

Abstract
Protein synthesis on the ribosome involves successive rapid recruitment of cognate aminoacyl-tRNAs and rejection of the much more numerous incorrect near- or non-cognates. The principal feature of translation elongation is that at every step, many incorrect aa-tRNAs unsuccessfully enter the A site for each cognate accepted. Normal levels of translational accuracy require that cognate tRNAs have relatively similar acceptance rates by the ribosome. To achieve that, tRNAs evolved to compensate for differences in amino acid properties and codon–anticodon strength that affect acceptance. Part of that response involved tRNA posttranscriptional modifications, which can affect tRNA decoding efficiency, accuracy, and structural stability. The most intensively modified regions of the tRNA are the anticodon loop and structural core of the tRNA. Anticodon loop modifications directly affect codon–anticodon pairing and therefore modulate accuracy. Core modifications have been thought to ensure consistent decoding rates principally by stabilizing tRNA structure to avoid degradation; however, degradation due to instability appears to only be a significant issue above normal growth temperatures. We suspected that the greater role of modification at normal temperatures might be to tune tRNAs to maintain consistent intrinsic rates of acceptance and peptide transfer and that hypomodification by altering these rates might degrade the process of discrimination, leading to increased translational errors. Here, we present evidence that most tRNA core modifications do modulate the frequency of misreading errors, suggesting that the need to maintain accuracy explains their deep evolutionary conservation.
Keywords: translational misreading, protein synthesis, ribosome, tRNA core modification, Saccharomyces cerevisiae
INTRODUCTION
Accuracy in protein synthesis is central to the maintenance of cellular homeostasis. Errors in the production of proteins can result in dire effects, including loss of protein function or toxicity resulting from protein aggregation (Drummond and Wilke 2009). Because of the seriousness of these effects, accuracy mechanisms have evolved to limit errors and quality control mechanisms to limit their effect (Wohlgemuth et al. 2011; Steiner and Ibba 2019; D'Orazio and Green 2021). Errors can result from misinterpretation of genetic instructions during any step in this process, including transcription, posttranscriptional processing, protein synthesis, posttranslational processing, protein folding, and protein degradation (Drummond and Wilke 2009). Not all errors impair cellular function; in fact, errors known or thought to expand the range of polypeptides produced can be exploited to the cell's benefit (Ribas de Pouplana et al. 2014).
Protein synthesis occurs on the ribosome, a complex ribozyme that interprets each nucleic acid sense codon to define a particular amino acid. An aminoacyl-tRNA binds to the mRNA, forming a 3-nt codon–anticodon pair placing the amino acid in the peptidyl transferase center of the ribosome, where it is added to the growing peptide chain. Errors arise either when the wrong amino acid is attached to the tRNA (misacylation) or when the ribosome accepts a tRNA that imperfectly pairs with the codon (mistranslation). Misacylation is an enzyme–substrate interaction error between aminoacyl-tRNA synthetases and their substrates. Synthetases have evolved active sites that preferentially bind correct amino acids and, where structural similarity is too high among a group of amino acids, eliminate incorrect ones by proofreading (Mohler and Ibba 2017).
Avoiding mistranslation errors is more problematic, because decoding occurs at a single complex active site in the ribosome, the decoding center, that must distinguish between one or more correct (cognate) and approximately 45 incorrect (near- or non-cognate) tRNAs. The large mass excess of incorrect tRNAs and the obligate structural homogeneity of all tRNAs (for review, see Giege et al. 2012) complicate error correction. As with synthetases, the ribosome increases accuracy by rejecting incorrect tRNA by proofreading. The ribosome exploits general structural rules to distinguish between correct and incorrect complexes. In the simplest terms, the ribosomal A site, at which successive aminoacyl-tRNAs are selected, distinguishes between tRNAs that form codon–anticodon complexes consisting of canonical Watson–Crick pairs or, at the third position, specific noncanonical “wobble” pairs (Rodnina et al. 2017). Complexes requiring base pairs deviating from these forms tend to be nearly quantitatively rejected.
The simplicity of this description belies the complexity of the discrimination step. Genetic and biochemical analysis has shown that posttranscriptional modification of the tRNA is critical for tRNA selection accuracy. Most clearly, modifications of the tRNA anticodon and adjacent nucleotides fine-tune codon–anticodon interactions to ensure efficient cognate decoding (Suzuki 2021). Counterintuitively, some anticodon loop modifications actually increase the frequency of misreading (El Yacoubi et al. 2011; Lamichhane et al. 2013; Manickam et al. 2016; Joshi et al. 2018). Apparently, some modifications that have evolved to stabilize cognate decoding also stabilize near-cognate codon–anticodon complexes, which results in more frequent errors. Presumably, selection for efficient cognate recognition was more powerful in these cases than selection to reduce errors. So, modifications can have a variety of effects on decoding accuracy, either increasing or decreasing error frequencies depending on the need to support decoding in general or to suppress errors specifically.
Recent work has demonstrated in vivo effects of modifications outside the anticodon loop, most of which target residues that are part of the structural core of the tRNA (Fig. 1), within which are concentrated nucleotides involved in tertiary interactions, or nucleotides immediately adjacent to tertiary pairs (Motorin and Helm 2010; Phizicky and Alfonzo 2010; Lorenz et al. 2017). Some of these core modifications have been shown to stabilize tRNAs to degradation in a temperature-sensitive fashion (Alexandrov et al. 2006). For example, at the elevated temperature of 37° the lack of two Saccharomyces cerevisiae modification enzymes, Trm4 and Trm8, which eliminates m7G46 and m5C49, causes rapid degradation of by the rapid tRNA decay (RTD) pathway (Alexandrov et al. 2006). This temperature-sensitive stability effect, however, is unlikely to explain the widespread evolution of these modifications across all taxa. Partly, this is because the effect is isoacceptor specific; lack of Trm4/Trm8 has no effect on several other tRNAs sharing the same modifications as (Alexandrov et al. 2006; Chernyakov et al. 2008). Because mistranslation frequency sensitively responds to changes in competition between cognate and near-cognate tRNAs for the ribosomal decoding site, core hypomodification changing the relative abundance of cognate and near-cognate tRNAs could increase or decrease misreading errors. On the other hand, loss of core modifications could affect misreading frequency by altering relative tRNA efficiency in some other way, for example by changing relative aminoacylation efficiency or by sequestering tRNAs away from the translation machinery. Alternatively, lack of modification could alter the intrinsic decoding efficiency of a tRNA by altering the structure or flexibility of the tRNA, which could alter the dynamics of its interaction with the ribosomal A site.
FIGURE 1.

(A) The location of highly conserved nucleotides shown on a cloverleaf diagram of a generic S. cerevisiae tRNA with bases involved in core interactions shown in black. (B) Conserved tertiary interactions shown in an L-shape diagram are indicated by dotted lines between interacting nucleotides. Non-dashed lines indicate phosphodiester links between adjacent nucleotides in the primary sequence. The five regions of a standard tRNA are indicated.
We have developed sets of mutant reporter genes that provide accurate in vivo measures of all possible mistranslation errors by individual tRNAs (Kramer and Farabaugh 2007; Kramer et al. 2010; Manickam et al. 2014; Joshi et al. 2018). These reporters exploit mutations targeting active site residues of reporter enzymes. The mutant enzymes have activity as much as 106-fold below wild type. The mutant codon includes one change from the wild-type codon, allowing the normally decoding tRNA to misread the mutant codon by near-cognate decoding involving one mismatched nucleotide pair, producing low amounts of fully functional protein. We have demonstrated that the activity of these mutant enzymes is a measure of the misreading error frequency for the wild-type tRNA at the near-cognate mutant codon (Kramer and Farabaugh 2007; Kramer et al. 2010; Manickam et al. 2014; Joshi et al. 2018).
The advantages of this approach are several. The error frequencies are physiologically relevant because they occur in the living cell. The protein produced by these errors has the wild-type structure, so the chance that errors are underestimated due to degradation of the mistranslated protein is eliminated. The activities produced from the mutant mRNAs are highly reproducible and quantitative, providing a highly significant estimate of any alterations to misreading frequency caused by mutations affecting various components of the translation system. Control experiments have demonstrated that the activities are not due to extraneous effects, including transcriptional errors, protein modification, or alterations in plasmid or protein stability.
