

Abstract
Schistosomiasis is a disease affecting >200 million people worldwide, but its treatment relies on a single agent, praziquantel. To investigate new avenues for schistosomiasis control, we have conducted the first systematic analysis of bromodomain-containing proteins (BCPs) in a causative species, Schistosoma mansoni. Having identified 29 putative bromodomains (BRDs) in 22 S. mansoni proteins, we selected SmBRD3, a tandem BRD-containing BCP that shows high similarity to the human bromodomain and extra terminal domain (BET) family, for further studies. Screening 697 small molecules identified the human BET BRD inhibitor I-BET726 as a ligand for SmBRD3. An X-ray crystal structure of I-BET726 bound to the second BRD of SmBRD3 [SmBRD3(2)] enabled rational design of a quinoline-based ligand (15) with an ITC Kd = 364 ± 26.3 nM for SmBRD3(2). The ethyl ester pro-drug of compound 15 (compound 22) shows substantial effects on sexually immature larval schistosomula, sexually mature adult worms, and snail-infective miracidia in ex vivo assays.
Introduction
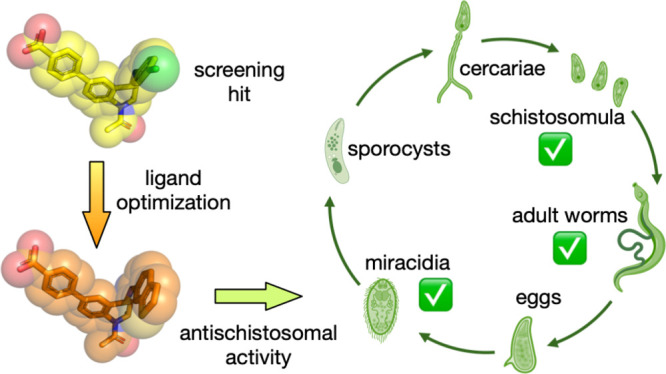
Epigenetic processes link changes in gene expression that do not result from alterations in the genetic code to phenotypic diversity in a population. The blood fluke parasite Schistosoma mansoni, which is responsible for the devastating neglected tropical disease schistosomiasis, adopts phenotypically diverse developmental forms as it progresses through its complex lifecycle (Figure 1A). However, the molecular processes that control these changes are poorly understood.1 As these phenotypic changes must occur without alteration of the parasite’s inherited genome, the involvement of epigenetic processes and cellular machinery is certain.2 At the molecular level, covalent modification of DNA and histone proteins modulate chromatin structure and recruit transcriptional machinery to modulate gene expression.3 The proteins that add these modifications are called epigenetic writers and those that remove them are called epigenetic erasers. A third class of proteins, known as epigenetic readers, bind to the covalent modifications on DNA or histones and recruit transcriptional machinery to that site.4 Bromodomain (BRD)-containing proteins (BCPs) are a particular class of epigenetic readers that bind to acetylated lysine (KAc) residues on histones, and many other proteins.5 Human BRDs are a therapeutic target for many oncology indications,6 but little is known about the role of BRDs in parasites,7 and almost nothing is known about their role in S. mansoni.8−10
Figure 1.

(A) Simplified cartoon of the S. mansoni lifecycle. (B) Domain architecture representation of the 22 putative S. mansoni BCPs classified by family (further defined in the Methods section).16 Where no family can be defined, the BCP is labeled “currently unclassified”. Gene ID (Smp; according to the S. mansoni v7 genome annotation; IDs remain stable in the v10 assembly), common name (in parentheses) and total protein length (in number of amino acids–aa) are all indicated. (C) Heat map illustration of bcp abundances across the S. mansoni lifecycle (derived from RNA-Seq meta data); SmBRD3 (Smp_147950) is highlighted in orange. Blue colors indicate higher abundance and red colors indicate lower abundance. Abbreviations: Mir: miracidia; Spo: sporocyst; Cerc: cercaria; Som: schistosomula.
Functional studies of schistosome BCPs will not only enrich our understanding of parasite development, but also reveal new targets for control of a disease that contributes to 200000 human deaths per year, increases the risk of developing certain types of cancers, modulates the immune system in infected individuals leading to reduced efficacy of vaccines, and is responsible for millions of disability adjusted life years lost in affected communities.11 Due to limited control options as well as its public health and zoonotic importance, the WHO has targeted schistosomiasis for elimination by 2030 in their most recent roadmap for ‘Ending the Neglect to Attain the Sustainable Development Goals’.12 Achieving this ambitious agenda will require basic investigations of schistosome biology to expose parasite vulnerabilities that, in turn, guide and target the development of novel therapeutic interventions.
Here, we report the first systematic study of BCPs in S. mansoni. Using a combination of sequence similarity and domain-based searches, applied previously to the characterization of schistosome histone methyltransferases and histone demethylases,13 we have identified 22 BCPs in S. mansoni containing 29 distinct BRDs. We have focused our investigation on SmBRD3 (Smp_147950) due to its relatively high temporal expression when compared to most other SmBCPs and similarity to the human bromodomain and extra terminal domain (BET) family of BCPs. SmBRD3 shares 37.5% sequence identity with human BRD3 (HsBRD3) and, similar to the BET BCPs, possesses two BRDs, SmBRD3(1) and SmBRD3(2). Using differential scanning fluorimetry (DSC) and isothermal titration calorimetry (ITC), we identified a number of human bromodomain ligands that bind to either SmBRD3(1) or SmBRD3(2). An X-ray crystal structure of I-BET726 bound to SmBRD3(2) allowed the structure-based optimization of this compound series to give the quinoline-derived compound 15, which has a Kd value of 364 ± 26.3 nM for SmBRD3(2). In a variety of whole organism phenotypic assays, the ethyl ester prodrug of compound 15, compound 22, demonstrated profound effects on both larval schistosomula and sexually reproductive adult worms. Interestingly, compound 22 also prevents the developmental transformation of snail-infective miracidia into asexually proliferative sporocysts. This suggests a role for SmBRD3 in the parasite developmental life cycle, mirroring the role of the BET bromodomains in humans.14,15 Our use of a structure- and target-based approach has enabled the rapid development of high affinity ligands for SmBRD3. While further work is required to determine the selectivity of the compounds, the development of chemical tools with defined cellular targets paves the way for contemporary medicinal chemistry techniques to be applied to S. mansoni.
Results
Identification of 22 Putative Bromodomain-Containing Proteins in S. mansoni
While BRDs have previously been identified in the S. mansoni histone acetyltransferases (HATs) SmGCN5,17SmCBP1 and SmCBP2,18,19 there has been no systematic search for these protein modules in the parasite proteome more generally. Additionally, while there have been some studies in which human BRD ligands have been assessed for their phenotypic effects on S. mansoni,20 there has not been a rational design approach taken to identifying and optimizing ligands for S. mansoni BRDs. Using a combination of genome- and domain-based bioinformatic searches of the S. mansoni genome, we identified 29 putative BRDs found in 22 distinct BCPs (Figure 1B). This differentially expressed gene family (Figure 1C) includes the previously characterized SmGCN5,17SmCBP1 and SmCBP2 proteins,18,19 but also a number of previously uncharacterized BCPs. One BCP, that is highly expressed throughout the S. mansoni lifecycle, possesses 37.5% sequence identity to human BRD3 (HsBRD3) and 33.2% identity to human BRD4 (HsBRD4); we have annotated this BCP as S. mansoni bromodomain-containing protein 3 (SmBRD3; Smp_147950; UniProt: G4 V8 V7), in analogy to HsBRD3. Sequence analysis shows that, similar to its human homologue, SmBRD3 possesses two bromodomains [SmBRD3(1) and SmBRD3(2)] that are located toward the N-terminus of the protein (Figure 1B), and which are separated by approximately 100 residues (Figure 2).
Figure 2.

(A) Cartoon of the SmBRD3(1) (green) and SmBRD3(2) (white) structures predicted using AlphaFold.21 (B) Multiple sequence alignment of the SmBRD3(1,2) construct, SmBRD3, HsBRD3, and HsBRD4. SmBRD3 has 36.64% and 34.69% sequence similarity with HsBRD3 and HsBRD4, respectively. The region identified as the first bromodomain is indicated with a green bar and the region identified as the second bromodomain is indicated with a white bar. Sequences were aligned using MUSCLE22 and the resulting multiple sequence alignment was visualized using ESPript 3 server.23 Vertical blue boxes indicate conserved residues, white letters in red boxes indicate strict identity and red letters in white boxes indicate similarity.
Expression of the SmBRD3 Bromodomains
Extensive structural studies on the human BET BCPs (including HsBRD3) have identified the residues that are important for its interactions with acetylated lysine (KAc) residues in target proteins. In HsBRD4(1), these are Y97 and N140; N140 forms a hydrogen bond directly with KAc, while Y97 interacts with KAc through a water-mediated hydrogen bond.24,25 The primary sequence of SmBRD(1) and SmBRD3(2) indicates that both of these BRDs possess equivalent residues to Y97 and N140, [Y67 & N110 in SmBRD3(1) and Y296 & N339 in SmBRD3(2)], indicating that they are canonical BRDs and that they can bind to KAc. A second region of the BET BRDs that is important for binding to KAc, and small molecule ligands, is the WFP shelf. Named after the residues that define it,26 this region binds to the lipophilic chain of a second KAc residue when bound to chromatin (e.g., PDB code: 3UVX).16 The lipophilic nature of this region has been exploited in the design of BRD4 ligands, with many high affinity compounds interacting with the WPF shelf. Interestingly, while SmBRD3(1) possesses the WPF residues (W56, P57, F58), these are replaced by an HFF motif (H280, F281, F282) in SmBRD3(2) perhaps indicating the ability of this BRD to recognize different residues, proteins, and/or small molecules from SmBRD3(1).
Development of biochemical/biophysical assays to identify SmBRD3 BRD ligands required the recombinant expression of SmBRD3(1), SmBRD3(2), and a construct that contained both bromodomains and the residues that link them [SmBRD3(1,2)]. While we were able to select and produce stable constructs of SmBRD3(2) and SmBRD3(1,2) in good yields (Figures S1–S5, Table S1), this was not possible for SmBRD3(1). Analysis of the full protein structure predicted by AlphaFold indicates that the helix comprising residues 617–638 and the subsequent disordered residues 639–660 form intraprotein interactions in the region between SmBRD3(1) and SmBRD3(2) (Figure S6A). We propose that these interactions are required to stabilize SmBRD3(1) (Figure S6B), and while they can be compensated for by other hydrophobic interactions in SmBRD3(1,2), this is not possible in SmBRD3(1) rendering it unstable when expressed alone.
Identification of Small Molecules That Bind to SmBRD3(1,2) and/or SmBRD3(2)
Using the SmBRD3(2) construct detailed above, we employed differential scanning fluorimetry (DSF)27 to screen 697 compounds, comprising 153 compounds from the PPI-net library, 160 compounds from our in-house library of human and Trypanosoma cruzi BRD ligands, and 384 compounds from the Maybridge Fragment Library against SmBRD3(2).28−37 Analysis of ΔTm values plotted against −log(SD) reveals a number of hits, including the known human BET bromodomain ligand, I-BET726 (Figure 3A).38 Selected compounds were then assessed against SmBRD3(1,2), and interestingly the human BET bromodomain ligands, OXFBD02 (1)29 and (+)-JQ1 (5),39 were observed to stabilize SmBRD3(1,2) but not SmBRD3(2), implying that these compounds bind preferentially to SmBRD3(1) (Figure 3B). Conversely, BI-2536 (7)40 and I-BET726 (8)38 stabilize both SmBRD3(1,2) and SmBRD3(2), suggesting that these compounds either bind to both SmBRD3 BRDs, or to SmBRD3(2) alone. Biophysical analysis using ITC (Table 1 and Table S2) confirmed the DFS results, and revealed that OXFBD02 (1) and (+)-JQ1 (5) have Kd values for SmBRD3(1,2) of 683 ± 166 and 445 ± 106 nM, respectively, with no binding detected to SmBRD3(2). The OXFBD02 (1) analogues OXFBD04 (4) and compound 3 showed the same behavior, but with a higher Kd value of 2950 ± 261 and 1750 ± 301 nM, respectively, for SmBRD3(1,2). I-BET726 (8) has a Kd = 1520 ± 520 nM for SmBRD3(1,2) and Kd = 1850 ± 361 nM, for SmBRD3(2) consistent with the idea that this compound either binds to both SmBRD3 BRDs or just to SmBRD3(2). I-BET151 (6) and BI-2536 (7) show similar behavior, but as their SmBRD3(2) affinities are lower, we progressed our studies with I-BET726 (8).
Figure 3.

