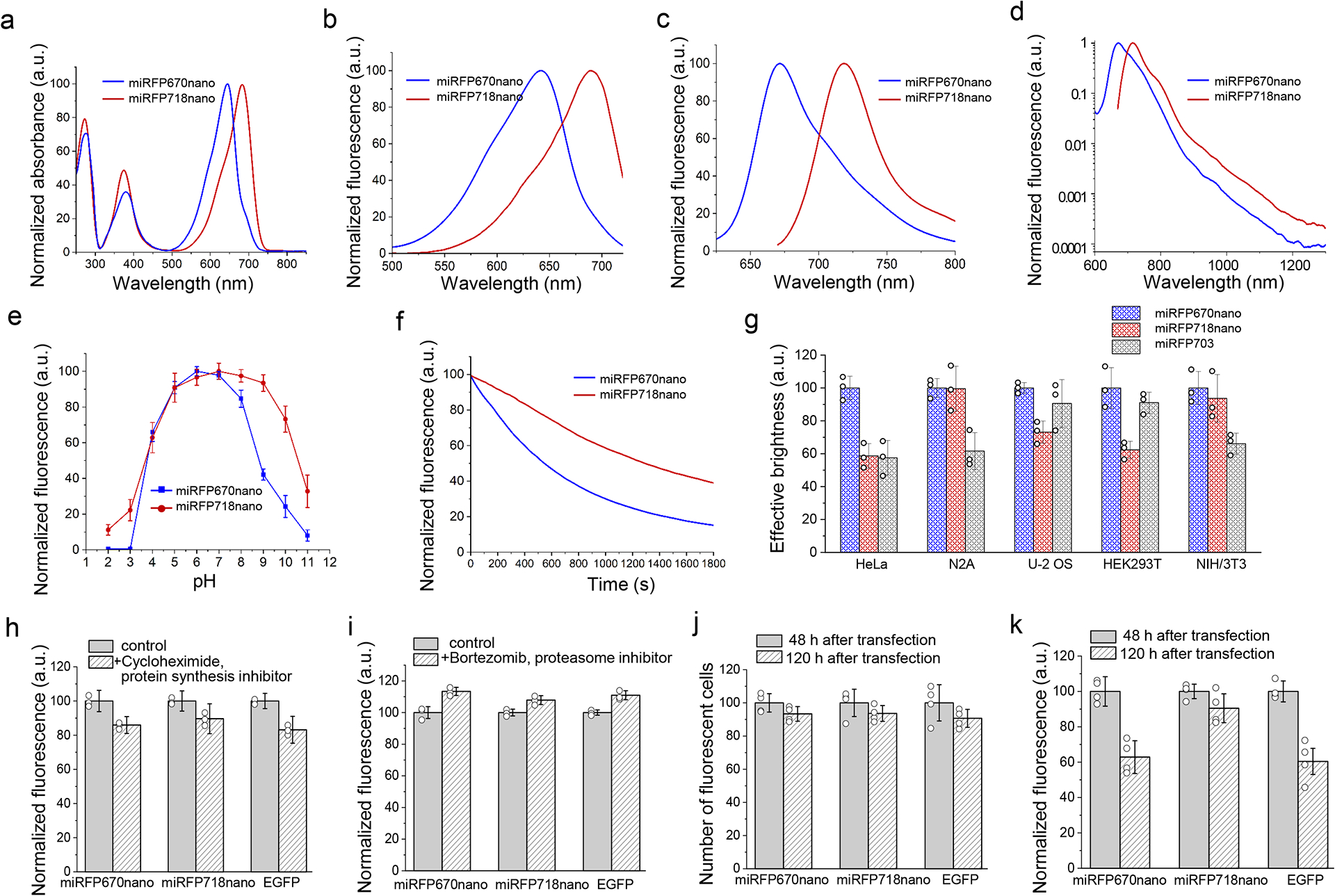
(a) Absorbance spectra. (b) Fluorescence excitation spectra (emission at 740 nm). (c, d) Fluorescence emission spectra (excitation at 580 nm for miRFP670nano and 650 nm for miRFP718nano). (e) pH dependencies of NIR fluorescence. Data are presented as mean values ± s.d. for n = 3 transfection experiments. In (a-e) spectroscopic measurements were performed in phosphate-buffered saline (PBS). (f) Photobleaching in live HeLa cells with 665/45 nm excitation filter. Light power density at the back aperture of the objective lens was 8.25 mW cm−2, resulting in 4.3 W cm−2 at the focal objective plane. Cells were incubated in a live cell imaging solution. (g) Cellular brightness of miRFP718nano, miRFP670nano and miRFP703 in live mammalian cells. Fluorescence was analyzed 72 h after transfection. The effective brightness of miRFP670nano was assumed to be 100% for each cell type. NIR fluorescence intensity was normalized to the excitation efficiency of each NIR FP by 640 nm laser, and to the emission spectrum of each FP in the emission filter. Gating was performed as shown in Supplementary Fig. 3. (h) Mean fluorescence intensity of HeLa cells transiently transfected with miRFP718nano, miRFP670nano and EGFP before and after 4 h of incubation with a protein synthesis inhibitor cycloheximide (20 μg/ml). Fluorescence was normalized to control conditions. (i) Mean fluorescence intensity of HeLa cells transiently transfected with miRFP718nano, miRFP670nano and EGFP before and after 4 h of incubation with an inhibitor of proteasome-dependent protein degradation bortezomib (10 μM). Fluorescence was normalized to control conditions. In (g, h, i) data are presented as mean values ± s.d. for n = 3 transfection experiments. (j) Mean fluorescence intensity of live HeLa cells transiently transfected with miRFP718nano miRFP670nano and EGFP 48 h and 120 h after transfection normalized to that at 48 h. (k) Number of FP expressing (fluorescent) HeLa cells transiently transfected with parental miRFP670nano, miRFP718nano or EGFP was calculated at 48 h and 120 h after transfection and normalized to that at 48 h. In (j, k) data are presented as mean values ± s.d. for n = 4 transfection experiments.
The fluorescence of miRFP718nano was stable over a wide pH range of 4.0–10.0. (Fig. 1e). miRFP718nano exhibited improved photostability, which was 2.3-fold higher than that of miRFP670nano and several-fold higher than the photostability of BphP-derived NIR FPs (Fig. 1f and Supplementary Table 1)10, 11.