In the work described here, we made use of two reporter systems based on Lys 529 of Photinus pyralis (firefly) luciferase (Fluc) and Glu 537 of Escherichia coli β-galactosidase. Two S. cerevisiae Lys tRNAs, which have anticodons UUU and CUU, normally decode AAA/AAG, and their most frequent misreading errors occur at the stop codon UAG (2.1 × 10−3), and the Arg AGG codon (8.4 × 10−4) (Kramer et al. 2010). These values are over 10-fold and fourfold above the background for the assay. Errors at the Asn codons AAU and AAC are 1.6 × 10−4 and 1.8 × 10−4 near the background for the assay (Kramer et al. 2010). However, various treatments that increase errors cause a significant increase over this activity, which is not true for non-error-prone codon mutants (Kramer et al. 2010). The most frequent errors by GAA/GAG decoding and GAG decoding in S. cerevisiae occur at the Gly codon GGA (1.8 × 10−4), intermediate at the Asp codons GAU and GAC (1.4 × 10−4 and 1.1 × 10−4), and least on the Gly codon GGG (2.7 × 10−5) (Joshi et al. 2018). However, unlike the luciferase system, the background in the assay for β-galactosidase is very low, about 2 × 10−6 (Joshi et al. 2018), so all these mutants produce activities very significantly above background.
We have used these in vivo mistranslation reporters to directly measure errors in protein primary structure to determine if loss of core modifications can alter misreading error frequency. We confirm that lack of the tRNA core modifications can either increase or decrease translational accuracy depending on identity of the tRNA and the codon being read. However, lack of modifications does so without affecting tRNA stability, aminoacylation or subcellular localization. These results show that core modifications modulate intrinsic decoding efficiency of the error-prone and their competing cognate tRNAs tested here. The fact that the same modifications occur to different degrees across the tRNA complement implies that they may play a general role in maintaining normal translational accuracy at other codons. More extensive analysis will be required to test this putative universal role.
RESULTS
Use of enzyme-based translational misreading reporters to test the role of core tRNA modifications on the frequency of misreading errors in vivo
To measure the effect of loss of modifications of both the cognate and near-cognate tRNAs, we tested the effect on accuracy of all viable mutants that eliminate modifications of Lys, Glu, Arg, Asn, Gly, and Asp tRNAs (Fig. 2; Supplemental Table S1). The fact that the errors occur in competition with the cognate tRNAs for the mutant codon is important in thinking about the effect of loss of modification. Where the modification is missing only from either the cognate or the error-prone near-cognate tRNA, the effect on accuracy should reflect a change in the efficiency of decoding by that affected tRNA. For example, where the modification is lost from the cognate tRNA, a finding of increased misreading most likely results from reduced efficiency of decoding by the cognate tRNA, allowing more frequent decoding by the error-prone tRNA. In a similar case where errors are reduced, it implies increased efficiency of decoding by the cognate tRNA. In cases where both the error-prone near-cognate and competing cognate tRNAs can carry these core modifications, the lack of modification of either of these tRNAs could be the cause of the change in error frequency.
FIGURE 2.


Posttranscriptional modifications of tRNAs implicated in misreading of error-prone codons. (A) Cloverleaf depiction of potential misreading tRNAs for the P. pyralis (firefly) luciferase Lys529 and E. coli β-galactosidase Glu537 reporter systems. The locations of all known modifications are labeled with the enzyme(s) responsible (e.g., Dus1), and the modification. The gene copy number for each tRNA, roughly proportional to tRNA concentration (Percudani et al. 1997) is shown. (B) Cloverleaf depiction of competing cognate tRNAs for Arg, Asn, Gly, and Asp codons subject to errors by misreading tRNAs. Core modification symbols: (D) dihydrouridine, (Y) pseudouridine, () N2,N2-dimethylguanosine, (m5U) 5-methyluridine, (m5C) 5-methylcytidine, (m7G) 7-methylguanosine, (m1G) 1-methylguanosine, (m2G) 2-methylguanosine, (Cm) 2-O-methylcytidine, (m1A) 1-methyladenosine. Anticodon loop modification symbols: (mcm5s2U) 5-methoxycarbonylmethyl-2-thiouridine, (t6A) N6-threonylcarbamoyladenosine, (m1G) 1-methylguanosine, (m3C) 3-methylcytidine.
Changes in the efficiency of translation resulting in altered misreading frequencies can arise from several causes. Loss of modification could destabilize the tRNA by making it subject to rapid tRNA decay resulting in reduced steady-state levels of the affected tRNA (Alexandrov et al. 2006). Hypomodified tRNAs may undergo retrograde transport to the nucleus for repair; the inability to repair in the affected tRNA may cause it to be sequestered there (Kramer and Hopper 2013). Alternatively, lack of modification could slow maturation or aminoacylation of the affected tRNA, again resulting in reduced availability (Lorenz et al. 2017). Finally, modifications are known to alter physical characteristics of the tRNA including altering its overall fold, so lack of a modification could reduce the proportion of the tRNAs having the stable folded structure or could alter the dynamics of its flexibility (Uhlenbeck and Schrader 2018). Such effects might reduce the availability of mature functional tRNAs or could alter the kinetics of acceptance or rejection of the tRNA by the ribosome. Under any of these models, loss of tRNA modification could either increase or decrease misreading error frequencies, depending on which tRNA is more affected and the effect of the loss of modification on the biochemical pathway. The goal of this work is to determine if hypomodification affects accuracy and which of these effects can explain observed changes in accuracy.
Loss of core modifications generally decrease tRNALys nonsense readthrough frequency
Misreading of termination codons, also known as nonsense readthrough, is a special case of misreading errors. Rather than competing against cognate tRNAs, at the termination codon UAG a near cognate tRNA misreads in competition with the peptide release factors (RF) responsible for promoting termination. RF efficiency is unaffected by loss of tRNA modification, so any change in accuracy caused by loss of modification must reflect altered efficiency of near-cognate decoding by the error-prone tRNA. The errors measured in our reporter system unlike other readthrough reporters require readthrough by a tRNA inserting the normal amino acid. In the case of the dual luciferase reporter with Lys codon 529 replaced by UAG, that activity requires misreading by tRNALys.
Misreading frequency, indicated by the average enzyme activity of the mutant protein relative to wild type, was determined in a wild type and 14 congenic modification mutants. Misreading of UAG as Lys was reduced in 12 cases, and in eight cases that reduction was statistically significant. This supports the conclusion that the presence of these modifications generally improves near-cognate decoding by tRNALys. The isoacceptors for Lys are , encoded by 14 gene copies, and , encoded by seven (Chan and Lowe 2016). The steady-state level of a tRNA is roughly proportional to its gene copy number (Percudani et al. 1997), so the ratio of the two tRNAs is expected to be approximately 2:1. The tRNA gene copy number makes tRNALys among the most abundant tRNAs in S. cerevisiae. The tRNAs are distinguishable by modification; two of the tested modification enzymes (Trm4 and Trm8) target but not , two enzymes (Trm10 and Trm11) target only and eight other enzymes target both. Trm10 modifies G9 of to m1G (Swinehart et al. 2013), but a second modification, m2G (Smith et al. 1973), is introduced by a second unknown enzyme. We have studied the effect of lack of Trm10 on this tRNA because its lack was shown to substantially reduce m1G modification (Swinehart et al. 2013). Only Dus4 targets neither tRNA, providing a control for the specificity of the effect.
Our assay of misreading of UAG by tRNALys showed that nonsense readthrough errors are generally less frequent in the absence of individual core modifications (Fig. 3A; Supplemental Table S2). Lack of any of the four modifications that target only one of the two Lys isoacceptors significantly reduced activity, suggesting that both tRNAs contribute to mistranslation errors at UAG codons. Lack of four enzymes targeting both tRNAs (Dus1, Dus2, Dus3, and Pus1) caused a similar significant decrease in activity. The magnitude of the effect varied from 2.0- to 4.6-fold and averaged 2.8-fold. These are highly physiologically relevant increases; by comparison, in a genetic background carrying a characterized yeast ribosomal inaccuracy mutation, RPS23A-K62R (Alksne et al. 1993), errors at UAG by tRNALys increased only 40% and increased an average of 1.9-fold on the four most error-prone codons (Joshi et al. 2018). The enzyme activity in the absence of Dus4 was identical to the wild type, consistent with its failure to modify either tRNA. We observed a genetic interaction between strains lacking Trm4 and Trm8 in that lacking either significantly decreased activity, but the double mutant had activity indistinguishable from the congenic wild type. It may be significant that these two enzymes modify residues 46 and 48 of the extra loop of , implying a functional interaction between the two modifications in this region.
