

Abstract
Numerous degenerative diseases are characterized by the aberrant polymerization and accumulation of specific proteins. These proteopathies include neurological disorders such as Alzheimer’s disease, Parkinson’s disease, Huntington’s disease and the prion diseases, in addition to diverse systemic disorders, particularly the amyloidoses. The prion diseases have been shown to be transmissible by an alternative conformation of the normal cellular prion protein. Other proteopathies have been thought to be non-transmissible, but there is growing evidence that some systemic and cerebral amyloidoses can be induced by exposure of susceptible hosts to cognate molecular templates. The mechanistic similarities among these diseases provide unprecedented opportunities for elucidating the induction of protein misfolding and assembly in vivo, and for developing an integrated therapeutic approach to degenerative proteopathies.
Introduction
In a remarkable variety of neurological and systemic disorders, specific proteins accumulate within cells and tissues, usually as a result of a change in protein conformation that renders the molecules prone to self-aggregation and resistant to clearance. These conformational diseases, or ‘proteopathies’, comprise systemic amyloidoses in addition to neurodegenerative conditions that are marked by the buildup of characteristic proteins in the brain, such as Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, and the prion diseases [1–4]. In this article, we consider the mechanistic commonalities among seemingly distinct protein-based diseases, and in particular emerging evidence that some proteopathies can be induced in animal models by exposure to exogenous material. We argue that an understanding of the earliest events that induce protein misconformation and aggregation in vivo will yield more focused strategies for discovering treatments for these devastating diseases.
Induction of prion diseases
The prion diseases, although rare, have attracted special attention because of their lethality and unorthodox transmissibility. They include Creutzfeldt-Jakob disease, kuru, fatal familial insomnia and Gerstmann-Straussler-Scheinker Syndrome in humans, and several diseases in nonhuman species, the best known being scrapie in sheep, bovine spongiform encephalopathy (BSE) in cattle, transmissible mink encephalopathy, and chronic wasting disease in deer and elk [5]. The prion diseases are typified pathologically by spongiform degeneration, astrocytosis, neuron loss, and the accumulation of aberrantly folded forms of the prion protein (PrP) in specific brain regions [2,6] (Figure 1).
Figure 1.
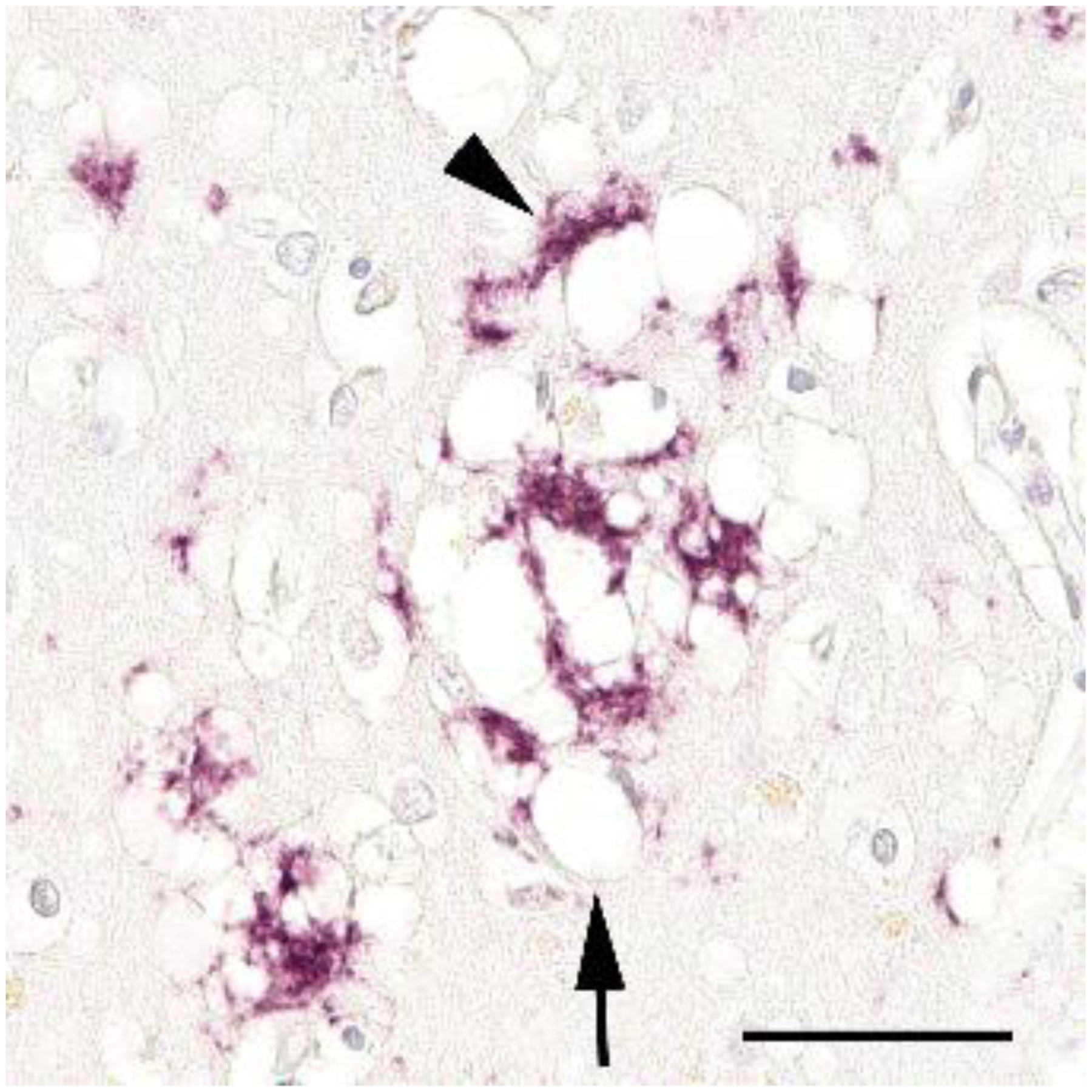
Spongiform degeneration (arrow) and the accumulation of prion protein (arrowhead) are consistent pathological features of prion disease, along with astrocytosis and neuronal degeneration. Shown is a section of neocortex from a patient who died of idiopathic Creutzfeldt-Jakob disease, labeled using the anti-PrP antibody 3F4 (Nissl counterstain). Bar = 50μm.