In transiently transfected HeLa, N2a, U-2 OS, HEK293T and NIH/3T3 cells, expressing miRFP670nano, miRFP718nano and miRFP703, miRFP718nano outperformed BphP-derived miRFP709 by 1.3-fold and has brightness comparable with that of spectrally-close miRFP703 (Fig. 1g, Supplementary Fig. 3 and Supplementary Table 1). In addition to cell lines, primary cortical rat neurons transfected with miRFP718nano exhibited bright fluorescence (Supplementary Fig. 4).
The protein stability tests with cycloheximide, an inhibitor of translational elongation, and with bortezomib, an inhibitor of proteasome-dependent protein degradation, both showed that miRFP718nano similar to EGFP and miRFP670nano is highly stable in mammalian cells (Fig. 1h,i). The comparison of brightness and percentage of fluorescent cells 48 h and 120 h after transfection further confirmed that miRFP718nano had high protein stability (Fig. 1j,k). Similar to the miRFP670nano, in transiently transfected HeLa cells miRFP718nano fluorescence reached a maximum at 72 h after transfection (Supplementary Fig. 5).
miRFP718nano performed well as a fluorescent probe in various N- and C-terminal, as well as internally labeled fusion proteins (Supplementary Fig. 6). Co-expression of the miRFP670nano and miRFP718nano fusions, enabled the clear separation of miRFP670nano and miRFP718 fluorescence signals for two-color NIR imaging (Supplementary Fig. 7).
We calculated that the Förster radius for the miRFP670nano–miRFP718nano FRET pair is 7.9 nm. Further evaluation of miRFP718nano as a FRET acceptor for miRFP670nano in a biosensor for c-Jun N-terminal kinase (JNK) activity12 (Supplementary Fig. 8a) showed that miRFP670nano and miRFP718nano formed an efficient NIR FRET pair (Supplementary Fig. 8b,c), which, similarly to BphP-based NIR FPs6, 13–16, can be used to engineer various fully NIR biosensors.
To gain insights into the structural basis of the miRFP718nano properties, we determined its crystal structure at 1.7 Å resolution (Extended Data Fig. 1, Supplementary Note 2, Supplementary Tables 1 and 2). The RMS deviation between miRFP718nano and miRFP670nano8 superimposed by Cα atoms does not exceed 0.74 Å. Both proteins have a similar GAF fold with the N- and C- termini positioned close to each other (Extended Data Fig. 1a–e,g). This fold is similar to that of the GAF domains of BphP-based miRFP709 linked to the adjacent PAS domain (Extended Data Fig. 1c,f)17.
The crystal structure of miRFP718nano showed that its chromophore forms a covalent thioether bond between the C32 atom of BV ring A and the rationally introduced Cys57 (Extended Data Fig. 1b,e,j). In contrast, the BV chromophore of miRFP670nano is connected to the conserved Cys86 by the C31 atom (Extended Data Fig. 1a,d,k). The BV chromophore of miRFP718nano also has a chemical structure, featuring a double bond between C2 and C3 atoms, absent in parental miRFP670nano8, thus extending the chromophore conjugation system. This chromophore was never observed before in CBCR-derived NIR FPs.
To demonstrate SWIR imaging’s advantages over NIR-I imaging4, 18–20, we first characterized miRFP718nano’s SWIR performance on bacteria phantoms (Supplementary Fig. 9). NIR-I (750/40 nm emission) and SWIR (1050 nm long-pass emission) imaging were individually performed with the same excitation at 660 nm. SWIR imaging could better maintain the spatial resolution and image contrast in a scattering medium (Supplementary Fig. 10).
We then validated in vivo performance of miRFP718nano-enabled SWIR imaging by tracking miRFP718nano-expressing bacteria motility in the mouse digestion model21. Both NIR-I and SWIR images showed the migration of the bacteria (Supplementary Fig. 11a), but NIR-I images were blurred. By contrast, SWIR images had much more confined signals inside the stomach and along the intestine (Supplementary Fig. 11b). We observed an approximately 3-cm-long migration path within 60 min, consistent with previous reports22, 23.
We next evaluated SWIR imaging in mammalian cells, including miRFP670nano- and miRFP718nano-expressing HEK293T cells. Whereas both cell types had similar NIR-I fluorescence levels, miRFP718nano-expressing cells had 2-fold stronger SWIR fluorescence (Supplementary Fig. 12a). In NIR-I images, the spatial resolution degraded from 0.1 mm at 0-mm depth to 1.14 mm at 3-mm depth and 1.96 mm at 5-mm depth (Supplementary Fig. 12b). By contrast, in SWIR images, the spatial resolution was 0.4 mm at 3-mm depth and 0.8 mm at 5-mm depth, approximately 65% and 61% better than NIR-I images (Supplementary Fig. 12c).
Next, we compared miRFP718nano- and miRFP670nano3-expressing HEK293T cells (Supplementary Figs. 13a–c). miRFP718nano-expressing cells had 2.3-fold stronger SWIR signals than miRFP670nano3-expressing cells, as well as higher image contrast and less scattering-induced blurring (Supplementary Fig. 12a, Supplementary Fig. 13a). We have quantified SWIR and NIR-I emission ratio (Supplementary Fig. 13d). miRFP718 has the highest SWIR/NIR-I emission ratio, which is >3-fold as large as that of miRFP670nano and miRFP670nano3.
We next injected miRFP718nano- or miRFP670nano-expressing HEK293T cells into the mouse mammary glands at ~1.5 mm depth (Supplementary Fig. 14a, b). SWIR imaging had better detection sensitivity than NIR-I imaging for both types of cells (Supplementary Fig. 14c). We increased the exposure time for low cell numbers, which benefited SWIR imaging but not NIR-I imaging due to the strong NIR-I autofluorescence (Supplementary Fig. 14a, b). SWIR imaging of miRFP718nano-expressing HEK293T cells had the highest sensitivity under all conditions (Supplementary Fig. 14d). Even with the lowest cell number of 1×104, SWIR image of miRFP718nano-expressing cells had an SBR of ~3.7, which was three times better than NIR-I image.