FIGURE 3.

Effect of lack of modification on misreading errors by tRNALys. The activity of the P. pyralis (firefly) luciferase with each of three error-prone codon mutants of the Lys529 codon; P. pyralis luciferase activity is calculated relative to a R. reniformis luciferase control. (A) Errors at the UAG nonsense codon, (B) errors at the AGG Arg codon, (C) errors at the AAU or AAC Asn codons. Activities, in relative light units, are shown for the congenic wild type and all modification mutations affecting at least one of the tRNAs involved in misreading. The presence (+) or absence (−) of each modification is indicated below each graph. Inset images above the graphs show the predicted codon–anticodon pairing of the mRNA codon (above) and misreading tRNA anticodon (below). The pairing symbols indicate the type of nucleotide pair formed: “|”, Watson–Crick pair; “•”, standard wobble; “°”, Watson–Crick mimic mispair. Error bars are the standard error of the mean. Asterisks indicate the probability of lack of a difference from the wild-type control-based calculated as described in Materials and Methods, (*) P < 0.05; (**) P < 0.01; (***) P < 0.001.
Overall, the conclusion of this experiment is that the presence of the core modifications of either tRNALys isoacceptor improve the decoding efficiency of the error-prone tRNA, allowing it to compete with RF to decode the nonsense codon, resulting in increased misreading of the nonsense codon in the wild-type strain.
Presence of core modifications tends to decrease translational errors by tRNALys at sense codons
Most misreading errors occur at sense codons and the frequency of those errors may be influenced by changes in the decoding efficiency of either the cognate or error-prone near-cognate tRNA. To test the effect of loss of modification enzymes on misreading at sense codons, we measured misreading frequency by tRNALys at the error-prone sense codons AGG, AAU, and AAC. In S. cerevisiae, the Arg codon AGG is decoded by , a low abundance tRNA encoded by a single structural gene, and the Asn codons AAU and AAC are decoded by , a high abundance tRNA encoded by 10 structural genes (Chan and Lowe 2016). We determined the frequency of errors at these three codons in the wild type and the same 14 congenic modification mutant backgrounds tested above (Fig. 3B,C; Supplemental Table S2).
The striking difference between these data and those for readthrough of the UAG nonsense codon is that in general, the loss of individual modifications tends to increase translational misreading frequency. For misreading errors by tRNALys at the Arg codon AGG, error frequencies are significantly increased in five of the 14 mutant backgrounds. The effect on errors at the synonymous AAU and AAC codons are similar in all 14 mutant backgrounds, consistent with decoding by the same isoacceptor, and errors are significantly increased in five (for AAU) or four (for AAC) of the 14 backgrounds. The absence of a modification enzyme in 10 of the 14 cases caused a significant increase in misreading of at least one of the three sense codons tested. The increase in enzyme activity varied from 1.4- to 3.0-fold and averaged 1.9-fold. The error frequency at these sense codons was slightly reduced in only several backgrounds, most notably for trm4Δ trm8Δ (averaging 1.3-fold), but these effects were not statistically significant. However, again the lack of both Trm4 and Trm8 reversed the significant 1.5-fold increase in errors at AGG in the absence of Trm4 alone, consistent with a genetic interaction between the modifications located close together in the extra loop.
The data on readthrough of UAG demonstrate clearly that absence of core modifications generally reduced near-cognate decoding efficiency by tRNALys. The data for the three sense codons in contrast showed a general trend toward increased misreading, which might seem contradictory. However, the frequency of misreading in a mutant depends on how much, if at all, the lack of a modification alters the intrinsic decoding efficiency of the cognate or the error-prone near-cognate tRNA on the codon being tested. If the relative activity results in an increase in the efficiency of near-cognate decoding relative to cognate, then misreading errors would increase. If the efficiency of cognate decoding relative to near-cognate increases, errors should decrease. Our finding a general effect of increasing errors on AGG, AAU, and AAC in this error reporter system suggests that in general the decoding efficiency of and/or increases relative to the efficiency of at the AGG Arg codon and at the AAU/AAC Asn codons. The competition between cognate and near-cognate decoding could be different at other hypothetical error-prone codons. The important point is that the core modifications do play a role in the result of that competition in terms of error frequency.
Core modification either limits or promotes misreading by tRNAGlu
We extended these results by testing the effect of core modifications on misreading by tRNAGlu using an error reporter system based on an active site amino acid of E. coli β-galactosidase, Glu 537 (Manickam et al. 2016). In S. cerevisiae, GAA/GAG decoding is encoded by 14 genes and GAG decoding is encoded by two (Chan and Lowe 2016). The isoacceptor is therefore likely responsible for most decoding of GAA/GAG. tRNAGlu misreads the Asp codons GAU/GAC in competition with , a high abundance tRNA encoded by 16 structural genes, and misreads the Gly codons GGA/GGG in competition with and , low abundance tRNAs encoded by three and two structural genes, respectively. The result of this analysis was very different than for tRNALys. The frequency of misreading errors by at these four error-prone codons varied widely among the modification mutant strains (Fig. 4; Supplemental Table S3), and rather than having a consistent effect, core modifications of tRNAGlu either increase or decrease accuracy depending on which codons are misread. We tested the effect of mutations that eliminate each of the eleven enzymes that target or at least one of the cognate tRNAs for GGA, GGG, GAU, and GAC (Fig. 2). Errors by were significantly different from in the congenic wild-type strain in seven of the eleven modification mutant strains tested; only the mutant strains lacking Trm4, Trm10, Trm11, or Trm13 showed no effect. Significantly, for each modification mutant strain showing a significant difference from wild type, the difference was always in the same direction; in no case did lack of a particular modification result in opposite effects based on codon misread. Lack of four enzymes (Dus1, Dus4, Pus1, and Trm2) caused increased misreading varying from 1.4- to 4.4-fold and averaging 2.2-fold. The lack of three others (Dus2, Pus4, Pus7) caused decreased misreading varying from 1.4- to 4.0-fold and averaging 2.0-fold. Lack of five enzymes showed strongly significant and consistent effects on at least three of the four codons tested (Dus4, Pus1, Pus4, Pus7, and Trm2), all of which modify but in six of 15 cases do not modify the competing cognate tRNA.
FIGURE 4.

Effect of lack of modification on misreading errors by tRNAGlu shown as in Figure 3. (A) Errors at the GGA Gly codon, (B) errors at the GGG Gly codon, (C) errors at the GAU or GAC Asp codons.
Changes in aminoacyl-tRNA abundance do not explain the error phenotype of lack of core modification
Previous work on core modification has focused on their effect on tRNA stability and steady-state tRNA abundance. For example, in trm4Δ trm8Δ double mutant strains lacking both m7G46 and m5C several mature tRNAs are rapidly deacylated and degraded by the rapid tRNA decay (RTD) pathway (Alexandrov et al. 2006; Chernyakov et al. 2008). Degradation is accelerated at 37°C, above the optimal growth temperature of 30°C. Selective destabilizing of normally modified tRNAs would reduce their availability for translation, shifting the competition between cognate and near-cognate tRNAs at an error-prone codon. Depending on the relative effect on cognate and near-cognate tRNAs, the result could either increase or decrease misreading. Our misreading assays were performed at 28°C where hypomodified tRNAs are minimally subject to RTD, if at all. This implies that instability might be an unlikely explanation of the accuracy effect. However, studies on instability of hypomodified tRNAs have not been performed on the tRNAs addressed in this work, so it is conceivable that they are destabilized even at this lower temperature. To determine if tRNA depletion by RTD is the cause of the changes in misreading we have identified, we measured the steady-state levels of relevant tRNAs. We focused this analysis on five mutant strains that showed highly significant effects on misreading efficiency, including those lacking Pus1, Trm2, Trm4, Trm8, and Trm4/Trm8. Pus1 and Trm2 demonstrated the highest level of misreading at GAU/GAC and GGA, respectively. The lack of the enzymes Trm4 and Trm8 showed a striking genetic interaction where lack of either enzyme caused 2.8- and 3.4-fold reductions in errors by tRNALys on UAG, but in the absence of both enzymes the error frequency was indistinguishable from the wild type. With this level of effect, we expected to easily visualize any putative change in tRNA stability responsible.
