

Abstract
Transcriptional deregulation is a hallmark of many cancers and is exemplified by genomic amplifications of the MYC family of oncogenes, which occur in at least 20% of all solid tumors in adults. Targeting of transcriptional cofactors and the transcriptional cyclin-dependent kinase (CDK9) has emerged as a therapeutic strategy to interdict deregulated transcriptional activity including oncogenic MYC. Here, we report the structural optimization of a small molecule microarray hit, prioritizing maintenance of CDK9 selectivity while improving on-target potency and overall physicochemical and pharmacokinetic (PK) properties. This led to the discovery of the potent, selective, orally bioavailable CDK9 inhibitor 28 (KB-0742). Compound 28 exhibits in vivo antitumor activity in mouse xenograft models and a projected human PK profile anticipated to enable efficacious oral dosing. Notably, 28 is currently being investigated in a phase 1/2 dose escalation and expansion clinical trial in patients with relapsed or refractory solid tumors.
Introduction
Cyclin-dependent kinases (CDKs) are a family of serine/threonine kinases responsible for cell cycle progression (CDK1, 2, 4, 6) and transcriptional regulation (CDK7, 8, 9, 12, 13).1 As both cell cycle control and transcription are recurrently altered in cancer, the essential regulatory roles of CDKs in these processes make them attractive targets for pharmacological intervention.2,3 Initial therapeutic strategies sought to target several CDKs with pan-CDK inhibitors such as alvocidib and seliciclib.4 These inhibitors exhibited mixed clinical success due to challenges associated with their polypharmacology, including toxicity related to cell cycle CDK inhibition. As an alternative approach, isoform-selective CDK inhibitors have potential to be safer and more effective, as evidenced by CDK4/6 inhibitors such as abemaciclib, palbociclib, and ribociclib in the treatment of estrogen receptor-positive/human epidermal growth factor receptor 2-negative (ER+/HER2−) metastatic breast cancer.5,6 Despite these successes with CDK inhibition as targeted cancer therapy, there remains significant unmet need in other tumor types with evident transcriptional deregulation, such as triple-negative breast cancer (TNBC), which is highly MYC dependent.7,8
Cyclin-dependent kinase 9 (CDK9) has emerged as an attractive CDK target owing to its role in potentiating oncogenic transcription programs.9−13 CDK9 is a transcription-regulating CDK that acts as a subunit of the positive transcription elongation factor b (P-TEFb).14 P-TEFb is recruited to the genome by transcription factors and other components of the transcription machinery, where it is sequestered in an inactive ribonucleotide protein complex (7SK snRNP).15 Its activation is facilitated through multiple molecular mechanisms, including recruitment of specific transcription factors such as MYC, transcription elongation-promoting complexes such as those including bromodomain-containing protein 4 (BRD4) and the super elongation complex (SEC), and by splicing factors and co-transcriptional RNA processing events.16−19 Activated CDK9 within the P-TEFb complex phosphorylates multiple transcription substrates—most notably the serine 2 (Ser2) residue of the carboxy-terminal domain (CTD) heptad repeats of RNA polymerase II (RNAP II). Phosphorylation of RNAP II Ser2 is an evolutionarily conserved and rate-limiting requirement for productive transcription elongation and mRNA processing.20−22 Although CDK9 is an essential gene and broadly required for global transcription, multiple studies have implicated highly transcribed, short half-life genes such as MYC and MCL1 as being uniquely sensitive to CDK9 inhibition.17,18,23
The MYC family of protooncogenes (MYC, MYCN, and MYCL) includes the most commonly amplified genes in cancer and is associated with greater tumor aggressiveness across tumor types.25,26 These genes encode basic helix–loop–helix (bHLH) transcription factors involved in the regulation of cell growth, proliferation, and apoptosis. MYC forms a heterodimer with MAX, another bHLH transcription factor, and together they bind E-box consensus sequences (CACGTG) to effect target gene expression.27 Transcriptional initiation begins with MYC engaging E-box sites and recruiting the RNAP II complex, which is subsequently paused for further complex assembly.18,28 CDK9 releases this paused state by phosphorylating the RNAP II Ser2 and allowing transcriptional elongation to proceed, making CDK9 an attractive target to attenuate unchecked transcriptional addiction to deregulated MYC. There are several CDK9 inhibitors, each with varying CDK selectivity profiles, currently being evaluated in the clinic, including atuveciclib, VIP152, AZD4573, PRT2527, and GFH009, that aim to attenuate transcriptional deregulation.29,30 Interestingly, all of these compounds rely on intravenous (i.v.) administration.
Using our small molecule microarray (SMM) platform, we screened a library of small molecules for binding to transcriptional complexes in a cell lysate context. This led to identification of a CDK9 inhibitor hit molecule, 1 (KI-Arv-03), that exhibited excellent kinome and CDK isoform selectivity.31 Herein we report a medicinal chemistry effort to optimize this hit, prioritizing maintenance of CDK9 selectivity while improving the on-target potency and overall physicochemical and pharmacokinetic (PK) properties. This allowed us to advance the initial SMM hit to the potent, selective, orally bioavailable CDK9 inhibitor 28 (KB-0742). Compound 28 exhibits in vivo antitumor activity in multiple mouse xenograft models, including aggressive MYC-driven TNBC, a toxicology profile supporting clinical dosing, and projected human PK expected to enable efficacious oral dosing. Preclinical studies have advanced 28 to the clinic, where it is currently being investigated in a phase 1/2 dose escalation and cohort expansion study in participants with relapsed or refractory solid tumors or non-Hodgkin lymphoma (NCT04718675).
Results and Discussion
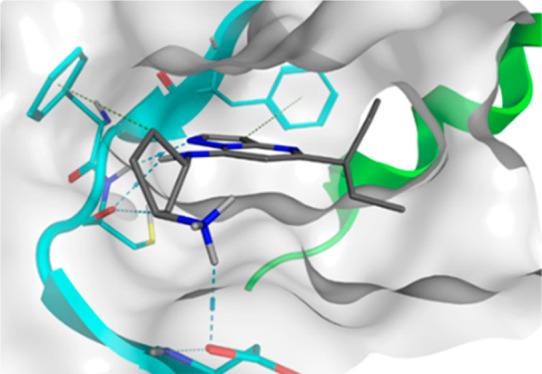
Previous work has described the identification of compound 1 via SMM screening of cell lysates and the discovery that this compound inhibited CDK9 with high isoform selectivity.32 Additional kinetic studies demonstrated ATP competition, suggesting that compound 1 binds in the catalytic cleft of CDK9. We therefore leveraged a structure-based drug design approach to further enhance the binding and physicochemical characteristics of the initial hit toward the ATP-competitive binding site. Docking of 1 into the ATP-binding site of CDK9/cyclin T1 from crystal structure PDB:3MY1 suggested binding in the catalytic cleft in a type I fashion (Figure 1).33
Figure 1.

(A) Structure of 1 and (B) predicted docking in the CDK9/cyclinT1 ATP-competitive binding site from crystal structure PDB: 3MY1.
The compound is positioned with the amino-pyrazolopyrimidine core poised in a H-bond acceptor/donor orientation that interacts with hinge residue Cys106 (NH and CO). This fixed hinge-binding of the core structure then positioned the terminal primary amine in the proximity of solvent-exposed residues Glu107 and Asp109 for a H-bond donation or ionic interaction with the respective backbone carbonyl and Asp109 side chain. The 5-propyl moiety of the pyrazolopyrimidine points to a hydrophobic patch formed by the Leu156 side chain.
We sought to exploit this docking model to identify essential interactions that would impact potency and selectivity. Beginning with a minimal pharmacophore structure–activity relationship (SAR) campaign, we hypothesized that modification of three key regions of compound 1, namely the C-3 (R3), C-5 (R1), and C-7 (R2) positions, would provide directional guidance for these early SAR efforts (Table 1 and Table 2). Using the hydrophobic isopropyl group as a standardized tool unit, we investigated the spatial requirements of the R2 group within the exit vector of the ATP-competitive binding site, specifically focusing on ligand interactions with Asp109 and Glu107 residues (Table 1). Shrinking the amino-cyclopentane ring of 1 to cyclobutane (compound 2) resulted in a minimal decrease in potency against CDK9/cyclin T1. Amino-azetidine analogue 3 lost 5-fold potency. Removing an H-bond donor by methylating the azetidine (compound 4) eliminated a Glu107 interaction and introduced hydrophobicity in the solvent-exposed exit vector, having a detrimental effect on potency. Oxetane moieties, as with 5, exhibited potency similar to that of the azetidine (3) and further reinforced that the introduction of the methyl, rather than H-bond donor omission, drove the observed potency loss. Extension off the cyclobutane ring with a hydroxyl group as shown in 6 delivered an IC50 = 183 nM biochemical response, reinstating some lost potency as compared to that with cyclobutane only. Alkylation of the hydroxyl group had minimal effect, as illustrated by ethereal compound 7.
Table 1. Structure and Activitya,b against CDK Enzymes 1–9 of Compounds 2–26.
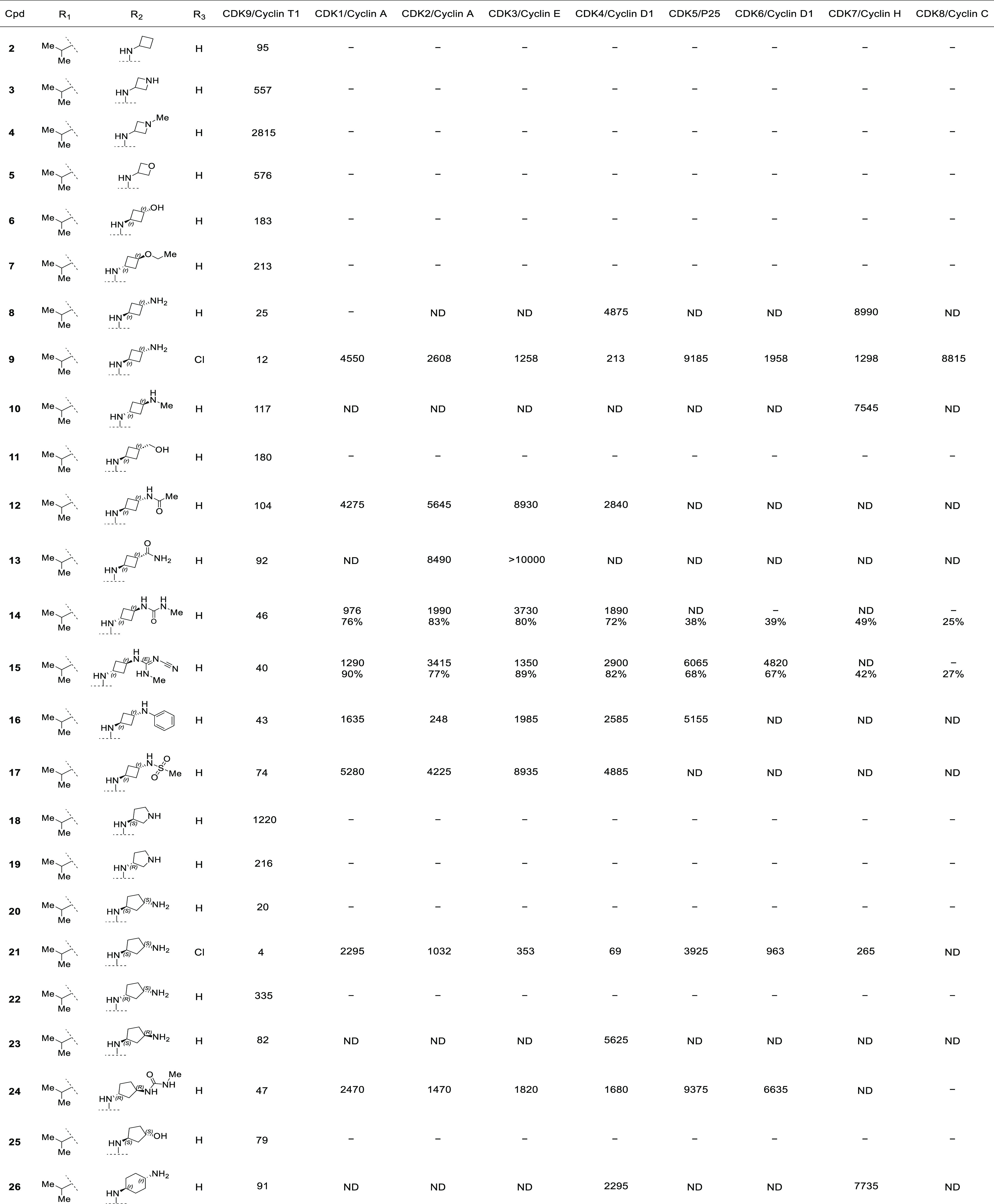
Geometric means of two IC50 determinations per compound as shown in nM at 10 μM [ATP].
Percent inhibition shown as geometric mean of two single-dose 10 μM compounds at CDK9/cyclin T1 Km of 10 μM [ATP]. ND: IC50 not determined from collected data; − : no data collected.
Table 2. Structure and Activitya,b against CDK Enzymes 1–9 of Compounds 1 and 27–37.
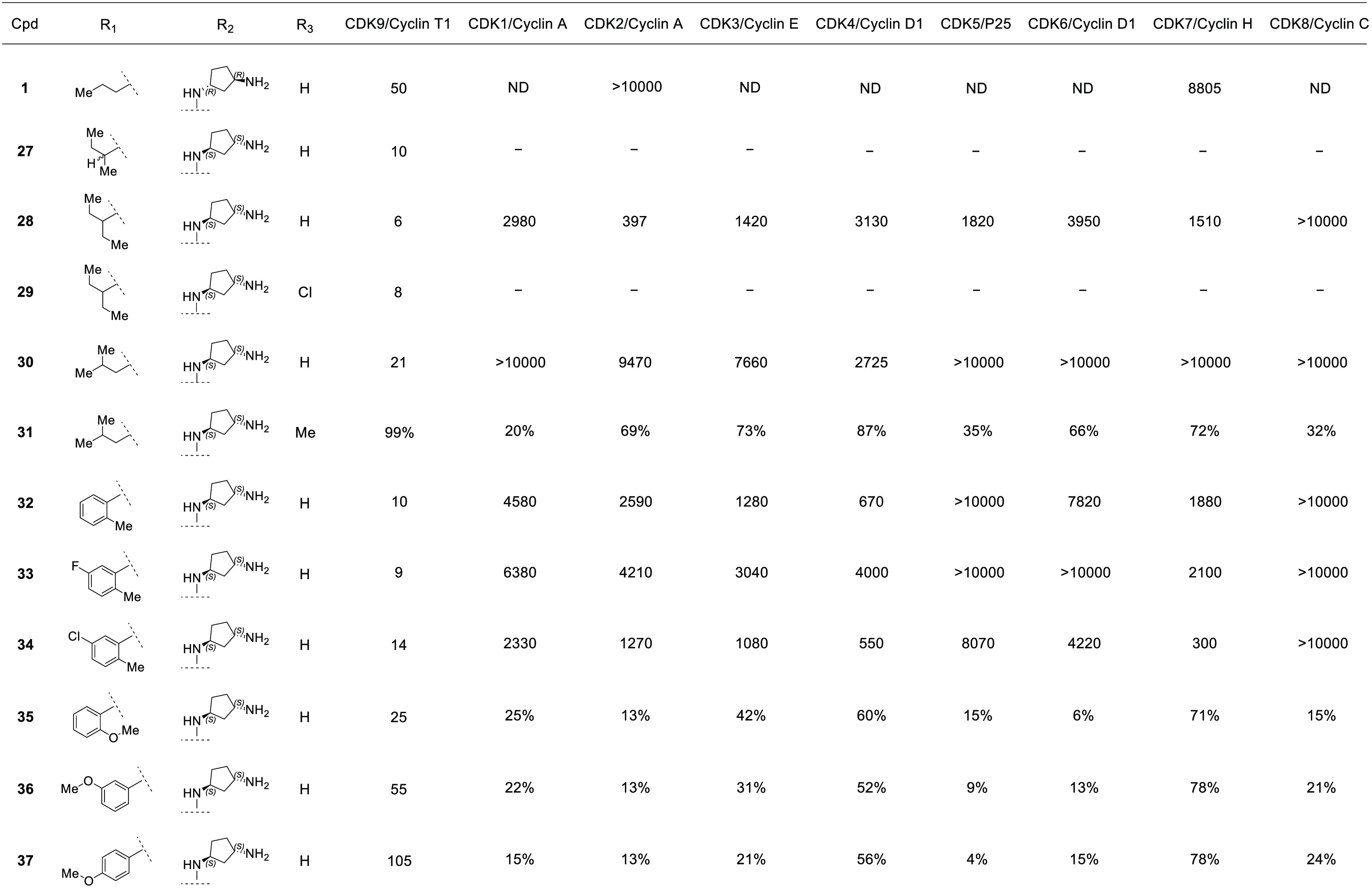
Geometric means of two IC50 determinations per compound as shown in nM at 10 μM [ATP].
Percent inhibition shown as geometric mean of two single-dose 10 μM compound at CDK9/cyclin T1 Km of 10 μM [ATP]. ND: IC50 not determined from collected data. ‘-‘: no data collected.
Installation of a pendant amine, as in 8, instead of a hydroxyl (6) enhanced potency by approximately 7-fold; we speculate that 8 allows for a more optimal interaction with Asp109 and Glu107 residues. Analogue 8 was then submitted to a CDK panel and displayed activity on CDK4 and CDK7, with 195-fold and 360-fold selectivity for CDK9, respectively. An analogue with a chlorine atom at the 3-position of the pyrazolopyrimidine (9) was more potent on CDK9 (12 nM) but lost selectivity, particularly on CDK4, where the fold selectivity dropped from 195-fold to 18-fold. Monomethylation analogue 10 lost potency as compared to 8, demonstrating the need for two hydrogen atoms capable of H-bonding in the space between Asp109 and Glu107.
Changing the distance and electronic requirements of the ability to hydrogen bond with Asp109 and Glu107 residues within R2 had mixed effects. Extending the HBD/HBA group further toward the solvent with a hydroxy-methylene unit, as in 11, approximated its non-methylene analogue 6. The acetamide 12 and amide 13 saw little change relative to the monomethylated amine form 10, suggesting that electronically manipulating the HBA ability on the nitrogen alone has little effect on potency. This idea was further enforced with methyl sulfonamide analogue 17, which showed only a minor improvement of potency and a similar selectivity profile. However, the methyl urea 14 saw an increase in potency to 46 nM and a ≥21-fold selectivity toward the reference CDK panel. We suggest that both urea N–H groups can productively H-bond with Asp109. A similar observation can be seen with cyano-guanidine 15, which improved the selectivity to ≥32-fold over other CDKs tested. Aniline 16 was equally potent to both cyano-guanidine and urea moieties but was less selective against CDK2.
