

Abstract
The central nervous system (CNS) antigen-presenting cell (APC) that primes anti-tumor CD8+ T-cell responses remains undefined. Elsewhere in the body, the conventional dendritic cell 1 (cDC1) performs this role. However, steady-state brain parenchyma cDC1 are extremely rare; cDC localize to the choroid plexus and dura. Thus, whether the cDC1 play a function in presenting antigen derived from parenchymal sources in the tumor setting remains unknown. Using preclinical glioblastoma models and cDC1-deficient mice, we explored the presently unknown role of cDC1 in CNS anti-tumor immunity. We determined that, in addition to infiltrating the brain tumor parenchyma itself, cDC1 prime neoantigen-specific CD8+ T-cells against brain tumors and mediate checkpoint blockade-induced survival benefit. We observed that cDC, including cDC1, isolated from the tumor, the dura, and the CNS-draining cervical lymph nodes (cLNs) harbored a traceable fluorescent tumor antigen. In patient samples, we observed several APC subsets (including the CD141+ cDC1 equivalent) infiltrating glioblastomas, meningiomas, and dura. In these same APC subsets, we identified a tumor-specific fluorescent metabolite of 5-aminolevulinic acid, which fluorescently labeled tumor cells during fluorescence-guided glioblastoma resection. Together, these data elucidate the specialized behavior of cDC1 and suggest cDC1 play a significant role in CNS anti-tumor immunity.
INTRODUCTION
Glioblastoma (GBM) is the most common central nervous system (CNS) primary malignancy and remains inexorably lethal. The standard of care, involving surgery, chemotherapy, and radiation treatment, leads to a median survival of only 15–20 months [1]. Because immunotherapy has revolutionized treatment of other cancers, there has been significant interest in applying immune-based treatments to patients with GBM, especially given a growing understanding that the CNS is not as immunoprivileged as long perceived [2]. However, despite numerous clinical trials, there remain no FDA-approved immunotherapies for GBM [3, 4].
Several critical issues underlie the difficulty of harnessing immune-potentiating therapies to treat GBM. First, GBM represents a severe example of cancer immunoediting in patients [5] and exhibits substantial evidence of immunosuppression. A range of immunologic deficits have been described in GBM patients, including lymphopenia [6], intrinsic immune suppression by tumor cells [7, 8], and overexpression of checkpoint molecules such as PD-L1 [9, 10], among others [11, 12], suggesting a “cold” immune phenotype. Secondly, because GBM tumors are molecularly heterogeneous and are comprised of several subclonal populations [13], T-cell antigen targets are not uniformly expressed in the tumor mass. Our incomplete understanding of the development of CNS immune responses also remains a significant barrier to successful immunotherapy in the brain. Specifically, the mechanistic underpinnings of antigen presentation leading to T-cell priming of brain tumor-specific T-cells remain undefined. Unlike other sites in the body, the brain parenchyma does not harbor dendritic cells (DCs) in the resting state [14–16]. Microglia and border-associated macrophages instead comprise the putative CNS antigen-presenting cells (APCs) [14]. Furthermore, the brain does not harbor classical lymphatic and secondary lymphoid structures [14, 15, 17]. However, the characterization of lymphatic channels within the dura mater of the cerebral meninges has provided new insights into APCs may encounter antigen originating from the brain parenchyma and potentially traffic that antigen to draining lymph nodes [18–21]. Finally, evidence suggesting that conventional (c)DC are important in mounting autoimmune responses in the brain has been reported, with experiments demonstrating that cDC depletion attenuates experimental autoimmune encephalitis (EAE) [22] and that restricting MHC-II expression to cDC subsets is sufficient to permit EAE progression [23]. Although these foundational observations highlight the importance of the anatomic basis for lymphatic drainage in the CNS and suggest that cDC contribute to inflammation in the brain, the specific role cDC play in this process remains unclear. Cell migration from the CNS to cervical lymph nodes has been reported [24–27], but these experiments used exogenously introduced monocyte-derived DCs. Thus, there remains a lack of understanding regarding the cellular basis for endogenous antigen presentation in CNS anti-tumor immunity.
Herein, we characterized the role of the cDC1 subset during the endogenous immune response to brain tumors. Elsewhere in the body, cDC1 phagocytize antigen, migrate to LNs, and cross-present antigen to prime CD8+ T-cell responses [28–32]. Although cDC1 are required for effective immune responses against a range of tumor types [28, 29, 31, 33–37], it is unclear whether these cells perform a similarly obligate role in the immune response to CNS tumors. In this study, we demonstrate that cDC1 are required both to mount neoantigen-specific immune responses and to respond to checkpoint blockade in wild-type, syngeneic orthotopic models of GBM. We also observed tumor antigen-containing cDC1 within the tumor, the dura, and cLNs. We determined that chemokine receptor 7 (CCR7) was required for DC to traffic tumor antigen from the CNS to cLNs and that clonal expansion of adoptively transferred tumor antigen-specific CD8+ T cells takes place within these LNs before expanding to other anatomic locations, which required both CCR7 and cDC1 for normal kinetics. In patients, we observed cDC, including the analogous cDC1 subset, infiltrating tumors, as well as the dura. Finally, from patients in which 5-aminolevulinic acid (5-ALA) was employed to facilitate fluorescence-guided GBM resection [38–41], we identified the presence of its fluorescent metabolite protoporphyrin-IX (PPIX) [42, 43] within tumor-infiltrating cDC and monocytes, but not in tumor-infiltrating T cells nor in matched peripheral APC counterparts. Together, these findings clarify the unique behavior of cDC1 in the CNS and demonstrate that cDC1 play a critical role in the CNS anti-tumor immune responses.
MATERIALS AND METHODS
Study design
The objective of this study was to investigate the nature of antigen presentation and the role of cDC1 in CNS anti-tumor immunity. We designed and performed experiments in cellular immunology, and employed preclinical models of GBM, as well as genotypes of mice deficient in cDC1, cell migration, or with lymphatics and/or cDC1 specifically labeled. We used previously characterized neoantigens and model antigens, as discussed, to study antigen-specific responses and kinetics of clonal expansion of adoptively transferred T cells. We also studied APC infiltration and antigen uptake by tumor-infiltrating APCs in patient tumors.
Human studies
All study participants were neurosurgical patients at Barnes-Jewish Hospital with grade 4 glioblastomas or meningiomas confirmed by pathology of the resected specimens. All meningiomas were resected without 5-ALA. A subset of GBM tumors were resected using 5-ALA. For patients whose tumors were resected with 5-ALA, 5-ALA (Gleolan®) was administered in a dose of 20mg/kg 2–4 hours before the induction of anesthesia. Any combination of tumor, dura, and blood were collected when specified. Experiments were performed in accordance with ethical standards set forth in the 1964 Declaration of Helsinki. Written informed consent for specimen inclusion in this study was obtained using Washington University School of Medicine Institutional Review Board-approved protocols (#20111101 and #202107071).
Human tumor and dura preparation
Human tumors and dura were stored on ice after resection. On the same day they were processed by masserating and incubating in 10–20mL of 2 mg/mL collagenase A (Roche) and 2 mg/mL collagenase D (Roche) in 10% heat-inactivated FBS (Gibco) and IMDM (Gibco) overnight at 37ºC, with trituration every few hours for dura disaggregation in particular. The next morning, single-cell suspensions were filtered and prepared for flow cytometry as described below.
Mice
All animal experiments were approved by the Washington University Animal Studies Committee. Male and female mice 6–16 weeks of age were used for all experiments. Wild type C57BL/6 mice were purchased from Taconic Biosciences (Hudson, NY). C57BL/6 IRF8+32kb−/− and SNX22GFP/GFP mice were obtained from Dr. Kenneth Murphy (Washington University in St. Louis). SNX22GFP/+ F1 mice were used for experiments, with cDC1-GFP expression confirmed by flow cytometry. For experiments with GFP-labeled cDC1 and Tomato-labeled lymphatic vessels, we crossed SNX22GFP/GFP C57BL/6 mice to Prox1-Cre-tdTomato+/+ C57BL/6 mice, obtained from Dr. Gwendalyn Randolph (Washington University in St. Louis). We treated the F1s with intraperitoneal tamoxifen (50µg/g, three times a week, for two weeks with a stock solution of 10mg/mL tamoxifen (Sigma) dissolved in sunflower oil (Sigma)) as described [44] to induce Tomato expression in lymphatic vessels, which was confirmed by examining whole dura with 2-photon microscopy. C57BL/6 CCR7−/− [45], OT-I transgenic [46], and CD45.1+ congenic [47] mice were purchased from Jackson Laboratories. All mice were housed in accordance with IACUC standards.
Cell lines
All cells for experiments were used one to five passages after thawing. CT2A cells were obtained from Dr. Peter Fecci in 2016 (Duke University), and GL261 cells were obtained from the National Cancer Institute Tumor Repository in 2014. GL261 and CT2A cell lines were subjected to whole exome and RNA sequencing to profile, characterize, and validate them [48, 49]. U343 cells were obtained from The Cancer Cell Line Encyclopedia at the Broad Institute in 2014 [50]. All cell lines were expanded and frozen at early aliquots, and each were cultured for less than a total cumulative time of six months from the time of acquisition to the time of each experiment. GL261-OFP cells were generated by transducing parent cells with mOrange2 (obtained from Dr. Robert Schreiber, Washington University in St. Louis). GL261-zsGreen and CT2A-zsGreen cells were generated by transducing parent cells with zsGreen (obtained from Dr. David DeNardo, Washington University in St. Louis). CT2A-mFlt3L cells were generated by transducing parent cells with murine Flt3L (SinoBiological catalog # MG51113-UT). GL261-OVA (ovalbumin) cells were generated by transducing parent cells with ovalbumin cloned from pcDNA3-OVA (Addgene plasmid # 64599). Transduction was performed as described [51]. Briefly, 293T cells were transfected with pLX304 vector plasmid along with Δ8.9 and VSV-G packaging plasmids (Addgene) using FuGENE® transfection reagent (Promega) according to manufacturer’s instructions. Two days later, viral supernatant was pushed through a 0.45µM PVDF syringe filter (Sigma), combined with polybrene transfection reagent (Sigma) for a final concentration of 8µg/mL and added to target cells. This was repeated the following day. After two rounds of transduction and recovery, target cells were selected Blasticidin (Sigma): 3µg/mL for CT2A, 10µg/mL for GL261 until untransduced control cells had died. Retroviral transductions were done with the pBABE backbone plasmid, pCL-Ampho packaging plasmid, and 2µg/mL puromycin selection. All cell lines were tested for mycoplasma regularly using an ATCC Universal Mycoplasma Detection Kit. All cells were cultured at 37ºC, 5% CO2 in D10 (DMEM (Gibco) with 10% heat-inactivated FBS (Gibco), 1% penicillin/streptomycin (Gibco), 1% minimum essential amino acids (Gibco), 1% L-glutamine, and 1% sodium pyruvate (Gibco)). Cells were harvested at 70%-90% confluency to inject intracranially.
Intracranial injections
50,000 tumor cells in a volume of 5µL PBS (Gibco) were injected 2mm to the right and 2mm posterior of bregma, at a depth of 3.5mm using a Stoelting stereotactic headframe. Tumors were analyzed at fourteen days unless stated otherwise. Sham surgeries consisted of the same protocol (including 5µL injection of PBS) minus the tumor cells. Sham mice were harvested at the same time as their tumor-bearing counterparts.
Survival studies
Age- and sex-matched wild-type (WT) and IRF8+32kb−/− C57BL/6 mice were intracranially injected with 50,000 untransduced GL261 cells as described above. At days 3, 5, 7, and 14, mice were administered by intraperitoneal injection of either PBS-vehicle or anti-PD-L1 diluted in PBS (Leinco Technologies, Inc. Clone 10F.9G2) at a dose of 200 µg/mouse in a volume of 100 µL. Beginning at 10 days after tumor cell injection, mice were monitored every day for signs of illness and euthanized when moribund.
Adoptive transfer model and OT-I T-cell division
CD45.1+ mice were crossed to CD45.2+ OT-I mice. For adoptive transfer experiments, 5x105 GL261 or GL261-OVA cells were injected intracranially. Four days later, CD8+ T cells were isolated from a CD45.1 x OT-I F1 mouse spleens using an EasySep™ Mouse CD8a Positive Selection Kit II (Stem Cell), and cells were CFSE (BioLegend) labeled (10 minutes at room temperature, 5µM), recovered in R10 media (RPMI-1640, 1% L-glutamine, 1% penicillin/streptomycin, 1% minimum essential amino acids, 10% heat-inactivated FBS (all Gibco) supplemented with β-mercaptoethanol (55µM, Sigma) at 37ºC/5%CO2, washed in PBS, and adoptively transferred via tail vein into recipient mice (5x105 OT-I cells per mouse). At days 3 and 6 post adoptive transfer, tissues from mice were harvested and prepared for flow cytometry as indicated below. The CFSE-high gate was based on the CFSE dilution (or absence thereof) of OT-I cells adoptively transferred into mice bearing untransduced GL261 brain tumors which didn’t express ovalbumin. The CFSE-low gate was based on the CFSE dilution demonstrated by maximally divided OT-I T cells infiltrating tumors at the day 6 post-transfer, and the CFSE-low gate was constructed to encompass the majority of terminally divided OT-I T cells within tumors.
