

Abstract
Since a 25% mortality rate occurred in critical Coronavirus disease 2019 (COVID‐19) patients, investigating the potential drivers remains to be important. Here, the authors applied Weighted Gene Co‐expression Network Analysis to identify the potential drivers in the blood samples of multiple COVID‐19 expression profiles. The authors found that the darkslateblue module was significantly correlated with critical COVID‐19, and Gene Ontology analysis indicated terms associated with the inflammation pathway and apoptotic process. The authors intersected differentially expressed genes, Maximal Clique Centrality calculated hub genes, and COVID‐19 related genes in the Genecards dataset, and two genes, toll‐like receptor 5 (TLR5) and acyl‐CoA synthetase long chain family member 1 (ACSL1), were screened out. The Gene Set Enrichment Analysis further supports their core role in the inflammatory pathway. Furthermore, the cell‐type identification by estimating relative subsets of RNA transcript demonstrated that TLR5 and ACSL1 were associated with neutrophil enrichment in critical COVID‐19 patients. Collectively, the aurthors identified two hub genes that were strongly correlated with critical COVID‐19. These may help clarify the pathogenesis and assist the immunotherapy development.
Keywords: bioinformatics, COVID‐19, data analysis, hub genes
TLR5 and ACLS1 might play important roles in critical COVID‐19 patients, and they were associated with neutrophil enrichment. These findings may help clarify the pathogenesis and assist the immunotherapy development.

1. INTRODUCTION
Coronavirus disease 2019 (COVID‐19) is a severe infectious disease that has strained healthcare systems all over the world. Over 439 million confirmed COVID‐19 cases, leading to over 5.98 million deaths, which was released by the World Health Organization on 01 March 2022 (https://www.worldometers.info/coronavirus/). Upon infection, patients presented with heterogeneous clinical manifestations with differential disease severity: severe, moderate, mild, and asymptomatic [1, 2]. The severe patients account for a small proportion of COVID‐19 while the mortality rate of severe patients is 25.7% on average and even 37.7% in China [3]. Thus, access to explored pathogenesis and improved therapies remains an unmet need for severe COVID‐19 patients.
Research on critical COVID‐19 were mainly focused on the inflammation and immune dysregulation. Elevated levels of C‐reactive protein and inflammatory cytokines, such as interleukin (IL)‐2, 5, 6, 7, 10, 13 and TNF‐α level etc., were found in severe COVID‐19 patients [4, 5, 6]. Anti‐IL‐6 receptor inhibitor tocilizumab could attenuate COVID‐19‐related acute respiratory distress syndrome, and anti‐IL‐13 treatment could significantly alleviate the mortality and disease severity of SARS‐CoV‐2‐infected mice models [7, 8]. Immune dysregulation was also significantly correlated with COVID‐19 severity. Decreased CD4+ T cells and CD8+ T cells, natural killer (NK) cells [4, 9], hyperactivated neutrophils [2, 10] etc. were also found to be a hallmark of disease severity. Despite the increased understanding of critical COVID‐19, the lack of effective hub genes for therapy limits the prevention and treatment.
Weighted gene co‐expression network analysis (WGCNA) is a powerful method to explore the correlation of gene expression patterns from large heterogeneous mRNA expression data sets [11, 12, 13]. According to the similarity of expression pattern, WGCNA can transform gene expression data into potentially biologically associated modules and reveal the potential relationships between the modules and clinical traits [14]. This method is of great importance in identifying the potential hub genes or therapeutic targets for diseases.
Here, we apply WGCNA to construct a gene co‐expression network and identify the significant modules using the data from GSE172114 [2], intending to dig out more potential hub genes and therapeutic targets for the critical COVID‐19 patients.