To validate miRFP718nano for monitoring signaling pathways in vivo, we engineered a miRFP718nano-based NF-κB transcription reporter, a key regulator of inflammatory responses. After activation NF-κB binds to the consensus sequence on DNA (NF-κB response element)24. We constructed the inflammation reporter containing five copies of an NF-κB response element that drives the miRFP718nano transcription upon NF-κB activation (Fig. 2a).
Figure 2. miRFP718nano as a reporter of inflammation.
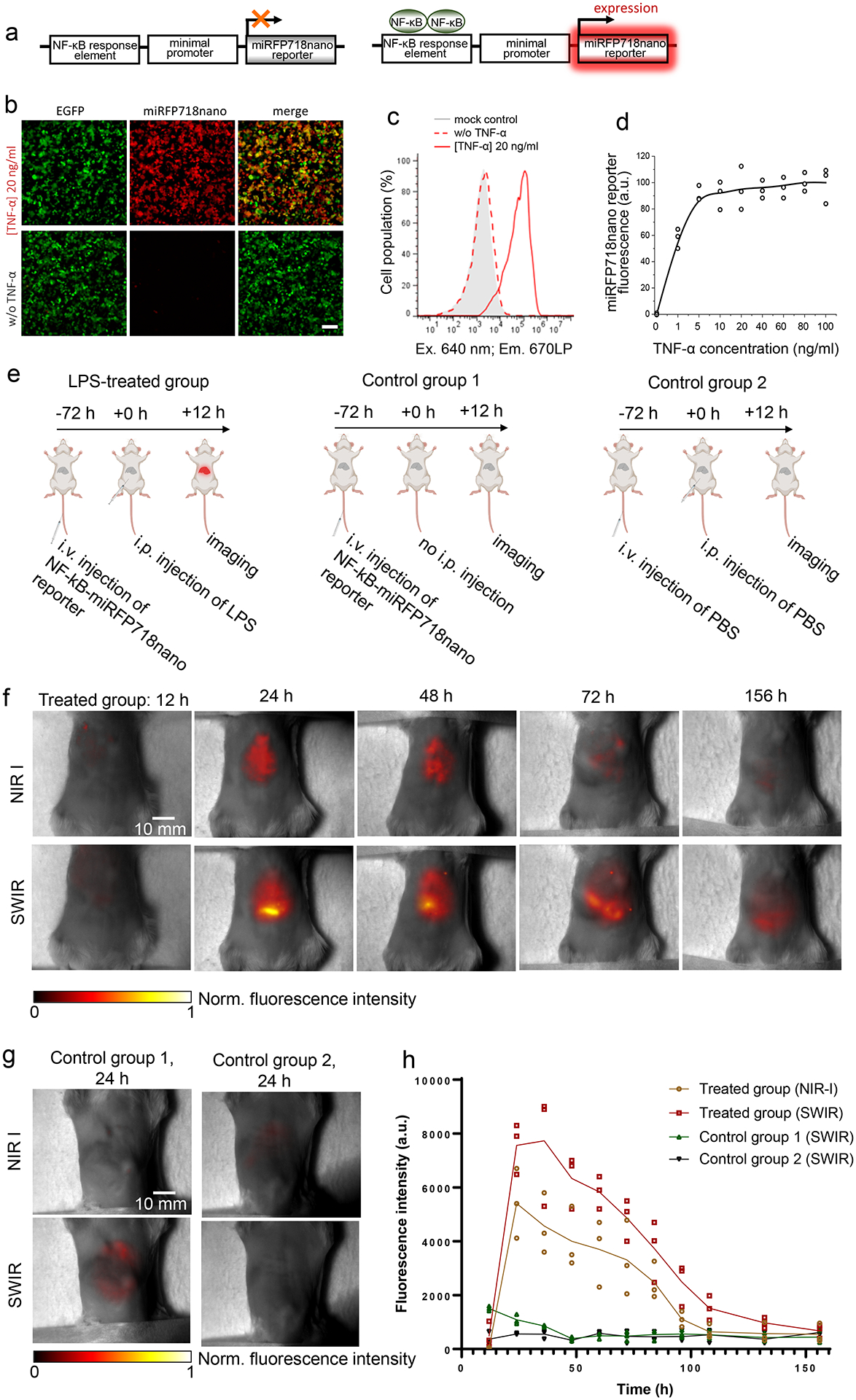
(a) Schematic representation of miRFP718nano-based reporter of inflammation. (b) HEK293T cells transfected with the miRFP718nano-based reporter of inflammation and cotransfected with pEGFP-N1 (1:10), 20 h after stimulation with TNF-α (20 ng/ml) (top row) or non-treated (bottom row). For imaging of miRFP718nano and EGFP, 685/20 nm excitation and 725/40 nm emission, and 485/20 nm excitation and 525/30 nm emission filters were used, respectively. Representative images of two experiments are shown. Scale bar, 100 μm. (c) TNF-α dose-response in HEK293T cell transfected with miRFP718nano reporter of inflammation, measured by flow cytometry. Data are presented as mean values ± s.d. for n = 3 transfection experiments. Gating was performed as shown in Supplementary Fig. 3. (d) Mean fluorescence intensity of HEK293T cells transiently transfected with miRFP718nano reporter of inflammation 20 h after stimulation with TNF-α (20 ng/ml). (e) The schematics of developing the mouse model injected with miRFP718nano reporter plasmid and LPS, as well as the two control models with only the plasmid or only PBS. (f) Representative NIR-I and SWIR images of the LPS-treated group at different time points after the LPS injection. (g) Representative NIR-I and SWIR images of the two control groups at 24 h. (h) The time course of NIR-I and SWIR signals in the liver region of the three animal groups (n=3 for each group).
To study the efficiency of the engineered NF-κB reporter, we transiently transfected HEK293T cells with a miRFP718nano-based reporter. Non-stimulated cells showed weak or no miRFP718nano expression (Fig. 2b, bottom), indicating a low background activity. In contrast, cells stimulated with 20 ng/ml TNF-α demonstrated strong miRFP718nano fluorescence (Fig. 2b,c). Stimulation of cells with different amounts of TNF-α showed that 5 ng/ml TNF-α was sufficient to fully activate the reporter while 1 ng/ml TNF-α caused more than 50% of activation (Fig. 2d).
