

Abstract
Pharmacometric models were used to investigate the utility of biomarkers in predicting the efficacy (Crohn's Disease Activity Index [CDAI]) of brazikumab and provide a data‐driven framework for precision therapy for Crohn's disease (CD). In a phase IIa trial in patients with moderate to severe CD, treatment with brazikumab, an anti‐interleukin 23 monoclonal antibody, was associated with clinical improvement. Brazikumab treatment effect was determined to be dependent on the baseline IL‐22 (BIL22) or baseline C‐reactive protein (BCRP; predictive biomarkers), and placebo effect was found to be correlated with the baseline CDAI (a prognostic biomarker). A maximal total inhibition on CDAI input function of 50.6% and 42.4% was predicted for patients with extremely high BIL22 or BCRP, compared to a maximal total inhibition of 20.9% and 17.8% for patients with extremely low BIL22 or BCRP, respectively, which were mainly due to the placebo effect. We demonstrated that model‐derived baseline biomarker levels that achieve 50% of maximum unbound systemic concentration of 22.8 pg/mL and 8.03 mg/L for BIL22 and BCRP as the cutoffs to select subpopulations can effectively identify high‐response subgroup patients with improved separation of responders when compared to using the median values as the cutoff. This work exemplifies the utility of pharmacometrics to quantify biomarker‐driven responses in biologic therapies and distinguish between predictive and prognostic biomarkers, complementing clinical efforts of identifying subpopulations with higher likelihood of response to brazikumab.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
A major challenge for biologics therapy in inflammatory bowel disease is the variability of clinical response after treatment, and personalized treatment is seen as an important way in maximizing the response to therapy.
WHAT QUESTION DID THIS STUDY ADDRESS?
A pharmacometrics approach showed how brazikumab treatment of patients with Crohn's disease (CD) can potentially be personalized based on baseline levels of IL‐22 and C‐reactive protein.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
The model suggests high responses would be observed in brazikumab treatment of patients with CD with high baseline serum IL‐22 or C‐reactive protein, whereas those with low baseline serum IL‐22 or C‐reactive protein may consider alternative treatments because their responses are not very different from placebo.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
This work demonstrates that pharmacometrics can be used to investigate and characterize the utility of biomarkers to predict response to biologic therapies and distinguish between predictive and prognostic biomarkers in a quantitative manner, thereby adding much value to clinical development.
INTRODUCTION
Crohn's disease (CD) and ulcerative colitis (UC) are the principal types of inflammatory bowel diseases (IBDs) affecting both adults and children. CD can cause inflammation in any part of the gastrointestinal tract and clinical presentations include abdominal pain, diarrhea and the formation of fistulas, strictures, and abscesses. 1 , 2 Treatment options include chemical steroids, immunosuppressives, such as azathioprine and methotrexate, and biologics, such as infliximab, adalimumab, certolizumab, ustekinumab, and vedolizumab, which have been shown to be effective for moderate to severe CD by targeting specific pathways implicated in CD. 3
When patients initiate treatment for CD, therapy is individualized based on clinical presentation, medical history, prior treatments, severity of disease, etc. 4 , 5 , 6 Either “bottom up” (step up) or “top down” approaches may be used. In bottom‐up therapy, when patients fail to respond or are intolerant to the chemical agents, they switch to the alternative treatment option of biologics. 4 , 5 , 6 However, there can be 10%–30% of primary nonresponders and about 30%–40% of secondary nonresponders that fail to achieve sustained clinical response to biologics. 7 The cause of the nonresponse can be due to drug‐related factors, such as neutralizing antibodies, altered clearance of drug or possibly biological escape mechanisms, or factors unrelated to the drug, including absence of active inflammation, concurrent infection, or septic complications. 7 The investigation of factors that influence response or failure will help identify the most appropriate biological regimen for individual patients with CD. 7 Among all the factors, clinical or biological factors that correlate with the disease prognosis or the response to therapy have been brought attention for further investigation, such as baseline Crohn's Disease Activity Index (BCDAI) and baseline C‐reactive protein (BCRP). 8 , 9 , 10 , 11 , 12 , 13 , 14
Brazikumab (MEDI2070, AMG139) a human monoclonal antibody (mAb) under clinical development for CD and UC, selectively targets the p19 subunit of interleukin‐23 (IL‐23), a pro‐inflammatory cytokine implicated in the pathogenesis of both diseases. 2 , 15 IL‐23 induces the downstream production of IL‐22, which is significantly increased in CD and correlates with disease activity. 16 A phase IIa clinical study investigating the efficacy of brazikumab in patients with CD demonstrated an association between baseline serum IL‐22 (BIL22) levels and the therapeutic effect of brazikumab: higher BIL22 was associated with an increased likelihood of response compared to the placebo group, suggesting that BIL22 may be a potential predictive biomarker to select patients who most likely will benefit from brazikumab treatment. 2
C‐reactive protein (CRP) is an acute phase protein produced by hepatocytes in response to inflammatory conditions, and its production is stimulated by active IBD‐associated cytokines. 17 The use of BCRP to predict treatment response has produced conflicting results. BCRP levels were higher in primary nonresponders than sustained responders following the treatment with infliximab. 11 However, in a different study of infliximab in patients with CD, more subjects with high BCRP (≥0.7 mg/dL) maintained remission through 54 weeks of infliximab therapy than subjects with low BCRP (<0.7 mg/dL, 44.8% vs. 22.0%, p value = 0.012). 12 Similarly, higher responses to the treatment with ustekinumab were also observed with patients with high BCRP levels (≥10 mg/L). 14 Besides therapeutic response, an inverse relationship between BCRP levels and placebo response was found among patients with only placebo treatment in the studies of certolizumab and ustekinumab. 13 , 14
Crohn's Disease Activity Index (CDAI) is a score that combines weighted scores of clinical and laboratory variables to estimate disease severity. 18 It is composed of eight subjective and objective variables of disease, including diarrhea, abdominal pain, general well‐being, extra‐intestinal manifestations, anti‐diarrheal use, abdominal mass, hematocrit, and weight. 18 CDAI scores of less than 150 indicate clinical remission or inactive disease and scores over 450 indicate severely active disease. In a previous study of certolizumab pegol, patients with high BCDAI (>292) had low response in both treatment and placebo arms (p value < 0.05). 8 In another retrospective study of ustekinumab, BCDAI scores were significantly lower in the subject group that achieved clinical remission than the group that failed to achieve it (p value = 0.03). 9 In addition, high BCDAI scores were associated with lack of treatment response to adalimumab, and the need of a more frequent dosing of adalimumab was discussed. 10 Overall, the underlying relationship of BCDAI and treatment response has not been quantitatively studied before. A quantitative relationship between BCDAI and therapeutic effect and/or disease progression is needed to demonstrate the role of BCDAI as a predictive or prognostic factor.