According to the prion hypothesis of infectivity, normal PrP (PrP cellular, or PrPc) assumes an anomalous, β-sheet-rich conformation (PrP scrapie, or PrPSc) that initiates and sustains the replication of the pathogenic molecule in vivo [7,8] by a mechanism termed permissive templating [9] (Figure 2). Unlike conventional infectious illnesses, which require the initial invasion of a microorganism, prion diseases also can arise de novo in both hereditary and idiopathic forms. In these instances, a mutation or a stochastic event, respectively, is thought to trigger the misfolding and polymerization of endogenously produced PrPc [2,7,9], although the participation of an exogenous factor in the induction of idiopathic prion disease has not been ruled out. Indeed, the unequivocal identification of the infectious agent in the prion diseases has been a fascinating and contentious area of research for many decades [10]. The experimental transmission of mammalian prionosis typically is accomplished by exposing the recipient to material from prion-laden tissue. The efficiency of disease induction is governed by route of administration, dose, and various host-specific and donor-specific factors [6–8,10–15] (Figure 3), all of which must be considered when assessing the inducibility of proteopathies [16].
Figure 2.
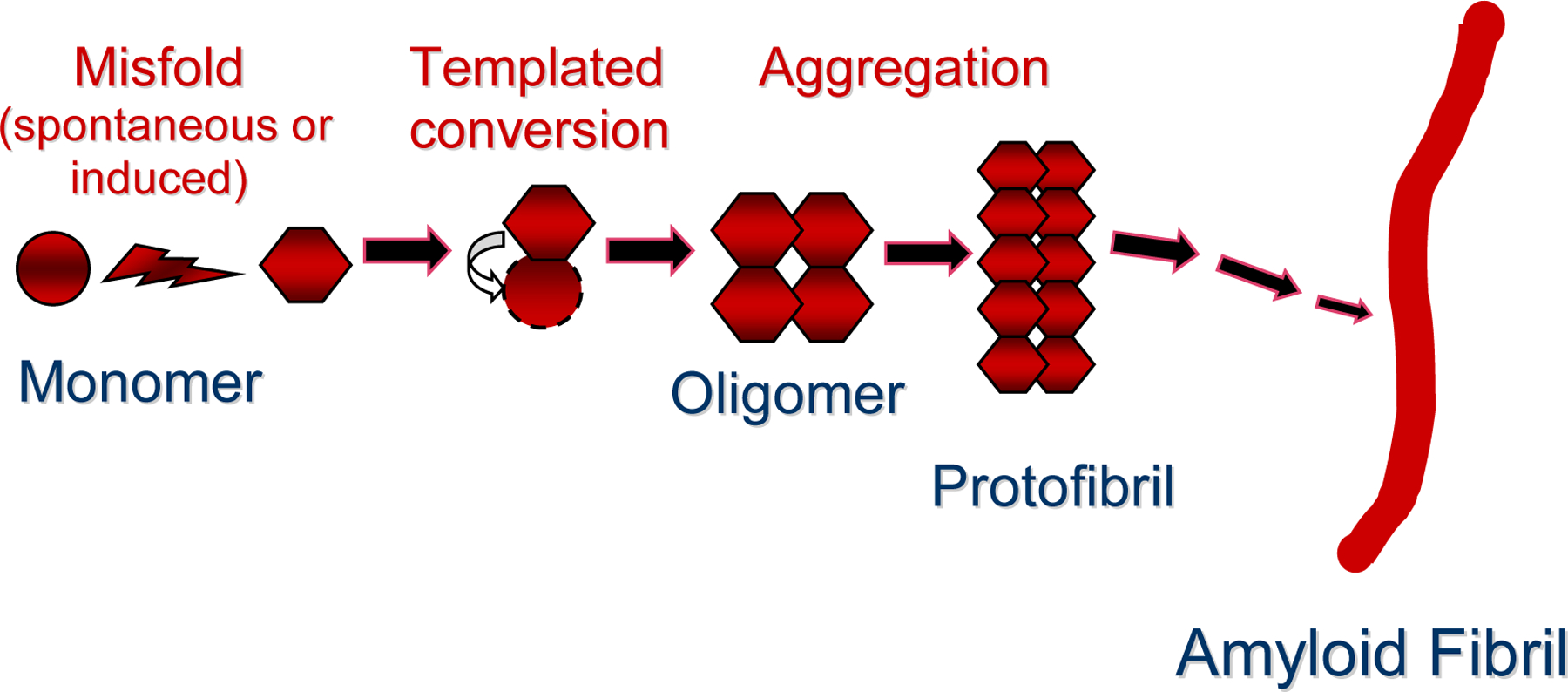
A hypothetical pathway leading from normally folded, monomeric protein to multimeric assemblies such as small oligomers, protofibrils and amyloid fibrils. In this instance, a particular monomeric protein (circle) assumes an atypical β-sheet-rich fold (hexagon), either as a stochastic or seeded event. This corrupted protein then impels the templated misconformation and consequent self-assembly of endogenously produced, cognate proteins. Multimeric protein aggregates can exist in multiple 3D forms consisting of various numbers of monomers; it is likely that multimers can themselves feed back into the proteopathic cascade as seeds. ‘Strain’ differences in inducibility appear to be coded in subtle conformational variations in proteins. There might be several pathways leading to different higher order assemblies. The biological activity of specific multimers, and the conditions that favor each step in the pathogenic sequence in vivo, remain incompletely understood.
Figure 3.