The relevant tRNAs for these studies include three potential misreading tRNAs (, , and ) and six competing cognate tRNAs (, , , , and ). We obtained eight single-stranded 5′ biotin-conjugated DNA probes specific to these tRNAs suitable for visualization by a chemiluminescent detection system. The probes are complementary to the region from position 32 to 58 of the mature tRNAs. To determine the specificity of the probes, we performed Southern blotting against a combination of all nine full-length DNA copies of the mature tRNAs (Supplemental Fig. S1), finding that all probes strongly recognized the corresponding sequence with little or no cross reaction with other targets.
Triplicate biological replicates of total RNA from the five modification mutant strains were separated by denaturing polyacrylamide gel electrophoresis, blotted to positively charged nylon membranes (Amersham) and hybridized to biotin-labeled probes specific for the nine tRNAs and 5S rRNA as a loading control. The hybridized probes were visualized as described in Materials and Methods. As shown in Figure 5 (and quantified in Supplemental Table S4) there was no significant variation in steady-state amounts of any of the tRNAs after adjusting for loading differences using the 5S rRNA control signal. We conclude that mRNA stability changes cannot explain the large effect of hypomodification on misreading frequencies in these mutant strains.
FIGURE 5.
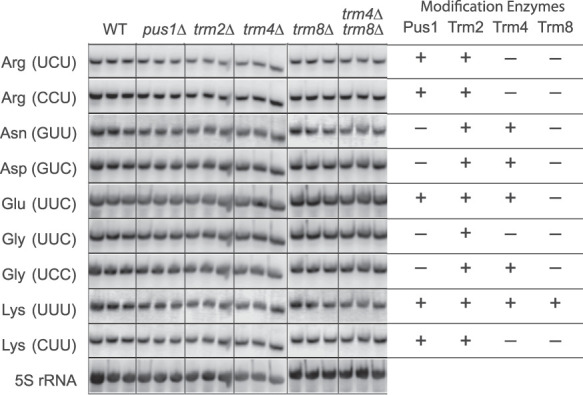
Northern blotting to determine relative tRNA concentration for all tRNAs implicated in errors tested in the wild type, and five mutant backgrounds showing highly significant error frequencies. Each column, labeled by strain, shows the results of RNA preparations in biological triplicate and successively probed with oligonucleotides specific to the tRNAs identified on the left by amino acid and anticodon. The last line represents the result of probing with an oligonucleotide specific to the 5S rRNA as a loading control.
Another possible explanation for the effect of hypomodification on misreading frequency is reduced aminoacylation of the hypomodified tRNA. Previous work has shown that the lack of the combination of Trm4 and Trm8 modification enzymes, which results in loss of m5C and m2G from positions in the extra loop, caused both a significant reduction in aminoacylation of at the permissive temperature of 28°C and a large and rapid loss of aminoacylation when shifted to 37°C (Alexandrov et al. 2006). However, this effect was specific to and no similar effects have been demonstrated for other tRNAs modified by both Trm4 and Trm8. We have extended that analysis to modification by Pus1 and Trm2 and have repeated the analysis of the effect of Trm4, Trm8 or both.
Total RNA purified in acid conditions, which maintains the tRNA acyl bond (Chernyakov et al. 2008), were separated by acidic acrylamide gel electrophoresis, blotted, and probed as described above. An aliquot of each acidic RNA was deacylated by incubation at pH 9.0, which breaks the acyl bond (Chernyakov et al. 2008). As shown in Figure 6, deacyl-tRNA runs slightly ahead of aminoacyl-tRNA providing a marker for the location of deacyl-tRNA in the acid purified RNAs. In these experiments, there was no evidence of deacyl-tRNA for any of the nine tRNAs tested either in the wild type or in the absence of any of the modification enzymes tested. The clearest results are with and , but the separation of acyl and deacyl species is less clear for , but even in this case there is no evidence for a band in the untreated sample corresponding to the deacyl-tRNA in the treated sample. This experiment confirms previous experiments showing no loss of aminoacyl- in the absence of Trm4 and Trm8 (Alexandrov et al. 2006). This result suggests that the changes in error frequency in the absence of Pus1, Trm2, Trm4, and Trm8 also does not appear to result from loss of aminoacylation of any of the tRNAs involved in the misreading studied here.
FIGURE 6.
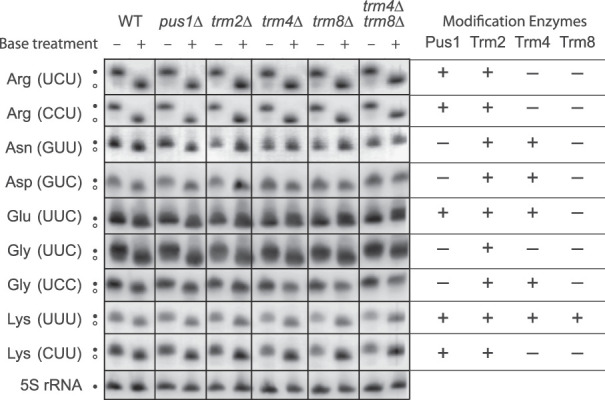
Northern analysis of RNA preparations made in acidic conditions to preserve aminoacyl linkages to tRNAs. Columns labeled “+” show separation of RNA treated with base to remove aminoacyl groups; those labeled “−” show separation of acidic RNA aminoacyl groups intact. Aminoacyl-tRNAs in the “−” columns are shifted upward relative to deacyl-tRNAs in the “+” column. Rows are identified as in Figure 5, and the modification enzymes that target each tRNA in the wild-type strain are indicated on the right (“+”, modified; “−” unmodified.)
A third way that aa-tRNA availability could be altered by modification is if hypomodified tRNAs were to become sequestered away from the translational machinery, by retrograde transport to the nucleus (Shaheen and Hopper 2005; Takano et al. 2005). Aberrant tRNAs are imported into the nucleus by a process dependent on the Mtr10 protein, a β-importin nuclear import receptor, where they are subject to degradation (Shaheen and Hopper 2005). Kramer and Hopper (2013) showed that hypomodified tRNAs, including , accumulate in an mtr10Δ mutant background. Despite our showing that there is no change to steady-state levels of hypomodified tRNAs in the modification mutant backgrounds, it could be that they undergo retrograde transport but evade degradation. This could lead to a reduction in cytoplasmic aa-tRNAs that could explain the misreading effects. Under this hypothesis, there would be no change in tRNA amounts, just a change in subcellular location. To test this idea, therefore, we introduced an mtr10Δ mutation into the five strongly affected mutant strains and repeated the reporter assays. Figure 7 (and as quantified in Supplemental Table S5) shows that there is no significant effect of the mtr10Δ mutation on any of the strains tested, showing that retrograde transport to the nucleus does not cause aa-tRNAs to be sequestered away from the translational machinery.
FIGURE 7.

Effect of lack of Mtr10 β-importin nuclear import receptor on errors in wild type and five mutant backgrounds showing highly significant error frequencies. Activities shown for modification mutant strains, labeled on the x-axis, expressing (“MTR10+”) or lacking Mtr10 (“mtr10Δ”). None of the differences between matched strains with and without Mtr10 were significant.
These data eliminate the hypotheses that availability of aa-tRNAs is reduced in the modification mutants tested either by degradation of hypomodified tRNAs, failure to fully aminoacylate them, or sequestration in the nucleus away from the translation machinery. The remaining hypothesis to explain the effect of hypomodification is that tRNA recognition of error-prone codons by cognate or near-cognate tRNAs is reduced by hypomodification leading to changes in competition for misreading error frequency.
