

Abstract
Background:
Treatment of chronic hepatitis C virus (HCV) infection with direct-acting antivirals (DAAs) for 6 weeks achieves sustained virologic response (SVR) rates of 95% in some patients. If effective, shorter therapeutic courses could improve adherence and treatment costs.
Objective:
To determine factors predictive of SVR to 4 weeks of DAA treatment in patients with stage F0 to F2 liver fibrosis.
Design:
Open-label, nonrandomized, phase 2a trial. (ClinicalTrials.gov: NCT01805882)
Setting:
Single-center.
Patients:
50 treatment-naive and predominantly African American patients with HCV genotype 1 infection and early-stage liver fibrosis were sequentially enrolled into 2 treatment groups.
Intervention:
25 participants received a 3-drug regimen consisting of ledipasvir and sofosbuvir plus GS-9451 for 4 weeks, and 25 received a 4-drug regimen consisting of ledipasvir, sofosbuvir, GS-9451, and GS-9669 for 4 weeks.
Measurements:
The primary efficacy end point was SVR12 (HCV RNA level below the lower limit of quantification at posttreatment week 12).
Results:
Forty percent (10 of 25) (95% CI, 21% to 61%) of patients in the 3-drug group and 20% (5 of 25) (CI, 7% to 41%) of those in the 4-drug group achieved SVR12. Exploratory analysis suggested that lower baseline HCV viral load, younger age, and HCV genotype 1b were associated with SVR12. Ten patients had baseline HCV variants conferring greater than 20-fold resistance in vitro to at least 1 study DAA; all had viral relapse. Forty-eight percent (12 of 25) of patients receiving the 3-drug regimen and 72% (18 of 25) of those receiving the 4-drug regimen had adverse events, most of which were mild. One participant was lost to follow-up.
Limitation:
Nonrandomized study design and small sample of patients with early-stage fibrosis.
Conclusion:
Combination DAA therapy with 3 or 4 drugs for 4 weeks was well-tolerated but resulted in limited cure rates.
Primary Funding Source:
National Institute of Allergy and Infectious Diseases, National Cancer Institute, and Clinical Center Intramural Program; supported in part by a cooperative research and development agreement between the National Institutes of Health and Gilead Sciences.
EDITORS’ NOTES
Context
In hepatitis C virus (HCV) infection, 6 weeks of therapy with direct-acting antivirals (DAAs) has been shown to result in sustained virologic response. A shorter regimen, if effective, could simplify therapy, avoid toxicity, and decrease drug costs.
Contribution
In a small clinical trial, 4-week therapy with ledipasvir and sofosbuvir plus 1 or 2 investigational agents resulted in suboptimal sustained viral suppression.
Caution
Patients were not randomly assigned, and the study was unblinded.
Implication
Additional clinical trials must be conducted to determine whether duration of DAA therapy for HCV infection can be shortened.
Hepatitis C virus (HCV) is a major cause of chronic liver disease and the leading cause of liver transplantation in developed countries (1, 2). With the approval of novel combinations of direct-acting antivirals (DAAs), several interferon-free treatment options are now available. Infection with HCV can now be treated with just 8 to 24 weeks of therapy, which has achieved sustained virologic response (SVR) rates as high as 91% to 100% in phase 3 clinical trials (3, 4).
Although regimens using only DAAs have shown markedly improved efficacy and safety, the cost of the drugs is substantial (5). Efforts to delineate patients who respond to shorter durations of therapy could help to determine the optimal combination of DAAs for different patient populations, which could in turn increase access to and uptake of these medications. However, the ideal regimen and duration for various subgroups have not yet been defined. In a recent preliminary study, we added a third potent antiviral—GS-9451 (an NS3/4A protease inhibitor) or GS-9669 (a nonnucleoside NS5B polymerase inhibitor)—to ledipasvir and sofosbuvir. With both 3-DAA combinations, an SVR rate of 95% (19 of 20 patients) was achieved in noncirrhotic patients with HCV genotype 1 infection after just 6 (rather than 12) weeks of treatment (6). A more rapid HCV viral kinetic decline was observed in patients treated with ledipasvir, sofosbuvir, and GS-9451 compared with ledipasvir and sofosbuvir only, suggesting that certain combinations of antivirals may hasten HCV eradication and permit shorter treatment courses (6).
In this study, we evaluated the efficacy, safety, and tolerability of ledipasvir, sofosbuvir, and GS-9451 with and without GS-9669 for 4 weeks in patients with chronic HCV genotype 1 infection. In addition, we examined whether characteristics of DAA regimens and host or viral factors previously overcome by duration or potency of multiple DAAs could be important determinants of response to shortened treatment duration. Finally, we targeted this evaluation to a predominantly urban African American population to improve the applicability of research findings in a group disproportionately affected by the HCV epidemic in the United States (7).
Methods
Patients
Patients were enrolled at a single site, the National Institutes of Health (NIH) Clinical Center in Bethesda, Maryland. Enrollment and follow-up data from January 2014 to May 2015 are reported here. Eligible participants were aged 18 years or older and had chronic HCV genotype 1 infection (serum HCV RNA level ≥2000 IU/mL), no prior treatment history, and stage F0 to F2 liver fibrosis. Fibrosis staging was determined by liver biopsy within 2 years of when the participant first received study medications or by a combination of the FibroSURE test (cutoff, <0.48) and the aspartate aminotransferase–platelet ratio (APRI) (cutoff, <1) within 6 months of screening. Full eligibility criteria are included in the Appendix (available at www.annals.org). Written informed consent was obtained from all participants at screening and enrollment.
Study Design
Study participants were sequentially enrolled into 2 groups, a design chosen to evaluate safety and efficacy in the 3-drug group before the 4-drug group, which could not be accomplished by randomization. Patients were contacted for screening and receipt of study drugs on the basis of the order of initial contact with the study team and fulfillment of eligibility requirements, respectively. Neither participants nor investigators were blinded. In the first group, 25 patients were treated for 4 weeks with ledipasvir, sofosbuvir, and GS-9451. In the second group, 25 patients were treated for 4 weeks with ledipasvir, sofosbuvir, GS-9451, and GS-9669. Sofosbuvir (400 mg) and ledipasvir (90 mg) were administered as a single combination tablet taken once daily, and GS-9451 (80 mg) and GS-9669 (250 mg) were administered as single tablets taken once daily. The protocol permitted participants for whom study treatment failed the option of re-treatment with the current standard of care.
Study Oversight
This study was approved by the Institutional Review Board of the National Institute of Allergy and Infectious Diseases (NIAID) and was conducted in accordance with good clinical practice guidelines, the Declaration of Helsinki, and regulatory requirements. The Office of Clinical Research Policy and Regulatory Operations (OCRPRO) of the NIAID served as the study sponsor and medical monitor.
Efficacy Assessments
Plasma HCV RNA levels were measured at day 0; week 2; week 4; and posttreatment weeks 2, 4, 12, and 24 using the RealTime HCV Assay (Abbott), with a lower limit of quantification (LLOQ) of 12 IU/mL. Serum HCV RNA levels, which were used for the primary outcome analysis, were also measured at selected time points using the COBAS TaqMan HCV RNA test, version 2.0 (Roche), with an LLOQ of 25 IU/mL.
Safety Assessments
Adverse events and clinical laboratory results were recorded throughout the study. Adverse events were graded from 1 (mild) to 4 (severe) according to the NIAID Division of AIDS toxicity table, version 1.0 (8).
Interferon-λ3 and Interferon-λ4 Genotyping
Interferon-λ3 (IFNL3), also known as IL28B, and interferon-λ4 (IFNL4) are genes whose phenotypes are associated with response to interferon-based treatment (9). Whole blood was collected using PAXgene Blood DNA Tubes (QIAGEN) and was stored at −80 °C until the time of DNA extraction with the PAXgene Blood DNA Kit (PreAnalytiX). IFNL3 and IFNL4 genotypes were determined using the 5′ nuclease assay with IFNL3 and IFNL4 allele-specific TaqMan probes (Applied Biosystems TaqMan allelic discrimination kit) and the Applied Biosystems 7500 Real-Time PCR System. Genotyping of variants at the rs12979860 (IFNL3/IL28B) and rs368234815 (IFNL4) loci was performed with custom TaqMan assays as previously described (10).
Clinical End Points
The primary efficacy end point was the proportion of participants with a serum HCV RNA level below the LLOQ at 12 weeks after completion of treatment (SVR12). The primary safety end point was the frequency and severity of adverse events. Secondary end points included the proportion of participants with unquantifiable levels of HCV RNA at specified time points, treatment discontinuations due to adverse events and laboratory changes, and evaluation of HCV resistance mutations at baseline in all patients and at viral relapse in applicable patients. Other uncompleted secondary end points are not reported. Data through posttreatment week 12 are included.
Deep Sequencing
Deep sequencing of the HCV NS3/NS4, NS5A, and NS5B genes (≥5000 reads) was performed by DDL Diagnostic Laboratory (Rijswijk, the Netherlands) to identify resistance-associated variants (RAVs). Sequencing was completed using samples collected at baseline in all patients and at the time of virologic failure in those who had relapse. All 3 regions were amplified by reverse transcriptase polymerase chain reaction using genotype-specific primers. Polymerase chain reaction products were further deep-sequenced by using Illumina MiSeq technology as previously described (10).
Statistical Analysis
The primary efficacy and safety end points were based on an intention-to-treat analysis and included all patients who received at least 1 dose of study medication. Patients with missing primary end point data were considered to have had treatment failure. A sample size of 25 per group provided a 93% probability of observing at least 1 participant with an adverse event that occurred in at least 10% of patients. This sample size also permitted an SVR12 estimate with a lower 95% confidence bound of 86% (assuming all 25 patients achieved SVR12) or 80% (assuming 24 of 25 achieved SVR12).
