

Abstract
Background:
To identify periods of heightened susceptibility to the association of secondhand tobacco smoke (SHS) exposure with cardiometabolic (CM) risk at age 12 years.
Methods:
We used data from 212 adolescents from the HOME Study, a prospective pregnancy and birth cohort in Cincinnati, OH. Using multiple informant models, we estimated associations of maternal serum cotinine (mean of concentrations at 16 and 26 weeks of pregnancy) and children’s serum cotinine concentrations (mean of concentrations at ages 1, 2, 3, and 4 years) with a CM risk summary score constructed of five risk components measured at age 12 years. We determined if these associations differed for pre- and postnatal exposure periods, and adolescent’s sex.
Results:
We found some evidence that the cotinine-outcome associations differed by exposure period and sex. Postnatal, but not prenatal, cotinine was associated with higher CM risk scores and individual CM risk component values (interaction p-values = 0.04 to 0.35). Each 10-fold increase in postnatal cotinine was associated with 0.57 (95% CI: 0.32, 1.45), 0.09 (95% CI: 0.13, 0.31), 0.14 (−0.08, 0.35), 0.07 (95% CI: 0.34, 0.48), and 0.11 (95% CI: 0.04, 0.27) higher CM risk, HOMA-IR, TG to HDL-C ratio, leptin to adiponectin ratio, and visceral fat area. Postnatal cotinine was associated with higher visceral fat area among females but not males (sex × period × cotinine interaction p-value = 0.01).
Conclusions:
Serum cotinine concentrations during the postnatal period had greater influence on adolescent’s CM risk compared to the prenatal period, and these associations may be sex-specific. This study reinforces the need for ongoing public health interventions to minimize children’s exposure to SHS.
Keywords: Secondhand tobacco smoke, Cotinine, Adolescents, Cardiometabolic risk factors
1. Introduction
Cardiovascular disease (CVD) is the leading cause of death and disability worldwide. In 2018, CVD accounted for approximately 31% of all recorded deaths in the USA (Virani et al., 2021). Tobacco smoke exposure, including secondhand tobacco smoke (SHS), is a major risk factor for CVD (Virani et al., 2021). In a meta-analysis, Lv and others found that SHS exposure, self-reported or objectively measured with cotinine, was associated with 23% increased risk of CVD, including stroke and coronary heart disease (myocardial infarction, acute coronary syndrome, or other ischemic heart diseases) (Lv et al., 2015).
Metabolic syndrome (MetS) is a multifactorial disorder characterized by physiologically altered cardiometabolic (CM) risk components that tend to cluster together, such as central obesity, impaired glucose metabolism, dyslipidemia, and elevated blood pressure (BP) (Expert panel on integrated guidelines, 2011; Mottillo et al., 2010; Grundy et al., 2004). MetS, which often emerges in childhood, is associated with increased risk of coronary heart disease, stroke, and diabetes (Jacobs et al., 2022a; Alberti et al., 2009). Although the prevalence of MetS among US adolescents has remained stable during the last two decades (Liu et al., 2022), the prevalences of central obesity and hyperglycemia have increased.
Nicotine exposure could alter central and peripheral sympathetic norepinephrine systems, key regulators of appetite and adipose metabolism (Chen et al., 2019). Alterations in peripheral signals of satiety and adiposity, which arise from adipose, liver, pancreas, and gastrointestinal tissues, could be altered by the effect of SHS constituents on glucose, peptides (leptin, adiponectin, insulin, cholecystokinin, and tumor necrosis factor alpha (TNF-α)), lipid-derived molecules (prostaglandins, triglycerides (TG), low density lipoprotein (LDL)), eicosanoids, and fatty acids during early development (Jo et al., 2002; Nagel et al., 2009).
During the last three decades, results of studies examining the impact of SHS exposure on adolescent CM risk (Chen et al., 2019; Weitzman et al., 2005; Li et al., 2020) have been inconclusive; this could be explained by differences in the definition of MetS and in the individual CM risk components analyzed. Moreover, no studies have investigated periods of heightened susceptibility to SHS exposure during pregnancy and childhood on CM risk in adolescents. This is critical as MetS begins early in life and increases the risk of all-cause mortality, CVD morbidity and mortality, and type 2 diabetes mellitus in adulthood (Eisenmann, 2008).
To fill this gap in knowledge, we estimated associations of early life exposure to SHS, using pre- and postnatal serum cotinine concentrations, with CM risk in adolescents by using a CM risk score based on five CM risk components to identify periods of heightened susceptibility to the adverse effects of SHS exposure.
2. Methods
2.1. Study participants
We used data from a prospective pregnancy and birth cohort, the Health Outcomes and Measures of the Environment (HOME) Study. The HOME Study recruited pregnant women between 2003 and 2006, and conducted follow-up visits with the mothers (or children’s caregivers) and their children through age 12 years (Braun et al., 2017). The principal objective of the HOME Study was to evaluate the association of pre- and postnatal exposure to environmental toxicants with health and neurobehavioral outcomes in infants and children. The inclusion criteria were: ≥18 years old, between 13 and 19 weeks of gestation, residing in a house built before 1978 within the study area, HIV-negative, not taking thyroid or epilepsy medication and not undergoing chemotherapy or radiation therapy. From March 2003 to January 2006, we recruited 468 pregnant women living in a five county region of the Cincinnati, Ohio metropolitan area and Northern Kentucky. Sixty-seven pregnant women dropped out during the run-in phase of a randomized controlled trial of residential lead and injury hazard nested within the cohort although some rejoined the study at later visits (Braun et al., 2018).
We conducted visits at the delivery hospital, the study clinic, and participants homes when children were approximately age 1 day, 4 weeks, and 1, 2, 3, 4, 5, 8 and 12 years. A detailed description of participant’s characteristics, follow-up, and study measures at the 12-year visit are described elsewhere (Braun et al., 2020a). Among the 441 children who were eligible for the 12-year study visit, 256 adolescents completed the visit; singleton children who had at least one serum cotinine concentration measurement during pregnancy or childhood, at least one CM risk component measured during adolescence, and complete covariate information were included in the final analytic sample (N = 212).
The institutional review boards (IRBs) at Cincinnati Children’s Hospital Medical Center (CCHMC) and participating delivery hospitals approved the HOME Study protocols. Brown University and the Centers for Disease Control and Prevention (CDC) IRBs deferred to the CCHMC IRB. All mothers provided informed consent for themselves and their children at all visits; children provided informed assent at the 12-year study visit.
2.2. Prenatal and postnatal tobacco smoke exposure assessment
Trained phlebotomists collected maternal venous blood samples at 16 and 26 weeks of pregnancy and child venous blood samples at 1, 2, 3 and 4 years via venipuncture. The samples were stored at or below −80 °C until analysis. To assess SHS exposure, serum cotinine concentrations were quantified by trained technicians at the CDC using high performance liquid chromatography tandem mass spectrometry (Bernert et al., 2009). The assay limit of detection threshold for cotinine was 0.015 ng/mL.
A total of 251 and 232 mothers had at least one serum cotinine measurement at 16 and 26 weeks of pregnancy, and 186, 149, 157, and 127 children had measurements at age 1, 2, 3, and 4, respectively. We used mean prenatal and mean postnatal serum cotinine concentrations given the high within-person correlation of serum cotinine concentrations during pregnancy or early childhood. The ICC between repeated maternal cotinine concentrations (N = 220 mothers) was 0.96 (95% CI 0.94, 0.97); the ICC between the four childhood cotinine concentrations (N = 65 children) was 0.95 (95% CI 0.93, 0.97). Thus, we averaged available maternal serum cotinine concentrations (range = 1–2 measures), and child’s serum cotinine concentrations (range = 1–4 measures) to assess pre- and postnatal exposure, respectively, in further analysis.
2.3. Adolescent’s CM risk outcomes
We measured all CM risk components at the 12-year study visit (Braun et al., 2020a). We obtained an overnight-fasting blood sample from adolescents via venipuncture. We measured serum glucose (mg/dL), insulin (mIU/L), TG (mg/dL), HDL-C (mg/dL), leptin (ng/mL), and adiponectin (ng/mL) concentrations using valid immunoassays. Trained laboratory technicians in the Cincinnati Children’s Hospital Medical Center NIH-funded Clinical Translational Research Center Core Laboratory conducted all assays; based on 22 blinded duplicates, the coefficient of variations for glucose, HDL-C, TG, insulin, leptin, and adiponectin were 2.0%, 3.0%, 6.5%, 9.5%, 11.1% and 13.1%, respectively. We obtained three sitting BP measurements, each 1 min apart, with an oscillometric monitor (Dinamap Pro100) (Gillman and Cook, 1995; Perloff et al., 1993). Visceral fat area was measured by dual x-ray absorptiometry (DXA, Hologic Horizon Densitometer) (Wang et al., 2010).