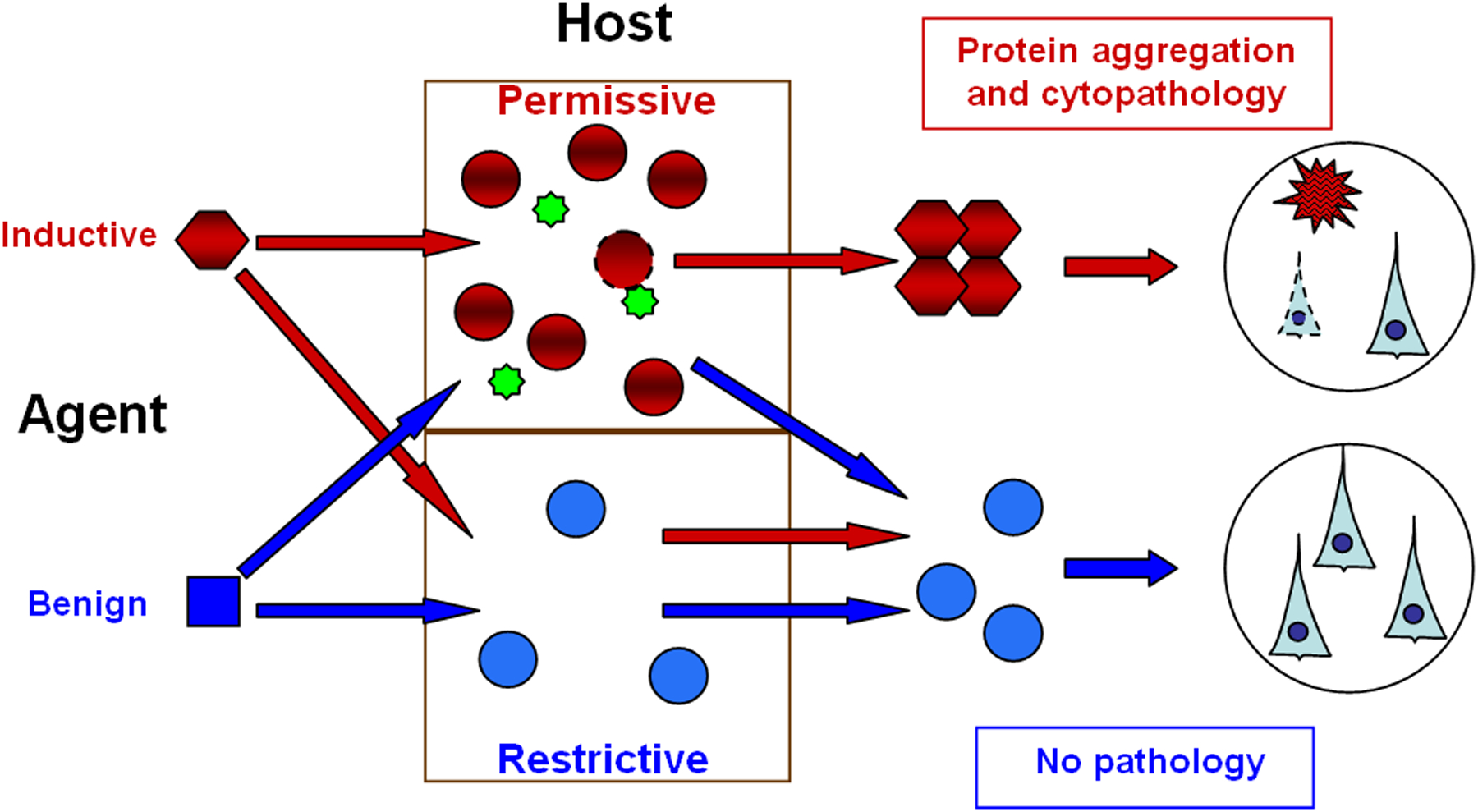
The interaction between the agent and host in the induction of disease by a proteopathic agent. By definition, a benign protein (blue square) is incapable of transmitting disease. An inductive agent (red hexagon) is pathogenic only in the context of a permissive environment (top). Such environments might include: A configuration of the cognate protein that is conducive to templating (red circles); increased production or sequestration of the cognate protein by the host; and/or the presence of essential cofactor(s) (green stars). A restrictive host environment, by contrast, might consist of a protein configuration that is resistant to templating (blue circles, bottom), sub-optimal protein levels, and/or the absence of cofactors. The role of soluble oligomeric species in seeding-induced aggregation and cytopathology remains to be defined.
Applying the prion model of induction to other proteopathies
In the mid-1800’s, Rudolf Virchow first employed the term ‘amyloid’, meaning ‘starch-like’, to describe accumulations of an unusual substance in animal organs that stained in a similar way to some constituents of plants. Today, ‘amyloid’ is generally used to describe fibrillar aggregates of particular proteins that have assumed a non-native, β-sheet-rich configuration. More than 20 proteins are known to form disease-related amyloid deposits in vivo, each having a unique amino acid sequence and yielding a characteristic disease phenotype [17]. Under permissive conditions, vulnerable proteins misfold and aggregate into polymeric fibrils and soluble oligomers. The appearance and secondary structure of each form are highly similar among proteins, regardless of the primary amino acid sequence of the starting protein [15,18,19]. Just as the probability of spontaneous misfolding varies among proteins [15], the susceptibility of proteins to permissive templating is likely to differ, such that some native polypeptides are more readily converted by misfolded cognate proteins acting as seeds, and thus are more apt to be transmissible.
Non-prion cerebral amyloidoses have been considered to be non-transmissible [20], but paradigms similar to those developed to study the transmission of prion diseases suggest that diverse conformational disorders can be induced in animal models by seeding-like mechanisms [21,22]. The inducible proteopathies include systemic amyloid A (AA) amyloidosis, systemic senile (apolipoprotein AII) amyloidosis, and cerebral amyloid-β (Aβ) amyloidosis. Although these proteopathies differ clinically and pathologically from the prionoses, the model systems used to investigate protein seeding are illuminating both the requisite host factors and the properties of the inducing material that are important for this unconventional mode of infectivity. They also provide a test-bed for potential therapeutic interventions. Analysis of the mechanisms by which heterologous proteins are impelled to aggregate in living organisms thus could yield fruitful insights into this surprisingly wide-ranging pathogenic process.