We next investigated 5-membered rings, beginning with 3-aminopyrrolidines 18 and 19. Interestingly, the (R)-enantiomer was approximately 6-fold more potent than the (S)-enantiomer. Switching back to the diaminocyclopentane series, as similarly seen in the initial SMM hit (1), we surprisingly found the (S,S)-configuration (20) to be exceptionally potent, rivaling that of diaminocyclobutane compound 8. That same analogue with a chlorine atom attached to the 3-position of the pyrazolopyrimidine core (21) was even more potent at 4 nM against CDK9 but lost selectivity, particularly against CDKs 3, 4, and 7. Neither the (R,S)- nor (S,R)-diastereomers (22 and 23, respectively) were as potent as 20 against CDK9. The methylurea variation of the 5-membered-ring series (24) was similarly potent as the 4-membered-ring comparator (14), with a ≥31-fold selectivity toward the reference CDK panel. Hydroxylated aminocyclopentane 25 was more potent than its cyclobutane counterpart 6 but failed to show enhanced improvements over the diaminocyclobutane (8) or diaminocyclopentane (20) analogues. The diaminocyclohexane (26) demonstrated a decreased potency as compared to its cyclobutane (8) and cyclopentane (20) comparators, implying a ring size boundary for analogue binding. Based on our results, we believe that any R2 substitution that interacts with both Asp109 and Glu107 improves the hinge binding of the amino-pyrazolopyrimidine core and leads to increased biochemical potency. R2 substituents that bind only one residue (especially Asp109) tend to demonstrate decreased biochemical potency, presumably due to a weaker or more transient interaction of the amino-pyrazolopyrimidine core with hinge residue Cys106.
After assessing the tested R2 variations, we found the (S,S)-diaminocyclopentane series to be optimal. We next evaluated different R1 groups attached to the 5-position of the pyrazolopyrimidine core (Table 2). Based on our model, these variations would point to a hydrophobic patch within the CDK9/cyclin T1 formed by the Leu156 side chain. We embraced our minimal pharmacophore approach and incrementally enlarged the isopropyl group of 20. A single methyl group extension to make the 2-butyl compound 27 as a mixture of diastereomers at the pseudo-benzylic carbon was tested and displayed enhanced potency,with a biochemical IC50 = 10 nM. An additional methyl to make symmetric 3-pentyl 28 delivered a 6 nM compound. A subsequent CDK selectivity panel demonstrated that this compound possessed a ≥66-fold selectivity toward CDK9 compared to others tested. The 3-chloro variant (29) did not show significantly improved activity against CDK9. Further alkyl variants, including 2-methylpropyl (compounds 30 and 31), failed to show improvement beyond 28.
We next evaluated aryl R1 groups and found ortho-substitution, especially with smaller groups such as methyl (32), to maintain potency and overall selectivity comparable to that of 28. This substitution trend is further exemplified with ortho-, meta-, and para-anisole analogues 35, 36, and 37, respectively. Interestingly, fluorinated tolyl compound 33 had a biochemical IC50 of 9 nM against CDK9 and an excellent selectivity profile, with ≥233-fold selectivity toward CDK9 over other tested CDKs. Switching to chlorine, as in compound 34, maintained CDK9 potency compared to its fluorine counterpart 33 but significantly lost selectivity toward CDKs 4 and 7. Given its biochemical potency on CDK9 and selectivity within the tested CDK panel, 28 was chosen for further evaluation. The stereochemical requirement of the diamino-cyclopentane was assessed as illustrated in Table 3, showcasing the (S,S) configuration as the most potent configuration. Each isomer was then subjected to selectivity profiling. Table 4 illustrates the exquisite selectivity profile of compound 28 within the tested CDKs (selectivity profiling of compounds 38, 39, and 40 can be found on page S32 of the Supporting Information). We docked the four isomers into the ATP-competitive binding pocket of CDK9 and found the associated strain energy, respective to each isomer, to scale almost linearly according to potency, with the (S,S) configuration being the most energetically favored (Figure 2).
Table 3. Diamino-cyclopentane Stereochemical Comparison against CDK9/Cyclin T1.

Enzymatic assay conducted at 10 μM ATP concentration (CDK9/cyclin T1 at Km).
Table 4. CDK Selectivity Profile of Compound 28.
| Enzymea | [ATP] (μM) | IC50(nM) |
|---|---|---|
| CDK9/Cyclin T1 | 10 | 6 |
| CDK1/Cyclin A | 10 | 2980 |
| CDK2/Cyclin A | 10 | 397 |
| CDK3/Cyclin E | 100 | 1420 |
| CDK4/Cyclin D1 | 100 | 3130 |
| CDK5/P25 | 10 | 1820 |
| CDK6/Cyclin D1 | 100 | 3950 |
| CDK7/Cyclin H | 50 | 1510 |
| CDK8/Cyclin C | 10 | >10000 |
| CDK12/Cyclin K | 30 | 589 |
| CDK13/Cyclin K | 5 | 372 |
| CDK14 (PFTK1)/Cyclin Y | 15 | 6250 |
| CDK16/Cyclin Y | 10 | 1580 |
| CDK17/Cyclin Y (PCTK2) | 20 | 2150 |
| CDK18/Cyclin Y | 20 | 1060 |
| CDK19/Cyclin C | 20 | >10000 |
Enzymatic assay conducted at Km (μM) ATP concentration.
Figure 2.

Computational model highlighting the associated strain energy of diamino-cyclopentane diastereomers within the ATP-competitive binding site. (a) Overlay of diamino-cyclopentane diastereomers 28, 38, 39, and 40 within the ATP-competitive binding site of CDK9/cyclin T1. (b) Predicted strain energy (kcal/mol) plotted against the IC50 of each diastereomer.
Co-crystallization of compound 28 with CDK9/cyclin T1 confirmed ligand binding as predicted in our model, albeit at 3.8 Å resolution (Figure 3). Regardless of the low resolution, the co-crystal structure provided a partial density to confirm our predicted binding mode of compound 28 (Figure 2). In brief, the amino-pyrazolopyrimidine core of 28 aligns to hinge Cys106 of the ATP-competitive binding pocket as postulated; the terminal amine of the diamino-cyclopentane moiety interacts with Asp109 in a HBD manner; and the 3-pentyl group points to a hydrophobic patch formed by the Leu156 side chain.
Figure 3.
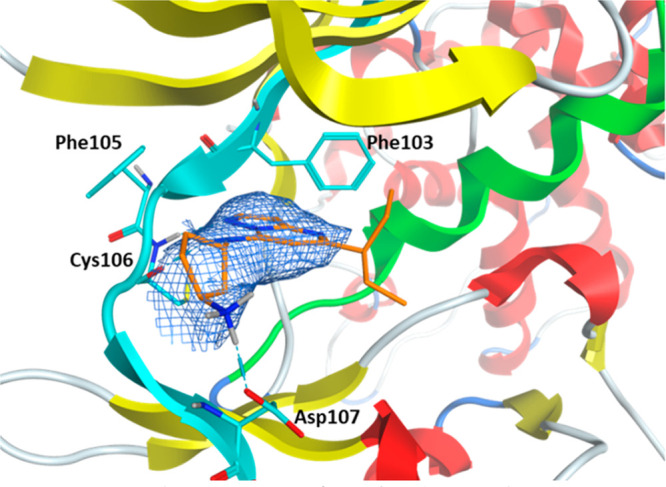
Crystal structure of 28 bound to the complex of CDK9 (cyan) and cyclin T1 (gray) (PDB: 8K5R). The ligand carbon atoms are shown in orange.
Compound 28 was further profiled in a kinase panel of 631 kinases (375 wild-type, 232 mutant, and 24 atypical kinases). It was found to retain selectivity as compared to SMM hit 1, displaying enhanced potency toward CDK9/cyclin T1 with greater than 66-fold selectivity over all CDKs profiled and greater than 100-fold selectivity against cell-cycle CDKs 1, 3, 4, 5, and 6 (Supporting Information, pages S11–S32).32
The cellular activity of 28 was evaluated in vitro using TNBC cell culture models utilizing an assay that leverages fluorescently labeled antibodies and high-content fluorescence imaging to measure cell proliferation, apoptosis, and cell-cycle arrest in a multiplexed format. Compound 28 was found to inhibit cellular growth, displaying cytostatic (GI50 values ranging from 530 nM to 1 μM) and cytotoxic (IC50 values ranging from 600 nM to 1.2 μM) effects among the TNBC cell lines tested (Table 5). Notably, 28 potently induced apoptosis in 4 of 5 tested lines.
Table 5. Summary of Compound 28’s Activity in TNBC Cell Linesa.
|
Cell countb |
Apoptosisc |
||||
|---|---|---|---|---|---|
| Cell line | GI50(μM) | IC50(μM) | 5-Fold Induction (μM) | Emax | dG1/S cell cycle block (μM) |
| BT-20 | 0.60 | 0.97 | 7.73 | 6.22 | 0.95 |
| BT-549 | 0.80 | 0.94 | 0.74 | 7.99 | 1.42 |
| MDA-MB-231 | 0.80 | 0.88 | 0.64 | 32.4 | 1.78 |
| MT-3 | 0.53 | 0.60 | 0.67 | 15.1 | 1.16 |
| Hs 578T | 1.06 | 1.23 | 0.78 | 7.24 | 2.13 |
BT-20, BT-549, MDA-MB-231, MT-3, and Hs 578T were treated with compound 28 in a serial dilution over 10 concentrations with a maximum of 0.1% DMSO over 72 h.
Cell proliferation was measured by the fluorescence intensity of an incorporated nuclear dye.
Apoptosis was measured by the fluorescence intensity of a fluorescently labeled antibody to activated caspase 3. The output is shown as a fold increase of apoptotic signal over vehicle background normalized to the relative cell count in each well. The concentration of test compound that caused a 5-fold induction in the caspase-3 signal is reported, indicating a significant apoptosis induction.
Cell cycle arrest was measured by labeling of mitotic cells with a fluorescently labeled antibody to phosphorylated histone H3.
In addition, the physicochemical and ADME properties of 28 were evaluated (Table 6). Compound 28 possesses a molecular weight, logD, and solubility parameters within the acceptable ranges for drug-like oral compounds. The compound performed well in a Caco-2 assay at 2, 10, and 50 μM, demonstrating permeability with an efflux ratio ranging from 1.48 to 1.85. Inhibition assays against a panel of five cytochrome P450 enzymes (CYP3A4, CYP2D6, CYP2C9, CYP2C19, and CYP1A2) indicated no inhibition of CYP3A4, CYP2D6, CYP2C9, and CYP2C19 isoforms (IC50 > 30 μM) and weak activity against CYP1A2 (IC50 = 15.3 μM). There was no observed IC50 shift in time-dependent inhibition (TDI) of the six tested cytochrome P450 isoforms (CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4) in the presence or absence of NADPH at 0 or 30 min. Compound 28 demonstrated low intrinsic clearance (Clint) in mouse, rat, dog, and human microsomes and low to moderate clearance in hepatocytes, resulting in t1/2 ≥ 2 h across these assays. Compound 28 exhibited a high free fraction in mouse and human plasma protein binding assays and was stable in plasma of all tested species beyond 360 min.
Table 6. Physical and In Vitro ADME Properties of Compound 28.
| Physical Properties | |
|---|---|
| Molecular weight (g/mol) | 287 |
| Kinetic solubility, pH = 7.4 (μM)a | 191 |
| LogD, pH = 7.4a | 0.9 |
| TPSA (Å2) | 68 |
| In Vitro ADMEa | |
|---|---|
| Caco-2 Papp A-B (10–6 cm/s)b | 95.7–84.2 |
| Caco-2 Papp B-A/A-Bb | 1.48–1.85 |
| CYP3A4/2D6/2C9/2C19/1A2 IC50 (μM) | >30/>30/>30/>30/15.3 |
| PPB m/r/d/h (% bound) | 48/41/72/38 |
| Plasma stability t1/2 m/r/d/h (min) | >360/2803/>360/>360 |
| Microsome stability m/r/d/h (% remaining 180 min) | 87.4/81.3/68.8/92.5 |
| Microsome Clint m/r/d/h (mL/min/kg)c | 7.0/5.5/5.5/0.93 |
| Microsome stability t1/2 m/r/d/h (min) | 775/510/283/1341 |
| Hepatocyte Clint m/r/d/h (mL/min/kg)d | 44/62/38/1.6 |
| Hepatocyte stability t1/2 m/r/d/h (min) | 374/118/195/2339 |
Values are the geometric means of at least two replicates.
Compounds were incubated at 2, 10, and 50 μM in cultured Caco-2 cells.
Plasma protein binding were determined at 0.1, 1, and 10 μM. Data shown is the average across the tested concentrations.
Liver microsome intrinsic clearance. Intrinsic clearance measured from fresh hepatocytes and cryopreserved human hepatocytes. PPB: plasma protein binding; Clint: intrinsic clearance; m/r/d/h: mouse/rat/dog/human.
Consistent with its favorable physicochemical and in vitro ADME properties, compound 28 exhibited good overall PK profiles in mouse, rat, and dog (Table 7). Plasma half-lives (T1/2) following intravenous dosing were 1.2 h in mouse, 2.4 h in rat, and 4.7 h in dog. The plasma clearance was low to moderate relative to hepatic blood flow in all preclinical species. Apparent steady state volumes of distribution (Vd,ss) varied slightly across species, with values ranging from 3.9 L/kg in mouse to 7.3 L/kg in rat. The compound showed oral bioavailability following p.o. administration of a solution formulation in mouse (33%) and rat (84.5%), and high oral bioavailability in dog (>100%). A combination of in vitro–in vivo extrapolation and allometric methods was utilized for the projection of human PK parameters for 28, with and without plasma protein binding corrections.34−36 Based on a 70 kg body weight, the mean predicted human plasma clearance (Cl) and Vd,ss are 2.0 mL/min/kg and 6.0 L/kg, respectively, together yielding an estimated terminal half-life of approximately 35 h, assuming one-compartment pharmacokinetics (half-life = [ln 2] × Vss/Cl). The predicted human oral bioavailability, derived from nonclinical species, is 75%. The projected long plasma half-life should enable 28 to achieve sustained exposures while avoiding high peak (Cmax) concentrations (Figure 4) as we explore dose escalation to a pharmacologically active dose in the clinic.
Table 7. Pharmacokinetic Parameters of Compound 28 in Mouse, Rat, and Dog.
| Species | T1/2(i.v., h) | Clint(i.v.,mL/min/kg) | Vd,ss(i.v.,L/kg) | Cmax(p.o., ng/mL) | AUC (p.o., ng·h/mL) | F (%) |
|---|---|---|---|---|---|---|
| Mousea | 1.2 | 23.4 | 3.9 | 501 | 1700 | 33 |
| Ratb | 2.4 | 51.7 | 7.3 | 411 | 3591 | 84.5 |
| Dogc | 4.7 | 13.4 | 4.5 | 494 | 4007 | >100 |
Vehicle used in intravenous and oral administration studies: EtOH, PEG400, HPβCD, 1/2/7 (v/v/v) at 1.0 mg/kg 28 for i.v. and 20 mg/kg 28 for p.o.
Vehicle used in intravenous and oral administration studies: 2.0 mg/kg 28 at pH 3.0 in 50 mM citrate buffer for i.v. and 10 mg/kg 28 at pH 3.0 in 50 mM citrate buffer for p.o.
Vehicle used in intravenous and oral administration studies: 0.50 mg/kg 28 in saline for i.v. and 2.5 mg/kg 28 in saline for p.o. T1/2: half-life; Clint: intrinsic clearance; Vd,ss: volume of distribution; F: bioavailability
Figure 4.
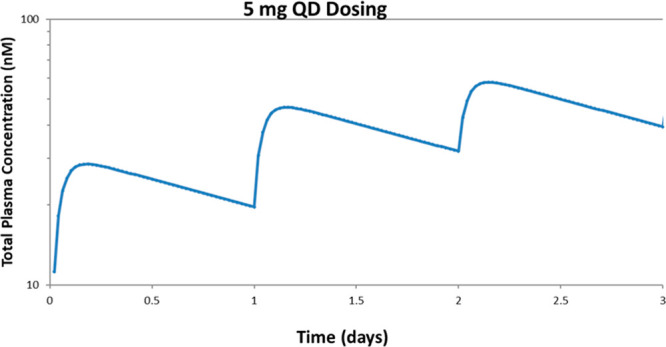
Simulated human pharmacokinetic (PK) profile for oral dosing of compound 28 at 5 mg daily with a clearance of 2 mL/min/kg and a volume distribution projection of 6 L/kg, giving a mean projected drug half-life of 35 h. Assumptions include PK linearity, one compartment, and 75% bioavailability.
Next, we sought to assess the pharmacodynamic (PD) effects and efficacy of 28 on in vivo tumor growth in MYC-amplified TNBC patient-derived xenograft (PDX) models. To assess the on-target PD of 28, we examined MYC levels and phosphorylation of RNAP II Ser 2 (pSER2) as downstream indicators of CDK9 activity in xenograft tumor samples. Both MYC and pSER2 were significantly reduced by 2 h post-terminal dose of 28 while plasma concentration of 28 was still high (Figure 5), consistent with CDK9 inhibition. By 8 h post-administration, both MYC and pSER2 levels were no longer significantly different from vehicle. This observation combined with the low plasma levels of compound 28 suggests target engagement with downstream effects on MYC and pSER2.
Figure 5.

Compound 28 decreased the levels of MYC (a) and pSER2 (b) in TNBC PDX CTG-1017 subcutaneous tumors. Tumor-bearing Athymic Nude-Foxn1nu mice were collected at 2 and 8 h post-terminal dose of either vehicle or 60 mg/kg 28 p.o. using an intermittent dosing schedule of 3-days on, 4-days off for up to 4 weekly cycles. Tumor lysates were prepared, and levels of MYC (a) and pSER2 (b) were determined using their respective HTRF assays. The plasma concentration of 28 was 846 ng/mL (2.9 μM) and 73 ng/mL (0.25 μM) at 2 and 8 h post-dose, respectively. Data are presented as mean ± SEM, n = 5. *P < 0.05; **P < 0.005; ***P < 0.0005; ****P < 0.0001. hr: hour; HTRF: homogeneous time-resolved fluorescence; LLoQ: lower limit of quantification; ns: not significant.
We evaluated in vivo anti-tumor activity of 28 in human MYC-amplified PDX models, focusing on TNBC cell lines CTG-1017 (Figure 6), CTG-0869, and CTG-0437 (Supporting Information, page S34). Athymic Nude-Foxn1nu mice bearing established subcutaneous PDX tumors were treated with vehicle, standard of care (SOC) chemotherapeutics, or 28. Tumor growth inhibition by 28 was observed in all models, with tumor growth inhibition by 28 being comparable to SOC for both CTG-1017 and CTG-0437 models, highlighting that the observed on-target PD effects correspond with significant antitumor effects in vivo.
Figure 6.

In vivo tumor growth inhibition for compound 28 in the TNBC model CTG-1017. Athymic Nude-Foxn1nu mice bearing established subcutaneous PDX tumors (mean starting tumor volume of 157 mm3) were treated with vehicle (saline), 28, or SOC chemotherapeutics (cisplatin and gemcitabine). Vehicle and 28 at 60 mg/kg were administered orally using an intermittent dosing schedule of 3-days on, 4-days off for up to 4 weekly cycles (day 30 TGI = 82%, p = 0.0001 vs vehicle). SOC were administered as follows: cisplatin 5 mg/kg i.p. Q7D×3 + gemcitabine 100 mg/kg i.p. Q7D×3. The corresponding mean body weight over time graph can be found in the Supporting Information.