We constructed a continuous CM risk summary score for each adolescent, with higher values indicative of a worse MetS profile compared to lower values (Li et al., 2020). We calculated CM risk score (N = 190) by summing age- and sex-standardized z-scores for five individual CM risk components (Supplemental Table 1).
Homeostatic model assessment for insulin resistance (HOMA-IR = fasting insulin × fasting glucose/405) (Matthews et al., 1985)
TG to HDL-C ratio
Leptin to adiponectin ratio
Average of the 2nd and 3rd measures of systolic BP (SBP) (Lacruz et al., 2017)
Visceral fat area (cm2)
We ran linear regression models of each individual CM risk component (dependent variable) with age and sex as predictors to derive the age- and sex-specific z-scores after standardization of the residuals (Eisenmann, 2008). We standardized BP z-scores by sex, age, and height according to the Fourth Report on the Diagnosis, Evaluation, and Treatment of High Blood Pressure in children and adolescents (Expert panel on integrated guidelines, 2011). HOMA-IR, TG to HDL-C ratio, leptin to adiponectin ratio, and visceral fat area were not normally distributed (right skewed) and thus, we log2-transformed them before standardization.
2.4. Covariates
We relied on previous literature and directed acyclic graphs (DAGs) (Werlinger and Cáceres, 2018) to identify potential confounders for serum cotinine concentrations and adolescent’s CM risk (Supplemental Figs. 1 and 2). Trained research staff collected sociodemographic covariates using standardized computer-assisted interviews including maternal age, race/ethnicity, marital status and education at baseline. During the first three years of life, research staff collected data on breastfeeding duration with standardized interviewer-administered surveys (Romano et al., 2016). Staff abstracted data on maternal blood pressure during pregnancy and infant’s sex from medical records and used self-reported weight and height (or imputed weight if missing) to calculate maternal pre-pregnancy BMI (van der Laan et al., 2007). Study participants rated their pubertal status using Tanner stage (I–V) based on pubic hair development at the 12-year study visit (Yayah Jones et al., 2021).
Trained research staff at Cincinnati Children’s Clinical Translational Research Center Bionutrition Core collected 24-h food recalls from adolescents (2 weekdays and 1 weekend day). We analyzed food recalls using the Nutrition Data systems for Research software and foods database (University of Minnesota, MN) and calculated the Healthy Eating Index (HEI) score (2010), a measure of conformance with US federal dietary guidance (Guenther et al., 2013). We assessed adolescent’s physical activity level by administering the validated Physical Activity Questionnaire for Older Children and calculating the activity summary score (Kowalski and Faulkner, 1997).
2.5. Statistical analysis
We described mothers’ and adolescents’ covariate characteristics, the distribution of adolescents’ CM risk scores and their individual components stratified by sex, and the distribution of maternal and child’s serum cotinine concentrations, including the number of active smoking and nonsmoking mothers who were exposed to SHS. We log10-transformed the average pre- and postnatal cotinine concentrations to reduce the potential influence of outliers, since cotinine distribution was right-skewed.
Using multiple informant models, we estimated covariate-adjusted differences in CM risk score, and individual components associated per 1-unit increase in prenatal and postnatal log10-transformed serum cotinine concentrations (beta coefficients and 95% CI). Details on the multiple informant method have been previously described (Buckley et al., 2019; Sanchez et al., 2011). Briefly, the multiple informant method can be used with repeated and sparsely sampled environmental exposure measures at different time points to test whether the estimates for exposure-outcome association are equal across different time points (Buckley et al., 2019; Sanchez et al., 2011). There are various methods to jointly estimate the regression models for each time point: maximum likelihood and generalized estimating equations (GEE). We chose the last approach since it takes into account within-individual correlation across the different time points, and it works with the entire sample even if some individual data are missing. We created two estimates, one for prenatal exposure period (mean maternal serum cotinine concentrations at 16 and 26 weeks of pregnancy) and another for postnatal exposure period (mean child’s serum cotinine concentrations at ages 1, 2, 3, and 4 years). The multiple informant model included exposure period, exposure period × continuous cotinine and covariates, allowing us to test whether cotinine-outcome associations differed between prenatal and postnatal exposure periods. The null hypothesis is that the associations are the same across the two time periods, and we considered those with interaction p-values <0.05 as evidence that the prenatal and postnatal cotinine-outcome associations differed.
We also examined the potential modifying effects of adolescent’s sex using a three-way interaction term of exposure period × cotinine × adolescent’s sex.
We adjusted all multiple informant models for maternal age, race/ethnicity, marital status, education, pre-pregnancy BMI, pregnancy induced hypertensive disorders, and breastfeeding duration; and adolescent’s sex, puberty and age.
Finally, we conducted several sensitivity analyses by further adjusting for adolescent’s HEI 2010 total scores, physical activity summary scores, both HEI and physical activity scores, and excluding mothers who were active smokers during pregnancy (serum cotinine concentrations ≥3 ng/mL at 16 or 26 weeks of gestation (Benowitz et al., 2009)) and those with pregnancy induced hypertensive disorders (maternal SBP ≥140 mmHg and/or DBP ≥90 mmHg after 20 weeks of pregnancy (Chobanian et al., 2003)).
We performed all these analyses using R statistical software, version 4.1.2 (R Core Team, Vienna, Austria).
3. Results
The characteristics of the final sample (N = 212) did not differ from those of the total HOME Study participants at the 12-year study visit (N = 256) (Table 1). The mean age of the 212 adolescents was 12.4 years (SD: 0.67, range: 11–14); 53% were females, and 89% were stage ≥2 for pubic hair development (Table 1). On average, mothers were age 29.4 years at delivery, 59% were non-Hispanic white, 93% had > high school education, 57% were overweight or obese before pregnancy, and 42% breastfed their children for ≥6 months (Table 1).
Table 1.
Characteristics and CM risk scores of the HOME Study participants at the 12-year study visit.
| HOME Study participants (12-year study visit) | Final analysis participants | CM Risk Score | ||
|---|---|---|---|---|
|
|
|
|
||
| N (%) | N (%) | N | Mean (SD) | |
|
| ||||
| Overall | 256 (100) | 212 (82.81) | 190 | 0.17 (3.43) |
| MATERNAL CHARACTERISTICS | ||||
| Age (years) | ||||
| 18–25 | 58 (25.11) | 51 (24.06) | 44 | 0.13 (2.97) |
| >25–29 | 60 (25.97) | 57 (26.89) | 49 | 0.51 (4.07) |
| >29–34 | 79 (34.20) | 72 (33.96) | 56 | −0.24 (3.15) |
| >34 | 34 (14.72) | 32 (15.09) | 26 | 0.32 (3.71) |
| Race/ethnicity | ||||
| White, non-Hispanic | 155 (60.55) | 126 (59.43) | 113 | −0.31 (3.36) |
| Black, non-Hispanic | 88 (34.38) | 73 (34.43) | 67 | 1.12 (3.46) |
| All others | 13 (5.09) | 13 (6.13) | 10 | −0.72 (3.01) |
| Education | ||||
| Some high school | 21 (8.50) | 15 (7.08) | 41 | 1.32 (3.22) |
| High school or GED | 31(12.55) | 30 (14.15) | 34 | −0.17 (3.24) |
| Some college or technical school | 72 (29.15) | 64 (30.19) | 53 | −0.69 (3.78) |
| Bachelor’s degree or more | 123 (49.80) | 103 (48.59) | 56 | 0.29 (3.19) |
| Prepregnancy BMI (kg/m2) | ||||
| Underweight/normal (<24.9) | 98 (41.88) | 91 (42.93) | 71 | −0.67 (3.06) |
| Overweight (25.0–29.9) | 76 (32.48) | 68 (32.06) | 62 | −0.28 (3.60) |
| Obese (≥30) | 60 (25.64) | 53 (25.00) | 45 | 1.80 (3.26) |
| Marital status | ||||
| Married | 146 (62.77) | 136 (64.15) | 108 | −0.21 (3.46) |
| Not married, living with someone | 28 (12.12) | 23 (10.85) | 21 | 0.01 (3.23) |
| Not married, living alone | 57 (24.68) | 53 (25.00) | 46 | 0.83 (3.20) |
| Pregnancy induced hypertensive disorders | ||||
| Yes | 4 (1.64) | 3 (1.42) | 2 | −0.16 (0.58) |
| No | 240 (98.36) | 209 (98.58) | 180 | 0.16 (3.47) |
| Breastfeeding duration (months) | ||||
| <6 | 143 (59.83) | 123 (58.02) | 108 | 0.24 (3.23) |
| ≥6 | 96 (40.17) | 89 (41.98) | 71 | −0.12 (3.73) |
| ADOLESCENT CHARACTERISTICS | N (%) N (%) | N | Mean (SD) | |
| Sex | ||||
| Female | 143 (55.86) | 113 (53.30) | 103 | 0.08 (3.79) |
| Male | 113 (44.14) | 99 (46.70) | 87 | 0.28 (2.97) |
| Pubic hair | ||||
| Stage 1 | 26 (10.32) | 24 (11.32) | 17 | −0.64 (3.51) |
| Stage 2 | 64 (25.40) | 55 (25.94) | 50 | −0.42 (3.67) |
| Stage 3 | 72 (28.57) | 65 (30.66) | 53 | 0.26 (3.36) |
| Stage 4 | 55 (21.83) | 40 (18.87) | 40 | 0.96 (3.46) |
| Stage 5 | 35 (13.89) | 28 (13.21) | 29 | 0.59 (2.90) |
Abbreviations: CM, cardiometabolic; GED, General Educational Development; BMI, body mass index.