(A) Scatter plot showing the thermal shift (ΔTm) values of the 697 compounds screened against SmBRD3(2), plotted against–log(SD). ΔTm correlates with compound affinity for SmBRD3(2) and higher values of–log(SD) correspond to greater confidence in the ΔTm value quoted. The dark gray dot shows the ΔTm value for BI2536, which was used as a positive control. The black dots show compounds that have a ΔTm value >2 °C, which we typically consider to be a hit, and the red dot shows the ΔTm value for I-BET726, and the orange dot shows the ΔTm value for I-BET151. Reported ΔTm values are the mean of two replicates. (B) Thermal shift (ΔTm) values of compounds 1, 5–8 against SmBRD3(1,2) (black bars) and SmBRD3(2) (gray bars). Compounds were tested at a concentration of 50 μM (n = 3), error bars show standard deviation (SD). (C)Chemical structures of compounds 1–8.
Table 1. SmBRD3(1,2) and SmBRD3(2) ITC Kd and pKd Values for Compounds 1–8a.

A color scale is used with darker purple representing lower Kd values. Values are n = 1 ± error of the curve fit. ITC traces are shown in Figures S26–S33.
Given the similarity of SmBRD3 and the human BET BRDs, we proposed that I-BET726 (8) would occupy the KAc binding pocket of SmBRD3(2) when bound. To probe this assumption, we obtained an X-ray crystal structure of I-BET726 (8) bound to SmBRD3(2) (Figure 4). This is the first X-ray crystal structure of a BRD from S. mansoni (PDB code: 7AMC) and it demonstrates that the overall fold of SmBRD3(2) is very similar to that of HsBRD4(1) (Figure 4A). The Tyr (Y97 and Y139) and Asn (N140) residues that are important for KAc in HsBRD4(1)24 are conserved in SmBRD3(2). N339 forms a hydrogen bond with the carbonyl oxygen atom of I-BET726 (8) and Y296 forms a water-mediated hydrogen bond with the same atom (Figure 4B). While HsBRD4(1) and HsBRD4(2) possess the WPF shelf, these residues are replaced by H280, F281, and F282 in SmBRD3(2) (Figure 4C). This change results in the formation of a narrow channel, the HFF cleft, which is defined by F281, E344, V345, and V348, and in which the chlorophenyl group of I-BET726 resides (Figure 4D).
Figure 4.

X-ray crystal structure of I-BET726 (8) bound to SmBRD3(2) (PDB code: 7AMC). (A) Overall fold of SmBRD3(2) (white cartoon) is similar to that of HsBRD4(1) (blue cartoon; PDB code: 4BJX). (B) X-ray crystal structure of I-BET726 (8) bound to SmBRD3(2) (cartoon and carbon = white; PDB code: 7AMC) overlaid with the X-ray crystal structure of HsBRD4(1) (carbon = blue; PDB code: 4BJX). This comparison shows that the NYY motif, which is important for KAc- and KAc-mimic binding, is conserved between the proteins. (C) WPF shelf region of HsBRD4(1) carbon = blue; PDB code: 4BJX) is altered to an HFF motif in SmBRD3(2) (cartoon and carbon = white; PDB code: 7AMC). (D) HFF residues result in a narrow channel, the HFF cleft, that is defined by F281, E344, V345, and V348. The chlorophenyl group of I-BET726 partly occupies the HFF cleft.
X-ray Crystal Structure of I-BET726 Bound to SmBRD3(2)
Despite the SmBRD3(2) affinity of I-BET726 (8), the occupancy of the HFF cleft by the chlorophenyl group does not appear to be optimal. As the affinity of many BET bromodomain ligands results from the binding of lipophilic groups to the WPF shelf,26,41 we reasoned that developing I-BET726 derivatives that can better occupy the HFF cleft would result in compounds with higher affinity for SmBRD3(2). Therefore, compounds 9–14 were designed, which incorporate a variety of [6.5]-fused ring systems (Figure 5). The quinoline derivative 15 was designed to probe the SmBRD3 affinity of a [6.6]-fused bicycle. The synthesis of these compounds was based, in part, on the work of Gosmini et al.38 and Shadrick et al.42 with the appropriate bicyclic bromides employed in the Buchwald–Hartwig coupling. Full details are provided in the Supporting Information.
Figure 5.
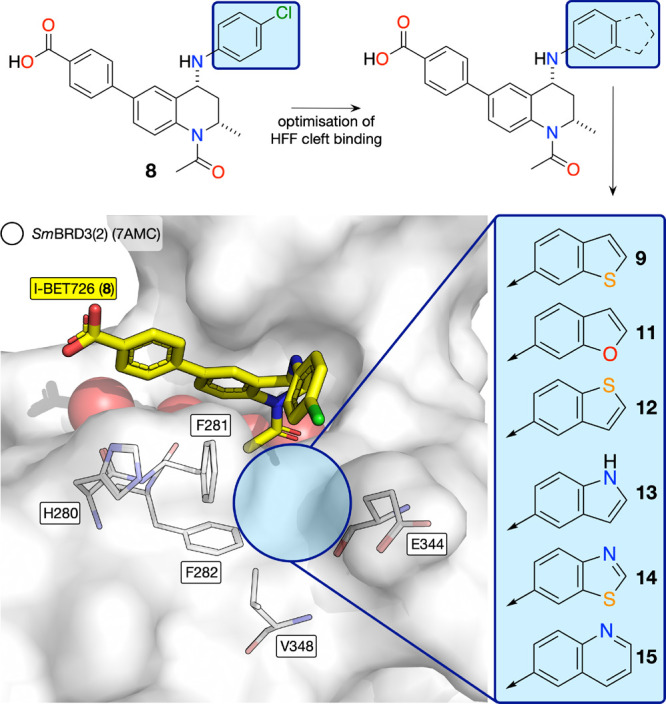
Design strategy that we employed to identify SmBRD3(2) ligands that occupy the HFF cleft more effectively than the chlorophenyl moiety of I-BET726 (8). We proposed that a bicyclic group replacing the cholorophenyl group would result in more effective HFF occupancy and therefore higher SmBRD3(2) affinity.
Structure-Based Optimization of Small Molecule Ligands for SmBRD3(2)
The benzothiophene-based compound 9 and its enantiomer, 10, were synthesized first. Analysis using ITC showed that compound 9 has a Kd value of 701 ± 64.9 nM, while compound 10 showed no detectable binding (Table 2). This observation is in line with data obtained on human bromodomains, where the enantiomer of an I-BET726 derivative showed reduced binding to HsBRD4(1).38 To determine whether the increase in SmBRD3(2) affinity displayed by compound 9 resulted from greater occupancy of the HFF cleft, we obtained an X-ray crystal structure of this compound bound to SmBRD3(2) (Figure 6, PDB code: 7AMH).
Table 2. SmBRD3(2) Kd Values of I-BET726 (8) and Compounds 9–15 Determined Using ITCd.

Single value ± error of the curve fit.
Mean value of 3 repeats ± s.e.m.
Mean value of 2 repeats ± s.e.m.
A colour scale is used with darker purple representing lower Kd values. ITC traces are shown in Figures S33–S47.
Figure 6.

X-ray crystal structure of compound 9 bound to SmBRD3(2) (cartoon and carbon = white, PDB code: 7AMH). (A) Overlay of 7AMH with 7AMC shows that I-BET726 (8) and 9 bind in the same orientation to SmBRD3(2) and make the same interactions with Y296, Y338, and N339. (B) Benzothiophene group of 9 occupies the HFF cleft more fully than the chlorophenyl group of I-BET726 (8).
This crystal structure shows that compound 9 binds to SmBRD3(2) in the same orientation to I-BET726 (8) and forms the same interactions with N339 and Y296 (Figure 6A). The benzothiophene moiety is observed to occupy the HFF cleft, as proposed (Figure 6B), and this interaction could contribute to the increased SmBRD3(2) affinity exhibited by compound 9. Encouraged by these data, we synthesized compounds 11–15, designed to probe the structure–activity relationship (SAR) of the HFF cleft, and evaluated their affinity for SmBRD3(2) using ITC. All compounds show higher SmBRD3(2) affinity compared to I-BET726 (8), with those compounds containing heteroatoms directed to the solvent (12, 13, 15) preferred over those with heteroatoms oriented toward the protein (9, 11, 14). Of the compounds evaluated, the quinoline derivative 15 possessed the highest SmBRD3(2) affinity (ITC Kd = 364 ± 26.3 nM), which we attribute to effective occupancy of the HFF cleft by the [6.6]-fused ring system. We note that compounds 9 and 15 also show high affinity for HsBRD4(1) (Table 2), which is not a problem for our studies here, but this activity would need to be absent from SmBRD3(2) ligands intended as treatments for schistosomiasis. The ethyl esters of compounds 9–15 (compounds 16–22) were also evaluated for their SmBRD3(2) affinity using ITC. In all cases no binding could be detected. To determine whether this observation resulted from low affinity or poor solubility, we analyzed the solubility (phosphate buffered saline at pH 7.4) and chromLogD values of the benzothiophene- and benzothiazole-based acids 9 and 14, and the benzothiophene- and quinoline-based esters 17 and 22 (Table S4). The acids 10 and 14 showed solubility of >259 μM and >260 μM, respectively, under these conditions; compound 10 has a chromLogD of 2.83 and 14 has a chromLogD of 1.78. The esters 17 and 22, however, have lower solubility of only 1 and 13 μM, respectively. The chromLogD value for 17 is 7.61 and for 22 the value is 5.95, indicating that these compounds are more lipophilic than the acid derivatives, as expected. These data suggest that we are unable to measure the SmBRD3(2) affinity of the ethyl esters due to poor solubility and we cannot be certain whether these compounds have any affinity for SmBRD3(2).
Encouraged by the above data and the broad expression of smbrd3 across the parasite’s lifecycle (Figure 1C), we evaluated the effects of these compounds on sexually immature larval schistosomula, sexually mature adult worms, and snail-infective miracidia, using a series of ex vivo assays. We were concerned that the carboxylic acid-containing I-BET726 analogues might not show high parasite permeability, and therefore we also evaluated their ethyl ester precursors, which we reasoned could act as prodrugs to release the SmBRD3(2)-binding ligand in situ.
Assessing the Effects of SmBRD3(2) Ligands on Schistosomula
A high-throughput imaging platform that quantifies both phenotype and motility ex vivo, was used to screen compounds against schistosomula; their effects were assessed as previously described.20 Briefly, the schistosomula stage of S. mansoni was obtained by mechanical transformation of cercariae, dispensed into 384-well tissue culture plates, and dosed with 20 μM (Figure 7) or 10 μM (Figure S48) of each compound. Following 72 h coincubation of the schistosomula and compound, the plate was subjected to high content imaging to quantify the effects of each compound. As expected, no carboxylic acid-containing I-BET726 analogues showed substantial activity on schistosomula phenotype and motility metrics, which we attributed to low permeability resulting from higher polarity (determined by chromLogD, see above). However, the majority of their ethyl ester derivatives were classified as hits. The quinoline derivative 22, and the benzothiazole derivative 21, showed the greatest effects on schistosomula phenotype and motility, while the benzofuran derivative 18 also had substantial effects on motility (Figure 7 and Figure S48). The carboxylic acid counterparts of these compounds all have affinity for SmBRD3(2). However, compound 10, which is the enantiomer of 9 showed no binding to SmBRD3(1) or SmBRD3(2), and the corresponding ethyl ester, 17, did not have substantial effects on either phenotype or motility. This result suggests a link between the phenotypic effects and inhibition of SmBRD3(2) function. In contrast, of the SmBRD3(1) binding compounds evaluated (orange, Figure 7A), only (+)-JQ1 (2) showed activity ex vivo, which is in line with previous reports.20 The phenotype and movement EC50 values of the more active compounds, 18 and 20-22 are in the low micromolar range (Table 3), but there is only moderate correlation with the SmBRD3(2) Kd values of the corresponding carboxylic acids. This reflects the range of characteristics that affect the activity of these compounds on the live schistosomula, including permeability and metabolic stability.
Figure 7.

Effect of compounds 1–5 and 8–22 on the (A) phenotype and (B) motility of schistosomula (at 20 μM in 0.625% DMSO, following 72 h incubation). Negative (0.625% DMSO) and positive (10 μM auranofin in 0.625% DMSO) controls are included in each drug screen (4–5 in total, two technical replicates each). The compound score is shown as gray dots, and whiskers represent the average score and standard deviation across the screens. Hit threshold is delineated by the vertical dashed red lines in the graphs; −0.15 and −0.35 for phenotype and motility scores, respectively. Orange: SmBRD3(1) selective compounds and Blue: SmBRD3(2) selective compounds. (C) Structure of compounds 1, 2, and 9–22.
Table 3. SmBRD3(2) Kd Values (ITC) for the Carboxylic Acids 11 and 13–15 and Schistosomula Phenotype and Movement EC50 Values for the Corresponding Ethyl Esters 18 and 20–22d.

Mean value of 3 repeats ± s.e.m.
95% Confidence Interval (CI).
Mean value of 2 repeats ± s.e.m.
A color scale is used with darker purple representing lower Kd values or lower EC50 values.