Conclusion
From discovery hit 1, we report the optimization of an SMM screening hit using a structure-based drug design approach that led to the discovery of clinical candidate 28 (KB-0742), a potent, selective, orally bioavailable CDK9 inhibitor. Compound 28 has pharmacological and physicochemical properties and exhibits in vivo antitumor activity in TNBC mouse xenograft models. In vitro, 28 reduced MYC and pSER2 in tumor lysates in a concentration-dependent manner and substantially inhibited in vivo tumor growth at the same dose that reduced levels of these PD markers. Further, 28 has a PK profile that projects sustained human exposure conducive to efficacious oral dosing in the clinic. These findings have advanced 28 to the clinic in an ongoing phase 1/2 dose escalation and expansion trial in patients with relapsed or refractory solid tumors or non-Hodgkin’s lymphoma (NCT04718675).
Experimental Section
Synthetic Chemistry
The syntheses of compounds 2–40 is described in Schemes 1–5 below and Schemes S3–S6 of the Supporting Information. The general approach focuses on modifications at three key carbon centers. The C-3 position (Figure 7) was functionalized through oxidative halogenation and potential further elaboration using Suzuki coupling chemistry. As a means to conserve the amino-pyrazolopyrimidine hinge binding motif, nucleophilic aromatic substitution of substituted primary amines with 5,7-dichloro-pyrazolo[1,5-a]pyrimidine gave C-7 substituted 5-chloro-pyrazolopyrimidines. Installation of the aryl R1 groups on the C-5 position also employed a Suzuki coupling of aryl boronic acids with 5-chloro-pyrazolo[1,5-a]pyrimidine intermediates. Alternatively, when hydrophobic moieties at the C-5 position were alkyl, a Masamune reaction accessed requisite β-ketoesters for a subsequent condensation of amino-pyrazole and deoxychlorination to deliver the 7-chloro-pyrazolo[1,5-a]pyrimidine intermediates. These compounds were then subjected to nucleophilic aromatic substitution with primary amines. Depending on the nature of each compound, deprotection and further amidation would deliver final targets.
Scheme 1. Synthesis of Four-Membered-Ring Minimal Pharmacophore Derivatives through Amination of Chloro-pyrazolo[1,5-a]pyrimidine 43.

Reagents and conditions: (a) 1H-pyrazol-5-amine, AcOH, reflux; (b) POCl3, pyridine, DMAP, MeCN, reflux; (c) RNH2, K2CO3, MeCN, reflux; (d) HCl, 1,4-dioxane, 0 °C; (e) LiAlH4, THF, 0–80 °C; (f) Ghaffar–Parkins catalyst, H2O, THF, 70 °C.
Scheme 5. Synthesis of 1,3-Diaminocyclopentane Isomers via Chloro-pyrazolo[1,5-a]pyrimidine 49.

Reagents and conditions: (a) 1H-pyrazol-5-amine, AcOH, reflux; (b) POCl3, pyridine, DMAP, MeCN, reflux; (c) K2CO3, MeCN, reflux; (d) HCl, 1,4-dioxane, 0 °C.
Figure 7.
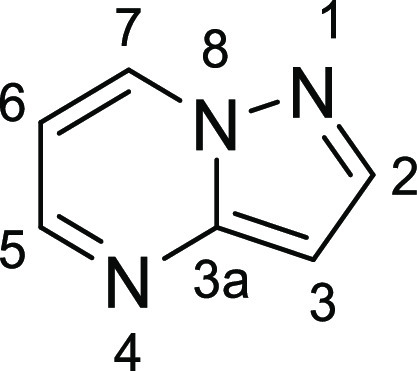
Numbering convention used for pyrazolo[1,5-a]pyrimidine core.
Compounds 2–8, 10, 11, 13, and 16 (Scheme 1) commenced from a condensation of amino-pyrazole with ethyl 4-methyl-3-oxopentanoate (41) (see Supporting Information for synthesis) to create intermediate pyrazolo[1,5-a]pyrimidine-7(4H)-one (42). Subsequent deoxychlorination staged intermediate (43) for a parallel medicinal chemistry effort focused on a four-membered-ring series installed via nucleophilic aromatic substitution. Compounds 2, 5–7, 11, and 16 were delivered directly. Unmasking of compounds 3 and 8 occurred after the Boc-deprotection of their precursor intermediates 3a and 8a, respectively. Reduction of 3a and 8a with LiAlH4 gave methylated analogues 4 and 10. 13a was converted to the carboxamide (13) using the Ghaffar–Parkins catalyst.
Compound 8 was then leveraged to access elaborated pendant amines (Scheme 2). Direct acylation of 8 delivered the amide 12. Methyl-urea compound 14 was created by treating 8 with S-methyl methylaminoethanethioate. Construction of tool compound 15 arose via exposure of 8 with diphenyl N-cyanocarbonimidate to build the intermediate phenyl-isourea (not shown), which was subsequently treated with methyl amine. Direct treatment of 8 with methylsulfonyl chloride gave sulfonamide 17.
Scheme 2. Modular Elaboration of Aminocyclobutane 8.

Reagents and conditions: (a) AC2O, pyridine; (b) S-methyl methylaminomethanethioate, 1,4-dioxane, H2O, 65 °C; (c) diphenyl N-cyanocarbonimidate, iPrOH, 70 °C; (d) MeNH2, iPrOH, 70 °C; (e) MeSO2Cl, NaHCO3, DIPEA, CH2Cl2.
The synthesis of five- and six-membered-ring compounds (Scheme 3) utilized a similar approach starting with chloro-pyrazolo[1,5-a]pyrimidine intermediate 43 to construct compounds 19–26. Nucleophilic aromatic substitution delivered 25 and Boc-protected intermediates 18a–20a, 22a–24a, and 26a from their corresponding primary amine building blocks. Subsequent deprotection with hydrochloric acid in dioxane provided the targeted analogues 18–20, 22, 23, 24b, and 26 in a rapid fashion. Furthermore, creation of methyl urea compound 24 arose from treatment of 24b with S-methyl N-methylcarbamothioate.
Scheme 3. Synthesis of Five- and Six-Membered-Ring Minimal Pharmacophore Derivatives through Amination of Chloro-pyrazolo[1,5-a]pyrimidine 43.

Reagents and conditions: (a) RNH2, K2CO3, MeCN, reflux; (b) HCl, 1,4-dioxane, 0 °C; (c) S-methyl N-methylcarbamothioate, 1,4-dioxane, H2O, 65 °C.
An alternative synthetic strategy was employed (Scheme 4) when the proposed analogues contained an aryl group at position 5 of the pyrazolo[1,5-a]pyrimidine core (compounds 32–37). Exploration of various aryl SAR trends started with a nucleophilic aromatic substitution of commercially available 5,7-dichloro-pyrazolo[1,5-a]pyrimidine (44) material with tert-butyl ((1S,3S)-3-aminocyclopentyl)carbamate to give compound 45. Subsequent Suzuki–Miyaura coupling with a targeted grouping of aryl boronic acids gave penultimate compounds 32a and 34a–37a. Deprotection then accessed final targets 32 and 34–37. In the case of 5-fluoro-2-methylphenyl substitution (33), the deprotection occurred prior to the coupling of the corresponding aryl boronic acid with intermediate 46.
Scheme 4. Suzuki–Miyaura Coupling Route to 5-Aryl-Substituted Pyrazolo[1,5-a]pyrimidine Analogues.

Reagents and conditions: (a) N1-Boc-((1S,3S)-3-aminocyclopentyl), TEA, MeCN, 50 °C; (b) N1-Boc-(1S,3S)-3-aminocyclopentyl), K2CO3, MeCN, reflux; (c) HCl, EtOAc; (d) ArB(OH)2, Pd2(dba)3, XantPhos, K2CO3, 1,4-dioxane, 100 °C; (e) 4 N HCl, 1,4-dioxane; (f) TFA, CH2Cl2 (g) ArB(OH)2, Pd(dppf)Cl2, K2CO3, 1,4-dioxane, H2O; (h) ArB(OH)2, Pd(PPh3)4, K3PO4, DMF.
Stereoisomers 28 and 38–40 (Scheme 5) were made starting from a condensation of amino-pyrazole with ethyl 4-ethyl-3-oxohexanoate (47) to create intermediate pyrazolo[1,5-a]pyrimidin-7-(4H)-one (48) similar to what was described in Scheme 1. Deoxychlorination of 48 gave compound 49, a compound positioned for subsequent nucleophilic aromatic substitution by the four possible stereoisomers of N-Boc-diamino-cyclopentane. Deprotection of intermediate amine-protected analogues 28a and 38a–40a delivered targeted stereoisomers 28 and 38–40.
General Chemistry
All reactions were carried out under an inert gas atmosphere (nitrogen or argon). Commercial reagents and anhydrous solvents were used without further purification. Removal of volatile organic solvents was performed by rotary evaporation, and residual trace solvents were removed via a high-vacuum manifold. Flash chromatography was performed on Combiflash Next Gen 100 from Teledyne ISCO using Agela Claricep 4–40g Si C-series silica gel 230–400 mesh cartridges. All reported yields are isolated yields. Preparative reversed-phase high-pressure liquid chromatography (prep-HPLC) was performed using an Agilent 1200 series instrument equipped with a Waters XBridge C18 OBD Prep Column (10 μm, 150 mm × 50 mm i.d.), eluting with a binary solvent of H2O and acetonitrile (ACN) using gradient elution (15–45% acetonitrile in a 10 mM ammonium bicarbonate solution in water, 8 min gradient), and detected via UV 220 nm. All assayed compounds were of ≥95% purity as determined by HPLC. UV detection at 220 nm was performed using an Agilent 1200 series with diode array detector (DAD) equipped with one of the following three columns: Phenomenex Gemini NX-C18 (3 μm, 100 mm × 4.6 mm i.d.), Agilent Zorbax Extend-C18 (5 μm, 150 mm × 4.6 mm i.d.), or Phenomenex Luna Omega Polar C18 (3 μm, 100 mm × 4.6 mm i.d.). A general method was employed: mobile phase, A = ACN, B = 10 mM NH4OAc in H2O; gradient, 5–95% A (0.0–18 min); flow rate = 1.0 mL/min; inj. vol. = 2.0 μL. Low-resolution liquid chromatography mass spectra (LCMS) were determined via a low-resolution electrospray ionization (ESI) source using any one of the following instruments and column combinations: Agilent 1260 Infinity II UPLC attached with Agilent 6110B single quad mass detector using a YMC Triart C18 column (3 μm, 33 mm × 2.1 mm); Waters Acquity UPLC attached with Waters ZQ SQ mass detector using a YMC Triart C18 column (3 μm, 33 mm × 2.1 mm); or Agilent 1260 Infinity II UPLC attached with Agilent 6110B single quad mass detector using a Phenomenex Kinetex C18 column (5 μm, 50 mm × 2.1 mm i.d.). High-resolution mass spectra (HRMS) were obtained on a Shimadzu Prominence HPLC coupled with an Applied Biosystems API2000/2000 Trap mass detector configured with an ESI source using a Waters Xbridge C18/Agilent Zorbax C18 column (5 μm, 50 mm × 4.6 mm i.d.). 1H NMR spectra were obtained on either a Bruker 400 MHz Ultra shield or Bruker 400 MHz Ascend spectrometer equipped with an Avance Neo, Avance III, or Avance III-HD console. Chemical shifts (δ) are reported in parts per million (ppm) relative to residual undeuterated solvent as an internal reference. The following abbreviations were used to label peak multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, quin = quintet; dd = doublet of doublets, dt = doublet of triplets, dtd = doublet of triplet of doublets, m = multiplet, br = broad.
5-Isopropyl-4H-pyrazolo[1,5-a]pyrimidin-7-one (42)
A mixture of ethyl 4-methyl-3-oxo-pentanoate (900 mg, 5.69 mmol) and 1H-pyrazol-5-amine (473 mg, 5.69 mmol) in glacial acetic acid (10 mL) was heated under reflux for 3 h. The solvent was evaporated in vacuo, and the residue was treated with ethyl acetate and filtered to give compound 42 (650 mg, 3.67 mmol, 64.5% yield) as a yellow solid. LCMS (ESI) m/z: 177.8 [M+H]+.
7-Chloro-5-isopropylpyrazolo[1,5-a]pyrimidine (43)
A stirred solution of 42 (100 mg, 0.560 mmol) in POCl3 (1.06 mL, 11.3 mmol) was heated to reflux for 2 h. The reaction mixture was cooled to rt, excess reagent was removed in vacuo, and the residue was treated with ice–water. The product was extracted from an aqueous mixture by DCM. The organic layer was separated, dried over anhyd. Na2SO4, and purified by Combiflash column chromatography (silica gel, 230–400 mesh), eluting with 10% ethyl acetate in hexanes to give compound 43 (65 mg, 0.332 mmol, 58.9% yield) as a light-yellow liquid. LCMS (ESI) m/z: 195.9 [M+H]+.
tert-Butyl ((1R,4R)-4-((5-Isopropylpyrazolo[1,5-a]pyrimidin-7-yl)amino)cyclohexyl)carbamate (26a)
A stirred solution of 43 (75 mg, 0.380 mmol), tert-butyl ((1R,4R)-4-aminocyclohexyl)carbamate (98.6 mg, 0.460 mmol), and K2CO3 (106 mg, 0.770 mmol) in MeCN (10 mL) was heated to reflux for 16 h. The reaction mixture was cooled and filtered. The filtrate was concentrated in vacuo, and the residue was purified by Combiflash column chromatography, eluting with 25% ethyl acetate in hexanes to give compound 26a (100 mg, 0.268 mmol, 69.9% yield) as a white solid. LCMS (ESI) m/z: 374.2 [M+H]+.
(1R,4R)-N1-[4-[(5-Isopropylpyrazolo[1,5-a]pyrimidin-4-ium-7-yl)amino]cyclohexyl]ammonium dichloride (26)
To compound 26a (80 mg, 0.210 mmol) was added 4 M HCl in dioxane (2 mL, 0.210 mmol), and the mixture was stirred at rt for 2 h. The reaction mixture was evaporated in vacuo to give compound 26 (64.8 mg, 0.18 mmol, 83.9% yield) as an off-white solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.24 (1 H, s), 6.53 (2 H, d, J = 16.3 Hz), 4.06–3.86 (1 H, m), 3.25–2.84 (2 H, m), 2.11–1.86 (4 H, m), 1.70 (2 H, q, J = 12.0 Hz), 1.54 (2 H, d, J = 11.9 Hz), 1.31 (6 H, d, J = 6.8 Hz). LCMS (ESI) m/z: 274.2 [M+H]+.
tert-Butyl N-[(1R,3S)-3-[(5-Isopropylpyrazolo[1,5-a]pyrimidin-7-yl)amino]cyclopentyl]carbamate (23a)
A stirred solution of compound 43 (75 mg, 0.380 mmol), tert-butyl N-[(1R,3S)-3-aminocyclopentyl]carbamate (92.1 mg, 0.460 mmol), and K2CO3 (159 mg, 1.15 mmol) in MeCN (10 mL) was heated to reflux for 16 h. The reaction mixture was filtered, concentrated in vacuo, and purified by Combiflash column chromatography (silica gel, 230–400 mesh), eluting with 25% ethyl acetate in hexanes to give compound 23a (95 mg, 0.264 mmol, 68.9% yield) as a clear sticky liquid. LCMS (ESI) m/z: 359.9 [M+H]+.
(1S,3R)-N3-(5-Isopropylpyrazolo[1,5-a]pyrimidin-7-yl)cyclopentane-1,3-diamine (23)
To 23a (75 mg, 0.210 mmol) was added 4 M HCl in dioxane (2 mL, 0.210 mmol) at 0 °C and stirred at rt for 2 h. The reaction mixture was evaporated in vacuo, and the residue was dissolved in MeOH and passed through a PL-HCO3MP SPE 200 mg/6 mL cartridge, eluting with MeOH, to give compound 23 (30.8 mg, 0.116 mmol, 55.5% yield) as a light-yellow gum. 1H NMR (400 MHz, DMSO-d6) δ ppm 7.97 (1 H, s), 6.28 (1 H, s), 6.04 (1 H, s), 4.21–4.05 (1 H, m), 3.43–3.35 (1 H, m), 2.96–2.84 (1 H, m), 2.27–2.10 (1 H, m), 2.10–1.92 (1H, m), 1.89–1.70 (2 H, m), 1.59–1.41 (2 H, m), 1.20 (6 H, d, J = 6.8 Hz). LCMS (ESI) m/z: 259.8 [M+H]+.
tert-Butyl N-[(1S,3R)-3-[(5-Isopropylpyrazolo[1,5-a]pyrimidin-7-yl)amino]cyclopentyl]carbamate (22a)
A stirred solution of compound 43 (75 mg, 0.380 mmol), tert-butyl N-[(1S,3R)-3-aminocyclopentyl]carbamate (92.1 mg, 0.460 mmol), and K2CO3 (158 mg, 1.15 mmol) in MeCN (10 mL) was heated to reflux for 16 h. The reaction mixture was filtered, concentrated in vacuo, and purified by Combiflash column chromatography (silica gel, 230–400 mesh), eluting with 25% ethyl acetate in hexanes to give compound 22a (95 mg,0.264 mmol, 68.9% yield) as a clear sticky liquid. LCMS (ESI) m/z: 360.0 [M+H]+.
(1R,3S)-N3-(5-Isopropylpyrazolo[1,5-a]pyrimidin-7-yl)cyclopentane-1,3-diamine (22)
To compound 22a (80 mg, 0.220 mmol) was added 4 M HCl in dioxane (2.mL, 0.220 mmol) at 0 °C and stirred at rt for 2 h. The reaction mixture was evaporated in vacuo, and the residue was dissolved in MeOH and passed through a PL-HCO3 MP SPE 200 mg/6 mL cartridge, eluting with MeOH to give compound 22 (26.03 mg,0.098 mmol, 43.97% yield) as a light-yellow gum. 1H NMR (400 MHz, DMSO-d6) δ ppm 7.97 (1 H, d, J = 1.4 Hz), 6.28 (1 H, d, J = 2.2 Hz), 6.04 (1 H, s), 4.16–4.08 (1 H, m), 3.37 (1 H, t, J = 5.5 Hz), 2.96–2.84 (1 H, m), 2.25–2.13 (1 H, m), 2.08–1.92 (1 H, m), 1.90–1.72 (2 H, m), 1.58–1.40 (2 H, m), 1.21 (6 H, d, J = 6.8 Hz). LCMS (ESI) m/z: 259.8 [M+H]+.