The CM risk score was higher among adolescents who were at more advanced pubertal stage at age 12 years, born to mothers who were non-Hispanic Black, aged 26 to 29 or >35 years, or had ≤ high school education, pre-pregnancy obesity (BMI ≥30 kg/m2) and breastfed for <6 months compared to other groups (Table 1).
Generally, glucose, insulin, TG, HDL-C, and BP values were typical for 12 year old adolescents; however, 10% of adolescents had elevated glucose (>100 mg/dL), 16% decreased HDL-C (<40 mg/dL), and 33% borderline/high TG (>90 mg/dL) (Expert panel on integrated guidelines, 2011). Compared to males, females had higher mean HOMA-IR, leptin to adiponectin ratio, but lower mean TG to HDL-C ratio, and visceral fat area (Supplemental Table 1). HOMA-IR was moderately correlated with visceral fat area (rho = 0.60) (Fig. 1).
Fig. 1.
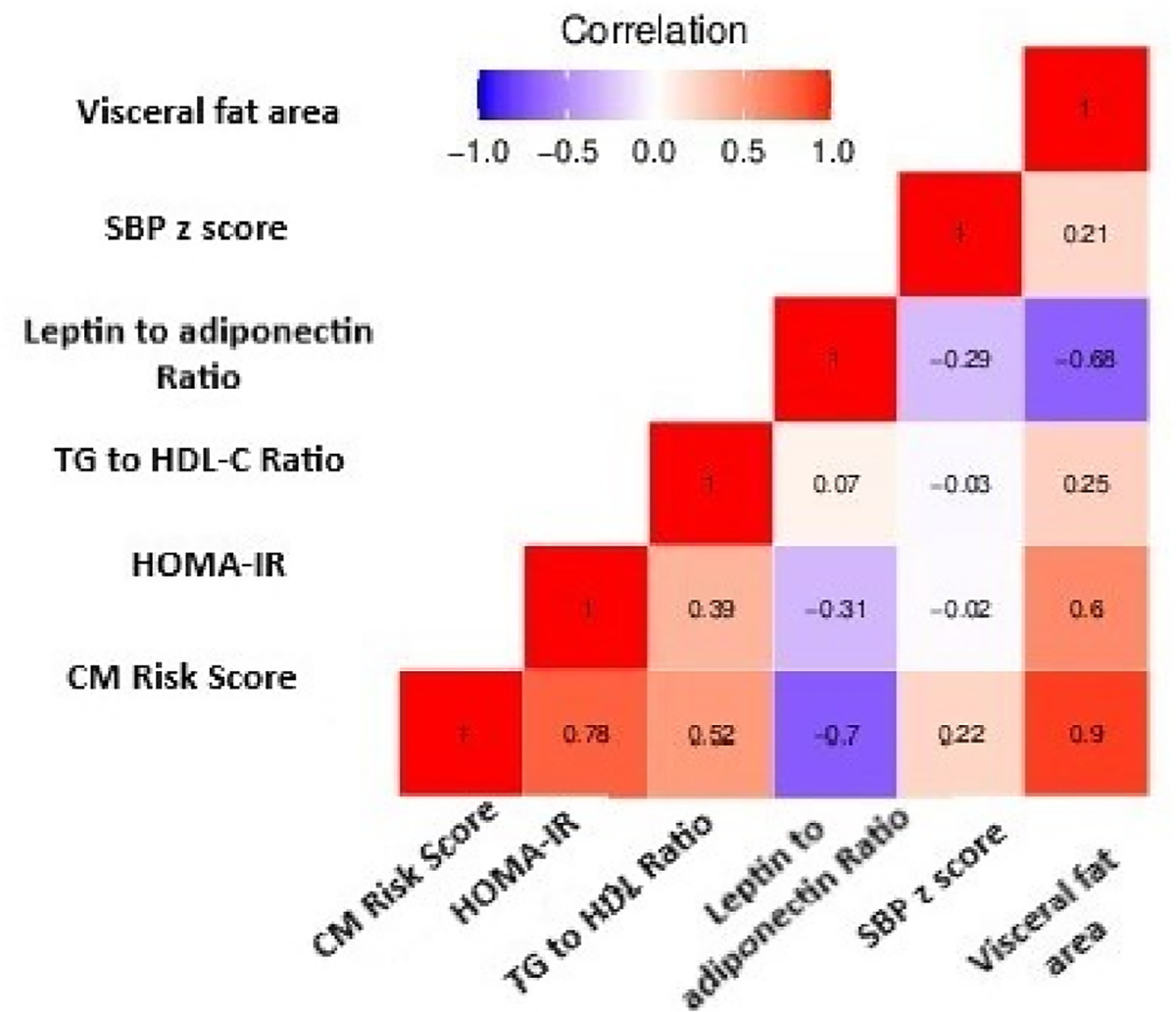
Spearman correlation coefficients between cardiometabolic components-related outcomes at age 12 years.
Thirty-two (14.8%) mothers had mean serum cotinine concentrations indicative of active smoking during pregnancy (≥3 ng/ml). Median maternal prenatal serum cotinine concentrations were similar to children’s median postnatal concentrations (0.04 vs. 0.05 ng/ml, respectively) (Supplemental Table 3). Girls’ median postnatal cotinine concentrations were similar to boys’ concentrations (0.06 vs. 0.04 ng/ml) (Supplemental Table 2 and Fig. 2). Serum cotinine concentrations were moderately correlated with each other during the prenatal period and postnatal period (rho = 0.76) and the ICC between pre- and postnatal concentrations was 0.84 (95% CI 0.81, 0.88).
Fig. 2.