2. MATERIALS AND METHODS
2.1. Gene expression dataset processing
The RNA‐seq data of the whole blood RNA samples from 46 Critical and 23 non‐critical COVID‐19 patients were collected from the Gene Expression Omnibus (GEO) dataset as the discovery cohort based on the original grouping (GSE172114, “critical” referred to the group of patients in the intensive care unit (ICU) under mechanical ventilation and “non‐critical” referred to the group of patients in a non‐critical care ward) [15]. The RNA‐seq data of the whole blood RNA samples from 8 severe forms and 90 moderate forms of COVID‐19 patients were collected to verify the expression of the hub genes based on the original grouping (GSE178967, “moderate” referred to the patients not requiring hospitalisation and “severe” referred to the patients hospitalised and requiring oxygen supplementation/admitted to the ICU and placed on mechanical ventilation/not considered a candidate for ICU treatment and with fatal outcome) [16]. Finally, we used the keyword “COVID‐19” to search the Genecards database (https://www.genecards.org/) to identify the associated genes.
2.2. Weighted gene co‐expression network analysis
The R package WGCNA [14] was used to evaluate the GSE172114 expression matrix. We exacted the clinical severity from the original set as the input data for WGCNA analysis. A soft‐threshold power of 18 was used for the analysis to achieve approximate scale‐free topology (R 2 > 0.8) [17]. Next, the topological overlap matrix (TOM) and dissimilarity TOM were created by TOM similarity and dissimilarity modules. The minimum module size was set to 30, and the dynamic shear trees were used to identify the modules. Then, we calculated the association between the module membership values and gene significance values. |GS| > 0.3 and |MM| > 0.7 was used to filter hub genes (the script was attached in the Supplementary Materials). Gene pairs with weight >0.1 were put into the Cytoscape software for the co‐expression network construction. CytoHubba [18] was used to select the hub genes in the key module. Top 10 genes with higher maximal clique centrality (MCC) values were screened [19].
2.3. Differentially expressed gene analysis
GSE172114 expression matrix was used to analyse the differentially expressed genes (DEGs) via the R limma package (http://www.bioconductor.org/packages/release/bioc/html/limma.html) (Smyth, 2004). |Foldchange| > 2 and the False Discovery Rate < 0.05 were used as the selection criteria [19]. Moreover, the expression of these hub genes between the severe form and moderate form of COVID‐19 patients with significantly differential expression was further validated in the GSE178967 expression matrix.
2.4. Gene ontology and gene set enrichment analysis
Gene ontology (GO) analysis was performed to identify the enriched pathway of the selected darkslateblue module. We conducted a gene set enrichment analysis (GSEA) using hallmarks (h.all.v7.4.symbols.gmt). The results with |NES| (normalised enrichment score) > 1, p‐value <0.05 were considered to be significant [20].
2.5. Evaluation of immune cell abundance
Cell‐type identification by estimating relative subsets of RNA transcript (https://cibersort.stanford.edu/) [21] is an algorithmic tool based on gene expression profiles to calculate abundances of member cell types from mixed cell populations. It was employed to reveal the proportion of 22 types of immune cells in each sample of GSE172114.
3. RESULTS
3.1. Weighted gene co‐expression network analysis to dig potential hub genes of critical Coronavirus disease 2019 patients
Figure 1 shows the overall study design. The samples in GSE172114 were divided into two groups (46 critical and 23 non‐critical COVID‐19 patients). To identify the key modules and hub genes related to the severe form of COVID‐19, we conducted WGCNA using the gene expression matrix of GSE172114. The hierarchical clustering revealed the potential differences between the critical and non‐critical COVID‐19 patients (Figure 2a). The soft threshold was set to 18 with the scale‐free topology fitting index reaching 0.84 (Figure 2b,c). The dynamic shear tree's merged shear height was 0.25, and a total of 20 modules were identified in our results with the minimum number of genes in each network module set to 30 (Figure 2d). The most strongly correlated positive module was darkslateblue for the critical COVID‐19 patients, which was chosen as the critical module (Figure 2e,f). In this module, we applied |GS| > 0.3 and |MM| > 0.7 as the criteria to screen for the essential genes, and we found 2156 genes in the critical trait. The GO biological process analysis demonstrated that these essential genes were enriched in the inflammation pathway and apoptotic process (Figure 2g).
FIGURE 1.