(1S,3S)-N3-(5S-butylpyrazolo[1,5-a]pyrimidin-7-yl)cyclopentane-1,3-diamine (27)
To compound 27e (105 mg, 0.280 mmol; see Supporting Information for synthesis) was added 4 M HCl in dioxane (2 mL, 0.280 mmol), and the mixture was stirred at rt for 2 h. The reaction mixture was evaporated in vacuo, and the residue was dissolved in MeOH and passed through PL-HCO3 MP SPE 200 mg/6 mL cartridge, eluting with MeOH to give compound 27 (20.6 mg,0.075 mmol, 26.7% yield) as a light-yellow gum. 1H NMR (400 MHz, DMSO-d6) δ ppm 7.99 (1 H, d, J = 2.0 Hz), 7.44 (1 H, d, J = 6.9 Hz), 6.30 (1 H, d, J = 2.0 Hz), 6.00 (1 H, s), 4.24 (1 H, q, J = 7.2 Hz), 3.50–3.39 (1 H, m), 2.76–2.59 (1 H, m), 2.48–2.26 (2 H, m), 2.28–2.14 (1 H, m), 1.99–1.83 (2 H, m), 1.84–1.61 (3 H, m), 1.62–1.47 (1 H, m), 1.41–1.25 (1 H, m), 1.21 (3 H, d, J = 6.9 Hz), 0.82 (3 H, t, J = 7.3 Hz). LCMS (ESI) m/z: 273.8 [M+H]+.
tert-Butyl ((1r,3r)-3-((5-Isopropylpyrazolo[1,5-a]pyrimidin-7-yl)amino)cyclobutyl)carbamate (8a)
A stirred solution of compound 43 (75 mg, 0.380 mmol), tert-butyl ((1r,3r)-3-aminocyclobutyl)carbamate (85.7 mg, 0.460 mmol), and K2CO3 (158 mg, 1.15 mmol) in MeCN (10 mL) was heated to reflux for 16 h. The reaction mixture was filtered and concentrated in vacuo, and the residue was purified by Combiflash column chromatography (silica gel, 230–400 mesh), eluting with 25% ethyl acetate in hexanes to give compound 8a (90 mg,0.261 mmol, 68.0% yield) as a white solid. LCMS (ESI) m/z: 346.1 [M+H]+.
(1r,3r)-N1-(5-Isopropylpyrazolo[1,5-a]pyrimidin-7-yl)cyclobutane-1,3-diamine (8)
To compound 8a (75.mg, 0.220 mmol) was added 4 M HCl in dioxane (2.mL, 0.220 mmol) at 0 °C and stirred at rt for 2 h. The reaction mixture was monitored by TLC (100% ethyl acetate, product Rf = 0.1, and SM Rf = 0.8). The reaction mixture was evaporated in vacuo, and the residue was dissolved in MeOH and passed through by PL-HCO3 MP SPE 200 mg/6 mL cartridge, eluting with MeOH, to give compound 8 (32.01 mg,0.125 mmol, 57.62% yield) as a light-yellow gum. 1H NMR (400 MHz, DMSO-d6) δ 8.00 (1 H, d, J = 2.2 Hz), 6.30 (1 H, d, J = 1.7 Hz), 5.82 (1 H, s), 4.32–4.24 (1 H, m), 3.57–3.42 (1 H, m), 2.95–2.83 (1 H, m), 2.43–2.31 (2 H, m), 2.19–2.10 (2 H, m), 1.20 (6 H, d, J = 6.8 Hz). LCMS (ESI) m/z: 246.4 [M+H]+.
tert-Butyl N-[(1S,3S)-3-[(5-isopropylpyrazolo[1,5-a]pyrimidin-7-yl)amino]cyclopentyl]carbamate (20a)
A stirred solution of compound 43 (75 mg, 0.380 mmol), tert-butyl ((1S,3S)-3-aminocyclopentyl)carbamate (92.13 mg, 0.460 mmol), and K2CO3 (159 mg, 1.15 mmol) in MeCN (10 mL) was heated to reflux for 16 h. The reaction mixture was filtered, concentrated in vacuo, and purified by Combiflash column chromatography (silica gel, 230–400 mesh), eluting with 25% ethyl acetate in hexanes to give compound 20a (100 mg,0.278 mmol, 72.6% yield) as a white solid. LCMS (ESI) m/z: 360.1 [M+H]+.
(1S,3S)-N3-(5-Isopropylpyrazolo[1,5-a]pyrimidin-7-yl)cyclopentane-1,3-diamine (20)
To compound 20a (80 mg, 0.220 mmol) was added 4 M HCl in dioxane (2.mL, 0.220 mmol) at 0 °C, and the mixture was stirred at rt for 2 h. The reaction mixture was evaporated in vacuo, and the residue was dissolved in MeOH and passed through a PL-HCO3 MP SPE 200 mg/6 mL cartridge, eluting with MeOH to give compound 20 (31.02 mg, 0.116 mmol, 52.14% yield) as a light-yellow gum. 1H NMR (400 MHz, DMSO-d6) δ ppm 7.98 (1 H, s), 6.32–6.26 (1 H, m), 6.03 (1 H, s), 4.43–4.07 (1 H, m), 3.52–3.34 (1 H, m), 2.95–2.86 (1 H, m), 2.29–2.11 (1 H, m), 2.08–1.75 (3 H, m), 1.71–1.56 (1 H, m), 1.41–1.28 (1 H, m), 1.21 (6 H, d, J = 6.9 Hz). LCMS (ESI) m/z: 260.2 [M+H]+.
N-Cyclobutyl-5-isopropylpyrazolo[1,5-a]pyrimidin-7-amine (2)
A stirred solution of compound 43 (100 mg, 0.480 mmol), cyclobutanamine (40.7 mg, 0.570 mmol), and K2CO3 (197 mg, 1.43 mmol) in MeCN (10 mL) was heated to reflux for 16 h. The reaction mixture was concentrated under reduced pressure, and then water (50 mL) was added and extracted with ethyl acetate (20 mL × 2). The combined organic layers were dried under anhyd. Na2SO4 and concentrated in vacuo. The crude product was purified by Combiflash column chromatography (silica, 230–400 mesh), eluting with 20% ethyl acetate in hexanes to give compound 2 (50 mg, 0.213 mmol, 44.8% yield) as an off-white solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.00 (1 H, d, J = 2.0 Hz), 7.87 (1 H, d, J = 7.3 Hz), 6.30 (1 H, d, J = 2.2 Hz), 5.98 (1 H, s), 4.29–4.14 (1 H, m), 2.96–2.87 (1 H, m), 2.43–2.31 (2 H, m), 2.28–2.13 (2 H, m), 1.86–1.63 (2 H, m), 1.22 (6 H, d, J = 6.9 Hz). LCMS (ESI) m/z: 231.0 [M+H]+.
(1S,3S)-N3-(5-Isobutylpyrazolo[1,5-a]pyrimidin-7-yl)cyclopentane-1,3-diamine (30)
To compound 30e (120 mg, 0.320 mmol; see Supporting Information for synthesis) was added 4 M HCl in dioxane (2 mL, 0.320 mmol) at 0 °C and stirred at rt for 2 h. The reaction mixture was evaporated in vacuo. The residue was dissolved in MeOH and passed through PL-HCO3MP SPE 500 mg/6 mL cartridge, eluting with MeOH to give compound 30 (69.1 mg,0.250 mmol, 77.9% yield) as a light-brown sticky gum. 1H NMR (400 MHz, CD3OD) δ ppm 7.96 (1 H, d, J = 2.0 Hz), 6.30 (1 H, d, J = 2.0 Hz), 6.03 (1 H, s), 4.39–4.27 (1 H, m), 3.75–3.63 (1 H, m), 2.58 (2 H, d, J = 7.3 Hz), 2.46–2.32 (1 H, m), 2.32–2.18 (1 H, m), 2.20–1.99 (3 H, m), 1.89–1.74 (1 H, m), 1.68–1.54 (1 H, m), 0.97 (6 H, d, J = 6.6 Hz). LCMS (ESI) m/z: 273.7 [M+H]+.
(1S,3S)-N3-(3-Chloro-5-isopropyl-pyrazolo[1,5-a]pyrimidin-7-yl)cyclopentane-1,3-diamine (21)
To 21a (85 mg, 0.220 mmol; see Supporting Information for synthesis) was added 4 M HCl in dioxane (2 mL, 0.220 mmol) at 0 °C and stirred at rt for 2 h. The reaction mixture was evaporated in vacuo. The residue was dissolved in MeOH and passed through PL-HCO3 MP SPE 500 mg/6 mL cartridge, eluting with MeOH to give compound 21 (27.0 mg, 0.0907 mmol, 42.0% yield) as an off-white sticky solid. 1H NMR (400 MHz, CD3OD) δ ppm 7.95 (1 H, s), 6.09 (1 H, s), 4.39–4.27 (1 H, m), 3.62–3.48 (1 H, m), 3.08–2.94 (1 H, m), 2.43–2.30 (1 H, m), 2.23–2.10 (1 H, m), 2.08–1.89 (2 H, m), 1.81–1.67 (1 H, m), 1.57–1.43 (1 H, m), 1.33 (6 H, d, J = 6.9 Hz). LCMS (ESI) m/z: 293.8 [M+H]+.
(1r,3r)-N1-(3-Chloro-5-isopropyl-pyrazolo[1,5-a]pyrimidin-7-yl)cyclobutane-1,3-diamine (9)
To compound 9a (65 mg, 0.170 mmol; see Supporting Information for synthesis) was added 4 M HCl in dioxane (2 mL, 0.170 mmol) at 0 °C and stirred at rt for 2 h. The reaction mixture was evaporated in vacuo. The residue was dissolved in MeOH and passed through PL-HCO3 MP SPE 500 mg/6 mL cartridge, eluting with MeOH to give compound 9 (26.1 mg,0.093 mmol, 54.2% yield) as an off-white sticky solid. 1H NMR (400 MHz, CD3OD) δ ppm 7.97 (1 H, s), 5.89 (1 H, s), 4.44–4.32 (1 H, m), 3.77–3.66 (1 H, m), 3.08–2.93 (1 H, m), 2.55–2.43 (2 H, m), 2.41–2.29 (2 H, m), 1.31 (6 H, d, J = 6.9 Hz). LCMS (ESI) m/z: 279.9 [M+H]+.
5-Isopropyl-N-(oxetan-3-yl)pyrazolo[1,5-a]pyrimidin-7-amine (5)
A stirred solution of compound 43 (100 mg, 0.480 mmol), oxetan-3-amine (41.8 mg, 0.570 mmol), and K2CO3 (197 mg, 1.43 mmol) in MeCN (10 mL) was heated to reflux for 16 h. The reaction mixture was concentrated under reduced pressure, and then water (50 mL) was added and extracted with ethyl acetate (20 mL × 2). The combined organic layers were dried under anhyd. Na2SO4 and concentrated in vacuo. The crude product was purified by Combiflash column chromatography (silica, 230–400 mesh), eluting with 20% ethyl acetate in hexanes to give compound 5 (90 mg, 0.384 mmol, 80.6% yield) as an off-white solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.44 (1 H, d, J = 6.2 Hz), 8.05 (1 H, d, J = 1.7 Hz), 6.34 (1 H, d, J = 1.8 Hz), 5.90 (1 H, s), 5.00–4.88 (1 H, m), 4.86 (2 H, t, J = 6.9 Hz), 4.74 (2 H, t, J = 6.1 Hz), 2.98–2.83 (1 H, m), 1.22 (6 H, d, J = 6.8 Hz). LCMS (ESI) m/z: 233.0 [M+H]+.
tert-Butyl (3S)-3-[(5-isopropylpyrazolo[1,5-a]pyrimidin-7-yl)amino]pyrrolidine-1-carboxylate (18a)
A stirred solution of compound 43 (200 mg, 1.02 mmol), tert-butyl (3S)-3-aminopyrrolidine-1-carboxylate (228 mg, 1.23 mmol), and K2CO3 (423 mg, 3.07 mmol) in MeCN (20 mL) was heated to reflux for 16 h. The reaction mixture was concentrated under reduced pressure, and then water (50 mL) was added and extracted with ethyl acetate (20 mL × 2). The combined organic layers were dried under anhyd. Na2SO4 and concentrated in vacuo. The crude product was purified by Combiflash column chromatography (silica, 230–400 mesh), eluting with 20% ethyl acetate in hexanes to give compound 18a (240 mg, 0.695 mmol, 68% yield) as a pale-yellow sticky compound. LCMS (ESI) m/z: 345.8 [M+H]+.
5-Isopropyl-N-[(3S)-pyrrolidin-3-yl]pyrazolo[1,5-a]pyrimidin-7-amine Hydrochloride (18)
To compound 18a (250 mg, 0.690 mmol) was added 4 M HCl in dioxane (3 mL, 0.690 mmol), and the mixture was stirred at rt for 2 h. The reaction mixture was evaporated in vacuo, and the resulting solid was triturated with pentane (3 × 5 mL) and decanted. The solid obtained was dried in vacuo to give compound 18 (160 mg, 0.565 mmol, 81.6% yield) as a pale-yellow solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 9.80–9.44 (1 H, m), 9.35–8.98 (1 H, m), 8.30 (1 H, s), 6.57 (2 H, s), 4.94–4.66 (1 H, m), 3.61–3.37 (4 H, m), 3.30–3.25 (1 H, m), 3.20–3.12 (1 H, m), 2.42–2.28 (1 H, m), 2.23–2.07 (1 H, m), 1.35 (6 H, d, J = 6.9 Hz). LCMS (ESI) m/z: 246.4 [M+H]+.
tert-Butyl (3R)-3-[(5-isopropylpyrazolo[1,5-a]pyrimidin-7-yl)amino]pyrrolidine-1-carboxylate (19a)
A stirred solution of compound 43 (200 mg, 1.02 mmol), tert-butyl (3R)-3-aminopyrrolidine-1-carboxylate (228 mg, 1.23 mmol), and K2CO3 (423 mg, 3.07 mmol) in MeCN (20 mL) was heated to reflux for 16 h. The reaction mixture was concentrated under reduced pressure, and then water (50 mL) was added and extracted with ethyl acetate (20 mL × 2). The combined organic layers were dried under anhyd. Na2SO4 and concentrated in vacuo. The crude mixture was purified by Combiflash column chromatography (silica, 230–400 mesh), eluting with 20% ethyl acetate in hexanes to give compound 19a (250 mg, 0.724 mmol, 70.8% yield) as a pale-yellow sticky liquid. LCMS (ESI) m/z: 346.4 [M+H]+.
tert-Butyl (3R)-3-[(5-isopropylpyrazolo[1,5-a]pyrimidin-7-yl)amino]pyrrolidine-1-carboxylate (19)
To compound 19a (248 mg, 0.720 mmol) was added 4 M HCl in dioxane (3 mL, 12 mmol), and the mixture was stirred at rt for 2 h. The reaction mixture was evaporated in vacuo, and the resulting solid was triturated with pentane (3 × 5 mL) and decanted. The remaining solid was dried in vacuo to give compound 19 (150 mg, 0.530 mmol, 73.87% yield) as a pale-yellow solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.29 (1 H, s), 6.59–6.51 (2 H, m), 4.91–4.72 (1 H, m), 3.62–3.53 (1 H, m), 3.49–3.37 (2 H, m), 3.34–3.23 (1 H, m), 3.17–3.05 (1 H, m), 2.46–2.32 (1 H, m), 2.22–2.08 (1 H, m), 1.33 (6 H, d, J = 6.9 Hz). LCMS (ESI) m/z: 245.9 [M+H]+.
tert-Butyl 3-[(5-Isopropylpyrazolo[1,5-a]pyrimidin-7-yl)amino]azetidine-1-carboxylate (3a)
A stirred solution of compound 43 (200 mg, 1.02 mmol), tert-butyl 3-aminoazetidine-1-carboxylate (211 mg, 1.23 mmol), and K2CO3 (423 mg, 3.07 mmol) in MeCN (20 mL) was heated to reflux for 16 h. The reaction mixture was concentrated under reduced pressure, and then water (50 mL) was added and extracted with ethyl acetate (30 mL × 2). The combined organic layers were dried under anhyd. Na2SO4 and concentrated in vacuo. The crude material was purified by Combiflash column chromatography (silica, 230–400 mesh), eluting with 20% ethyl acetate in hexanes to give compound 3a (240 mg, 0.724 mmol, 70.8% yield) as a pale-yellow sticky compound. LCMS (ESI) m/z: 332.3 [M+H]+.
N-(Azetidin-3-yl)-5-isopropylpyrazolo[1,5-a]pyrimidin-7-amine (3)
To compound 3a (240 mg, 0.690 mmol) was added 4 M HCl in dioxane (3 mL, 0.690 mmol), and the mixture was stirred at rt for 2 h. The reaction mixture was evaporated in vacuo and triturated with pentane (3 × 5 mL). The crude solid was purified by prep-HPLC followed by lyophilization to give compound 3 (55 mg, 0.236 mmol, 34.2% yield) as an off-white solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.26 (6 H, d, J = 6.9 Hz), 2.90–2.97 (1 H, m), 3.64–3.73 (2 H, m), 3.80 (2 H, t, J = 7.6 Hz), 4.57 (1 H, q, J = 7.0 Hz), 5.91 (1 H, s), 6.30 (1 H, s), 7.5–7.61 (1 H, m), 7.98 (1 H, s). LCMS (ESI) m/z: 231.9 [M+H]+.
5-Isopropyl-N-(1-methylazetidin-3-yl)pyrazolo[1,5-a]pyrimidin-7-amine (4)
A solution of compound 3a (180 mg, 0.540 mmol) in THF (15 mL) was cooled to 0 °C. Then LiAlH4 (61.8 mg, 1.63 mmol) was added portion-wise. After the complete addition, the reaction mixture was stirred at 80 °C for 4 h. The reaction mixture was quenched carefully with an aq. Na2SO4 solution and extracted with ethyl acetate (3 × 40 mL). The combined organic layers were dried under anhyd. Na2SO4 and concentrated in vacuo to give the crude product. This crude product was purified by prep-HPLC followed by lyophilization to give compound 4 (50 mg, 0.201 mmol, 37.0% yield) as a white, sticky solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.22 (6 H, d, J = 6.9 Hz), 2.27 (3 H, s), 2.84–2.97 (1 H, m), 3.11 (2 H, t, J = 6.9 Hz), 3.69 (2 H, t, J = 6.7 Hz), 4.22–4.35 (1 H, m), 5.98 (1 H, s), 6.32 (1 H, s), 8.02–8.05 (2 H, m). LCMS (ESI) m/z: 246.1 [M+H]+.
(1r,3r)-3-[(5-Isopropylpyrazolo[1,5-a]pyrimidin-7-yl)amino]cyclobutanol (6)
A stirred solution of compound 43 (100 mg, 0.510 mmol), (1r,3r)-3-aminocyclobutan-1-ol hydrochloride (75.8 mg, 0.610 mmol), and K2CO3 (212 mg, 1.53 mmol) in MeCN (10 mL) was heated to reflux for 16 h. The reaction mixture was concentrated under reduced pressure, and then water (50 mL) was added and extracted with ethyl acetate (30 mL × 2). The combined organic layers were dried under anhyd. Na2SO4 and concentrated in vacuo. The crude material was purified by Combiflash column chromatography (silica, 230–400 mesh), eluting with 20% ethyl acetate in hexanes to give the crude product. The crude product was re-purified by prep-HPLC followed by lyophilization to give compound 6 (35 mg, 0.142 mmol, 27.8% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.25–1.28 (6 H, m), 2.31–2.38 (2 H, m), 2.45–2.47 (2 H, m), 2.91–2.93 (1 H, m), 4.23–4.33 (1 H, m), 4.37–4.45 (1 H, m), 4.77 (1 H, d, J = 5.2 Hz), 5.82 (1 H, s), 6.26–6.32 (1 H, m), 7.31–7.34 (1 H, m), 7.95–7.98 (1 H, m). LCMS (ESI) m/z: 247.2 [M+H]+.