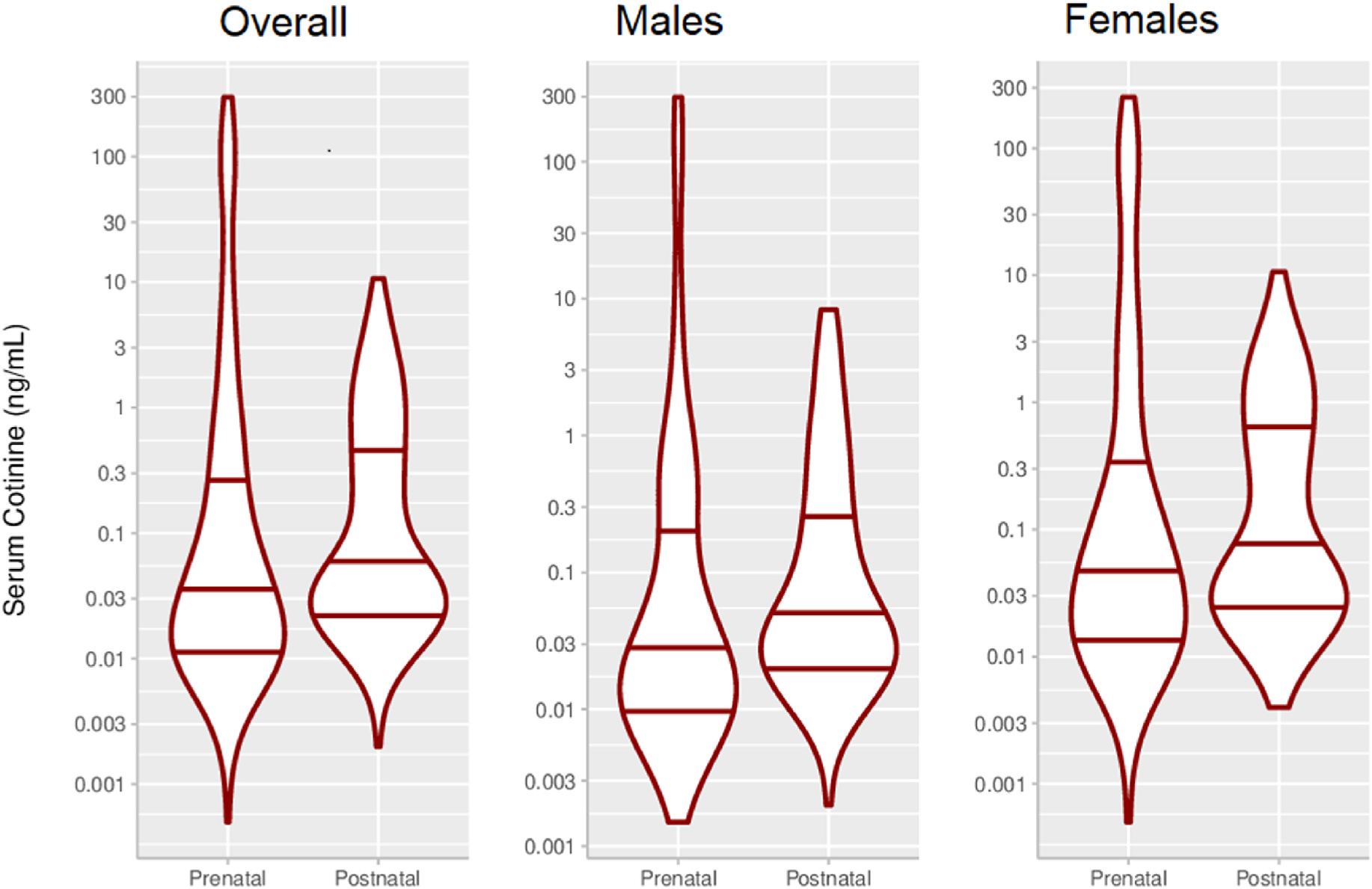
Violin plots of prenatal and postnatal mean serum cotinine concentrations (ng/mL)a.
After adjusting for covariates, associations of serum cotinine concentrations with CM risk score and visceral fat area at age 12 years differed by exposure period (exposure period × cotinine interaction terms: <0.05) (Supplemental Table 3 and Fig. 3). Unlike the prenatal cotinine concentrations, postnatal cotinine concentrations were associated with higher CM risk scores and visceral fat.
Fig. 3.
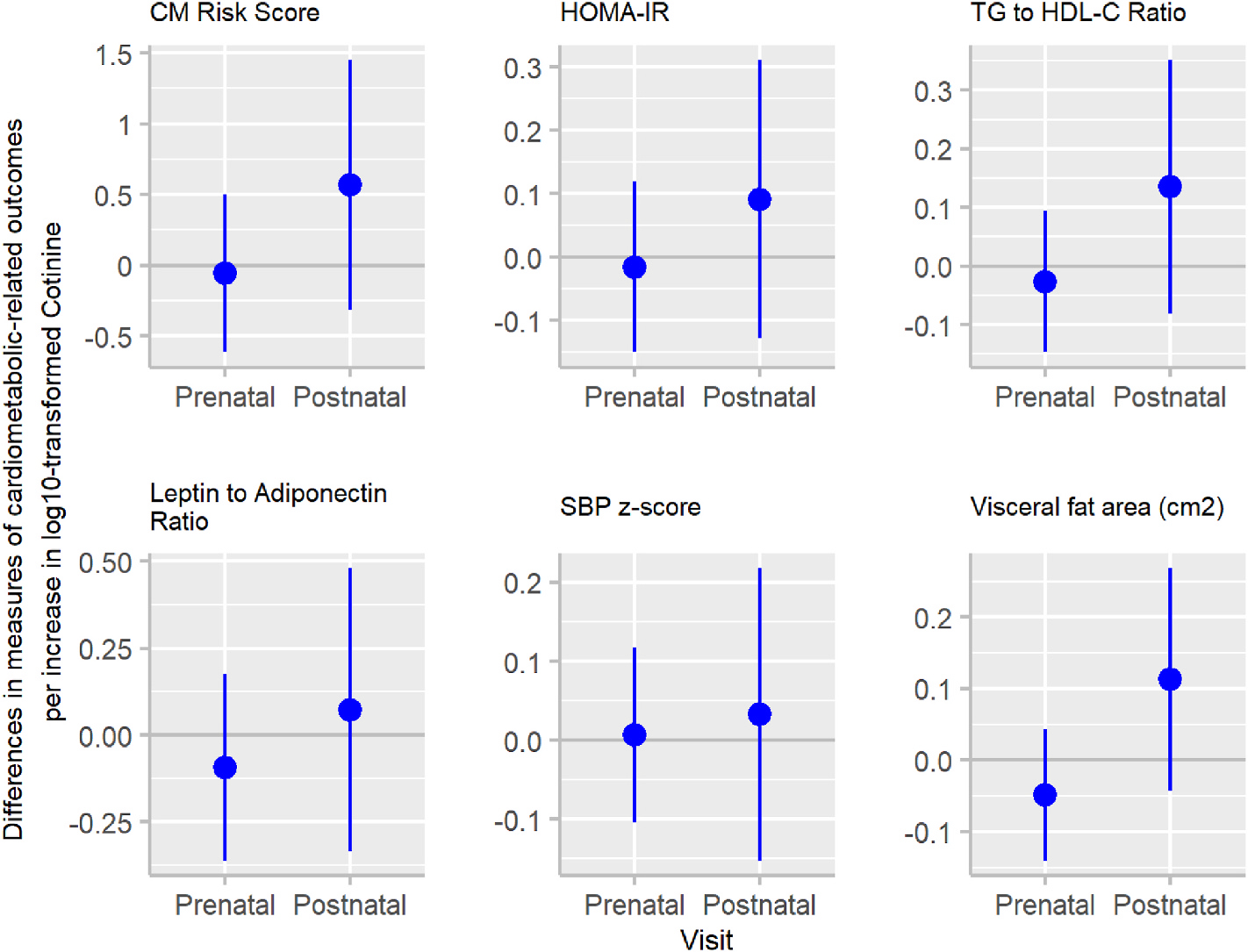
Adjusted associations (beta coefficients (95% CI)) of log10-transformed mean prenatal and mean postnatal serum cotinine concentrations (ng/mL) with cardiometabolic (CM) risk outcomes at age 12 yearsa.
For the three-way interaction term for exposure period × serum cotinine × adolescent’s sex from the multiple informant model, we observed some evidence of sex-specific associations of serum cotinine concentrations with visceral fat area (p-value: 0.01) (Supplemental Table 4 and Fig. 4). This was largely driven by the stronger association of postnatal mean concentrations with visceral fat area in females (0.27; 95%CI: 0.10, 0.45) compared to males (−0.09; 95%CI: 0.25, 0.07). Postnatal cotinine was associated with greater HOMA-IR, TG to HDL-C ratio and leptin to adiponectin ratio among males, compared to females (Supplemental Table 4). Prenatal cotinine concentrations were not associated with CM risk scores in either sex.
Fig. 4.