To monitor the NF-κB activity in vivo, we adapted a gene delivery approach in the liver based on hydrodynamic transfection25. After 72 h, we induced acute inflammation with an intraperitoneal injection of E. coli lipopolysaccharide (LPS). The first control group received NF-kB-based miRFP718nano reporter plasmid through the tail vein but did not receive an LPS injection. The second control group received two injections of PBS instead of the reporter plasmid and LPS (Fig. 2e). For the LPS-treated group, in vivo results showed clear fluorescence of miRFP718nano in the liver, which peaked at 24 h after the LPS injection, in both NIR-I and SWIR images (Fig. 2f). No significant fluorescence emission was observed in the two control groups (Fig. 2g and Supplementary Fig. 15). The fluorescence signals gradually decreased over 6.5 days, reflecting the progress of LPS-induced inflammation (Fig. 2h). As expected, SWIR images provided stronger signals as well as better contrast (Supplementary Fig. 16). Moreover, SWIR images were able to detect weak NF-κB activity at the late stages of the inflammation.
In conclusion, employing a structure-based design followed by directed molecular evolution, we have developed the first 17 kDa red-shifted NIR FP, miRFP718nano. miRFP718nano brightly fluoresces in both NIR-I and SWIR windows, has high protein stability and performs well as terminal and internal protein tags. Spectral properties of miRFP718nano enable its use as a FRET acceptor for blue-shifted 17 kDa miRFP670nano, resulting in the smallest fully NIR FRET pair. The structure of miRFP718nano and its BV chromophore revealed the basis of its red shift and explained the effect of rational design. This rational design could be further applied to other CBCRs. CBCRs demonstrate a large spectral diversity and among them, CBCRs with native red-shifted fluorescence are of particular interest7, 26. Further efforts, directed at the discovery of more red-shifted CBCRs and applying described strategy should result in NIR FPs with significantly SWIR-shifted fluorescence.
Comparison of miRFP718nano in the NIR-I and SWIR range demonstrated its superior performance in the SWIR window, such as larger penetration depth, higher spatial resolution and contrast, and improved detection sensitivity. We showed miRFP718nano-based SWIR imaging on several animal models. More generally, we demonstrated that the long off-peak fluorescence emission tail of red-shifted NIR FP enables advanced SWIR deep-tissue imaging. Unlike in the NIR-I window, biological tissues have relatively weak autofluorescence and optical scattering in the SWIR window, and thus increasing the exposure time can improve the image contrast without strong background signals or blurring the targets. Compared with NIR fluorescent dyes, miRFP718nano can be specifically expressed in tissues through genetic encoding, does not need complex exogenous delivery, and has high biocompatibility for longitudinal studies.
Methods
Mutagenesis and directed molecular evolution.
Genes encoding miRFP670nano, miRFP718nano and their mutants were cloned into a pBAD/His-B vector (Thermo-Fisher Scientific) by using the KpnI and EcoRI restriction sites. All oligonucleotide primers for PCR amplification and saturated mutagenesis were purchased from Biomers (Supplementary Table 4). LMG194 (Invitrogen/Thermo-Fisher Scientific) was used as host E.coli cells. For BV productions LMG194 was transformed with a pWA23h plasmid encoding heme oxygenase from Bradyrhizobium ORS278 under rhamnose promoter10. Random mutagenesis was performed with a GeneMorph II random mutagenesis kit (Agilent Technologies). Saturated and site-specific mutagenesis was performed by overlap-extension PCR. Resulted mixtures of mutated genes were cloned into pBAD/His-B vector and electroporated into LMG194 cells, transformed with a pWA23h plasmid. Bacterial libraries of mutants contained 107– 108 clones. FACS-screening of mutant libraries was performed with an Influx cell sorter (BD Biosciences), using a 640 nm excitation laser and 725/40 nm emission filter. For sorting, the bacteria were grown in an LB medium, containing carbenicillin and kanamycin (100 μg/mL, Thermo-Fisher Scientific) and supplemented with 0.02% rhamnose (Santa Cruz) and 0.005% arabinose (Calbiochem) for 5 h at 37°C and then overnight at 22°C. Before sorting bacterial cells were pelleted, washed and diluted with ice-cold phosphate-buffered saline (PBS) to an optical density of 0.03 at 600 nm. The brightest collected bacterial cells were incubated in SOC medium for 1 h at 37°C and then plated on LB/ampicillin/kanamycin Petri dishes supplemented with 0.005% arabinose and 0.02% rhamnose overnight at 37°C. Screening of the brightest clones was performed with a Leica M205 fluorescence stereomicroscope equipped with a CCD camera (Tucsen), using 700/20 nm excitation and 730 nm long-pass emission filters (Chroma). To exclude blue-shifted mutants, 615/30 nm excitation and 670/30 nm emission filters (Chroma) were used. The fluorescence spectra of selected clones were then measured using a Cary Eclipse fluorimeter (Agilent Technologies) and around 30 brightest red-shifted clones were subcloned into a pcDNA3.1 plasmid (Invitrogen/Thermo-Fisher Scientific) and tested in transiently transfected HeLa cells.
Protein expression and characterization.
Proteins were expressed in LMG194 bacterial cells, cotransformed with pWA23h plasmid, as described above for sorting of libraries of mutants. Protein purification was performed with Ni-NTA agarose (Qiagen). For elution, PBS containing 100 mM EDTA was used.
For the recording of fluorescence spectra, the Cary Eclipse fluorimeter (Agilent Technologies) was used. Absorbance spectra were recorded with a Hitachi U-2000 spectrophotometer. The extinction coefficient of miRFP718nano was determined as a ratio between the maximum absorbance of the main peak at the Q band and the side peak at the Soret band assuming that the extinction coefficient at the Soret band corresponds to 39,900 M−1cm−1 10. The fluorescence quantum yield of miRFP718nano was determined using miRFP709 as standard. The pH stability was studied using a series of Hydrion buffers (Micro Essential Laboratory). Data fitting and statistical analysis were performed using OriginPro 2021b v.9.8.5.212 (OriginLab) and Excel v.15.36 (Microsoft) software.