Overall study design. Weighted gene co‐expression network analysis (WGCNA), gene ontology (GO), maximal clique centrality (MCC), differentially expressed genes (DEGs), Gene Set Enrichment Analysis (GSEA), cell‐type identification by estimating relative subsets of RNA transcript (CIBERSORT).
FIGURE 2.

Weighted gene co‐expression network analysis (WGCNA) analysis in GSE172114 and key module identification. (a) The sample‐trait clustering heatmap. (b) The scale‐free topology model fit index analysis for soft threshold powers and the mean connectivity analysis for soft threshold powers. (c) Scale‐free topology fitting graph. (d) Dynamic shearing tree merging similar module genes. (e) Module‐trait correlation heatmap. (f) Scatter plot for correlation between module membership in darkslateblue module and gene significance for the critical trait. (g) Gene ontology (GO) enrichment analysis of the hub genes in darkslateblue module.
3.2. Identification of hub genes
Gene pairs with weight >0.1 were put into the Cytoscape, and the top 10 hub genes in this network were identified by the cytoHubba plugin via MCC values (Figure 3a). The differential gene analysis demonstrated that 916 significant DEGs with 675 upregulated genes and 241 downregulated genes in the critical group compared with the non‐critical group. The Venn diagram demonstrated that toll‐like receptor 5 (TLR5) and acyl‐CoA synthetase long chain family member 1 (ACSL1) were intersected both in the DEGs and genecards dataset associated with COVID‐19 (Figure 3b). Toll‐like receptor 5 and ACSL1 were upregulated with the adjusted p < 0.001. Moreover, TLR5 and ACSL1 were upregulated in the whole blood samples of severe forms of COVID‐19 patients compared with not‐severe forms (p = 0.021, p = 0.027, respectively) (Figure 3c,d). These results suggest that TLR5 and ACSL1 may serve as hub genes in the critical COVID‐19 patients.
FIGURE 3.

Identification of the hub gene. (a) Top 10 hub genes identification by maximal clique centrality (MCC). (b) Venn diagram screening for key genes. (c) Expression of acyl‐CoA synthetase long chain family member 1 (ACSL1) in the additional Coronavirus disease 2019 (COVID‐19) dataset GSE178967. (d) Expression of toll‐like receptor 5 (TLR5) in the additional COVID‐19 dataset GSE178967.
3.3. Gene set enrichment analysis analysis between high and low toll‐like receptor 5 and acyl‐CoA synthetase long chain family member 1 groups
We used the binary classification to divide the samples of GSE172114 into two groups based on the expression level of TLR5 and ACSL1, respectively (34 high expression samples and 33 low expression samples). Hallmark datasets is one of the most widely used and comprehensive databases for GSEA, which could better represent a wider range of biological processes/diseases [22]. In our study, we applied hallmark datasets for investigation. The GSEA results showed significant hallmark pathways associated with high TLR5 and ACSL1, including IL6/JAK/STAT3 pathway, TNFA pathway via NFKB, and inflammatory response (Figure 4a–f).
FIGURE 4.

Gene set enrichment analysis (GSEA) enrichment analysis. (a) High expression of acyl‐CoA synthetase long chain family member 1 (ACSL1) could upregulate the IL6/JAK/STAT3 signalling pathway. (b) High expression of ACSL1 could upregulate the TNFA signalling pathway via NFKB. (c) High expression of ACSL1 could upregulate the inflammatory response signalling pathway. (d) High expression of toll‐like receptor 5 (TLR5) could upregulate the TNFA signalling pathway via NFKB. (e) High expression of TLR5 could upregulate the IL6/JAK/STAT3 signalling pathway. (f) High expression of TLR5 could upregulate the inflammatory response signalling pathway.
3.4. Immune cell subtypes between high and low toll‐like receptor 5 and acyl‐CoA synthetase long chain family member 1 groups
To explore the immunological changes between the high and low TLR5 and ACSL1 groups, we utilised cell‐type identification by estimating relative subsets of RNA transcript to evaluate the immune cell infiltration on GSE172114 (Figure 5a–d). Interestingly, we found a significant decline of naïve CD4 T cells, resting‐memory CD4 T cells, monocytes but a significant augment of neutrophils in the high TLR5 and ACSL1 group. Moreover, a decline of the resting NK cell was also found in the high TLR5 group.