Assessing the Effects of SmBRD3(2) Ligands on Adult S. mansoni Worms
The ex vivo screens of these small molecules on adult schistosome pairs broadly mirrors the results of the phenotypic and motility assays performed on schistosomula, with the ethyl esters of the I-BET726 derivatives again showing the greatest effects on the adult worms. The benzothiophene (16), benzofuran (18), indole (20), benzothiazole (21), and quinoline (22) derivatives all showed substantial effects on worm movement (Figure 8A); however, none of the compounds were lethal to the parasite. Compounds that affect worm motility are also associated with reduced oviposition (Figure 8B) and decreased worm pairing (Figure 8C). It is notable that compound 10 does not bind to SmBRD3(2), but its ethyl ester (17) showed some modest effects on worm movement and egg count (Figure 8A,B), but not on worm pairing (Figure 8C).
Figure 8.

Effect of compounds 1–5 and 8–22 on adult S. mansoni motility, pairing, and egg production. (A) Worm movement recordings following 72 h compound-treatment (20 μM in 0.2% DMSO). Negative (0.2% DMSO) and positive (20 μM Praziquantel – PZQ in 0.2% DMSO) controls are included in each screen (n = 2 independent compound screens, 2 technical replicate each). The gray bars represent the mean worm movement (+ standard deviation). (B) At 72 h, in vitro laid eggs (IVLEs) were collected and enumerated. For each compound tested, individual egg counts are represented in a scatter plot; the purple line represent the mean trend across the treatments. (C) Effect of compound treatment on worm pairing, with 1 and 0 corresponding to 100% and 0% of parasite pairs following 72 h drug-treatment, respectively, (mean of n = 2 compound screens).
Assessing the Effects of SmBRD3(2) Ligands on the Transformation of Miracidia to Sporocysts
As smbrd3 is most abundantly expressed in miracidia and sporocysts (Figure 1C), we were intrigued to investigate whether our SmBRD3 BRD ligands affect ex vivo miracidia-to-sporocyst transformation. This process occurs naturally in the intermediate host snail and is critical for lifecycle progression. Two SmBRD3(2) ligand pro-drugs compounds 22 and 16 showed concentration-dependent inhibition of the miracidia to sporocyst transformation (Figure 9 and Figure S49). Interestingly, compound 22 is the ethyl ester of compound 15, which has the highest affinity for SmBRD3(2) (Table 2). Most other SmBRD3(2) ligands had no effect on the transformation. The negative control, 17, and (+)-JQ1 were toxic at the higher concentrations (Figure S49). While OXFBD02 and OXFBD04 showed modest effects on the miracidia to sporocyst transformation at a concentration of 25 μM, the other SmBRD3(1)-selective ligands generally had no effect in this assay (Figure S49).
Figure 9.
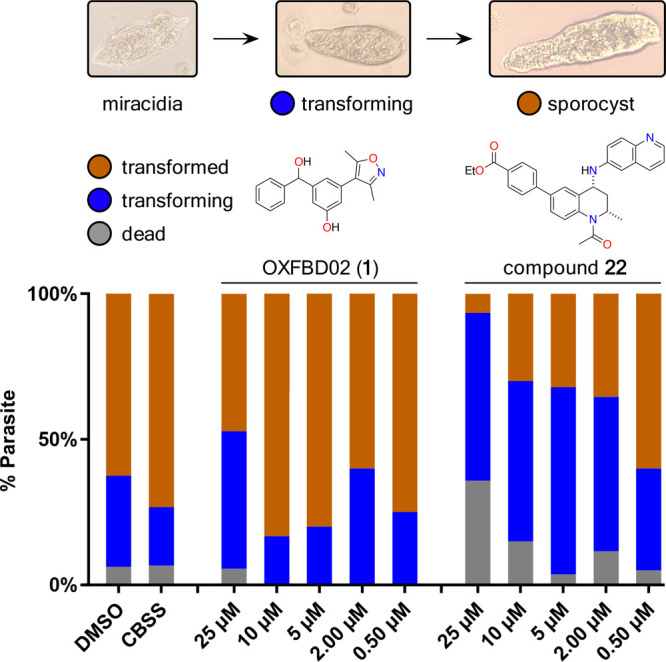
Differential activity of compound OXFBD02 (1) and compound 22 on ex vivo miracidia to sporocyst transformation. Miracidia were exposed to the selected compounds in a dose–response titration (CBSS containing 25, 10, 5, 2, or 0.5 μM in 1% DMSO). Dead parasites (in gray), transforming miracidia (in blue), and fully transformed sporocysts (in brown) were enumerated after 48 h (scored as percentage of parasite population, % Parasite). Each titration point was assessed in three independent experiments (two technical replicates per data point) and compared to parasites cultured in CBSS with 1% DMSO (controls) at a constant temperature of 26 °C, in the dark.
Determining the Permeability of Acids and Ethyl Esters in Adult Worms
As the low solubility of the ethyl ester derivatives prevented us from determining their affinity for SmBRD3(2) using ITC, it is possible that these compounds are themselves inhibitors of SmBRD3(2). Alternatively, the ethyl esters could function as pro-drugs that release the corresponding acid in the parasite. To investigate compound permeability, adult worms were treated with 20 μM of the acid 15 or the ethyl ester 22 for 24 h, and then the media exchanged three times. The worms were then lysed and the levels of the acid 15 or the ethyl ester 22 were determined using LCMS (Figure S50). High quantities of the ethyl ester 22 were found in the worms treated with this compound, while lower quantities of the acid 15 were found in the worms treated with 15 (Figure S51). Interestingly, a similar amount of the acid 15 was also observed in the worms treated with the ester 22, indicating that it can be converted from the ester to the acid in the worm. However, some ester hydrolysis was also seen in media alone (Figure S52). These data confirm the high worm permeability of the ethyl ester 22 and demonstrate that it is possible for this compound to be converted to the corresponding acid 15. Using this approach, it was also shown that OXFBD03 (2) is rapidly deacetylated to give OXFBD02 (1) in media in the presence or absence of worms. As both 1 and 2 have similar affinities for SmBRD3(1,2), this does not affect the ability of the compound to interact with this protein once in the parasite.
Discussion
The complete reliance on praziquantel to treat schistosomiasis represents a global vulnerability in the sustainable control of this neglected disease, especially in the case that praziquantel-resistant schistosomes develop. In the absence of a vaccine, and with very little new chemical matter being progressed into late-stage preclinical investigations, this situation presents a substantial challenge for the teams of medicinal chemists and parasitologists seeking to identify praziquantel replacements as part of collaborative drug discovery initiatives. Historically, the main approach to identifying antischistosomal compounds has relied on phenotypic screening, where the cellular target of the compound is (at least initially) unknown. More recently, efforts have been made to identify S. mansoni targets that can be modulated for therapeutic benefit or to repurpose molecules that target a specific class of human proteins.43 As a digenetic parasite, S. mansoni has adopted a lifecycle comprised of distinct morphological forms to maximize survival in a range of harsh environments, including the intermediate host snail, the definitive mammalian host, and fresh-water bodies. The phenotypic changes that accompany lifecycle progression occur without alteration of its genetic code, suggesting that the parasite’s epigenetic machinery plays a key role in these processes. We reasoned, therefore, that BRD-containing epigenetic regulators could be therapeutically interesting targets for the development of antischistosomal compounds. Having identified 29 BRDs in S. mansoni, we have begun a systematic investigation into their function. Our approach of focusing on SmBRD3, which is the S. mansoni BRD that is most similar to the human BET BCPs, was validated by the identification of high affinity ligands for both SmBRD3(1) and SmBRD3(2).
Following identification of ligands for both SmBRD3(1) and SmBRD3(2), a structure-based optimization allowed us to develop the quinoline derivative 15, which has a Kd value of 364 ± 26.3 nM for SmBRD3(2). The ethyl ester derivatives of this compound, 22, had strong effects on schistosomula phenotype and movement. It also reduced adult worm movement, pairing, and egg production (the lifecycle stage responsible for schistosomiasis pathology and transmission). Compound 22 additionally showed a substantial and concentration-dependent effect on the ability of miracidia to transform into sporocysts; this was likely due to high expression of smbrd3 in these lifecycle states (Figure 1C). At concentrations as low as 2.00 μM, the majority of the miracidia were prevented from transforming into sporocysts. It is notable that most SmBRD3(1) ligands did not show effects against schistosomula, miracidia, or adult worms. Given that both compound 22 and OXFBD03 (2) can be delivered to worms, these results suggest that SmBRD3(1) and SmBRD3(2) have different functions, and consequently their selective inhibition results in different phenotypes.
Conclusions
In conclusion, our data strongly indicate that inhibition of the SmBRD3 BRDs has significant effects on three S. mansoni life stages, and studies are ongoing to link the detrimental effects on S. mansoni lifecycle progression and survival to SmBRD3 inhibition. The high affinity small molecule SmBRD3 ligands we have identified provide a firm foundation for the development of further molecular tools that will enable us to investigate the link between SmBRD3 BRD inhibition and the phenotypes observed, providing unprecedented insight into the role of BRDs and epigenetics in the S. mansoni lifecycle. The identification of a new S. mansoni target that is ligandable and important for parasite survival is a major advancement in the search for novel antischistosomal targets and drugs.
Chemistry Experimental Section
Reagents and solvents used were of commercially available reagent grade quality from Sigma-Aldrich, Fluorochem, Alfa Aesar, Merck, Acros Organics, Apollo Scientific, Fisher Scientific, or Fluka and used without purification unless otherwise stated. All nonaqueous reactions requiring anhydrous conditions were carried out in a flame-dried flask under an inert atmosphere of argon or nitrogen unless otherwise stated. Anhydrous solvents were obtained from an MBRAUN MB5 Solvent Purification System and stored over 3 Å molecular sieves under an inert argon atmosphere. Concentration in vacuo refers to removal of solvent on a Buchi rotary evaporator under reduced pressure in a water bath at 40 °C. Celite refers to Celite 545 filter aid, treated with sodium carbonate, flux-calcined (Sigma-Aldrich). Brine refers to a saturated aqueous solution of sodium chloride. Petroleum ether refers to the fraction in the boiling point range 40–60 °C.
1H NMR spectra were measured on a Bruker AVIII HD 400 (400 MHz), AVII 500 (500 MHz), AVIIIHD 600 (600 MHz), or NEO 600 (600 mHz) spectrometer in the stated solvents as a reference for the internal deuterium lock. The chemical shift data for each signal are given as δ in units of parts per million (ppm) relative to tetramethylsilane (TMS) where δ(TMS) = 0.00. The spectra are calibrated using the solvent peak with the data provided by Fulmer et al.44 The multiplicity of each signal is indicated by s (singlet); d (doublet); t (triplet); q (quartet); m (multiplet); sp (septet); or combinations thereof. The number of protons (n) for a given resonance signal is indicated by nH. Where appropriate, coupling constants (J) are quoted in Hz, recorded to the nearest 0.1 Hz. Spectra were assigned using COSY, HSQC, and HMBC experiments as necessary. Relative stereochemistry was assigned via NOESY experiments.
13C NMR spectra were measured on a Bruker AVII 500 (126 MHz), AVIIIHD 600 (151 MHz), or NEO 600 (151 MHz) spectrometer in the stated solvents as a reference for the internal deuterium lock. The chemical shift data for each signal are given as δ in units of parts per million (ppm) relative to tetramethylsilane (TMS) where δ(TMS) = 0.00. The spectra are calibrated using the solvent peak with the data provided by Fulmer et al.44 The chemical shift is quoted to 1 decimal places, unless two different shifts are indistinguishable, when the shifts are quoted to 2 decimal places. Spectra were assigned using HSQC and HMBC as necessary.
11B NMR spectra were measured on a Bruker AVIII HD 400 (128 MHz) spectrometer in the stated solvents as a reference for the internal deuterium lock. The chemical shift data for each signal are given as δ in units of parts per million (ppm). The spectra are uncalibrated. Coupling constants (J) are quoted in Hz, recorded to the nearest 0.1 Hz.
Mass spectra were acquired on either an Agilent 6120 (low resolution), Waters LCT Premier XE benchtop, Waters LCT Premier benchtop orthogonal acceleration time-of-flight LC-MS system (low resolution), or Bruker microToF spectrometer (high resolution) using electrospray ionization (ESI) from solutions of methanol or acetonitrile. m/z values are reported in Daltons and followed by their percentage abundance in parentheses.
Melting points were determined using a Griffin capillary tube melting point apparatus or Leica Galen III hot stage microscope and are uncorrected. The solvent(s) from which the sample was crystallized is given in parentheses.
Specific optical rotations were measured using a Schmidt Haensch Unipol polarimeter, using a sodium lamp at 589 nm and a path length of 1.0 dm. The concentration (c) is expressed in g/100 mL (equivalent to g/0.1 dm3). Specific rotations are denoted [α]TD and are given in implied units of 10–1 deg cm2g–1 (where T = ambient temperature in °C).