Although various efforts have been made to identify and validate the biomarkers for treatment response and disease prognosis, the clinical usage of biomarkers has been very challenging due to the lack of well‐characterized, quantitative relationships among biomarker levels, treatment response, and disease progression. This challenge creates a perfect scenario that pharmacometrics could exert its capability of detecting an effect with higher power because it could integrate all data collected over the time course, characterize pharmacokinetics (PKs), treatment response, and disease progression at the same time, and account for multiple sources of variability and study‐specific artifacts (compliance, assay variability, covariate effect, and within‐individual variability), and PK/exposure‐dependent variability in response. 19 , 20 , 21 , 22 With these advantages over traditional statistical analysis, pharmacometric modeling has been commonly used during clinical development in characterizing the efficacy/safety response of candidate drugs. In this work, we used pharmacometric models to investigate the relationship among PK exposure, baseline biomarkers, and efficacy, and establish cutoff values of BIL22 and BCRP to identify patients who are more likely to respond to brazikumab, in comparison to that using the median values as the cutoffs. 2 With this pharmacometrics approach, we characterized the interplay among PKs, biomarkers, and efficacy of brazikumab, and quantified the impact of the biomarkers on overall response, thus laying a data‐driven framework for precision medicine in CD.
METHODS
Study population and trial design
The analysis data was derived from a phase Ib study and a previously published phase IIa study, which were registered at clinicaltrial.gov (NCT01258205 and NCT01714726, respectively). 2 , 15 The phase Ib study was a randomized, double‐blind, placebo‐controlled, sequential multiple‐ascending dose design in 34 subjects. The study was designed to evaluate the safety, tolerability, PKs, pharmacodynamics, and effects on disease activity of brazikumab. Investigational product doses ranged from 70 mg to 700 mg q4w; routes of administration were subcutaneous (s.c.) injections or intravenous (i.v.) infusion. Extensive sampling was conducted after the first and third doses.
The phase IIa study was a two‐part study comprising a 12‐week, double‐blind, placebo‐controlled, treatment period followed by a 100‐week, open‐label, treatment period to evaluate the short‐term efficacy and the short‐ and long‐term safety of brazikumab in 119 patients with moderate to severe active CD who have failed or are intolerant to anti‐TNFα therapy. Placebo or 700 mg of brazikumab was administered as an i.v. infusion over a period of at least 60 min on day 1 and day 29 during the 12‐week induction period, whereas subjects enrolled in the open‐label period received s.c. injections of 210 mg brazikumab q4w from week 12 through week 112. Sparse sampling was conducted before or after the dose during both the double‐blinded and open‐label periods. The detailed design and protocol for the phase IIa trial has been reported. 2 , 15
Model building and data analysis
A nonlinear mixed effects modeling approach was utilized for all the PK and biomarker/efficacy analysis using NONMEM (ICON Development Solutions, version 7.3), PsN (Perl‐speaks‐NONMEM; https://uupharmacometrics.github.io/PsN/, version 4.6.0), pirana, and the first‐order conditional estimation method with interaction (FOCE‐I). R (www.r‐project.org, version 3.3.1) was used to assemble the NONMEM data file, run simulations, and create graphical outputs. 23 , 24 , 25
PK model development
Using all PK data that was derived from the phase Ib and IIa studies, including the open‐label period, one‐ and two‐compartment models with first‐order absorption from the dosing site of s.c. administration of brazikumab, and linear or nonlinear elimination from the central compartment were explored to describe the PK profiles of brazikumab.
Covariate relationship identification
The identification of covariates was undertaken using “Stepwise Covariate Model‐Building” (SCM) using PsN and NONMEM. This method involved stepwise testing of linear and dichotomous relationships on categorical covariates, and linear, hockey‐stick, and exponential relationships on continuous covariates in a forward inclusion procedure (change in objective function value Δ[OFV] >3.84, p < 0.05 for 1 degree of freedom [DF]). Covariates, including subject population (healthy subjects vs. patients with CD), sex, race, age, body weight, baseline albumin (BALB), and body mass index, were screened in the SCM. The detected covariate effects were kept in the final model if the relationship is qualitatively meaningful and clinically significant with a cutoff of 20%.
Efficacy model development
The population efficacy model was built using data from the double‐blind placebo‐controlled period of the phase IIa trial. An indirect response (IDR) model with inhibition on disease production rate (k in) was used to describe the longitudinal CDAI score data. Biomarker‐dependent drug effects on k in were evaluated using both linear and/or nonlinear functions. The temporal relationship of the placebo effect was explored using a linear, step, or mono‐exponential decay function. Biological and clinical markers, including BIL‐22, BCRP, BFCP, BIL17, BLCN2, BMIP3A, BALB, and BCDAI scores, were tested as predictive or prognostic biomarkers in the efficacy model.
Modeling interindividual variabilities
The interindividual variability for PK parameters were assumed to be log‐normally distributed, that is, , where θ ij is the jth parameter for the ith individual, θ TVj is the typical value of the jth parameter for the population, and η i,j is assumed to follow a normal distribution of mean zero and variance of for the jth parameter. BCDAI in the efficacy model was also assumed to be log‐normally distributed. The placebo effect, however, is modeled to have an additive variability (i.e., PLAC i = PLACTV + η i ), with the assumption that the placebo effect in any individual may be higher or lower than the typical value of the placebo effect by an equal probability.
A proportional residual error model was found to best fit the PK data and can be described as where Cobs i (t) and Cpred i (t) is the observed and predicted serum concentration of the ith individual at time t, respectively, and the residual error ε i is assumed to follow a normal distribution with mean zero and variance of σ 2. A combined model of proportional and additive residual error was found to best fit the efficacy data and can be described as , where Aobs i (t) and Apred i (t) is the observed and predicted CDAI score of the ith individual at time t, respectively, and the residual error ε1 i and ε2 i are assumed to follow a normal distribution with mean zero and variance of σ12 and σ22, respectively.
Model evaluation and selection
During the model‐building process, successful minimizations, the likelihood OFV (change in OFV > 3.84, p < 0.05 for 1 DF), and goodness‐of‐fit plots were used to evaluate the intermediate models. These include plots of population and individual predictions versus observed data, and conditional weighted residuals versus time. Visual predictive checks (VPCs) were also used at the end of every key stage to compare the observed data with model‐simulated concentration‐time profiles and the associated medians and 95% prediction intervals.
RESULTS
Demographics and characteristics of study subjects
The population PK analysis included data pooled from a total of 34 and 119 subjects from the phase Ib and IIa studies, respectively, whereas the biomarker/efficacy analysis included only data from the phase IIa study of brazikumab. The demographics of the study population are summarized in Table 1. Briefly, among the subjects (4 women and 30 men) in the phase Ib study, four subjects had mild to severe CD, whereas the remaining 30 subjects were healthy volunteers (HVs). For the phase IIa study, all the subjects (74 women and 45 men) had moderate to severe active CD who failed or were intolerant to anti‐TNFα therapy. The median values for BCDAI, BIL22, and BCRP at baseline of all patients with CD in the phase IIa study were 317, 15.6 pg/mL, and 15.7 mg/L, respectively (as measured at day 0 before treatment). The other disease‐related biological characteristics of the patients in the phase IIa study are summarized in Table 1. These clinical or biological biomarkers were screened to identify the relationship between biomarkers and efficacy of brazikumab using the pharmacometric modeling approach. Subjects in the treatment and placebo arms had similar baseline characteristics. Although one subject in the treatment arm had extremely high BIL22 (711 pg/mL) and BCRP (212.8 mg/L), this outlier does not affect the overall distribution of these characteristics.
TABLE 1.