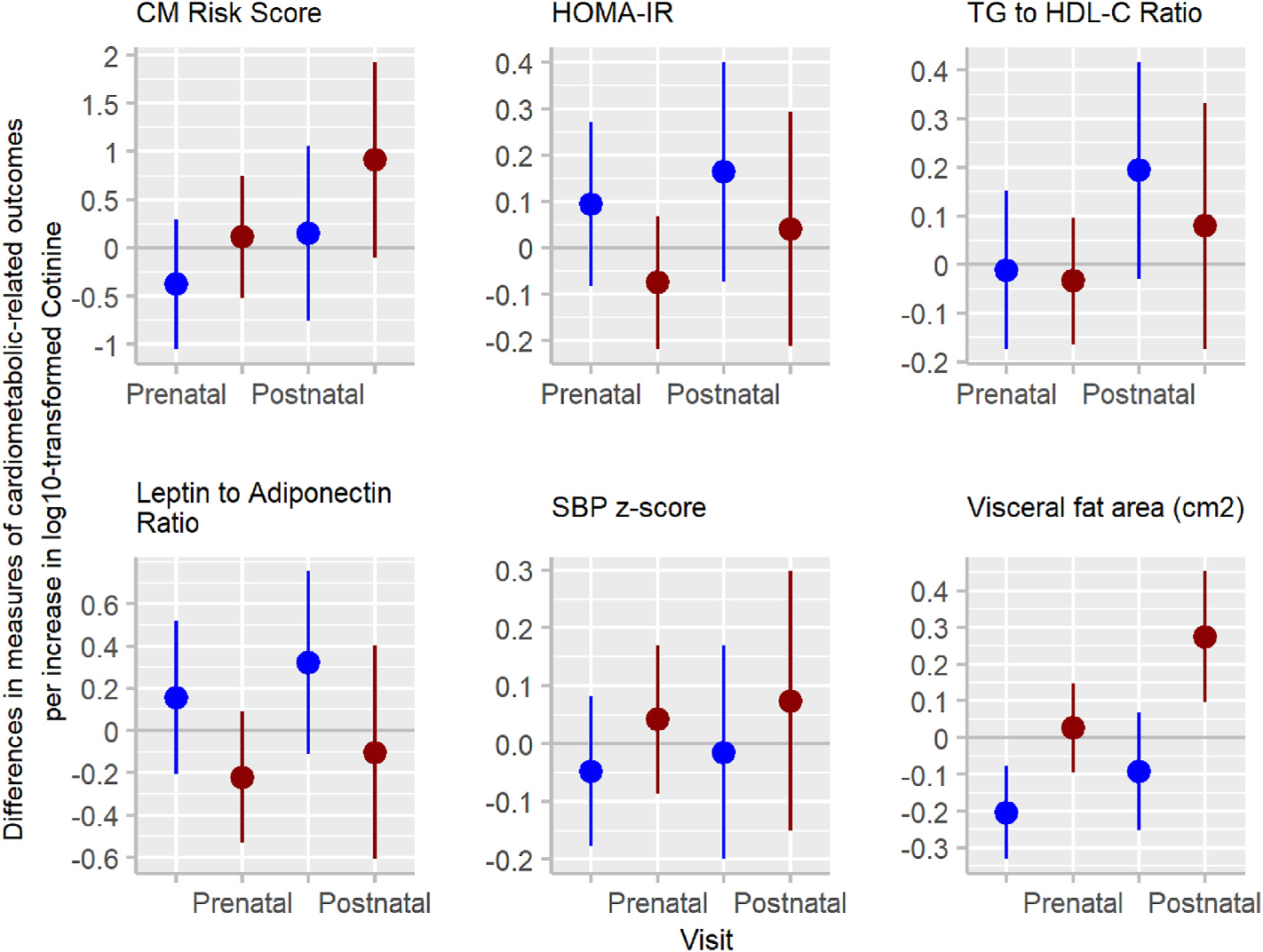
Adjusted associations (beta coefficients (95% CI)) of log10-transformed mean prenatal and mean postnatal serum cotinine concentrations (ng/mL) with cardiometabolic (CM) risk outcomes at age 12 years by adolescent’s sexa.
In secondary analyses, excluding mothers who smoked during pregnancy (N = 32), or further adjusting for HEI scores, physical activity scores, or both, associations of cotinine with CM risk score and visceral fat area did not meaningfully change in magnitude; however, exposure period × serum cotinine interaction term was not significant (p-value: ≥0.05). After excluding those mothers with pregnancy induced hypertensive disorders (N = 4), results did not meaningfully change (Supplemental Table 5).
4. Discussion
This is the first study to assess the associations of repeated measures of serum cotinine concentrations from pregnancy to age 4 years with CM risk at age 12 years. We found some evidence that the cotinine-outcome associations differed by exposure period and it may be sex-specific. Postnatal cotinine concentrations were suggestively associated with higher CM risk score at 12 years. We found no evidence that prenatal cotinine concentrations were associated with CM risk components. The sex-specific association for cotinine and overall score was driven by visceral fat area; females exposed to SHS had higher increase than males.
In 1986, Barker reported the influence of adverse intrauterine conditions on the development of adult’s CVD (Wilson, 1999). Since then, inconsistent results have been found when assessing the effects of prenatal or postnatal SHS exposure on the CM risk score and some individual risk components in children and adolescents (Weitzman et al., 2005; Stevens et al., 2018; Behl et al., 2013; Davis et al., 2016; Ebrahimi et al., 2019; Kelishadi et al., 2016; Groner et al., 2017; Merianos et al., 2017; Cajachagua-Torres et al., 2022). Differences among studies could be related to participants’ characteristics (age, race/ethnicity, hormones and body composition), definition, intensity and timing of SHS exposure, method to measure exposure (self-report vs. biomarkers), biomarkers cut-points to classify exposure, and varying concentration ranges, and CM risk assessment (MetS definition, risk components included, measurement methods, and cut-points used to define increased concentrations in children) (Stevens et al., 2018; Thiering et al., 2011; Staiano and Katzmarzyk, 2012; Salvador et al., 2008; Yahia et al., 2017).
The prenatal, early childhood, and adolescent periods have been proposed as critical periods for the development of obesity and related CM disorders. We found evidence that exposure to SHS in early childhood may have greater influence on adolescent’s CM risk than prenatal exposure. Exposure to SHS is linked to oxidative stress, lipoprotein modification, abnormal lipid metabolism, vascular inflammation, endothelial dysfunction, platelet activation, and cardiovascular function impairment (Ebrahimi et al., 2019; Raupach et al., 2006). Oxidative stress seems to be crucial in the fetal programming of metabolic disorders and CVD, as it is associated with insulin resistance, higher levels of BP, serum TG and leptin, and lower levels of serum HDL-C and adiponectin (Salvador et al., 2008; Rodríguez-Rodríguez et al., 2018; Bodea and Popa, 2015; Fonseca, 2005; Furukawa et al., 2004).
Our findings showed that the exposure period-specific association for serum cotinine concentrations and overall CM risk score was driven by visceral fat. However, when we excluded smoking mothers or further adjusted for HEI scores or HEI scores and physical activity, associations of cotinine with CM risk score and visceral fat area were less precise and the interaction p-values were >0.05.
We did not find evidence in this study that higher prenatal serum cotinine concentrations increased CM risk. Postnatal serum cotinine concentrations were associated with higher CM risk components during adolescence, specifically TG to HDL-C ratio, visceral fat area and HOMA-IR. Higher levels of serum TG and lower levels of HDL-C have been found in previous studies among adolescents exposed to postnatal SHS exposure, compared to those unexposed (Weitzman et al., 2005; Ebrahimi et al., 2019; Miyamura et al., 2021; Moskowitz et al., 1999; Le-Ha et al., 2013). Interestingly, when we excluded smoking mothers during pregnancy, associations between postnatal cotinine and TG to HDL-C ratio differed by exposure period in our study (interaction p-value: 0.04), with a higher increase among those adolescents exposed to postnatal exposure, compared to prenatal exposure. Considering that multiple informant models do not adjust for SHS exposure at other time, postnatal findings could reflect consistent and longer postnatal exposure to SHS since birth. Thus, after adjusting for all the covariates, with the exception of pubertal status, we observed that log10-transformed mean postnatal serum cotinine concentrations were higher among those children exposed to SHS during pregnancy, compared to those unexposed (difference between postnatal cotinine concentrations of those children exposed vs. unexposed during the prenatal period: 0.36; 95%CI: 0.28, 0.44; p-value <0.0001).
Insulin resistance plays a role in initiating and perpetuating the pathologic manifestations of MetS (Heindel et al., 2017). A prior study reported an adverse association between self-reported postnatal SHS exposure and 10-year-old children’s HOMA-IR (Thiering et al., 2011). To our knowledge, our study is the second to use HOMA-IR as an indicator of insulin resistance (Matthews et al., 1985; Antuna-Puente et al., 2011), rather than measuring levels of fasting insulin or glucose (Weitzman et al., 2005; Stevens et al., 2018; Kelishadi et al., 2016; Groner et al., 2017), when examining SHS exposure and CM outcomes in children.
Associations of postnatal cotinine with leptin to adiponectin ratio and SBP, were weaker than other CM risk components. We only identified one study that included leptin and adiponectin concentrations for assessing the association between postnatal SHS exposure at home and CM risk (Nagel et al., 2009). They found that SHS exposure was associated with higher leptin and lower adiponectin concentrations in children aged 10 years. Further studies should consider the leptin to adiponectin ratio, since this is a better predictor of insulin resistance and MetS, compared to individual adipocytokines (Srikanthan et al., 2016). For the association between cotinine and BP, prior meta-analyses have shown that pre- and postnatal SHS exposure could increase SBP figures during childhood and adolescence (Aryanpur et al., 2019; Brion et al., 2008; Nordenstam, 2021); however, the included studies were heterogeneous in terms of recruitment, country, definition of SHS exposure, participants’ characteristics, adjustment variables, number of BP measures, and measurement method. Considering that arterial hypertension is an increasingly common condition among children (Song et al., 2019), and the leading modifiable risk factor for CVD and premature death worldwide (Global, 2018a, 2018b), future cohort studies are warranted to verify whether early exposure to SHS increases BP figures later in life, during childhood or adolescence (Basile, 2002).