Induction of amyloid A amyloidosis
Under chronic inflammatory conditions that increase the hepatic production of amyloid A protein, the levels of amyloid A rise dramatically in blood, and this protein accumulates as amyloid fibrils in systemic organs, including the kidneys, liver and spleen [23]. With time, the burgeoning amyloid load triggers the impairment or failure of organ function. In animal models, administration of a systemic inflammatory stimulus (such as silver nitrate) eventually causes amyloid A deposition, but the process is slow. If an extract from organs rich in amyloid A protein – termed amyloid enhancing factor (AEF) - is administered along with the inflammatory agent, amyloidogenesis is strikingly accelerated [23–25]. The identity of this ‘factor’ remained uncertain until recently, when a purified fraction of splenic extract corresponding to aggregated amyloid A protein was shown to seed amyloid A deposition in vivo [25]. Although the isolated fraction probably contained small amounts of other, unidentified material [21], a compelling case can be made that aggregated amyloid A protein per se is the active component of AEF. Interestingly, amyloid A amyloidosis also can be elicited, although less potently, by diverse exogenous substances that are high in amyloid-type β-sheet content, including silk fibrils, the yeast prion Sup35, and bacterial curli fimbriae [26]. All of these proteins possess elements of amyloid structure. The corruption of certain normal proteins by heterologous ‘seeds’ suggests a tantalizing link to environmental factors, an issue that warrants further study.
Induction of apolipoprotein AII amyloidosis
Apolipoprotein AII (ApoAII) is an abundant, yet poorly understood, apolipoprotein [27] that can deposit spontaneously as amyloid fibrils in aged mice [28] and in a hereditary human disease caused by a stop-codon mutation in the apoAII gene [29]. Mouse senile amyloidosis entails the accumulation of ApoAII in systemic organs, a process that can be stimulated by peripheral injection of ApoAII fibrils isolated from affected liver [28]. ApoAII also induces amyloid disease when introduced into the gastrointestinal tract via gavage or in drinking water, and might even be transmitted to cage-mates by the ingestion of feces containing ApoAII fibrils [30]. When fibrils of a strongly amyloidogenic sequence-variant of the protein (ApoAII[C]) are injected into mice expressing a more resistant protein subtype (ApoAII[B]), the normally refractory mice produce conformationally altered ApoAII[B]-amyloid that has become highly amyloidogenic [31]. Denaturation of the ApoAII fibrillar extracts abolishes their ability to initiate disease [28,32], implicating ApoAII conformation in the induction of ApoAII amyloidosis. Like amyloid A amyloidosis, ApoAII amyloidosis is most potently stimulated by the same protein, though it also can be seeded in vivo by other substances. For example, in ApoAII[C]-expressing mice, ApoAII[C] fibrils are the most effective seed, but β-sheet rich fibrils from heterogeneous sources (including amyloid A and synthetic Aβ) also promote ApoAII deposition, albeit with reduced efficacy [33].
Amyloid A and ApoAII amyloidoses affect systemic organs that are supplied by fenestrated capillaries, which facilitate the entry of large molecules such as proteins into the tissues. By contrast, the most salient clinical manifestations of prion disease result from the proliferation of prions within the central nervous system, where the blood-brain barrier helps to protect the brain from many exogenous agents. Prions, probably with the aid of immune cells, are able to circumvent this obstacle [11]. Even so, the most effective way to transmit prion disease is by direct inoculation of the agent into the brain. As we will now review, emerging data suggest that Aβ-amyloidosis, perhaps the most common age-associated cerebral proteopathy, also can be stimulated in experimental models by the intracerebral injection of diseased tissue extracts.
Induction of Aβ proteopathy
Aβ is a minor proteolytic cleavage product of the Aβ-precursor protein (βAPP), a ubiquitous, type-1 transmembrane protein that is abundant in brain. Aβ, like other proteopathic molecules, is liable to misconformation and aggregation into macromolecular assemblies such as oligomers and amyloid fibrils. Aggregated Aβ constitutes the cores of senile plaques, and forms deposits in the walls of brain blood vessels known as cerebral β-amyloid angiopathy. In humans and several other mammalian species, the probability of developing Aβ lesions in the brain increases considerably in old age [34,35].
Substantial genetic, biochemical and pathologic evidence supports a primary role of aberrant Aβ in the genesis of Alzheimer’s disease (AD) [4], although some cognitively normal humans and all nonhuman species fail to acquire the full phenotype of AD, despite sometimes copious Aβ in the brain. The reasons for the apparent resistance to AD in animals that generate the identical, human-type Aβ-sequence remain uncertain. It is possible that large, extracellular Aβ aggregates (i.e. senile plaques) are relatively benign, and that, instead, cytotoxicity is mediated mainly by disease-specific oligomeric assemblies of Aβ; another possibility, which is not necessarily exclusive, is that the intracellular milieu is in some way more conducive to cytopathology in AD than in resistant organisms (Box 1). Interestingly, Aβ also is implicated in inclusion body myopathy, a degenerative muscle disorder characterized by the intracellular buildup of Aβ and βAPP fragments in myocytes [36].
Box 1. Cytotoxic mechanisms of pathogenic proteins.
Increasing evidence suggests that different pathogenic proteins damage and kill cells by similar mechanisms. In each case, oligomeric assemblies recently have emerged as prime suspects [50,58,59], and cell membranes are the sites where much of the molecular mischief occurs. Although attention initially focused on the plasma membrane, these proteins also can aggregate intracellularly, resulting in impaired organelle function and cell death [60,61]. Aβ, PrP and other aggregation-prone proteins possess amphipathic properties that facilitate their interactions with lipid membranes (particularly within lipid rafts [62–64]), where hydrogen peroxide production, lipid peroxidation [65–67] and disruption of cellular ion homeostasis [68–71] might be important steps in the neurotoxic cascade. In addition, other chemical changes might occur during the aggregation of the misfolded peptide [72,73]. Specific metal ions such as iron, copper and zinc can promote the multimerization of pathogenic proteins and the generation of reactive oxygen species in neurodegenerative diseases [74–78]. The age-associated increase in oxidative stress [62,79,80] and protein accumulation [81–83] might explain why spontaneous proteopathies are typically age-related, often with incubation periods of many years.