Demographic summary of the subjects in phase Ib and phase IIa trials.
| Variable | Phase Ib study | Phase IIa study (Double‐blind) | |
|---|---|---|---|
| Study arm | – | Treatment arm | Placebo arm |
| Type of data | PK | PK, biomarkers, efficacy | |
| Number of subjects | 34 (30 healthy volunteers, 4 Crohn's disease patients) | 59 (59 Crohn's disease patients) | 60 (60 Crohn's disease patients) |
| Duration of study | 36 weeks | 12‐week induction, 100‐week open‐label | |
|
Samples per subject Median (min, max) |
PK: 21 (14, 21) |
PK: 9 (1, 10) Efficacy: 5 (1, 5) |
PK: 9 (1, 10) Efficacy: 5 (3, 5) |
| Female, N (%) | 4 (11.8%) | 37 (62.7%) | 37 (61.7%) |
| Dose/route, N (%) | |||
| 70 mg i.v. | 6 (17.6%) | – | – |
| 210 mg i.v. | 8 (23.5%) | – | – |
| 210 mg s.c. | 6 (17.6%) | – | – |
| 420 mg i.v. | 6 (17.6%) | – | – |
| 700 mg i.v. | 8 (23.5%) | 59 (49.6%) | – |
| Placebo i.v | – | – | 60 (50.4%) |
| Age, years | |||
| Median (min, max) | NA | 34 (18, 58) | 36 (19, 61) |
| Weight, kg | |||
| Median (min, max) | NA | 65.8 (44.0, 158.8) | 69.3 (46, 112.0) |
| Height, cm | |||
| Median (min, max) | NA | 169.5 (155.0, 188.0) | 168.0 (151.1, 187.0) |
| Race, N (%) | |||
| White | NA | 55 (93.2%) | 56 (93.3%) |
| Black | 4 (6.8%) | 2 (3.3%) | |
| Other | 2 (3.3%) | ||
| BCDAI score | |||
| Median (min, max) | ND | 330 (222, 439) | 304 (221, 450) |
| BIL22, pg/mL | |||
| Median (min, max) | ND | 15.9 (1.00, 711) | 14.1 (1.00, 170) |
| BCRP, mg/L | |||
| Median (min, max) | ND | 18.2 (0.32, 212.8) | 9.55 (0.44, 110.4) |
| BFCP, μg/g | |||
| Median (min, max) | ND | 628.5 (58.0, 1086) | 639.0 (48.0, 2469) |
| BIL17, ng/dL | |||
| Median (min, max) | ND | 0.48 (0.11, 2.76) | 0.48 (0.09, 2.25) |
| BLCN2, ng/mL | |||
| Median (min, max) | ND | 215.4 (102.7, 434.0) | 214.2 (77.7, 434.0) |
| BMIP3A, pg/mL | |||
| Median (min, max) | ND | 22.7 (5.5, 224.1) | 22.7 (4.9, 139.1) |
| BALB, mg/dL | |||
| Median (min, max) | ND | 39.0 (32.0, 46.0) | 40.0 (33.0, 50.0) |
Abbreviations: % percent of the total subjects; BCDAI, baseline Crohn's Disease Activity Index; BIL22, baseline IL‐22; FCP, fecal calprotectin; LCN2, lipcalin‐2; MIP3A, macrophage inflammatory protein‐3; N, number of subjects; NA, not available; ND, not determined; PK, pharmacokinetic.
Pharmacometric modeling
By combining the data from both phase Ib and phase IIa studies, the PK profiles are presented depending on study populations and administration routes (Figure 1a). Based on the raw PK data collected in the phase Ib study, we observed a linear elimination in HVs within the dose range of brazikumab (70–700 mg i.v.). Additionally, we were able to identify that the patients with CD had a faster clearance (CL) than HVs, as reported in other studies 14 , 15 , 16 (Figure 1b).
FIGURE 1.
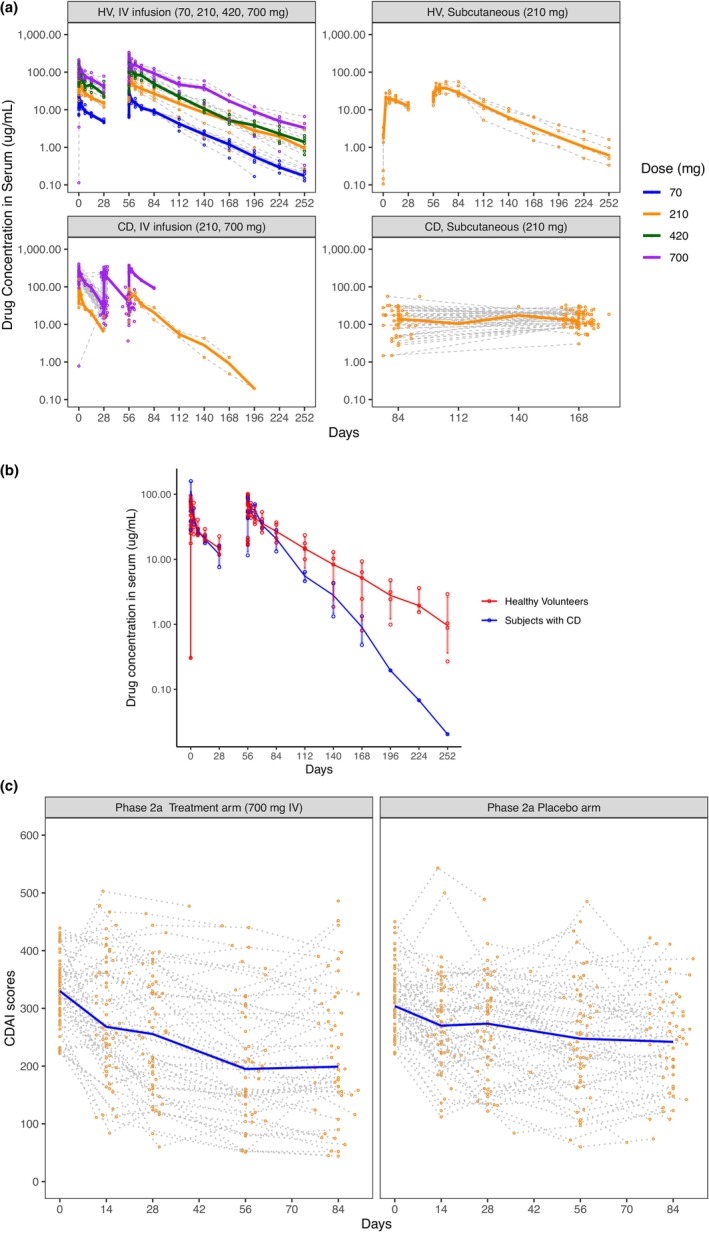
PK and efficacy raw data profiles of subjects in phase Ib and IIa trials. (a) PK raw data profiles of HVs and patients with CD in phase Ib and IIa trials. The PK data are stratified by the subject populations and the route of drug administration. The data of HV are all from the phase Ib study with three doses in total, q4w, at a large dose range (70–700 mg). All of the data of patients with CD with intravenous (i.v.) infusion at 210 mg and part of the data of patients with CD with i.v. infusion at 700 mg are also from the phase Ib study. All data of patients with CD with subcutaneous (s.c.) injection at 210 mg and most of the data of patients with CD with s.c. injection at 700 mg were from the phase IIa study, which investigated only two i.v. doses at week 0 and week 4 during the double‐blinded period and 26 s.c. q4w doses between week 12 and week 112 during the open‐label period. The solid lines represent the median value of the concentrations within the same dose and route arm over the treatment time. The gray dashed lines represent individual concentrations over time. (b) PK elimination profiles of HVs and patients with CD in a phase Ib trial. The solid lines represent the median value of the concentrations within HVs or subjects with CD over the treatment time. (c) Efficacy raw data profiles of patients with CD in a phase IIa trial double‐blinded period. The efficacy data are stratified by the treatment arms. The solid blue lines represent the median value of the CDAI scores within the treatment or placebo arm over the treatment time. The gray dashed lines represent individual CDAI scores over time. CD, Crohn's disease; CDAI, Crohn's Disease Activity Index; HV, healthy volunteers; PK, pharmacokinetic.