FIGURE 5.
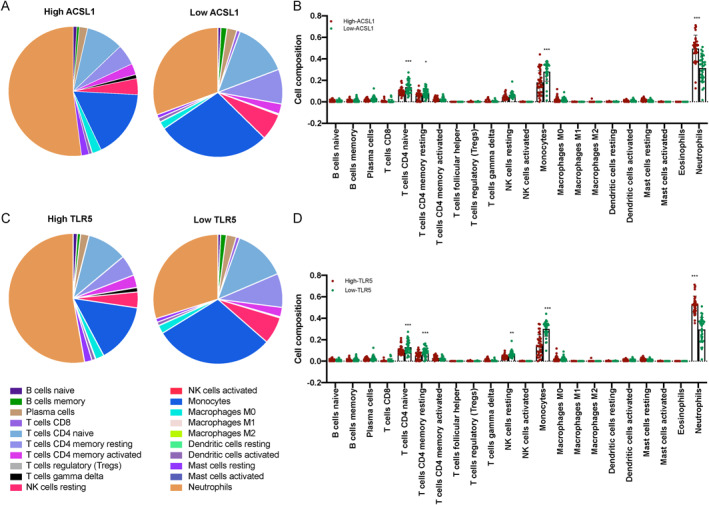
22 immune cells analysis via cell‐type identification by estimating relative subsets of RNA transcript (CIBERSORT). (a) The mean proportion of 22 immune cells in the high acyl‐CoA synthetase long chain family member 1 (ACSL1) and low ACSL1 groups. (b) The histogram shows the cell compositions of 22 immune cells in the high ACSL1 and low ACSL1 groups. (c) The mean proportion of 22 immune cells in the high toll‐like receptor 5 (TLR5) and low TLR5 groups. (d) The histogram shows the cell compositions of 22 immune cells in the high TLR5 and low TLR5 groups.
4. DISCUSSION
Coronavirus disease 2019 is a serious pandemic caused by severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) [23]. Among these, the critical COVID‐19 is the most severe type and its mortality rate was extremely high (more than 25%). Therefore, it is imperative to identify the potential hub genes to improve the therapeutic regimen and prognosis of critical COVID‐19 patients.
Several studies have applied WGCNA to identify the hub genes COVID‐19 patients, including the hub genes in the nasopharyngeal swabs between normal subjects and COVID‐19 patients, critical genes of age‐related module in the peripheral blood mononuclear cell of COVID‐19 patients, and key genes in the platelets of between health controls and COVID‐19 patients [24, 25, 26]. These studies explored the key genes of COVID‐19 from a variety of perspectives. However, the potential hub genes in critical COVID‐19 patients remain largely unclear. In our study, we applied WGCNA to construct the co‐expression modules associated with critical COVID‐19 using the GSE172114 dataset to identify the driver genes. The hierarchical clustering of the WGCNA indicated that there existed some patients in the two groups with similar gene expression patterns. We think that it is reasonable for patients suffering from the same disease. The following analysis demonstrated that the darkslateblue module was significantly positively correlated with the critical COVID‐19 trait, and the GO biological process analysis of the essential genes in this module indicated the enrichment of inflammation pathway and apoptotic process, which are the important processes that participated in the pathogenesis of COVID‐19 [4, 5, 6, 27, 28]. Finally, two candidate hub genes, ACSL1 and TLR5, were screened out to be the intersection of MCC calculated hub genes, DEGs, and genecards dataset associated with COVID‐19.