Analytical High-Performance Liquid Chromatography (HPLC)
Method 1 was carried out on a PerkinElmer Flexar system with a Binary LC Pump and UV/vis LC Detector, with detection at 254 nm, using a Dionex Acclaim reverse phase 120 column (C18, 5 μm, 120 Å, 4.6 × 150 mm) which was employed using a flow rate of 1.5 mL min–1; [95:5 H2O: MeCN → 5:95 H2O: MeCN: H2O with 0.1% TFA modifier, 10 min; 5 min hold; 1.5 mL min–1]. Method 2 was carried out on an Agilent 1260 Infinity II system, with detection at 254 nm, using a Poroshell 120 EC-C18 column [4 μM, 4.6 × 100 mm]; [95:5 H2O: MeCN → 5:95 H2O: MeCN with 0.1% FA modifier, 10 min; 5 min hold; 1 mL min–1]. The purity of all biologically tested compounds was ≥95% as determined using the HPLC methods above.
Semipreparative high-performance liquid chromatography (HPLC) was carried out an a Agilent 1260 Infinity II with an Agilent 5 Prep C18 column [5 μM, 21.2 × 50 mm] [95:5 H2O:MeCN % FA modifier (1 min), 95:5 H2O:MeCN → 5:95 H2O:MeCN with 0.1% FA modifier (10 min), 5 min hold; 20 mL min–1].
Chiral high-performance liquid chromatography (HPLC) was carried out on a PerkinElmer Flexar system with a Binary LC Pump and UV/vis LC Detector or on a Thermofisher/Dionex Ultimate 3000 system comprising of a LPG-3400SD pump, WPS-3000SL autosampler, TCC-3000SD column compartment fitted with the appropriate Daicel Chiralpak column (dimensions: 0.46 cm o̷ × 25 cm) and corresponding guard column (0.4 cm o̷ × 1 cm), and a DAD3000 diode array detector, both at 1 mL min–1. The solvent system and column used for the compound are stated, where appropriate, and UV absorbance for both methods was measured at 254 nm.
A CAD solubility assay was carried out by Physchem Team, Discovery Analytical, NCE Molecular Discovery at GSK. 5 μL of 10 mM DMSO stock solution was diluted to 100 μL with pH 7.4 phosphate buffered saline, equilibrated for 1 h at room temperature, and filtered through Millipore MultiscreenHTS-PCF filter plates (MSSL BPC). The filtrate was quantified using a suitably calibrated charged aerosol detector.45
Lipophilicity: a chromLogD assay was carried out by the Physchem Team, Discovery Analytical, NCE Molecular Discovery at GSK. The chromatographic hydrophobicity index (CHI)46,47 values were measured using reversed phase HPLC column (50 × 2 mm 3 μM Gemini NX C18, Phenomenex, UK) with fast acetonitrile gradient at starting mobile phase of pHs 2, 7.4, or 10.5. CHI values were derived directly from the gradient retention times using calibration parameters for standard compounds. The CHI value approximates to the volume % organic concentration when the compound elutes. CHI was linearly transformed into a chromLogD48 value by least-squares fitting of experimental CHI values to calculated cLogP values for over 20000 research compounds.
Isopropyl Carbamate (S1)
Using a modified version of the procedure reported by Laurin et al.,37 trifluoroacetic acid (TFA) (17.0 mL, 222 mmol, 1.7 equiv) was added to a solution of isopropyl alcohol (10.0 mL, 131 mmol, 1.0 equiv) and NaOCN (12.6 g, 195 mmol, 1.5 equiv) in toluene (40 mL) cooled to 0 °C. The resulting suspension was warmed to rt and stirred for 4 h. After this time, H2O (100 mL) was added, and the mixture was extracted with EtOAc (2 × 150 mL). The combined organic components were washed with brine (150 mL), dried over MgSO4, filtered, and concentrated in vacuo to yield colorless crystals of isopropyl carbamate (S1) (10.7 g, 79%), which was used without further purification: Rf 0.31 (20% EtOAc/petroleum ether); mp 82–84 °C (from EtOAc) [lit.49 88–90 °C, lit.50 89–93 °C, lit.37 90–92 °C]; 1H NMR (400 MHz, CDCl3) δH 4.89 (1H, sp, J 6.3), 4.58 (2H, br s), 1.24 (6H, d, J 6.3); LRMS m/z (ESI+) 126 ([M + Na]+, 20%). These data are in good agreement with the literature values.37,49,50
Isopropyl (E)-But-2-enoylcarbamate (S2)
Using a procedure reported by Shadrick et al.,42 crotonoyl chloride (4.8 mL, 50 mmol, 1.1 equiv) was added to a solution of compound S1 (4.6 g, 45 mmol, 1.0 equiv) in anhydrous THF (43 mL) at −78 °C followed by LiHMDS (1 M in THF, 90 mL, 90 mmol, 2.0 equiv). The reaction mixture was warmed to rt and was stirred for 16 h. After this time, the reaction was quenched by the addition of chilled saturated NH4Cl(aq) (50 mL). The resulting solution was extracted with EtOAc (3 × 50 mL). The combined organic components were washed with brine (50 mL), dried over MgSO4, filtered, and concentrated in vacuo. The crude material was purified using flash column chromatography (0–20% EtOAc/petroleum ether) to give compound S2 as a colorless solid (4.7 g, 61%): Rf 0.39 (17% EtOAc/petroleum ether); mp 75–78 °C (from EtOAc) [lit.42 91 °C, lit.37 72–74 °C]; v̅max (neat)/cm–1 3285 (N–H, w), 2980 (C–H, s), 1765 (C=O, s), 1682 (C=O, w), 1647 (C=C, s); 1H NMR (400 MHz, CDCl3) δH 7.23 (1H, s), 7.13 (1H, dq, J 15.3, 6.9); 6.87 (1H, dq, J 15.3, 1.7), 4.99 (1H, sp, J 6.3), 1.94 (3H, dd, J 6.9, 1.7), 1.29 (6H, d, J 6.3); 13C NMR (151 MHz, CDCl3) δC 166.1, 151.5, 146.4, 123.1, 70.4, 21.9, 18.5; LRMS m/z (ESI+) 194 ([M + Na]+, 100%); HRMS m/z (ESI+) [found: 194.0786, C8H13O3NNa, requires [M + Na]+ 194.0788]. These data are in good agreement with the literature values.37,38,42
Isopropyl (S)-(3-(4′-Bromoanilinyl)butanoyl)carbamate (S3a)
(R)-BINAP(OTf)2(H2O)2Pd (728 mg, 0.685 mmol, 0.06 equiv) was added to a solution of compound S2 (2.00 g, 11.7 mmol, 1.0 equiv) in anhydrous degassed toluene (36 mL), and the resulting suspension was stirred at rt for 20 min. 4-Bromoaniline (3.54 g, 20.6 mmol, 1.8 equiv) was then added and the reaction mixture was stirred for a further 21 h. After this time, the reaction mixture was concentrated in vacuo and purified using flash column chromatography (0–20% EtOAc/petroleum ether) to give S3a as a colorless solid (3.82 g, 95%): Rf 0.16 (20% EtOAc/petroleum ether); [α]25D = −6.7 (c 1.0, MeOH) [lit.37 −16.1 (c 1.0, CHCl3)]; mp 146–149 °C (from EtOAc) [lit.42 131 °C, lit.37 128–130 °C]; v̅max (neat)/cm–1 3266 (N–H, w), 1748 (C=O, s), 1178 (C–O, m); 1H NMR (400 MHz, CDCl3) δH 7.36 (1H, br s), 7.26–7.21 (2H, m), 6.52–6.48 (2H, m), 5.02–4.92 (1H, m), 4.04–3.94 (1H, m), 3.94–3.84 (1H, m), 3.09 (1H, dd, J 16.0, 5.9), 2.89 (1H, dd, J 16.0, 5.9), 1.30–1.26 (9H, m); 13C NMR (151 MHz, CDCl3) δC 172.8, 151.4, 145.9, 132.2, 115.5, 109.5, 70.7, 46.2, 42.0, 21.9, 20.8; LRMS m/z (ESI–) 341 ([M79Br–H+]−, 100%), 343 ([M81Br–H+]−, 81%); HRMS m/z (ESI+) [Found: 343.0647, C14H20O3N279Br, requires [M + H]+ 343.0652]; Chiral HPLC, Chiral AD-H column (80:20 heptane/ethanol, 0.1% DEA, 1.0 mL min–1), retention time = 11.0 min (S3a, 96.7% UV), retention time = 16.3 min [opposite enantiomer (S3b), 3.3% UV]; 93% e.e. These data are in good agreement with the literature values.37,38,42
Isopropyl ((2S,4R)-6-Bromo-2-methyl-1,2,3,4-tetrahydroquinolin-4-yl)carbamate (S4a)
Sodium borohydride (229 mg, 6.05 mmol, 0.75 equiv) was added to a solution of compound S3a (2.77 g, 8.07 mmol, 1.0 equiv) in EtOH (143 mL), cooled to a temperature below −10 °C followed by addition of MgCl2·6H2O (1.81 g, 8.89 mmol, 1.1 equiv) in water (14.3 mL). The reaction mixture was stirred at a temperature below 0 °C for 2 h and then warmed to rt and stirred for 1 h. The resulting suspension was poured into a mixture of citric acid (60 mL, 0.5 M in water), HCl(aq) (205 mL, 1 M), and CH2Cl2 (200 mL) and left to stir for 1 h. After this time, the layers were separated, the aqueous layer was extracted with CH2Cl2 (3 × 150 mL), and the combined organic components were dried over MgSO4, filtered, and concentrated in vacuo to give compound S4a as a colorless solid (2.60 g, 98%): Rf 0.68 (30% EtOAc/toluene); [α]25D = +12.9 (c 1.0, MeOH) [lit.37 – 14.9 (c 1.0, CHCl3)], mp 130–136 °C (from toluene) [lit.42 167 °C, lit.37 148–150 °C]; v̅max (neat)/cm–1 3309 (N–H, m), 2980 (C–H, s), 1684 (C = O, s); 1H NMR (400 MHz, D6-DMSO) δH 7.38 (1H, d, J 9.1), 7.02 (1H, dd, J 8.6, 2.4), 6.95–6.92 (1H, m), 6.41 (1H, d, J 8.6), 5.89 (1H, br s), 4.87–4.79 (1H, m), 4.77–4.67 (1H, m), 3.48–3.39 (1H, m), 1.95–1.85 (1H, m), 1.49–1.40 (1H, m), 1.23 (3H, d, J 6.2), 1.20 (3H, d, J 6.2), 1.11 (3H, d, J 6.2); 13C NMR (151 MHz, D6-DMSO) δC 156.4, 144.2, 129.9, 128.6, 124.5, 115.6, 106.7, 66.1, 46.2, 40.1, 36.5, 22.0, 21.5; LRMS m/z (ESI+) 327 ([M+H+]+, 73%); HRMS m/z (ESI+) [Found: 327.0703, C14H20O2N279Br, requires [M + H]+ 327.0703]. Chiral AD-H column (90:10 heptane:ethanol, 0.1% DEA, 1.0 mL min–1), retention time = 8.1 min (S4a, 94.3% UV), retention time = 11.5 min (opposite enantiomer (S4b), 5.7% UV); 89% e.e. These data are in good agreement with the literature values.37,38,42
Isopropyl ((2S,4R)-1-Acetyl-6-bromo-2-methyl-1,2,3,4-tetrahydroquinolin-4-yl)carbamate (S5a)
Compound S4a (2.18 g, 6.36 mmol, 1.0 equiv) was dissolved in CH2Cl2 (60 mL) under N2 at room temperature. Pyridine (1.66 mL, 20.5 mmol, 3.2 equiv) was added followed by dropwise addition of acetyl chloride (0.75 mL, 10.5 mmol, 1.7 equiv). The reaction mixture was stirred for 40 min. The reaction mixture was partitioned between EtOAc (250 mL) and saturated NaHCO3(aq) (250 mL). The aqueous layer was extracted with EtOAc (3 × 200 mL) and the combined organic components washed with water and brine, dried over Na2SO4, filtered, and then concentrated in vacuo to give compound S5a as a brown solid, which was judged to be pure enough to use in the next step without further purification (2.10 g, 90%). Rf 0.27 (50% EtOAc/petroleum ether); [α]25D = +209.9 (c 1.0, MeOH) [lit.37 + 298.4 (c 1.0, CHCl3)]; mp 105–106 °C (from EtOAc) [lit.42 163 °C, lit.37 141–143 °C]; v̅max (neat)/cm–1 3334 (N–H, w), 1688 (C=O, s), 1660 (C=O, s); 1H NMR (400 MHz, D6-DMSO) δH 7.63 (1H, d, J 8.6), 7.47 (1H, dd, J 8.4, 2.3), 7.30 (1H, d, J 8.4), 7.21 (1H, d, J 2.3), 4.87–4.79 (1H, m), 4.67–4.56 (1H, m), 4.40–4.31 (1H, m), 2.44 (1H, ddd, J 12.7, 8.6, 4.3), 2.05 (3H, s), 1.25 (3H, d, J 6.3), 1.22 (3H, d, J 6.3), 1.20–1.14 (1H, m), 1.01 (3H, d, J 6.3); 13C NMR (151 MHz, D6-DMSO) δC 168.4, 155.7, 139.2, 135.5, 129.6, 128.2, 125.6, 117.9, 67.3, 47.0, 46.7, 39.8 (hidden by D6-DMSO multiplet, assigned using HSQC), 22.6, 22.0, 21.3; LRMS m/z (ESI)+ 391 ([M79Br+Na]+, 100%); HRMS m/z (ESI+) [Found: 369.0803, C16H22O3N279Br, requires [M + H]+ 369.0808]. These data are in good agreement with the literature values.37,38,42
Ethyl 4-((2S,4R)-1-Acetyl-4-((isopropoxycarbonyl)amino)-2-methyl-1,2,3,4-tetrahydroquinolin-6-yl)benzoate (S6a)