The PK data were adequately described by a two‐compartment model with first‐order absorption from the dosing site of s.c. administration of brazikumab and linear CL from the central compartment within the dose range investigated (Figures 1 and 2, Supplementary Material Equations 1–6 in Appendix S1). In the final PK model with covariate analysis, CL was identified to be negatively dependent on BALB. As BALB increased by 10%, CL decreased by 11.8% for both HVs and patients with CD. For subjects that had the same level of BALB, HVs had a 36.2% lower CL compared to patients with CD. Male subjects had a higher volume of distribution for the central compartment than female subjects (3.97 vs. 3.27 L). All three covariate relationships were identified with statistical significance (p value < 0.01). The final PK model parameters are shown in Table 2.
FIGURE 2.
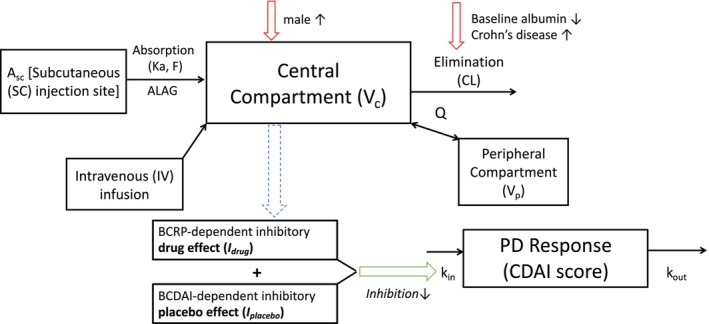
Schematic structure of final PK and biomarker/efficacy models. The PK can be described using a two‐compartment model and is linked to an indirect response model where the total inhibitory effect accounts for the contribution from a placebo effect and drug effect. The drug effect, in turn, depends on the baseline level of the serum IL‐22 or CRP biomarker described by a Hill function . A c and A p are the amount of brazikumab present in the central and peripheral compartments respectively. Similarly, V c and V p are the volumes of distribution of the central and peripheral compartments respectively. A SC represents the amount of brazikumab administered at the subcutaneous injection site and ka and F are the first‐order absorption rate constant and bioavailability respectively. CL is the clearance of brazikumab from the central compartment while Q is the intercompartmental clearance. k in is input function for the CDAI scores while k out is the elimination rate. CL is dependent on BALB and disease status of the subjects and V c is dependent on the gender of the subjects. (a) BIL22‐dependent efficacy model structure. (b) BCRP‐dependent efficacy model structure. CD, Crohn's disease; PK, pharmacokinetic.
TABLE 2.
Final population PK and biomarker/efficacy model parameter estimates.
| PK model | ||
|---|---|---|
| Parameter, unit | Parameter estimates (RSE%) | Interindividual variability (RSE%) |
| CL in female patients with CD, a L/d | 0.26 (5%) | 0.131 (33%) |
| Effect of baseline albumin on CL | −1.32 (40%) | – |
| Effect of health status on CL | −0.362 (10%) | – |
| V c b in female subjects, L | 3.27 (5%) | 0.0502 (18%) |
| Effect of male gender on V c | 0.214 (37%) | – |
| V p, L | 2.64 (8%) | – |
| Q, L/d | 0.412 (19%) | – |
| k a, d−1 | 0.286 (11%) | – |
| T lag, d | 0.0296 (14%) | – |
| F | 0.88 (5%) | – |
| Proportional error | 24.9% (9%) | – |
| Biomarker/efficacy models | ||
|---|---|---|
| Parameter, unit | Parameter estimates (RSE%) | |
| IL‐22 | CRP | |
| Half‐life HL, day | 11.6 (33%) | 11.7 (8%) |
| Baseline CDAI | 318 (3%) | 318 (2%) |
| Inhibitory placebo effect | 0.209 (7%) | 0.178 (16%) |
| I max | 0.297 (30%) | 0.246 (10%) |
| IB50, pg/mL for IL‐22 and mg/L for CRP | 22.8 (10%) | 8.03 (10%) |
| Hill coefficient γ | 20 FIX | 2.07 FIX |
| IIV of BCDAI score (RSE%) | 0.0106 (74%) | 0.00997 (35%) |
| IIV of placebo effect (RSE%) | 0.0509 (26%) | 0.0519 (4%) |
| Correlation between IIV of BCDAI score and placebo effect | −100% | −100% |
| Proportional error, efficacy | 0.116 (42%) | 0.117 (9%) |
| Additive error, efficacy | 38.9 (28%) | 38.9 (8%) |
BCDAI, baseline Crohn's Disease Activity Index; CD, Crohn's disease; CDAI, Crohn's Disease Activity Index; CL, clearance; F, bioavailability; IB50, 50% of maximum unbound systemic concentration; I max, maximum inhibition; IIV, interindividual variability; k a, absorption rate constant; PK, pharmacokinetic; Q, intercompartmental clearance; RSE, relative standard error; V c, central volume of distribution; T lag, absorption lag time; V p, peripheral volume of distribution
Typical value for CL = 0.26 × (1–0.362 × health status) × (baseline albumin/39)−1.32 × exp (η1); for health status, patients with CD = 0, and healthy subjects = 1.
Typical value for V c = 3.27 × (1 + 0.214 × gender) × exp (η2); for gender, female = 0, and male = 1.
Because brazikumab is an mAb specifically binding to IL‐23 and blocking the binding of IL‐23 to its receptor, an IDR model with inhibition of k in was chosen rather than an IDR model with stimulation of the first‐order rate for the elimination of the response (k out), to reflect the mechanism of action and better describe the observed longitudinal CDAI scores (Figure 1c, Equations (1), (2), (3), (4)). Data from the open‐label period were not included due to potential influence of unblinding on the clinical readout from the open‐label period. The placebo effect () was first estimated using the placebo arm data (Figure 1c right). The placebo effect was found to be a constant value as the best fit over the time in the double‐blind period (Equation 4).
| (1) |
| (2) |
| (3) |
| (4) |
CDAI is the CDAI scores over time; k in is the zero‐order production rate of the response; k out is the first‐order rate for the elimination of the response; I total is the total inhibitory effect accounting for the placebo effect (I placebo) and/or the drug effect (I drug); for the data from only the placebo arm, I drug is fixed as 0; and HL is the half‐life to describe the remission rate.