(1S,3S)-3-[(5-Isopropylpyrazolo[1,5-a]pyrimidin-7-yl)amino]cyclopentanol (25)
A stirred solution of compound 43 (100 mg, 0.510 mmol), (1S,3S)-3-aminocyclopentanol hydrochloride (84.4 mg, 0.610 mmol), and K2CO3 (212 mg, 1.53 mmol) in MeCN (10 mL) was heated at 80 °C for 16 h. The reaction mixture was concentrated under reduced pressure, and then water (50 mL) was added and extracted with ethyl acetate (30 mL × 2). The combined organic layers were dried under anhyd. Na2SO4 and concentrated in vacuo. The crude material was purified by Combiflash column chromatography (silica, 230–400 mesh), eluting with 20% ethyl acetate in hexanes to give compound 25 (50 mg, 0.188 mmol, 36.7% yield) as a colorless sticky liquid. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.24 (6 H, d, J = 6.8 Hz), 1.48–1.56 (1 H, m), 1.59–1.69 (1 H, m), 1.84–2.04 (3 H, m), 2.15–2.26 (1 H, m), 2.87–2.99 (1 H, m), 4.19–4.30 (2 H, m), 4.57–4.62 (1 H, m), 6.03 (1 H, s), 6.29–6.32 (1 H, m), 7.50 (1 H, d, J = 7.9 Hz), 7.99–8.00 (1 H, m). LCMS (ESI) m/z: 261.3 [M+H]+.
(1r,3r)-N1-(5-Isopropylpyrazolo[1,5-a]pyrimidin-7-yl)-N3-methylcyclobutane-1,3-diamine (10)
A solution of compound 8a (100 mg, 0.290 mmol) in THF (10 mL) was cooled to 0 °C, and LiAlH4 (32.9 mg, 0.870 mmol) was added portion-wise. After complete addition, the reaction mixture was stirred at 80 °C for 16 h. Then the reaction mixture was quenched carefully with cold aq. Na2SO4 solution and extracted with ethyl acetate (3 x× 40 mL). The combined organic layers were dried over anhyd. Na2SO4 and concentrated in vacuo to give a crude material. This crude material was purified by prep-HPLC followed by lyophilization to give compound 10 (30 mg, 0.115 mmol, 39.9% yield) as an off-white solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.26 (6 H, d, J = 6.9 Hz), 1.89–1.95 (1 H, br), 2.21–2.28 (5 H, m), 2.29–2.40 (2 H, m), 2.91–2.93 (1 H, m), 3.27–3.36 (1 H, m), 4.28 (1 H, q, J = 6.7 Hz), 5.83 (1 H, s), 6.29 (1 H, s), 7.34 (1 H, br), 7.97 (1 H, s). LCMS (ESI) m/z: 260.0 [M+H]+.
(1r,3r)-N-[3-[(5-Isopropylpyrazolo[1,5-a]pyrimidin-7-yl)amino]cyclobutyl]methanesulfonamide (17)
To a stirred solution of compound 8 (200 mg, 0.630 mmol) in DCM (5 mL) was added NaHCO3 (264 mg, 3.14 mmol), stirred for 30 min, extracted with DCM, and then washed with water and brine to give the free amine. To the free amine in DCM (5 mL) were added DIPEA (0.16 mL, 0.940 mmol) and methanesulfonyl chloride (0.07 mL, 0.940 mmol) at 0 °C, and the mixture was stirred for 6 h. The reaction mixture was washed with water and brine and dried over anhyd. Na2SO4. The crude product was purified by prep HPLC to give compound 17 (73.6 mg, 0.22 mmol, 36.1% yield) as a white solid. 1H NMR (400 MHz, CDCl3) δ ppm 1.30 (6 H, d, J = 6.9 Hz), 2.51–2.68 (4 H, m), 2.89–3.05 (4 H, m), 4.24–4.30 (2 H, m), 4.87 (1 H, d, J = 7.4 Hz), 5.64 (1 H, s), 6.40 (1 H, d, J = 5.4 Hz), 6.44 (1 H, s), 7.95 (1 H, s). LCMS (ESI) m/z: 324.0 [M+H]+.
(1r,3r)-[3-[(5-Isopropylpyrazolo[1,5-a]pyrimidin-7-yl)amino]cyclobutyl]methanol (11)
A stirred solution of compound 43 (100 mg, 0.510 mmol), (1r,3r)- (3-aminocyclobutyl)methanol hydrochloride (84.4 mg, 0.610 mmol), and K2CO3 (212 mg, 1.53 mmol) in MeCN (10 mL) was heated to reflux for 16 h. The reaction mixture was concentrated under reduced pressure, and then water (25 mL) was added and extracted with ethyl acetate (30 mL × 2). The combined organic layers were dried under anhyd. Na2SO4 and concentrated in vacuo. The crude material was purified by Combiflash column chromatography (silica, 230–400 mesh), eluting with 20% ethyl acetate in hexanes to give compound 11 (90 mg, 0.34 mmol, 66.4% yield) as a sticky liquid. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.26 (6 H, d, J = 6.9 Hz), 2.24–2.34 (4 H, m), 2.41–2.43 (1 H, m), 2.91–2.93 (1 H, m), 3.56 (2 H, t, J = 5.7 Hz), 4.20–4.24 (1 H, m), 4.28 (1 H, br), 5.85 (1 H, s), 6.29 (1 H, s), 7.32 (1 H, br), 7.97 (1 H, s). LCMS (ESI) m/z: 260.8 [M+H]+.
(1r,3r)-N-[3-[(5-Isopropylpyrazolo[1,5-a]pyrimidin-7-yl)amino]cyclobutyl]acetamide (12)
To a stirred solution of compound 8 (100 mg, 0.410 mmol) in pyridine (6 mL) was added acetic anhydride (0.05 mL, 0.490 mmol), and the mixture was stirred overnight. The reaction mixture was concentrated, partitioned between ethyl acetate and water, and extracted. The organic layers were washed with water and brine, dried over anhyd. Na2SO4, and evaporated in vacuo to give the crude product. The crude product was purified by prep-HPLC to give compound 12 (5 mg, 0.017 mmol, 4.2% yield) as an off-white solid. 1H NMR (400 MHz, CDCl3) δ ppm 7.94 (1 H, d, J = 2.0 Hz), 6.41 (1 H, d, J = 2.0 Hz), 5.63 (1 H, s), 4.54 (1 H, t, J = 6.8 Hz), 4.38–4.13 (1 H, m), 3.14–2.81 (1 H, m), 2.66–2.43 (3 H, m), 2.00 (3 H, s), 1.30 (6 H, d, J = 6.9 Hz). LCMS (ESI) m/z: 288.0 [M+H]+.
(1r,3r)-3-[(5-Isopropylpyrazolo[1,5-a]pyrimidin-7-yl)amino]cyclobutanecarbonitrile (13a)
A stirred solution of compound 43 (300 mg, 1.53 mmol), (1r,3r)-3-aminocyclobutanecarbonitrile hydrochloride (244 mg, 1.84 mmol), and K2CO3 (635 mg, 4.6 mmol) in MeCN (30 mL) was heated at reflux temperature for 16 h. The reaction mixture was concentrated in vacuo, and then water (50 mL) was added and extracted with ethyl acetate (20 mL × 2). The combined organic layers were dried under anhyd. Na2SO4 and concentrated in vacuo. The crude material was purified by Combiflash column chromatography (silica, 230–400 mesh), eluting with 20% ethyl acetate in hexanes to give compound 13a (300 mg,1.175 mmol, 76.6% yield) as an off-white solid. LCMS (ESI) m/z: 256.1 [M+H]+.
(1r,3r)-3-[(5-Isopropylpyrazolo[1,5-a]pyrimidin-7-yl)amino]cyclobutanecarboxamide (13)
To a stirred solution of compound 13a (70 mg, 0.270 mmol) in THF (2 mL) and water (0.01 mL, 0.550 mmol) was added hydrido(dimethylphosphinous acid-kP)[hydrogen bis(dimethylphosphinito-kP)]platinum(II) (Ghaffar–Parkins catalyst) (11.8 mg, 0.030 mmol) and stirred at 70 °C for 12 h. The reaction mixture was then diluted with water (20 mL) and extracted with ethyl acetate (25 mL). The combined ethyl acetate layers were washed with a water layer, dried over anhyd. Na2SO4, and concentrated in vacuo. The crude material was purified by Combiflash column chromatography (silica, 230–400 mesh), eluting with 10% MeOH–DCM to give compound 13 (35 mg, 0.127 mmol, 46.3% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.22 (6 H, d, J = 6.9 Hz), 2.36–2.48 (2 H, m), 2.52–2.54 (2 H, m), 2.86–3.01 (2 H, m), 4.22–4.36 (1 H, m), 5.85 (1 H, s), 6.29–6.35 (1 H, m), 6.81–6.87 (1 H, m), 7.29 (1 H, s), 7.94–8.05 (2 H, m). LCMS (ESI) m/z: 273.8 [M+H]+.
(1r,3r)-N1-(5-Isopropylpyrazolo[1,5-a]pyrimidin-7-yl)-N3-phenylcyclobutane-1,3-diamine (16)
A stirred solution of compound 43 (90 mg, 0.460 mmol), (1r,3r)-N1-phenylcyclobutane-1,3-diamine hydrochloride (110 mg, 0.550 mmol), and K2CO3 (190 mg, 1.38 mmol) in MeCN (9 mL) was heated to reflux for 16 h. The reaction mixture was concentrated under reduced pressure, and then water (50 mL) was added and extracted with ethyl acetate (30 mL × 2). The combined organic layers were dried under anhyd. Na2SO4 and concentrated in vacuo. The crude product was purified by Combiflash column chromatography (silica, 230–400 mesh), eluting with 30% ethyl acetate in hexanes to give compound 16 (50 mg, 0.153 mmol, 33.4% yield) as a pale-yellow solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.22 (6 H, d, J = 6.9 Hz), 2.28–2.39 (2 H, m), 2.55–2.66 (2 H, m), 2.86–2.98 (1 H, m), 3.97 (1 H, d, J = 5.9 Hz), 4.27–4.37 (1 H, m), 5.87 (1 H, s), 5.98 (1 H, d, J = 5.8 Hz), 6.33 (1 H, s), 6.47–6.59 (3 H, m), 7.08 (2 H, t, J = 7.7 Hz), 8.00–8.09 (2 H, m). LCMS (ESI) m/z: 322.0 [M+H]+.
(1r,3r)-N-(3-Ethoxycyclobutyl)-5-isopropylpyrazolo[1,5-a]pyrimidin-7-amine (7)
A stirred solution of compound 43 (100 mg, 0.510 mmol), (1r,3r)-3-ethoxycyclobutanamine hydrochloride (93 mg, 0.610 mmol), and K2CO3 (212 mg, 1.53 mmol) in MeCN (10 mL) was heated to reflux for 16 h. The reaction mixture was then filtered and concentrated under reduced pressure. The crude product was purified by prep-HPLC to give compound 7 (36.5 mg, 0.133 mmol, 25.9% yield) as a colorless, sticky gum. 1H NMR (400 MHz, CDCl3) δ ppm 1.21–1.26 (3 H, m), 1.30 (6 H, d, J = 6.8 Hz), 2.34–2.41 (2 H, m), 2.49–2.61 (2 H, m), 2.91–3.04 (1 H, m), 3.44 (2 H, q, J = 7.0 Hz), 4.20–4.31 (2 H, m), 5.69 (1 H, s), 6.31 (1 H, d, J = 5.7 Hz), 6.42 (1 H, s), 7.94 (1 H, s). LCMS (ESI) m/z: 274.8 [M+H]+.
tert-Butyl N-[(1R,3R)-3-[(5-Isopropylpyrazolo[1,5-a]pyrimidin-7-yl)amino]cyclopentyl]carbamate (24a)
A stirred solution of compound 43 (0.35 g, 1.79 mmol), tert-butyl N-[(1R,3R)-3-aminocyclopentyl]carbamate (0.4 g, 2 mmol), and K2CO3 (0.78 g, 5.64 mmol) in MeCN (17.5 mL) was heated to reflux for 16 h. The reaction mixture was concentrated under reduced pressure, and then water (50 mL) was added and extracted with ethyl acetate (30 mL × 2). The combined organic layers were dried under anhyd. Na2SO4 and concentrated in vacuo. This crude material was purified by Combiflash column chromatography (silica, 230–400 mesh), eluting with 30% ethyl acetate in hexanes to give compound 24a (450 mg, 1.25 mmol, 70.0% yield) as a white solid. LCMS (ESI) m/z: 360.0 [M+H]+.
(1R,3R)-N3-(5-Isopropylpyrazolo[1,5-a]pyrimidin-7-yl)cyclopentane-1,3-diamine (24b)
To a stirred solution of compound 24a (0.45 g, 1.25 mmol) in dioxane (0.5 mL) at 0 °C was added 4 M HCl in dioxane (20 mL, 1.25 mmol) and stirred at 0 °C for 30 min. The reaction mixture was then stirred at rt for 6 h. After completion of the reaction, the reaction mixture was concentrated in vacuo, and the residue was diluted with CH2Cl2 (50 mL). The organic layer was washed with sat. NaHCO3 (10 mL) solution and brine (10 mL). The organic layer was then dried over anhyd. Na2SO4 and concentrated in vacuo to give crude compound 24b (200 mg, 0.771 mmol, 61.6% yield) as a gum. LCMS (ESI) m/z: 259.9 [M+H]+.
1-[(1R,3R)-3-[(5-Isopropylpyrazolo[1,5-a]pyrimidin-7-yl)amino]cyclopentyl]-3-methylurea (24)
In a sealed tube, a stirred solution of compound 24b (170 mg, 0.660 mmol) in 1,4-dioxane (2 mL), water (1 mL), and S-methyl N-methylcarbamothioate (220 mg, 2.09 mmol) was heated at 65 °C for 12 h. The progress of the reaction was monitored by LCMS. After completion of the reaction, the solvent was evaporated under reduced pressure, and the crude material was purified by prep-HPLC to give compound 24 (70 mg, 0.220 mmol, 33.6% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 7.98 (1 H, d, J = 2.08 Hz), 7.54 (1 H, d, J = 7.6 Hz), 6.28 (1 H, d, J = 2.18 Hz), 5.98 (1 H, s), 5.94 (1 H, d, J = 7.24 Hz) 5.58–5.55 (1 H, m), 4.18–4.11 (1 H, m), 4.07–4.01 (1 H, m), 2.95–2.84 (1 H, m), 2.51 (3 H, d, J = 4.6 Hz), 2.19–2.08 (1 H, m), 2.03–1.93 (2 H, m), 1.85–1.73 (1 H, m), 1.75–1.61 (1 H, m), 1.43–1.29 (1 H, m), 1.21 (6 H, d, J = 6.8 Hz). – δ ppm 7.97 (1 H, d, J = 2.08 Hz), 6.31 (1 H, d, J = 2.04 Hz), 6.00 (1 H, s), 4.29–4.18 (2 H, m), 3.01–2.91 (1 H, m), 2.69 (3 H, s), 2.36–2.32 (1 H, m), 2.25–1.95 (3 H, m), 1.82–1.69 (1 H, m), 1.64–1.50 (1 H, m), 1.32 (6 H, d, J = 6.9 Hz). LCMS (ESI) m/z: 317.0 [M+H]+.
2-Cyano-1-((1r,3r)-3-((5-Isopropylpyrazolo[1,5-a]pyrimidin-7-yl)amino)cyclobutyl)-3-methylguanidine (15)
To a solution of compound 15a (70 mg, 0.180 mmol; see Supporting Information) in isopropyl alcohol (3.5 mL) in a sealed tube was added methanamine (338 mg, 3.59 mmol) in 33% aq. isopropanol at rt, and then the mixture was heated at 70 °C for 4 h. The progress of the reaction was monitored by TLC and LCMS. After completion of the reaction, the solvent was evaporated in vacuo, and the crude product was purified by Combiflash column chromatography (silica, 230–400 mesh), eluting with 5% MeOH in CH2Cl2 to give compound 15 (31.47 mg, 0.096 mmol, 53.20% yield) as an off-white solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.03 (1 H, d, J = 1.92 Hz), 7.98 (1 H, d, J = 6.08 Hz), 7.17 (1 H, d, J = 6.5 Hz), 6.90 (1 H, br s), 6.33 (1 H, d, J = 2.0 Hz), 5.84 (1 H, s), 4.28–4.24 (1 H, m), 4.19–4.14 (1 H, m), 2.99–2.87 (1 H, m), 2.70 (3 H, d, J = 4.5 Hz), 2.59–2.56 (2 H, m), 2.46–2.39 (2 H, m), 1.23 (6 H, d, J = 6.8 Hz). 1H NMR (400 MHz, CD3OD): δ ppm 7.99 (1 H, s), 6.33 (1 H, s), 5.87 (1 H, s), 4.41–4.29 (2 H, m), 3.02–2.90 (1 H, m), 2.83 (3 H, s), 2.62–2.57 (4 H, m), 1.31 (6 H, d, J = 7.0 Hz). LCMS (ESI) m/z: 327.5 [M+H]+.
(1S,3S)-N3-(5-Isobutyl-3-methylpyrazolo[1,5-a]pyrimidin-7-yl)cyclopentane-1,3-diamine (31)
To a stirred solution of compound 31c (60 mg, 0.150 mmol; see Supporting Information) in 1,4-dioxane (2 mL) was added 4 M HCl-dioxane (1 mL, 4 mmol) at 0 °C and stirred at rt for 2 h. The reaction mixture was evaporated in vacuo, and the resulting solid was triturated with pentane (3 × 2 mL) and ether (2 × 2 mL). The solid was dried in vacuo and lyophilized to give compound 31 (8.2 mg, 0.028 mmol, 18.2% yield) as an off-white solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 7.84 (1 H, s), 7.37 (1 H, d, J = 7.4 Hz), 5.95 (1 H, s), 4.27–4.18 (1 H, m), 3.48–3.45 (2 H, m), 2.13–2.07 (5 H, m), 1.99–1.94 (2 H, m), 1.85 (3 H, s), 1.83–1.72 (1 H, m), 1.70–1.63 (1 H, m), 1.38–1.33 (1 H, m), 0.92 (6 H, d, J = 6.6 Hz). LCMS (ESI) m/z: 288.3 [M+H]+.
tert-Butyl N-[(1S,3S)-3-[(5-chloro-pyrazolo[1,5-a]pyrimidin-7-yl)amino]cyclopentyl]carbamate (45)
A stirred solution of 5,7-dichloro-pyrazolo[1,5-a]pyrimidine 44 (2 g, 10.64 mmol), tert-butyl ((1S,3S)-3-aminocyclopentyl)carbamate (2.34 g, 11.7 mmol), and K2CO3 (4.4 g, 31.91 mmol) in MeCN (20 mL) was heated to reflux for 16 h. The reaction mixture was filtered and concentrated under reduced pressure, and the crude material was purified by Combiflash column chromatography (silica, 230–400 mesh), eluting with 30% ethyl acetate in hexanes to give compound 45 (1.9 g, 5.36 mmol, 50.4% yield) as a white solid. LCMS (ESI) m/z: 352.1 [M+H]+.
tert-Butyl N-[(1S,3S)-3-[(5-chloro-pyrazolo[1,5-a]pyrimidin-7-yl)amino]cyclopentyl]carbamate (45)
A solution of tert-butyl ((1S,3S)-3-aminocyclopentyl)carbamate (3.83 g, 19.12 mmol), 5,7-dichloro-pyrazolo[1,5-a]pyrimidine 44 (3.6 g, 19.1 mmol), and NEt3 (2.68 mL, 19.1 mmol) in MeCN (40 mL) was stirred at 50 °C for 4 h. TLC (petroleum ether/ethyl acetate = 1:1, Rf = 0.6) indicated starting material was consumed completely and one new spot formed. The reaction mixture was concentrated in vacuo, and the residue was purified by Combiflash column chromatography (silica gel, 230–400 mesh), eluting with 0–50% ethyl acetate/petroleum ether gradient at 50 mL/min to give compound 45 (6.8 g,17.4 mmol, 91.0% yield) as a yellow gum. 1H NMR (400 MHz, CDCl3) δ ppm 7.96 (1 H, d, J = 2.6 Hz), 6.43 (1 H, d, J = 2.0 Hz), 5.93 (1 H, s), 4.55 (1 H, br s), 4.24–4.16 (1 H, m), 2.43–2.34 (1 H, m), 2.32–2.22 (1 H, m), 2.19–2.02 (2 H, m), 1.74–1.72 (1 H, m), 1.65–1.52 (3 H, m), 1.46 (9 H, s).
tert-Butyl N-[(1S,3S)-3-[[5-(3-methoxyphenyl)pyrazolo[1,5-a]pyrimidin-7-yl]amino]cyclopentyl]carbamate (36a)
A stirred mixture of compound 45 (80 mg, 0.230 mmol), (3-methoxyphenyl)boronic acid (69.1 mg, 0.450 mmol), and K2CO3 (94.3 mg, 0.680 mmol) in 1,4-dioxane (5 mL) was degassed for 30 min using argon, and then Pd2(dba)3 (20.8 mg, 0.020 mmol) and Xantphos Gen 3 (38.5 mg, 0.050 mmol) were added. The reaction mixture was heated to 100 °C for 16 h. After completion of the reaction, the reaction mixture was filtered through a pad of Celite and purified by Combiflash column chromatography (silica, 230–400 mesh), eluting with 30% ethyl acetate in hexanes to give compound 36a (60 mg, 0.139 mmol, 61.3% yield) as a yellow, sticky liquid. LCMS (ESI) m/z: 424.0 [M+H]+.