Acyl‐CoA synthetase long chain family member 1 is an isozyme of the long‐chain fatty‐acid‐coenzyme A ligase family and is associated with lipid metabolism, fatty acid uptake, and inflammation [29, 30, 31, 32]. The abnormal expression of ACSL1 was identified in various disease, including cancer [33], virus infection [34, 35], diabetes [32], sepsis [29], osteoporosis [36], non‐alcoholic fatty liver disease [37] etc. In our results, we found that ACSL1 significantly upregulated in the whole blood cells of critical COVID‐19 patients. Moreover, it was identified as a hub gene of critical COVID‐19 traits, and the high expression of ACSL1 was correlated with more severe inflammation, especially neutrophil infiltration, in severe COVID‐19 patients. Consistent with our findings, the ACSL1 inhibitor triacsin C was reported to be effective against different viruses and proposed to be a promising drug to tackle SARS‐CoV‐2 at the cell level [34, 38, 39]. We hypothesised that the inhibitor of ACSL1 might be an effective therapy for the critical COVID‐19 patients.
Toll‐like receptor 5 is one of the pattern recognition receptors of great importance in innate immunity by eliciting the first line of defence against invading pathogens [40]. This pathway has been identified as an immunotherapeutic target for a lot of antibacterial or antiviral drug development [41]. An early TLR5 activation could enhance the immunogenicity for immunotherapeutic development, which could be found after the vaccination of SARS‐COV‐2 [42, 43, 44]. Therefore, it was proposed that immunomodulation via the activation of TLR5 might be an innovative approach to fight COVID‐19 [45]. However, in our study, we identified TLR5 as a hub gene for critical COVID‐19, and high TLR5 expression corresponded to higher cell composition of neutrophils, which indicated more severe inflammation in the critical COVID‐19 patients. According to the previous research, TLR5 was elevated in the liver of hyperammonaemia rat model and TLR5 silencing could ameliorate HA‐induced liver injury via the inhibition of apoptosis, oxidative stress, and inflammation responses [46]. The ligand of TLR5, flagellin, could trigger the innate immune responses of cardiac and acutely depress the myocardial contractility and the deficiency of TLR5 could diminish doxorubicin‐induced acute cardiotoxicity in mice [47, 48]. Toll‐like receptor 5 was suggested to be a potential key factor in inflammation‐induced bone erosions in diseases, such as rheumatoid arthritis, reactive arthritis, and periodontitis [49]. These studies were consistent with our results. Thus, we hypothesised that targeting TLR5 may decrease the inflammation and alleviate the symptoms of the critical COVID‐19 patients even though the early activation of TLR5 might minimise SARS‐COV‐2 replication.
Our study contains several limitations. First, this study focused on the transcriptome analysis of whole blood cells. Adding data from the lung and bronchoalveolar lavage fluid could make the results more comprehensive and conclusive. Second, a more precise value of ACSL1 and TLR5 in critical COVID‐19 patients needs to be further clarified with deeper functional experiments.
5. CONCLUSION
Using WGCNA‐based analysis methods, we discovered two hub genes, ACSL1 and TLR5, related to the characteristics of critical COVID‐19 patients, which might be involved in the pathogenesis and become potential targets for subsequent treatment explorations in critical COVID‐19 patients.
AUTHOR CONTRIBUTIONS
Luoyi Wang: Data curation; formal analysis; funding acquisition; investigation; methodology; project administration; resources; software; supervision; validation; visualisation; writing – original draft; writing – review & editing. Zhaomin Mao: Data curation; formal analysis; funding acquisition; investigation; methodology; resources; software; supervision; validation; writing – original draft; writing – review & editing. Fengmin Shao: Conceptualisation; funding acquisition; project administration; supervision; writing – original draft; writing – review & editing.
CONFLICT OF INTEREST STATEMENT
The authors declare that they have no competing interests.
Supporting information
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to express our appreciation to “Henan Provincial People's Hospital” and “The First Affiliated Hospital of Zhengzhou University” for their effort.
Wang, L. , Mao, Z. , Shao, F. : Identification of toll‐like receptor 5 and acyl‐CoA synthetase long chain family member 1 as hub genes are correlated with the severe forms of COVID‐19 by Weighted gene co‐expression network analysis. IET Syst. Biol. 17(6), 327–335 (2023). 10.1049/syb2.12079
Luoyi Wang and Zhaomin Mao contribute equally to this study.