Baseline demographic characteristics were compared using t tests for continuous outcomes and the chi-square or Fisher exact test for categorical outcomes. Confidence intervals for SVR12 were calculated using exact binomial distribution. In an exploratory analysis, treatment outcomes were compared by host and viral factors using t tests for continuous variables and chi-square or Fisher exact tests for dichotomous variables. Analyses were performed using PRISM 6.0 (GraphPad Software); S-Plus 8.0 (CANdiensten); and SAS, version 9.3 (SAS Institute).
Role of the Funding Source
The NIAID served as the study sponsor, and the OCRPRO of the NIAID was involved in the review and approval of the study (via the usual peer-review process) as well as the management of the study. The OCRPRO had no role in the study design; collection, analysis, or interpretation of the data; preparation, review, or approval of the manuscript; or the decision to submit the manuscript for publication. Investigators from the NIH and the University of Maryland had full access to all data in the study and performed data collection, review, and analysis. Gilead Sciences provided the study drug and collaborated on study design and analysis. All sponsors were able to provide comments on the manuscript, but the primary and corresponding authors made the final decisions on inclusion of edits and submission for publication.
Results
Sixty-seven participants were screened, and 50 were enrolled in the study. Seventeen patients were excluded because of active comorbid illness, advanced liver disease, or inability to attend frequent trial visits or stop using contraindicated medication. Forty-nine of 50 patients completed 4 weeks of treatment with study medications, and primary end point results were obtained for them. One patient in the 4-drug group was lost to follow-up after her day-7 visit and was therefore considered to have treatment failure.
Baseline Characteristics of Study Participants
Baseline characteristics were similar among participants in both treatment groups (Table 1). Overall, patients predominantly were African American (76% [38 of 50]) and male (72% [36 of 50]), had the IL28B non-CC genotype (90% [44 of 49]) or the IFNL4 non-TT/TT genotype (94% [46 of 49]), and were infected with HCV genotype 1a (68% [34 of 50]). Forty-two percent (21 of 50) of the participants had plasma HCV RNA levels greater than 6 × 106 IU/mL at baseline.
Table 1.
Baseline Demographic and Clinical Characteristics*
| Characteristic | Ledipasvir + Sofosbuvir + GS-9451 (n = 25) | Ledipasvir + Sofosbuvir + GS-9451 + GS-9669 (n = 25) |
|---|---|---|
| Mean age (SD), y | 54 (10) | 58 (8) |
| Male | 17 (68) | 19 (76) |
| Race† | ||
| African American | 19 (76) | 19 (76) |
| White | 5 (20) | 6 (24) |
| Ethnicity† | ||
| Hispanic | 1 (4) | 0 (0) |
| Non-Hispanic | 24 (96) | 25 (100) |
| Mean BMI (SD), kg/m2 | 28 (5) | 27 (5) |
| HCV genotype | ||
| 1a | 18 (72) | 16 (64) |
| 1b | 7 (28) | 9 (36) |
| HCV RNA level >6 × 106 IU/mL IFNL3/IL28B genotype‡ | 11 (44) | 10 (40) |
| CC | 3 (12) | 2 (8) |
| CT | 18 (72) | 14 (56) |
| TT | 4 (16) | 8 (32) |
| IFNL4 genotype‡ | ||
| TT/TT | 2 (8) | 1 (4) |
| ΔG/TT | 18 (72) | 15 (60) |
| ΔG/ΔG | 5 (20) | 8 (32) |
| Knodell HAI, METAVIR, or FibroSURE score§ | ||
| 0 | 5 (20) | 8 (32) |
| 1 | 14 (56) | 12 (48) |
| 2 | 6 (24) | 4 (16) |
| 3 | 0 (0) | 1 (4)∥ |
| Presence of any NS3, NS5A, or NS5B RAV | 10 (40) | 10 (40) |
| Presence of NS3, NS5A, or NS5B RAV with >20-fold resistance | 3 (12) | 7 (28) |
BMI = body mass index; HAI = Histology Activity Index; HCV = hepatitis C virus; RAV = resistance-associated variant.
Values are numbers (percentages) unless otherwise indicated.
Self-reported.
Could not be typed in 1 patient.
In the 3-drug group, fibrosis staging was done with the FibroSURE test/aspartate aminotransferase–platelet ratio in 8 patients (32%), the Knodell HAI system in 15 patients (60%), and the METAVIR system in 2 patients (8%). In the 4-drug group, staging was done with the FibroSURE test/aspartate aminotransferase–platelet ratio in 5 patients (20%), the Knodell HAI system in 16 patients (64%), and the METAVIR system in 3 patients (12%). For fibrosis scores reported as a range, the maximum value in the range was used.
One patient with stage 2 fibrosis by the METAVIR system was considered eligible before enrollment but was deemed to have stage 3 fibrosis when the slide was reread at the National Institutes of Health using the Knodell HAI system.
Virologic Response
Of the patients treated with ledipasvir, sofosbuvir, and GS-9451, 40% (10 of 25) (95% CI, 21% to 61%) achieved SVR12. Viral relapse occurred in 60% (15 of 25) (Figure 1). Of the patients treated with ledipasvir, sofosbuvir, GS-9451, and GS-9669, 20% (5 of 25) (CI, 7% to 41%) achieved SVR12. Seventy-six percent (19 of 25) had viral relapse, and 4% (1 of 25) were lost to follow-up (Figure 1 and Appendix Table 1, available at www.annals.org).
Figure 1. Proportion of patients with viral load below the LLOQ, by treatment week.
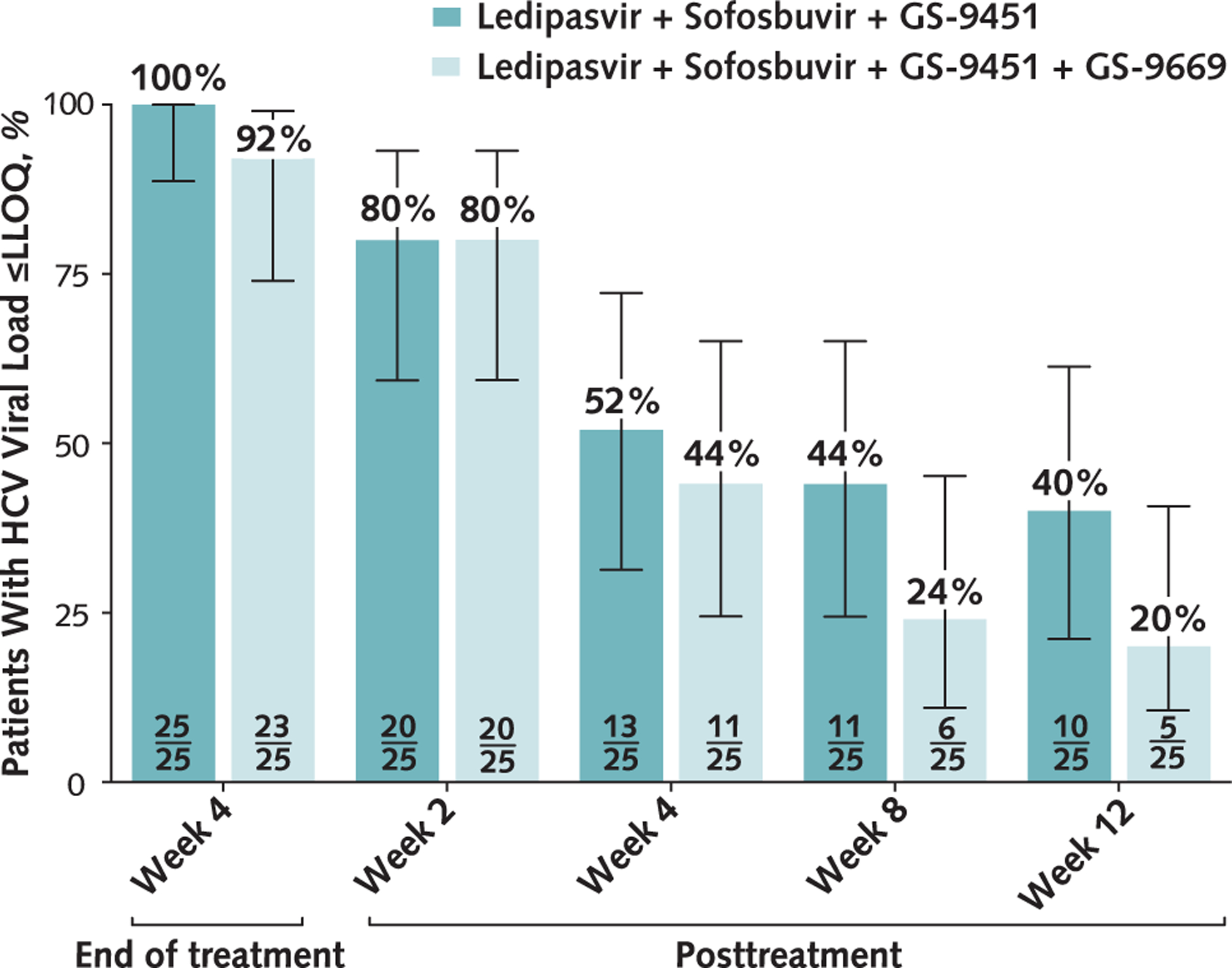
One patient in the 4-drug group who was lost to follow-up was counted as 1 of the 2 patients who did not achieve viral load below the LLOQ at week 4 (end of treatment). HCV = hepatitis C virus; LLOQ = lower limit of quantification.
All participants (25 of 25) enrolled in the 3-drug treatment group had unquantifiable levels of HCV RNA at the end of treatment (week 4) (Figure 1). After completion of treatment, viral relapse occurred in 20% (5 of 25) of patients at week 2, 28% (6 of 25) at week 4, 8% (2 of 25) at week 8, and 4% (1 of 25) at week 12. One patient had no quantifiable HCV RNA at posttreatment week 2 but was lost to follow-up until posttreatment week 24, when viral relapse was identified.
Ninety-two percent (23 of 25) of patients enrolled in the 4-drug group had unquantifiable levels of HCV RNA at week 4 (Figure 1). Eight percent (2 of 25) did not achieve viral load below the LLOQ at the end of treatment; 1 patient was lost to follow-up, and a second patient had quantifiable levels of HCV RNA. After completion of treatment, viral relapse occurred in 12% (3 of 25) of patients at week 2, 36% (9 of 25) at week 4, 20% (5 of 25) at week 8, and 4% (1 of 25) at week 12.