Flt3L treatment
C57BL/6 mice were injected subcutaneously in the flank with 1x106 CT2A-mFlt3L cells, such that the transgenic CT2A-Flt3L flank tumors themselves were the source of systemic Flt3L, and the dura could be analyzed in the presence or absence of Flt3L produced by the CT2A-Flt3L transgenic flank tumors compared to untransduced CT2A controls, similar to previously described [15]. As a negative control, mice were injected subcutaneously with 1x106 untransduced CT2A cells. Dura from mice was harvested 2–3 weeks post-transplant of tumor cells, when flank tumors had reached 1–2 cm in diameter as measured with a caliper every 2–3 days, taking the control and Flt3L mice contemporaneously.
Tissue Preparation
Intracranial tumors from mice were analyzed 14 days after injection, unless otherwise specified. For a given study, up to and including the following tissues were harvested: tumors, superficial cervical lymph nodes, deep cervical lymph nodes, a non-draining contralateral inguinal lymph node, dura, and/or spleens. Lymph nodes, dura, and tumors were mechanically dissociated between two frosted slides and digested in 1 mg/mL collagenase A (Roche) in a solution of 2% heat-inactivated FBS (Gibco) in RMPI-1640 (Gibco) for 20 minutes at 37ºC/5% CO2. Suspensions were washed and red blood cells lyced with ACK buffer (Lonza) as necessary. Mouse and human brain tumors were separated from myelin using a 22.5% Percoll™ solution (Fisher) and centrifuged at room temperature for 15 minutes at 500 x g (acceleration 9, deceleration 5). Mononuclear cells were separated from spleens by first dissociating spleens between a frosted slide and then centrifuged using a Ficoll™ gradient (Fisher) at room temperature for 20 minutes at 400 x g. We retained the cells in the buffy coat for experiments. In preparation for flow cytometry, cells were suspended in MACS buffer (0.5% BSA (Fisher), 2 mM EDTA (Fisher) in PBS).
ELISPOT
Tumor single-cell suspensions harvested 14 days after injection were separated from myelin using a 22.5% Percoll™ (Fisher) solution and subjected to ACK buffer to lyse red blood cells as described above. CD8+ T cells were isolated with an EasySep™ Mouse CD8a Positive Selection Kit II (Stem Cell), counted, and plated with naïve splenocytes. 50,000 CD8+ T cells were plated with 125,000 naïve splenocytes with or without mutant-Imp3 (Imp3 D81N mutation) (mImp3) peptide at a concentration of 10µM (peptide sequence: AALLNKLYA, synthesized by Peptide 2.0) overnight on a pre-coated murine IFNγ detection plate (Cellular Technologies Limited) and analyzed with an ImmunoSpot plate reader (Cellular Technologies Limited). As additional controls for IFNγ release, we plated 125,000 naïve splenocytes ± mImp3 peptide ± concavalin A at a concentration of 1µg/well (Invivogen).
Flow cytometry and tetramer detection
Cell suspensions from tissues were filtered, subjected to Fc block (mouse: BioLegend; human: StemCell), and stained with surface antibodies for >20 minutes on ice. Cells were suspended in MACS buffer as indicated above. Data were acquired on a BD LSRFortessa™ X-20 flow cytometer and analyzed using FlowJo. The full antibody panel is listed in Supplementary Table S1. The mImp3 (AALLNKLYA)/H-2Db tetramer was generated by the Andrew M. and Jane M. Bursky Center for Immunology and Immunotherapy Programs Immune Monitoring lab (Washington University in St. Louis) as described [48]. For zsGreen experiments, the 488B/FITC channel was used, with zsGreen+ tumor cells as the compensation control. Tumors derived with untransduced tumor cells were used to assess the baseline fluorescence of immune cells isolated from that particular tumor type, and to determine where to draw the positive gate for zsGreen expression by a particular immune cell. Gating strategies are shown in Supplementary Figure S1A-F (mice) and Supplementary Figure S2A (human) and are defined in Supplementary Table S2.
5-aminolevulinic acid (5-ALA) flow cytometry
5-ALA uptake in U343 cells was assessed by flow cytometry. U343 cells were incubated with 1 mM 5-aminolevulinic acid hydrochloride (Sigma) or vehicle (H2O) as a negative control, trypsinized, and subjected to flow cytometry. In GBM patients whose tumors were resected using 5-ALA, and who had signed written consent to a Washington University School of Medicine Institutional Review Board-approved protocol (#202107071), resected tumors and peripheral blood were studied. Tumor specimens were prepared as described above. Peripheral blood mononuclear cells (PBMCs) were isolated by centrifuging blood on a Ficoll™ gradient, followed by buffy coat harvest. The 5-ALA metabolite, protoporphyrin IX (PPIX), was assessed in the BV650/405C channel, and the unstained tumor cell suspension was used as the compensation control for PPIX+ cells. The thresholds for PPIX+ versus PPIX− populations was determined by comparing tumor-infiltrating APC against their matched peripheral counterpart, which were PPIX−.
2-Photon Microscopy
Mice were overdosed by intraperitoneal injection with avertin (Sigma) at a dose of 250mg/kg and perfused with ~ 20mL of ice-cold PBS until the liver blanched over five minutes using a syringe. The cranial cap was removed with dura still attached to skull, which was then fixed in ice cold 4% paraformaldehyde/30% sucrose in PBS and incubated with shaking overnight. In a subset of mice, blood vessels were labeled 5 minutes prior to perfusion by injecting intravenously (IV) with 594-lectin [Lycopersicon Esculentum (Tomato) Lectin (LEL, TL), DyLight® 594, Vector Laboratories]. For brain sections, harvested brains were cut with a vibrotome after fixation. Fixed tissues were glued to a cover slip with superglue (Loctite) and immersed in PBS for imaging. Images were collected using a custom Leica SP8 two-photon microscope (Leica Microsystems, Wetzlar, Germany) equipped with a 25x 0.95 NA water immersion objective, and two femtosecond-pulsing tunable Ti:Sapphir lasers (Mai Tai HP DeepSee and InSight DS+), both from Spectra-Physics (Mountain View, CA, USA). GFP, mOrange, and TdTomato were excited at a wavelength of 925 nm, whereas Dylight 594 and Dylight 649 were excited at 830 nm. Fluorescence emission was guided directly to four external detectors in dendritic arrangement [two hybrid and two classical photomultiplier tubes (PMTs)]. For signal separation, three dichroic beam splitters (Semrock, Rochester, NY, USA) were used. To separate GFP, mOrange, Dylight 594, and the SHG (second-harmonic generation), the three cutoff wavelengths were 358 nm, 538 nm, and 593 nm, respectively. The separation of GFP, tdTomato, DyLight 649, and the SHG was obtained with cutoff wavelengths of 458 nm, 560 nm, and 652 nm. Images were processed and rendered with Imaris cell imaging software (Oxford Instruments).
Statistical analysis
Students T-tests were used to analyze differences between groups. Survival statistics were analyzed with log-rank tests. One-tailed T test with Welch’s correction was used to analyze zsGreen data, in which unequal standard deviations were anticipated between groups, and the signal difference between groups was directionally restricted. Unpaired T tests were employed when one mouse was compared to another. Paired T tests were used when individual organs within a mouse were compared against one another, or when cell types within an individual patient were simultaneously analyzed and compared. Grubbs outlier test was used when noted. p < 0.05 was considered significant, and statistical analysis were performed with GraphPad Prism 9. Specific statistical tests used for each experiment are outlined in figure legends.
Data and materials availability:
All data needed to evaluate the conclusions of this paper are present in the main paper or in the Supplementary Materials. Any cell lines generated are available upon request.
RESULTS
cDC1 are recruited to the CNS tumor microenvironment and mediate protection by checkpoint blockade
We first investigated whether cDC were recruited to the brain tumor microenvironment in orthotopic, syngeneic murine GBM models. We injected C57BL/6-derived GL261 or CT2A cells into the cerebral hemispheres of wild-type mice and identified DC subsets (CD45+F4/80−I-Ab+CD11c+), including cDC1 (additionally gated on XCR1+SIRPα−Ly-6C−; Supplementary Figure S1A). DC infiltration into both types of brain tumors was much greater than in sham controls (Figure 1A).
Figure 1. cDC1 infiltrate mouse GBM and mediate benefit from anti-PD-L1 therapy.
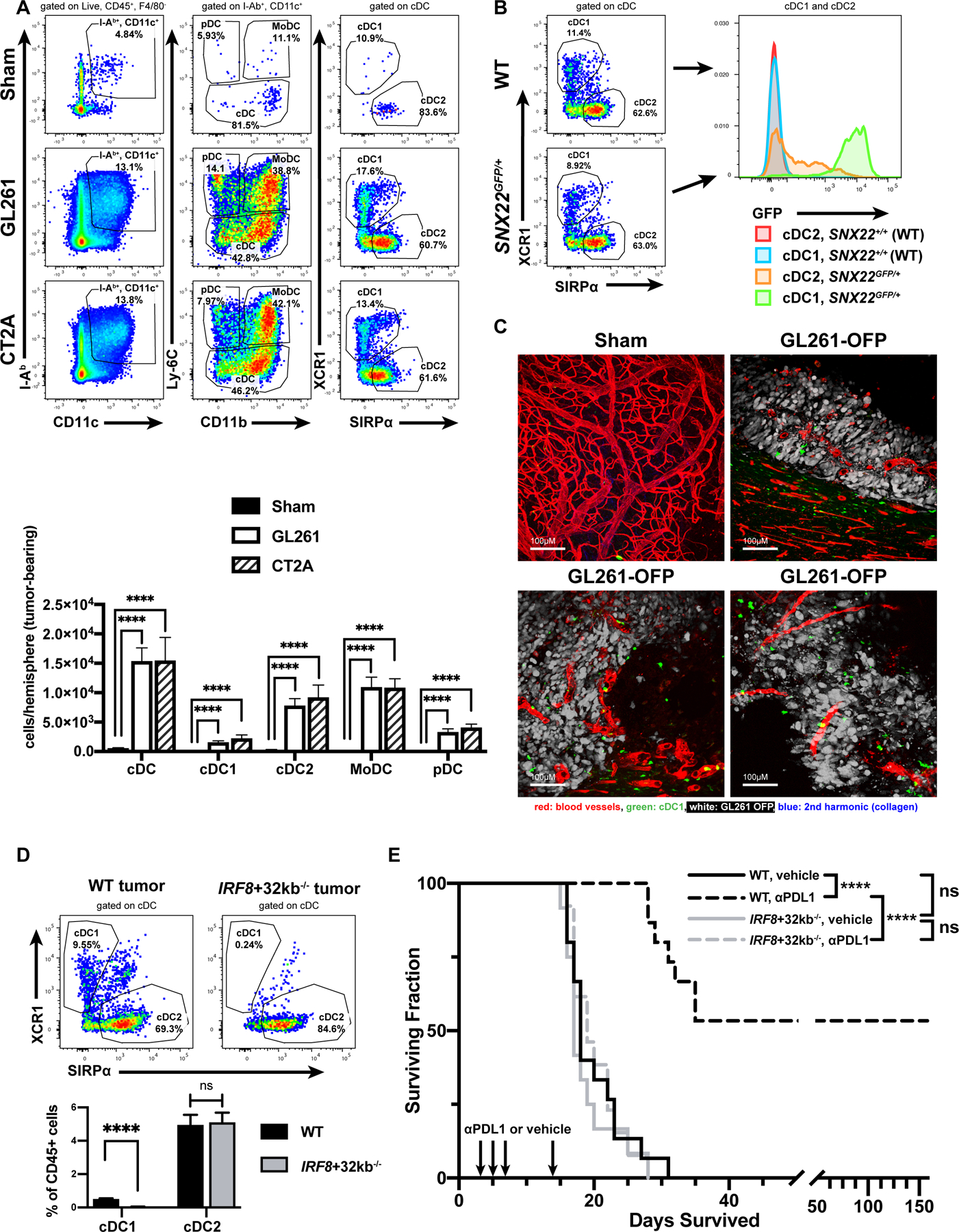
(A) Sham-treated controls (20 mice) or mice with GL261 (28 mice) or CT2A (6 mice) brain tumors assessed by flow cytometry. (B) GFP expression in cDC1/cDC2 infiltrating GL261 brain tumors in SNX22GFP/+ mice (3 mice). (C) 2-photon microscopy of various regions in sham-injected brains (5 mice) or GL261-OFP brain tumors (9 mice). Scale bar: 100 µm. (D) cDC1/cDC2 fraction of CD45+ cells in wild-type (16 mice) vs. IRF8+32kb−/− (15 mice) GL261 brain tumors. (E) Survival of vehicle- or anti-PD-L1-treated WT or IRF8+32−/− mice (WT/vehicle: 15 mice, WT/anti-PD-L1: 15 mice, IRF8+32−/−/vehicle: 12 mice, and IRF8+32−/−/anti-PD-L1: 13 mice). Data are represented as mean +/- SEM of at least three independent experiments. ****p<0.0001, ns: not significant. Differences in DCs analyzed with unpaired, two-tailed T tests. Survival analyzed with log-rank test between individual groups.