The observed efficacy data (longitudinal CDAI scores) of the brazikumab treatment arm was included to establish a PK/efficacy relationship (Figure 1c left). The trend between the predicted average brazikumab concentrations and changes of CDAI scores at week 8 with or without outliers excluded (two subjects had average exposures far above the others) both indicated the higher the average exposure, the less the response, which did not imply a clinically meaning relationship (Figure S1). Given only one dose level (700 mg i.v.) was investigated in the phase IIa study and the range of average brazikumab concentrations was narrow (the 5th and 95th percentiles of the average exposures: 43.7–106.1 ug/mL), the concentration dependent‐drug effect was not explored further. Instead, other confounding factors were expected to play a major role in influencing the therapeutic effect (e.g., prognostic or predictive biomarker). As such, concentration dependent‐drug effect for the dose level of 700 mg i.v. was considered as constant and cannot be estimated in the presence of other confounding factors. The concentration‐dependent drug effect was incorporated with the predictive biomarker‐dependent drug effect as part of (Figure 2, blue dash arrow). Statistical correlations and mathematical functions, such as linear, loglinear, maximum effect, and sigmoidal functions, were used to explore the relationships between the biomarkers of interest listed in Table 2 and drug and/or placebo effect. A minimal OFV of the model and pharmacologically meaningful parameter estimates were considered as the criteria to select the final model.
Among all the biological or clinical biomarkers, BIL22, BCRP, and BCDAI were the three biomarkers identified with statistical significance based on the screening criteria. Final models included BCDAI‐correlated placebo effect plus BIL22‐ or BCRP‐dependent drug effect (Figure 2). Pearson correlation was used to describe the relationship between BCDAI and model‐estimated placebo effect (p value < 0.001; Figure 3, Figure S2), whereas a sigmoidal function was used to describe the quantitative relationships between BIL22 or BCRP and drug effect (Equation 5).
| (5) |
I max is the maximum inhibition on the CDAI production rate dependent on the baseline biomarker level caused by the drug effect; IB50 is the baseline level of biomarker that would achieve 50% of I max; γ is the Hill coefficient to describe the steepness of the biomarker‐dependent drug effect; and C B is the baseline biomarker level of BIL22 or BCRP.
FIGURE 3.
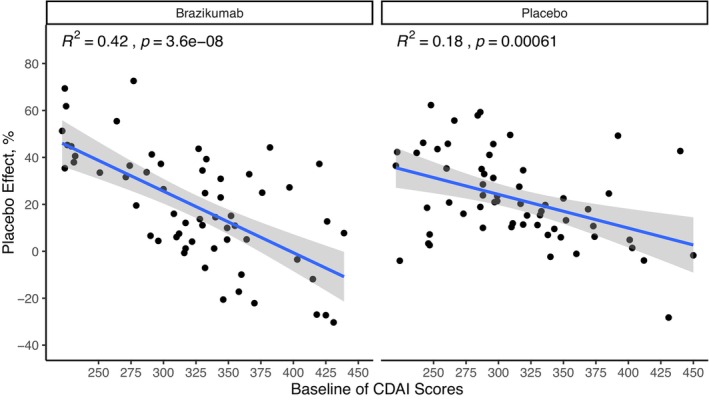
Statistical correlation between BCDAI and placebo effect of patients with the treatment of brazikumab or placebo in the final BIL22‐dependent efficacy model. BCDAI, baseline Crohn's Disease Activity Index; CDAI, Crohn's Disease Activity Index.
In the final BIL22‐ and BCRP‐dependent efficacy models, the placebo effect was estimated to inhibit k in by 20.9% and 17.8% and the correlations between individual BCDAI and placebo effect of all the subjects from placebo and treatment arms were both 100% (p value < 0.05; Table 2). Drug effect was found to be significantly related to BIL22 (p value < 0.01) or BCRP (p value < 0.05). In the final BIL22‐dependent efficacy model, the typical value of I max on k in caused by drug effect was 29.7%, the biomarker level achieving IB50 was 22.8 pg/mL and the steepness of the sigmoidal curve (γ) was 20 (fixed in the final model due to large uncertainty in the initial model estimation). In the final BCRP‐dependent efficacy model, the typical values of I max, IB50, and γ were 24.6%, 8.03 mg/L, and 2.07 (fixed in the final model due to large uncertainty in the initial model estimation), respectively.
Throughout the process of model development, the PK and biomarker/efficacy models were evaluated based on goodness‐of‐fit plots and VPCs. Prediction‐corrected VPC for the final population PK model was conducted and stratified based on subject populations and routes of drug administration in the phase Ib and IIa trials (Figure S3a). Similarly, VPCs were conducted for the final BIL22‐dependent and BCRP‐dependent efficacy models individually and stratified based on study arms in the phase IIa trial (Figure S3b,c).
The biomarker/efficacy relationship parameters in the final models, including BCDAI‐dependent placebo effect and BIL22‐ or BCRP‐dependent drug effect, were estimated with good precision. Re‐sampled parameters using the final model estimates demonstrate that for subjects with low BIL22 (far below 22.8 pg/mL), the population mean value of the maximal total inhibition (I total) is 20.9%, much lower compared to 50.6% of I total for subjects with high BIL22 (far above 22.8 pg/mL; Figure 4a). Similarly, the treatment of brazikumab could lead to an I total as low as 17.8% for subjects with low BCRP (far below 8.03 mg/L), compared to 42.4% for subjects with high BCRP (far above 8.03 mg/L; Figure 4b).
FIGURE 4.
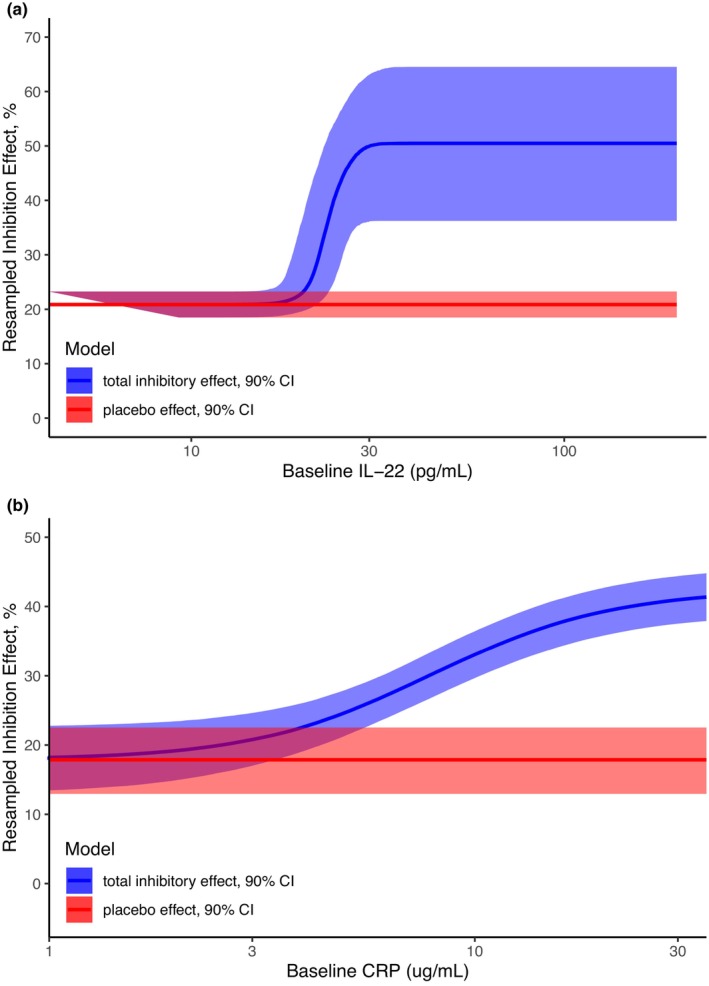
Re‐sampled relationship between the decrease in CDAI scores as a function of the baseline level of IL‐22 (a) and CRP (b) based on the efficacy model. the placebo effect is shown as the red ribbon (red line representing the typical placebo effect and shaded area representing the 90% confidence interval [CI]), while the total inhibitory effect from both the placebo effect and drug effect is shown as the blue ribbon (blue line representing the typical total inhibitory effect and shaded area representing the 90% confidence interval of the drug effect together with the typical placebo effect). This CI is derived from 5000 simulations using the parameters in the covariance matrix output from NONMEM. (a) BIL22‐dependent inhibitory effect. (b) BCRP‐dependent inhibitory effect. CDAI, Crohn's Disease Activity Index; CI, confidence interval.