At week 2, patients in the 3-drug group had a mean decrease in HCV RNA level (±SE) of 4.5 ± 0.8 log10 IU/mL, and those in the 4-drug group had a mean decrease of 4.7 ± 0.5 log10 IU/mL. Decreases in HCV viral load were also similar between groups at week 4.
Analysis of Viral Mutations Associated With Resistance to DAAs Used in the Study
To evaluate whether baseline RAVs affect treatment outcomes, baseline frequencies of RAVs in each treatment cohort were determined by deep sequencing (Appendix Table 2, available at www.annals.org). Among the 50 total patients in both groups, 1 patient in the 4-drug group was lost to follow-up and had the NS3 R155K variant at baseline, and another patient in the 4-drug group had assay failure at baseline. Of the remaining 48 patients who completed the study and were included in the baseline and relapse RAV analyses, 33 had HCV genotype 1a infection and 15 had genotype 1b infection. Among the 33 patients with genotype 1a infection, 10 had the NS3 Q80K variant at baseline, which confers a 3-fold reduced susceptibility to GS-9451. Sixty percent (6 of 10) of patients with this variant and 87% (20 of 23) without it had viral relapse after 4 weeks of 3- or 4-drug treatment. At baseline, NS3 RAVs (R155K and D168E) and/or NS5A RAVs (L31M, Y93H, and Y93N) were observed in 3 of 25 and 7 of 23 patients in the 3- and 4-drug groups, respectively. These NS3 and NS5A RAVs confer a greater than 20-fold reduced susceptibility to GS-9451 and ledipasvir, respectively. Viral relapse occurred in 100% (10 of 10) of patients with these pretreatment RAVs versus 63% (25 of 40) of those without high-level RAVs (55% [12 of 22] in the 3-drug group and 72% [13 of 18] in the 4-drug group). The M423I RAV in the NS5B region confers an approximate 4.6-fold resistance to GS-9669 in vitro and was found in 1 patient in the 4-drug group, who had viral relapse after 4 weeks of treatment. No variants conferring resistance to sofosbuvir were observed.
Frequencies of RAVs among patients with viral relapse were also determined, and RAVs were classified as emerged or enriched. Emerged RAVs were not present at baseline but were detected at relapse, whereas enriched RAVs were present at a higher frequency after relapse than at baseline. Among 34 patients with viral relapse after completion of therapy, none had emerged NS3 or NS5B RAVs; however, 85% (29 of 34) had emerged (58% [21 of 34]), maintained (9% [3 of 34]), or enriched (15% [5 of 34]) NS5A RAVs. Fifteen percent (5 of 34) had no resistance in any of the 3 genes.
Treatment Outcomes, by Host and Viral Factors
The mean age of patients who achieved SVR12 was 51.1 years versus 57.7 years for those with viral relapse (P = 0.02). Fifty percent (8 of 16) of patients infected with HCV genotype 1b achieved SVR12 compared with 21% (7 of 34) of those with genotype 1a infection (P = 0.049) (Figures 2 and 3). Forty-five percent (13 of 29) of Article 02: Untitled patients with a baseline HCV viral load less than 6 × 106 IU/mL achieved SVR12 compared with 10% (2 of 21) of those with a viral load greater than 6 × 106 IU/mL (P = 0.007). Finally, 38% (15 of 40) of patients without pretreatment RAVs that conferred a greater than 20-fold resistance to study drugs achieved SVR12, whereas all 10 patients with such RAVs had treatment failure (P = 0.022) (Appendix Table 3, available at www.annals.org). In the subset of patients (treated with either regimen) who were infected with HCV genotype 1b, had an HCV viral load less than 6 × 106 IU/mL, and had no baseline RAVs, 75% (6 of 8) achieved SVR (Appendix Figures 1 and 2, available at www.annals.org).
Figure 2. HCV viral load, by baseline HCV RNA level and RAV subgroup, in patients treated with ledipasvir and sofosbuvir plus GS-9451.
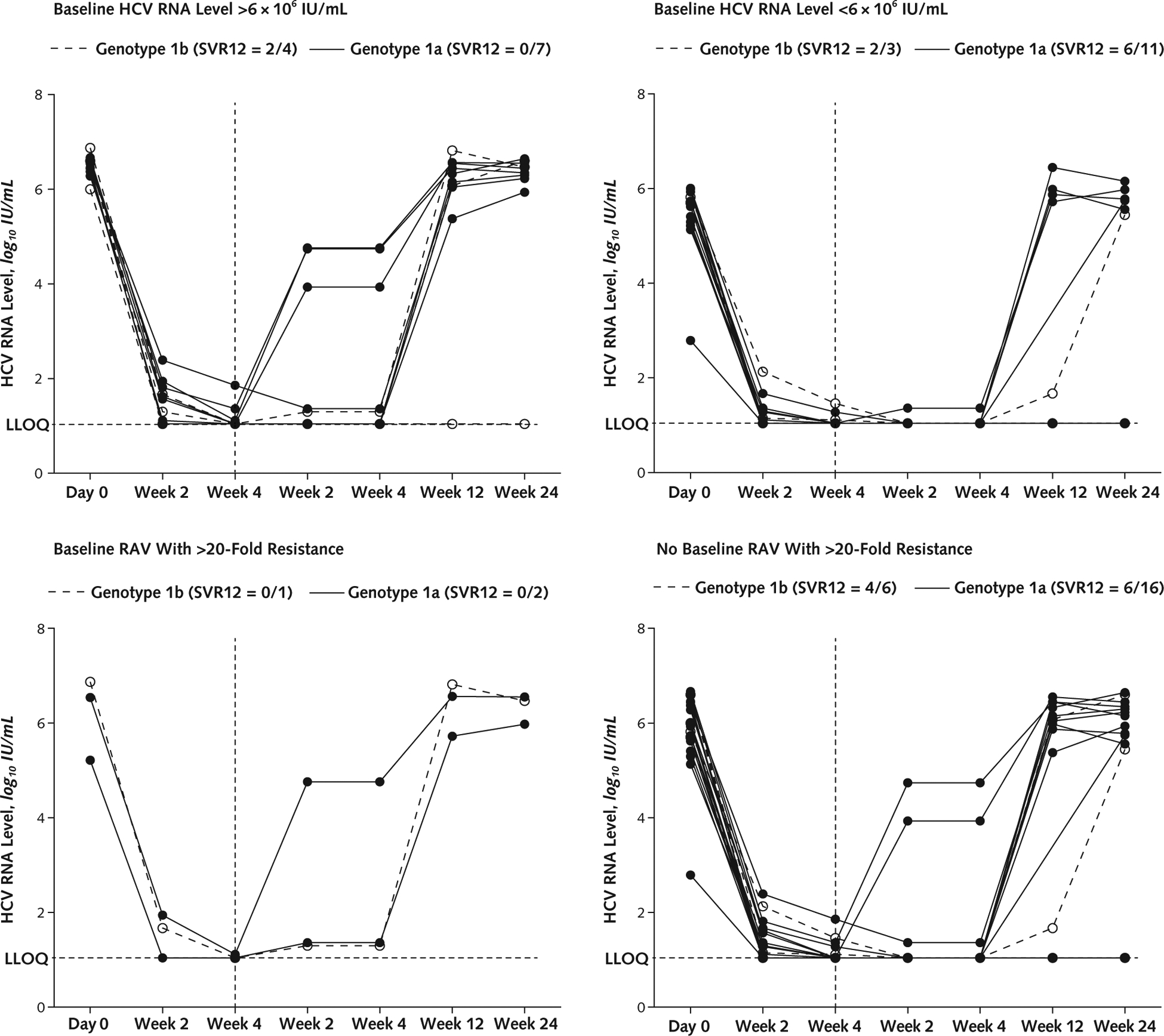
Vertical dashed lines indicate end of treatment. HCV = hepatitis C virus; LLOQ = lower limit of quantification; RAV = resistance-associated variant; SVR12 = HCV RNA level below LLOQ at posttreatment week 12.
Figure 3. HCV viral load, by baseline HCV RNA level and RAV subgroup, in patients treated with ledipasvir and sofosbuvir plus GS-9451 with or without GS-9669.