To date, there is no evidence that AD per se is transmissible, but several laboratories have begun to explore the possibility that Aβ deposition, one pathological hallmark of the disease, can be induced by exogenous seeding in animal models. The first attempts to transmit cerebral Aβ-amyloidosis employed nonhuman primates [37], which have a human-like Aβ sequence and naturally develop β-amyloid deposits (but not AD) in old age. In these experiments, senile plaques and Aβ-angiopathy were induced in young marmosets by the intracerebral inoculation of Aβ-rich brain homogenates, but a limitation of this paradigm is that the lesions do not materialize for several years in marmosets [37–39]. Mice have a much shorter life-span than do primates, and although wild-type mice do not manifest Aβ-amyloidosis due to idiosyncrasies in the murine Aβ sequence, several lines of mice that are transgenic for human βAPP develop plaques and β-amyloid angiopathy with age (e.g., [40,41]). In young βAPP-transgenic mice, cerebral Aβ-amyloidosis can be seeded by dilute cortical extracts from autopsied AD patients within the span of only a few months [22,42]. Interestingly, Aβ-rich brain extracts from βAPP-transgenic mice produce Aβ-seeding similar to that achieved using cortical material from humans [43], indicating that the inducing agent is not uniquely present in the human brain. Extracts from young murine or human brains that are devoid of Aβ-lesions have no effect in transgenic mice, and seeding does not occur when Aβ-rich extract is injected into wild-type mice, which produce a non-polymerogenic form of Aβ [42,43].
The evidence increasingly implicates exogenous Aβ itself as a crucial element in the seeding phenomenon. However, many important questions remain to be addressed, and the precise nature of the agent remains to be defined. To date, synthetic Aβ fibrils, in concentrations similar to those in brain extracts, have not been demonstrated to stimulate the endogenous generation of β-amyloid pathology in transgenic mice, suggesting that intrinsic properties of the Aβ peptide, or brain-specific cofactors in the extract, are needed. In this regard, it is useful to note that most attempts to transmit prion disease by in vitro-generated, recombinant prion protein have failed [15,44]. Even when successful, recombinant PrP is inefficient compared to prionotic brain extracts [12]. One possibility is that multimeric proteins can assume different 3D configurations, or ‘strains’, depending on the conditions under which they are formed. Indeed, Aβ, like PrP and other proteins, can form distinct strains that differ both in their structure and cytotoxicity [45–49]. As with prions, conformational strain differences also might influence the efficiency (and possibly the eventual phenotype) of Aβ-seeding. This question can be addressed experimentally using suitable animal models, in the context of newly emerging tools for analyzing disease-related conformations of Aβ in Alzheimer’s disease [50–52].
Inducible proteopathies: some caveats
The transmission of prion diseases is relatively unambiguous because the clinical manifestations (ultimately death) are particularly obvious [2,5]. By contrast, the neurologic consequences of cerebral Aβ-amyloidosis, especially in non-human species, often are more subtle and variable than those of the prionoses [22,38,39,42]. As a result, the effects of ‘infection’ might be relatively difficult to discern in some proteopathies, at least from a functional standpoint. This matter is complicated by the fact that monkeys and βAPP-transgenic mice spontaneously generate Aβ-pathology with age; hence, it is likely that Aβ seed-rich tissue extracts accelerate amyloidogenesis by supplementing (or anticipating) endogenously generated Aβ-seeds [22]. Analogously, AEF greatly accelerates amyloid A amyloidogenesis, even though systemic inflammation alone eventually results in amyloid disease [23].
For several reasons, then, the non-prion proteopathies might not be communicable in exactly the same sense as are prionoses. However, we contend that the ability of diseased tissue extracts to augment the pathogenesis of diverse proteopathies in vulnerable hosts indicates that, at the molecular level, the prion model of permissive templating has parallels in other protein conformational disorders. The concept of inducible proteopathies also accommodates the heterologous induction of a conformational change in a susceptible protein by different molecules that share critical structural features [19,21,33,53–55]. In this regard, the potential for cross-seeding by nanoscale-organized structures such as amyloid fibrils [56] might require careful toxicological assessment for some applications of nanotechnology [57] (Box 2). A fuller understanding of proteopathic induction in vivo, and of the common cellular and molecular mechanisms by which aggregation-prone proteins propagate misfolding and exert their toxicity, might lead to unified strategies for deciphering the ontogeny of a number of seemingly disparate disorders.
Box 2. Nanotoxic?
The repeating molecular structure of amyloid fibrils is attracting interest in the rapidly expanding field of nanotechnology. Engineering of nanomaterials and nanomachines in sizes ranging from smaller than antibodies up to viruses (1 – 100 nm) is a feasible objective. A variety of medical applications take advantage of the ability of nanoparticles to deliver drug cargoes across biological membranes or to control the release of their contents [84]. Early nanomaterials were spawned by the microelectronics industry, which built small devices on a massive scale. Organic polymers soon were adapted to take advantage of their variable structures and easily accessible chemistry for customizing properties. The quasi-crystalline and controllable self-assembly of amyloidogenic proteins has suggested that amyloid fibrils and other higher-order protein assemblies might be useful as nanomaterials.