DISCUSSION
Posttranscriptional modifications of tRNAs outside the anticodon loop are concentrated in the structural core (Fig. 1), many of them directly altering nucleotides involved in tertiary interactions (Motorin and Helm 2010; Phizicky and Alfonzo 2010). tRNA modifications, including those to the structural core, are generally nonessential despite their deep evolutionary conservation (e.g., Pichard-Kostuch et al. 2023). Of the 62 S. cerevisiae genes encoding proteins required for nucleotide modification (Phizicky and Hopper 2010), however, five are essential (Anderson et al. 1998; Gerber and Keller 1999; Gu et al. 2003); loss of five causes slow growth (Carbone et al. 1991; Bjork et al. 2001; Pintard et al. 2002; El Yacoubi et al. 2009) or temperature-sensitive growth (Johansson and Bystrom 2004). Arguably, the fact that so many genes are nonessential might suggest that the function of individual modifications is relatively unimportant during growth under optimal conditions. The fact that lack of some combinations of modification genes is synthetically lethal (Tong et al. 2001; Purushothaman et al. 2005) suggests that modifications may play redundant essential roles. The finding that tRNAs lacking core modifications can be subject to rapid tRNA degradation (RTD) suggested that their essential role might be to stabilize tRNAs against degradation (Alexandrov et al. 2006). However, high levels of RTD for most singly hypomodified tRNAs occur at temperatures elevated beyond normal yeast growth conditions. We speculated that some other role might help explain the durable conservation of these modifications.
Here, we show that maintenance of translational accuracy is a biologically relevant effect of core modification that could help explain their evolutionary conservation. We found that the lack of any of the core modification enzymes, except for Trm13, altered accuracy of tRNALys or tRNAGlu on at least one error-prone codon. Lack of some modifications affected errors on nearly all such codons and highly significantly. Errors by tRNALys were affected on at least one codon by lack of every enzyme and for tRNAGlu on all but Trm4, Trm10, Trm11, and Trm13. Lack of modification consistently decreased errors by tRNALys at the UAG nonsense codon but tended to increase errors on the three sense codons, AGG, AAU, and AAC, although often affecting only a subset of these codons. For example, Dus2 (D20) modifies tRNALys and the three competing cognate tRNAs, but its absence increases errors on AGG but not AAU or AAC. In contrast, lack of modification of tRNAGlu or of its competing cognate tRNAs either increased or decreased misreading, but for each modification the effect was consistent across all codons affected. For example, lack of Pus4 (Ψ55) resulted in a highly significant decrease in misreading on all four error-prone codons, but lack of Pus1 (Ψ27, Ψ28) resulted in a highly significant increase in all misreading errors. These data suggest that the presence of modifications of tRNALys tended to increase its near-cognate decoding to a greater extent than that of the competing cognate tRNAs but the modifications of tRNAGlu inconsistently affected this competition.
The difference between tRNALys misreading of nonsense and sense codons suggests that though the modifications stabilize tRNALys near-cognate decoding, as shown by the UAG data, they stabilize cognate decoding by the competing tRNAs more strongly. The AGG-decoding tRNAArg is extremely low abundance, which might suggest the need to increase the stability of its association with its codon. The AAU/AAC decoding tRNAAsn like tRNALys is a particularly weak tRNA, requiring adjacent A•U codon–anticodon pairs, again suggesting the need to stabilize cognate decoding. Because of the differential effect on cognate and near-cognate tRNAs, the effect is a measurable increase in errors as the bias against near-cognate decoding is reduced by hypomodification.
The codon specificity of the lack of modification on errors raises the question of the mechanistic origin of the effect. What type of mechanism could explain that specificity? We tested three mechanisms, all of which could differentially affect hypomodified cognate and near-cognate tRNAs: tRNA degradation, altered aminoacylation efficiency and changes in subcellular localization by retrograde transport of the hypomodified tRNAs to the nucleus. We tested these models either biochemically or genetically and found no evidence supporting any of the mechanisms. Lack of modifications are known to induce rapid tRNA decay at 37°C, consistent with the reduction of tRNA thermal stability (Alexandrov et al. 2006). Consistent with this result, our northern analysis showed unchanged steady-state levels at 28°C for all the tRNAs relevant to hypomodification-induced misreading errors, arguing against errors being modulated by tRNA degradation. In the absence of degradation, the availability of aminoacyl-tRNA could vary if hypomodification interfered with aminoacylation. Aminoacylation of some hypomodified tRNAs does decline at 37°C (Chernyakov et al. 2008); however, as with tRNA instability, we showed that in all modification mutants all tRNAs tested were fully aminoacylated at 28°C, arguing against hypomodification explaining our misreading results. The cell has multiple quality control mechanisms to ensure only mature tRNAs are available for cytoplasmic protein synthesis. Retrograde transport of 5′ and 3′ extended tRNAs or hypomodified tRNAs could target tRNAs lacking modifications to return to the nucleus and if the defect cannot be corrected, sequester them there. An mtr10Δ mutation that inactivates the receptor required for retrograde transport would reverse sequestration. We found mtr10Δ had no effect in misreading modification mutant backgrounds, so retrograde transport also cannot explain our misreading results.
The remaining hypothesis is that the lack of core modifications directly alters the decoding efficiency of the tRNAs. Anticodon loop modifications (Hagervall et al. 1998; Manickam et al. 2016; Joshi et al. 2018) either decrease or increase decoding activity of the tRNA depending on the details of the codon•anticodon interaction. Core modifications, though they do not directly affect the codon•anticodon complex, might do so as well. Loss of core modifications reduced misreading of the UAG nonsense codon, suggesting a general reduction in decoding efficiency. We have no equivalent readthrough assay for since UAA or UAG mutants of our reporter system shows background activity (Joshi et al. 2018). We can, however, assess hypomodification effects on near-cognate decoding by where the competing cognate tRNA is not targeted for modification. This is true for two modifications, Pus1 (Ψ27) and Dus4 (D20a). Pus1 modifies but none of the competing cognates. Lack of Ψ27 significantly increases errors by on each of its four error-prone codons, suggesting that Ψ27 reduces near-cognate decoding efficiency. Similarly, the absence of Dus4 resulted in significantly increased misreading of three codons decoded by tRNAs lacking D20a (GAU, GAC, and GGG) but had no effect on GGA, decoded by D20a-containing . This again suggests that D20a reduces near-cognate decoding efficiency on the three affected codons.
Core modifications are known to improve tRNA thermal stability and stabilize the tertiary fold required for the tRNA L-shape structure of tRNA (for reviews, see Motorin and Helm 2010; Phizicky and Alfonzo 2010; Lorenz et al. 2017). Core hypomodification leading to RTD at elevated temperatures presumably results from loss of these stabilizing effects of core modifications. Significantly, modifications that modulate misreading errors also affect stability and tertiary folding. For example, the presence of each of the error modulating modifications D20A, T54, and Ψ55 increases the melting temperatures of E. coli (Nomura et al. 2016). Formation of the L-shape involves tertiary interactions between the two halves of the molecule, the D and T hairpins (for reviews, see Motorin and Helm 2010; Phizicky and Alfonzo 2010; Lorenz et al. 2017). In vitro interaction assay between synthetic tRNA half molecules showed that singly the core modifications T54, Ψ55, and 5mC49, all located in the T hairpin, increased the biomolecular affinity constant for interaction between the two halves (Nobles et al. 2002). The effect of T54 may reflect the formation of the T54•m1A58 pair internal to the T loop whereas the effect of Ψ55 is longer range, as part of the two stabilizing base pairs (Ψ55•G18, C56•G19) between the T and D loops.
Uhlenbeck and Schrader (2018) have proposed that tRNAs have been “tuned” by evolution to minimize differences in the efficiency of their use by the translation machinery. They made this comment in the context of design of tRNAs to insert unnatural amino acids, but the insight is very relevant to the issue of translational accuracy. They noted that tRNAs differ in two important ways. First, each tRNA carries a different amino acid and differences in its physical nature can affect, for example, the affinity of the aminoacyl-tRNA with the elongation factor EF-Tu (EF-1A in eukaryotes) (Dale et al. 2004). Below optimal affinities based on amino acid identity cause reduced translational efficiency. Especially weaker complexes are recruited to the A site more slowly, and for stronger complexes dissociation of the EF-Tu after GTP hydrolysis is slower, blocking rapid peptide transfer (Schrader et al. 2011). Importantly for this work, because there is little modification of the EF-1A interaction region, tRNA modifications have little effect on the affinity of aminoacyl-tRNAs for EF-Tu (Asahara and Uhlenbeck 2005).