Protein crystallization.
For crystallization, miRFP718nano was equilibrated in 20 mM Tris-HCl, 300 mM NaCl at pH 8.0 buffer and concentrated to 27.8 mg ml−1. Initial crystallization conditions were found with the NT8 crystallization robot (Formulatrix) using Hampton Research, Jena Bioscience, and Molecular Dimensions screens. The conditions were further optimized with additive screens. The best crystals could be obtained from 12.6% PEG 6000, 0.1 M lithium sulfate, 0.07 M citric acid buffer pH 3.5, and 2.1% D-sorbitol. The crystals suitable for X-ray data collection were grown by the hanging-drop vapor diffusion method. In the large-scale crystallization experiment, 2 μl of the protein solution was mixed with 2 μl of the reservoir solution and incubated against 500 ml of the same reservoir solution at 20°C for a week.
Determination of protein structure.
X-ray data were acquired on SER-CAT 22-ID and 22-BM beamline stations (Advanced Photon Source, Argonne National Laboratory). Before data collection, the crystals were flash-frozen in a 100 K nitrogen gas stream. Diffraction images were processed with HKL200027. The statistics and data processing are given in Supplementary Table 2. The structure was solved by the molecular replacement method with MOLREP v.11.028 using the structure of miRFP670nano (PDB ID: 6MGH) as a search model. To remove model bias, the structure was rebuilt with ARP/wARP model building and density improvement software29. The structure refinement was carried out with REFMAC530 (CCP4 suite) v.8.0.005 and PHENIX.REFINE31 (PHENIX suite) v.1.8.4–1496 programs. A realspace model correction and structure validation were performed with COOT v.0.9.8.132. The refinement statistics are given in Supplementary Table 3.
Construction of mammalian plasmids.
To construct plasmids encoding miRFP718nano or its mutants, the respective genes were cloned into the pcDNA3.1 plasmid (Invitrogen/Thermo Fisher Scientific) by using the KpnI and EcoRI restriction sites.
To engineer plasmids for protein tagging and labeling of intracellular structures, the gene encoding miRFP718nano was swapped with the gene encoding miRFP670nano either as C- (for α-tubulin, actin, myosin and vesicular protein clathrin), N-terminal fusions (for lysosomal membrane glycoprotein LAMP1, mitochondria, histone H2B), or internal fusions (for β2 adrenergic receptor and G-protein α subunit). To engineer a JNK activity NIR-biosensor plasmids, a gene encoding miRFP720 was replaced with the gene encoding miRFP718nano by NotI/XbaI sites in the reported previously NIR FRET biosensors of JNK kinase8. To construct a miRFP718nano-based reporter of inflammation, the gene encoding miRFP718nano was swapped with a gene encoding NLucP in the pNL3.2.NF-κB-RE[NlucP/NF-κB-RE/Hygro] vector (Promega).
Mammalian cells and transfection.
HeLa (CCL-2), N2A (CCL-131), U-2 OS (HTB-96), HEK293T (CRL-3216) and NIH3T3 (CRL-1658) cells were obtained from the ATCC. Cells were cultured in a DMEM medium supplemented with 10% FBS, 0.5% penicillin-streptomycin and 2 mM glutamine (Invitrogen/Thermo-Fisher Scientific) at 37°C in a humidified 5% CO2 atmosphere. For fluorescence microscopy, cells were plated in 35 mm glass-bottom Petri dishes (Greiner Bio-One International). Transient transfections were performed using polyethylenimine33.
Neuronal culture and transfection.
Primary embryonic rat cortical neurons were prepared in the Neuronal Cell Culture Unit of the University of Helsinki. The embryos staged at E17–18 from the female rats were used. Animals were kept in standard conditions. All animal work was performed under the ethical guidelines of the European convention and regulations of the Ethics Committee for Animal Research of the University of Helsinki. Cells were plated at a density of 500,000–700,000 per 35 mm glass-bottom dish, coated with Poly-L-Lysine (0.01 mg/ml) (Merck). Neurons were grown at 37°C and 5% CO2 in a neurobasal medium (Gibco) supplemented with B27 (Invitrogen/Thermo-Fisher Scientific), L-glutamine (Invitrogen/Thermo-Fisher Scientific), and penicillin-streptomycin (Lonza). Cultured neurons were transfected with pcDNA plasmids encoding miRFP718nano at 4–5 days in vitro (DIV) using Effectene Transfection Reagent (Qiagen) and imaged 48 h after transfection.
NF-kB-based transcription reporter of inflammation.
HEK293T cells were transfected with plasmids encoding reporter of inflammation and EGFP (1:10) and stimulated with TNF-α in indicated concentrations. Measurement of miRFP718nano fluorescence was performed by flow cytometry and fluorescent microscopy 20 h after stimulation.
Wide-field fluorescence microscopy.
Live cells were imaged with an Olympus IX81 inverted epifluorescence microscope equipped with a Xenon lamp (Lambda LS, Sutter), 60× 1.35 NA oil objective lens (UPlanSApo, Olympus) and an ORCA-Flash4.0 V3 camera (Hamamatsu). During imaging, HeLa cells were incubated in a cell imaging solution (Life Technologies-Invitrogen) and kept at 37°C. Neurons were imaged in cultured media. The microscope was operated with a SlideBook v.6.0.8 software (Intelligent Imaging Innovations).
To separately image miRFP670nano and miRFP18nano, two filter sets (605/30 nm exciter with 667/30 nm emitter, and 685/20 nm exciter with 725/40 nm emitter) (Chroma) were used. For imaging of miRFP718nano 685/20 nm excitation and 725/40 nm emission filters were used. The data were analyzed using SlideBook v. 6.0.8 (Intelligent Imaging Innovations) and Fiji v.1.50b software.
For FRET images a 605/30 excitation filter and two emission filters (667/30 nm for miRFP670nano and 725/40 nm for miRFP718nano) were used. Emission ratios were obtained by calculating background-subtracted 605/30 nm and 725/40 nm image intensities divided by background-subtracted 605/30 nm and 667/30 nm image intensities. Images were obtained using ImageJ v.1.51u. Intensity-modulated display mode was generated with a full-spectrum lookup table. Time-course ratio measurements were normalized to baseline prestimulation values. Before imaging cells were starved for 6 h with DMEM medium (Gibco/Thermo-Fisher Scientific).