The rush to nanotechnology has raised concerns about the largely untested toxicological and environmental hazards of nanomaterials. Little is known about how they interact with biological materials and how they are transported, modified, or degraded. The templating nature of amyloid-like assemblies discussed in this article is an example of a nanoscale property that could have health-related implications. The cross-seeding observed for a variety of amyloids [26,33] and the ability of materials such as silk to accelerate amyloid fibril formation by the amyloid A protein [26,85] suggest a need to evaluate the toxicology of nanoscale assemblies. A natural substrate candidate for seeding by amyloid-like structures is the amyloid A protein, which increases dramatically as a normal physiological response to routine inflammatory stimuli and in certain medical conditions [23]. Other misfolding proteins, such as those involved in the chronic neurodegenerative diseases, theoretically could be templated by exposure to amyloid-like materials that penetrate the blood-brain barrier. It is also worth noting that nanomaterials, such as fullerene (buckyballs), might be employed therapeutically to inhibit abnormal protein assembly [86]. In any case, nanotoxicological issues deserve a prominent place in current and future nanomedicine initiatives.
Concluding remarks
The weight of evidence now supports the concept that exogenous, structurally complementary molecules can induce specific diseases of protein conformation and assembly in animals. Key objectives for future research are to define, at the molecular level, how disease originates de novo in both the sporadic and the hereditary proteopathies, to establish the structural idiosyncrasies of agents that act as corruptive protein templates, and to elucidate the cytotoxic mechanisms of protein aggregates. Finally, it is essential to determine the genetic, biochemical and physiological characteristics of the host that regulate the permissiveness of templating in protein deposition disorders.
Acknowledgments
We gratefully acknowledge helpful discussions with John Hardy, Ingo Autenrieth, Rolf Warzok, Margaret Walker and Rebecca Rosen. This work was supported by grants from the Woodruff Foundation, NIH RR-00165, by the Sanders-Brown Center on Aging and Chandler Medical Center of the University of Kentucky, by the National Institute on Aging Intramural Research Program of the NIH, and by the Alzheimer’s Association.
References
- 1.Walker LC and LeVine H (2000) The cerebral proteopathies: neurodegenerative disorders of protein conformation and assembly. Mol Neurobiol 21, 83–95 [DOI] [PubMed] [Google Scholar]
- 2.Prusiner SB (2001) Shattuck lecture--neurodegenerative diseases and prions. N Engl J Med 344, 1516–1526 [DOI] [PubMed] [Google Scholar]
- 3.Carrell RW and Lomas DA (2002) Alpha1-antitrypsin deficiency--a model for conformational diseases. N Engl J Med 346, 45–53 [DOI] [PubMed] [Google Scholar]
- 4.Hardy J and Selkoe DJ (2002) The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356 [DOI] [PubMed] [Google Scholar]
- 5.Johnson RT (2005) Prion diseases. Lancet Neurol 4, 635–642 [DOI] [PubMed] [Google Scholar]
- 6.McKintosh E et al. (2003) Prion diseases. J Neurovirol 9, 183–193 [DOI] [PubMed] [Google Scholar]
- 7.Prusiner SB et al. (1999) The Prion Diseases. In Alzheimer Disease (Terry RD et al., eds.), pp. 161–179, Lippincott Williams and Wilkins [Google Scholar]
- 8.Weissman C (2004) The state of the prion. Nat Rev Microbiol 2, 861–871 [DOI] [PubMed] [Google Scholar]
- 9.Hardy J (2005) Expression of normal sequence pathogenic proteins for neurodegeneration contributes to disease risk: “Permissive templating” as a general disease mechanism of neurodegeneration. Biochem Soc Trans 33, 578–581 [DOI] [PubMed] [Google Scholar]
- 10.Van Everbroeck B et al. (2002) Transmissible spongiform encephalopathies: the story of a pathogenic protein. Peptides 23, 1351–1359 [DOI] [PubMed] [Google Scholar]
- 11.Aguzzi A et al. (2003) Immune system and peripheral nerves in propagation of prions to CNS. Br Med Bull 66, 141–159 [DOI] [PubMed] [Google Scholar]
- 12.Legname G et al. (2004) Synthetic mammalian prions. Science 305, 673–676 [DOI] [PubMed] [Google Scholar]
- 13.Chesebro B et al. (2005) Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science 308, 1420–1421 [DOI] [PubMed] [Google Scholar]
- 14.Castilla J et al. (2005) In vitro generation of infectious scrapie prions. Cell 121, 195–206 [DOI] [PubMed] [Google Scholar]
- 15.Dobson CM (1999) Protein misfolding, evolution and disease. Trends Biochem Sci 24, 329–332 [DOI] [PubMed] [Google Scholar]
- 16.Walker L, LeVine H and Jucker M (2006) Koch’s postulates and infectious proteins. Acta Neuropath, in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Westermark P (2005) Aspects on human amyloid forms and their fibril polypeptides. Febs J 272, 5942–5949 [DOI] [PubMed] [Google Scholar]
- 18.Dobson CM (2003) Protein folding and misfolding. Nature 426, 884–890 [DOI] [PubMed] [Google Scholar]
- 19.Walker LC and LeVine H 3rd. (2002) Proteopathy: the next therapeutic frontier? Curr Opin Investig Drugs 3, 782–787 [PubMed] [Google Scholar]
- 20.Gajdusek DC (1994) Spontaneous generation of infectious nucleating amyloids in the transmissible and nontransmissible cerebral amyloidoses. Mol Neurobiol 8, 1–13 [DOI] [PubMed] [Google Scholar]
- 21.Sigurdsson EM et al. (2002) Infectivity of amyloid diseases. Trends Mol Med 8, 411–413 [DOI] [PubMed] [Google Scholar]
- 22.Walker LC et al. (2002) Exogenous induction of cerebral beta-amyloidosis in βAPP-transgenic mice. Peptides 23, 1241–1247 [DOI] [PubMed] [Google Scholar]