Contributor Information
Luoyi Wang, Email: wly318@sina.com.
Fengmin Shao, Email: fengminshao@126.com.
DATA AVAILABILITY STATEMENT
These data were derived from the following resources available in the GEO dataset (GSE172114 and GSE178967).
REFERENCES
- 1. Wu, Z. , McGoogan, J.M. : Characteristics of and important lessons from the coronavirus disease 2019 (Covid‐19) outbreak in China: summary of a report of 72314 cases from the Chinese center for disease control and prevention. JAMA 323(13), 1239–1242 (2020). 10.1001/jama.2020.2648 [DOI] [PubMed] [Google Scholar]
- 2. Carapito, R. , et al.: Identification of driver genes for critical forms of Covid‐19 in a deeply phenotyped young patient cohort. Sci. Transl. Med. eabj7521 (2021) [DOI] [PubMed] [Google Scholar]
- 3. Quah, P. , Phua, J. : Mortality rates of patients with Covid‐19 in the intensive care unit: a systematic review of the emerging literature. Crit. Care 24(1), 285 (2020). 10.1186/s13054-020-03006-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chen, G. , et al.: Clinical and immunological features of severe and moderate coronavirus disease 2019. J. Clin. Invest. 130(5), 2620–2629 (2020). 10.1172/jci137244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lucas, C. , et al.: Longitudinal analyses reveal immunological misfiring in severe Covid‐19. Nature 584(7821), 463–469 (2020). 10.1038/s41586-020-2588-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Huang, C. , et al.: Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395(10223), 497–506 (2020). 10.1016/s0140-6736(20)30183-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Michot, J.M. , et al.: Tocilizumab, an anti‐Il‐6 receptor antibody, to treat Covid‐19‐related respiratory failure: a case report. Ann. Oncol. 31(7), 961–964 (2020). 10.1016/j.annonc.2020.03.300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Donlan, A.N. , et al.: Il‐13 is a driver of Covid‐19 severity. JCI Insight 6(15) (2021). 10.1172/jci.insight.150107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Giamarellos‐Bourboulis, E.J. , et al.: Complex immune dysregulation in Covid‐19 patients with severe respiratory failure. Cell Host. Microbe. 27(6), 992–1000.e1003 (2020). 10.1016/j.chom.2020.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Meizlish, M.L. , et al.: A neutrophil activation signature predicts critical illness and mortality in Covid‐19. Blood Adv. 5(5), 1164–1177 (2021). 10.1182/bloodadvances.2020003568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pepe, J. , et al.: Characterization of extracellular vesicles in osteoporotic patients compared to osteopenic and healthy controls. J. Bone Miner. Res. 37(11), 2186–2200 (2022). 10.1002/jbmr.4688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Friedrich, M. , et al.: Il‐1‐Driven stromal‐neutrophil interactions define a subset of patients with inflammatory bowel disease that does not respond to therapies. Nat. Med. 27(11), 1970–1981 (2021). 10.1038/s41591-021-01520-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Panebianco, V. , et al.: Network analysis integrating microrna expression profiling with mri biomarkers and clinical data for prostate cancer early detection: a proof of concept study. Biomedicines 9(10), 1470 (2021). 10.3390/biomedicines9101470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Langfelder, P. , Horvath, S. : Wgcna: an R package for weighted correlation network analysis. BMC Bioinf. 9(1), 559 (2008). 10.1186/1471-2105-9-559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Carapito, R. , et al.: Identification of driver genes for critical forms of Covid‐19 in a deeply phenotyped young patient cohort. Sci. Transl. Med. 14(628), eabj7521 (2022). 10.1126/scitranslmed.abj7521 [DOI] [PubMed] [Google Scholar]