Consistent with prior studies (Staiano and Katzmarzyk, 2012; Cook et al., 2003), we found that males have higher CM risk score than females, driven mainly by TG to HDL-C ratio and visceral fat area (Staiano and Katzmarzyk, 2012). These sex differences may be explained by differences in body fat distribution, hormonal factors, stress level, sleeping patterns, sociodemographic characteristics, and eating behaviors (Staiano and Katzmarzyk, 2012; Björntorp, 1991; Tchernof and Després, 2013; Warkentin et al., 2020). However, when considering associations between cotinine and CM risk outcomes, our findings show that postnatal cotinine was associated with a higher increase among females compared to males. Modifications of the cotinine-CM risk outcome associations by adolescent’s sex were observed in previous studies, with males having a higher risk than females associated with SHS (Weitzman et al., 2005; Stevens et al., 2018; Miyamura et al., 2021). While differences by sex could be related to the level of SHS exposure or cotinine metabolism and clearance (Pirkle et al., 2006; Larson et al., 2011), males and females from our study had similar cotinine concentrations. However, tobacco smoke constituents could affect the endocrine system (gonadal, steroid, thyroid, and leptin hormones) or the growth and distribution of adipose tissue differentially in females vs. males (Miyamura et al., 2021; Tchernof and Després, 2013; Dietz, 1994; Wells, 2001; Braun et al., 2020b). Indeed, gonadal hormones may play a significant role in further modulating CM risk components beyond their effects on visceral fat accumulation and insulin resistance (Tchernof and Després, 2013).
This study has some limitations and strengths. First, we had a modest sample size that limited our ability to identify distinct periods of heightened susceptibility and sex-specific cotinine-outcome associations. Some of our findings should be interpreted cautiously; still, no studies have assessed the effects of SHS, measured with serum cotinine during both pregnancy and early childhood, on adolescents’ CM health. Unfortunately, we did not have more recent serum cotinine measures and it is possible that these might be more important than earlier measures. Second, considering that CM risk components tend to cluster together, and the lack of consensus both on the definition of MetS and on the appropriate cut-points to define increased concentrations of its components, we constructed a CM risk summary score, which is a good predictor of future CM health (Jacobs et al., 2022b), using five validated biomarkers (Nordenstam, 2021). Of note, we assumed that each individual component has equal weights/contribution to the overall CM risk, but it is possible that some of them – such as visceral fat area or HOMA-IR – are more important predictors of CVD risk (Salvador et al., 2008). Third, although the use of multiple informant models enable us to identify potential periods of heightened susceptibility and reduce the number of regression models, it does not adjust for SHS exposure at other time; considering the high correlation found between pre- and postnatal cotinine concentrations (ICC = 0.84; 95%CI: 0.81, 0.88), significant findings for postnatal cotinine could be just a reflection of the longer and consistent exposure to SHS after birth. Attrition over the course of follow-up is another potential limitation. Still, measured sociodemographic characteristics were not substantially different among participants that did and did not complete follow-up at 12 years (Braun et al., 2020a). Residual confounding by other factors associated with CM risk and tobacco smoke is a concern. However, we adjusted for an extensive set of covariates which included breastfeeding duration, adolescent’s pubertal status, diet quality, and physical activity. Cotinine concentrations could also reflect third-hand smoke (THS) exposure (inhalation, ingestion or dermal uptake transferred from nicotine residue accumulated in dust, objects, home surfaces and on smokers’ skin and clothes). Of note, we did not include questions to evaluate it; future studies should consider THS exposure since it could be more toxic than SHS (multiple entry routes, longer duration of exposure, and novel pollutants not found in SHS) (Mahabee-Gittens et al., 2018). Finally, our findings may not be generalizable to other populations; reassuringly, serum cotinine concentrations among HOME Study participants were very similar to those of other pregnant women and children in the USA during the time of enrolment and follow-up (Woodruff et al., 2011; Fourth Report on Human Exposure, 2021).
5. Conclusions
Our results suggest that tobacco smoke exposure during early childhood may have greater influence on adolescent’s CM risk than exposure during the prenatal period, and these associations may be sex-specific. Early identification and management of CM risk factors are warranted to reduce the onset or progression of CM disorders (Yahia et al., 2017). This study reinforces the need for ongoing public health interventions aiming at discouraging parents and caretakers from smoking, and minimizing their children’s exposure to SHS.
Supplementary Material
Acknowledgments
We thank the HOME Study staff, patient advisers and participants for their years of dedication to the study.
Funding sources
The HOME Study was funded by National Institutes of Environmental Health Sciences grants P01 ES011261, R01 ES014575 and R01 ES015517. The U.S. National Institutes (National Institute of Environmental Health Sciences), Environmental Protection Agency, Department of Housing and Urban Development, and Flight Attendant Medical Research Institute provided funding for the presented results in the HOME Study.
Abbreviations:
- CVD
Cardiovascular disease
- USA
United States of America
- SHS
Secondhand tobacco smoke
- MetS
Metabolic syndrome
- BP
Blood pressure
- TNF-α
Tumor necrosis factor alpha
- TG
Triglycerides
- LDL
Low density lipoprotein
- CM
Cardiometabolic
- HOME
Health Outcomes and Measures of the Environment
- HIV
Human immunodeficiency virus
- IRBs
Institutional review boards
- CCHMC
Cincinnati Children’s Hospital Medical Center
- CDC
Centers for Disease Control and Prevention
- 95% CI
95% confidence interval
- N
Sample size
- HDL-C
High density lipoprotein cholesterol
- NIH
National Institutes of Health
- DXA
Dual x-ray absorptiometry
- HOMA-IR
Homeostatic model assessment for insulin resistance
- DAGs
Directed acyclic graphs
- BMI
Body mass index
- HEI
Healthy Eating Index
- SBP
Systolic blood pressure
- THS
Third-hand smoke
Footnotes
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Credit author statement
Nerea Mourino: term; Conceptualization; Methodology; Formal analysis; Writing – original draft; Writing – review & editing; Visualization.Mónica Pérez-Ríos: term; Conceptualization; Methodology; Formal analysis; Data curation; Validation; Writing – original draft; Writing – review & editing; Visualization; Supervision.Kimberly Yolton: Conceptualization; Writing – review & editing.Bruce P. Lanphear: Conceptualization; Writing – review & editing.Aimin Chen: Conceptualization; Writing – review & editing.Jessie P. Buckley: Conceptualization; Writing – review & editing.Heidi J. Kalkwarf: Conceptualization; Writing – review & editing.Kim M. Cecil: Conceptualization; Writing – review & editing.Joseph M. Braun: term; Conceptualization; Methodology; Validation; Writing – original draft; Writing – review & editing; Visualization; Supervision.
This paper is part of the PhD work of Nerea Mourino.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.envres.2023.115572.
Data availability
Data will be made available on request.