To investigate whether the cutoff estimated through modeling can effectively identify subgroup subjects with higher response in the treatment arm, a t‐test was conducted to compare the change of CDAI score from week 0 to week 8 between biomarker subgroup subjects (biomarker high vs. biomarker low) using the model‐estimated IB50 or median of biomarker levels in the phase IIa study (Figure 5). 2 Using the IB50, comparing the mean value between subgroups, the change of CDAI scores at week 8 from week 0 shows a statistically significant difference between high and low biomarker groups for both BIL22 (high group vs. low group: −169.8 (60% relative standard deviation [RSD] vs. −72.3 [137% RSD], p value < 0.01) and BCRP (high group vs. low group: −131.5 [71% RSD] vs. − 29.9 [396% RSD], p value < 0.01) in the treatment arm. When using the median, the difference between high and lower biomarker group for the change of CDAI score at week 8 from week 0 is not significant for BIL22 (high group vs. low group: −123.0 [100% RSD] vs. −89.2 [108% RSD], p value = 0.29), but remains significant for BCRP (high group vs. low group: −147.7 [66% RSD] vs. −69.7 [154% RSD], p value <0.01). The difference in efficacy response between high and low biomarker groups for BIL22 is more significant when using the model‐derived IB50 as the cutoff than the median (p = 0.0026 vs. p = 0.29). The difference in efficacy response between high and low biomarker groups for BCRP is comparable between using IB50 as the cutoff and using median (p = 0.009 vs. p = 0.0084). However, the change of CDAI scores at week 8 from week 0 using both method is not statistically significantly different between high and low biomarker groups for both BIL22 and BCRP in the placebo arm (Figure S4). This comparison demonstrated that using pharmacometrics has the potential for improving power to detect the therapeutic effect of brazikumab by separating the high‐responders from the low‐ or nonresponders.
FIGURE 5.
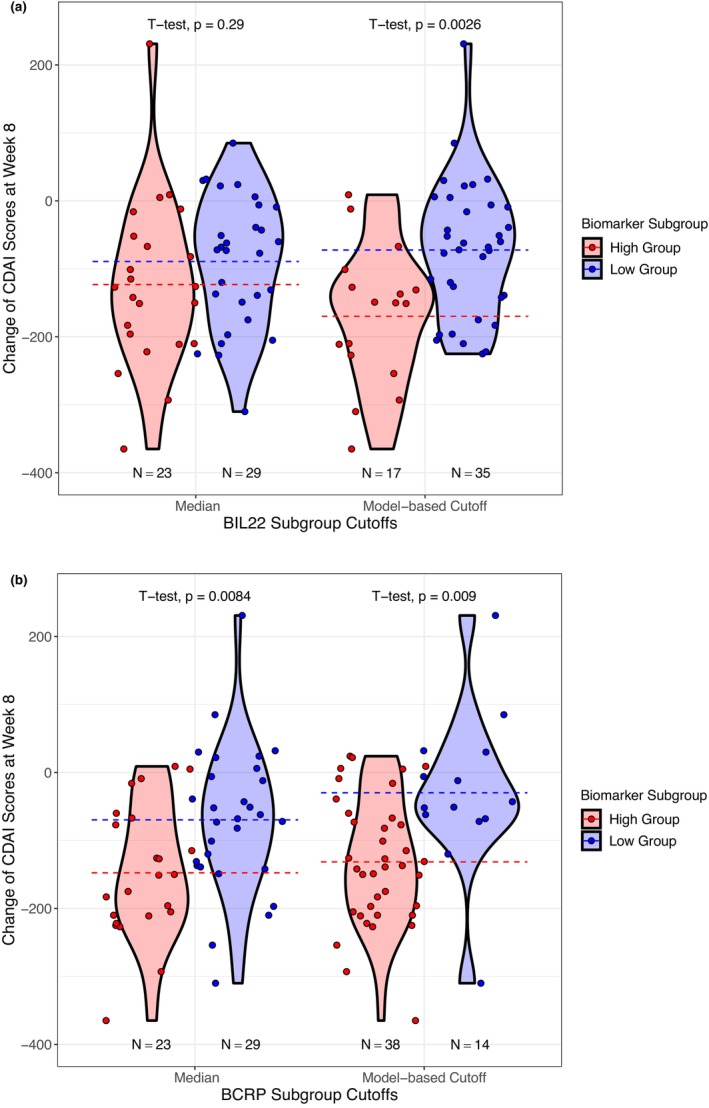
Statistical analysis of the difference in changes of CDAI scores at week 8 between high and low biomarker groups in the treatment arm. Among 59 patients who received brazikumab in the treatment arm, 52 patients had CDAI measurement around week 8. The p values are compared between the t‐tests using biomarker median and model‐based estimate (IB50) as the cutoff for BIL22 and BCRP individually. Dash lines are the means of the changes of CDAI scores at week 8 of each biomarker subgroup. (a) BIL22‐dependent efficacy response. (b) BCRP‐dependent efficacy response. CDAI, Crohn's Disease Activity Index.
DISCUSSION
In this work, we presented a pharmacometric framework for analyzing the clinical data by developing two biomarker/efficacy models to investigate the utility of biomarkers, specifically BIL22 and BCRP, in predicting efficacy response to brazikumab. This framework allows for a quantitative description of the relationship between biomarkers and efficacy response caused by drug effect and/or placebo effect, as well as random effects in the model parameters. This work exemplifies the value of pharmacometrics to define the cutoffs of BIL22 and BCRP for identifying patients with higher response to brazikumab and enable biomarker‐based precision medicine.
The PK data of brazikumab obtained from both phase Ib and IIa studies were best described using a two‐compartment model with first‐order absorption and elimination (Figures 1 and 2). The PK model showed a 36.2% slower clearance in HVs, compared with patients with CD at the same BALB level. Fecal loss of proteins due to the disruption of the mucosal lining of the gut wall (“leaky gut”) was speculated as the major factor contributing to increased drug clearance, as was shown in patients with severe IBD undergoing infliximab treatment. 26 Like several other biologics, BALB had a negative impact on the clearance of brazikumab. 27 Compared to female subjects, male subjects had a higher volume of distribution of the central compartment.