[(1S,3S)-3-[[5-(3-Methoxyphenyl)pyrazolo[1,5-a]pyrimidin-7-yl]amino]cyclopentyl]ammonium chloride (36)
To 36a (60 mg, 0.140 mmol) was added 4 M HCl in dioxane (2 mL, 0.140 mmol) at 0 °C and stirred at rt for 4 h. The reaction mixture was evaporated in vacuo and triturated with pentane to give compound 36 (30.1 mg, 0.083 mmol, 58.2% yield) as a light-yellow solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.09 (1 H, s), 7.98–7.93 (3 H, m), 7.73–7.69 (2 H, m), 7.44 (1 H, t, J = 7.6 Hz), 7.08 (1 H, d, J = 8.2 Hz), 6.67 (1 H, s), 6.49 (1 H, s), 4.66–4.59 (1 H, m), 3.89 (3 H, s), 3.83–3.78 (1 H, m), 2.34–2.32 (1 H, m), 2.22 (3 H, t, J = 7.1 Hz), 1.89–1.87 (1 H, m), 1.74 (1 H, s). LCMS (ESI) m/z: 324.0 [M+H]+.
tert-Butyl N-[(1S,3S)-3-[[5-(4-Methoxyphenyl)pyrazolo[1,5-a]pyrimidin-7-yl]amino]cyclopentyl]carbamate (37a)
A stirred solution of compound 45 (80 mg, 0.230 mmol), (4-methoxyphenyl)boronic acid (69.1 mg, 0.450 mmol), and K2CO3 (94.3 mg, 0.680 mmol) in 1,4-dioxane (5 mL) was degassed for 30 min using argon, and then Pd2(dba)3 (20.82 mg, 0.020 mmol) and Xantphos Gen 3 (38.5 mg, 0.050 mmol) were added. The reaction mixture was heated at 100 °C for 16 h. After completion of the reaction, the reaction mixture was filtered through a pad of Celite, and the filtrate was evaporated in vacuo. The crude mixture was purified by Combiflash column chromatography (silica, 230–400 mesh), eluting with 30–40% ethyl acetate in hexanes to give compound 37a (80 mg, 0.187 mmol, 82.1% yield) as a yellow sticky liquid. LCMS (ESI) m/z: 424.0 [M+H]+.
[(1S,3S)-3-[[5-(4-Methoxyphenyl)pyrazolo[1,5-a]pyrimidin-7-yl]amino]cyclopentyl]ammonium Chloride (37)
To compound 37a (80 mg, 0.190 mmol) was added 4 M HCl in dioxane (2 mL, 0.190 mmol) at 0 °C and then stirred at rt for 4 h. The reaction mixture was evaporated in vacuo and triturated with pentane to give compound 37 (30.9 mg, 0.085 mmol, 45.3% yield) as an off-white solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.15–8.11 (2 H, m), 8.07 (1 H, s), 8.03–7.95 (2 H, m), 7.80–7.74 (1 H, m), 7.08 (2 H, d, J = 8.0 Hz), 6.66 (1 H, s), 6.45 (1 H, s), 4.64 (1 H, d, J = 6.3 Hz), 3.87 (3 H, s), 3.83–3.78 (1 H, m), 2.35–2.32 (1 H, m), 2.26–2.18 (3 H, m), 1.93–1.83 (1 H, m), 1.77–1.72 (1 H, m). LCMS (ESI) m/z: 324.2 [M+H]+.
tert-Butyl N-[(1S,3S)-3-[[5-(2-methoxyphenyl)pyrazolo[1,5-a]pyrimidin-7-yl]amino]cyclopentyl]carbamate (35a)
A stirred solution of compound 45 (80 mg, 0.230 mmol), (2-methoxyphenyl)boronic acid (69.1 mg, 0.450 mmol), and K2CO3 (94.3 mg, 0.680 mmol) in 1,4-dioxane (5 mL) was degassed for 30 min, and Pd2(dba)3 (20.8 mg, 0.020 mmol) and Xantphos Gen 3 (38.5 mg, 0.050 mmol) were added. The reaction mixture was heated at 100 °C for 16 h. After completion of the reaction, the reaction mixture was filtered through a pad of Celite and purified by Combiflash column chromatography (silica, 230–400 mesh), eluting with 30–40% ethyl acetate in hexanes to give compound 35a (50 mg, 0.118 mmol, 51.9% yield) as a yellow sticky liquid. LCMS (ESI) m/z: 424.2 [M+H]+.
[(1S,3S)-3-[[5-(2-Methoxyphenyl)pyrazolo[1,5-a]pyrimidin-7-yl]amino]cyclopentyl]ammonium Chloride (35)
To compound 35a (50 mg, 0.120 mmol) was added 4 M HCl in dioxane (2 mL, 0.120 mmol) at 0 °C and then stirred at rt for 4 h. The reaction mixture was evaporated in vacuo and triturated with pentane to give compound 35 (23.7 mg, 0.065 mmol, 55.4% yield) as a light-brown solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.6 (1 H, m), 8.18–7.75 (4 H, m), 7.74 (1 H, d, J = 7.7 Hz), 7.53 (1 H, t, J = 7.9 Hz), 7.23 (1 H, d, J = 8.3 Hz), 7.13 (1 H, t, J = 7.5 Hz), 6.72 (1 H, s), 6.53 (1 H, s), 4.63 (1 H, s), 3.91 (3 H, s), 3.79 (1 H, s), 2.33–2.21 (4 H, m), 1.98–1.84 (1 H, m), 1.81–1.71 (1 H, m). LCMS (ESI) m/z: 324.4 [M+H]+.
Ethyl 4-Ethyl-3-oxo-hexanoate (47)
2-Ethylbutanoic acid (10 g, 86.1 mmol) was dissolved in THF (200 mL) and cooled to 0 °C. After stirring at 0 °C for 20 min, CDI (21.6 g, 133 mmol) was added portion-wise. The temperature was allowed to rise to rt, and the mixture was stirred at rt for 16 h. In a second reaction flask, MgCl2 (8.19 g, 86.1 mmol) and potassium 3-ethoxy-3-oxo-propanoate (22.7 g, 133 mmol) were mixed in THF (200 mL) and stirred under argon overnight at 50 °C. The resultant white suspension from the second reaction flask was cooled to rt, the first flask contents were added dropwise over 10 min, and the reaction mixture was stirred for 16 h at rt. A difficult to stir chewing gum-like solid appeared upon initial addition, but after several hours the reaction mixture became more homogeneous and easier to stir. The reaction mixture was concentrated to approximately a third of its final combined volume, taken up in an equal volume of sat. potassium bisulfate solution, and extracted twice with ethyl acetate. The combined organic layers were washed with sat. sodium bicarbonate solution, dried over anhyd. Na2SO4, and evaporated in vacuo. The crude material was purified by Combiflash column chromatography (silica, 230–400 mesh), eluting with ethyl acetate–hexane to give compound 47 (7.2 g, 38.7 mmol, 44.9% yield) as a transparent liquid.
5-(1-Ethylpropyl)-4H-pyrazolo[1,5-a]pyrimidin-7-one (48)
A mixture of compound 47 (7 g, 37.6 mmol) and 1H-pyrazol-5-amine (3.12 g, 37.6 mmol) in acetic acid (35 mL) was heated at 110 °C for 3 h. The solvent was evaporated in vacuo, and the residue was treated with ethyl acetate and filtered to give compound 48 (2.1 g, 9.96 mmol, 26.5% yield) as an off-white solid. LCMS (ESI) m/z: 206.4 [M+H]+.
7-Chloro-5-(1-ethylpropyl)pyrazolo[1,5-a]pyrimidine (49)
A stirred solution of compound 48 (300 mg, 1.46 mmol) in POCl3 (2.1 mL, 22.5 mmol) was heated to 100 °C for 4 h. The reaction mixture was brought to rt, and the excess reagent was removed in vacuo. The residue was treated with ice–water, and the chlorinated product was extracted from the aqueous mixture by DCM. The organic layer was separated, dried over anhyd. Na2SO4, and purified by Combiflash column chromatography (silica, 230–400 mesh), eluting with 10–20% ethyl acetate in hexanes to give 49 (180 mg, 0.797 mmol, 54.5% yield) as a light-yellow liquid. LCMS (ESI) m/z: 224.1 [M+H]+.
(1S,3S)-N3-[3-Chloro-5-(1-ethylpropyl)pyrazolo[1,5-a]pyrimidin-7-yl]cyclopentane-1,3-diamine Hydrochloride (29)
To a stirred solution of tert-butyl compound 29b (80 mg, 0.190 mmol; see Supporting Information) in 1,4-dioxane (6.40 mL) was added 4 M HCl–dioxane (0.5 mL, 2 mmol) at 0 °C and stirred at rt for 2 h. The reaction mixture was evaporated in vacuo, and the resulting solid was triturated with pentane (3 × 2 mL) and ether (2 × 2 mL). The solid was then dried in vacuo and lyophilized to give compound 29 (65 mg, 0.180 mmol, 95.0% yield) as an off-white solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 9.03–8.49 (1 H, m), 8.36–8.05 (4 H, m), 6.44 (1 H, s), 4.64 (1 H, s), 3.75–3.64 (2 H, m), 3.53–3.43 (1 H, m), 2.65–2.59 (1 H, m), 2.30–1.99 (4 H, m), 1.88–1.59 (6 H, m), 0.80 (6 H, t, J = 7.3 Hz). LCMS (ESI) m/z: 322.4 [M+H]+.
tert-Butyl N-[(1S,3S)-3-[[5-(o-tolyl)pyrazolo[1,5-a]pyrimidin-7-yl]amino]cyclopentyl]carbamate (32a)
To a solution of compound 45 (50 mg, 0.14 mmol), o-tolylboronic acid (38 mg, 0.28 mmol), K2CO3 (59 mg, 0.43 mmol), and Pd(dppf)Cl2 (2 mg, 0.01 mmol) were added 1,4-dioxane (2.0 mL) and water (0.2 mL). The mixture was degassed and purged with N2 for 3 times and then stirred at 90 °C for 4 h under N2. The reaction mixture was concentrated in vacuo, and the residue was purified by prep-TLC (SiO2, petroleum ether/ethyl acetate 1:1, Rf = 0.6) to give compound 32a (55 mg, 0.13 mmol, 94% yield) as a white solid. LCMS (ESI) m/z: 408.2 [M+H]+.
(1S,3S)-N3-[5-(o-Tolyl)pyrazolo[1,5-a]pyrimidin-7-yl]cyclopentane-1,3-diamine (32)
To a solution of compound 32a (100 mg, 0.25 mmol) in DCM (3.0 mL) was added TFA (1.0 mL, 11.6 mmol), and the mixture was stirred at 25 °C for 12 h. The resulting solution was cooled to 0 °C and basified by addition of aq. sat. NaHCO3. The reaction mixture was concentrated in vacuo, and the residue was purified by prep-HPLC (neutral condition) to give compound 32 (17.7 mg, 0.056 mmol, 23% yield) as a yellow solid. LCMS (ESI) m/z: 308.2 [M+H]+. 1H NMR (400 MHz, CD3OD) δ ppm = 8.06 (1 H, d, J = 2.4 Hz), 7.42–7.26 (4 H, m), 6.42 (1 H, d, J = 2.0 Hz), 6.19 (1 H, s), 4.35 (1 H, quin, J = 6.7 Hz), 3.61 (1 H, quin, J = 6.6 Hz), 2.76–2.70 (1 H, m), 2.43–2.42 (1 H, m), 2.45–2.30 (1 H, m), 2.24–2.14 (1 H, m), 2.13–2.03 (1 H, m), 2.03–1.93 (1 H, m), 1.88–1.69 (1 H, m). LCMS (ESI) m/z: 308.2 [M+H]+.
tert-Butyl N-[1S,3S)-3-[[5-(5-chloro-2-methylphenyl)pyrazolo[1,5-a]pyrimidin-7-yl]amino]cyclopentyl]carbamate (34a)
A mixture of compound 45 (50 mg, 0.14 mmol), (5-chloro-2-methyl-phenyl)boronic acid (72 mg, 0.43 mmol), K2CO3 (59 mg, 0.43 mmol), and Pd(dppf)Cl2 (10 mg, 0.01 mmol) in 1,4-dioxane (3 mL) and water (0.30 mL) was degassed and purged with N2 3 times, and then the mixture was stirred at 90 °C for 12 h under N2 atmosphere. The reaction mixture was concentrated under reduced pressure to give a residue which was purified by prep-TLC (SiO2, petroleum ether/ethyl acetate 1:1, Rf = 0.7) to give compound 34a (45 mg, 0.038 mmol, 26% yield) as a yellow solid. LCMS (ESI) m/z: 442[M+H]+.
(1S,3S)-N3-[5-(5-Chloro-2-methylphenyl)pyrazolo[1,5-a]pyrimidin-7-yl]cyclopentane-1,3-diamine (34)
To a solution of compound 34a (40 mg, 0.09 mmol) in DCM (3 mL) was added TFA (1.00 mL, 11.6 mmol). The mixture was stirred at 20 °C for 0.5 h. The reaction mixture was concentrated in vacuo to give a residue. The residue was dissolved in MeOH (2 mL) and basified to pH 8 by NH3·H2O (25% in H2O). Then the residue was purified by prep-HPLC (neutral conditions) to give compound 34 (9.85 mg, 0.0285 mmol, 31% yield) as a yellow solid. 1H NMR (400 MHz, CD3OD) δ ppm 8.07 (1 H, d, J = 2.2 Hz), 7.42 (1 H, d, J = 2.0 Hz), 7.38–7.29 (2 H, m), 6.44 (1 H, d, J = 2.2 Hz), 6.21 (1 H, s), 4.36 (1 H, q, J = 6.9 Hz), 3.70–3.54 (1 H, m), 2.44–2.30 (4 H, m), 2.27–2.15 (1 H, m), 2.14–1.94 (2 H, m), 1.87–1.74 (1 H, m), 1.59–1.46 (1 H, m). LCMS (ESI) m/z: 342.1 [M+H]+.
(1S,3S)-N3-(5-Chloro-pyrazolo[1,5-a]pyrimidin-7-yl)cyclopentane-1,3-diamine (46)
A solution of compound 45 (900 mg, 2.56 mmol) in HCl/EtOAC (15 mL, 60 mmol) was stirred at 25 °C for 12 h. The reaction mixture was concentrated and then reconstituted in DCM/MeOH (10:1, 15 mL). The mixture was basified by sat. NaHCO3 (∼50 mL) until pH ∼ 8 in an ice bath. The resulting mixture was extracted with DCM/MeOH (10:1,15 mL × 3). The combined organic layers were dried over Na2SO4 and concentrated to give compound 46 as a pale-yellow oil. LCMS (ESI) m/z: 252.0 [M+H]+.
(1S,3S)-N3-[5-(5-Fluoro-2-methylphenyl)pyrazolo[1,5-a]pyrimidin-7-yl]cyclopentane-1,3-diamine (33)
To a solution of compound 46 (50 mg, 0.20 mmol) in DMF (1 mL) were added (5-fluoro-2-methyl-phenyl)boronic acid (45.9 mg, 0.30 mmol), potassium phosphate (0.3 mL, 0.60 mmol), and tetraphenylphosphine palladium (23.0 mg, 0.02 mmol). The mixture was bubbled with nitrogen for 1 min and then stirred at 130 °C for 10 min in the microwave. LCMS showed that the starting material was completely consumed, and the desired mass was detected. The reaction mixture was filtered, and the filtrate was purified by prep-HPLC (Waters Xbridge Prep OBD C18 150 mm × 50 mm, 10 μm column; 15–45% acetonitrile in a 10 mM ammonium bicarbonate solution in water, 8 min gradient). Compound 33 (10.8 mg,0.03 mmol, 16.2% yield) was obtained as a white solid. LCMS (ESI) m/z 326.2 [M+H]+. 1H NMR (400 MHz, CD3OD) δ ppm 8.07 (1 H, d, J = 2.4 Hz), 7.33–7.31 (1 H, m), 7.18–7.15 (1 H, m), 7.09–7.08 (1 H, m), 6.44 (1 H, d, J = 2.4 Hz), 6.17 (1 H, s), 4.44–4.37 (1 H, m), 3.68–3.61 (1 H, m), 2.39–2.37 (1 H, m), 2.34 (1 H, s), 2.27–2.25 (1 H, m), 2.15–2.09 (1 H, m), 1.86–1.83 (1 H, m), 1.62–1.60 (1 H, m), 1.59–1.57 (1 H, m).
1-((1r,3r)-3-((5-Isopropylpyrazolo[1,5-a]pyrimidin-7-yl)amino)cyclobutyl)-3-methylurea (14).
S-Methyl N-methylcarbamothioate (300 mg, 2.85 mmol) was added to a stirred solution of compound 8 (200 mg, 0.82 mmol) in 1,4-dioxane (2.5 mL) and water (1.5 mL) in a sealed tube at rt. The reaction mixture was heated at 65 °C for 12 h. The progress of the reaction was monitored by LCMS. After completion of reaction, the reaction mixture was evaporated in vacuo and purified by Combiflash column chromatography (silica, 230–400 mesh), eluting with 5% MeOH in CH2Cl2 to give (100 mg, 0.323 mmol, 39.6% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.02 (1 H, d, J = 2.2 Hz), 7.96 (1 H, d, J = 6.1 Hz), 6.35–6.27 (2 H, m), 5.81 (1 H, s), 5.68 (1 H, d, J = 4.8 Hz), 4.22–4.10 (2 H, m), 2.98–2.86 (1 H, m), 2.54 (3 H, d, J = 4.6 Hz), 2.48–2.42 (2 H, m), 2.33–2.22 (2 H, m), 1.22 (6 H, d, J = 6.9 Hz). LCMS (ESI) m/z: 303.4 [M + H]+.
tert-Butyl N-[(1S,3S)-3-[[5-(1-Ethylpropyl)pyrazolo[1,5-a]pyrimidin-7-yl]amino]cyclopentyl]carbamate (28a).