A solution of compound S5a (1.10 g, 2.98 mmol, 1.0 equiv), {4-[(ethyloxy)carbonyl]-phenyl}boronic acid (0.609 g, 3.14 mmol, 1.05 equiv), and Pd(Ph3)4 (57 mg, 0.0493 mmol, 0.017 equiv) in DME (11.7 mL) was treated with an aqueous solution of Na2CO3 (2 M, 5.87 mL, 11.7 mmol, 3.9 equiv). The mixture was degassed and heated at 105 °C under argon for 2.5 h. After this time the mixture was cooled to rt and partitioned between EtOAc and water. The layers were separated, and the aqueous layer extracted with EtOAc (3 × 100 mL). The combined organic components were washed with water and brine, dried over Na2SO4, filtered, and concentrated in vacuo to give a gray-brown solid. This solid was redissolved in EtOAc and filtered through a silica plug eluting with a 1:1 EtOAc/petroleum ether mixture. The eluents containing the product were concentrated in vacuo to give S6a as a colorless solid (1.19 g, 91%). Rf 0.31 (25% acetone/petroleum ether); [α]25D = +272.6 (c 1.0, MeOH); mp 73–75 °C (from acetone); v̅max (neat)/cm–1 3324 (N–H, w), 1718 (C=O, s), 1695 (C=O, s), 1659 (C=O, s), 1609 (N–H, m); 1H NMR (400 MHz, D6-DMSO) δH 8.08–8.04 (2H, m), 7.83–7.78 (2H, m), 7.70 (1H, d, J 8.7), 7.65 (1H, dd, J 8.3, 2.1), 7.48–7.44 (2H, m), 4.89–4.81 (1H, m), 4.70–4.61 (1H, m), 4.48–4.40 (1H, m), 4.34 (2H, q, J 7.1), 2.48–2.44 (1H, m) (partially hidden by DMSO peak), 2.10 (3H, s), 1.34 (3H, t, J 7.1), 1.28–1.21 (6H, m), 1.21–1.16 (1H, m), 1.06 (3H, d, J 6.3); 13C NMR (151 MHz, D6-DMSO) δC 168.5, 165.5, 155.9, 144.3, 137.1, 136.4, 135.7, 129.9, 128.7, 126.7, 126.6, 125.5, 121.3, 67.2, 60.7, 47.1, 46.9, 40.1, 22.7, 22.04, 22.01, 21.4, 14.2; LRMS m/z (ESI+) 461 ([M+Na23]+, 100%); HRMS m/z (ESI+) [found: 439.2220, C25H31O5N2, requires [M + H]+ 439.2227]. The LRMS and 1H NMR data are in good agreement with the literature.38
Ethyl 4-((2S,4R)-1-Acetyl-4-amino-2-methyl-1,2,3,4-tetrahydroquinolin-6-yl)benzoate (S7a)
Compound S6a (500 mg, 1.14 mmol, 1.0 equiv) was added to a suspension of AlCl3 (760 mg, 5.70 mmol, 5.0 equiv) in anhydrous CH2Cl2 (9 mL) and the resulting solution stirred for 20 min at 0 °C. A solution of anhydrous NEt3 (1.9 mL, 13.7 mmol, 12 equiv) and anhydrous MeOH (2 mL) was added dropwise. The mixture was stirred for 20 min at 0 °C, then diluted with EtOAc (20 mL) and saturated Rochelle salt solution (40 mL), and stirred for 30 min. After this time, the solution was filtered through Celite, eluting with EtOAc, saturated aqueous NaHCO3, acetone, and MeOH. The organic components were removed from the filtrate in vacuo. The resulting solution was then diluted with EtOAc (50 mL) and the organic and aqueous components were partitioned. The aqueous components were extracted with EtOAc (3 × 50 mL). The organic components were the dried with Na2SO4, and concentrated in vacuo. The crude material was purified using silica gel flash column chromatography (0–10% MeOH/CH2Cl2) to give S7a as a colorless solid (339 mg, 84%). Rf 0.69 (10% MeOH/CH2Cl2); [α]25D = +315.0 (c 1.0, CHCl3); mp 38–45 °C (from EtOAc); v̅max (thin film)/cm–1 2978 (C–H, w), 1711 (C = O, s), 1645 (N–H, s); 1H NMR (400 MHz, CDCl3) δH 8.14–8.09 (2H, m), 7.77–7.75 (1H, m), 7.72–7.67 (2H, m), 7.53 (1H, dd, J 8.2, 1.8), 7.20 (1H, d, J 8.2), 4.91–4.78 (1H, m), 4.41 (2H, q, J 7.1), 3.80 (1H, dd, J 12.1, 4.4), 2.56 (1H, ddd, J 12.7, 8.6, 4.4), 2.16 (3H, s), 1.65 (2H, br s), 1.42 (3H, t, J 7.1), 1.20–1.11 (1H, m), 1.16 (3H, d, J 6.3); 13C NMR (151 MHz, CDCl3) δC 169.6, 166.6, 145.0, 140.7, 137.9, 136.5, 130.3, 129.5, 127.0, 126.3, 125.7, 121.6, 61.2, 47.8, 47.7, 44.7, 23.1, 21.6, 14.5; LRMS m/z (ESI+) 337 ([M–NH2+H]+, 25%); HRMS m/z (ESI+) [found: 353.1861, C21H25O3N2, requires [M + H]+ 353.1860]. The LRMS data are in good agreement with the literature.38
Isopropyl (R)-(3-(4′-Bromoanilinyl)butanoyl)carbamate (S3b)
The preparation of S3b was based upon a previously reported procedure.37,38,42 Compound S2 (161 mg, 0.940 mmol, 1.0 equiv) was dissolved in anhydrous, degassed toluene (3 mL). (S)-BINAP(OTf)2(H2O)2Pd (50 mg, 0.0470 mmol, 0.050 equiv) was added, and the resulting mixture was stirred for 20 min. 4-Bromoaniline (240 mg, 1.40 mmol, 1.5 equiv) was added then and the resulting solution was stirred for 6 h 25 min. After this time the solution was concentrated in vacuo and purified using silica gel flash column chromatography (0–50% EtOAc/petroleum ether) to give S3b as a colorless solid (291 mg, 90%): Rf 0.13 (20% EtOAc/petroleum ether); [α]25D = +7.45 (c 1.0, MeOH); mp 72–88 °C (from EtOAc) [lit.42 131 °C, lit.37 128–130 °C, (opposite enantiomer)]; 1H NMR (400 MHz, D6-DMSO) δH 10.47 (1H, s), 7.21–7.16 (2H, m), 6.54–6.48 (2H, m), 5.66 (1H, d, J 8.7), 4.84 (1H, sp, J 6.3), 3.85–3.71 (1H, m), 2.68 (1H, dd, J 15.7, 5.6), 2.45 (1H, dd, J 15.7, 7.5), 1.22 (6H, d, J 6.3), 1.12 (3H, d, J 6.4); LRMS m/z (ESI+) 365 ([M + Na]+, 64%); Chiral HPLC, Chiral AD-H column (80:20 heptane:ethanol, 0.1% DEA, 1.0 mL min–1), retention time = 11.0 min (opposite enantiomer (S3a), 7.7% UV), retention time = 16.3 min (S3b, 92.3% UV); 85% e.e. These data are in good agreement with the spectroscopic data for S3a.37,38,42
Isopropyl ((2R,4S)-6-Bromo-2-methyl-1,2,3,4-tetrahydroquinolin-4-yl)carbamate (S4b)
The preparation of S4b was based upon a previously reported procedure.37,38,42 Compound S3b (252 mg, 0.734 mmol, 1.00 equiv) was dissolved in EtOH (13 mL), and the resulting solution was cooled to below −10 °C. Sodium borohydride (21 mg, 0.555 mmol, 0.76 equiv) was added to this solution followed by a solution of MgCl2·6H2O (164 mg, 0.807 mmol, 1.1 equiv) in water (1.3 mL). The resulting suspension was poured into a mixture of citric acid (5.3 mL, 0.5 M), HCl(aq) (1 M, 18 mL), and CH2Cl2 (18 mL). The layers were separated and the aqueous components were extracted with CH2Cl2 (3 × 30 mL), dried over Na2SO4, filtered, and concentrated in vacuo to give S4b as a colorless solid (224 mg, 93%): Rf 0.68 (30% EtOAc/petroleum ether); mp 140–143 °C (from CH2Cl2) [lit.42 167 °C lit.37 148–150 °C, (opposite enantiomer)]; [α]25D = −7.8 (c 1.0, MeOH); 1H NMR (400 MHz, D6-DMSO) δH 7.38 (1H, d, J 9.1), 7.02 (1H, dd, J 8.6, 2.4), 6.96–6.93 (1H, m), 6.42 (1H, d, J 8.6), 4.87–4.79 (1H, m), 4.77–4.66 (1H, m), 3.44–3.39 (1H, m), 1.91 (1H, ddd, J 12.4, 5.8, 2.6), 1.49–1.37 (1H, m), 1.23 (3H, d, J 6.2), 1.20 (3H, d, J 6.2), 1.12 (3H, d, J 6.3); LRMS m/z (ESI+) 349 ([M + Na]+, 8%); Chiral HPLC, Chiral AD-H column (85:15 heptane/ethanol, 0.1% DEA, 1.0 mL min–1), retention time = 6.0 min (opposite enantiomer (S4a), 9.9% UV), retention time = 7.9 min (S4b, 90.1% UV); 80% e.e. These data are in good agreement with the nonstereospecific spectroscopic data for S4a.37,38,42
Isopropyl ((2R,4S)-1-Acetyl-6-bromo-2-methyl-1,2,3,4-tetrahydroquinolin-4-yl)carbamate (S5b)
The preparation of compound S5b was based upon a previously reported procedure.38,42 Pyridine (0.114 mL, 1.41 mmol, 3.0 equiv) was added to a solution of compound S4b (154 mg, 0.471 mmol, 1.0 equiv) dissolved in anhydrous CH2Cl2 (4.5 mL) under argon at rt. Acetyl chloride (537 μL, 0.753 mmol, 1.6 equiv) was added dropwise and the reaction mixture was left to stir for 1 h 10 min. After this time the mixture was partitioned with EtOAc (30 mL) and NaHCO3(aq) (30 mL). The aqueous layer was extracted with EtOAc (3 × 30 mL) and the combined organic components washed with H2O and brine, dried over Na2SO4, filtered, and concentrated in vacuo. The crude residue was purified using silca gel flash column chromatography (50–100% EtOAc/petroleum ether) to give S5b as a brown solid (152 mg, 88%): Rf 0.27 (50% EtOAc/petroleum ether); mp 127–129 °C (from EtOAc) [lit.42 163 °C, lit.37 141–143 °C, (opposite enantiomer)]; [α]25D = −214.1 (c 1.0, MeOH); 1H NMR (400 MHz, D6-DMSO) δH 7.63 (1H, d, J 8.6), 7.47 (1H, dd, J 8.4, 2.2), 7.30 (1H, d, J 8.4), 7.21 (1H, d, J 2.2), 4.91–4.75 (1H, m), 4.67–4.55 (1H, m), 4.40–4.30 (1H, m), 2.48–2.40 (1H, m), 2.05 (3H, s), 1.25 (3H, d, J 6.3), 1.22 (3H, d, J 6.3), 1.19–1.13 (1H, m), 1.01 (3H, d, J 6.3); LRMS m/z (ESI+) 391 ([M + Na]+, 100%). These data are in good agreement with the nonstereospecific spectroscopic data of S5a.37,38,42
Ethyl 4-((2R,4S)-1-Acetyl-4-((isopropoxycarbonyl)amino)-2-methyl-1,2,3,4-tetrahydroquinolin-6-yl)benzoate (S6b)
The preparation of compound S6b was based upon a previously reported procedure.38 Degassed Na2CO3(aq) (2 M, 4.7 mL, 9.40 mmol, 3.93 equiv) was added to a solution of compound S5b (879 mg, 2.39 mmol, 1.00 equiv) {4-[(ethyloxy)carbonyl]phenyl}boronic acid (485 mg, 2.50 mmol, 1.05 equiv), and Pd(PPh3)4 (41 mg, 0.0355 mmol, 0.015 equiv) in degassed DME (9.6 mL). The resulting solution was stirred at 105 °C for 17 h 15 min. After this time, the reaction mixture was cooled to rt and partitioned between EtOAc (20 mL) and water (20 mL). The layers were separated, and the aqueous layer extracted with EtOAc (3 × 50 mL). The combined organic components were washed with water (50 mL) and brine (50 mL), dried over Na2SO4, filtered, and concentrated in vacuo. The resulting gray-brown crude solid was redissolved in EtOAc and filtered through a plug of silica. The silica was washed with EtOAc and the filtrate concentrated in vacuo to give S6b as a brown solid (843 mg, 80%): Rf 0.31 (acetone:petroleum ether 1:3); mp 146–148 °C (from EtOAc) [opposite enantiomer: 73–75 °C (from acetone)]; [α]25D = −260.9 (c 1.0, MeOH); 1H NMR (400 MHz, D6-DMSO) δH 8.08–8.04 (2H, m), 7.83–7.77 (2H, m), 7.69 (1H, d, J 8.8), 7.65 (1H, dd, J 8.2, 2.2), 7.48–7.43 (2H, m), 4.90–4.80 (1H, m), 4.72–4.60 (1H, m), 4.49–4.39 (1H, m), 4.34 (2H, q, J 7.1), 2.48–2.44 (partially hidden by D6-DMSO peak) (1H, m), 2.10 (3H, s), 1.34 (3H, t, J 7.1), 1.27–1.22 (6H, m), 1.24–1.15 (1H, m), 1.06 (3H, d, J 6.4); LRMS m/z (ESI+) 461 ([M + Na]+, 100%). The LRMS and 1H NMR data are in good agreement with the literature values for S6a.38
Ethyl 4-((2R,4S)-1-Acetyl-4-amino-2-methyl-1,2,3,4-tetrahydroquinolin-6-yl)benzoate (S7b)
The preparation of compound S7b was based upon a previously reported procedure.38 AlCl3 (76 mg, 0.570 mmol, 5.0 equiv) was added to a solution of compound S6b (50 mg, 0.114 mmol, 1.0 equiv) dissolved in anhydrous CH2Cl2 (0.9 mL) at 0 °C, and the resulting suspension was stirred for 20 min. After this time, a solution of anhydrous NEt3 (0.2 mL, 1.4 mmol, 12 equiv) in dry MeOH (0.2 mL) was added dropwise and the resulting suspension was stirred for a further 1 h 30 min. The reaction mixture was then diluted with EtOAc (5 mL) and saturated solution of Rochelle’s salt (10 mL), and the solution left to stir for 30 min. The suspension was then filtered through Celite, eluting with EtOAc, saturated NaHCO3(aq), acetone, and MeOH. The organic components were removed in vacuo and the aqueous components partitioned between saturated NaHCO3(aq) (10 mL) and EtOAc (10 mL). The aqueous layer was washed with EtOAc (3 × 10 mL) and the combined organic components were dried over Na2SO4, filtered, and concentrated in vacuo. The crude residue was purified using silica gel flash column chromatography (0–10% MeOH/CH2Cl2) to give S7b as a colorless solid (33 mg, 82%). Rf 0.69 (10% MeOH/CH2Cl2); [α]25D = −264.3 (c 1.0, CHCl3); mp 120–125 °C (from toluene) [opposite enantiomer: 38–45 °C (from EtOAc)]; 1H NMR (400 MHz, CDCl3) δH 8.14–8.09 (2H, m), 7.78–7.76 (1H, m), 7.71–7.67 (2H, m), 7.53 (1H, dd, J 8.2, 1.9), 7.20 (1H, d, J 8.2), 4.91–4.78 (1H, m), 4.41 (2H, q, J 7.1), 3.81 (1H, dd, J 12.0, 4.4), 2.57 (1H, ddd, J 12.8, 8.7, 4.4), 2.16 (3H, s), 1.65 (2H, br s), 1.42 (3H, t, J 7.1), 1.23–1.18 (1H, m), 1.17 (3H, d, J 6.4); LRMS m/z (ESI+) 375 ([M + Na]+, 25%). The 1H NMR and LRMS data are in good agreement with the literature value for S7a.