References
- Alberti KG, Eckel RH, Grundy SM, Zimmet PZ, Cleeman JI, Donato KA, et al. , 2009. Harmonizing the metabolic syndrome: a joint interim statement of the international diabetes federation task force on epidemiology and prevention; national heart, lung, and blood Institute; American heart association; world heart federation; international atherosclerosis society; and international association for the study of obesity. Circulation 120 (16), 1640–1645. [DOI] [PubMed] [Google Scholar]
- Antuna-Puente B, Disse E, Rabasa-Lhoret R, Laville M, Capeau J, Bastard JP, 2011. How can we measure insulin sensitivity/resistance? Diabetes Metab. 37 (3), 179–188. [DOI] [PubMed] [Google Scholar]
- Aryanpur M, Yousefifard M, Oraii A, Heydari G, Kazempour-Dizaji M, Sharifi H, et al. , 2019. Effect of passive exposure to cigarette smoke on blood pressure in children and adolescents: a meta-analysis of epidemiologic studies. BMC Pediatr. 19 (1), 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basile JN, 2002. Systolic blood pressure. BMJ 325 (7370), 917–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behl M, Rao D, Aagaard K, Davidson TL, Levin ED, Slotkin TA, et al. , 2013. Evaluation of the association between maternal smoking, childhood obesity, and metabolic disorders: a national toxicology program workshop review. Environ. Health Perspect. 121 (2), 170–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benowitz NL, Bernert JT, Caraballo RS, Holiday DB, Wang J, 2009. Optimal serum cotinine levels for distinguishing cigarette smokers and nonsmokers within different racial/ethnic groups in the United States between 1999 and 2004. Am. J. Epidemiol. 169 (2), 236–248. [DOI] [PubMed] [Google Scholar]
- Bernert JT, Jacob P, Holiday DB, Benowitz NL, Sosnoff CS, Doig MV, et al. , 2009. Interlaboratory comparability of serum cotinine measurements at smoker and nonsmoker concentration levels: a round-robin study. Nicotine Tob. Res. 11 (12), 1458–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björntorp P, 1991. Metabolic implications of body fat distribution. Diabetes Care 14 (12), 1132–1143. [DOI] [PubMed] [Google Scholar]
- Bodea A, Popa A, 2015. Abdominal obesity – a cardiometabolic risk factor. Romanian Journal of Diabetes Nutrition and Metabolic Diseases 22. [Google Scholar]
- Braun JM, Kalloo G, Chen A, Dietrich KN, Liddy-Hicks S, Morgan S, et al. , 2017. Cohort profile: the health outcomes and measures of the environment (HOME) study. Int. J. Epidemiol. 46 (1), 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun JM, Hornung R, Chen A, Dietrich KN, Jacobs DE, Jones R, et al. , 2018. Effect of residential lead-hazard interventions on childhood blood lead concentrations and neurobehavioral outcomes: a randomized clinical trial. JAMA Pediatr. 172 (10), 934–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun JM, Buckley JP, Cecil KM, Chen A, Kalkwarf HJ, Lanphear BP, et al. , 2020a. Adolescent follow-up in the health outcomes and measures of the environment (HOME) study: cohort profile. BMJ Open 10 (5), e034838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun M, Klingelhöfer D, Oremek GM, Quarcoo D, Groneberg DA, 2020b. Influence of second-hand smoke and prenatal tobacco smoke exposure on biomarkers, genetics and physiological processes in children-an overview in research insights of the last few years. Int. J. Environ. Res. Publ. Health 17 (9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brion MJ, Leary SD, Lawlor DA, Smith GD, Ness AR, 2008. Modifiable maternal exposures and offspring blood pressure: a review of epidemiological studies of maternal age, diet, and smoking. Pediatr. Res. 63 (6), 593–598. [DOI] [PubMed] [Google Scholar]
- Buckley JP, Hamra GB, Braun JM, 2019. Statistical approaches for investigating periods of susceptibility in children’s environmental health research. Curr Environ Health Rep 6 (1), 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cajachagua-Torres KN, El Marroun H, Reiss IKM, Santos S, Jaddoe VWV, 2022. Foetal tobacco and cannabis exposure, body fat and cardio-metabolic health in childhood. Pediatr Obes 17 (3), e12863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HJ, Li GL, Sun A, Peng DS, Zhang WX, Yan YE, 2019. Age differences in the relationship between secondhand smoke exposure and risk of metabolic syndrome: a meta-analysis. Int. J. Environ. Res. Publ. Health 16 (8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chobanian AV, Bakris GL, Black HR, Cushman WC, Green LA, Izzo JL Jr., et al. , 2003. The seventh report of the joint national committee on prevention, detection, evaluation, and treatment of high blood pressure: the JNC 7 report. JAMA 289 (19), 2560–2572. [DOI] [PubMed] [Google Scholar]
- Cook S, Weitzman M, Auinger P, Nguyen M, Dietz WH, 2003. Prevalence of a metabolic syndrome phenotype in adolescents: findings from the third National Health and Nutrition Examination Survey, 1988–1994. Arch. Pediatr. Adolesc. Med. 157 (8), 821–827. [DOI] [PubMed] [Google Scholar]
- Davis CL, Tingen MS, Jia J, Sherman F, Williams CF, Bhavsar K, et al. , 2016. Passive smoke exposure and its effects on cognition, sleep, and health outcomes in overweight and obese children. Child. Obes. 12 (2), 119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietz WH, 1994. Critical periods in childhood for the development of obesity. Am. J. Clin. Nutr. 59 (5), 955–959. [DOI] [PubMed] [Google Scholar]
- Ebrahimi M, Aghdam MH, Qorbani M, Abbaspour Kaboodan F, Shafiee G, Khatami F, et al. , 2019. Passive smoking and cardiometabolic risk factors in Iranian children and adolescents: CASPIAN-V study. J. Diabetes Metab. Disord. 18 (2), 401–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenmann JC, 2008. On the use of a continuous metabolic syndrome score in pediatric research. Cardiovasc. Diabetol. 7, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Expert panel on integrated guidelines for cardiovascular health and risk reduction in children and adolescents: summary report. Pediatrics 128 (Suppl. 5), 2011, S213–S256. Suppl 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca VA, 2005. The metabolic syndrome, hyperlipidemia, and insulin resistance. Clin. Cornerstone 7 (2–3), 61–72. [DOI] [PubMed] [Google Scholar]
- (March 2021) Fourth Report on Human Exposure to Environmental Chemicals, Updated Tables, 2021. U.S.: Department of Health and Human Services, Centers for Disease Control and Prevention, Atlanta, GA. [Google Scholar]
- Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, et al. , 2004. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Invest. 114 (12), 1752–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillman MW, Cook NR, 1995. Blood pressure measurement in childhood epidemiological studies. Circulation 92 (4), 1049–1057. [DOI] [PubMed] [Google Scholar]
- Global, regional, 2018a. Global, regional, and, and national comparative risk assessment of 84 behavioural, environmental and occupational, and metabolic risks or clusters of risks for 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 392 (10159), 1923–1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Global, regional, 2018b. Global, regional, and, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 392 (10159), 1736–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groner JA, Huang H, Joshi MS, Eastman N, Nicholson L, Bauer JA, 2017. Secondhand smoke exposure and preclinical markers of cardiovascular risk in toddlers. J. Pediatr. 189, 155–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy SM, Brewer HB Jr., Cleeman JI, Smith SC Jr., Lenfant C, American Heart A, et al. , 2004. Definition of metabolic syndrome: report of the national heart, lung, and blood Institute/American heart association conference on scientific issues related to definition. Circulation 109 (3), 433–438. [DOI] [PubMed] [Google Scholar]
- Guenther PM, Casavale KO, Reedy J, Kirkpatrick SI, Hiza HAB, Kuczynski KJ, et al. , 2013. Update of the Healthy eating index: HEI-2010. J. Acad. Nutr. Diet. 113 (4), 569–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heindel JJ, Blumberg B, Cave M, Machtinger R, Mantovani A, Mendez MA, et al. , 2017. Metabolism disrupting chemicals and metabolic disorders. Reprod. Toxicol. 68, 3–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs DR Jr., Woo JG, Sinaiko AR, Daniels SR, Ikonen J, Juonala M, et al. , 2022a. Childhood cardiovascular risk factors and adult cardiovascular events. N. Engl. J. Med. 386 (20), 1877–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs DR Jr., Woo JG, Sinaiko AR, Daniels SR, Ikonen J, Juonala M, et al. , 2022b. Childhood cardiovascular risk factors and adult cardiovascular events. N. Engl. J. Med. 386 (20), 1877–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo YH, Talmage DA, Role LW, 2002. Nicotinic receptor-mediated effects on appetite and food intake. J. Neurobiol. 53 (4), 618–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelishadi R, Noori A, Qorbani M, Rahimzadeh S, Djalalinia S, Shafiee G, et al. , 2016. Are active and passive smoking associated with cardiometabolic risk factors in adolescents? The CASPIAN-III Study. Paediatr. Int. Child Health 36 (3), 181–188. [DOI] [PubMed] [Google Scholar]