- 16. Hu, Z. , et al.: Early immune markers of clinical, virological, and immunological outcomes in patients with Covid‐19: a multi‐omics study. Elife 11 (2022). 10.7554/elife.77943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jia, N.Y. , et al.: Weighted gene Co‐expression network analysis reveals different immunity but shared renal pathology between Iga nephropathy and lupus nephritis. Front. Genet. 12, 634171 (2021). 10.3389/fgene.2021.634171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chin, C.H. , et al.: Cytohubba: identifying hub objects and sub‐networks from complex interactome. BMC Syst. Biol. 8(Suppl 4), S11 (2014). 10.1186/1752-0509-8-s4-s11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shen, L. , et al.: Identification and validation of Ifi44 as key biomarker in lupus nephritis. Front. Med. 8, 762848 (2021). 10.3389/fmed.2021.762848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang, C. , Berndt‐Paetz, M. , Neuhaus, J. : A comprehensive bioinformatics analysis of notch pathways in bladder cancer. Cancers 13(12), 3089 (2021). 10.3390/cancers13123089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Newman, A.M. , et al.: Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12(5), 453–457 (2015). 10.1038/nmeth.3337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liberzon, A. , et al.: The molecular signatures database (Msigdb) hallmark gene set collection. Cell Syst. 1(6), 417–425 (2015). 10.1016/j.cels.2015.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Coronaviridae Study Group of the International Committee on Taxonomy of, V. : The species severe acute respiratory syndrome‐related coronavirus: classifying 2019‐Ncov and naming it sars‐Cov‐2. Nat. Microbiol. 5(4), 536–544 (2020). 10.1038/s41564-020-0695-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hu, R.W. , et al.: Identification of hub genes and molecular subtypes in Covid‐19 based on Wgcna. Eur. Rev. Med. Pharmacol. Sci. 25(20), 6411–6424 (2021) [DOI] [PubMed] [Google Scholar]
- 25. Lin, Y. , et al.: Weighted gene Co‐expression network analysis revealed T cell differentiation associated with the age‐related phenotypes in Covid‐19 patients. BMC Med. Genom. 16(1), 59 (2023). 10.1186/s12920-023-01490-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Alarabi, A.B. , et al.: Co‐expression analysis to identify key modules and hub genes associated with Covid‐19 in platelets. BMC Med. Genom. 15(1), 83 (2022). 10.1186/s12920-022-01222-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li, S. , et al.: Sars‐Cov‐2 triggers inflammatory responses and cell death through caspase‐8 activation. Signal Transduct. Targeted Ther. 5(1), 235 (2020). 10.1038/s41392-020-00334-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Leonardi, A.J. , Proenca, R.B. : Akt‐fas to quell aberrant T cell differentiation and apoptosis in Covid‐19. Front. Immunol. 11, 600405 (2020). 10.3389/fimmu.2020.600405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Roelands, J. , et al.: Long‐chain acyl‐Coa synthetase 1 role in sepsis and immunity: perspectives from a parallel review of public transcriptome datasets and of the literature. Front. Immunol. 10, 2410 (2019). 10.3389/fimmu.2019.02410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kanter, J.E. , Bornfeldt, K.E. : Inflammation and diabetes‐accelerated atherosclerosis: myeloid cell mediators. Trends Endocrinol. Metabol. 24(3), 137–144 (2013). 10.1016/j.tem.2012.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Al‐Rashed, F. , et al.: Tnf‐alpha induces a pro‐inflammatory phenotypic shift in monocytes through Acsl1: relevance to metabolic inflammation. Cell. Physiol. Biochem. 52(3), 397–407 (2019) [DOI] [PubMed] [Google Scholar]
- 32. Kanter, J.E. , et al.: Diabetes promotes an inflammatory macrophage phenotype and atherosclerosis through acyl‐Coa synthetase 1. Proc. Natl. Acad. Sci. U S A 109(12), E715–724 (2012). 10.1073/pnas.1111600109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Quan, J. , Bode, A.M. , Luo, X. : Acsl family: the regulatory mechanisms and therapeutic implications in cancer. Eur. J. Pharmacol. 909, 174397 (2021). 10.1016/j.ejphar.2021.174397 [DOI] [PubMed] [Google Scholar]