A study of tRNA mutant forms of E. coli identified many that minimized misreading of near-cognate codons (Shepotinovskaya and Uhlenbeck 2013). These hyperaccurate tRNAs result from changes in the core of the tRNA where modifications are concentrated. A canonical error-prone mutation, the G24A “Hirsh suppressor” of E. coli (Hirsh 1971; Cochella and Green 2005) also modifies the core region. Uhlenbeck and Schrader (2018) argue that the structure of the core, including its modifications, have coevolved with the anticodon to “tune” tRNAs with strong or weak anticodons to achieve a consistent efficiency of decoding. Under this model, increased or decreased errors result from shifting decoding efficiency away from that optimum.
Mutations affecting tRNA structural genes or tRNA modification enzymes have been identified as responsible for a wide range of human diseases (Tahmasebi et al. 2018; Suzuki 2021; Orellana et al. 2022). These are a subset of a larger group of diseases resulting from mutations affecting a variety of targets in the translational machinery including ribosomes, translation factors, aminoacyl-tRNA synthetases, translation elongation factors, the translation-associated integrated stress response, and the TORC1 kinase system, a global regulator of the translation machinery (Mills and Green 2017; Tahmasebi et al. 2018). A class of human diseases characterized by protein aggregation and neurodegeneration result from a loss of proteostasis, a network of factors that control protein synthesis, folding and degradation to maintain cellular health (Klaips et al. 2018). Our work here suggests that hypomodification-induced misreading results from a failure of the tuning that ensures consistent rates of decoding by all tRNAs.
MATERIALS AND METHODS
Strains, media, and misreading reporter assays
Yeast cells were grown on rich YPD medium or, to maintain transforming plasmids, on drop-out synthetic complete medium lacking uracil (USBiological). All experiments were performed on members of a set of S. cerevisiae congenic strains (Winston et al. 1995; Brachmann et al. 1998); they are listed in Supplemental Table S6. Derivatives carrying various mutations eliminating tRNA modification enzymes were obtained from commercial sources or were generated by insertional mutagenesis using the drug resistance cassettes, KanMX (Wach et al. 1994), NatMX, HphMX, or BleMX (Lorenz 2015). To avoid unwanted selection of external suppressors, mutants were obtained or created as heterozygous diploids from which appropriate haploids were derived by sporulation and dissection as described (Guthrie and Fink 1991). Accuracy reporter plasmids were used in this study to measure misreading error frequency by tRNALys, using the previously described dual luciferase reporter, pDB688 (Salas-Marco and Bedwell 2005), which comprises genes encoding Photinus pyralis (firefly) luciferase (Fluc) expressed as a translational fusion to downstream Renilla reniformis luciferase (Rluc) (Kramer et al. 2010), or misreading errors by tRNAGlu using the Escherichia coli lacZ gene encoding β-galactosidase from a derivative of the plasmid pMB38 (Belcourt and Farabaugh 1990), which expresses β-galactosidase from a fusion of lacZ to the amino-terminal 100 bp of the yeast HIS4 gene (Joshi et al. 2018). The generation of mutations altering codon Lys 529 of Fluc (Kramer et al. 2010) and Glu 537 of β-galactosidase (Manickam et al. 2014) has been reported. Transformation of yeast strains was performed as described (Gietz and Schiestl 2007), and from six to 18 independently transformed strains were assayed, each in triplicate technical replicates. The significance of differences in experimental results according to genetic background were determined by Student's t-test.
Northern blotting
Total RNA (nonaminoacylated) was purified from cells grown in YPD medium to midexponential phase as described (Chatterjee et al. 2017). To determine levels of aminoacylation, RNA was purified in acidic conditions, to preserve the acyl group, as described (Chernyakov et al. 2008). An amount of 20 µg of total nonaminoacylated RNA was separated by electrophoresis in 10% polyacrylamide gel (19:1 bis) containing 8 M urea and 1× TBE (0.9 M Tris-borate, 1 mM EDTA, pH 8.3. acidic RNA was separated on 6.5% polyacrylamide gel [19:1 bis] containing 8 M urea and 0.1 M NaOAc, pH 5 as described [Chernyakov et al. 2008]). After separation, the RNA was transferred to a Hybond N Membrane (Amersham Biosciences) in 1× TAE (40 mM Tris-acetate, 1 mM EDTA, pH 8.1, from a 50× stock) at 4°C in a Bio-Rad Trans-Blot apparatus for 3 h at 30 V. The RNA was then UV cross-linked to the membrane with 120,000 mjoules at 254 nm in a Stratagene UV cross-linker and probed as described (Wu et al. 2013) using digoxigenin-modified probes (Genewiz). The membrane was imaged using a C-DiGit Blot Scanner (LI-COR), and the signal intensities of various tRNA species were quantified using ImageJ software or by Image Studio (LI-COR) and normalized to the 5S RNA controls. To visualize multiple tRNAs from one preparation, the blot was stripped and reprobed, the last probe targeting the 5S control rRNA.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We would like to thank Dr. Eric Phizicky for the gift of modification deficient strains and for useful discussions of our results, and Mr. Grant Wunderlin for technical assistance. This work was supported by a grant from the National Science Foundation (grant numbers CBET 1805139 and MCB 1645795).
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079797.123.
MEET THE FIRST AUTHOR
Sima Saleh.

Meet the First Author(s) is an editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Sima Saleh is the first author of this paper, “Posttranscriptional modification to the core of tRNAs modulates translational misreading errors.” Sima is an Iranian-American scientist who returned to the U.S. to continue her education in molecular biology. She successfully completed her PhD in biology with a focus on tRNA posttranscriptional modifications from the University of Maryland Baltimore County (UMBC) in March 2023. She studied the role of tRNA core posttranscriptional modification on translational misreading errors.
What are the major results described in your paper and how do they impact this branch of the field?
tRNA body modifications are involved in translational accuracy of the tRNA. Most people think that the body modifications are just to stabilize the tRNA to degradation, but we show here that they are also involved in modulating translational errors. Lack of any of the core body modifications of tRNALys or tRNAGlu affects accuracy on at least one codon. Lack of modifications has codon-specific outcomes on the translation misreading errors and is not due to the tRNA degradation, decreased aminoacylation efficiency, or changes in the subcellular localization of tRNA. We suggest that these core body modifications maintain translational accuracy. These findings could be particularly valuable in the context of human disease research, especially concerning mutations in tRNA modification enzymes.
What led you to study RNA or this aspect of RNA science?
My passion for molecular biology and my interest in tRNA research align with the fascinating intricacies of this essential molecule in the protein synthesis process. tRNA has a unique structure, posttranscriptional modifications and interactions with ribosomes that make it a captivating subject of study. Furthermore, tRNA research can have practical applications in biotechnology and medicine. Advancement in the field of tRNA posttranscriptional modifications can lead to potential therapeutic developments.
If you were able to give one piece of advice to your younger self, what would that be?
I encourage everyone to “pursue perpetual learning.” The continuous expanding of knowledge, whether in the field of science or in new areas, keeps your mind active and equips you to adapt and thrive in an ever-changing world. The ongoing learning commitment not only fosters personal growth, but also empowers you to make meaningful contributions to your field.
Are there specific individuals or groups who have influenced your philosophy or approach to science?
My Principal Investigator, Dr. Philip Farabaugh, had a profound influence on my approach to science. Observing his work ethic, commitment to research, and perseverance in the face of challenges inspired dedication in my own scientific pursuit. Through discussion and feedback, he enhanced my critical thinking skills. Above all, Dr. Farabaugh instilled in me a love for science and a curiosity to explore new areas of research.
What are your subsequent near- or long-term career plans?