To visualize cDC1 in brain tumors, we employed the Snx22GFP/+ knock-in mouse, in which cDC1 specifically and constitutively express GFP [52]. In GL261 or CT2A brain tumors from Snx22GFP/+ mice, flow cytometry showed that cDC1 and cDC2 subsets infiltrated brain tumors, and GFP expression was restricted to the XCR1+/cDC1 subset (Figure 1B). We used 2-photon microscopy to precisely localize GFP-expressing brain tumor-associated cDC1. Whereas cDC1 were scarce within steady-state brain parenchyma, cDC1 infiltrated extravascular spaces in Snx22GFP/+ brain tumors (Figure 1C). These data demonstrate that DCs, including cDC1, infiltrate orthotopic GBM.
We next tested the hypothesis that cDC1 were required to mount an immune response against GL261 brain tumors. We employed cDC1-deficient IRF8+32kb−/− mice [53]. Compared to wild-type mice, the immune infiltrate within GL261 brain tumors in IRF8+32kb−/− brains lacked XCR1+ cDC1 (Figure 1D). Both the deep and superficial cervical (c)LNs were additionally deficient in cDC1 in IRF8+32kb−/− compared to wild-type mice with brain tumors (Supplementary Figure S3A-B). We next determined whether checkpoint blockade protection against brain tumors required cDC1, as anti-PD-1/PD-L1 treatment improves survival against GL261 [21, 49, 54, 55]. Consistently, anti-PD-L1 treatment increased the median and overall survival of wild-type GL261-bearing mice compared to vehicle controls; however, cDC1-deficient IRF8+32kb−/− mice experienced no treatment benefit compared to wild-type controls (Figure 1E). Wild-type and IRF8+32kb−/− vehicle controls exhibited no difference in median or overall survival. These data show that DCs, including the cDC1 subset, although scarce in steady-state brain parenchyma, are recruited to the brain tumor microenvironment, and that anti-PD-L1 checkpoint blockade responsiveness in this setting requires cDC1.
cDC1 prime CD8+ T-cell responses against glioblastoma
Because cDC1 can present antigen to prime T-cell responses, we determined the effects of cDC1 deficiency on T-cell composition within the GL261 brain tumors. Compared to wild-type mice, tumor-infiltrating lymphocytes (TIL) of cDC1-deficient IRF8+32kb−/− mice harbored fewer T cells, including both non-Treg CD4+ and CD8+ T cells (Figure 2A; Supplementary Figure S1B-C). In contrast, TIL from both genotypes harbored similar frequencies of CD4+ regulatory T cells (Tregs) (Figure 2A; Supplementary Figure S1C). A smaller proportion of CD8+ T cells from GL261 tumors in IRF8+32kb−/− mice expressed the functional markers granzyme B and PD-1 compared to wild-type mice (Figure 2B; Supplementary Figure S1D). These data show that GL261 tumors from IRF8+32kb−/− mice both harbor fewer T cells and that a smaller proportion of the infiltrating CD8+ T cells exhibit an activated phenotype.
Figure 2. cDC1 prime effector and neoantigen-specific CD8 T-cell responses against mouse GBM.
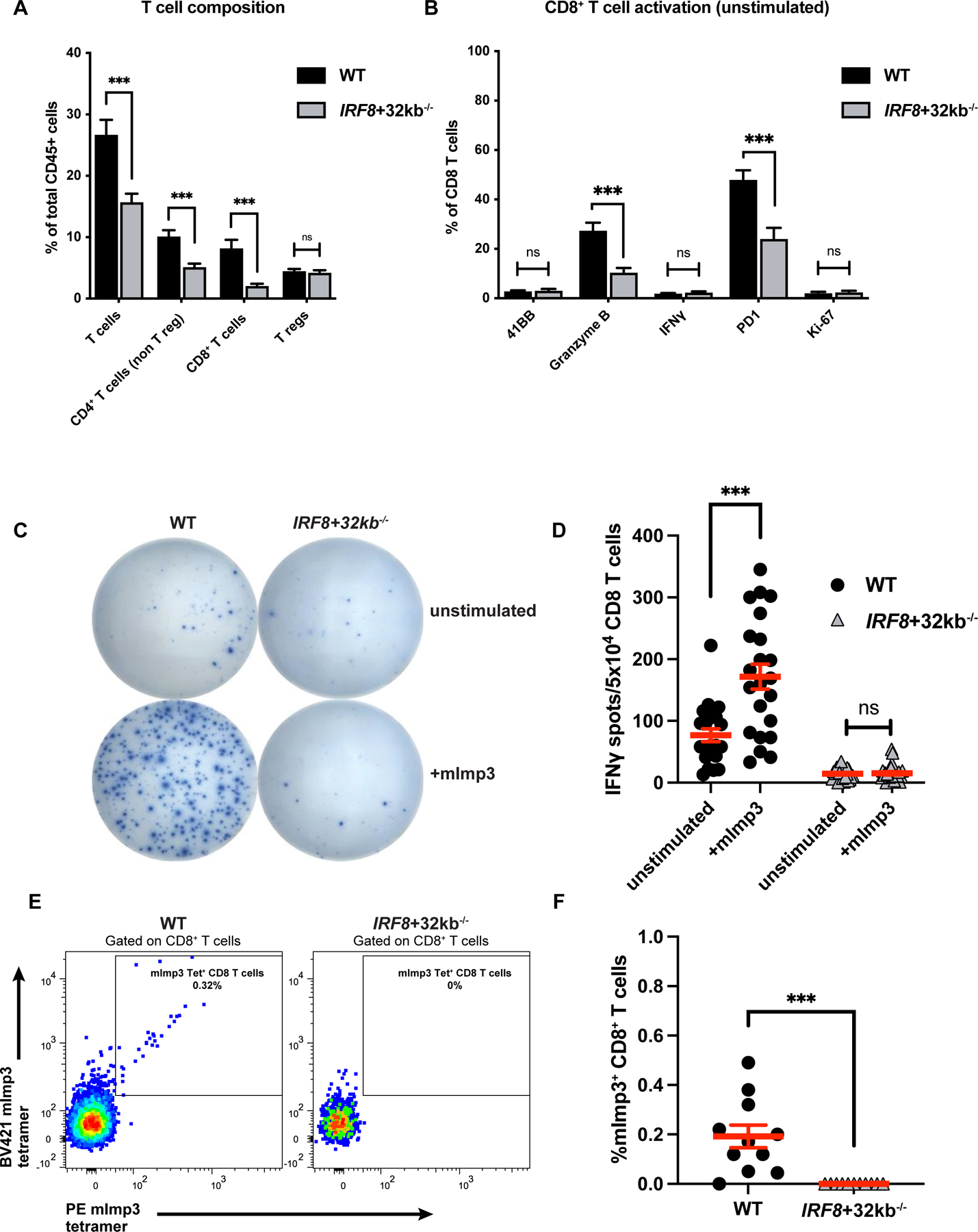
(A) T-cell composition of wild-type (WT) vs. IRF8+32kb−/− GL261 brain tumors assessed by flow cytometry. (B) CD8+ T-cell activation marker expression of WT (16 mice) vs. IRF8+32kb−/− (15 mice) GL261 brain tumors assessed by flow cytometry. (C-D) CD8+ T-cells were purified from tumors of WT (11 mice) and IRF8+32kb−/− (12 mice) mice bearing GL261 tumors and assessed by ELISPOT (IFNγ release) for mImp3 neoantigen-specific CD8+ T-cell responses. (C) Representative images and (D) quantification. (E-F) mImp3 neoantigen specific CD8+ T-cell responses assessed by tetramer staining and flow cytometry (11 WT and 11 IRF8+32kb−/− mice). (E) Representative histograms and (F) cumulative results. Data are represented as mean +/- SEM of at least three independent experiments. ***p <0.001, ns: not significant. Differences in cell infiltrate, tetramer binding, and IFNγ release analyzed with unpaired two-tailed T tests.
Although studies have demonstrated that cDC1 cross-present antigen to prime CD8+ T-cell responses in tumors outside the CNS [28, 29, 31, 33–37], it remains unclear whether they perform a similar function in brain tumors, particularly given their scarcity in steady-state brain parenchyma. Therefore, we tested whether cDC1 were required to prime neoantigen-specific CD8+ T cells against GL261 brain tumors. Previously, we found that mice harboring intracranial GL261 mount endogenous CD8+ T-cell responses against the H-2Db-restricted neoantigen, mutant-Imp3 (mImp3)[48]. In contrast to wild-type mice, we identified that equal numbers of CD8+ T cells isolated from cDC1-deficient IRF8+32kb−/− GL261 brain tumors had deficient interferon-gamma (IFNγ) production when stimulated ex vivo by APCs presenting mImp3 peptide by ELISPOT (Figure 2C-D), and further, completely lacked H-2Db-mImp3 tetramer+ CD8+ T cells (Figure 2E-F). These data show that cDC1 are required to prime effector CD8+ and neoantigen-specific CD8+ T-cell responses against GL261 brain tumors.
Tumor antigen-containing cDC1 infiltrate brain tumors and localize to cLNs
cDC1 can phagocytize antigen in the periphery and home to secondary lymphoid tissues to prime naive T cells. Although the brain parenchyma does not harbor conventional secondary lymphoid tissue, compelling data have implicated the extracranial cLNs as central to priming CNS antigen-specific T cells [19, 21, 24–26, 56]. We used flow cytometry to determine whether cDC1 phagocytized, retained, and trafficked fluorescent protein overexpressed by brain tumor cells, similar to previously employed approaches in pre-clinical melanoma models [29–31]. We created two transgenic glioma lines: GL261-zsGreen and CT2A-zsGreen. In mice, the tumor-derived zsGreen protein, phagocytosed by infiltrating immune cells, functions as a detectable surrogate for tumor antigen uptake and trafficking (Figure 3A). We used CT2A-zsGreen for most tumor antigen-tracking experiments because it most consistently retained zsGreen expression at the time of harvest. In intracranial CT2A-zsGreen tumors, we detected not only zsGreen-positive cDC1, but also zsGreen-positive cDC2, monocyte-derived DCs (MoDC), and plasmacytoid DCs (pDC) (Figure 3B; Supplementary Figure S1A), which were absent from non-fluorescent CT2A controls, suggesting that these populations phagocytose tumor-derived antigens in vivo. We next examined the ipsilateral cLNs to probe for extracranial zsGreen+ APCs. We identified migratory and resident zsGreen+ cDC1 in both the deep and the superficial cLNs (Figure 3C-D, Supplementary Figure S1E). A larger fraction of migratory cDC1 (CD103+CD8α−) were zsGreen+ compared to the resident cDC1 subset (CD8α+CD103−). We also identified zsGreen+ cDC2, MoDC, and pDC in the superficial cLNs and MoDC in the deep cLNs (Figure 3C-D; Supplementary Figure S4A-B). Although GL261-zsGreen tumors less reliably retained zsGreen expression in vivo, zsGreen was still detectable within a considerable proportion of the tumor-infiltrating DCs (Supplementary Figure S5A). We additionally detected a significant proportion zsGreen+ cDC2 in the deep cLNs, as well as cDC2, MoDC and pDC in the superficial cLNs compared to non-fluorescent controls (Supplementary Figure S5B-C). This extended our findings with CT2A-zsGreen brain tumors. These data show that cDC1, as well as other APC populations, harbor tumor-derived antigen within the tumor microenvironment and localize to cervical lymphoid tissue across different brain tumor preclinical models.
Figure 3. cDC1 derived from cervical lymph nodes harbor tumor-derived antigen in a CCR7-mediated manner.