- Kowalski KCCP, Faulkner RA, 1997. Validation of the physical activity Questionnaire for older children. Pediatr. Exerc. Sci. 9 (2), 174–186. [Google Scholar]
- Lacruz ME, Kluttig A, Kuss O, Tiller D, Medenwald D, Nuding S, et al. , 2017. Short-term blood pressure variability - variation between arm side, body position and successive measurements: a population-based cohort study. BMC Cardiovasc. Disord. 17 (1), 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson LR, Green GT, Cordell Hk, 2011. Children’s time outdoors: results and implications of the national kids survey. J. Park Recreat. Adm. 29, 20. [Google Scholar]
- Le-Ha C, Beilin LJ, Burrows S, Huang RC, Oddy WH, Hands B, et al. , 2013. Gender difference in the relationship between passive smoking exposure and HDL-cholesterol levels in late adolescence. J. Clin. Endocrinol. Metab. 98 (5), 2126–2135. [DOI] [PubMed] [Google Scholar]
- Li Y, Yang Z, Wang X, Gao D, Zou Z, Dong B, et al. , 2020. Association between maternal lifestyle and risk of metabolic syndrome in offspring-A cross-sectional study from China. Front. Endocrinol. 11, 552054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Ma J, Orekoya O, Vangeepuram N, 2022. Trends in Metabolic Syndrome Among US Youth. JAMA Pediatr. From 1999 to 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv X, Sun J, Bi Y, Xu M, Lu J, Zhao L, et al. , 2015. Risk of all-cause mortality and cardiovascular disease associated with secondhand smoke exposure: a systematic review and meta-analysis. Int. J. Cardiol. 199, 106–115. [DOI] [PubMed] [Google Scholar]
- Mahabee-Gittens EM, Merianos AL, Matt GE, 2018. Preliminary evidence that high levels of nicotine on children’s hands may contribute to overall tobacco smoke exposure. Tobac. Control 27 (2), 217–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC, 1985. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 28 (7), 412–419. [DOI] [PubMed] [Google Scholar]
- Merianos AL, Jandarov RA, Mahabee-Gittens EM, 2017. Secondhand smoke exposure and pediatric Healthcare visits and Hospitalizations. Am. J. Prev. Med. 53 (4), 441–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamura K, Nawa N, Isumi A, Doi S, Ochi M, Fujiwara T, 2021. The association of passive smoking and dyslipidemia among adolescence in Japan: results from A-CHILD study. J. Clin. Endocrinol. Metab. 106 (7), e2738–e2748. [DOI] [PubMed] [Google Scholar]
- Moskowitz WB, Schwartz PF, Schieken RM, 1999. Childhood passive smoking, race, and coronary artery disease risk: the MCV Twin Study. Medical College of Virginia. Arch. Pediatr. Adolesc. Med. 153 (5), 446–453. [DOI] [PubMed] [Google Scholar]
- Mottillo S, Filion KB, Genest J, Joseph L, Pilote L, Poirier P, et al. , 2010. The metabolic syndrome and cardiovascular risk a systematic review and meta-analysis. J. Am. Coll. Cardiol. 56 (14), 1113–1132. [DOI] [PubMed] [Google Scholar]
- Nagel G, Arnold FJ, Wilhelm M, Link B, Zoellner I, Koenig W, 2009. Environmental tobacco smoke and cardiometabolic risk in young children: results from a survey in south-west Germany. Eur. Heart J. 30 (15), 1885–1893. [DOI] [PubMed] [Google Scholar]
- Nordenstam F, 2021. Prenatal nicotine exposure was associated with long-term impact on the cardiovascular system and regulation-Review. Acta Paediatr. 110 (9), 2536–2544. [DOI] [PubMed] [Google Scholar]
- Perloff D, Grim C, Flack J, Frohlich ED, Hill M, McDonald M, et al. , 1993. Human blood pressure determination by sphygmomanometry. Circulation 88 (5 Pt 1), 2460–2470. [DOI] [PubMed] [Google Scholar]
- Pirkle JL, Bernert JT, Caudill SP, Sosnoff CS, Pechacek TF, 2006. Trends in the exposure of nonsmokers in the U.S. population to secondhand smoke. Environ. Health Perspect. 114 (6), 853–858, 1988–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raupach T, Schäfer K, Konstantinides S, Andreas S, 2006. Secondhand smoke as an acute threat for the cardiovascular system: a change in paradigm. Eur. Heart J. 27 (4), 386–392. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Rodríguez P, Ramiro-Cortijo D, Reyes-Hernández CG, López de Pablo AL, Gonzalez MC, Arribas SM, 2018. Implication of oxidative stress in fetaĺ programming of cardiovascular disease. Front. Physiol. 9, 602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano ME, Xu Y, Calafat AM, Yolton K, Chen A, Webster GM, et al. , 2016. Maternal serum perfluoroalkyl substances during pregnancy and duration of breastfeeding. Environ. Res. 149, 239–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvador J, Silva C, Pujante P, Frühbeck G, 2008. Abdominal obesity: an indicator of cardiometabolic risk. Endocrinol. Nutr. 55 (9), 420–432. [DOI] [PubMed] [Google Scholar]
- Sanchez BN, Hu H, Litman HJ, Tellez-Rojo MM, 2011. Statistical methods to study timing of vulnerability with sparsely sampled data on environmental toxicants. Environ. Health Perspect. 119 (3), 409–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song P, Zhang Y, Yu J, Zha M, Zhu Y, Rahimi K, et al. , 2019. Global prevalence of hypertension in children: a systematic review and meta-analysis. JAMA Pediatr. 173 (12), 1154–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srikanthan K, Feyh A, Visweshwar H, Shapiro JI, Sodhi K, 2016. Systematic review of metabolic syndrome biomarkers: a panel for early detection, management, and risk stratification in the west virginian population. Int. J. Med. Sci. 13 (1), 25–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staiano AE, Katzmarzyk PT, 2012. Ethnic and sex differences in body fat and visceral and subcutaneous adiposity in children and adolescents. Int. J. Obes. 36 (10), 1261–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens DR, Malek AM, Laggis C, Hunt KJ, 2018. In utero exposure to tobacco smoke, subsequent cardiometabolic risks, and metabolic syndrome among U.S. adolescents. Ann. Epidemiol. 28 (9), 619–624.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchernof A, Després JP, 2013. Pathophysiology of human visceral obesity: an update. Physiol. Rev. 93 (1), 359–404. [DOI] [PubMed] [Google Scholar]
- Thiering E, Brüske I, Kratzsch J, Thiery J, Sausenthaler S, Meisinger C, et al. , 2011. Prenatal and postnatal tobacco smoke exposure and development of insulin resistance in 10 year old children. Int. J. Hyg Environ. Health 214 (5), 361–368. [DOI] [PubMed] [Google Scholar]
- van der Laan MJ, Polley EC, Hubbard AE, 2007. Super Learner. Statistical Applications in Genetics and Molecular Biology, vol. 6. Article25. [DOI] [PubMed] [Google Scholar]
- Virani SS, Alonso A, Aparicio HJ, Benjamin EJ, Bittencourt MS, Callaway CW, et al. , 2021. Heart disease and stroke statistics-2021 update: a report from the American heart association. Circulation 143 (8), e254–e743. [DOI] [PubMed] [Google Scholar]
- Wang Z, Heymsfield SB, Chen Z, Zhu S, Pierson RN, 2010. Estimation of percentage body fat by dual-energy x-ray absorptiometry: evaluation by in vivo human elemental composition. Phys. Med. Biol. 55 (9), 2619–2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warkentin S, Santos AC, Oliveira A, 2020. Associations of appetitive behaviors in 7-year-old children with their cardiometabolic health at 10 years of age. Nutr. Metabol. Cardiovasc. Dis. 30 (5), 810–821. [DOI] [PubMed] [Google Scholar]
- Weitzman M, Cook S, Auinger P, Florin TA, Daniels S, Nguyen M, et al. , 2005. Tobacco smoke exposure is associated with the metabolic syndrome in adolescents. Circulation 112 (6), 862–869. [DOI] [PubMed] [Google Scholar]
- Wells JC, 2001. A critique of the expression of paediatric body composition data. Arch. Dis. Child. 85 (1), 67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werlinger F, Cáceres DD, 2018. [Directed acyclic graphs in statistical modelling of epidemiological studies]. Rev. Med. Chile 146 (7), 907–913. [DOI] [PubMed] [Google Scholar]
- Wilson J, 1999. The Barker hypothesis. An analysis. Aust. N. Z. J. Obstet. Gynaecol. 39 (1), 1–7. [DOI] [PubMed] [Google Scholar]
- Woodruff TJ, Zota AR, Schwartz JM, 2011. Environmental chemicals in pregnant women in the United States: NHANES 2003–2004. Environ. Health Perspect. 119 (6), 878–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahia N, Brown CA, Snyder E, Cumper S, Langolf A, Trayer C, et al. , 2017. Prevalence of metabolic syndrome and its individual components among midwestern university students. J. Community Health 42 (4), 674–687. [DOI] [PubMed] [Google Scholar]
- Yayah Jones NH, Khoury JC, Xu Y, Newman N, Kalkwarf HJ, Braun JM, et al. , 2021. Comparing adolescent self staging of pubertal development with hormone biomarkers. J. Pediatr. Endocrinol. Metab.34(12),1531–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.