A stirred solution of compound 49 (2.4 g, 10.73 mmol), tert-butyl ((1S,3S)-3-aminocyclopentyl)carbamate (2.36 g, 11.8 mmol) and K2CO3 (4.44 g, 32.19 mmol) in MeCN (25 mL) was heated to reflux for 16 h. The reaction mixture was cooled and filtered. The filtrate was concentrated in vacuo, and the residue was purified by Combiflash column chromatography, eluting with 30% ethyl acetate in hexanes to give compound 28a (3.6 g, 9.05 mmol, 84.3% yield) as an off-white solid. LCMS (ESI) m/z: 388.3 [M + H]+.
[(1S,3S)-3-[[5-(1-Ethylpropyl)pyrazolo[1,5-a]pyrimidin-4-ium-7-yl]amino]cyclopentyl]ammonium Dichloride (28).
To 28a (3.4 g, 8.77 mmol) in 1,4-dioxane (6.8 mL) was added 4 M HCl in dioxane (10.6 mL, 43.9 mmol) at 0 °C and stirred at rt for 4 h. The reaction mixture was evaporated in vacuo, triturated with pentane, and lyophilized from H2O to give compound 28 (3 g, 8.27 mmol, 94.3% yield) as an off-white sticky solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 9.13 (1 H, s), 8.40 (3 H, s), 8.20 (1 H, d, J = 2.1 Hz), 6.62 (1 H, s), 6.56 (1 H, d, J = 2.2 Hz), 4.90 (1 H, s), 3.77 (1 H, s), 2.86–2.74 (1 H, m), 2.38–2.14 (4 H, m), 1.97–1.73 (6 H, m), 0.88 (6 H, t, J = 7.4 Hz). LCMS (ESI) m/z: 288.2 [M + H]+.
tert-Butyl ((1R,3R)-3-((5-(Pentan-3-yl)pyrazolo[1,5-a]pyrimidin-7-yl)amino)cyclopentyl)carbamate (38a).
To a stirred solution of compound 49 (4.8 g, 21.5 mmol) in MeCN (50 mL) was added tert-butyl ((1R,3R)-3-aminocyclopentyl)carbamate (4.3 g, 21.5 mmol) and K2CO3 (8.89 g, 64.4 mmol) at 20 °C before the mixture was stirred at 80 °C for 12 h. The progress of the reaction was monitored by LCMS. The reaction mixture was cooled to 20 °C, poured into H2O (100 mL), and extracted with ethyl acetate (3 × 200 mL). The combined organic layers were washed with brine (300 mL), dried over anhyd. Na2SO4, filtered and concentrated under reduced pressure to give compound 38a (7.6 g, 19.6 mmol, 91.6% yield) as a yellow solid, which was used directly in the next step without further purification. LCMS (ESI) m/z: 388.3 [M + H]+.
(1R,3R)-N1-(5-(Pentan-3-yl)pyrazolo[1,5-a]pyrimidin-7-yl)cyclopentane-1,3-diamine (38).
To a solution of compound 38a (8.0 g, 20.6 mmol) in ethyl acetate (40 mL) was added HCl/ethyl acetate (40 mL, 614 mmol) and the mixture was stirred at 15 °C for 24 h. The progress of the reaction was monitored by LCMS. After completion of reaction, the reaction mixture was evaporated in vacuo dissolved in H2O (200 mL) and extracted with ethyl acetate (3 × 100 mL). The aqueous phase was adjusted to pH = 8 with ammonium hydroxide and extracted with CH2Cl2/MeOH (10:1, 3 × 300 mL). The combined organic layers were washed with brine (2 × 300 mL), dried over anhyd. Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography (ISCO 40 g silica, 0–10% MeOH in CH2Cl2, gradient over 20 min) to give compound 38 (5.1 g, 17.745 mmol, 86.0% yield) as a yellow solid. 1H NMR (400 MHz, CD3OD) δ ppm 7.98 (1 H, d, J = 2.0 Hz), 6.32 (1 H, d, J = 2.4 Hz), 6.02 (1 H, s), 4.35–4.28 (1 H, m), 3.59–3.53 (1 H, m), 2.51–2.49 (1 H, m), 2.49–2.47 (1 H, m), 2.16–2.01 (1 H, m), 2.00–1.97 (2 H, m), 1.77–1.70 (5 H, m), 1.52–1.47 (1 H, m), 0.84 (6 H, t, J = 7.6 Hz). LCMS (ESI) m/z: 288.2 [M + H]+.
tert-Butyl ((1S,3R)-3-((5-(Pentan-3-yl)pyrazolo[1,5-a]pyrimidin-7-yl)amino)cyclopentyl)carbamate (39a).
To a stirred solution of compound 49 (10.1 g, 44.9 mmol) in MeCN (150 mL) was added tert-butyl ((1R,3S)-3-aminocyclopentyl)carbamate (9.0 g, 44.9 mmol) and K2CO3 (18.6 g, 135 mmol) at 20 °C before the mixture was stirred at 80 °C for 16 h. The progress of the reaction was monitored by LCMS. The reaction mixture was cooled to 20 °C, poured into H2O (100 mL), and extracted with ethyl acetate (3 × 200 mL). The combined organic layers were washed with brine (300 mL), dried over anhyd. Na2SO4, filtered and concentrated under reduced pressure to give compound 39a (20 g, 51.6 mmol, >100.0% yield) as a pale, yellow gum, which was used directly in the next step without further purification. LCMS (ESI) m/z: 388.4 [M + H]+.
(1R,3S)-N1-(5-(Pentan-3-yl)pyrazolo[1,5-a]pyrimidin-7-yl)cyclopentane-1,3-diamine (39).
To a solution of compound 39a (20 g, 51.6 mmol) in ethyl acetate (100 mL) was added HCl/ethyl acetate (100 mL, 400 mmol) and the mixture was stirred at 20 °C for 10 h. The progress of the reaction was monitored by LCMS. After completion of reaction, the reaction mixture was poured into sat. NaHCO3 (200 mL) and extracted with ethyl acetate (200 mL) and CH2Cl2 (2 × 300 mL). The combined organic layers were washed with brine (50 mL), dried over anhyd. Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography (300 g Agela C18, 35–65% MeOH in NH3H2O, gradient over 55 min) to give compound 39 (8.5 g, 29.37 mmol, 57.3% yield) as a yellow gum. 1H NMR (400 MHz, CD3OD) δ ppm 7.96 (1 H, d, J = 1.8 Hz), 6.38–6.28 (1 H, m), 5.98 (1 H, s), 4.22–4.16 (1 H, m), 3.49–3.41 (1 H, m), 2.62–2.41 (2 H, m), 2.27–2.11 (1 H, m), 2.01 (1 H, qd, J = 6.8, 13.2 Hz), 1.94–1.79 (1 H, m), 1.77–1.58 (5 H, m), 1.57–1.41 (1 H, m), 0.82 (6 H, t, J = 7.2 Hz). LCMS (ESI) m/z: 288.2 [M + H]+.
tert-Butyl ((1R,3S)-3-((5-(Pentan-3-yl)pyrazolo[1,5-a]pyrimidin-7-yl)amino)cyclopentyl)carbamate (40a).
To a stirred solution of compound 49 (10.05 g, 44.94 mmol) in MeCN (150 mL) was added tert-butyl ((1S,3R)-3-aminocyclopentyl)carbamate (9.00 g, 44.94 mmol) and K2CO3 (18.60 g, 134.81 mmol) at 20 °C before the mixture was stirred at 80 °C for 15 h. The progress of the reaction was monitored by LCMS. The reaction mixture was cooled to 20 °C, poured into H2O (100 mL), and extracted with ethyl acetate (3 × 200 mL). The combined organic layers were washed with brine (300 mL), dried over anhyd. Na2SO4, filtered and concentrated under reduced pressure to give compound 40a (18.0 g, 46.5 mmol, 103.4% yield) as a yellow oil, which was used directly in the next step without further purification. LCMS (ESI) m/z: 388.2 [M + H]+.
(1S,3R)-N1-(5-(Pentan-3-yl)pyrazolo[1,5-a]pyrimidin-7-yl)cyclopentane-1,3-diamine (40).
To a solution of compound 40a (19.4 g, 49.9 mmol) in ethyl acetate (125 mL) was added HCl/ethyl acetate (125 mL, 499 mmol) and the mixture was stirred at 20 °C for 15 h. The progress of the reaction was monitored by LCMS. After completion of reaction, the reaction mixture was evaporated in vacuo dissolved in H2O (200 mL) and extracted with ethyl acetate (3 × 100 mL). The aqueous phase was adjusted to pH = 8 with ammonium hydroxide and extracted with CH2Cl2/MeOH (10:1, 3 × 300 mL). The combined organic layers were washed with brine (2 × 300 mL), dried over anhyd. Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography (ISCO 120 g silica, 0–10% MeOH in CH2Cl2, gradient over 20 min) to give compound 40 (11.0 g, 38.3 mmol, 76.7% yield) as a pale, yellow gum. 1H NMR (400 MHz, CD3OD) δ ppm 7.98 (1 H, d, J = 2.0 Hz), 6.33 (1 H, d, J = 2.4 Hz), 6.00 (1 H, s), 4.20–4.16 (1 H, m), 3.46–3.42 (1 H, m), 2.56–2.47 (2 H, m), 2.25–2.16 (1 H, m), 2.15–1.95 (1 H, m), 1.94–1.83 (1 H, m), 1.77–1.70 (4 H, m), 1.68–1.55 (1 H, m), 1.54–1.52 (1 H, m), 0.84 (6 H, t, J = 7.6 Hz). LCMS (ESI) m/z: 288.2 [M + H]+.
CDK Kinases Assay
In vitro kinase profiling of the CDK kinase panel was performed at Reaction Biology Corporation (www.reactionbiology.com, Malvern, PA) using the “HotSpot” assay platform. Assay performed as described.37 In summary, each CDK and cyclin pairing along with required cofactors was prepared in base reaction buffer [20 mM HEPES (pH 7.5), 10 mM MgCl2, 1 mM EGTA, 0.01% Brij35, 0.02 mg/mL BSA, 0.1 mM Na3VO4, 2 mM DTT, 1% DMSO]. Compounds in DMSO stock solutions are then acoustically delivered to the CDK/cyclin buffer suspension and incubated for 20 min before a mixture of ATP (Sigma, St. Louis MO) and32P-ATP (specific activity 0.01 μCi/μL final; PerkinElmer, Waltham MA) to a final concentration of 10 μM is added to initiate the reaction. The reactions are incubated for 2 h at rt whereupon reactions are spotted onto P81 ion exchange paper (Whatman Inc., Piscataway, NJ). The filters are washed extensively with 0.75% phosphoric acid, and the remaining radioactive phosphorylated substrate remaining on the paper was measured. Kinase activity data were expressed as the percent remaining kinase activity in test samples compared to that in vehicle (DMSO) reactions. IC50 values and curve fits were obtained using Prism4 Software (GraphPad). Kinome tree representations were prepared using Kinome Mapper (https://www.reactionbiology.com/resources/tools/kinase-mapper).
Ligand Docking Protocol
The crystal structure of CDK9 (PDB:3MY1) was used for this study. Structure preparation and molecular docking were performed by using MOE software. Protein structure was prepared using the MOE “Structure Preparation” protocol with all default parameters. Ligand, 28, was prepared using MOE and properly protonated. “Induced Fit” molecular docking protocol was used. Top five docking poses were kept and further analyzed.
Quantum Mechanics Calculation
The torsional profile between pyrazolo[1,5-a]pyrimidine ring and diamino-cyclopentane ring of docked pose for each diamino-cyclopentane diastereomers was analyzed using GAUSSIAN 16 software at the B3LYP/6-31G* level with PCM as solvent model.38 The energy difference between the docked conformer and the conformer with the lowest energy was defined as the molecular strain energy in this study.
X-ray Crystallography
The CDK9 and CyclinT1 constructs were designed as reported.39 GST-TEV-CDK9 (M1-T330, S7D, V8N, K44R, Y138F, K280A, D307E, N311E) and His-Flag-TEV-CyclinT1(M1-R259, R26A, Q77R, E96G, K106R, F241L) were cloned into the pFastBac1 vector. The baculovirus CDK9 and cyclinT1 was added in a 1:1 ratio to infect SF21 cells to express the CDK9/cyclin T1 complex. The cell pellet containing protein complex was loaded onto the Ni-NTA column and then the GST column followed by TEV cleavage to remove the GST-tag on CDK9 and the His tag on cyclin T1. The protein complex was further separated through reverse Nickel and GST column and purified by SEC column (Superdex 200 Increase 10/300 GL) in a final buffer 20 mM Tris-HCl (pH 8.0), 500 mM NaCl, 5 mM DTT. The protein complex was concentrated to 5.66 mg/mL and incubated with 28 on ice for 1 h before setting up crystallization trays. The co-crystals of CDK9-cyclin T1-28 were obtained from 0.2 M potassium citrate and 20% PEG 3350 before being harvested and flash-cooled in liquid nitrogen for data collection. A 3.75 Å dataset was collected on beamline BL45XU at Spring-8 synchrotron source, indexed and integrated with XDS,40 and scaled by aimless. The CDK9-CyclinT1 complex structure was solved by molecular replacement with Phaser module in the CCP4 package suite,41 using the coordinates of PDB 3BLH as the search model. Compound 28 was placed into the electron density based on the difference electron density maps. After multiple cycles of iterative refinement with Refmac module in the CCP4 package suite and manual adjustment in the Coot program,42 the complex structure was finally refined to Rwork/Rfree as 19.85%/24.99%, respectively. The refinement statistics are listed in Table S1.
OncoPanel Multiplexed Cytotoxicity Assay
On-cell compound profiling was performed at Eurofins Discovery Services (www.eurofinsdiscovery.com, St. Charles, MO) using the OncoPanel Multiplexed Cytotoxicity assay platform. Cells were grown in RPMI 1640, 10% FBS, 2 mM l-alanyl-l-glutamine, 1 mM Na pyruvate, or a special medium. Cells were seeded into 384-well plates and incubated in a humidified atmosphere of 5% CO2 at 37 °C. Compounds were added the day following cell seeding. At the same time, a time zero untreated cell plate was generated. After a 3-day incubation period, cells were fixed and stained with fluorescently labeled antibodies and nuclear dye to allow imaging of nuclei, apoptotic cells, and mitotic cells. Compound 28 was serially diluted in 2-fold steps from the highest test concentration specified in the above table and assayed over 10 concentrations with a maximum assay concentration of 0.1% DMSO. Automated fluorescence microscopy was carried out using a Molecular Devices ImageXpress Micro XL high-content imager, and images were collected with a 4× objective. 16-bit TIFF images were acquired and analyzed with MetaXpress 5.1.0.41 software. Cell proliferation was measured by the fluorescence intensity of the incorporated nuclear dye. The output is referred to as the relative cell count, where the measured nuclear intensity is transformed to the percent of control.
Xenograft Tumor Models
Athymic Nude-Foxn1nu immunocompromised female mice between 5 and 8 weeks of age were implanted subcutaneously into the left flank with TNBC tumor model fragments. After tumors grew to 150–300 mm3, mice (n = 10/group) were administered vehicle p.o., 28 at 60 mg/kg p.o. QD×1 followed by (QD×3 on, QD×4 off) for 4 cycles and standard of care [Carboplatin 50 mg/kg i.p. Q14×2 + Paclitaxel 10 mg/kg i.v. Q14×2 or Cisplatin 5 mg/kg i.p. Q7D×3 + Gemcitabine 100 mg/kg i.p. Q7D×3 or Carboplatin 40 mg/kg i.p. Q7D×3 + Gemcitabine 100 mg/kg i.p. Q7D×3 for 3 cycles]. All experimental procedures were performed according to the guidelines of the Institutional Animal Care and Use Committee (IACUC) of Champions Oncology. After inoculation, the animals were checked daily for morbidity and mortality. At the time of routine monitoring, the animals were checked for any effects of tumor growth and treatments on normal behavior such as mobility, food and water consumption, tumor volume, body weight gain/loss (body weights and tumor volumes were measured twice weekly), eye/hair matting, and any other abnormal effect. Death and observed clinical signs were recorded on the basis of the numbers of animals within each subset. Tumor volume (TV) was calculated using the formula (0.52[length × width2]). Inhibition of tumor growth (TGI) was determined by calculating the percent TGI with the following formula: (100% × [1 – (final TV × initial TV of a treated group)/(final TV – initial TV of the control group)]). Treatment started on Day 0. Statistical analyses comparing all groups were performed by one-way ANOVA followed by Tukey’s multiple comparisons test (GraphPad Prism 8.4.3). P-values <0.05 are statistically significant.
Analysis of RNAP II (Ser2/5) and MYC in Tumor Samples
Tumors were harvested at 2 h and at 8 h post-dose. Tumors were bisected: half of the tumor was flash frozen, placed on dry ice, and stored at −80 °C, and the other half was fixed in 10% neural-buffered formalin for 18–24 h and then transferred to 70% ethanol at rt until paraffin was embedded. Tumors <250 mm3 were processed as a single snap-frozen sample. Phosphorylation of the RNAP II large subunit POLR2A (Ser2/5) and MYC levels were measured via Homogeneous Time Resolved Fluorescence (HTRF) available from Cisbio (www.cisbio.com, Bedford, MA). Frozen tumor tissue was homogenized by using a mortar and pestle and lysed according to the manufacturer’s protocol. Protein concentrations were measured using a Pierce BCA Protein assay kit (www.thermofisher.com, Waltham, MA). Optimized protein concentration (1–2 mg/mL) was added to the appropriate well of a white-walled 384-well plate. HTRF antibodies [Anti-Human c-Myc-Eu Cryptate Antibody/Anti-Human c-Myc-d2 Antibody and Anti-Human pRNA (S2/S5)-Eu Cryptate Antibody/Anti-Human pRNA (S2/S5)-d2 Antibody] were then combined and diluted into detection buffer, and 4 μL was added to each well. The plate was then incubated at rt out of light for 3 h, before being read on the Envision platform (www.perkinelmer.com, Waltham, MA).
Acknowledgments
This manuscript is dedicated to the memory of John C. Martin for his support of Kronos Bio. We thank Christopher Dinsmore and Zhihua Ma for their enriching editorial comments and suggestions. Christopher M. Wilfong and Angela N. Koehler provided insightful discussions. We thank all current and past members of the CDK9 program. Crystal Kraft from MJH Life Sciences assisted with editorial review and formatting. Zhijia Lv of Wuxi Biortus Biosciences Co., Ltd. assisted with X-ray co-crystal collection and structural elucidation.