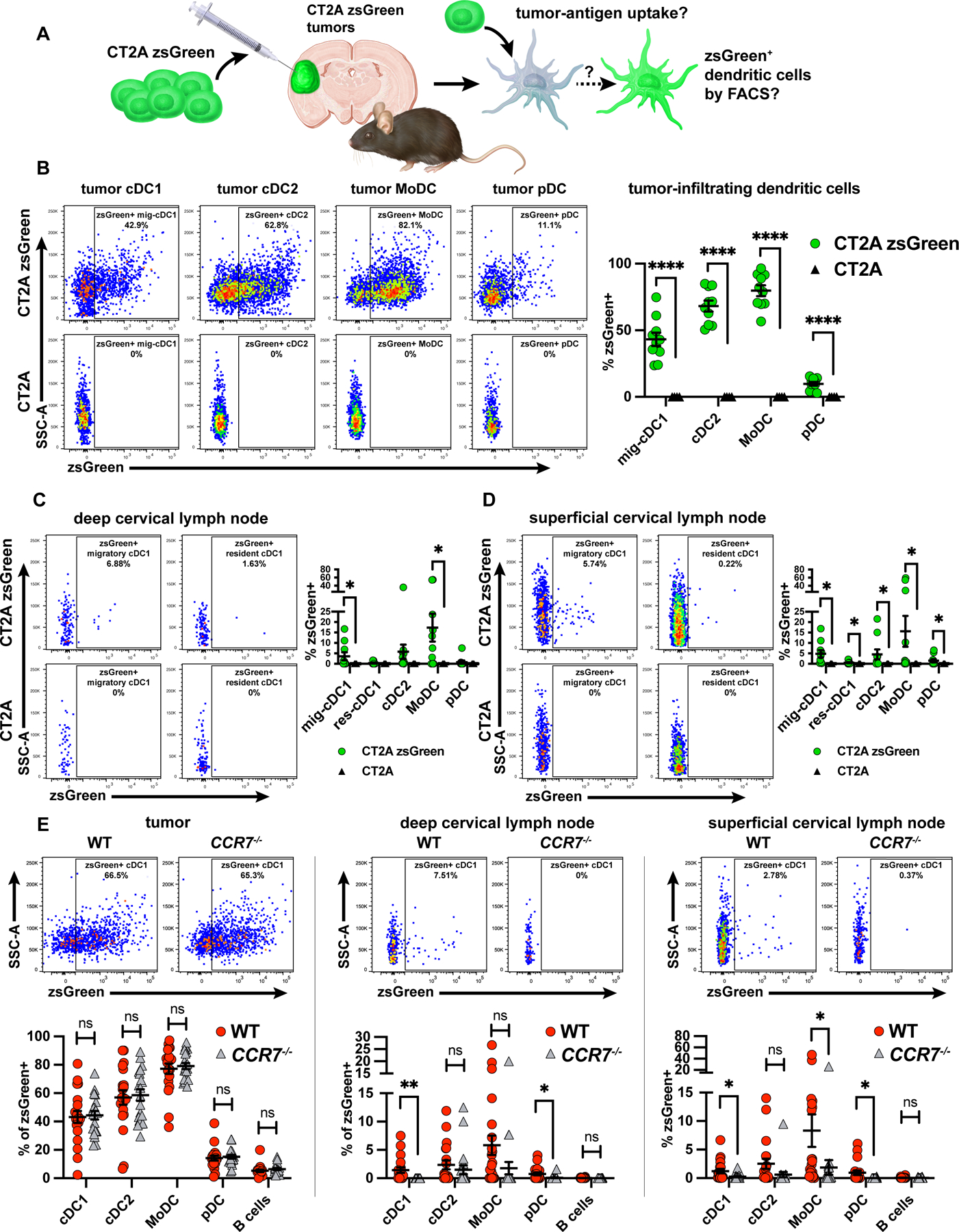
(A) zsGreen-labeled tumor cells were used in an orthotopic model. DCs were assessed for zsGreen uptake via flow cytometry. (B-D) zsGreen phagocytosis by DCs at day 14 post-injection (B) CT2A-zsGreen (10 mice) or CT2A tumors (4 mice) with associated (C) deep cervical lymph nodes and (D) superficial cervical lymph nodes. (E-G) zsGreen phagocytosis by DCs at day 12 post-injection (E) CT2A-zsGreen tumors of wild-type (18 mice) vs. CCR7−/− mice (18 mice) with associated (F) deep cervical lymph nodes, and (G) superficial cervical lymph nodes. Data are represented as mean +/- SEM of at least three independent experiments. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Differences in %zsGreen+ analyzed with unpaired one-tailed T test with Welch’s correction. Grubb’s outlier test in (E) used to test for and exclude an outlier from both the WT and the CCR7−/− genotypes.
Given our findings regarding the importance of cDC1 in mounting CNS anti-tumor immunity, we set out to investigate the requirement of cDC1 for trafficking tumor antigen from brain tumors to draining cLNs. Although cDC1 were deficient IRF8+32kb−/− mice, cDC2, MoDC, and pDC phagocytosed and trafficked tumor antigen from tumors to superficial and deep cLNs in equivalent proportions between wild-type and IRF8+32kb−/− mice (Supplementary Figure S6A-B). These data demonstrate that although cDC1 may be required to mount CNS antitumor immunity, they are not required to traffic tumor antigen from brain tumors to draining cLNs.
Having observed zsGreen+ cLN-associated cDC1, we asked how tumor-derived antigen was trafficked from the CNS to cLNs. We hypothesized that brain tumor-associated antigens could be either actively trafficked by cellular transport within APCs via CCR7-mediated chemotaxis [57] or passively transferred from the brain to the cLN via acellular mechanisms. Therefore, we tested if zsGreen+ cDC1 required CCR7 to localize to cLNs. Because CCR7−/− mice lack migratory DCs in LNs, we did not distinguish between migratory and resident cDC1 in our analysis and assessed total cDC1. We intracranially injected CT2A-zsGreen into both wild-type and CCR7−/− mice and monitored the immune cell zsGreen signal in brain tumors and cLNs. We observed equivalent zsGreen uptake by tumor-infiltrating cDC1 in both wild-type and CCR7−/− mice, suggesting that cDC1 were able to infiltrate brain tumors and phagocytose tumor antigen independent of genotype (Figure 3E, left). However, in CCR7−/− mice we observed a significantly attenuated zsGreen signal in deep and superficial cLN cDC1 and pDC, and in superficial cLN MoDC compared to wild-type controls (Figure 3E, middle and right). Small but equal fractions of B cells (CD45+F4/80−I-Ab+CD11c−B220+Ly-6C−) were zsGreen+ in cLNs of wild-type and CCR7−/− mice (Figure 3E, middle and right), which poses passive cell migration-independent trafficking as a minor contributing mechanism of CNS lymphatic drainage as B cells enter lymph nodes through high endothelial venules rather than migrating from the periphery [58, 59]. Additionally, the zsGreen signal was incompletely diminished in DCs from CCR7−/− cLNs, further suggesting that CCR7-independent passive drainage functions to traffic a small fraction of tumor antigen to draining cLNs. Together with our observation that cDC1 are required to generate brain tumor-specific T-cell responses, these results suggest that cDC1 perform this function, at least in part, by phagocytizing tumor-associated material within the brain and trafficking it to the LNs in a CCR7/cell migration-dependent manner.
Dura-associated cDC1 undergo dynamic changes in response to glioblastoma
Previous work has identified the dura, the outer meningeal layer covering the brain, as an immunologically dynamic structure that harbors cDC1, cDC2, pDC, macrophages, T cells, and B cells in both mice [14, 23] and humans [60]. This contrasts with the resting-state brain parenchyma, which is devoid of all leukocytes except microglia and border-associated macrophages [14]. Additional experiments have demonstrated that Flt3L stimulation expands dura-associated DCs [14, 15]. Further work has also shown that the dura harbors a network of lymphatic vessels [18–21, 61, 62], which not only drain cerebrospinal fluid and antigens to the deep cLNs but also proliferate in response to VEGF-C stimulation to potentiate CNS antitumor immunity [21]. Given these findings, we set out to further characterize how the dura might support CNS antitumor immunity, focusing specifically on DCs and cDC1. We evaluated cDC1 localization, the dynamics of DC populations during antitumor responses, as well as the antigen presentation capacity of dura-associated cDC during brain tumor growth. We first investigated localization of dura-associated GFP+cDC1 in SNX22GFP/+ mice by 2-photon microscopy. In control and GL261-OFP-bearing SNX22GFP/+ mice, the dura harbored extravascular cDC1 (Figure 4A). We hypothesized that brain tumors would drive increased dura-infiltrating cDC1. Compared to sham-injected mice, whole dura from intracranial GL261-bearing mice had increased numbers of cDC1, cDC2, MoDC, and pDC by flow cytometry (Figure 4B), although the dura harbored considerably fewer DCs than tumors themselves. We resected the dura surrounding the injection site to avoid skewing data by inadvertently attributing tumor populations to the dura.
Figure 4. DCs are harbored by dura and dura lymphatics and undergo dynamic changes in response to intracranial tumors.
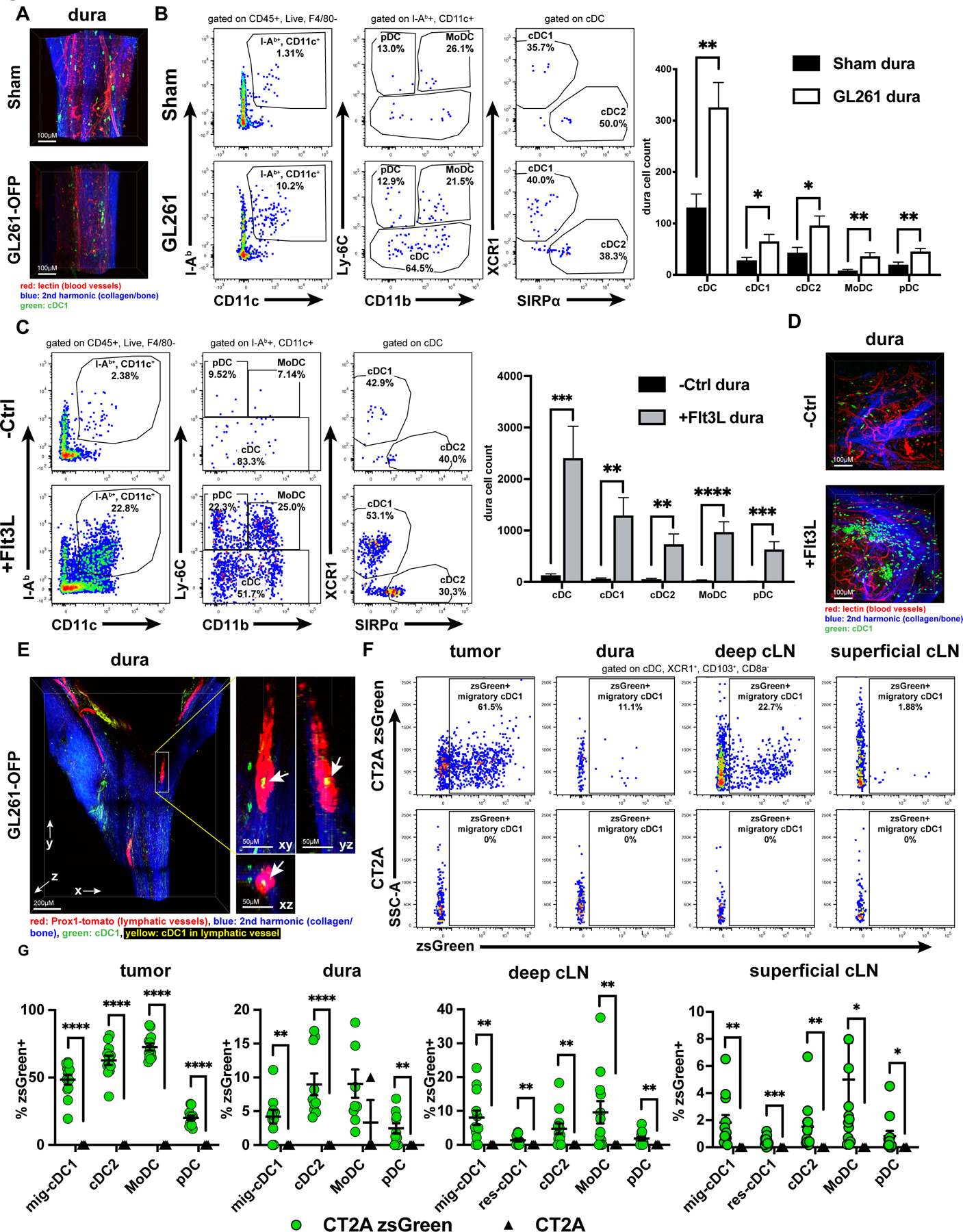
(A) 2-photon microscopy of SNX22GFP/+ mouse dura along the superior sagittal sinus from sham (6 mice) vs. GL261-OFP (6 mice) intracranially injected mice. Scale bar: 100µM. (B) Dura DCs assessed and quantified by flow cytometry in sham-injected (21 mice) vs. GL261-injected (28 mice) brains and (C) in control (-Ctrl, 13 mice) vs. Flt3L-treated (12 mice). (D) 2-photon microscopy of SNX22GFP/+ mouse dura from -Ctrl (6 mice) vs. Flt3L-treated (6 mice). Scale bar: 100µM. (E) Dura from intracranial GL261-OFP bearing SNX22GFP/+/Prox1-Cre-tdTomato+/- (tamoxifen-induced) (5 mice) examined to assess localization of GFP+ cDC1 with respect to tdTomato+ lymphatic vessels. Scale bar: 200µM. Zoomed, scale bar: 50µM. 3-D image on left with XY/YZ/XZ planes derived from the 3-D image on right. Arrowheads point to GFP (green)/tdTomato (red) overlap (resulting in yellow), indicating cDC1 within lymphatic vessel. (F) Dura-associated zsGreen+ migratory cDC1 assessed by flow cytometry at day 7 post-injection, and (G) quantified across all DC subsets CT2A-zsGreen (12 mice) or CT2A (3 mice) brain tumors. Dura samples with CD45−/zsGreen+ cells were assumed to be contaminated by tumor infiltrate and were excluded from analysis. Data are represented as mean +/- SEM of at least three independent experiments. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Differences in DCs (B-C) analyzed with unpaired two-tailed T tests. Differences in %zsGreen+ (G) analyzed with unpaired one-tailed T test with Welch’s correction.