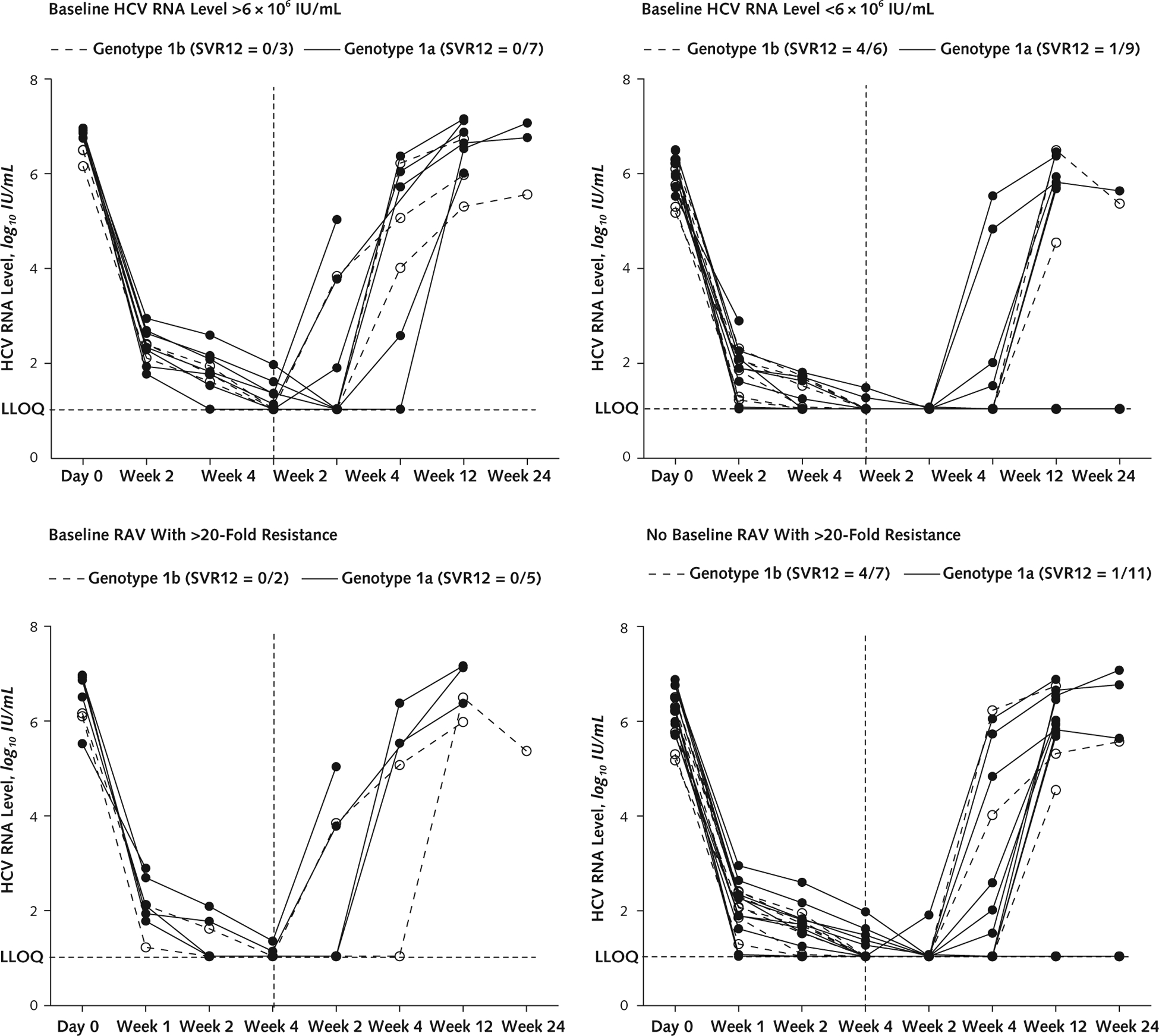
Vertical dashed lines indicate end of treatment. HCV = hepatitis C virus; LLOQ = lower limit of quantification; RAV = resistance-associated variant; SVR12 = HCV RNA level below LLOQ at posttreatment week 12.
Safety
Forty-eight percent (12 of 25) of patients receiving the 3-drug regimen and 72% (18 of 25) of those receiving the 4-drug regimen had adverse events, most of which were mild. The most common adverse events were fatigue, diarrhea, and headache (Table 2). Two serious adverse events occurred: vertigo for 3 days in a patient with a heart rate of 57 beats/min who was not receiving antiarrhythmic agents, and angioedema that resolved after the patient discontinued use of lisinopril. Both events occurred in patients receiving the 4-drug regimen but were deemed to be unrelated to the study drugs. No patient discontinued therapy because of an adverse event, and no deaths occurred in the study.
Table 2.
Adverse Events and Laboratory Abnormalities During Treatment*
| Variable | Ledipasvir + Sofosbuvir + GS-9451 (n = 25) | Ledipasvir + Sofosbuvir + GS-9451 + GS-9669 (n = 25) |
|---|---|---|
| Any adverse event during treatment† | 12 (48) | 18 (72) |
| Any serious adverse event during treatment | 0 (0) | 2 (8) |
| Discontinuations due to adverse events | 0 (0) | 0 (0) |
| Adverse events occurring in ≥ 10% of patients‡ | ||
| Constipation | 3 (12) | 2 (8) |
| Nausea | 0 (0) | 6 (24) |
| Fatigue | 4 (16) | 4 (16) |
| Diarrhea | 2 (8) | 5 (20) |
| Headache | 1 (4) | 6 (24) |
| Dry mouth | 4 (16) | 2 (8) |
| Dizziness | 1 (4) | 3 (12) |
| Abdominal pain | 0 (0) | 3 (12) |
| Any grade 3 laboratory abnormality during treatment | 2 (8) | 2 (8) |
| Hypophosphatemia | 1 (4) | 0 (0) |
| Blood amylase level increased | 1 (4) | 0 (0) |
| Hyperglycemia | 0 (0) | 2 (8) |
Values are numbers (percentages).
Includes time receiving study medication and 30 d after discontinuation.
Refers to patients with events.
No grade 4 laboratory abnormalities occurred. Four patients had grade 3 laboratory abnormalities (2 with a history of type 2 diabetes mellitus had hyperglycemia, 1 had asymptomatic hypophosphatemia after completion of treatment, and 1 had an asymptomatic elevation in blood pancreatic amylase levels after completion of treatment).
DISCUSSION
In this proof-of-concept study involving treatment-naive noncirrhotic patients with chronic HCV genotype 1 infection, 4 weeks of treatment with ledipasvir, sofosbuvir, and GS-9451 with or without GS-9669 was well-tolerated; however, only 30% (15 of 50) of patients achieved SVR12. Sixty-eight percent (34 of 50) of patients had viral relapse, the majority (68% [23 of 34]) by posttreatment week 4. Thus, a treatment duration of 4 weeks with 3 or 4 potent DAAs is not sufficient to cure HCV infection in most patients.
A previous study showed that the addition of a third drug to ledipasvir and sofosbuvir for 6 weeks resulted in SVR12 in 39 of 40 patients, suggesting that regimens shorter than 8 to 12 weeks may be highly effective for some patients with chronic HCV infection (6). Given the high cost of DAAs, the medical care required, and the improvements in patient adherence with reductions in treatment duration (11), attempts to evaluate shorter courses of therapy could have important clinical and public health implications. Hence, we explored predictors of achievement of SVR in patients treated with multiple DAA combinations for 4 weeks.
Our results suggest that younger age, infection with HCV genotype 1b, baseline viral load less than 6 × 106 IU/mL, and absence of an RAV conferring a greater than 20-fold in vitro resistance were associated with SVR12 with 4 weeks of therapy (Appendix Figures 1 and 2). This is similar to the findings of another study that evaluated 4 weeks of therapy with a different combination of DAAs (12). This population should be evaluated in larger studies to identify the subsets of patients with acceptable response rates to a 4-week treatment regimen, thus resulting in a shorter and probably more cost-effective strategy for treatment of HCV infection.
Factors associated with treatment success that were observed in this study are consistent with those from previous studies (Figures 2 and 3). The favorable association between HCV genotype 1b infection and SVR12 has been observed in trials of sofosbuvir and other DAA-only regimens and with interferon-containing regimens (13–16). This may be related to host–virus interactions; a higher resistance barrier for genotype 1b than for genotype 1a, making RAVs more likely to develop to genotype 1a; or the differential efficacy of the DAAs used in this study in patients with genotype 1b infection. In the HCV replicon system, the mean effective concentrations for ledipasvir, sofosbuvir, GS-9669, and GS-9451 are lower against genotype 1b than genotype 1a. In addition, low baseline HCV viral load has been reported to be a predictor of treatment success during 8-week regimens of ledipasvir and sofosbuvir (3). Among patients with a high baseline viral load, a 12-week duration results in higher SVR12 rates, suggesting that patients with higher levels of circulating virus may require longer-duration therapy for eradication. In our study, the NS3/4A RAV Q80K did not seem to have a negative effect on treatment outcomes with GS-9451. However, 20% of patients had a baseline RAV conferring a greater than 20-fold resistance to ledipasvir or GS-9451 (6), and none of these patients achieved SVR12. In contrast, preexisting RAVs did not preclude treatment success in phase 3 trials of ledipasvir and sofosbuvir for 8 to 24 weeks, although NS5A RAVs were more common in patients with viral relapse than in those who achieved SVR (3, 4). In a recent study of a 6-week regimen similar to the one evaluated here (6), 8 of 40 patients had baseline RAVs conferring greater than 20-fold resistance, and all but 1 achieved SVR12 (17).
Our data suggest that for most patients, a treatment duration longer than 4 weeks is required to achieve SVR. However, some patients are capable of achieving SVR with 4 weeks of therapy, which could be attributable to the presence of several favorable factors, such as early fibrosis, low viral burden, and absence of RAVs.
Although the addition of a third DAA seemed to improve SVR rates in a prior study of 6-week regimens (6, 18), we found no evidence that the addition of a fourth drug to a potent 4-week regimen with maximal suppression of HCV enhanced the likelihood of achieving SVR12. Furthermore, the lack of resistance in the NS3 and NS5B regions suggests that relapse was primarily due to the need for a longer duration of viral suppression rather than viral resistance. A minimal duration of viral suppression may be necessary to eliminate all infected hepatocytes in most patients.
Limitations of our study include its sequential, nonrandomized enrollment and the fact that it was a single-center trial. In addition, our small patient sample limits confidence in the estimates of efficacy and limited our ability to perform multivariate analyses of factors related to treatment response. Patients with stage 3 or 4 liver disease were not included because other studies have shown that these patients may be more difficult to treat or may require therapy lasting longer than 4 weeks (10).
In conclusion, treatment for 4 weeks with the all-oral combination DAA regimens used in this study seems to have high tolerability but limited response in achieving SVR in noncirrhotic treatment-naive patients with HCV genotype 1 infection.
Annals of Internal Medicine Junior Investigator Awards.
Annals of Internal Medicine and the American College of Physicians recognize excellence among internal medicine trainees and junior investigators with annual awards for original research and scholarly review articles published in Annals in each of the following categories:
Most outstanding article with a first author in an internal medicine residency program or general medicine or internal medicine sub-specialty fellowship program
Most outstanding article with a first author within 3 years following completion of training in internal medicine or one of its subspecialties
Selection of award winners will consider the article’s novelty, methodological rigor, clarity of presentation, and potential to influence practice, policy, or future research. Judges will include Annals Editors and representatives from Annals’ Editorial Board and the American College of Physicians’ Education/Publication Committee.