The efficacy data from the phase IIa study was described in two indirect response models, of which the placebo effect was correlated with BCDAI and the drug effect was dependent on BIL22 or BCRP in a sigmoidal function (Figures 1c and 2). In the relationship between BIL22 and drug effect, a value of γ around 20 can be easily affected in the estimation step and thus results in a large uncertainty in the initial estimation, and it was fixed as 20 in the final biomarker/efficacy model. This large γ value indicates a steep relationship, which approximates a dichotomous function for the biomarker‐dependent drug effect, suggesting that at the dose of 700 mg i.v., brazikumab would lead to roughly 50.6% inhibition for patients with BIL22 level above the IB50 (22.8 pg/mL), whereas for patients with a BIL22 level much lower than 22.8 pg/mL, brazikumab had minimal total inhibition of 20.9% (Figure 4a). This is consistent with the previous publication for the phase IIa study, where patients with higher BIL22 tended to have a higher remission in terms of the reduction in CDAI scores, of which the median of BIL22 (15.6 pg/mL) was used arbitrarily as the cutoff. 2 The t‐test result with the IB50 as the cutoff for subgrouping the subjects showed a more significant difference in the change of CDAI score at week 8 between the high and low biomarker groups, in comparison to that with the median as the cutoff (p value: 0.0026 vs. 0.29; Figure 5a). Therefore, our model‐based analysis provides a data driven cutoff value of BIL22 (IB50) to distinguish the high‐responders from the low‐ or nonresponders to brazikumab with improved precision than the traditional statistical analysis. In the BCRP‐dependent efficacy model, although the value of γ in the initial estimation is moderate, it still caused large uncertainty in the estimation of the steepness of the biomarker/drug effect relationship. As such, the value of γ was fixed as the initial estimate of 2.07 (Table 2). The BCRP‐dependent drug effect implies that brazikumab could lead to a total inhibition between 17.8% and 42.4% depending on the individual BCRP level at the dose of 700 mg i.v. (Figure 4b). This relationship is consistent with the findings of ustekinumab, which also targets IL‐23, that subjects with high BCRP (≥10 mg/L) had a high response to the treatment. 14 Although there is not a steep relationship between BCRP and drug effect in our model‐based analysis, the t‐test using the IB50 (8.03 mg/L) as the cutoff showed that the difference between the high and low biomarker groups had similar significance as that using the median (15.7 mg/L) as the cutoff (p value: 0.009 vs. 0.0084; Figure 5b). Neither IB50‐ nor median‐based cutoff of BIL22 and BCRP resulted in statistical significance in the comparison between the high and low biomarker groups in the t‐test of the placebo arm (Figure S4). In the phase IIa study, fewer subjects (17 vs. 23) were in the BIL22 high biomarker group when using IB50 of 22.8 pg/mL as the cutoff than using the median of 15.6 pg/mL as the cutoff, whereas more subjects (38 vs. 23) were in BCRP biomarker high group when using the IB50 of 8.03 mg/L as the cutoff than using the median of 15.7 mg/L as the cutoff. Adding both BIL22 and BCRP into the biomarker/efficacy model did not result in a more statistically significant relationship between biomarkers and CDAI scores, possibly due to the small sample size of the treatment arm and that BIL22 and BCRP are 60% correlated (p value = 5.332 × 10−13). Overall, total sample size included for this analysis is quite low, and this work is considered as exploratory. The cutoff estimation will be further optimized using more data in larger phase IIb or phase III studies. Patient selection using this approach should also consider other factors, such as the risk–benefit profile for brazikumab in the particular patient population.
By using a pharmacometrics approach, we were able to identify a negative correlation between BCDAI and placebo effect, and the latter has a typical value of 20.9% and 17.8% in the final BIL22‐dependent and BCRP‐dependent efficacy models, respectively (Table 2). The negative correlations between BCDAI and placebo effect is supported by the findings from several studies where patients with higher BCDAI had lower clinical response in the treatment arm and/or placebo arm. 8 , 9 , 10 The BCDAI‐correlated placebo effect and the sigmoidal relationships of biomarker‐dependent drug effect identified in the pharmacometric models implies that for those patients with extremely low baseline biomarker levels of BIL22 or BCRP, the efficacy is mainly driven by the placebo effect (Figure 4). Indeed, for a subpopulation that had BIL22 or BCRP below the IB50 estimates, the mean reduction of CDAI score from baseline was only 72.3 or 29.9, contrasted with 169.8 or 131.5 for the subpopulation that had BIL22 or BCRP above the IB50 estimates, respectively (Figure 5).
Five focus areas of the challenges in IBD research, which include preclinical human IBD mechanisms, environmental triggers, novel technologies, precision medicine, and pragmatic clinical research, were brought up in 2019. 28 Precision medicine, as one of the five focus areas, is to utilize specific clinical and biological characteristics to predict the course of disease development and treatment outcome in order to optimize clinical care. 28 The precision medicine approach involves stratifying patients into distinct subgroups using clinical or biological characteristics and determining the optimal treatment strategies. 15 Therefore, identifying such clinical or biological characteristics and determining the optimal cutoff to stratify patients can be critical in the process of drug development and clinical practice. In this work, we utilized a pharmacometrics approach to screen a series of clinical or biological characteristics, identify predictive or prognostic biomarkers to understand the relationships between these characteristics and clinical response, and predict the course of disease development and treatment outcomes. The advantages of pharmacometrics over traditional statistical analysis commonly used during clinical development provided us a tool with higher power to detect the biomarker‐dependent effect by integrating the longitudinal data over the time course while taking into account of the variability of multiple sources, including drug effect and placebo effect. In addition, this tool was also able to differentiate between predictive biomarkers (BIL22 and BCRP) and prognostic biomarkers (BCDAI) by quantifying biomarker‐dependent effect associated with placebo and drug treatment, respectively. The model‐estimated cutoffs of BIL22 and BCRP to identify patients with higher response will help in the design of subsequent phase IIb and phase III clinical trials with a selected subpopulation of patients and increase the power to detect the efficacy leading to a greater probability of success for the clinical development of brazikumab. Potentially, this knowledge will also help identify the patient population that is most likely to benefit from brazikumab.
In summary, this work demonstrates that pharmacometrics can be used to quantitatively evaluate potential predictive and prognostic biomarkers for biologic therapies for CD. Optimization of the biomarker cutoffs by pharmacometric analysis will potentially increase the power of detecting the efficacy of brazikumab, improve the probability of clinical trial success, and accomplish the precision medicine objective of identifying the patients most likely to benefit from treatment.
AUTHOR CONTRIBUTIONS
N.Z., M.L.C., and R.S. wrote the manuscript. N.Z., M.L.C., and R.S. designed the research. N.Z., M.L.C., B.S., and R.S. performed the research. N.Z., M.L.C., R.S., R.F., L.K.R., I.V., B.S. P.Z.B., and J.L. analyzed the data.
FUNDING INFORMATION
The study was supported by Catalyst Award for Therapeutic Project between March 1, 2016 and June 30, 2019.
CONFLICT OF INTEREST STATEMENT
N.Z., J.L., P.Z.B., B.S., I.V., L.K.R., and R.F. were employees of AstraZeneca and own stock in AstraZeneca at the time the study was conducted or the manuscript was being prepared. All other authors declared no competing interests for this work.
Supporting information
Appendix S1
Zhang N, Chan ML, Li J, et al. Combining pharmacometric models with predictive and prognostic biomarkers for precision therapy in Crohn's disease: A case study of brazikumab. CPT Pharmacometrics Syst Pharmacol. 2023;12:1945‐1959. doi: 10.1002/psp4.13044
Nan Zhang and Ming Liang Chan contributed equally to this work.