Given prior studies demonstrating the existence of dura-associated Flt3L-responsive CD11c+I-Ab+ cells [15] or Flt3L-driven cDC1/cDC2 expansion when whole brain and surrounding meninges are examined together [14], we specifically examined the dynamics of dura-associated cDC1 in response to Flt3L stimulation. When we administered Flt3L by introduction of CT2A cells expressing mFlt3L into the flank, dura DC subsets (including cDC1) expanded, observed by flow cytometry (Figure 4C), as well as by 2-photon microscopy (Figure 4D, Supplementary Figure S7A). Notably, Flt3L induced greater expansion of dura DCs than intracranial GL261 brain tumors. These data show that dura-resident cDC1 expand in response to brain tumors or systemically administered Flt3L.
We next investigated the spatial relationship between cDC1 and the CNS-draining dura lymphatic vessels, which could presumably support DC migration from the brain parenchyma to cLNs. We crossed the SNX22GFP/GFP mouse with the Prox1-Cre-tdTomato+/+ mouse, which expresses tdTomato specifically in lymphatic vessels [44]. Tamoxifen-treated F1 mice have GFP+ cDC1 and tdTomato+ lymphatic vessels. Using 2-photon microscopy, we detected GFP+ cDC1 within the dura lymphatic vessels of mice which harbored GL261-OFP brain tumors (Figure 4E, Supplementary Figure S7B) or were sham-injected (Supplementary Figure S7C). These data show that cDC1 localize to the lumen of dura-lymphatic vessels in both tumor-bearing and steady-states.
Having identified that dura harbors cDC1 which can localize to lymphatic vessels, we tested if dura-associated cDC1 could acquire tumor-derived antigen from intraparenchymal brain tumors. We orthotopically injected CT2A-zsGreen tumors into the brain parenchyma of mice and monitored the zsGreen APC signal 7 days post-injection in tumors, dura, and cLNs by flow cytometry. To avoid contamination of the dura with underlying parenchymal zsGreen+ tumor cells and tumor-infiltrating immune cells, we (1) selected an early time point to ensure that tumors were small and did not abut the dura and (2) resected the dura surrounding the prior injection site to avoid potential contamination of the dura samples by adjacent tumor and immune cells. We observed zsGreen+ migratory cDC1 (in addition to cDC2, pDCs, and MoDCs) in tumors, dura, superficial and deep cLNs, as well as resident cDC1 in the cLNs (Figure 4F-G). We did not observe CD45−zsGreen+ cells in the dura, which indicates that our dura samples lacked tumor contaminant. Together, these data show that dura-associated, Flt3L-responsive cDC1 can localize to dura lymphatic vessels and, along with other dura-associated APC, harbor tumor antigen from intraparenchymal tumors.
CD8+ T-cell priming occurs in cLNs and requires cDC1 and CCR7
Having established that tumor antigen-containing DCs can be found in the dura, as well as the superficial and the deep cLNs, we examined where T-cell priming occurred in response to CNS tumors, as this remains incompletely characterized. Several observations support a role for cLNs in CNS pathology: (1) ligation of deep cLNs attenuates VEGF-C-mediated brain tumor rejection [21]; (2) lymphadenectomy of cLNs ameliorates disease burden in rodent models of EAE [63–65]; and (3) ablation of meningeal lymphatics decreases antigen-specific T-cell/CD11c+ cell interactions in cLNs of mice with EAE [62]. Because we detected tumor antigen-containing APCs in several anatomic sites, we hypothesized that CD8+ T-cell priming could occur within tumors, dura, cLNs, or the spleen. To this end, we tracked cell division in vivo of OT-I T cells adoptively transferred into mice bearing intracranial GL261 tumors expressing full-length ovalbumin (GL261-OVA). OT-I T cells recognize the H-2Kb-restricted OVA antigen SIINFEKL [46] and proliferate upon being primed. We injected GL261-OVA into the brains of CD45.2+ C57BL/6 hosts. Four days post, we intravenously injected CFSE-labeled, CD45.1+CD45.2+ OT-I CD8+ T cells. We harvested tumors, dura, ipsilateral superficial and deep cLNs, spleens, and non-CNS-draining contralateral inguinal LNs at days 3 and 6 post-transfer to evaluate CFSE dilution in OT-I CD8+ T cells (Figure 5A-C). We distinguished CFSE-low, CFSE-mid, and CFSE-high OT-I CD8+ T cells, which we envisaged to reflect terminal effector, initially primed, or unprimed OT-I cells, respectively.
Figure 5. Clonal Expansion of OT-I CD8+ T-cells occurs primarily in extracranial CNS-draining cervical lymph nodes.
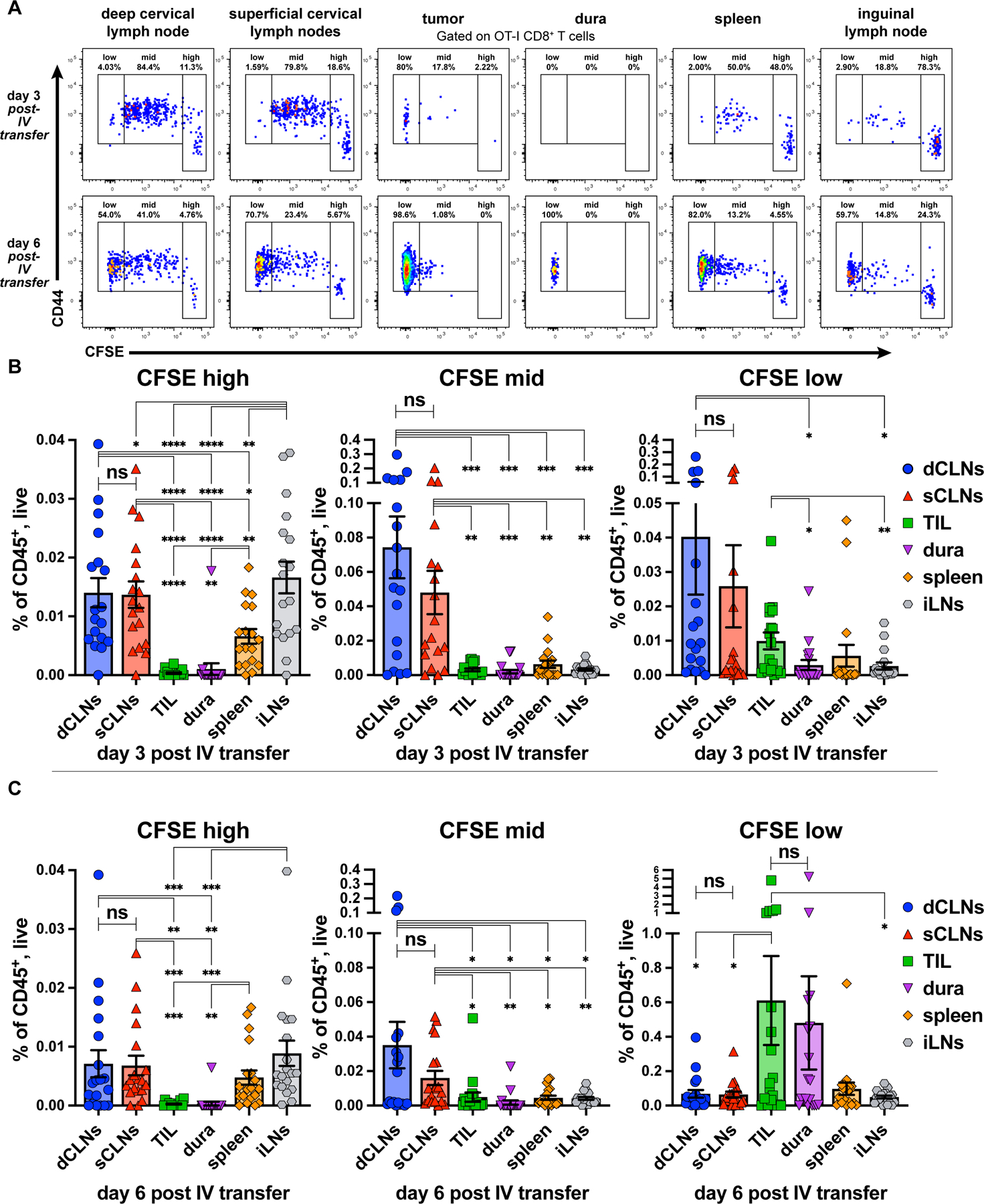
(A) CD44 expression and CFSE dilution of OT-I CD8+ T-cells in ipsilateral deep cervical lymph nodes (dCLNs), ipsilateral superficial cervical lymph nodes (sCLNs), tumors, dura (with tumor-abutting region resected), spleen, and non-draining contralateral inguinal lymph node (iLNs) assessed by flow cytometry at day 3 (18 mice) and day 6 (19 mice) post adoptive IV transfer. Quantitation of CFSE-high, -mid, and -low OT-I CD8+ T-cells at (B) day 3 and (C) day 6 post-transfer. Data are represented as mean +/- SEM of at least three independent experiments. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, ns: not significant. Differences between organs for OT-I CFSE compared with paired two-tailed T tests.
We harvested tissues at days 3 and 6 post-transfer to capture snapshots of clonal expansion evolution across tissues. At these timepoints, we observed the greatest numbers of CFSE-high/unprimed OT-I T cells (Supplementary Figure S1F) in LNs, regardless of location, with fewer in the spleen, and almost none in the tumor or dura (Figure 5B-C, left), commensurate with our understanding that naive T cells circulate between lymphoid organs before activating and dividing. At both 3 and 6 days post-transfer, the CNS-draining superficial and deep cLNs harbored significantly more CFSE-mid/initially primed OT-I T cells than other tissues (Figure 5B-C, middle), consistent with our understanding that clonal expansion first occurs in proximal draining LNs. At day 3 post-transfer, CFSE-low/effector OT-I T cells occupied the greatest fraction of CD45+ cells in the deep cLNs, but were also in the superficial cLNs and TIL, albeit at lower numbers (Figure 5B, right). However, by day 6 post-transfer, TIL harbored the greatest fraction CFSE-low/effector OT-I T cells compared to all other sites except the dura (Figure 5C, right). This underscores our understanding that the tumor is both the source of antigen and effector site for OT-I T cells.
As a control, we compared CFSE dilution of adoptively transferred OT-I cells from the same tissues in mice that harbored parental GL261 or GL261-OVA brain tumors and observed that mice harboring GL261-OVA tumors had significantly more divided OT-I cells in CNS-draining cLNs, as well as in the spleen, and inguinal LNs at day 3 post-transfer compared to GL261 controls (Supplementary Figure S8A-B; top and left, respectively). At day 6 post-transfer, these same differences held, but expanded to include tumors (Supplementary Figure S8A-B; bottom and right, respectively). We observed similar patterns of expansion of OT-I cells in CT2A-OVA tumors vs. untransduced CT2A tumors, although the expansion was weaker and somewhat delayed compared to that seen in GL261-OVA tumors (Supplementary Figure S9A-B), which may be due to the immune-suppressive effects of CT2A. Together, these data support a model in which CD8+ T cells are primed against brain tumors in the CNS-draining cLNs, and subsequently home to the CNS to mount anti-tumor responses. Additionally, although tumors are a primary site of effector function, the dura also harbors effector T cells.
Given our observations that cDC1 are required to prime neoantigen-specific T-cell responses against CNS tumors, and that CCR7 is required to traffic antigen from brain tumors to draining cLNs, we next investigated their role in clonal expansion of adoptively transferred OT-I T cells in mice with GL261-OVA brain tumors. Compared to wild-type mice, CCR7−/− and IRF8+32kb−/− mice had decreased clonal expansion of OT-I T cells in cLNs and spleens 3 days post-transfer (Supplementary Figure S10A, 1st, 2nd, and 5th graph from left). In contrast, we observed minimal differences in clonal expansion between wild-type, CCR7−/−, and IRF8+32kb−/− mice at day 6 post-transfer (Supplementary Figure S10B). These data collectively demonstrate that CD8+ T-cell priming and clonal expansion against brain tumor-specific antigens occurs in the cLNs and that absence of cell migration to lymph nodes and cDC1 deficiency lead to delayed, but not completely absent clonal expansion in this setting.