- 23.Rocken C and Shakespeare A (2002) Pathology, diagnosis and pathogenesis of AA amyloidosis. Virchows Arch 440, 111–122 [DOI] [PubMed] [Google Scholar]
- 24.Kisilevsky R and Fraser PE (1997) A beta amyloidogenesis: unique, or variation on a systemic theme? Crit Rev Biochem Mol Biol 32, 361–404 [DOI] [PubMed] [Google Scholar]
- 25.Lundmark K et al. (2002) Transmissibility of systemic amyloidosis by a prion-like mechanism. Proc Natl Acad Sci U S A 99, 6979–6984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lundmark K et al. (2005) Protein fibrils in nature can enhance amyloid protein A amyloidosis in mice: Cross-seeding as a disease mechanism. Proc Natl Acad Sci U S A 102, 6098–6102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martin-Campos JM et al. (2004) Apolipoprotein A-II, genetic variation on chromosome 1q21-q24, and disease susceptibility. Curr Opin Lipidol 15, 247–253 [DOI] [PubMed] [Google Scholar]
- 28.Higuchi K et al. (1998) Fibrilization in mouse senile amyloidosis is fibril conformation-dependent. Lab Invest 78, 1535–1542 [PubMed] [Google Scholar]
- 29.Benson MD et al. (2001) A new human hereditary amyloidosis: the result of a stop-codon mutation in the apolipoprotein AII gene. Genomics 72, 272–277 [DOI] [PubMed] [Google Scholar]
- 30.Xing Y et al. (2001) Transmission of mouse senile amyloidosis. Lab Invest 81, 493–499 [DOI] [PubMed] [Google Scholar]
- 31.Xing Y et al. (2002) Induction of protein conformational change in mouse senile amyloidosis. J Biol Chem 277, 164–169 [DOI] [PubMed] [Google Scholar]
- 32.Zhang H et al. (2006) Transmissibility of mouse AApoAII amyloid fibrils: inactivation by physical and chemical methods. FASEB J [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 33.Fu X et al. (2004) Induction of AApoAII amyloidosis by various heterogeneous amyloid fibrils. FEBS Lett 563, 179–184 [DOI] [PubMed] [Google Scholar]
- 34.Warzok RW et al. (1998) Apolipoprotein E4 promotes incipient Alzheimer pathology in the elderly. Alzheimer Dis Assoc Disord 12, 33–39 [DOI] [PubMed] [Google Scholar]
- 35.Walker LC (2000) Cerebral amyloid angiopathy in aged dogs and nonhuman primates. In Cerebral Amyloid Angiopathy in Alzheimer’s Disease and Related Disorders (Verbeek MM et al. , eds.), pp. 313–324, Kluwer [Google Scholar]
- 36.Askanas V and Engel WK (2005) Molecular pathology and pathogenesis of inclusion body myositis. Microsc Res Tech 67, 114–120 [DOI] [PubMed] [Google Scholar]
- 37.Baker HF et al. (1994) Induction of beta (A4)-amyloid in primates by injection of Alzheimer’s disease brain homogenate. Comparison with transmission of spongiform encephalopathy. Mol Neurobiol 8, 25–39 [DOI] [PubMed] [Google Scholar]
- 38.Maclean CJ et al. (2000) Naturally occurring and experimentally induced beta-amyloid deposits in the brains of marmosets (Callithrix jacchus). J Neural Transm 107, 799–814 [DOI] [PubMed] [Google Scholar]
- 39.Ridley RM et al. (2005) Very long term studies of the seeding of beta-amyloidosis in primates. J Neural Transm [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 40.Callahan MJ et al. (2001) Augmented senile plaque load in aged female beta-amyloid precursor protein-transgenic mice. Am J Pathol 158, 1173–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Herzig MC, Van Nostrand WE and Jucker M (2006) Mechanism of cerebral β-amyloid angiopathy: murine and cellular models. Brain Pathol 16, 40–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kane MD et al. (2000) Evidence for seeding of β-amyloid by intracerebral infusion of Alzheimer brain extracts in β-amyloid precursor protein-transgenic mice. J Neurosci 20, 3606–3611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meyer-Luehmann M et al. (2003). Initiation of amyloid deposition in APP23 transgenic mice. Program No. 731.11, Abstract Viewer/Itinerary Planner. New Orleans, LA: Society for Neuroscience. [Google Scholar]
- 44.Hill AF et al. (1999) Protease-resistant prion protein produced in vitro lacks detectable infectivity. J Gen Virol 80, 11–14 [DOI] [PubMed] [Google Scholar]
- 45.Petkova AT et al. (2005) Self-propagating, molecular-level polymorphism in Alzheimer’s beta-amyloid fibrils. Science 307, 262–265 [DOI] [PubMed] [Google Scholar]
- 46.Del Mar C et al. (2005) Structure and properties of alpha-synuclein and other amyloids determined at the amino acid level. Proc Natl Acad Sci U S A 102, 15477–15482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Heise H et al. (2005) Molecular-level secondary structure, polymorphism, and dynamics of full-length alpha-synuclein fibrils studied by solid-state NMR. Proc Natl Acad Sci U S A 102, 15871–15876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dzwolak W et al. (2005) Ethanol-perturbed amyloidogenic self-assembly of insulin: looking for origins of amyloid strains. Biochemistry 44, 8948–8958 [DOI] [PubMed] [Google Scholar]
- 49.Piccini A et al. (2005) β-Amyloid is different in normal aging and in Alzheimer disease. J Biol Chem 34, 186–192 [DOI] [PubMed] [Google Scholar]
- 50.Kayed R et al. (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300, 486–489 [DOI] [PubMed] [Google Scholar]
- 51.Klunk WE et al. (2005) Binding of the positron emission tomography tracer Pittsburgh compound-B reflects the amount of amyloid-beta in Alzheimer’s disease brain but not in transgenic mouse brain. J Neurosci 25, 10598–10606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Klunk WE et al. (2003) The binding of 2-(4’-methylaminophenyl)benzothiazole to postmortem brain homogenates is dominated by the amyloid component. J Neurosci 23, 2086–2092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.O’Nuallain B et al. (2004) Seeding specificity in amyloid growth induced by heterologous fibrils. J Biol Chem 279, 17490–17490 [DOI] [PubMed] [Google Scholar]