Ethyl 4-{(2S,4R)-1-Acetyl-4-[(1-benzothiophen-6-yl)amino]-2-methyl-1,2,3,4-tetrahydroquinolin-6-yl}benzoate (16)
Compound S7a (100 mg, 0.284 mmol, 1.0 equiv), 6-bromobenzothiophene (121 mg, 0.568 mmol, 2 equiv), BrettPhos (49 mg, 0.0913 mmol, 0.32 equiv), BrettPhos Pd G3 (21 mg, 0.0232 mmol, 0.08 equiv), and NaOtBu (33 mg, 0.343 mmol, 1.2 equiv) were added to a dry microwave vial and the vial was purged with argon. Degassed anhydrous toluene (1.7 mL) was added and the resulting suspension was stirred for 15 h at 70 °C. After this time the suspension was filtered through Celite, eluting with toluene and Et2O, and the filtrate was concentrated in vacuo. The resulting residue was redissolved in EtOAc (50 mL) and washed with H2O (40 mL) and brine (40 mL). The organic components were dried over Na2SO4, filtered, and concentrated in vacuo. The crude material was purified using silica gel flash column chromatography (0–60% EtOAc/petroleum ether) to give 16 as a yellow solid (120 mg, 87%, 94.8% purity by HPLC). To achieve a higher purity, 50 mg of 16 was further purified using semipreparative HPLC and lyophilized to give 16 as an off-white solid (>99.9% purity by HPLC):Rf 0.43 (10% EtOAc/CH2Cl2); [α]25D = +338.1 (c 1.0, CHCl3); mp 78–84 °C (from CHCl3); v̅max (thin film)/cm–1 3350 (N–H, w), 1711 (C=O, s), 1646 (C=O, s), 1606 (s), 1487 (s), 1277 (s); 1H NMR (400 MHz, CD3CN) δH 8.09–7.89 (2H, m), 7.66–7.57 (4H, m), 7.56–7.54 (1H, m), 7.37 (1H, d, J 8.2), 7.18 (1H, dd, J 5.4, 0.8), 7.16–7.12 (2H, m), 6.91 (1H, dd, J 8.7, 2.2), 4.97 (1H, d, J 8.0), 4.86–4.73 (1H, m), 4.40 (1H, ddd, J 12.2, 8.0, 4.2), 4.31 (2H, q, J 7.1), 2.71 (1H, ddd, J 12.6, 8.5, 4.2), 2.17 (3H, s), 1.33 (3H, t, J 7.1), 1.35–1.24 (1H, m), 1.14 (3H, d, J 6.3); 13C NMR (151 MHz, CD3CN) δC 170.0, 166.9, 146.8, 145.7, 142.8, 139.5, 138.2, 137.6, 132.5, 130.8, 130.4, 127.9, 127.7, 126.5, 125.1, 124.6, 123.3, 122.5, 114.7, 104.6, 61.8, 50.7, 48.6, 41.6, 23.4, 21.7, 14.6; LRMS m/z (ESI+) 485 ([M + H]+, 2.6%); HRMS m/z (ESI+) [found: 485.18976, C29H29O3N232S, requires [M + H]+ 485.18934]; HPLC (Method 1), retention time = 12.4 min, > 99.9%; Chiral HPLC, Chiral AD-H column (40:60 hexane/IPA, 1.0 mL min–1), retention time = 7.7 min (16, 91.4% UV), retention time = 10.1 min (opposite enantiomer (17), 8.6% UV); 83% e.e.
4-{(2S,4R)-1-Acetyl-4-[(1-benzothiophen-6-yl)amino]-2-methyl-1,2,3,4-tetrahydroquinolin-6-yl}benzoic Acid (9)
Compound 16 (55 mg, 0.113 mmol, 1 equiv) was dissolved in EtOH (0.71 mL). NaOH(aq) (2 M, 565 μL, 1.13 mmol, 10 equiv) was added over a period of 1 h and the resulting solution was left to stir at rt for 3 h. The solution was then acidified to ∼pH 3 with 1 M HCl(aq) and diluted with H2O (5 mL) and EtOAc (10 mL). The aqueous components were then extracted with EtOAc (3 × 10 mL). The combined organic components were dried over Na2SO4, filtered, and concentrated in vacuo to give 9 as a yellow solid (45 mg, 87%, 94.7% purity by HPLC). To achieve higher purity, 9 was purified using semipreparative HPLC and lyophilized to give 9 as an off-white solid: Rf 0.29 (50% EtOAc/petroleum ether, 1% acetic acid); [α]25D = +308.8 (c 0.66, CHCl3); mp 115–120 °C (from CHCl3); v̅max (thin film)/cm–1 3362 (N–H, w), 3034 (O–H, w), 1704 (C=O, m), 1606 (s), 1488 (m); 1H NMR (400 MHz, CD3CN) δH 8.02–7.94 (2H, m), 7.66–7.58 (4H, m), 7.55 (1H, d, J 1.4), 7.38 (1H, d, J 8.2), 7.19 (1H, dd, J 5.5, 0.7), 7.16–7.11 (2H, m), 6.92 (1H, dd, J 8.6, 2.2), 4.86–4.73 (1H, m), 4.41 (1H, dd, J 12.1, 4.1), 2.71 (1H, ddd, J 12.5, 8.6, 4.1), 2.18 (3H, s), 1.36–1.24 (1H, m), 1.14 (3H, d, J 6.3); 13C NMR (151 MHz, CD3CN) δC 170.1, 167.6, 146.8, 145.8, 142.8, 139.6, 138.3, 137.6, 132.5, 131.2, 130.0, 127.9, 127.7, 126.6, 125.1, 124.6, 123.3, 122.5, 114.7, 104.6, 50.7, 48.7, 41.6, 23.4, 21.7; LRMS m/z (ESI–) 455 ([M–H]−, 100%); HRMS m/z (ESI+) [Found: 457.15808, C29H29O3N232S, requires [M + H]+ 457.15804]; HPLC (Method 2) retention time = 9.18 min, 99.0%; Chiral HPLC, Chiral AD-H column (50:50 hexane/IPA, 1.0 mL min–1), retention time = 7.3 min (9, 91.2% UV), retention time = 11.0 min (opposite enantiomer (10), 8.8% UV); 82% e.e.
Ethyl 4-{(2R,4S)-1-Acetyl-4-[(1-benzothiophen-6-yl)amino]-2-methyl-1,2,3,4-tetrahydroquinolin-6-yl}benzoate (17)
Compound S7b (140 mg, 0.398 mmol, 1.0 equiv), 6-bromobenzo[b]thiophene (169 mg, 0.793 mmol, 2.0 equiv), BrettPhos (68 mg, 0.127 mmol, 0.32 equiv), BrettPhos Pd G3 (29 mg, 0.032 mmol, 0.080 equiv), and NaOtBu (46 mg, 0.479 mmol, 1.2 equiv) were added to a dry microwave vial, which was subsequently purged with argon. Anhydrous, degassed toluene (2.4 mL) was added, and the resulting suspension was stirred for 15 h at 70 °C. After this time, the suspension was filtered through Celite, eluting with toluene and Et2O, and the filtrate was concentrated in vacuo. The resulting residue was redissolved in EtOAc (50 mL) and washed with H2O (40 mL) and brine (40 mL). The organic components were dried over Na2SO4, filtered, and concentrated in vacuo. The crude material was purified using silica gel flash column chromatography (0–60% EtOAc/petroleum ether) to give 17 as a yellow solid (132 mg, 68%, 96.2% purity by HPLC). To achieve higher purity 66 mg of compound 17 was further purified using semipreparative HPLC, and lyophilized to give 17 as an off-white solid (>99.9% purity by HPLC): Rf 0.43 (10% EtOAc/CH2Cl2); [α]25D = −285.9 (c 1.0, CHCl3); mp 102–105 °C (from CHCl3) [opposite enantiomer: 78–84 °C (from CHCl3)]; 1H NMR (400 MHz, CD3CN) δH 8.03–7.94 (2H, d, J 8.0), 7.65–7.57 (4H, m), 7.57–7.54 (1H, m), 7.38 (1H, d, J 8.2), 7.19 (1H, dd, J 5.4, 0.7), 7.16–7.13 (2H, m), 6.91 (1H, dd, J 8.6, 2.2), 4.97 (1H, d, J 8.0), 4.87–4.73 (1H, m), 4.41 (1H, ddd, J 12.1, 8.0, 4.1), 4.31 (2H, q, J 7.1), 2.71 (1H, ddd, J 12.5, 8.6, 4.1), 2.18 (3H, s), 1.34 (3H, t, J 7.1), 1.35–1.25 (1H, m), 1.14 (3H, d, J 6.3); 13C NMR (151 MHz, CD3CN) δC 170.0, 167.0, 146.8, 145.7, 142.8, 139.5, 138.3, 137.6, 132.5, 130.9, 130.4, 127.9, 127.7, 126.5, 125.1, 124.6, 123.3, 122.5, 114.7, 104.6, 61.8, 50.7, 48.7, 41.6, 23.4, 21.7, 14.6; LRMS: m/z (ESI+) 507 ([M + Na]+, 28%); HPLC (Method 1), retention time = 12.4 min, > 99.9%; Chiral HPLC, Chiral AD-H column (40:60 hexane/IPA, 1.0 mL min–1), retention time = 7.2 min (opposite enantiomer (16), 5.6% UV), retention time = 10.1 min (17, 94.4% UV); 89% e.e.