REFERENCES
- 1. Harlan WR, Meyer A, Fisher J. Inflammatory bowel disease: epidemiology, evaluation, treatment, and health maintenance. N C Med J. 2016;77(3):198‐201. [DOI] [PubMed] [Google Scholar]
- 2. Sands BE, Chen J, Feagan BG, et al. Efficacy and safety of MEDI2070, an antibody against interleukin 23, in patients with moderate to severe Crohn's disease: a phase 2a study. Gastroenterology. 2017;153(1):77‐86.e6. [DOI] [PubMed] [Google Scholar]
- 3. Neurath MF. Cytokines in inflammatory bowel disease. Nat Rev Immunol. 2014;14(5):329‐342. [DOI] [PubMed] [Google Scholar]
- 4. Mould D, Upton R, Wojciechowski J. Pharmacokinetics‐based dosing strategies for therapeutic proteins in inflammatory bowel disease. In: Zhou H, Mould D, eds. Quantitative Pharmacology and Individualized Therapy Strategies in Development of Therapeutic Proteins for Immune‐Mediated Inflammatory Diseases. Wiley; 2019. [Google Scholar]
- 5. Dignass A, Van Assche G, Lindsay JO, et al. The second European evidence‐based consensus on the diagnosis and management of Crohn's disease: current management. J Crohns Colitis. 2010;4(1):28‐62. [DOI] [PubMed] [Google Scholar]
- 6. Travis SP, Stange EF, Lémann M, et al. European evidence based consensus on the diagnosis and management of Crohn's disease: current management. Gut. 2006;55(Suppl 1):i16‐i35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. D'Haens GR, Panaccione R, Higgins PD, et al. The London position statement of the world congress of gastroenterology on biological therapy for IBD with the European Crohn's and colitis organization: when to start, when to stop, which drug to choose, and how to predict response? Am J Gastroenterol. 2011;106(2):199‐212. quiz 213. [DOI] [PubMed] [Google Scholar]
- 8. Hanauer SB, Panes J, Colombel JF, Bloomfield R, Schreiber S, Sandborn WJ. Clinical trial: impact of prior infliximab therapy on the clinical response to certolizumab pegol maintenance therapy for Crohn's disease. Aliment Pharmacol Ther. 2010;32(3):384‐393. [DOI] [PubMed] [Google Scholar]
- 9. Miyazaki T, Watanabe K, Kojima K, et al. Efficacies and related issues of Ustekinumab in Japanese patients with Crohn's disease: a preliminary study. Digestion. 2020;101(1):53‐59. [DOI] [PubMed] [Google Scholar]
- 10. Sandborn WJ, Colombel JF, Schreiber S, et al. Dosage adjustment during long‐term adalimumab treatment for Crohn's disease: clinical efficacy and pharmacoeconomics. Inflamm Bowel Dis. 2011;17(1):141‐151. [DOI] [PubMed] [Google Scholar]
- 11. Magro F, Rodrigues‐Pinto E, Santos‐Antunes J, et al. High C‐reactive protein in Crohn's disease patients predicts nonresponse to infliximab treatment. J Crohns Colitis. 2014;8(2):129‐136. [DOI] [PubMed] [Google Scholar]
- 12. Reinisch W, Wang Y, Oddens BJ, Link R. C‐reactive protein, an indicator for maintained response or remission to infliximab in patients with Crohn's disease: a post‐hoc analysis from ACCENT I. Aliment Pharmacol Ther. 2012;35(5):568‐576. [DOI] [PubMed] [Google Scholar]
- 13. Schreiber S, Rutgeerts P, Fedorak RN, et al. A randomized, placebo‐controlled trial of certolizumab pegol (CDP870) for treatment of Crohn‘s disease. Gastroenterology. 2005;129(3):807‐818. [DOI] [PubMed] [Google Scholar]
- 14. Toedter GP, Blank M, Lang Y, Chen D, Sandborn WJ, de Villiers WJS. Relationship of C‐reactive protein with clinical response after therapy with Ustekinumab in Crohn's disease. Am J Gastroenterol. 2009;104(11):2768‐2773. [DOI] [PubMed] [Google Scholar]
- 15. Ma C, Jairath V, Khanna R, Feagan BG. Investigational drugs in phase I and phase II clinical trials targeting interleukin 23 (IL23) for the treatment of Crohn's disease. Expert Opin Investig Drugs. 2018;27:1‐12. [DOI] [PubMed] [Google Scholar]
- 16. Schmechel S, Konrad A, Diegelmann J, et al. Linking genetic susceptibility to Crohn's disease with Th17 cell function: IL‐22 serum levels are increased in Crohn's disease and correlate with disease activity and IL23R genotype status. Inflamm Bowel Dis. 2007;14(2):204‐212. [DOI] [PubMed] [Google Scholar]
- 17. Iskandar HN, Ciorba MA. Biomarkers in inflammatory bowel disease: current practices and recent advances. Transl Res. 2012;159(4):313‐325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Best WR, Becktel JM, Singleton JW, Kern F Jr. Development of a Crohn's disease activity index. National cooperative Crohn's disease study. Gastroenterology. 1976;70(3):439‐444. [PubMed] [Google Scholar]
- 19. Lalonde RL, Kowalski KG, Hutmacher MM, et al. Model‐based drug development. Clin Pharmacol Therapeut. 2007;82(1):21‐32. [DOI] [PubMed] [Google Scholar]
- 20. Pinheiro J, Bornkamp B, Glimm E, Bretz F. Model‐based dose finding under model uncertainty using general parametric models. Stat Med. 2014;33(10):1646‐1661. [DOI] [PubMed] [Google Scholar]
- 21. Chan PL, Nutt JG, Holford NH. Pharmacokinetic and pharmacodynamic changes during the first four years of levodopa treatment in Parkinson's disease. J Pharmacokinet Pharmacodyn. 2005;32(3–4):459‐484. [DOI] [PubMed] [Google Scholar]
- 22. Gobburu JV, Lesko LJ. Quantitative disease, drug, and trial models. Annu Rev Pharmacol Toxicol. 2009;49:291‐301. [DOI] [PubMed] [Google Scholar]
- 23. Keizer RJ, Karlsson MO, Hooker A. Modeling and simulation workbench for NONMEM: tutorial on Pirana, PsN, and Xpose. CPT Pharmacometrics Syst Pharmacol. 2013;2:e50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jonsson EN, Karlsson MO. Xpose‐‐an S‐PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput Methods Programs Biomed. 1999;58(1):51‐64. [DOI] [PubMed] [Google Scholar]
- 25. Keizer RJ, van Benten M, Beijnen JH, Schellens JH, Huitema AD. Piraña and PCluster: a modeling environment and cluster infrastructure for NONMEM. Comput Methods Programs Biomed. 2011;101(1):72‐79. [DOI] [PubMed] [Google Scholar]
- 26. Brandse JF, van den Brink GR, Wildenberg ME, et al. Loss of infliximab into feces is associated with lack of response to therapy in patients with severe ulcerative colitis. Gastroenterology. 2015;149(2):350‐355.e2. [DOI] [PubMed] [Google Scholar]
- 27. Bajaj G, Suryawanshi S, Roy A, Gupta M. Evaluation of covariate effects on pharmacokinetics of monoclonal antibodies in oncology. Br J Clin Pharmacol. 2019;85(9):2045‐2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Denson LA, Curran M, McGovern DPB, et al. Challenges in IBD research: precision medicine. Inflamm Bowel Dis. 2019;25(Suppl 2):S31‐S39. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1