Papers published in the year following submission are eligible for the award in the year of publication. First author status at the time of manuscript submission will determine eligibility. Authors should indicate that they wish to have their papers considered for an award when they submit the manuscript, and they must be able to provide satisfactory documentation of their eligibility if selected for an award. Announcement of awards for a calendar year will occur in January of the subsequent year. We will provide award winners with a framed certificate, a letter documenting the award, and complimentary registration for the American College of Physicians’ annual meeting.
Please refer questions to Mary Beth Schaeffer at mschaeffer@acponline.org or visit www.annals.org/public/juniorinvestigatoraward.aspx.
Acknowledgment:
The authors thank the following persons for their contributions: Erin Rudzinski and Susan Vogel, RN, BSN (clinical monitoring support); Judith Starling, PharmD (pharmacy); Jerome Pierson, PhD, and John Tierney, BSN, MPM (regulatory support); Marc Teitelbaum, MD, CPI (sponsor medical monitor); Mary Hall (protocol support); Cathy Rehm and Sara Jones (laboratory support); and Richard Kwan, PA, Maryellen McManus, RN, and Senora Mitchell (clinic support).
Financial Support: This project was funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract HHSN261200800001E. This research was supported in part by the National Institute of Allergy and Infectious Diseases and by the German Research Foundation clinical research unit KFO 129. Study medications were provided by Gilead Sciences, and the study was partially funded by a cooperative research and development agreement between the National Institutes of Health and Gilead Sciences. Drs. Osinusi and Mo are employees of Gilead Sciences and provided commentary to the manuscript.
Appendix: Inclusion and Exclusion Criteria
Main Inclusion Criteria
Aged 18 years or older at screening
- Female study participants with childbearing potential (as defined below) and all male participants must be willing to practice either
- abstinence from sexual intercourse, or
-
one or more forms of effective barrier contraception throughout dosing and for 30 days after the last dose. This cannot include hormonal contraception for female participants.Effective forms of barrier contraception include
- a male condom with spermicide, or
-
use by female sexual partner of a female condom with spermicide.Nonchildbearing potential (i.e., physiologically incapable of becoming pregnant) includes any woman who
- has had a hysterectomy,
- as had a bilateral oophorectomy (ovariectomy),
- is postmenopausal (a demonstration of a total cessation of menses for ≥1 year), or
- has had a bilateral tubal ligation or fallopian tube inserts.
- Chronic infection with HCV genotype 1, as documented by at least 1 measurement of serum HCV RNA of at least 2000 IU/mL during screening and at least 1 of the following:
- A positive anti-HCV antibody, HCV RNA, or HCV genotype test result at least 12 months before the baseline (day 0) visit, plus current positive HCV RNA and anti-HCV antibody test results.
- Positive HCV RNA and anti-HCV antibody test results, plus liver biopsy consistent with chronic HCV infection or liver biopsy performed before enrollment with evidence of chronic HCV infection, such as the presence of fibrosis.
-
Group A may include up to 20% of participants with compensated cirrhosis. Groups B and C may only include participants without cirrhosis.
Cirrhosis is defined as 1 of the following:- Any biopsy showing cirrhosis.
-
A FibroSURE score of at least 0.75 and an APRI greater than 2 performed within 12 months of screening.Liver imaging within 6 months of day 0 to exclude hepatocellular carcinoma is required in patients with cirrhosis.
Absence of cirrhosis is defined as 1 of the following:- A liver biopsy performed within 36 calendar months of screening showing absence of cirrhosis.
-
A FibroSURE score less than 0.48 and APRI less than 1 performed within 6 months of screening.In the absence of a definitive diagnosis of presence or absence of cirrhosis by the above criteria, a liver biopsy is required.
Ability to communicate effectively with the study investigator and other key personnel.
Willing to give written informed consent and comply with the study restrictions and requirements.
Opioid-dependent persons must be participating in a supervised treatment program.
Participants must have an external primary care physician (outside of the Clinical Center and the NIH) for their medical management.
Main Exclusion Criteria
- Current or prior history of any of the following:
- Clinically significant illness (other than HCV infection) or any other major medical disorder that may interfere with treatment, assessment, or adherence to the protocol; participants currently under evaluation for a potentially clinically significant illness (other than HCV infection) are also excluded.
- Gastrointestinal disorder or postoperative condition that could interfere with the absorption of the study drug.
- Poor venous access interfering with required study blood collection.
- Clinical hepatic decompensation (i.e., ascites, encephalopathy, or variceal hemorrhage).
- Solid organ transplantation.
- Significant pulmonary disease, significant cardiac disease, or porphyria.
- Unstable psychiatric disease (participants with psychiatric illness that is well-controlled on a stable treatment regimen or currently not requiring medication may be included).
- Any malignancy or its treatment that in the opinion of the principal investigator (PI) may cause ongoing interference with host immunity; participants under evaluation for malignancy are not eligible.
- Significant drug allergy (such as anaphylaxis or hepatotoxicity).
- Substance abuse, which in the opinion of the investigator is likely to interfere with medication adherence or study compliance.
- Lactose allergy; patients with lactose intolerance will be evaluated on a case-by-case basis.
Positive test results at screening for hepatitis B virus (HBV) surface antigen, IgM antibody subclass to hepatitis B core antigen, HBV DNA (if medically indicated), or anti-HIV antibody.
Prior exposure to any DAAs for HCV infection.
History of clinically significant chronic liver disease due to other cause (e.g., hemochromatosis, auto-immune hepatitis, Wilson disease, [H9251]1-antitrypsin deficiency, alcoholic liver disease, moderate or more severe nonalcoholic steatohepatitis, and toxin exposures).
Use of herbal/natural remedies for potential benefit to the liver within 21 days of day 0.
History of ascites, variceal hemorrhage, hepatic encephalopathy, or conditions consistent with decompensated liver disease.
Screening or baseline electrocardiography with clinically significant findings or a personal/first-degree relative history of torsade de pointes.
- Abnormal hematologic and biochemical parameters at screening, including:
- Neutrophil count less than 750 cells/mm3.
- Hemoglobin level less than 9 g/dL. If hemoglobin level is less than 11 g/dL (for women) or less than 12 g/dL (for men), other causes of anemia should be excluded as medically indicated.
- Platelet count of 50 000 cells/mm3 or lower.
- Estimated glomerular filtration rate less than 50 mL/min/1.73 m2.
- Alanine aminotransferase or aspartate aminotransferase level at least 10 times the upper limit of normal (ULN).
- Serum lipase level at least 1.5 times the ULN at screening or during the screening period.
- Total bilirubin level at least 2.0 times the ULN, except in participants with Gilbert syndrome.
- Albumin level of 3.0 g/dL or lower in patients without cirrhosis (≤2.8 g/dL in cirrhotic patients).
Poorly controlled diabetes mellitus indicated by hemoglobin A1c level greater than 9% at screening.
Donation or loss of blood greater than 400 mL within 8 weeks before the first dose of the study drugs.
Known hypersensitivity to GS-5885, GS-7977, GS-9669, GS-9451, or formulation excipients.
Pregnant or breastfeeding women.
- Need for the following medications from 21 days before the start of study drugs through the end of treatment (unless otherwise specified in Tables 8 to 10 of protocol):
- Hematologic stimulating agents (e.g., erythropoiesis-stimulating agents, granulocyte colony-stimulating factor; thrombopoietin mimetics).
- Chronic systemic immunosuppressants, including but not limited to corticosteroids (prednisone equivalent of >10 mg/d for >2 weeks), azathioprine, or monoclonal antibodies (e.g., infliximab).
- Investigational agents or devices for any indication.
- Medications for disease conditions excluded from the protocol (e.g., active cancer or transplantation) are not listed under this concomitant medication section and are disallowed in the study.
- Concomitant use of certain medications or herbal/natural supplements per PI discretion expected to result in pharmacokinetic interactions resulting in increases or decreases in exposure of study drug(s) as listed in Tables 7 to 9 of the protocol.
Appendix Table 1.