- 54.Lewis J et al. (2001) Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 293, 1487–1491 [DOI] [PubMed] [Google Scholar]
- 55.Gotz J et al. (2001) Formation of neurofibrillary tangles in P301L tau transgenic mice by Abeta 42 fibrils. Science 293, 1491–1495 [DOI] [PubMed] [Google Scholar]
- 56.Hamada D et al. (2004) Engineering amyloidogenicity towards the development of nanofibrillar materials. Trends Biotechnol 22, 93–97 [DOI] [PubMed] [Google Scholar]
- 57.Nel A et al. (2006) Toxic potential of materials at the nanolevel. Science 311, 622–627 [DOI] [PubMed] [Google Scholar]
- 58.Walsh DM et al. (2002) Amyloid-beta oligomers: their production, toxicity and therapeutic inhibition. Biochem Soc Trans 30, 552–557 [DOI] [PubMed] [Google Scholar]
- 59.Lacor PN et al. (2004) Synaptic targeting by Alzheimer’s-related amyloid beta oligomers. J Neurosci 24, 10191–10200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Walsh DM and Selkoe DJ (2004) Deciphering the molecular basis of memory failure in Alzheimer’s disease. Neuron 44, 181–193 [DOI] [PubMed] [Google Scholar]
- 61.Glabe CC (2005) Amyloid accumulation and pathogenesis of Alzheimer’s disease: significance of monomeric, oligomeric and fibrillar Abeta. Subcell Biochem 38, 167–177 [DOI] [PubMed] [Google Scholar]
- 62.Cutler RG et al. (2004) Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc Natl Acad Sci U S A 101, 2070–2075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Naslavsky N et al. (1999) Sphingolipid depletion increases formation of the scrapie prion protein in neuroblastoma cells infected with prions. J Biol Chem 274, 20763–20771 [DOI] [PubMed] [Google Scholar]
- 64.Gellermann GP et al. (2005) Raft lipids as common components of human extracellular amyloid fibrils. Proc Natl Acad Sci U S A 102, 6297–6302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mattson MP and Goodman Y (1995) Different amyloidogenic peptides share a similar mechanism of neurotoxicity involving reactive oxygen species and calcium. Brain Res 676, 219–224 [DOI] [PubMed] [Google Scholar]
- 66.Mattson MP (1998) Modification of ion homeostasis by lipid peroxidation: roles in neuronal degeneration and adaptive plasticity. Trends Neurosci 21, 53–57 [DOI] [PubMed] [Google Scholar]
- 67.Milhavet O et al. (2000) Prion infection impairs the cellular response to oxidative stress. Proc Natl Acad Sci U S A 97, 13937–13942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mattson MP et al. (1992) beta-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci 12, 376–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Demuro A et al. (2005) Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J Biol Chem 280, 17294–17300 [DOI] [PubMed] [Google Scholar]
- 70.Kagan BL et al. (2004) Amyloid peptide channels. J Membr Biol 202, 1–10 [DOI] [PubMed] [Google Scholar]
- 71.Kayed R et al. (2004) Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J Biol Chem 279, 46363–46366 [DOI] [PubMed] [Google Scholar]
- 72.Wogulis M et al. (2005) Nucleation-dependent polymerization is an essential component of amyloid-mediated neuronal cell death. J Neurosci 25, 1071–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tabner BJ et al. (2005) Hydrogen peroxide is generated during the very early stages of aggregation of the amyloid peptides implicated in Alzheimer disease and familial British dementia. J Biol Chem 280, 35789–35792 [DOI] [PubMed] [Google Scholar]
- 74.Atwood CS et al. (2004) Copper mediates dityrosine cross-linking of Alzheimer’s amyloid-beta. Biochemistry 43, 560–568 [DOI] [PubMed] [Google Scholar]
- 75.Tabner BJ et al. (2005) Protein aggregation, metals and oxidative stress in neurodegenerative diseases. Biochem Soc Trans 33, 1082–1086 [DOI] [PubMed] [Google Scholar]
- 76.Maynard CJ et al. (2005) Metals and amyloid-beta in Alzheimer’s disease. Int J Exp Pathol 86, 147–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cuajungco MP et al. (2005) Amyloid-beta metal interaction and metal chelation. Subcell Biochem 38, 235–254 [DOI] [PubMed] [Google Scholar]
- 78.Gaeta A and Hider RC (2005) The crucial role of metal ions in neurodegeneration: the basis for a promising therapeutic strategy. Br J Pharmacol 146, 1041–1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Barja G (2004) Free radicals and aging. Trends Neurosci 27, 595–600 [DOI] [PubMed] [Google Scholar]
- 80.Balaban RS et al. (2005) Mitochondria, oxidants, and aging. Cell 120, 483–495 [DOI] [PubMed] [Google Scholar]
- 81.Grune T et al. (2004) Decreased proteolysis caused by protein aggregates, inclusion bodies, plaques, lipofuscin, ceroid, and ‘aggresomes’ during oxidative stress, aging, and disease. Int J Biochem Cell Biol 36, 2519–2530 [DOI] [PubMed] [Google Scholar]
- 82.Chondrogianni N and Gonos ES (2005) Proteasome dysfunction in mammalian aging: Steps and factors involved. Exp Gerontol 40, 931–938 [DOI] [PubMed] [Google Scholar]
- 83.Keller JN et al. (2004) Autophagy, proteasomes, lipofuscin, and oxidative stress in the aging brain. Int J Biochem Cell Biol 36, 2376–2391 [DOI] [PubMed] [Google Scholar]
- 84.Chithrani BD, Ghazani AA and Chan WC (2006) Determining the size and shape dependence of gold nanoparticle uptake into mammalian cells. Nano Lett 6, 662–668 [DOI] [PubMed] [Google Scholar]
- 85.Kisilevsky R et al. (1999) New clothes for amyloid enhancing factor (AEF): silk as AEF. Amyloid 6, 98–106 [DOI] [PubMed] [Google Scholar]
- 86.Kim JE and Lee M (2003) Fullerene inhibits β-amyloid peptide aggregation. Biochem Biophys Res Comm 303, 576–579 [DOI] [PubMed] [Google Scholar]