Glossary
Abbreviations Used
- ACN
acetonitrile
- ADME
absorption, distribution, metabolism, and excretion
- anhyd.
anhydrous
- aq.
aqueous
- ATP
adenosine 5′-triphosphate
- BSA
bovine serum albumin
- CDI
1,1′-carbonyldiimidazole
- CDK
cyclin-dependent kinase
- DCM
dichloromethane
- DIPEA
N,N-diisopropylethylamine
- DMF
N,N-dimethylformamide
- DMSO
dimethyl sulfoxide
- DTT
dithiothreitol
- EGTA
ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid
- ESI
electrospray ionization
- GCMS
gas chromatography mass spectrometry
- GST
glutathione S-transferase
- HBD/HBA
hydrogen-bond donor/hydrogen-bond acceptor
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HPLC
high-pressure liquid chromatography
- HRMS
high-resolution mass spectrometry
- i.p.
intraperitoneal
- i.v.
intravenous
- LCMS
liquid chromatography mass spectrometry
- NBS
N-bromosuccinimide
- NCS
N-chlorosuccinimide
- PARP
poly(ADP-ribose) polymerase
- PD
pharmacodynamic
- PK
pharmacokinetic
- p.o.
oral
- Rf
retention factor
- RNAP II
RNA polymerase II
- SAR
structure–activity relationship
- sat.
saturated
- SEC
size exclusion chromatography
- SMM
small molecule microarray
- TEA
triethanolamine
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
- TLC
thin-layer chromatography
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.3c01233.
X-ray crystal data collection and refinement statistics of compound 28 bound to ATP-competitive binding site of CDK9 in complex with cyclin T1; experimental procedures for the synthesis of select reagents and intermediates; kinome selectivity of compound 28 as determined by Kinase Hotspot; stereochemical effect on CDK panel selectivity among analogues 28, 38, 39, and 40; plotted viability effects of 28 on TNBC cell lines; OncoPanel Multiplexed Cytotoxicity Assay control compound data; and two additional TNBC PDX models treated with 28 (PDF)
Molecular formula strings containing chemical structures and biochemical characterization of relevant compounds (CSV)
Accession Codes
The coordinates of compound 28 with CDK9 in complex with cyclin T1 have been deposited in RCSB Protein Data Bank (PDB) under accession code8K5R.
Author Contributions
The manuscript was written through contributions of all authors. All authors were employees, consultants or contractors of Kronos Bio, Inc. during the scope of the described work and have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Ghafouri-Fard S.; Khoshbakht T.; Hussen B. M.; Dong P.; Gassler N.; Taheri M.; Baniahmad A.; Dilmaghani N. A. A Review on the Role of Cyclin Dependent Kinases in Cancers. Cancer Cell Int. 2022, 22 (1), 325. 10.1186/s12935-022-02747-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradner J. E.; Hnisz D.; Young R. A. Transcriptional Addiction in Cancer. Cell 2017, 168 (4), 629–643. 10.1016/j.cell.2016.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaub F. X.; Dhankani V.; Berger A. C.; Trivedi M.; Richardson A. B.; Shaw R.; Zhao W.; Zhang X.; Ventura A.; Liu Y.; Ayer D. E.; Hurlin P. J.; Cherniack A. D.; Eisenman R. N.; Bernard B.; Grandori C.; et al. Pan-Cancer Alterations of the MYC Oncogene and Its Proximal Network across the Cancer Genome Atlas. Cell Syst. 2018, 6 (3), 282–300.e2. 10.1016/j.cels.2018.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapenna S.; Giordano A. Cell Cycle Kinases as Therapeutic Targets for Cancer. Nat. Rev. Drug Discov 2009, 8 (7), 547–566. 10.1038/nrd2907. [DOI] [PubMed] [Google Scholar]
- Braal C. L.; Jongbloed E. M.; Wilting S. M.; Mathijssen R. H. J.; Koolen S. L. W.; Jager A. Inhibiting CDK4/6 in Breast Cancer with Palbociclib, Ribociclib, and Abemaciclib: Similarities and Differences. Drugs 2021, 81 (3), 317–331. 10.1007/s40265-020-01461-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marra A.; Curigliano G. Are All Cyclin-Dependent Kinases 4/6 Inhibitors Created Equal?. Npj Breast Cancer 2019, 5 (1), 27. 10.1038/s41523-019-0121-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt A. C.; Goel S. Cellular Mechanisms Underlying Response and Resistance to CDK4/6 Inhibitors in the Treatment of Hormone Receptor-Positive Breast Cancer. Breast Cancer Res. 2022, 24 (1), 17. 10.1186/s13058-022-01510-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Z.; Tian L.; Qiang T.; Li J.; Xing Y.; Ren X.; Liu C.; Liang C. From Structure Modification to Drug Launch: A Systematic Review of the Ongoing Development of Cyclin-Dependent Kinase Inhibitors for Multiple Cancer Therapy. J. Med. Chem. 2022, 65 (9), 6390–6418. 10.1021/acs.jmedchem.1c02064. [DOI] [PubMed] [Google Scholar]
- Cidado J.; Boiko S.; Proia T.; Ferguson D.; Criscione S. W.; San Martin M.; Pop-Damkov P.; Su N.; Roamio Franklin V. N.; Sekhar Reddy Chilamakuri C.; D'Santos C. S.; Shao W.; Saeh J. C.; Koch R.; Weinstock D. M.; Zinda M.; Fawell S. E.; Drew L. AZD4573 Is a Highly Selective CDK9 Inhibitor That Suppresses MCL-1 and Induces Apoptosis in Hematologic Cancer Cells. Clin. Cancer Res. 2020, 26 (4), 922–934. 10.1158/1078-0432.CCR-19-1853. [DOI] [PubMed] [Google Scholar]
- Morales F.; Giordano A. Overview of CDK9 as a Target in Cancer Research. Cell Cycle 2016, 15 (4), 519–527. 10.1080/15384101.2016.1138186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey J.; Deckwerth T. L.; Kerwin W. S.; Casalini J. R.; Merrell A. J.; Grenley M. O.; Burns C.; Ditzler S. H.; Dixon C. P.; Beirne E.; Gillespie K. C.; Kleinman E. F.; Klinghoffer R. A. Voruciclib, a Clinical Stage Oral CDK9 Inhibitor, Represses MCL-1 and Sensitizes High-Risk Diffuse Large B-Cell Lymphoma to BCL2 Inhibition. Sci. Rep-uk 2017, 7 (1), 18007. 10.1038/s41598-017-18368-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lücking U.; Scholz A.; Lienau P.; Siemeister G.; Kosemund D.; Bohlmann R.; Briem H.; Terebesi I.; Meyer K.; Prelle K.; Denner K.; Bömer U.; Schäfer M.; Eis K.; Valencia R.; Ince S.; von Nussbaum F.; Mumberg D.; Ziegelbauer K.; Klebl B.; Choidas A.; Nussbaumer P.; Baumann M.; Schultz-Fademrecht C.; Rühter G.; Eickhoff J.; Brands M. Identification of Atuveciclib (BAY 1143572), the First Highly Selective, Clinical PTEFb/CDK9 Inhibitor for the Treatment of Cancer. ChemMedChem 2017, 12 (21), 1776–1793. 10.1002/cmdc.201700447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond J. R.; Boni V.; Lim E.; Nowakowski G.; Cordoba R.; Morillo D.; Valencia R.; Genvresse I.; Merz C.; Boix O.; Frigault M. M.; Greer J. M.; Hamdy A. M.; Huang X.; Izumi R.; Wong H.; Moreno V. First-in-Human Dose Escalation Study of Cyclin-Dependent Kinase-9 Inhibitor VIP152 in Patients with Advanced Malignancies Shows Early Signs of Clinical Efficacy. Clin. Cancer Res. 2022, 28 (7), 1285. 10.1158/1078-0432.CCR-21-3617. [DOI] [PubMed] [Google Scholar]
- Anshabo A. T.; Milne R.; Wang S.; Albrecht H. CDK9: A Comprehensive Review of Its Biology, and Its Role as a Potential Target for Anti-Cancer Agents. Front. Oncol. 2021, 11, 678559 10.3389/fonc.2021.678559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara R. P.; Bacon C. W.; D’Orso I. Transcription Elongation Control by the 7SK SnRNP Complex: Releasing the Pause. Cell Cycle 2016, 15 (16), 2115–2123. 10.1080/15384101.2016.1181241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S.; Coutinho-Mansfield G.; Wang D.; Pandit S.; Fu X.-D. The Splicing Factor SC35 Has an Active Role in Transcriptional Elongation. Nat. Struct Mol. Biol. 2008, 15 (8), 819–826. 10.1038/nsmb.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake D. R.; Vaseva A. V.; Hodge R. G.; Kline M. P.; Gilbert T. S. K.; Tyagi V.; Huang D.; Whiten G. C.; Larson J. E.; Wang X.; Pearce K. H.; Herring L. E.; Graves L. M.; Frye S. V.; Emanuele M. J.; Cox A. D.; Der C. J. Application of a MYC Degradation Screen Identifies Sensitivity to CDK9 Inhibitors in KRAS-Mutant Pancreatic Cancer. Sci. Signal. 2019, 12 (590), eaav7259 10.1126/scisignal.aav7259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C.-H.; Lujambio A.; Zuber J.; Tschaharganeh D. F.; Doran M. G.; Evans M. J.; Kitzing T.; Zhu N.; de Stanchina E.; Sawyers C. L.; Armstrong S. A.; Lewis J. S.; Sherr C. J.; Lowe S. W. CDK9-Mediated Transcription Elongation Is Required for MYC Addiction in Hepatocellular Carcinoma. Genes Dev. 2014, 28 (16), 1800–1814. 10.1101/gad.244368.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itzen F.; Greifenberg A. K.; Bösken C. A.; Geyer M. Brd4 Activates P-TEFb for RNA Polymerase II CTD Phosphorylation. Nucleic Acids Res. 2014, 42 (12), 7577–7590. 10.1093/nar/gku449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall N. F.; Peng J.; Xie Z.; Price D. H. Control of RNA Polymerase II Elongation Potential by a Novel Carboxyl-Terminal Domain Kinase. J. Biol. Chem. 1996, 271 (43), 27176–27183. 10.1074/jbc.271.43.27176. [DOI] [PubMed] [Google Scholar]
- Rahl P. B.; Lin C. Y.; Seila A. C.; Flynn R. A.; McCuine S.; Burge C. B.; Sharp P. A.; Young R. A. C-Myc Regulates Transcriptional Pause Release. Cell 2010, 141 (3), 432–445. 10.1016/j.cell.2010.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCracken S.; Fong N.; Yankulov K.; Ballantyne S.; Pan G.; Greenblatt J.; Patterson S. D.; Wickens M.; Bentley D. L. The C-Terminal Domain of RNA Polymerase II Couples MRNA Processing to Transcription. Nature 1997, 385 (6614), 357–361. 10.1038/385357a0. [DOI] [PubMed] [Google Scholar]
- Gregory G. P.; Hogg S. J.; Kats L. M.; Vidacs E.; Baker A. J.; Gilan O.; Lefebure M.; Martin B. P.; Dawson M. A.; Johnstone R. W.; Shortt J. CDK9 Inhibition by Dinaciclib Potently Suppresses Mcl-1 to Induce Durable Apoptotic Responses in Aggressive MYC-Driven B-Cell Lymphoma in Vivo. Leukemia 2015, 29 (6), 1437–1441. 10.1038/leu.2015.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S. K.; Lewis B. A.; Levens D. MYC: A Complex Problem. Trends Cell Biol. 2023, 33 (3), 235–246. 10.1016/j.tcb.2022.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang C. V. MYC on the Path to Cancer. Cell 2012, 149 (1), 22–35. 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwell T. K.; Kretzner L.; Blackwood E. M.; Eisenman R. N.; Weintraub H. Sequence-Specific DNA Binding by the c-Myc Protein. Science 1990, 250 (4984), 1149–1151. 10.1126/science.2251503. [DOI] [PubMed] [Google Scholar]
- Hirose Y.; Ohkuma Y. Phosphorylation of the C-Terminal Domain of RNA Polymerase II Plays Central Roles in the Integrated Events of Eucaryotic Gene Expression. J. Biochem. 2007, 141 (5), 601–608. 10.1093/jb/mvm090. [DOI] [PubMed] [Google Scholar]
- Toure M. A.; Koehler A. N. Addressing Transcriptional Dysregulation in Cancer through CDK9 Inhibition. Biochemistry 2023, 62 (6), 1114–1123. 10.1021/acs.biochem.2c00609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z.; Wang T.; Wang C.; Fan Y. CDK9 Inhibitors in Cancer Research. RSC Med. Chem. 2022, 13 (6), 688–710. 10.1039/D2MD00040G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richters A.; Doyle S. K.; Freeman D. B.; Lee C.; Leifer B. S.; Jagannathan S.; Kabinger F.; Koren J. V.; Struntz N. B.; Urgiles J.; Stagg R. A.; Curtin B. H.; Chatterjee D.; Mathea S.; Mikochik P. J.; Hopkins T. D.; Gao H.; Branch J. R.; Xin H.; Westover L.; Bignan G. C.; Rupnow B. A.; Karlin K. L.; Olson C. M.; Westbrook T. F.; Vacca J.; Wilfong C. M.; Trotter B. W.; Saffran D. C.; Bischofberger N.; Knapp S.; Russo J. W.; Hickson I.; Bischoff J. R.; Gottardis M. M.; Balk S. P.; Lin C. Y.; Pop M. S.; Koehler A. N. Modulating Androgen Receptor-Driven Transcription in Prostate Cancer with Selective CDK9 Inhibitors. Cell Chem. Biol. 2021, 28 (2), 134–147.e14. 10.1016/j.chembiol.2020.10.001. [DOI] [PubMed] [Google Scholar]
- Richters A.; Doyle S. K.; Freeman D. B.; Lee C.; Leifer B. S.; Jagannathan S.; Kabinger F.; Koren J. V.; Struntz N. B.; Urgiles J.; Stagg R. A.; Curtin B. H.; Chatterjee D.; Mathea S.; Mikochik P. J.; Hopkins T. D.; Gao H.; Branch J.; Xin H.; Westover L.; Bignan G. C.; Rupnow B. A.; Karlin K. L.; Olson C. M.; Westbrook T. F.; Vacca J.; Wilfong C. M.; Trotter B. W.; Saffran D. C.; Bischofberger N.; Knapp S.; Russo J. W.; Hickson I.; Bischoff J. R.; Gottardis M. M.; Balk S. P.; Lin C. Y.; Pop M. S.; Koehler A. N. Modulating Androgen Receptor-Driven Transcription in Prostate Cancer with Selective CDK9 Inhibitors. Cell Chem. Biol. 2021, 28, 134. 10.1016/j.chembiol.2020.10.001. [DOI] [PubMed] [Google Scholar]
- Baumli S.; Endicott J. A.; Johnson L. N. Halogen Bonds Form the Basis for Selective P-TEFb Inhibition by DRB. Chem. Biology 2010, 17 (9), 931–936. 10.1016/j.chembiol.2010.07.012. [DOI] [PubMed] [Google Scholar]
- Jones R. D.; Jones H. M.; Rowland M.; Gibson C. R.; Yates J. W. T.; Chien J. Y.; Ring B. J.; Adkison K. K.; Ku M. S.; He H.; Vuppugalla R.; Marathe P.; Fischer V.; Dutta S.; Sinha V. K.; Björnsson T.; Lavé T.; Poulin P. PhRMA CPCDC Initiative on Predictive Models of Human Pharmacokinetics, Part 2: Comparative Assessment of Prediction Methods of Human Volume of Distribution. J. Pharm. Sci. 2011, 100 (10), 4074–4089. 10.1002/jps.22553. [DOI] [PubMed] [Google Scholar]
- Liu D.; Song H.; Song L.; Liu Y.; Cao Y.; Jiang J.; Hu P. A Unified Strategy in Selection of the Best Allometric Scaling Methods to Predict Human Clearance Based on Drug Disposition Pathway. Xenobiotica 2016, 46 (12), 1105–1111. 10.1080/00498254.2016.1205761. [DOI] [PubMed] [Google Scholar]
- Ring B. J.; Chien J. Y.; Adkison K. K.; Jones H. M.; Rowland M.; Jones R. D.; Yates J. W. T.; Ku M. S.; Gibson C. R.; He H.; Vuppugalla R.; Marathe P.; Fischer V.; Dutta S.; Sinha V. K.; Björnsson T.; Lavé T.; Poulin P. PhRMA CPCDC Initiative on Predictive Models of Human Pharmacokinetics, Part 3: Comparative Assessement of Prediction Methods of Human Clearance. J. Pharm. Sci. 2011, 100 (10), 4090–4110. 10.1002/jps.22552. [DOI] [PubMed] [Google Scholar]
- Anastassiadis T.; Deacon S. W.; Devarajan K.; Ma H.; Peterson J. R. Comprehensive Assay of Kinase Catalytic Activity Reveals Features of Kinase Inhibitor Selectivity. Nat. Biotechnol. 2011, 29 (11), 1039–1045. 10.1038/nbt.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H.; Li X.; Caricato M.; Marenich A. V.; Bloino J.; Janesko B. G.; Gomperts R.; Mennucci B.; Hratchian H. P.; Ortiz J. V.; Izmaylov A. F.; Sonnenberg J. L.; Williams-Young D.; Ding F.; Lipparini F.; Egidi F.; Goings J.; Peng B.; Petrone A.; Henderson T.; Ranasinghe D.; Zakrzewski V. G.; Gao J.; Rega N.; Zheng G.; Liang W.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Throssell K.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M. J.; Heyd J. J.; Brothers E. N.; Kudin K. N.; Staroverov V. N.; Keith T. A.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A. P.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Millam J. M.; Klene M.; Adamo C.; Cammi R.; Ochterski J. W.; Martin R. L.; Morokuma K.; Farkas O.; Foresman J. B.; Fox D. J.. Gaussian 16; Gaussian, Inc.: Wallingford, CT, 2016. [Google Scholar]
- Barlaam B.; Casella R.; Cidado J.; Cook C.; De Savi C.; Dishington A.; Donald C. S.; Drew L.; Ferguson A. D.; Ferguson D.; Glossop S.; Grebe T.; Gu C.; Hande S.; Hawkins J.; Hird A. W.; Holmes J.; Horstick J.; Jiang Y.; Lamb M. L.; McGuire T. M.; Moore J. E.; O’Connell N.; Pike A.; Pike K. G.; Proia T.; Roberts B.; San Martin M.; Sarkar U.; Shao W.; Stead D.; Sumner N.; Thakur K.; Vasbinder M. M.; Varnes J. G.; Wang J.; Wang L.; Wu D.; Wu L.; Yang B.; Yao T. Discovery of AZD4573, a Potent and Selective Inhibitor of CDK9 That Enables Short Duration of Target Engagement for the Treatment of Hematological Malignancies. J. Med. Chem. 2020, 63 (24), 15564–15590. 10.1021/acs.jmedchem.0c01754. [DOI] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr. Sect. D: Biol. Crystallogr. 2010, 66 (Pt 2), 125–132. 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G. W.; McCoy A.; McNicholas S. J.; Murshudov G. N.; Pannu N. S.; Potterton E. A.; Powell H. R.; Read R. J.; Vagin A.; Wilson K. S. Overview of the CCP4 Suite and Current Developments. Acta Crystallogr. Sect. D 2011, 67 (4), 235–242. 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and Development of Coot. Acta Crystallogr. Sect. D 2010, 66 (4), 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.