Individual Patient Characteristics and Treatment Response
| Patient | Treatment Response | Age, y | Sex | Race | BMI, kg/m2 | IFNL3/IL28B Genotype | IFNL4 Genotype | Baseline LDL Cholesterol Level, mg/dL | Baseline Hemoglobin A1c Level, % | Baseline IP-10 Level | HCV Genotype | Baseline HCV Viral Load, IU/mL | Presence of Any NS3, NS5A, or NS5B RAV | Presence of Any NS3, NS5A, or NS5B RAV With >20-Fold Resistance |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ledipasvir + sofosbuvir + GS-9451 | ||||||||||||||
| 1 | REL | 57 | Male | African American | 28.77 | CT | dG/TT | 50 | 6 | 556 | 1a | 1 700 000 | No | No |
| 2 | SVR12 | 62 | Female | White | 31.2 | CT | dG/TT | 116 | 5.1 | 346 | 1a | 1 020 000 | Yes | No |
| 3 | REL | 63 | Female | African American | 38.52 | CT | dG/TT | 97 | 5.9 | 1185 | 1a | 7 270 000 | Yes | No |
| 4 | REL | 50 | Male | African American | 30.4 | CT | dG/TT | 89 | 5.3 | 444 | 1a | 3 980 000 | No | No |
| 5 | SVR12 | 52 | Male | African American | 37.61 | TT | dG/dG | 76 | 7.2 | 809 | 1a | 1 200 000 | No | No |
| 6 | SVR12 | 30 | Male | White | 33.5 | CT | dG/TT | 95 | 5.1 | 349 | 1a | 859 000 | Yes | No |
| 7 | SVR12 | 62 | Male | African American | 27.79 | CT | dG/TT | 126 | 5.4 | 913 | 1a | 169 000 | Yes | No |
| 8 | SVR12 | 55 | Female | African American | 25.95 | CT | dG/TT | 89 | 5.8 | 2232 | 1b | 1 750 000 | No | No |
| 9 | REL | 48 | Male | African American | 28.27 | CT | dG/TT | 35 | 4.4 | 1105 | 1a | 8 130 000 | No | No |
| 10 | REL | 58 | Male | African American | 25.8 | CT | dG/TT | 167 | 5.4 | 1505 | 1a | 2 980 000 | Yes | Yes |
| 11 | REL | 61 | Male | African American | 23.81 | CT | dG/TT | 73 | 6.1 | 498 | 1a | 5 140 000 | No | No |
| 12 | REL | 58 | Male | African American | 24.37 | CT | dG/TT | 68 | 6 | 1192 | 1b | 23 200 000 | No | No |
| 13 | REL | 63 | Male | African American | 31 | CT | dG/TT | 107 | 6.4 | 1093 | 1a | 29 600 000 | Yes | No |
| 14 | REL | 48 | Male | African American | 23.15 | TT | dG/dG | 52 | 5.3 | 1041 | 1a | 14 200 000 | Yes | Yes |
| 15 | SVR12 | 54 | Male | African American | 30.45 | CT | dG/TT | 86 | 7.4 | 887 | 1a | 875 000 | Yes | No |
| 16 | SVR12 | 55 | Female | African American | 20.63 | CT | dG/TT | 133 | 5.3 | 951 | 1b | 6 540 000 | No | No |
| 17 | REL | 60 | Male | African American | 27.26 | TT | dG/dG | 102 | 5.8 | 474 | 1a | 7 940 000 | Yes | No |
| 18 | REL | 56 | Male | African American | 27.93 | CC | dG/dG | 111 | 5.6 | 880 | 1a | 7 470 000 | No | No |
| 19 | SVR12 | 51 | Female | African American | 28.77 | CT | dG/TT | 82 | 5.9 | 900 | 1a | 4 830 000 | No | No |
| 20 | SVR12 | 51 | Female | White | 18.12 | CC | TT/TT | 172 | 5.3 | 280 | 1b | 1 300 000 | No | No |
| 21 | REL | 70 | Female | White | 29.88 | CT | dG/TT | 87 | 5.4 | 1457 | 1b | 1 880 000 | No | No |
| 22 | SVR12 | 25 | Male | Multiple | 25.35 | CC | TT/TT | 125 | 4.9 | 395 | 1b | 79 110 000 | No | No |
| 23 | REL | 59 | Male | African American | 28.2 | TT | dG/dG | 58 | 5.4 | 976 | 1b | 28 700 000 | Yes | Yes |
| 24 | REL | 56 | Female | African American | 35.52 | CT | dG/TT | 96 | 4.7 | 1694 | 1a | 9 150 000 | No | No |
| 25 | REL | 42 | Male | White | CT | dG/TT | 80 | 5.8 | 365 | 1a | 1 380 000 | No | No | |
| Ledipasvir + sofosbuvir + GS-9451 + GS-9669 | ||||||||||||||
| 1 | REL | 63 | Female | African American | 26.98 | TT | dG/dG | 81 | 8.3 | 964 | 1b | 38 400 000 | No | No |
| 2 | REL | 60 | Male | White | 26.72 | CT | dG/TT | 67 | 5 | 1094 | 1a | 9 680 000 | No | No |
| 3 | REL | 58 | Male | African American | 26.78 | 63 | 5.7 | 3511 | 1a | 52 500 000 | No | No | ||
| 4 | REL | 65 | Female | White | 23.31 | CT | dG/TT | 80 | 5.5 | 703 | 1a | 3 040 000 | No | No |
| 5 | SVR12 | 57 | Female | White | 22.23 | TT | dG/dG | 89 | 5.2 | 290 | 1b | 4 920 000 | No | No |
| 6 | REL | 65 | Male | African American | 31.82 | TT | dG/dG | 30 | 6.3 | 710 | 1a | 54 200 000 | Yes | Yes |
| 7 | REL | 65 | Male | African American | 24.24 | TT | dG/dG | 108 | 9 | 2111 | 1a | 11 600 000 | Yes | Yes |
| 8 | REL | 61 | Male | African American | 28.09 | CT | dG/TT | 91 | 5.7 | 272 | 1b | 4 920 000 | Yes | Yes |
| 9 | REL | 63 | Male | African American | 26.45 | CT | dG/TT | 104 | 5.5 | 382 | 1a | 34 900 000 | Yes | Yes |
| 10 | REL | 37 | Male | White | 31.88 | CT | dG/TT | 129 | 4.8 | 339 | 1a | 3 090 000 | No | No |
| 11 | SVR12 | 60 | Male | African American | 26.29 | TT | dG/dG | 61 | 5 | 906 | 1b | 1 200 000 | No | No |
| 12 | REL | 49 | Male | White | 25.51 | CT | dG/TT | 104 | 5 | 301 | 1b | 7 230 000 | No | No |
| 13 | REL | 54 | Male | African American | 30.53 | TT | dG/dG | 96 | 5.4 | 678 | 1a | 3 940 000 | Yes | Yes |
| 14 | REL | 68 | Male | African American | 27.52 | CT | dG/TT | 161 | 5.6 | 1711 | 1a | 14 800 000 | No | No |
| 15 | SVR12 | 49 | Male | African American | 32.33 | CT | dG/TT | 65 | 6.5 | 548 | 1b | 1 420 000 | No | No |
| 16 | REL | 69 | Male | African American | 26.67 | CT | dG/TT | 45 | 4.8 | 491 | 1a | 1 120 000 | No | No |
| 17 | REL | 58 | Male | African American | 24.92 | TT | dG/dG | 105 | 4.3 | 674 | 1a | 1 130 000 | Yes | No |
| 18 | REL | 65 | Male | African American | 34.06 | CT | dG/TT | 94 | 5.9 | 1054 | 1b | 10 700 000 | Yes | Yes |
| 19 | REL | 57 | Male | African American | 32.9 | CC | TT/TT | 163 | 6.6 | 310 | 1a | 3 450 000 | No | No |
| 20 | REL | 62 | Male | African American | 28.83 | CC | dG/TT | 94 | 5.5 | 680 | 1a | 616 000 | Yes | No |
| 21 | Lost to follow-up | 56 | Female | African American | 29.17 | TT | dG/dG | 94 | 6.3 | 321 | 1a | 297 000 | Yes | Yes |
| 22 | REL | 40 | Female | White | 15.1 | CT | dG/TT | 54 | 4.7 | 277 | 1b | 191 000 | No | No |
| 23 | REL | 54 | Male | African American | 22.61 | CT | dG/TT | 45 | 5.2 | 939 | 1a | 10 900 000 | Yes | No |
| 24 | SVR12 | 46 | Male | African American | 22.82 | CT | dG/TT | 98 | 5.8 | 831 | 1a | 1 810 000 | No | No |
| 25 | SVR12 | 57 | Female | African American | 37.28 | CT | dG/TT | 148 | 8.2 | 2099 | 1b | 3 560 000 | No | No |
BMI = body mass index; HCV = hepatitis C virus; IP = inducible protein; LDL = low-density lipoprotein; RAV = resistance-associated variant; REL = relapse; SVR12 = HCV RNA level below the lower limit of quantification at posttreatment week 12.
Appendix Table 2.
Baseline HCV Resistance Mutations
| Patient | Genotype | NS3/4 Mutation (% Population) | NS5A Mutation (% Population) | NS5B Mutation (% Population) | |||
|---|---|---|---|---|---|---|---|
| Baseline | Relapse | Baseline | Relapse | Baseline | Relapse | ||
| Ledipasvir + sofosbuvir + GS-9451 | |||||||
| 1 | 1a | None | None | None | Q30R (20%), L31M (71%) | None | None |
| 2 | 1a | Q80K (>99%) | * | None | * | None | * |
| 3 | 1a | Q80K (>99%) | None | None | None | None | None |
| 4 | 1a | None | None | None | None | None | None |
| 5 | 1a | None | * | None | * | None | * |
| 6 | 1a | Q80K (>99%) | * | None | * | None | * |
| 7 | 1a | Q80K (>99%) | * | None | * | None | * |
| 8 | 1b | None | * | None | * | None | * |
| 9 | 1a | None | None | None | L31M (99%) | None | None |
| 10 | 1a | R155K (>99%) | R155K (>99%) | None | L31M (>99%) | None | None |
| 11 | 1a | None | None | None | None | None | None |
| 12 | 1b | None | None | None | L31V (6%), L31M (46%), L31I (48%) | None | None |
| 13 | 1a | Q80K (>99%) | None | None | Q30H (>99%), Y93H (>99%) | None | None |
| 14 | 1a | None | None | L31M (65.8%) | L31M (>99%) | None | None |
| 15 | 1a | Q80K (>99%) | * | None | * | None | * |
| 16 | 1b | None | * | None | * | None | * |
| 17 | 1a | Q80K (>99%) | None | None | Q30R (30%), L31M (70%) | None | None |
| 18 | 1a | None | None | None | Q30R (28%), L31M (71%) | None | None |
| 19 | 1a | None | * | None | * | None | * |
| 20 | 1b | None | * | None | * | None | * |
| 21 | 1b | None | None | None | None | None | None |
| 22 | 1b | None | * | None | * | None | * |
| 23 | 1b | None | None | L31M (94.1%) | L31M (>99%), Y93H (>99%) | None | None |
| 24 | 1a | None | None | None | Q30R (11%), Q30K (13%), L31V (2%), L31M (75%) | None | None |
| 25 | 1a | None | None | None | L31V (>99%) | None | None |
| Ledipasvir + sofosbuvir + GS-9451 + GS-9669 | |||||||
| 1 | 1b | Assay failed | Q80K (>99%) | Assay failed | Q24K (>99%), R30T (>99%), L31V (>99%), Y93H (>99%) | Assay failed | None |
| 2 | 1a | None | None | None | M28T (1%), Q30H (3%), Q30R (57%), L31M (34%), S38F (2%) | None | None |
| 3 | 1a | None | None | None | L31M (>99%) | None | None |
| 4 | 1a | None | None | None | Q30R (1%), Q30H (23%) | None | None |
| 5 | 1b | None | * | None | * | None | * |
| 6 | 1a | None | None | L31M (1.9%) | L31M (99%) | None | None |
| 7 | 1a | None | None | L31M (14.3%) | L31M (>99%) | None | None |
| 8 | 1b | D168E (4.5%) | None | L31M (8.15%) Y93H (>99%) | Y93H (>99%) | None | None |
| 9 | 1a | None | None | Y93N (45.0%) | Y93N (>99%) | None | None |
| 10 | 1a | None | None | None | Q30R (>99%) | None | None |
| 11 | 1b | None | * | None | * | None | * |
| 12 | 1b | None | None | None | Y93H (>99%) | None | None |
| 13 | 1a | Q80K (>99%) | None | L31M (>99%) | L31M (>99%) | None | None |
| 14 | 1a | None | None | None | K24R (90%), L31M (>99%) | None | None |
| 15 | 1b | None | * | None | * | None | * |
| 16 | 1a | None | None | None | Q30R (89%) | None | None |
| 17 | 1a | Q80K (>99%) | Q80K (>99%) | None | None | None | None |
| 18 | 1b | None | None | Y93H (>99%) | Y93H (>99%) | None | None |
| 19 | 1a | None | None | None | M28T (2%), Q30H (46%), Q30R (54%) | None | None |
| 20 | 1a | Q80K (>99%) | Q80K (>99%) | None | Q30H (1%) | None | None |
| 21 | 1a | R155K (41.2%) | LTF | None | LTF | None | LTF |
| 22 | 1b | None | None | None | Y93H (1%),Y93C (2%) | None | None |
| 23 | 1a | None | None | None | Q30H (1%), Q30R (11%), L31M (84%) | M423I (>99%) | M423I (>99%) |
| 24 | 1a | None | * | None | * | None | * |
| 25 | 1b | None | * | None | * | None | * |
HCV = hepatitis C virus; LTF = lost to follow-up.