Photobleaching measurements were performed in live HeLa cells 48 h after the transfection using a 60× 1.35NA oil objective lens (UPlanSApo, Olympus). Obtained raw data were normalized to corresponding absorbance spectra and extinction coefficients of the miRFPnanos, the spectrum of the Xenon lamp and the transmission of the 665/45 nm excitation filter. Photobleaching was performed using a live-cell imaging solution without antifade reagents (Invitrogen/Thermo-Fisher Scientific).
Flow cytometry.
Flow cytometry analysis was performed using an Accuri C6 flow cytometer (BD Biosciences). Before analysis, cells were washed and diluted in cold PBS to a density of 500,000 cells per ml. At least 50,000 cells per sample were recorded. The fluorescence intensity of miRFP718nano, miRFP670nano and miRFP703 and expressing cells were analyzed using the 640 nm excitation laser and 670 nm LP emission filters. EGFP fluorescence was analyzed using a 488 nm excitation laser and 510/15 nm emission filter. Supplementary Fig. 3 exemplifies the gating strategy. Flow cytometry data were collected using BD Accuri C6 v.1.0.264.21 software. The data were analyzed using FlowJo v.7.6.2 software.
Fluorescence emission spectroscopy.
Fluorescence emission spectra were acquired on an Edinburgh FLS920 fluorescence spectrometer equipped with a 450W Xenon arc lamp (Edinburgh Xe900 450) as the excitation light source and an extended-red sensitive PMT (Hamamatsu R2658P, spectral range: 200–1010 nm) and NIR-PMT (Hamamatsu H10330–75, spectral range: 950–1700 nm) for detection. The excitation wavelength was set at 630 nm for miRFP703, miRFP709 and miRFP718nano, and at 580 nm for miRFP670nano.
NIR-I and SWIR fluorescence imaging.
Shown in Supplementary Figure 9, a lab-made SWIR imaging system used a high-sensitivity InGaAs camera that has a broad detection spectrum from 600 nm to 1.7 μm (Ninox 640 II, Raptor Photonics). The camera has a pixel size of 15 μm and a maximum frame rate of 120 Hz. The camera was equipped with a VIS-SWIR objective lens with an F-number of 1.8 (RPL-OESWIRECON14mmx1.8A, Raptor Photonics). The excitation light source was a 665 nm laser diode (M660L4, Thorlabs). The surface exciton light intensity was ~50 mW/cm2 for all the NIR-I and SWIR imaging in this study. The camera exposure time was 10 ms for NIR-I imaging and 30 ms for SWIR imaging, if not stated otherwise. The fluorescence emission filters were 750/40 nm (FB750–40, Thorlabs) for NIR-I imaging and 1050 nm longpass (FEL1050, Thorlabs) for SWIR imaging. A dichroic mirror with the cut-off wavelength at 730 nm (DMLP730B, Thorlabs) is used for both the NIR-I and SWIR imaging.
Data analysis in NIR-I and SWIR imaging.
The raw white-light and fluorescence images were acquired by using Micro-Manager and saved as TIFF files. Further data processing was performed using MATLAB v.2019b. The image fusion of the white-light image and the fluorescence image was done by adjusting the transparency of the fluorescence image on top of the white-light image. Before the image fusion, a threshold of three times the noise level was applied to the fluorescence images to remove the background. The signal-to-background ratio in Supplementary Fig. 12d was computed by identifying the cell regions (the ‘signal’) and cell-free regions (the ‘background’) from the white-light image. The fluorescence signal intensity in Figure 2h was computed by averaging all the image pixels that were three times above the noise level. All the quantitative analyses with multiple animals were shown as the mean together with the individual data points.
Animal preparation for the NIR-I and SWIR imaging.
Female BALB/c mice (8–16 weeks old, 20–30 g) were used for all the NIR-I and SWIR imaging. For animal experiments, the sample size included 3 mice per group for each experiment. Animals were maintained on a 12 h light/dark cycle in a facility supplied with fresh air with humidity equivalent to outside ambient air. The mice were on an autofluorescence-free diet for at least 14 days before the imaging. Three days before the imaging, the abdominal region of the mice was shaved using hair-removal cream. During the imaging, the mice were anesthetized by inhaling isoflurane (1–1.5% v/v) and the body temperate was kept at 37°C by using an electrical heating pad placed underneath. All the experimental procedures were approved by the IACUC of the Duke University.
Injecting HEK293T cells into the mammary fat pad.
Mice were anesthetized with 2% isoflurane in 30% oxygen balanced with nitrogen through a face mask and kept in a supine position. The fourth nipple skin was gently held up using a fine tweezer to form a tent shape, and a 27 gauge needle was inserted into the skin next to the nipple until the needle bevel was in. After 20 μl miRFP670nano- or miRFP718nano-expressing HEK293T cell solution was slowly injected, the tweezer was switched to hold the skin around the needle to keep the hole closed while the needle was withdrawn. Then the contralateral fourth mammary fat pad was injected using the same procedure.
NF-kB-based transcription reporter of LPS-induced inflammation in mice.
We followed a similar protocol adapted from reference25. Briefly, the mice were anesthetized by inhaling isoflurane (1–1.5% v/v). Hydrodynamic injection of the NF-kB-miRFP718nano reporter plasmid (10%, v/wt) was performed via the tail vein. For a 20-gram mouse, 2.0 mL of plasmid solution (10 μg/mL) was injected. The entire solution volume was injected in less than 10 s. The mice were then released to the cages for recovery. After 72 h, 100 μL of LPS dissolved in PBS (4 mg/kg body weight) was administered by intraperitoneal injection at the mammary gland region. The fluorescence imaging started 12 h after the LPS administration and continued for 6.5 days.
Extended Data
Extended Data Figure 1. Structure of miRFP718nano and its red-shifted chromophore.