Currently, I am a postdoctoral research fellow at the Center for Biologics Evaluation and Research in the Food and Drug Administration (FDA). My near-term objective is to advance my research and contribute to scientific knowledge through publishing. But, I also hope to gain expertise in regulatory affairs and drug/biologics safety regulations. My long-term career plan is to either take a leadership role in the FDA where I can influence policy and regulatory decisions, or to transition to industry where I can leverage my regulatory expertise in pharmaceutical or food companies.
REFERENCES
- Alexandrov A, Chernyakov I, Gu W, Hiley SL, Hughes TR, Grayhack EJ, Phizicky EM. 2006. Rapid tRNA decay can result from lack of nonessential modifications. Mol Cell 21: 87–96. 10.1016/j.molcel.2005.10.036 [DOI] [PubMed] [Google Scholar]
- Alksne LE, Anthony RA, Liebman SW, Warner JR. 1993. An accuracy center in the ribosome conserved over 2 billion years. Proc Natl Acad Sci 90: 9538–9541. 10.1073/pnas.90.20.9538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson J, Phan L, Cuesta R, Carlson BA, Pak M, Asano K, Bjork GR, Tamame M, Hinnebusch AG. 1998. The essential Gcd10p–Gcd14p nuclear complex is required for 1-methyladenosine modification and maturation of initiator methionyl-tRNA. Genes Dev 12: 3650–3662. 10.1101/gad.12.23.3650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asahara H, Uhlenbeck OC. 2005. Predicting the binding affinities of misacylated tRNAs for Thermus thermophilus EF-Tu·GTP. Biochemistry 44: 11254–11261. 10.1021/bi050204y [DOI] [PubMed] [Google Scholar]
- Belcourt MF, Farabaugh PJ. 1990. Ribosomal frameshifting in the yeast retrotransposon Ty: tRNAs induce slippage on a 7 nucleotide minimal site. Cell 62: 339–352. 10.1016/0092-8674(90)90371-K [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjork GR, Jacobsson K, Nilsson K, Johansson MJ, Bystrom AS, Persson OP. 2001. A primordial tRNA modification required for the evolution of life? EMBO J 20: 231–239. 10.1093/emboj/20.1.231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachmann CB, Davies A, Cost GJ, Caputo E, Li J, Hieter P, Boeke JD. 1998. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14: 115–132. [DOI] [PubMed] [Google Scholar]
- Carbone ML, Solinas M, Sora S, Panzeri L. 1991. A gene tightly linked to CEN6 is important for growth of Saccharomyces cerevisiae. Curr Genet 19: 1–8. 10.1007/BF00362080 [DOI] [PubMed] [Google Scholar]
- Chan PP, Lowe TM. 2016. GtRNAdb 2.0: an expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res 44: D184–D189. 10.1093/nar/gkv1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee K, Majumder S, Wan Y, Shah V, Wu J, Huang HY, Hopper AK. 2017. Sharing the load: Mex67-Mtr2 cofunctions with Los1 in primary tRNA nuclear export. Genes Dev 31: 2186–2198. 10.1101/gad.305904.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernyakov I, Whipple JM, Kotelawala L, Grayhack EJ, Phizicky EM. 2008. Degradation of several hypomodified mature tRNA species in Saccharomyces cerevisiae is mediated by Met22 and the 5′-3′ exonucleases Rat1 and Xrn1. Genes Dev 22: 1369–1380. 10.1101/gad.1654308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochella L, Green R. 2005. An active role for tRNA in decoding beyond codon:anticodon pairing. Science 308: 1178–1180. 10.1126/science.1111408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale T, Sanderson LE, Uhlenbeck OC. 2004. The affinity of elongation factor Tu for an aminoacyl-tRNA is modulated by the esterified amino acid. Biochemistry 43: 6159–6166. 10.1021/bi036290o [DOI] [PubMed] [Google Scholar]
- D'Orazio KN, Green R. 2021. Ribosome states signal RNA quality control. Mol Cell 81: 1372–1383. 10.1016/j.molcel.2021.02.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond DA, Wilke CO. 2009. The evolutionary consequences of erroneous protein synthesis. Nat Rev Genet 10: 715–724. 10.1038/nrg2662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Yacoubi B, Lyons B, Cruz Y, Reddy R, Nordin B, Agnelli F, Williamson JR, Schimmel P, Swairjo MA, de Crecy-Lagard V. 2009. The universal YrdC/Sua5 family is required for the formation of threonylcarbamoyladenosine in tRNA. Nucleic Acids Res 37: 2894–2909. 10.1093/nar/gkp152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Yacoubi B, Hatin I, Deutsch C, Kahveci T, Rousset JP, Iwata-Reuyl D, Murzin AG, de Crecy-Lagard V. 2011. A role for the universal Kae1/Qri7/YgjD (COG0533) family in tRNA modification. EMBO J 30: 882–893. 10.1038/emboj.2010.363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber AP, Keller W. 1999. An adenosine deaminase that generates inosine at the wobble position of tRNAs. Science 286: 1146–1149. 10.1126/science.286.5442.1146 [DOI] [PubMed] [Google Scholar]
- Giege R, Juhling F, Putz J, Stadler P, Sauter C, Florentz C. 2012. Structure of transfer RNAs: similarity and variability. Wiley Interdiscip Rev RNA 3: 37–61. 10.1002/wrna.103 [DOI] [PubMed] [Google Scholar]
- Gietz RD, Schiestl RH. 2007. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc 2: 31–34. 10.1038/nprot.2007.13 [DOI] [PubMed] [Google Scholar]
- Gu W, Jackman JE, Lohan AJ, Gray MW, Phizicky EM. 2003. tRNAHis maturation: an essential yeast protein catalyzes addition of a guanine nucleotide to the 5′ end of tRNAHis. Genes Dev 17: 2889–2901. 10.1101/gad.1148603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie C, Fink G. 1991. Guide to yeast genetics and molecular biology. Academic Press, San Diego. [Google Scholar]
- Hagervall TG, Pomerantz SC, McCloskey JA. 1998. Reduced misreading of asparagine codons by Escherichia coli tRNALys with hypomodified derivatives of 5-methylaminomethyl-2-thiouridine in the wobble position. J Mol Biol 284: 33–42. 10.1006/jmbi.1998.2162 [DOI] [PubMed] [Google Scholar]
- Hirsh D. 1971. Tryptophan transfer RNA as the UGA suppressor. J Mol Biol 58: 439–458. 10.1016/0022-2836(71)90362-7 [DOI] [PubMed] [Google Scholar]
- Johansson MJ, Bystrom AS. 2004. The Saccharomyces cerevisiae TAN1 gene is required for N4-acetylcytidine formation in tRNA. RNA 10: 712–719. 10.1261/rna.5198204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi K, Bhatt MJ, Farabaugh PJ. 2018. Codon-specific effects of tRNA anticodon loop modifications on translational misreading errors in the yeast Saccharomyces cerevisiae. Nucleic Acids Res 46: 10331–10339. 10.1093/nar/gky664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaips CL, Jayaraj GG, Hartl FU. 2018. Pathways of cellular proteostasis in aging and disease. J Cell Biol 217: 51–63. 10.1083/jcb.201709072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer E, Farabaugh P. 2007. The frequency of translational misreading errors in E. coli is largely determined by tRNA competition. RNA 13: 87–96. 10.1261/rna.294907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer EB, Hopper AK. 2013. Retrograde transfer RNA nuclear import provides a new level of tRNA quality control in Saccharomyces cerevisiae. Proc Natl Acad Sci 110: 21042–21047. 10.1073/pnas.1316579110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer EB, Vallabhaneni H, Mayer LM, Farabaugh PJ. 2010. A comprehensive analysis of translational missense errors in the yeast Saccharomyces cerevisiae. RNA 16: 1797–1808. 10.1261/rna.2201210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamichhane TN, Blewett NH, Crawford AK, Cherkasova VA, Iben JR, Begley TJ, Farabaugh PJ, Maraia RJ. 2013. Lack of tRNA modification isopentenyl-A37 alters mRNA decoding and causes metabolic deficiencies in fission yeast. Mol Cell Biol 33: 2918–2929. 10.1128/MCB.00278-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz A. 2015. New cassettes for single-step drug resistance and prototrophic marker switching in fission yeast. Yeast 32: 703–710. 10.1002/yea.3097 [DOI] [PubMed] [Google Scholar]