- 34. Xia, H. , Zhang, Z. , You, F. : Inhibiting Acsl1‐related Ferroptosis restrains murine coronavirus infection. Viruses 13(12), 2383 (2021). 10.3390/v13122383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang, Q. , et al.: Acsl1 inhibits Alv‐J replication by Ifn‐ signaling and Pi3k/Akt pathway. Front. Immunol. 12, 774323 (2021). 10.3389/fimmu.2021.774323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li, L. , et al.: Integrative analysis reveals key Mrnas and lncrnas in monocytes of osteoporotic patients. Math. Biosci. Eng. 16(5), 5947–5971 (2019). 10.3934/mbe.2019298 [DOI] [PubMed] [Google Scholar]
- 37. Dongiovanni, P. , et al.: Beta‐klotho gene variation is associated with liver damage in children with Nafld. J. Hepatol. 72(3), 411–419 (2020). 10.1016/j.jhep.2019.10.011 [DOI] [PubMed] [Google Scholar]
- 38. Dechandt, C.R.P. , et al.: Triacsin C reduces lipid droplet formation and induces mitochondrial biogenesis in primary rat hepatocytes. J. Bioenerg. Biomembr. 49(5), 399–411 (2017). 10.1007/s10863-017-9725-9 [DOI] [PubMed] [Google Scholar]
- 39. Santos‐Beneit, F. , et al.: A metabolic modeling approach reveals promising therapeutic targets and antiviral drugs to combat Covid‐19. Sci. Rep. 11(1), 11982 (2021). 10.1038/s41598-021-91526-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Takeda, K. , Kaisho, T. , Akira, S. : Toll‐like receptors. Annu. Rev. Immunol. 21(1), 335–376 (2003). 10.1146/annurev.immunol.21.120601.141126 [DOI] [PubMed] [Google Scholar]
- 41. Mifsud, E.J. , Tan, A.C.L. , Jackson, D.C. : Tlr agonists as modulators of the innate immune response and their potential as agents against infectious disease. Front. Immunol. 5, 79 (2014). 10.3389/fimmu.2014.00079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kim, E. , et al.: Microneedle array delivered recombinant coronavirus vaccines: immunogenicity and rapid translational development. EBioMedicine 55, 102743 (2020). 10.1016/j.ebiom.2020.102743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bhattacharya, M. , et al.: Development of epitope‐based peptide vaccine against novel coronavirus 2019 (Sars‐Cov‐2): immunoinformatics approach. J. Med. Virol. 92(6), 618–631 (2020). 10.1002/jmv.25736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chowdhury, U.N. , et al.: Effects of bacille calmette guerin (Bcg) vaccination during Covid‐19 infection. Comput. Biol. Med. 138, 104891 (2021). 10.1016/j.compbiomed.2021.104891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chakraborty, C. , et al.: Consider Tlr5 for new therapeutic development against Covid‐19. J. Med. Virol. 92(11), 2314–2315 (2020). 10.1002/jmv.25997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yan, J. , et al.: Tlr5 silencing reduced hyperammonaemia‐induced liver injury by inhibiting oxidative stress and inflammation responses via inactivating Nf‐Kappab and Mapk signals. Chem. Biol. Interact. 299, 102–110 (2019). 10.1016/j.cbi.2018.11.026 [DOI] [PubMed] [Google Scholar]
- 47. Rolli, J. , et al.: Bacterial flagellin triggers cardiac innate immune responses and acute contractile dysfunction. PLoS One 5(9), e12687 (2010). 10.1371/journal.pone.0012687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ma, Z.G. , et al.: Toll‐like receptor 5 deficiency diminishes doxorubicin‐induced acute cardiotoxicity in mice. Theranostics 10(24), 11013–11025 (2020). 10.7150/thno.47516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kassem, A. , et al.: Tlr5, a novel mediator of innate immunity‐induced osteoclastogenesis and bone loss. FASEB J 29(11), 4449–4460 (2015). 10.1096/fj.15-272559 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
These data were derived from the following resources available in the GEO dataset (GSE172114 and GSE178967).