4-{(2R,4S)-1-Acetyl-4-[(1-benzothiophen-6-yl)amino]-2-methyl-1,2,3,4-tetrahydroquinolin-6-yl}benzoic Acid (10)
Compound 17 (67 mg, 0.138 mmol, 1 equiv) was dissolved in EtOH (0.86 mL), NaOH(aq) (2 M, 695 μL, 1.39 mmol, 10 equiv) was added over 1 h, and the resulting solution was left to stir at rt for 3 h. After this time, the solution was acidified to approximately pH 3 with 1 M HCl(aq) and diluted with H2O (5 mL) and EtOAc (10 mL). The aqueous components were then extracted with EtOAc (3 × 10 mL). The combined organic components were dried over Na2SO4, filtered, and concentrated in vacuo. The crude material was purified using semipreparative HPLC, and lyophilized to give 10 as an off-white solid (20 mg, 32%): Rf 0.63 (50% EtOAc/petroleum ether, 1% acetic acid); [α]25D = −247.7 (c 0.47, CHCl3); mp 118–123 °C (from CH2Cl2) [opposite enantiomer: 115–120 °C]; 1H NMR (400 MHz, CD3CN) δH 8.02–7.95 (2H, m), 7.66–7.58 (4H, m), 7.55 (1H, d, J 1.4), 7.38 (1H, d, J 8.2), 7.19 (1H, d, J 5.5, 0.7), 7.16–7.13 (2H, m), 6.92 (1H, dd, J 8.6, 2.2), 4.86–4.73 (1H, m), 4.41 (1H, dd, J 12.1, 4.1), 2.71 (1H, ddd, J 12.5, 8.6, 4.1), 2.18 (3H, s), 1.30 (1H, m), 1.15 (3H, d, J 6.3); 13C NMR (151 MHz, CD3CN) δC 170.1, 167.6, 146.8, 145.8, 142.8, 139.6, 138.2, 137.7, 132.5, 131.2, 130.0, 127.9, 127.7, 126.6, 125.1, 124.6, 123.3, 122.5, 114.7, 104.6, 50.7, 48.7, 41.6, 23.4, 21.7; LRMS: m/z (ESI+) 479 ([M + Na]+, 25%); HPLC (Method 1), retention time = 10.1 min, 99.8%; Chiral HPLC, Chiral AD-H column (hexane/IPA, 1.0 mL min–1), retention time = 6.7 min (opposite enantiomer (9), 5.5% UV), Retention time = 9.8 min (10, 94.5% UV); 89% e.e.
Ethyl 4-{(2S,4R)-1-Acetyl-4-[(1-benzofuran-6-yl)amino]-2-methyl-1,2,3,4-tetrahydroquinolin-6-yl}benzoate (18)
Compound S7a (133 mg, 0.377 mmol, 1.0 equiv), 6-bromobenzofuran (149 mg, 0.756 mmol, 2.0 equiv), BrettPhos (65 mg, 0.121 mmol, 0.32 equiv), BrettPhos Pd G3 (27 mg, 0.0298 mmol, 0.079 equiv), and NaOtBu (44 mg, 0.458 mmol, 1.2 equiv) were added to a dry microwave vial, which was subsequently purged with argon. Degassed, anhydrous toluene (2.2 mL) was added, and the resulting suspension was stirred for 15 h at 70 °C. After this time, the suspension was filtered through Celite, eluting with toluene and Et2O, and the filtrate concentrated in vacuo. The resulting residue was redissolved in EtOAc (50 mL) and washed with H2O (40 mL) and brine (40 mL). The organic components were dried over Na2SO4, filtered, and concentrated in vacuo. The crude material was purified using silica gel flash column chromatography (0–50% EtOAc/petroleum ether) to give 18 as a yellow solid (120 mg, 68%, 94.6% purity by HPLC). To achieve a higher purity, 60 mg of 18 was purified using semipreparative HPLC, and lyophilized to give 18 as a light-green solid: Rf 0.17 (30% EtOAc/petroleum ether); [α]25D = +258.7 (c 1.0, CHCl3); mp 84–87 °C (from CHCl3); v̅max (thin film)/cm–1 3358 (N–H, w), 1713 (C = O, s), 1629 (s), 1608 (s), 1488 (s); 1H NMR (400 MHz, CD3CN) δH 8.04–7.89 (2H, m), 7.65–7.58 (3H, m), 7.57–7.55 (1H, m), 7.48 (1H, d, J 2.2), 7.40–7.34 (2H, m), 6.82–6.80 (1H, m), 6.78 (1H, dd, J 8.3, 2.1), 6.68 (1H, dd, J 2.2, 1.0), 4.93 (1H, d, J 8.1), 4.84–4.74 (1H, m), 4.43–4.35 (1H, dd, J 9.1, 4.4), 4.32 (2H, q, J 7.1), 2.71 (1H, ddd, J 12.5, 8.5, 4.1), 2.17 (3H, s), 1.34 (3H, t, J 7.1), 1.34–1.24 (1H, m), 1.14 (3H, d, J 6.3); 13C NMR (151 MHz, CD3CN) δC 170.0, 167.0, 157.7, 147.5, 145.7, 143.9, 139.6, 138.2, 137.6, 130.9, 130.4, 127.8, 127.7, 126.5, 123.4, 122.5, 119.1, 112.4, 107.4, 95.4, 61.8, 50.8, 48.7, 41.6, 23.4, 21.7, 14.6; LRMS m/z (ESI+) 469 ([M + H]+, 11%); HRMS m/z (ESI+) [Found: 469.21239, C29H29O4N2, requires [M + H]+ 469.21218]; HPLC (Method 1), retention time = 11.9 min, 99.7%; Chiral HPLC, Chiral AD-H column (80:20 hexane/IPA, 1.0 mL min–1), retention time = 19.7 min (18, 91.7% UV), retention time = 28.0 min (opposite enantiomer, 8.3% UV); 83% e.e.
4-((2S,4R)-1-Acetyl-4-[(1-benzofuran-6-yl)amino]-2-methyl-1,2,3,4-tetrahydroquinolin-6-yl)benzoic Acid (11)
Compound 18 (48 mg, 0.102 mmol, 1.0 equiv) was dissolved in EtOH (0.63 mL). NaOH(aq) (2 M, 0.512 mL, 1.02 mmol, 10 equiv) was added portion-wise over 30 min, and the resulting solution was stirred for 1 h. The solution was then acidified to ∼pH 5 using HCl(aq) (1 M), upon which a precipitate formed, and then extracted with EtOAc (3 × 10 mL). The organic components were dried over Na2SO4, filtered, and concentrated in vacuo. The crude material was purified using semipreparative HPLC and the fractions containing 11 were combined and lyophilized to give 11 as a colorless solid (9 mg, 20%): Rf 0.54 (10% MeOH/CH2Cl2); [α]25D = +177.8 (c 0.5, MeOH); mp 170–173 °C (from MeOH); v̅max (thin film)/cm–1 3404 (N–H, m), 1700 (C=O, m), 1628 (N–H, m); 1H NMR (600 MHz, MeOD) δH 8.06–7.95 (2H, m), 7.68–7.62 (2H, m), 7.59 (2H, d, J 8.4), 7.47 (1H, d, J 2.2), 7.40 (1H, d, J 8.1), 7.36 (1H, d, J 9.1), 6.83–6.72 (2H, m), 6.65 (1H, d, J 2.1), 4.93–4.81 (partially hidden by H2O peak) (1H, m), 4.33 (1H, dd, J 12.3, 4.1), 2.74 (1H, ddd, J 12.6, 8.6, 4.2), 2.26 (3H, s), 1.41–1.33 (1H, m), 1.20 (3H, d, J 6.4); 13C NMR (151 MHz, MeOD) δC 172.2, 170.4, 158.3, 147.8, 145.9, 143.8, 140.5, 139.3, 137.5, 131.7, 131.3, 127.9, 127.7, 126.8, 124.1, 122.3, 119.6, 112.3, 107.3, 95.7, 51.3, 49.6, 41.7, 23.0, 21.5; LRMS m/z (ESI–) 439 ([M-H]−, 77%); HRMS m/z (ESI+) [Found: 441.1812, C27H25O4N2, requires [M + H]+ 441.1809]; HPLC (Method 1), retention time = 9.7 min, 98.9%; Chiral HPLC, Chiral IG-3 column (70:30 hexane/IPA, 0.1% TFA, 1.0 mL min–1), retention time = 13.8 min (11, 92.5% UV), retention time = 18.4 min (opposite enantiomer, 7.5% UV); 85% e.e.
Ethyl 4-{(2S,4R)-1-Acetyl-4-[(1-benzothiophen-5-ylamino]-2-methyl-1,2,3,4-tetrahydroquinolin-6-yl}benzoate (19)
Compound S7a (100 mg, 0.284 mmol, 1.0 equiv), 5-bromobenzothiophene (121 mg, 0.568 mmol, 2.0 equiv), BrettPhos (49 mg, 0.0913 mmol, 0.32 equiv), BrettPhos Pd G3 (21 mg, 0.0232 mmol, 0.082 equiv), and NaOtBu (33 mg, 0.343 mmol, 1.2 equiv) were added to a dry microwave vial, and the vial was purged with argon. Degassed anhydrous toluene (1.7 mL) was added and the resulting suspension was stirred for 15 h at 70 °C. After this time, the suspension was filtered through Celite, eluting with toluene and Et2O, and the filtrate was concentrated in vacuo. The resulting residue was redissolved in EtOAc (50 mL) and washed with H2O (40 mL) and brine (40 mL). The organic components were dried over Na2SO4, filtered, and concentrated in vacuo. The crude material was purified using silica gel flash column chromatography (0–60% EtOAc/petroleum ether) to give 19 as a yellow solid (88 mg, 66%, 94.7% purity by HPLC). To achieve a higher purity, 44 mg of 19 was further purified using semipreparative HPLC and the fractions containing 19 lyophilized to give 19 as an off-white solid:Rf 0.34 (50% EtOAc/petroleum ether); [α]25D=+324.5 (c 1.0, CHCl3); mp 95–99 °C (from CHCl3); v̅max (thin film)/cm–1 3360 (N–H, w), 1713 (C = O, s), 1647 (N–H, s), 1606 (s), 1488 (s), 1278 (s); 1H NMR (400 MHz, CD3CN) δH 8.04–7.91 (2H, m), 7.68 (1H, d, J 8.7), 7.63–7.54 (4H, m), 7.43 (1H, d, J 5.4), 7.38 (1H, d, J 8.0), 7.13 (1H, d, J 5.5, 0.8), 7.10–7.08 (1H, m), 6.93 (1H, dd, J 8.7, 2.3), 4.87 (1H, d, J 8.0), 4.83–4.74 (1H, m), 4.40 (1H, ddd, J 12.1, 8.0, 4.1), 4.31 (2H, q, J 7.1), 2.72 (1H, ddd, J 12.5, 8.6, 4.1), 2.18 (3H, s), 1.34 (3H, t, J 7.1), 1.35–1.22 (1H, m), 1.14 (3H, d, J 6.3); 13C NMR (151 MHz, CD3CN) δC 170.0, 167.0, 146.7, 145.7, 142.2, 139.7, 138.2, 137.6, 130.9, 130.4, 130.0, 128.1, 127.9, 127.7, 126.5, 124.4, 123.8, 123.4, 115.1, 106.1, 61.8, 50.8, 48.7, 41.6, 23.4, 21.7, 14.6; LRMS m/z (ESI+) 485 ([M + H]+, 2.6%); HRMS m/z (ESI+) [Found: 485.18976, C29H29O3N232S, requires [M + H]+ 485.18934]; HPLC (Method 1), retention time = 12.3 min, > 99.9%; Chiral HPLC, Chiral AD-H column (50:50 hexane/EtOH, 1.0 mL min–1), retention time = 10.9 min (opposite enantiomer, 6.7% UV), retention time = 13.9 min (19, 93.3% UV); 87% e.e.