- Lorenz C, Lunse CE, Morl M. 2017. tRNA modifications: impact on structure and thermal adaptation. Biomolecules 7: 35. 10.3390/biom7020035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manickam N, Nag N, Abbasi A, Patel K, Farabaugh PJ. 2014. Studies of translational misreading in vivo show that the ribosome very efficiently discriminates against most potential errors. RNA 20: 9–15. 10.1261/rna.039792.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manickam N, Joshi K, Bhatt MJ, Farabaugh PJ. 2016. Effects of tRNA modification on translational accuracy depend on intrinsic codon-anticodon strength. Nucleic Acids Res 44: 1871–1881. 10.1093/nar/gkv1506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills EW, Green R. 2017. Ribosomopathies: there's strength in numbers. Science 358: eaan2755. 10.1126/science.aan2755 [DOI] [PubMed] [Google Scholar]
- Mohler K, Ibba M. 2017. Translational fidelity and mistranslation in the cellular response to stress. Nat Microbiol 2: 17117. 10.1038/nmicrobiol.2017.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motorin Y, Helm M. 2010. tRNA stabilization by modified nucleotides. Biochemistry 49: 4934–4944. 10.1021/bi100408z [DOI] [PubMed] [Google Scholar]
- Nobles KN, Yarian CS, Liu G, Guenther RH, Agris PF. 2002. Highly conserved modified nucleosides influence Mg2+-dependent tRNA folding. Nucleic Acids Res 30: 4751–4760. 10.1093/nar/gkf595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura Y, Ohno S, Nishikawa K, Yokogawa T. 2016. Correlation between the stability of tRNA tertiary structure and the catalytic efficiency of a tRNA-modifying enzyme, archaeal tRNA-guanine transglycosylase. Genes Cells 21: 41–52. 10.1111/gtc.12317 [DOI] [PubMed] [Google Scholar]
- Orellana EA, Siegal E, Gregory RI. 2022. tRNA dysregulation and disease. Nat Rev Genet 23: 651–664. 10.1038/s41576-022-00501-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percudani R, Pavesi A, Ottonello S. 1997. Transfer RNA gene redundancy and translational selection in Saccharomyces cerevisiae. J Mol Biol 268: 322–330. 10.1006/jmbi.1997.0942 [DOI] [PubMed] [Google Scholar]
- Phizicky EM, Alfonzo JD. 2010. Do all modifications benefit all tRNAs? FEBS Lett 584: 265–271. 10.1016/j.febslet.2009.11.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phizicky EM, Hopper AK. 2010. tRNA biology charges to the front. Genes Dev 24: 1832–1860. 10.1101/gad.1956510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichard-Kostuch A, Da Cunha V, Oberto J, Sauguet L, Basta T. 2023. The universal Sua5/TsaC family evolved different mechanisms for the synthesis of a key tRNA modification. Front Microbiol 14: 1204045. 10.3389/fmicb.2023.1204045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pintard L, Lecointe F, Bujnicki JM, Bonnerot C, Grosjean H, Lapeyre B. 2002. Trm7p catalyses the formation of two 2′-O-methylriboses in yeast tRNA anticodon loop. EMBO J 21: 1811–1820. 10.1093/emboj/21.7.1811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purushothaman SK, Bujnicki JM, Grosjean H, Lapeyre B. 2005. Trm11p and Trm112p are both required for the formation of 2-methylguanosine at position 10 in yeast tRNA. Mol Cell Biol 25: 4359–4370. 10.1128/MCB.25.11.4359-4370.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas de Pouplana L, Santos MA, Zhu JH, Farabaugh PJ, Javid B. 2014. Protein mistranslation: friend or foe? Trends Biochem Sci 39: 355–362. 10.1016/j.tibs.2014.06.002 [DOI] [PubMed] [Google Scholar]
- Rodnina MV, Fischer N, Maracci C, Stark H. 2017. Ribosome dynamics during decoding. Philos Trans R Soc Lond B Biol Sci 372: 20160182. 10.1098/rstb.2016.0182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salas-Marco J, Bedwell DM. 2005. Discrimination between defects in elongation fidelity and termination efficiency provides mechanistic insights into translational readthrough. J Mol Biol 348: 801–815. 10.1016/j.jmb.2005.03.025 [DOI] [PubMed] [Google Scholar]
- Schrader JM, Chapman SJ, Uhlenbeck OC. 2011. Tuning the affinity of aminoacyl-tRNA to elongation factor Tu for optimal decoding. Proc Natl Acad Sci 108: 5215–5220. 10.1073/pnas.1102128108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaheen HH, Hopper AK. 2005. Retrograde movement of tRNAs from the cytoplasm to the nucleus in Saccharomyces cerevisiae. Proc Natl Acad Sci 102: 11290–11295. 10.1073/pnas.0503836102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepotinovskaya I, Uhlenbeck OC. 2013. tRNA residues evolved to promote translational accuracy. RNA 19: 510–516. 10.1261/rna.036038.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CJ, Teh HS, Ley AN, D'Obrenan P. 1973. The nucleotide sequences and coding properties of the major and minor lysine transfer ribonucleic acids from the haploid yeast Saccharomyces cerevisiae S288C. J Biol Chem 248: 4475–4485. 10.1016/S0021-9258(19)43792-7 [DOI] [PubMed] [Google Scholar]
- Steiner RE, Ibba M. 2019. Regulation of tRNA-dependent translational quality control. IUBMB Life 71: 1150–1157. 10.1002/iub.2080 [DOI] [PubMed] [Google Scholar]
- Suzuki T. 2021. The expanding world of tRNA modifications and their disease relevance. Nat Rev Mol Cell Biol 22: 375–392. 10.1038/s41580-021-00342-0 [DOI] [PubMed] [Google Scholar]
- Swinehart WE, Henderson JC, Jackman JE. 2013. Unexpected expansion of tRNA substrate recognition by the yeast m1G9 methyltransferase Trm10. RNA 19: 1137–1146. 10.1261/rna.039651.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahmasebi S, Khoutorsky A, Mathews MB, Sonenberg N. 2018. Translation deregulation in human disease. Nat Rev Mol Cell Biol 19: 791–807. 10.1038/s41580-018-0034-x [DOI] [PubMed] [Google Scholar]
- Takano A, Endo T, Yoshihisa T. 2005. tRNA actively shuttles between the nucleus and cytosol in yeast. Science 309: 140–142. 10.1126/science.1113346 [DOI] [PubMed] [Google Scholar]
- Tong AH, Evangelista M, Parsons AB, Xu H, Bader GD, Page N, Robinson M, Raghibizadeh S, Hogue CW, Bussey H, et al. 2001. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 294: 2364–2368. 10.1126/science.1065810 [DOI] [PubMed] [Google Scholar]
- Uhlenbeck OC, Schrader JM. 2018. Evolutionary tuning impacts the design of bacterial tRNAs for the incorporation of unnatural amino acids by ribosomes. Curr Opin Chem Biol 46: 138–145. 10.1016/j.cbpa.2018.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wach A, Brachat A, Pohlmann R, Philippsen P. 1994. New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast 10: 1793–1808. 10.1002/yea.320101310 [DOI] [PubMed] [Google Scholar]
- Winston F, Dollard C, Ricupero-Hovasse SL. 1995. Construction of a set of convenient Saccharomyces cerevisiae strains that are isogenic to S288C. Yeast 11: 53–55. 10.1002/yea.320110107 [DOI] [PubMed] [Google Scholar]
- Wohlgemuth I, Pohl C, Mittelstaet J, Konevega AL, Rodnina MV. 2011. Evolutionary optimization of speed and accuracy of decoding on the ribosome. Philos Trans R Soc Lond B Biol Sci 366: 2979–2986. 10.1098/rstb.2011.0138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Huang HY, Hopper AK. 2013. A rapid and sensitive nonradioactive method applicable for genome-wide analysis of Saccharomyces cerevisiae genes involved in small RNA biology. Yeast 30: 119–128. 10.1002/yea.2947 [DOI] [PMC free article] [PubMed] [Google Scholar]