CD141+ cDC1 are detectable in human dura and brain tumors
Having identified cDC1 and additional cDC subsets in murine brain tumors and matched dura, we investigated whether human tumors and dura also harbored infiltrating DC populations. Although human DC subsets and functions are less well characterized than in mice, the human cDC1 parallel are CD141-expressing cDC. In both mice and humans, CD141+/cDC1 subset produces IL12 [66], cross-presents exogenous proteins to CD8+ T-cells [66], and expresses IRF8 [67], a critical regulatory factor required for mouse [53] and human [68] cDC1 development. We explored immune cell populations within the tumor and matched dura of patients. In addition to tumors, adjacent normal, non-tumor involved dura was also resected when indicated (Figure 6A). We performed flow cytometry on six matched tumor/dura specimens (five meningioma, one GBM) and on an additional twelve GBM tumor specimens (thirteen total-11 primary, 2 recurrent). In both GBM and meningiomas, we detected multiple human DC subsets within tumor and dura specimens, including cDC1-equivalent CD141+ cDC, cDC1-equivalent CD1c+ cDC, as well as CD14+ and CD16+ monocytes (Figure 6B-D, Supplementary Figure S2A). We identified cDC1, cDC2, CD14+ monocytes, CD16+ monocytes, CD4+ and CD8+ T cells in most of the GBM tumors that we analyzed (Figure 6C). Between GBM and meningiomas, the matched dura samples harbored similar fractions of each subset, although our analysis was limited to a single GBM specimen with matched dura resection (Figure 6E). These findings collectively show that human conventional DC subsets, as well as monocytes, are abundant in dura and tumors and across multiple brain tumor types, which suggests they may play a role in human CNS anti-tumor immunity.
Figure 6. DCs infiltrate human dura and tumors.
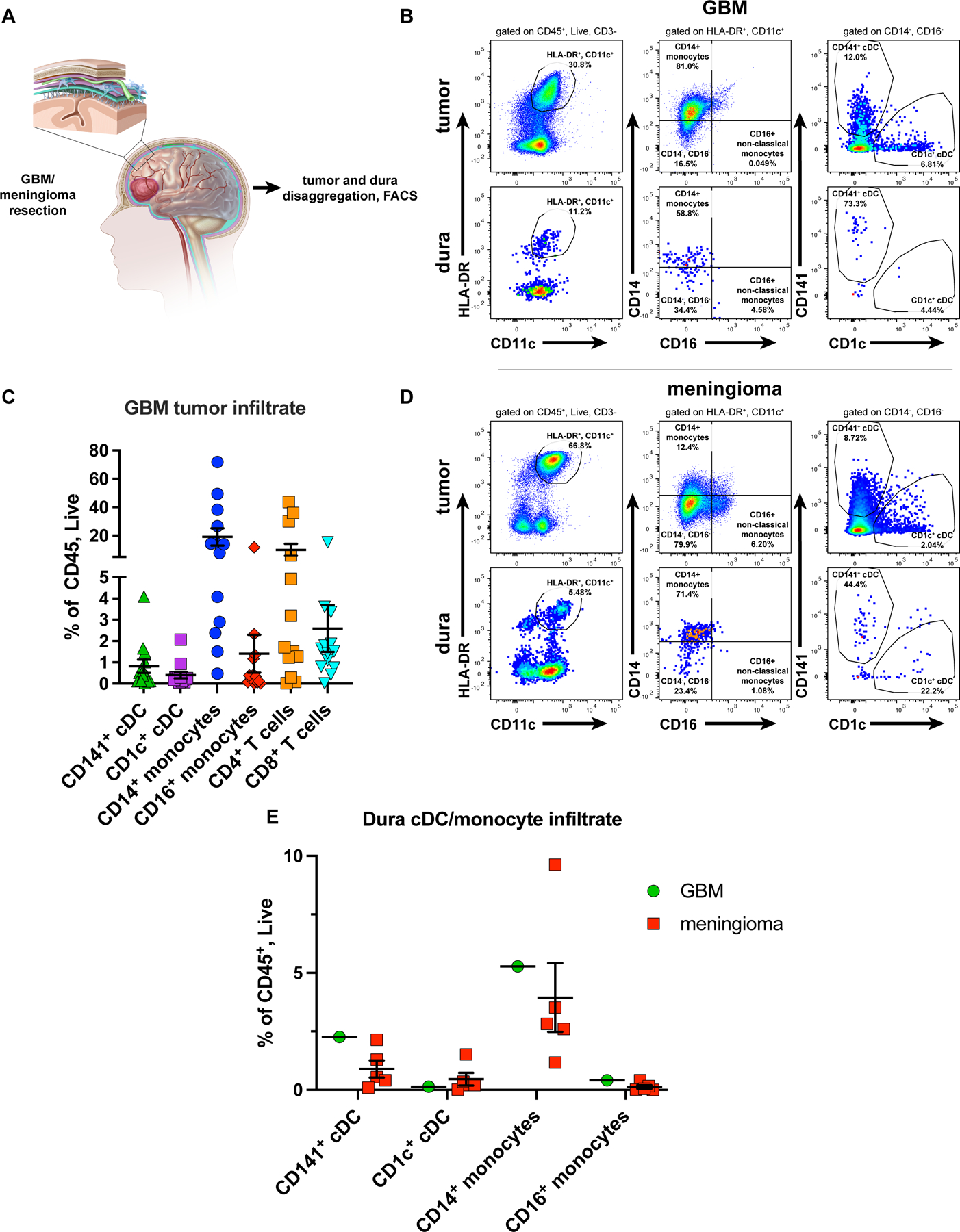
(A) Representative sketch of the resection of brain with meningioma/GBM and associated dura (also resected when clinically indicated). Dura and tumor infiltrating CD141+ cDC and CD1c+ cDC assessed by flow cytometry. (B) cDC in GBM and (C) associated immune cell composition (11 primary, 2 recurrent GBM tumors). (D) cDC in a meningioma. (E) DC and monocyte composition in a GBM-associated (1 specimen) dura sample vs. meningioma-associated (5 specimens) dura samples. Plotted are mean +/- SEM.
CD141+ cDC and other APCs phagocytose tumor-specific markers in GBM
We next evaluated cDC and APC subsets for ability to phagocytize GBM-derived material. Analogous to our studies tracking tumor-derived zsGreen, during GBM resection, the FDA-approved compound 5-aminolevulinic acid (5-ALA) can be used in “fluorescence-guided surgery” to fluorescently label tumors and distinguish them from normal brain [38–41]. After patients ingest 5-ALA preoperatively, the compound crosses the blood-brain barrier and is metabolized by target GBM cells to the fluorescent protoporphyrin IX (PPIX) [42], which specifically accumulates and is retained by GBM cells [42, 43]. Illuminating the tumor with blue light fluorescently excites PPIX, causing tumors to emit pink light (Figure 7A). We tested via flow cytometry whether APCs, including cDC, from GBMs resected using 5-ALA could acquire and retain tumor-derived PPIX (Figure 7B). We analyzed specimens both from 5-ALA-treated patients and U343 GBM cells exposed to 5-ALA. We determined that the PPIX signal was brightest in the BV650 channel (Supplementary Figure S11A). We also observed clearly defined PPIX-low and PPIX-high cell populations, which suggests a range of PPIX uptake and retention among tumor cells (Figure 7C, left). We thus used the BV650 channel to detect PPIX for further experiments. Compared to untreated U343 cells, 5-ALA treated U343 cells also fluoresced brightly in the BV650 channel (Figure 7C, right). We next analyzed via flow cytometry tumor and matched peripheral blood mononuclear cells (PBMCs) from 5-ALA-treated GBM patients to determine if APCs phagocytosed tumor-derived PPIX. Specifically, we addressed two central questions: (1) Do APCs phagocytose PPIX only in the tumor microenvironment and not in the periphery? (2) In tumors, do only phagocytic immune cells acquire and retain tumor-derived PPIX? In PBMCs, neither the CD3ε+ nor the CD3ε− fractions had a detectable PPIX signal. In contrast, whereas the CD3ε+ fraction of the tumor infiltrate was PPIX-negative, a majority of cells in the CD3ε− fraction were PPIX-positive (Figure 7D-E), demonstrating that PPIX-acquisition was both location-specific and immune cell type-specific. Compared to PBMCs, we observed PPIX within different APC subsets infiltrating tumors, including CD141+ cDC, CD1c+ cDC, CD14+ classical monocytes, and CD16+ non-classical monocytes across eight specimens: six primary and two recurrent GBMs (Figure 7F-G). Moreover, the PPIX-signal occurred in APCs but not T cells infiltrating the tumor (Figure 7H). Thus, multiple tumor-infiltrating APC subsets acquire and retain GBM-derived tracer. Together, these data show that PPIX-acquisition by CD45+ cells depends on both location and cell identity: only phagocytic immune cells (including DCs) within the tumor microenvironment, not the periphery, acquire PPIX.
Figure 7. APCs infiltrating human GBM uptake the tumor-specific reporter PPIX.
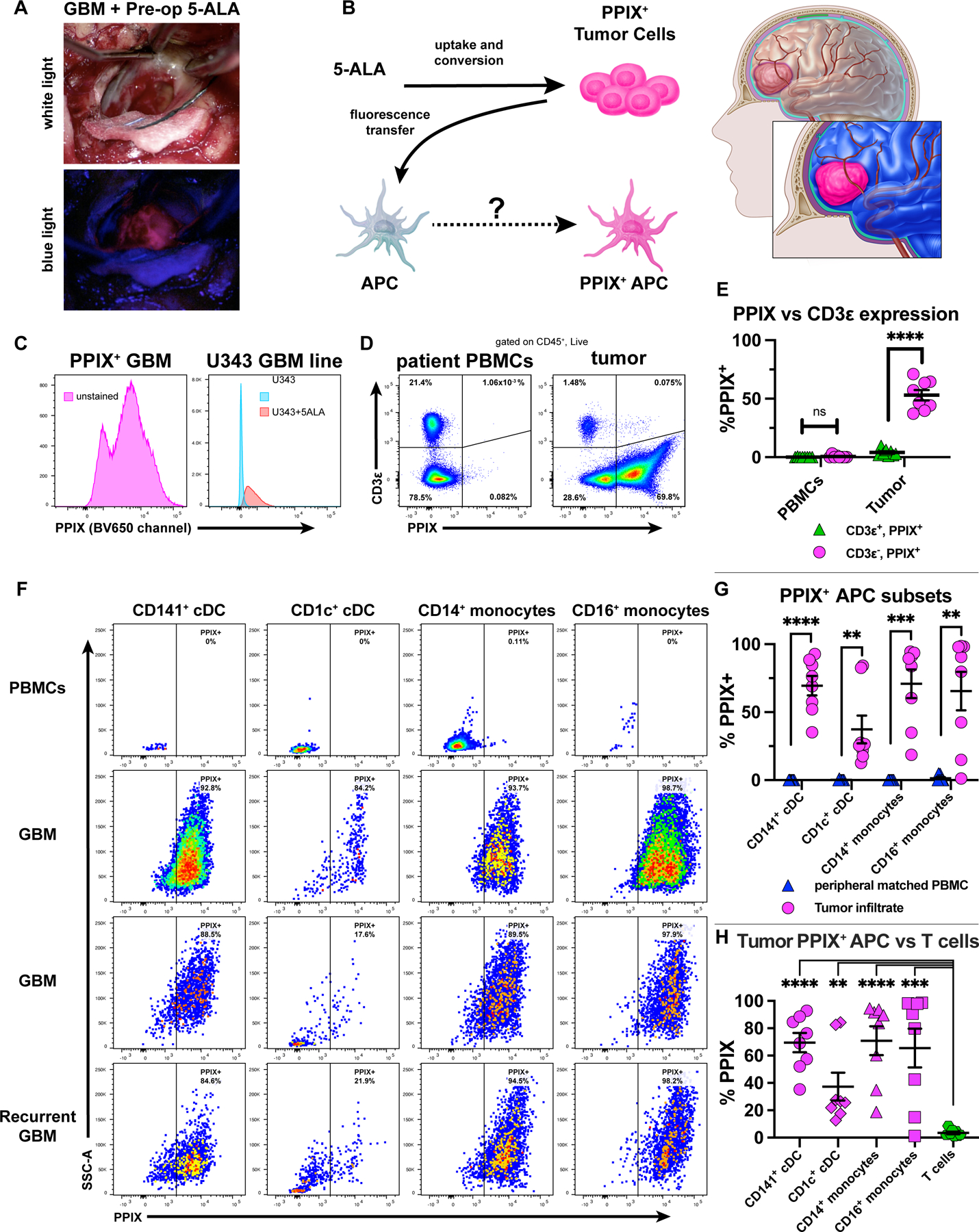
(A) GBM patient with pre-administered 5-ALA: tumor visualized under white (top) or blue (bottom) light. (B) PPIX as tumor antigen surrogate, traceable in tumor-infiltrating APCs. (C) PPIX expression in a bulk unstained tumor from a 5-ALA-resected GBM (left) or in U343 cells treated with 5-ALA (right) (representative of independent experiments). (D-E) CD3ε vs. PPIX expression of live/CD45+ cells from patient-derived PBMCs or resected GBM tumors. (F-G) PPIX+ APC subsets across 3 GBM tumors (two primary, one recurrent) compared to patient PBMCs. (H) Tumor-infiltrating PPIX+ APCs vs. T-cells. Data representative of eight patients (six primary, two recurrent) in which GBM and matched intraoperative PBMCs were taken. Plotted are mean +/- SEM. **p<0.01, ***p<0.001, ****p<0.0001, ns: not significant. Unpaired two-tailed T tests for (E) and (H). Paired two-tailed T tests for (G).