Patient achieved HCV RNA level below the lower limit of quantification at posttreatment week 12.
Appendix Table 3.
Host and Viral Factors, by SVR12
| Variable | Ledipasvir + Sofosbuvir + GS-9451 ± GS-9669 (n = 50) | ||
|---|---|---|---|
| SVR (n =15) | Relapse (n = 35) | P Value* | |
| Host factors | |||
| Mean age, y | 51.1 | 57.7 | 0.02 |
| Female, n (%) | 7 (47) | 7 (20) | 0.09 |
| African American, n (%) | 10 (67) | 28 (80) | 0.47 |
| Mean BMI, kg/m2 | 28.0 | 27.9 | 0.92 |
| IFNL3/IL28B CC genotype, n (%) | 2 (13) | 3 (9) | 0.63 |
| IFNL4 TT/TT genotype, n (%) | 2 (13) | 1 (3) | 0.21 |
| Mean baseline LDL cholesterol level, mg/dL | 104.1 | 88.0 | 0.12 |
| Mean baseline hemoglobin A1c level, % | 5.9 | 5.7 | 0.52 |
| Mean baseline IP-10 level | 849 | 914 | 0.74 |
| SVR by viral factors, n/N (%) by subgroup | |||
| HCV genotype 1b | 8/16 (50) | 0.049 | |
| HCV genotype 1a | 7/34 (21) | ||
| Baseline HCV VL <6 × 106 IU/mL (Roche) | 13/29 (45) | 0.007 | |
| Baseline HCV VL ≥6 × 106 IU/mL | 2/21 (10) | ||
| Week 2 HCV VL <12 IU/mL (Abbott) | 6/17 (35) | 0.70 | |
| Week 2 HCV VL ≥12 IU/mL | 9/33 (27) | ||
| Absence of any NS3, NS5A, or NS5B RAV | 11/29 (38) | 0.35 | |
| Presence of ≥1 NS3, NS5A, or NS5B RAV | 4/21 (19) | ||
| Absence of NS3, NS5A, or NS5B RAV with >20-fold resistance | 15/40 (38) | 0.022 | |
| Presence of NS3, NS5A, or NS5B RAV with >20-fold resistance | 0/10 | ||
BMI = body mass index; HCV = hepatitis C virus; IP = inducible protein; LDL = low-density lipoprotein; RAV = resistance-associated variant; SVR = sustained virologic response; SVR12 = HCV RNA level below the lower limit of quantification at posttreatment week 12; VL = viral load.
Calculated using t test for continuous variables and Fisher exact test for categorical variables. Boldface values are statistically significant (P < 0.05).
Appendix Figure 1.
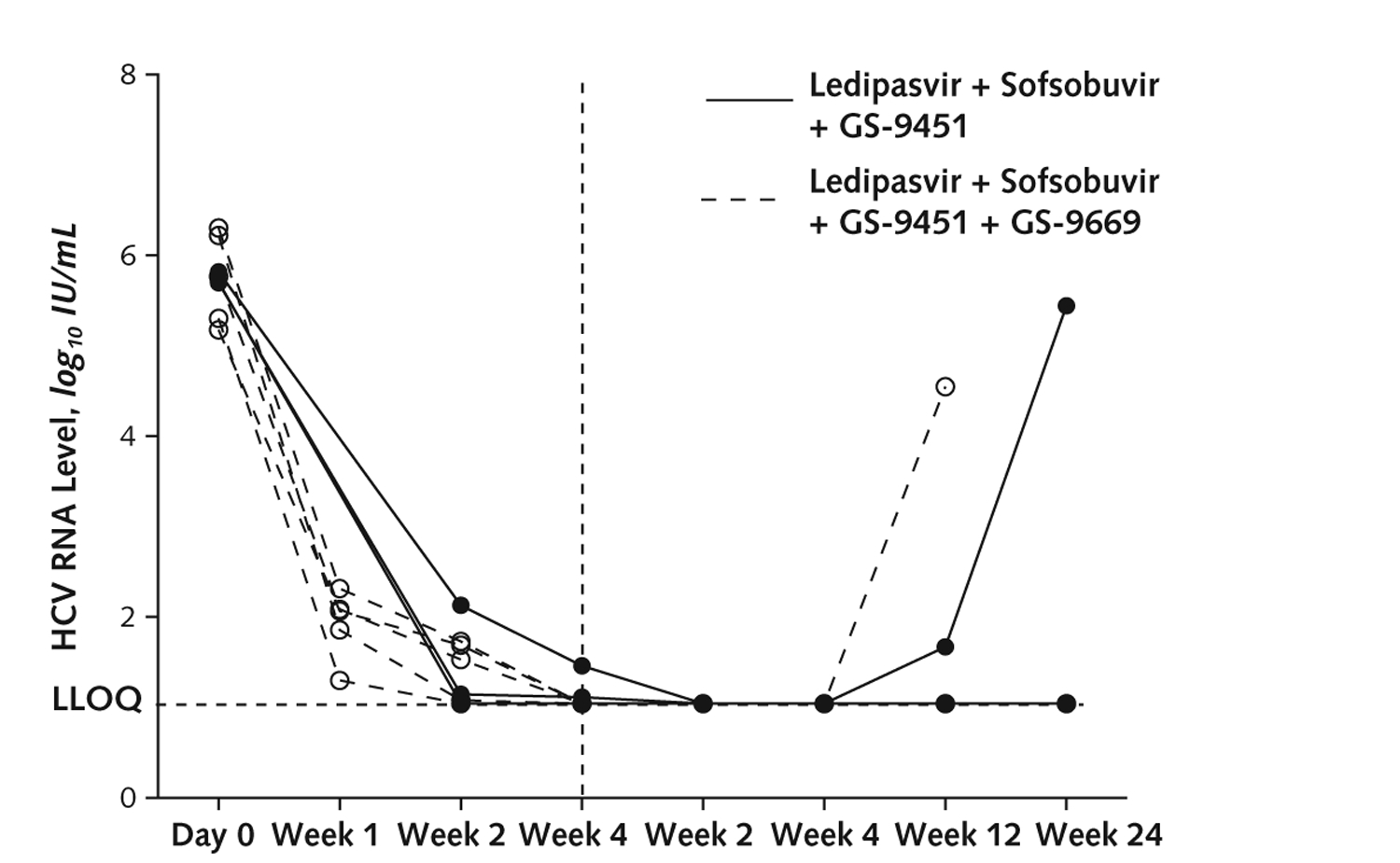
HCV viral load in patients with genotype 1b infection with low viral load and no RAV with >20-fold resistance at baseline.
Vertical dashed line indicates end of treatment. Low viral load is <6 × 106 IU/mL. HCV = hepatitis C virus; LLOQ = lower limit of quantification; RAV = resistance-associated variant.
Appendix Figure 2.