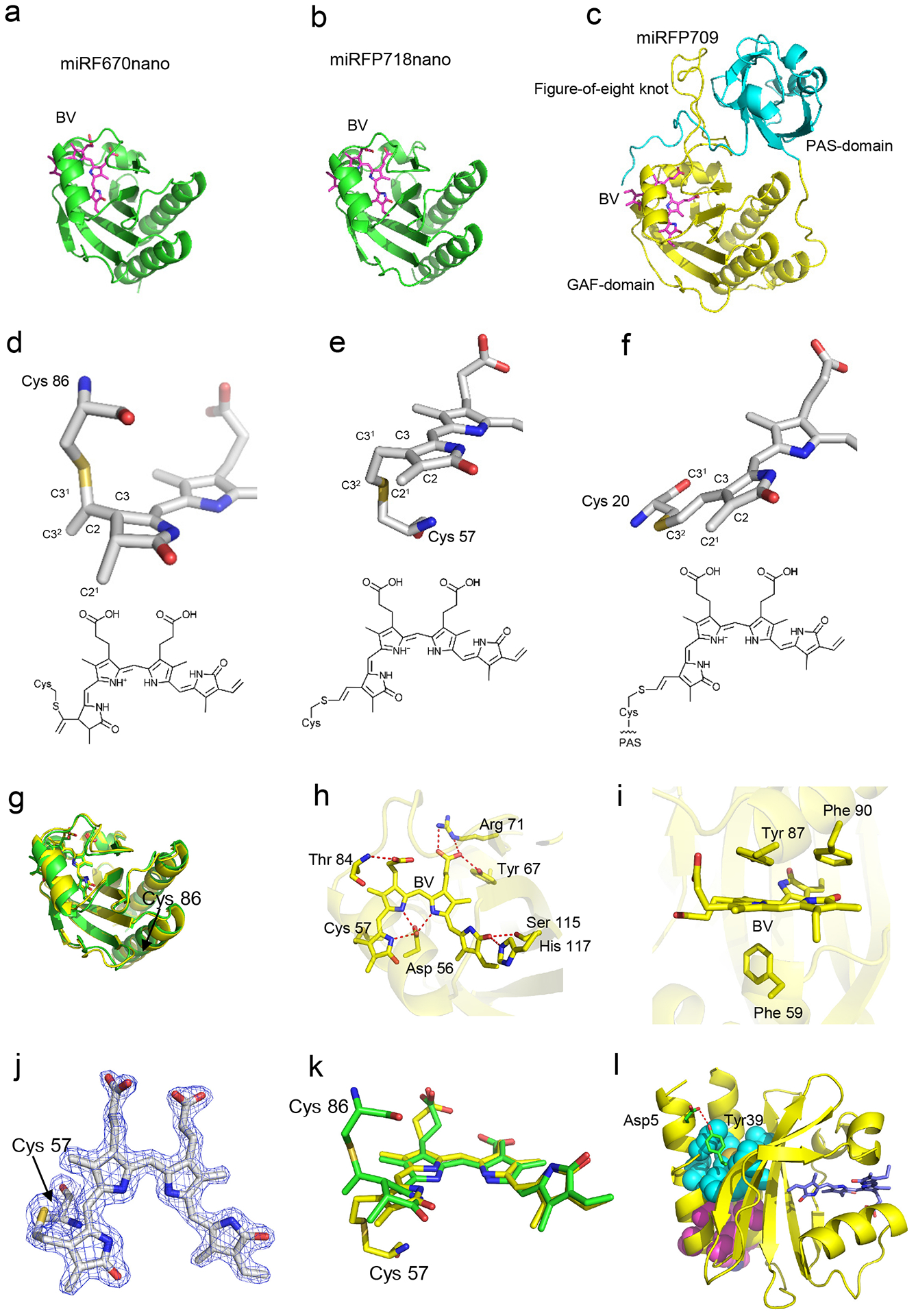
Overall structures of (a) miRFP670nano (PDB ID: 6MGH), (b) miRFP718nano, and (c) miRFP709 (PDB ID: 5VIQ). The biliverdin (BV) chromophores are shown in magenta. The PAS and GAF domains of miRFP709 are in cyan and yellow, respectively. The BV chromophores in (d) miRFP670nano, (e) miRFP718nano, and (f) miRFP709 bound to the respective Cys residues and their chemical formulas. Carbon, nitrogen, oxygen, and sulfur atoms are in white, blue, red, and yellow, respectively. Sticks representations show only rings A and B of the chromophores and Cys residues. In miRFP670nano, the BV chromophore (d) is bound to Cys86 via the C31 atom, miRFP718nano (e) and miRFP709 (f) have the same chromophore species bound to the Cys57 and Cys20, respectively. (g) Superposition of miRFP670nano (green) and miRFP718nano (yellow) structures. (h) miRFP718nano hydrogen bond network around the chromophores (i) Stacking interactions between the chromophores and the surrounding residues in miRFP718nano. (j) The chromophores of miRFP718nano bound to the respective Cys57 residues in the 2Fo-Fc electron density map. The map is countered at 2.0σ-levels. (k) Superposition of the chromophores in miRFP670nano (green) and miRFP718nano (yellow). (l) Stabilizing mutations and hydrophobic clusters in miRFP718nano. The residues forming H-bonds are shown in green, hydrophobic clusters (one formed by residues Leu8, Ile11, Val12, Val26, Ile104, Leu114, Met140 and the other by Val15, Phe18, Leu19, Trp128, Phe132, Leu133) are in cyan and magenta.
Supplementary Material
Acknowledgments
We thank N. Peitsaro and N. Aarnio from the Flow Cytometry Core Facility of the University of Helsinki for the technical assistance. This work was supported by grants from the US National Institutes of Health (EB028143, NS111039 and NS115581), Chan Zuckerberg Initiative (226178), Cancer Foundation Finland, and Magnus Ehrnrooth Foundation. S.P. was supported in part by the NIH Intramural Research Program for the Vaccine Research Center of the National Institute of Allergy and Infectious Diseases.
Footnotes
Code Availability
The miRFP718nano nucleotide sequence in GenBank is MW627296.1; link: https://www.ncbi.nlm.nih.gov/nuccore/MW627296.1 . The miRFP718nano structural data were deposited at the Protein Data Bank (PDB ID: 7LSD); link: https://www.rcsb.org/structure/7LSD .
Competing interests
The authors declare no competing interests.