DISCUSSION
Here, we described the functional importance of cDC1 in immune responses to GBM. We showed that cDC1, although devoid from the brain parenchyma in the steady-state, infiltrate GBM in two preclinical models, and that cDC1 are critically required for the development of functional neoantigen-specific CD8+ T-cell responses against GBM, and mediate checkpoint blockade responsiveness. Using a traceable fluorescent tumor antigen system, we observed that multiple DC subsets, including cDC1, acquire tumor-derived material and traffic it to both the superficial and deep cLNs in a CCR7-dependent manner. We also identified cDC1 within the dura, a subset of which localized to within dura lymphatic vessels. We additionally detected tumor antigen within dura-associated cDC, a presumed midpoint of cDC migrating between the brain-tumor parenchyma and cLNs. Based on the dynamics of expansion of adoptively transferred T cells specific to an ectopic GBM antigen, cLNs were the predominant location of priming immune responses to GBM, which required intact cell migration and cDC1 for normal kinetics. Finally, we extended our work in mice to human tumors and dura. Both tumors and dura harbored the mouse cDC1-equivalent CD141+ DCs, among other subsets. In GBM surgically resected using 5-ALA, infiltrating APCs acquired the fluorescent PPIX tumor antigen-surrogate.
Previous reports identified the critical role of cDC1 in anti-tumor immunity for a range of tumor types [28, 29, 31, 33–37]. Our findings extend the importance of the cDC1 to the antitumor immune response in the CNS. Recent work demonstrated that Batf3-dependent cDC1 mediated rejection of immunogenic GL261-FGL2−/− brain tumors [69]. One important caveat of our respective observations is that the GL261 orthotopic pre-clinical model is sensitive to checkpoint blockade therapy. This differs from human GBM, whereby checkpoint blockade therapy is only effective in limited settings [70, 71]. Nevertheless, this model is useful to define the elements required for effective CNS anti-tumor immunity. These data collectively suggest that cDC1 are required for broad CNS anti-tumor immunity, particularly to generate neoantigen-specific CD8+ Τ-cell responses. Further work is required to understand the requirements for antigen presentation to CD4+ T cells in this setting, and further, to characterize the necessity of cDC1 in additional types of GBM preclinical models. Nevertheless, despite the immunologically specialized nature of the CNS—absence of steady-state parenchyma DCs, lack of LNs in the parenchyma, the presence of cerebrospinal fluid, and the blood brain barrier, among other features—the dependence on the cDC1 subset for immune responses to cancer appears to be shared between the CNS and other extracranial tumor types.
CNS antigen presentation has been a topic of substantial investigation; unlike other sites, the only steady-state parenchymal leukocytes are microglia and border-associated macrophages (which localize to blood vessel basement membranes) [14]. They both present antigen but lack extracranial migratory capacity. In contrast, cDC can phagocytose antigen, migrate to secondary lymphoid tissues, and prime naive T cells. Although restricted to the choroid plexus and meninges in the steady-state [14, 15], cDC infiltrate the brain parenchyma during inflammation [16, 22, 23, 25, 26, 56, 72], which holds true in tumors. Previously, the Fabry group showed that intracerebrally injected monocyte-derived DCs could migrate to the cLNs [24]. Additional studies demonstrate that cDC depletion attenuates EAE disease severity [22], that MHCII restriction to cDC was sufficient to drive disease progression [23], or that partial depletion of cDC attenuates checkpoint blockade responsiveness against GL261 [73]. Nevertheless, these observations leave unanswered whether or how endogenous cDC1 drive CNS antitumor immunity. We demonstrate that cDC1 specifically function as a key CNS APC, by acquiring antigen and migrating extracranially to endogenously prime neoantigen-specific CD8+ T cells and also by mediating anti-PD-L1 responsiveness in the GL261 preclinical model.
The relationship between CNS pathophysiology and the role of cLNs has been supported by a growing body of work in different disease models [18, 19, 21, 63–65]. Given the importance of cLNs in CNS lymphatic drainage, we explored the role of DCs, with particular attention to antigen trafficking. Researchers have shown with fluorescent tracers that draining LN APCs (including cDC1/2 subsets, monocytes, and macrophages) contain tumor-associated fluorescent antigens in preclinical melanoma models [28–31]. Our studies mirrored this: cDC1 (predominantly migratory), cDC2, MoDCs, and pDC harbored zsGreen in both the superficial and deep cLNs, underscoring the tumor antigen trafficking capability of multiple DC subsets. We additionally characterized lymphatic drainage in the absence of cDC1 and found that cDC2 and other DC subsets were capable of trafficking tumor antigens, although they did not possess the necessary machinery to prime CD8+ T cells and stimulate CNS antitumor immunity. This could be for a variety of reasons [74], and further work will be necessary to clarify the mechanism(s).
We consistently observed that both the superficial and deep cLNs contained zsGreen+ cDC following intracranial injection with CT2A-zsGreen, consistent with prior studies demonstrating that deep [19], followed by superficial cLNs [18] can accumulate CNS-derived tracer. Herein, we observed more substantial antigen trafficking to deep cLNs at day 7, but no difference by day 14. Our data support a model whereby deep cLNs capture parenchymal antigens prior to superficial LNs. However, whereas the drainage route from the venous sinuses to the deep cLNs has been well characterized [18, 19], the drainage route to the superficial cLNs is not, meriting further study.
Parenchymal brain tumor antigens may drain to the cLNs via several potential routes, such as via active cell-mediated transport, LN APC capture of passively draining antigens, or via transfer of antigen-bearing exosomes. We used mice lacking CCR7, which cDC use to home from the periphery to draining LNs [57], to explore whether active cell-mediated migration was required for tumor antigen to be trafficked to the cLNs. Although tumors from both genotypes harbored tumor antigen-containing DCs, wild-type mice had a much larger fraction zsGreen-containing DCs in cLNs compared to the CCR7−/− mice. This suggests that antigen trafficking is predominantly an active process that requires intact CCR7-mediated cell migration, which has also been observed in cutaneous melanoma [29]. However, it is important to note that a small zsGreen signal was still observed in DCs derived from the LNs of CCR7−/− mice, which suggests that either passive drainage mechanisms or cell-mediated transit via other chemokine receptors may also contribute to the trafficking of tumor-derived material from the brain to the cLNs.
Complementing our CNS tumor antigen trafficking observations, we investigated where T cells were primed using the GL261-OVA/OT-I model antigen system. We observed OT-I CD8+ T-cell clonal expansion in cLNs, in contrast to the tumor and the dura, which harbored only effector T cells at the later timepoint, suggesting they were primed in the cLNs but had homed to the tumor and dura. We further observed that both CCR7-deficient and cDC1-deficient mice had delayed, but not deficient clonal expansion, which suggests two things: (1) that tumor antigen still passively drains to cLNs in sufficient amounts to eventually drive T-cell clonal expansion, despite not being carried by APCs, and (2) that cell types other the cDC1 can drive clonal expansion of adoptively transferred CD8+ T cells if given enough time. However, these data still underscore the importance of cell migration and cDC1 in priming an effective and early T-cell response against CNS tumors. Our observations extend those of previous work demonstrating the importance of cLN for disease progression in EAE models [63–65], and highlight the importance of the cLNs in priming a CNS immune responses. Our data support a model in which APCs carry tumor antigens from intracranial tumors to cLNs, where cDC1 specifically prime tumor-specific CD8+ T cells.
We extended our preclinical observations to patients. There has been significant work on the use of autologous cultured monocyte-derived DCs in GBM immunotherapy [75, 76]. Although the approaches are promising, instead of focusing on DCs as a therapeutic approach, we instead studied the behavior and function of endogenously arising cDC subsets in GBM patients. We identified CD141+ cDC1, CD1c+ cDC2, CD14+ monocytes, and CD16+ monocytes, as well as CD4+ and CD8+ T cells, in GBM, extending our observations that cDC infiltrate mouse GBM.
To our knowledge, the phenomenon of direct tumor antigen uptake by cDC has not been observed in human cancers. 5-ALA is selectively up-taken, metabolized into the PPIX fluorophore, and retained GBM tumor cells [42, 43], which allows the operator to more precisely discriminate glioblastoma from normal brain [38–42]. We leveraged this to probe for PPIX in various GBM-infiltrating DC and APC subsets. We observed PPIX specifically in tumor-associated APCs. Tumor-associated T cells were PPIX-deficient. Additionally, PPIX+ APC specifically localized to tumors, matched PBMC subsets obtained contemporaneously were PPIX-negative. These important controls established that the PPIX signal was dependent on immune cell identity and location.
Our traceable GBM tumor antigen data from mice (using zsGreen) and humans (using PPIX) pose several potential mechanisms of antigen acquisition by tumor-infiltrating APCs: (1) phagocytosis of GBM cells, (2) antigen-transfer to APCs via exosomes, (3) phagocytosis of debris released from dying tumor cells, or (4) ingestion from other currently undefined mechanisms. We observed large fractions (>75%) of PPIX+ DCs in human GBM, similar to mice (50%-75% of zsGreen+ DC). The fluorescent signal was not uniform between DC subsets in both settings. This implies: (1) that fluorophores with different molecular structures (one a heme-derivative, the other a protein) might be transferred into APCs by similar mechanisms, and (2) that differences in fluorescent percentage between different APC subsets might reflect functional differences. These findings suggest parallel mechanisms of tumor antigen-uptake exist in mice and humans. Further work should employ more granular analysis to characterize additional subsets that might acquire tumor antigen and should aim to identify molecular signatures of APC cell states pre- and post-fluorophore ingestion.
Growing work has identified the meninges as an immunologically dynamic structure that contains Flt3L-sensitive cDC and pDC [14, 15, 23, 60] and harbors a network of VEGF-C responsive lymphatic vessels [18–21, 61, 62] that drain cerebrospinal fluid and antigen to the deep cLNs and facilitate CNS anti-tumor immunity. We showed that brain tumors stimulate expansion of Flt3L-sensitive dendritic cell populations. We identified cDC1 in dura lymphatic vessels by 2-photon microscopy and observed that dura harbors tumor antigen-containing DCs. We translated our preclinical findings to human specimens and observed that the dura harbors both the CD141+ cDC1 and the CD1c+ cDC2 human equivalents. Additional studies are needed to define the functional contributions of dura-associated DCs, the interplay with the dura lymphatic network, and the anatomic pathways by which APC traffic parenchymal tumor antigens to the dura.
Taken together, these data show that CNS antitumor immunity requires cDC1, which infiltrate tumors, acquire antigen, and traffic to cLNs, with the dura playing a supportive role. Additional work will be needed to understand the anatomic basis of cDC1-associated antigen trafficking to the dura and LNs and to determine whether appropriately polarized cDC1 can be leveraged therapeutically in patients with GBM.
Supplementary Material
Synopsis:
Data show that cDC1 facilitate CNS anti-tumor immunity by capturing tumor antigens and trafficking them to the dura and CNS-draining cervical lymph nodes to prime CD8+ T-cell responses. DC antigen uptake in human glioblastoma is also demonstrated.
Acknowledgments:
We acknowledge the Immune Monitoring Lab (particularly Diane Bender) and Washington University Department of Pathology Flow Cytometry Core for flow cytometer and instrumentation support. We acknowledge David DeNardo, Gwendalyn Randolph, Josh Rubin, Morey Blinder, and Robert Schreiber for their incisive input. We thank Matthew Holt for his superb illustrations.
Funding:
National Institutes of Health NINDS grant R01NS112712 (GPD), Cancer Research Institute Lloyd J Old STAR Award (GPD), National Institutes of Health NCI grant F30CA236454 (JABK).
Footnotes
Conflicts of Interest: AHK has received research grants from Monteris Medical for a mouse laser therapy study as well as from Stryker and Collagen Matrix for clinical outcomes studies about a dural substitute, which have no direct relation to this study. All other authors declare they have no potential conflicts of interest.