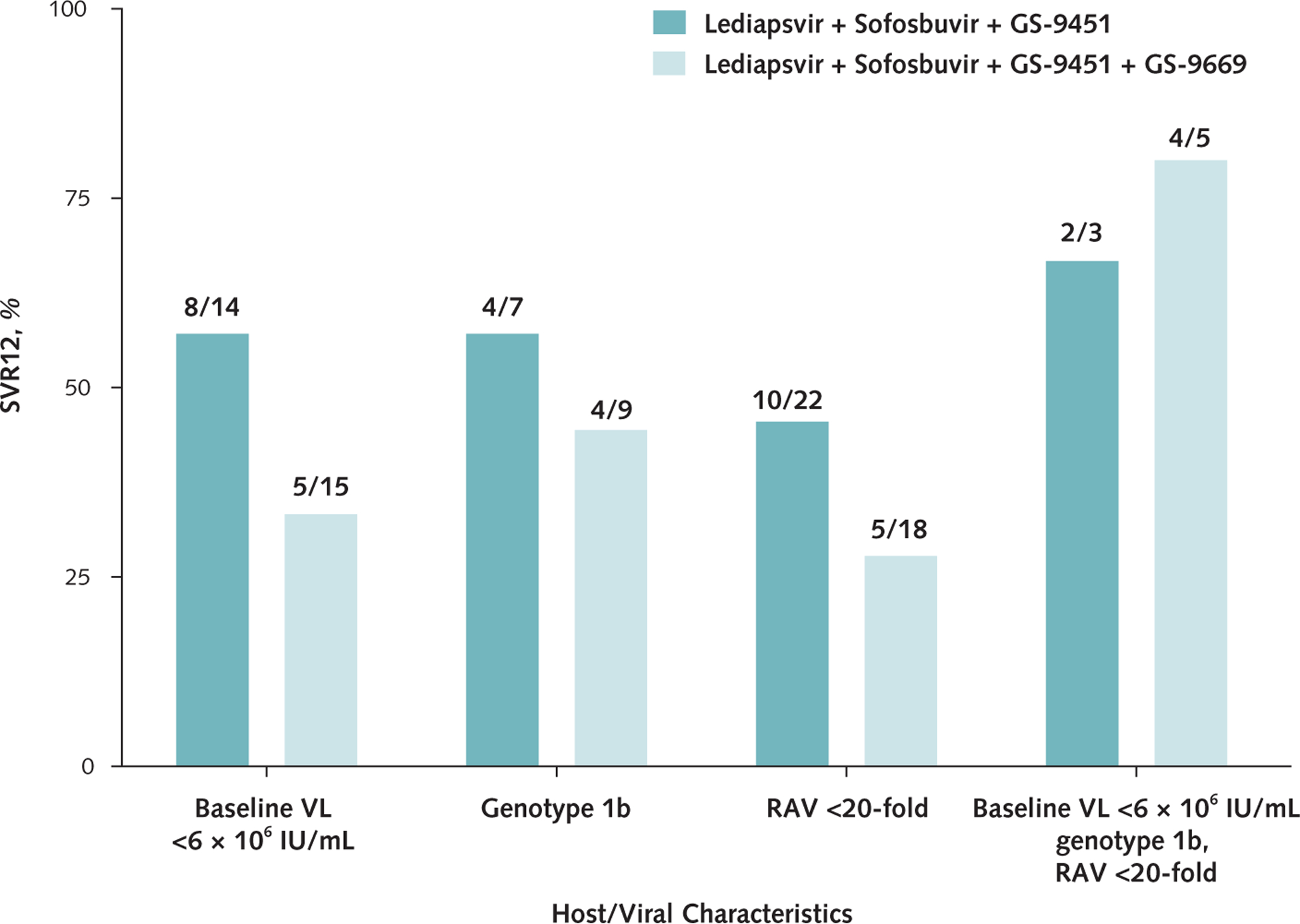
Sustained virologic response, by host or viral characteristic. HCV = hepatitis C virus; RAV = resistance-associated variant; SVR12 = HCV RNA level below lower limit of quantification at posttreatment week 12; VL = viral load.
Footnotes
Publisher's Disclaimer: Disclaimer: The content of this article does not necessarily reflect the views or policies of the U.S. Department of Health and Human Services, and the mention of trade names, commercial products, or organizations does not imply endorsement by the U.S. government.
Note: Drs. Kohli, Kattakuzhy, and Kottilil had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Reproducible Research Statement: Study protocol and data set: Available from Dr. Kattakuzhy (skattakuzhy@ihv.umaryland.edu). Statistical code: Not available.
Current author addresses and author contributions are available at www.annals.org.
Disclosures: Dr. Kohli reports a cooperative research and development agreement between the National Institutes of Health and Gilead Sciences during the conduct of the study. Ms. Nelson reports a cooperative research and development agreement between the National Institutes of Health and Gilead Sciences during the conduct of the study. Ms. Seamon reports a cooperative research and development agreement between the National Institutes of Health and Gilead Sciences during the conduct of the study. Dr. Meissner reports grants from Gilead Sciences outside the submitted work. Ms. Gross reports that she worked with the National Institutes of Health during the conduct of the study and owns stock in Merck, Pfizer, and Johnson & Johnson. Mr. Jolley reports that he previously owned stock in Gilead Sciences. Dr. Teferi reports grants and personal fees from Gilead Sciences outside the submitted work. Dr. Chavez reports personal fees from Gilead Sciences and Bristol-Myers Squibb outside the submitted work. Dr. Osinusi reports employment with Gilead Sciences outside the submitted work. Dr. Mo reports that she is an employee of and stockholder in Gilead Sciences. Authors not named here have disclosed no conflicts of interest. Disclosures can also be viewed at www.acponline.org/authors/icmje/ConflictOfInterestForms.do?msNum=M15-0642.
References
- 1.Ly KN, Xing J, Klevens RM, Jiles RB, Ward JW, Holmberg SD. The increasing burden of mortality from viral hepatitis in the United States between 1999 and 2007. Ann Intern Med. 2012;156:271–8. [DOI] [PubMed] [Google Scholar]
- 2.Davis GL, Alter MJ, El-Serag H, Poynard T, Jennings LW. Aging of hepatitis C virus (HCV)-infected persons in the United States: a multiple cohort model of HCV prevalence and disease progression. Gastroenterology. 2010;138:513–21, 521.e1–6. [DOI] [PubMed] [Google Scholar]
- 3.Kowdley KV, Gordon SC, Reddy KR, Rossaro L, Bernstein DE, Lawitz E, et al. ; ION-3 Investigators. Ledipasvir and sofosbuvir for 8 or 12 weeks for chronic HCV without cirrhosis. N Engl J Med. 2014;370: 1879–88. [DOI] [PubMed] [Google Scholar]
- 4.Afdhal N, Reddy KR, Nelson DR, Lawitz E, Gordon SC, Schiff E, et al. ; ION-2 Investigators. Ledipasvir and sofosbuvir for previously treated HCV genotype 1 infection. N Engl J Med. 2014;370:1483–93. [DOI] [PubMed] [Google Scholar]
- 5.Harvoni Pollack A., a hepatitis C drug from Gilead, wins F.D.A. approval. The New York Times. 10 October 2014:B2. Accessed at www.nytimes.com/2014/10/11/business/harvoni-a-hepatitis-c-drug-from-gilead-wins-fda-approval.html on 29 January 2015. [Google Scholar]
- 6.Kohli A, Osinusi A, Sims Z, Nelson A, Meissner EG, Barrett LL, et al. Virological response after 6 week triple-drug regimens for hepatitis C: a proof-of-concept phase 2A cohort study. Lancet. 2015;385: 1107–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Denniston MM, Jiles RB, Drobeniuc J, Klevens RM, Ward JW, McQuillan GM, et al. Chronic hepatitis C virus infection in the United States, National Health and Nutrition Examination Survey 2003 to 2010. Ann Intern Med. 2014;160:293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.National Institute of Allergy and Infectious Diseases. Division of AIDS Table for Grading the Severity of Adult and Pediatric Adverse Events, Version 1.0, December, 2004; Clarification August 2009 Bethesda, MD: U.S. Department of Health and Human Services; 2009. Accessed at http://rsc-beta.tech-res.com/Document/safetyandpharmacovigilance/Table_for_Grading_Severity_of_Adult_Pediatric_Adverse_Events.pdf on 27 January 2015. [Google Scholar]
- 9.Prokunina-Olsson L, Muchmore B, Tang W, Pfeiffer RM, Park H, Dickensheets H, et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat Genet. 2013;45:164–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Osinusi A, Meissner EG, Lee YJ, Bon D, Heytens L, Nelson A, et al. Sofosbuvir and ribavirin for hepatitis C genotype 1 in patients with unfavorable treatment characteristics: a randomized clinical trial. JAMA. 2013;310:804–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Petersen T, Gordon LA, Townsend K, Kohli A. Pill burden & treatment length reduce adherence to IFN-free hepatitis C therapy in an urban cohort [Abstract]. Presented at the 21st Conference on Retroviruses and Opportunistic Infections, Boston, Massachusetts, 3–6 March 2014. Abstract no. 667. [Google Scholar]
- 12.Lawitz E, Poordad F, Gutierrez JA, Evans B, Hwang P, Howe A, et al. C-SWIFT: grazoprevir (MK-5172) + elbasvir (MK-8742) + sofosbuvir in treatment-naive patients with hepatitis C virus genotype 1 infection, with and without cirrhosis, for durations of 4, 6, or 8 weeks. Presented at the 65th Annual Meeting of the American Association for the Study of Liver Diseases, Boston, Massachusetts, 7–11 November 2014. [Google Scholar]
- 13.Sciences Gilead. Harvoni U.S. prescribing information Foster City, CA: Gilead Sciences; 2014. [Google Scholar]
- 14.Zeuzem S, Dusheiko GM, Salupere R, Mangia A, Flisiak R, Hyland RH, et al. ; VALENCE Investigators. Sofosbuvir and ribavirin in HCV genotypes 2 and 3. N Engl J Med. 2014;370:1993–2001. [DOI] [PubMed] [Google Scholar]
- 15.Fried MW, Hadziyannis SJ, Shiffman ML, Messinger D, Zeuzem S. Rapid virological response is the most important predictor of sustained virological response across genotypes in patients with chronic hepatitis C virus infection. J Hepatol. 2011;55:69–75. [DOI] [PubMed] [Google Scholar]
- 16.Thompson AJ, Muir AJ, Sulkowski MS, Ge D, Fellay J, Shianna KV, et al. Interleukin-28B polymorphism improves viral kinetics and is the strongest pretreatment predictor of sustained virologic response in genotype 1 hepatitis C virus. Gastroenterology. 2010;139:120–9.e18. [DOI] [PubMed] [Google Scholar]
- 17.Sims Z, Kohli A, Bon D, Nelson A, Polis MA, Masur H, et al. Lack of impact of baseline mutations on HCV viral kinetics with multiple combination DAA therapy. Presented at HIV and Liver Disease 2014, Moran, Wyoming, 18–20 September 2014. [Google Scholar]
- 18.Gane EJ, Stedman CA, Hyland RH, Ding X, Svarovskaia E, Subramanian GM, et al. Efficacy of nucleotide polymerase inhibitor sofosbuvir plus the NS5A inhibitor ledipasvir or the NS5B nonnucleoside inhibitor GS-9669 against HCV genotype 1 infection. Gastroenterology. 2014;146:736–743.e1. doi: 10.1053/j.gastro.2013.11.007 [DOI] [PubMed] [Google Scholar]