Data availability
The data supporting the findings of this study are available within the article and its Supplementary Information. All other data that support the findings of the study are available from the corresponding author upon request. The major plasmids constructed in this study, their maps, and sequences are deposited to Addgene. For the structure of miRFP670nano PDB ID: 6MGH; link: https://www.rcsb.org/structure/6mgh . For the structure of miRFP709 PDB ID: 5VIQ; link: https://www.rcsb.org/structure/5VIQ .
References
- 1.Frangioni JV In vivo near-infrared fluorescence imaging. Current opinion in chemical biology 7, 626–634 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Scholkmann F et al. A review on continuous wave functional near-infrared spectroscopy and imaging instrumentation and methodology. Neuroimage 85, 6–27 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Welsher K et al. A route to brightly fluorescent carbon nanotubes for near-infrared imaging in mice. Nat Nanotechnol 4, 773–780 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith AM, Mancini MC & Nie S Bioimaging: second window for in vivo imaging. Nat Nanotechnol 4, 710–711 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feng Z et al. Perfecting and extending the near-infrared imaging window. Light Sci Appl 10, 197 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shcherbakova DM, Stepanenko OV, Turoverov KK & Verkhusha VV Near-Infrared Fluorescent Proteins: Multiplexing and Optogenetics across Scales. Trends Biotechnol 36, 1230–1243 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oliinyk OS, Chernov KG & Verkhusha VV Bacterial Phytochromes, Cyanobacteriochromes and Allophycocyanins as a Source of Near-Infrared Fluorescent Probes. Int J Mol Sci 18 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oliinyk OS, Shemetov AA, Pletnev S, Shcherbakova DM & Verkhusha VV Smallest near-infrared fluorescent protein evolved from cyanobacteriochrome as versatile tag for spectral multiplexing. Nat Commun 10, 279 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oliinyk OS et al. Single-domain near-infrared protein provides a scaffold for antigen-dependent fluorescent nanobodies. Nat Methods 19, 740–750 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shcherbakova DM et al. Bright monomeric near-infrared fluorescent proteins as tags and biosensors for multiscale imaging. Nat Commun 7, 12405 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu D et al. A naturally monomeric infrared fluorescent protein for protein labeling in vivo. Nat Methods 12, 763–765 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Komatsu N et al. Development of an optimized backbone of FRET biosensors for kinases and GTPases. Mol Biol Cell 22, 4647–4656 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shcherbakova DM Near-infrared and far-red genetically encoded indicators of neuronal activity. J Neurosci Methods 362, 109314 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shcherbakova DM, Cox Cammer N, Huisman TM, Verkhusha VV & Hodgson L Direct multiplex imaging and optogenetics of Rho GTPases enabled by near-infrared FRET. Nat Chem Biol 14, 591–600 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li L, Hsu HC, Verkhusha VV, Wang LV & Shcherbakova DM Multiscale Photoacoustic Tomography of a Genetically Encoded Near-Infrared FRET Biosensor. Adv Sci (Weinh) 8, e2102474 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shemetov AA et al. A near-infrared genetically encoded calcium indicator for in vivo imaging. Nat Biotechnol 39, 368–377 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baloban M et al. Designing brighter near-infrared fluorescent proteins: insights from structural and biochemical studies. Chem Sci 8, 4546–4557 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang B et al. A phosphorescent probe for in vivo imaging in the second near-infrared window. Nat Biomed Eng (2021). [DOI] [PubMed] [Google Scholar]
- 19.Jeong S et al. Multiplexed In Vivo Imaging Using Size-Controlled Quantum Dots in the Second Near-Infrared Window. Adv Healthc Mater 7, e1800695 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Robinson JT et al. In vivo fluorescence imaging in the second near-infrared window with long circulating carbon nanotubes capable of ultrahigh tumor uptake. J Am Chem Soc 134, 10664–10669 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hugenholtz F & de Vos WM Mouse models for human intestinal microbiota research: a critical evaluation. Cell Mol Life Sci 75, 149–160 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hamano N, Inada T, Iwata R, Asai T & Shingu K The alpha2-adrenergic receptor antagonist yohimbine improves endotoxin-induced inhibition of gastrointestinal motility in mice. Br J Anaesth 98, 484–490 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Wang D et al. Trans-illumination intestine projection imaging of intestinal motility in mice. Nat Commun 12, 1682 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu T, Zhang L, Joo D & Sun SC NF-kappaB signaling in inflammation. Signal Transduct Target Ther 2 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Osorio FG, de la Rosa J & Freije JM Luminescence-based in vivo monitoring of NF-kappaB activity through a gene delivery approach. Cell Commun Signal 11, 19 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fushimi K & Narikawa R Phytochromes and Cyanobacteriochromes: Photoreceptor Molecules Incorporating a Linear Tetrapyrrole Chromophore. Adv Exp Med Biol 1293, 167–187 (2021). [DOI] [PubMed] [Google Scholar]
- 27.Otwinowski Z & Minor W [20] Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276, 307–326 (1997). [DOI] [PubMed] [Google Scholar]
- 28.Vagin A & Teplyakov A MOLREP: an automated program for molecular replacement. Journal of Applied Crystallography 30, 1022–1025 (1997). [Google Scholar]
- 29.Lamzin VS, Perrakis A, Wilson KS in In International Tables for Crystallography, Vol. F: Crystallography of biological macromolecules. (ed. Arnold E HD, Rossmann MG) 525–528 (Kluwer Academic Publishers, The Netherlands; 2012). [Google Scholar]
- 30.Murshudov GN et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr 67, 355–367 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adams PD et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Emsley P, Lohkamp B, Scott WG & Cowtan K Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Longo PA, Kavran JM, Kim MS & Leahy DJ Transient mammalian cell transfection with polyethylenimine (PEI). Methods Enzymol 529, 227–240 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting the findings of this study are available within the article and its Supplementary Information. All other data that support the findings of the study are available from the corresponding author upon request. The major plasmids constructed in this study, their maps, and sequences are deposited to Addgene. For the structure of miRFP670nano PDB ID: 6MGH; link: https://www.rcsb.org/structure/6mgh . For the structure of miRFP709 PDB ID: 5VIQ; link: https://www.rcsb.org/structure/5VIQ .