References
- 1.Wen PY, et al. , Glioblastoma in Adults: A Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) Consensus Review on Current Management and Future Directions. Neuro Oncol, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Engelhardt B, Vajkoczy P, and Weller RO, The movers and shapers in immune privilege of the CNS. Nat Immunol, 2017. 18(2): p. 123–131. [DOI] [PubMed] [Google Scholar]
- 3.Maxwell R, Jackson CM, and Lim M, Clinical Trials Investigating Immune Checkpoint Blockade in Glioblastoma. Curr Treat Options Oncol, 2017. 18(8): p. 51. [DOI] [PubMed] [Google Scholar]
- 4.McGranahan T, et al. , Current State of Immunotherapy for Treatment of Glioblastoma. Curr Treat Options Oncol, 2019. 20(3): p. 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dunn GP, Fecci PE, and Curry WT, Cancer immunoediting in malignant glioma. Neurosurgery, 2012. 71(2): p. 201–22; discussion 222–3. [DOI] [PubMed] [Google Scholar]
- 6.Chongsathidkiet P, et al. , Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat Med, 2018. 24(9): p. 1459–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wainwright DA, et al. , IDO expression in brain tumors increases the recruitment of regulatory T cells and negatively impacts survival. Clin Cancer Res, 2012. 18(22): p. 6110–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ladomersky E, et al. , IDO1 Inhibition Synergizes with Radiation and PD-1 Blockade to Durably Increase Survival Against Advanced Glioblastoma. Clin Cancer Res, 2018. 24(11): p. 2559–2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brennan CW, et al. , The somatic genomic landscape of glioblastoma. Cell, 2013. 155(2): p. 462–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nduom EK, et al. , PD-L1 expression and prognostic impact in glioblastoma. Neuro Oncol, 2016. 18(2): p. 195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heimberger AB and Sampson JH, Immunotherapy coming of age: what will it take to make it standard of care for glioblastoma? Neuro Oncol, 2011. 13(1): p. 3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown NF, et al. , Harnessing the immune system in glioblastoma. Br J Cancer, 2018. 119(10): p. 1171–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schaettler MO, et al. , Characterization of the Genomic and Immunologic Diversity of Malignant Brain Tumors through Multisector Analysis. Cancer Discov, 2022. 12(1): p. 154–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mrdjen D, et al. , High-Dimensional Single-Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease. Immunity, 2018. 48(3): p. 599. [DOI] [PubMed] [Google Scholar]
- 15.Anandasabapathy N, et al. , Flt3L controls the development of radiosensitive dendritic cells in the meninges and choroid plexus of the steady-state mouse brain. J Exp Med, 2011. 208(8): p. 1695–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matyszak MK and Perry VH, The potential role of dendritic cells in immune-mediated inflammatory diseases in the central nervous system. Neuroscience, 1996. 74(2): p. 599–608. [DOI] [PubMed] [Google Scholar]
- 17.Waisman A, Liblau RS, and Becher B, Innate and adaptive immune responses in the CNS. Lancet Neurol, 2015. 14(9): p. 945–55. [DOI] [PubMed] [Google Scholar]
- 18.Louveau A, et al. , Structural and functional features of central nervous system lymphatic vessels. Nature, 2015. 523(7560): p. 337–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aspelund A, et al. , A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med, 2015. 212(7): p. 991–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ahn JH, et al. , Meningeal lymphatic vessels at the skull base drain cerebrospinal fluid. Nature, 2019. 572(7767): p. 62–66. [DOI] [PubMed] [Google Scholar]
- 21.Song E, et al. , VEGF-C-driven lymphatic drainage enables immunosurveillance of brain tumours. Nature, 2020. 577(7792): p. 689–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Giles DA, et al. , CNS-resident classical DCs play a critical role in CNS autoimmune disease. J Clin Invest, 2018. 128(12): p. 5322–5334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mundt S, et al. , Conventional DCs sample and present myelin antigens in the healthy CNS and allow parenchymal T cell entry to initiate neuroinflammation. Sci Immunol, 2019. 4(31). [DOI] [PubMed] [Google Scholar]
- 24.Karman J, et al. , Initiation of immune responses in brain is promoted by local dendritic cells. J Immunol, 2004. 173(4): p. 2353–61. [DOI] [PubMed] [Google Scholar]
- 25.Karman J, et al. , Dendritic cells amplify T cell-mediated immune responses in the central nervous system. J Immunol, 2006. 177(11): p. 7750–60. [DOI] [PubMed] [Google Scholar]
- 26.Zozulya AL, et al. , Intracerebral dendritic cells critically modulate encephalitogenic versus regulatory immune responses in the CNS. J Neurosci, 2009. 29(1): p. 140–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harris MG, et al. , Immune privilege of the CNS is not the consequence of limited antigen sampling. Sci Rep, 2014. 4: p. 4422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Broz ML, et al. , Dissecting the Tumor Myeloid Compartment Reveals Rare Activating Antigen-Presenting Cells Critical for T Cell Immunity. Cancer Cell, 2014. 26(6): p. 938. [DOI] [PubMed] [Google Scholar]
- 29.Roberts EW, et al. , Critical Role for CD103(+)/CD141(+) Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell, 2016. 30(2): p. 324–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Binnewies M, et al. , Unleashing Type-2 Dendritic Cells to Drive Protective Antitumor CD4(+) T Cell Immunity. Cell, 2019. 177(3): p. 556–571 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Salmon H, et al. , Expansion and Activation of CD103(+) Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity, 2016. 44(4): p. 924–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maier B, et al. , A conserved dendritic-cell regulatory program limits antitumour immunity. Nature, 2020. 580(7802): p. 257–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hildner K, et al. , Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science, 2008. 322(5904): p. 1097–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spranger S, Bao R, and Gajewski TF, Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature, 2015. 523(7559): p. 231–5. [DOI] [PubMed] [Google Scholar]
- 35.Spranger S, et al. , Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell, 2017. 31(5): p. 711–723 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fuertes MB, et al. , Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med, 2011. 208(10): p. 2005–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Diamond MS, et al. , Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med, 2011. 208(10): p. 1989–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pastor J, et al. , Role of intraoperative neurophysiological monitoring during fluorescence-guided resection surgery. Acta Neurochir (Wien), 2013. 155(12): p. 2201–13. [DOI] [PubMed] [Google Scholar]
- 39.Zhao S, et al. , Intraoperative fluorescence-guided resection of high-grade malignant gliomas using 5-aminolevulinic acid-induced porphyrins: a systematic review and meta-analysis of prospective studies. PLoS One, 2013. 8(5): p. e63682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li Y, et al. , Intraoperative fluorescence-guided resection of high-grade gliomas: a comparison of the present techniques and evolution of future strategies. World Neurosurg, 2014. 82(1–2): p. 175–85. [DOI] [PubMed] [Google Scholar]
- 41.Huang Z, et al. , Fluorescence-guided resection of brain tumor: review of the significance of intraoperative quantification of protoporphyrin IX fluorescence. Neurophotonics, 2017. 4(1): p. 011011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Collaud S, et al. , On the selectivity of 5-aminolevulinic acid-induced protoporphyrin IX formation. Curr Med Chem Anticancer Agents, 2004. 4(3): p. 301–16. [DOI] [PubMed] [Google Scholar]
- 43.Stummer W, et al. , In vitro and in vivo porphyrin accumulation by C6 glioma cells after exposure to 5-aminolevulinic acid. J Photochem Photobiol B, 1998. 45(2–3): p. 160–9. [DOI] [PubMed] [Google Scholar]
- 44.Bianchi R, et al. , A transgenic Prox1-Cre-tdTomato reporter mouse for lymphatic vessel research. PLoS One, 2015. 10(4): p. e0122976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hopken UE, et al. , The chemokine receptor CCR7 controls lymph node-dependent cytotoxic T cell priming in alloimmune responses. Eur J Immunol, 2004. 34(2): p. 461–70. [DOI] [PubMed] [Google Scholar]
- 46.Hogquist KA, et al. , T cell receptor antagonist peptides induce positive selection. Cell, 1994. 76(1): p. 17–27. [DOI] [PubMed] [Google Scholar]
- 47.Janowska-Wieczorek A, et al. , Platelet-derived microparticles bind to hematopoietic stem/progenitor cells and enhance their engraftment. Blood, 2001. 98(10): p. 3143–9. [DOI] [PubMed] [Google Scholar]
- 48.Johanns TM, et al. , Endogenous Neoantigen-Specific CD8 T Cells Identified in Two Glioblastoma Models Using a Cancer Immunogenomics Approach. Cancer Immunol Res, 2016. 4(12): p. 1007–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu CJ, et al. , Treatment of an Aggressive Orthotopic Murine Glioblastoma Model with Combination Checkpoint Blockade and a Multivalent Neoantigen Vaccine. Neuro Oncol, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barretina J, et al. , The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature, 2012. 483(7391): p. 603–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fu Y, et al. , GATA2 Regulates Constitutive PD-L1 and PD-L2 Expression in Brain Tumors. Sci Rep, 2020. 10(1): p. 9027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brahler S, et al. , Opposing Roles of Dendritic Cell Subsets in Experimental GN. J Am Soc Nephrol, 2018. 29(1): p. 138–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Durai V, et al. , Cryptic activation of an Irf8 enhancer governs cDC1 fate specification. Nat Immunol, 2019. 20(9): p. 1161–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reardon DA, et al. , Glioblastoma Eradication Following Immune Checkpoint Blockade in an Orthotopic, Immunocompetent Model. Cancer Immunol Res, 2016. 4(2): p. 124–35. [DOI] [PubMed] [Google Scholar]
- 55.Zhai L, et al. , Non-tumor cell IDO1 predominantly contributes to enzyme activity and response to CTLA-4/PD-L1 inhibition in mouse glioblastoma. Brain Behav Immun, 2017. 62: p. 24–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Karman J, et al. , Dendritic cells in the initiation of immune responses against central nervous system-derived antigens. Immunol Lett, 2004. 92(1–2): p. 107–15. [DOI] [PubMed] [Google Scholar]
- 57.Forster R, et al. , CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell, 1999. 99(1): p. 23–33. [DOI] [PubMed] [Google Scholar]
- 58.Stamper HB Jr. and Woodruff JJ, Lymphocyte homing into lymph nodes: in vitro demonstration of the selective affinity of recirculating lymphocytes for high-endothelial venules. J Exp Med, 1976. 144(3): p. 828–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Springer TA, Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell, 1994. 76(2): p. 301–14. [DOI] [PubMed] [Google Scholar]
- 60.Wang AZ, et al. , Single-cell profiling of human dura and meningioma reveals cellular meningeal landscape and insights into meningioma immune response. Genome Med, 2022. 14(1): p. 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Da Mesquita S, et al. , Functional aspects of meningeal lymphatics in ageing and Alzheimer’s disease. Nature, 2018. 560(7717): p. 185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Louveau A, et al. , CNS lymphatic drainage and neuroinflammation are regulated by meningeal lymphatic vasculature. Nat Neurosci, 2018. 21(10): p. 1380–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Furtado GC, et al. , Swift entry of myelin-specific T lymphocytes into the central nervous system in spontaneous autoimmune encephalomyelitis. J Immunol, 2008. 181(7): p. 4648–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Phillips MJ, Needham M, and Weller RO, Role of cervical lymph nodes in autoimmune encephalomyelitis in the Lewis rat. J Pathol, 1997. 182(4): p. 457–64. [DOI] [PubMed] [Google Scholar]
- 65.van Zwam M, et al. , Surgical excision of CNS-draining lymph nodes reduces relapse severity in chronic-relapsing experimental autoimmune encephalomyelitis. J Pathol, 2009. 217(4): p. 543–51. [DOI] [PubMed] [Google Scholar]
- 66.Poulin LF, et al. , Characterization of human DNGR-1+ BDCA3+ leukocytes as putative equivalents of mouse CD8alpha+ dendritic cells. J Exp Med, 2010. 207(6): p. 1261–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Murphy TL, et al. , Transcriptional Control of Dendritic Cell Development. Annu Rev Immunol, 2016. 34: p. 93–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hambleton S, et al. , IRF8 mutations and human dendritic-cell immunodeficiency. N Engl J Med, 2011. 365(2): p. 127–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yan J, et al. , FGL2 promotes tumor progression in the CNS by suppressing CD103(+) dendritic cell differentiation. Nat Commun, 2019. 10(1): p. 448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Johanns TM, et al. , Immunogenomics of Hypermutated Glioblastoma: A Patient with Germline POLE Deficiency Treated with Checkpoint Blockade Immunotherapy. Cancer Discov, 2016. 6(11): p. 1230–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cloughesy TF, et al. , Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med, 2019. 25(3): p. 477–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Clarkson BD, et al. , Innate-adaptive crosstalk: how dendritic cells shape immune responses in the CNS. Adv Exp Med Biol, 2012. 946: p. 309–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Garzon-Muvdi T, et al. , Dendritic cell activation enhances anti-PD-1 mediated immunotherapy against glioblastoma. Oncotarget, 2018. 9(29): p. 20681–20697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Allan RS, et al. , Migratory dendritic cells transfer antigen to a lymph node-resident dendritic cell population for efficient CTL priming. Immunity, 2006. 25(1): p. 153–62. [DOI] [PubMed] [Google Scholar]
- 75.Liau LM, et al. , First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J Transl Med, 2018. 16(1): p. 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mitchell DA, et al. , Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature, 2015. 519(7543): p. 366–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.