4-((2S,4R)-1-Acetyl-4-[(1-benzothiophen-5-yl)amino]-2-methyl-1,2,3,4-tetrahydroquinolin-6-yl)benzoic Acid (12)
Compound 19 (36 mg, 0.0769 mmol, 1 equiv) was dissolved in EtOH (0.46 mL). NaOH(aq) (2 M, 0.371 mL, 0.742 mmol, 10 equiv) was added over the course of 30 min, and the resulting solution was left to stir for 3 h at rt. After this time the solution was acidified to ∼pH 5, upon which a precipitate formed. EtOAc (10 mL) was added, upon which the precipitate redissolved, and the aqueous and organic components were separated. The aqueous components were extracted with EtOAc (3 × 10 mL), and the combined organic components were dried over Na2SO4, filtered, and concentrated in vacuo. The crude material was purified using semipreparative HPLC to give 12 as an off-white solid (14 mg, 41%): Rf 0.61 (50% EtOAc/petroleum ether, 1% acetic acid); [α]25D=+170.1 (c 0.37, CHCl3); mp 137–143 °C (from CHCl3); v̅max (thin film)/cm–1 3352 (N–H, w), 1710 (C=O, m), 1638 (m), 1605 (s), 1513 (m); 1H NMR (400 MHz, CD3CN) δH 8.03–7.95 (2H, m), 7.68 (1H, d, J 8.7), 7.64–7.59 (3H, m), 7.59–7.57 (1H, m), 7.44 (1H, d, J 5.4), 7.39 (1H, d, J 8.1), 7.14 (1H, dd, J 5.4, 0.8), 7.10–7.08 (1H, m), 6.94 (1H, dd, J 8.7, 2.3), 4.88–4.73 (1H, m), 4.40 (1H, dd, J 12.1, 4.1), 2.72 (1H, ddd, J 12.5, 8.5, 4.1), 2.18 (3H, s), 1.35–1.24 (1H, m), 1.15 (3H, d, J 6.3); 13C NMR (151 MHz, CD3CN) δC 170.1, 167.7, 146.7, 145.7, 142.2, 139.7, 138.2, 137.7, 131.2, 130.2, 130.0, 128.1, 127.9, 127.7, 126.5, 124.4, 123.8, 123.4, 115.1, 106.1, 50.8, 48.7, 41.6, 23.4, 21.7; LRMS: m/z (ESI+) 457 ([M + H]+, 100%); HRMS m/z (ESI–) [found: 455.14422, C27H23O332S, requires [M–H]− 455.14349]; HPLC (Method 1), retention time = 9.97 min, > 99.9%; Chiral HPLC, Chiral IG-3 column (70:30 hexane/IPA, 0.1% TFA, 1.0 mL min–1), retention time = 15.0 min (12, 92.35% UV), retention time = 18.5 min (opposite enantiomer, 7.65% UV); 85% e.e.
(1-(tert-Butoxycarbonyl)-1H-indol-5-yl)boronic Acid (S8)
Di-tert-butyl dicarbonate (542 mg, 2.48 mmol, 4.0 equiv) and DMAP (15 mg, 0.123 mmol, 0.2 equiv) were added to a solution of 5-indoylboronic acid (100 mg, 0.621 mmol, 1.0 equiv) in MeCN (2.8 mL). The resulting solution was stirred at rt for 1 h, after which time a solution of aqueous citric acid (0.5 M, 50 mL) was added, and the aqueous components were extracted with EtOAc (3 × 50 mL). The organic components were washed with brine (100 mL), dried over Na2SO4, filtered, and concentrated in vacuo. The crude material was purified using silica gel flash column chromatography (0–50% EtOAc/petroleum ether) to give S8 as an off-white solid (80 mg, 49%). Rf 0.48 (50% EtOAc/petroleum ether); mp 82–85 °C (from EtOAc); v̅max (thin film)/cm–1 2979 (C–H, w), 1734 (C=O, m), 1331 (m); 1H NMR (600 MHz, CDCl3) δH 8.55–8.54 (1H, m), 8.32–8.26 (1H, m), 8.26–8.23 (1H, m), 7.67 (1H, d, J 3.7), 6.74 (1H, d, J 3.7), 1.72 (9H, s); 13C NMR (151 MHz, CDCl3) δC 149.9, 138.2, 131.5, 130.6, 129.6, 126.4, 124.5, 114.9, 107.9, 84.1, 28.4; 11B NMR (128 MHz, CDCl3) δB 4.31; LRMS m/z (ESI+) 284 ([M + Na]+, 87%). The Rf, 1H and 13C data are in agreement with the literature values.51
tert-Butyl 5-{(2S,4R)-(1-Acetyl-6-[4-(ethoxycarbonyl)phenyl]-2-methyl-1,2,3,4-tetrahydroquinolin-4-yl)amino}-1H-indole-1-carboxylate (S9)
Compound S7a (47 mg, 0.133 mmol, 1 equiv) and boronic acid S8 (69 mg, 0.264 mmol, 2 equiv) were dissolved in anhydrous CH2Cl2 (1.0 mL) in a round-bottom flask containing activated 3 Å molecular sieves, connected to a drying tube containing P2O5, open to the atmosphere. NEt3 (55 μL, 0.395 mmol, 3 equiv) was added and the resulting solution was left to stir at rt for 15 h. The molecular sieves were removed using filtration, and the filtrate was concentrated in vacuo. The resulting residue was purified using silica gel flash column chromatography column (0–15% EtOAc/CH2Cl2) to give S9 as a colorless solid (40 mg, 53%): Rf 0.20 (10% EtOAc/CH2Cl2); [α]25D = +244.3 (c 1.0, CHCl3); mp 117–121 °C (from CH2Cl2); v̅max (thin film)/cm–1 3347 (N–H, w), 2978 (C–H, w), 1720 (C=O, s), 1653 (N–H, m); 1H NMR (400 MHz, CDCl3) δH 8.06–8.01 (2H, m), 8.00–7.91 (1H, m), 7.68–7.64 (1H, m), 7.58–7.49 (4H, m), 7.28–7.21 (1H, m), 6.80–6.79 (1H, m), 6.79–6.73 (1H, m), 6.40 (1H, d, J 3.6), 4.99–4.82 (1H, m), 4.37 (2H, q, J 7.1), 4.31 (1H, dd, J 12.0, 4.2), 3.84 (1H, br s), 2.72 (1H, ddd, J 12.5, 8.6, 4.2), 2.26 (3H, s), 1.65 (9H, s), 1.38 (3H, t, J 7.1), 1.36–1.27 (1H, m), 1.21 (3H, d, J 6.3);13C NMR (126 MHz, CDCl3) δC 169.6, 166.5, 149.9, 144.7, 143.2, 138.8, 137.9, 136.6, 131.9, 130.2, 129.4, 129.1, 127.0, 126.5, 126.0, 123.0, 116.1, 112.7, 107.0, 103.7, 83.4, 61.1, 51.0, 47.8, 41.4, 28.4, 23.3, 21.5, 14.5; LRMS m/z (ESI+) 568 ([M + H]+, 37%); HRMS m/z (ESI+) [found: 568.2803, C34H38O5N3, requires [M + H]+ 568.2806]; HPLC (Method 1), retention time = 13.1 min, 98.1%; Chiral HPLC, Chiral IG-3 column (50:50 hexane:IPA, 1.0 mL min–1), retention time = 11.2 min (S9, 92.0% UV), retention time = 17.0 (opposite enantiomer, 8.0% UV); 84% e.e.
Ethyl 4-{(2S,4R)-1-Acetyl-4-[(1H-indol-5-yl)amino]-2-methyl-1,2,3,4-tetrahydroquinolin-6-yl}benzoate (20)
TFA (0.8 mL) was added dropwise to a solution of S8 (85 mg, 0.150 mmol, 1 equiv) in CH2Cl2 (0.8 mL), and the resulting solution was stirred at room temperature for 30 min. After this time, the solution was diluted with EtOAc (15 mL) and neutralized with saturated NaHCO3(aq) (10 mL). The organic and aqueous components were separated, and the aqueous components were extracted with EtOAc (3 × 10 mL). The organic components were combined, dried over Na2SO4, filtered, and concentrated in vacuo. The crude material was purified using silica gel flash column chromatography (0–50% EtOAc/petroleum ether) to give 20 as a yellow solid (32 mg, 46%): Rf 0.28 (50% EtOAc/pentane); [α]25D = +280.4 (c 1.0, CHCl3); mp 104–108 °C (from EtOAc); v̅max (thin film)/cm–1 2975 (C–H, m), 1709 (C = O, m), 1647 (N–H, m); 1H NMR (600 MHz, CD3CN) δH 9.00 (1H, s), 8.00 (2H, d, J 8.2), 7.67–7.66 (1H, m), 7.63 (2H, d, J 8.2), 7.60 (1H, dd, J 8.2, 2.2), 7.37 (1H, d, J 8.2), 7.26 (1H, d, J 8.6), 7.13–7.10 (1H, m), 6.85–6.82 (1H, m), 6.75 (1H, dd, J 8.6, 2.3), 6.23–6.21 (1H, m), 4.81–4.74 (1H, m), 4.38–4.29 (3H, m), 2.71 (1H, ddd, J 12.4, 8.5, 4.0), 2.17 (3H, s), 1.34 (3H, t, J 7.1), 1.30–1.22 (1H, m), 1.14 (3H, d, J 6.3); 13C NMR (151 MHz, CD3CN) δC 170.0, 167.0, 145.8, 142.4, 140.5, 138.2, 137.5, 131.4, 130.9, 130.4, 130.0, 127.7, 126.3, 125.9, 123.7, 112.9, 112.9, 103.4, 101.6, 61.8, 51.6, 48.8, 41.7, 23.3, 21.8, 14.6; LRMS m/z (ESI+) 468 ([M + H]+, 13%); HRMS m/z (ESI+) [found: 468.2281, C29H30O3N3 requires [M + H]+ 468.2282]; HPLC (Method 1), retention time = 8.8 min, 97.5%; Chiral HPLC, Chiral IF-3 column (70:30 hexane/IPA, 1.0 mL min–1), retention time = 14.0 min (20, 95.45% UV), retention time = 16.7 (opposite enantiomer, 4.55% UV); 91% e.e.
4-{(2S,4R)-1-Acetyl-4-[(1H-indol-5-yl)amino]-2-methyl-1,2,3,4-tetrahydroquinolin-6-yl}benzoic Acid (13)
Compound 20 (27 mg, 0.0577 mmol, 1.00 equiv) was dissolved in EtOH (0.36 mL). NaOH(aq) (2 M, 289 μL, 0.577 mmol, 10.0 equiv) was added portion wise. A precipitate developed, which was redissolved with addition of further EtOH (0.76 mL), and the resulting solution was stirred for 3 h. Further NaOH(aq) (2 M, 578 μL, 1.16 mmol, 20.0 equiv) was added over a period of 1.5 h, after which time the reaction solution was diluted with CH2Cl2 (5 mL) and H2O (5 mL), and the aqueous and organic layers were separated. The aqueous components were acidified to approximately pH 3, then and extracted with EtOAc (3 × 10 mL). The organic components were dried over Na2SO4, filtered, and concentrated in vacuo. The crude material was purified using silica gel flash column chromatography (reverse phase, 5–40% MeCN/H2O) and the pure fractions lyophilized to give 13 as a yellow solid (8.2 mg, 32%): Rf 0.24 (50% MeCN/H2O; reverse phase); [α]25D = +197.0 (c 0.29, MeOH); mp 176–179 °C (lyophilized from MeCN/H2O); v̅max (thin film)/cm–1 3401 (N–H, br, m), 2924 (C–H, w), 1703 (C=O, s), 1631 (C=O, s);1H NMR (400 MHz, MeOD) δH 8.45 (1H, s), 8.01–7.96 (2H, m), 7.77–7.75 (1H, m), 7.63 (1H, dd, J 8.2, 2.2), 7.61–7.57 (2H, m), 7.38 (1H, d, J 8.2), 7.25 (1H, d, J 8.6), 7.11 (1H, d, J 3.1), 6.90–6.88 (1H, m), 6.79 (1H, dd, J 8.6, 2.2), 6.25 (1H, dd, J 3.1, 0.9), 4.90–4.78 (1H, hidden by H2O peak), 4.30 (1H, dd, J 12.2, 4.1), 2.74 (1H, ddd, J 12.5, 8.6, 4.1), 2.25 (3H, s), 1.37–1.25 (1H, m), 1.19 (3H, d, J 6.3); 13C NMR (151 MHz, MeOD) δC 172.2, 170.7, 145.8, 142.3, 141.1, 139.2, 137.5, 132.4, 131.9, 131.3, 130.3, 127.7, 126.5, 125.8, 124.3, 113.2, 112.8, 104.5, 101.5, 52.3, 49.6, 41.8, 23.0, 21.5; LRMS m/z (ESI–) 438 ([M–H]−, 8%); HRMS m/z (ESI–) [found: 438.1832, C27H2403N3 requires [M–H]− 438.1823]; HPLC (Method 1) Retention time = 6.9 min, 97.9%; Chiral HPLC, Chiral IG-3 column (70:30 hexane:IPA, 0.1% TFA, 1.0 mL min–1) Retention time = 15.4 min (13, 89.6% UV), Retention time = 19.8 (opposite enantiomer, 10.4% UV); 79% e.e.
Ethyl 4-{(2S,4R)1-Acetyl-4-[(1,3-benzothiazol-6-yl)amino]-2-methyl-1,2,3,4-tetrahydroquinolin-6-yl}benzoate (21)
Compound S7a (60 mg, 0.170 mmol, 1.0 equiv), 6-bromobenzothiazole (72 mg, 0.340 mmol, 2.0 equiv), BrettPhos (29 mg, 0.0540 mmol, 0.32 equiv), NaOtBu (20 mg, 0.208 mmol, 1.2 equiv), and BrettPhos Pd G3 (12 mg, 0.0132 mmol, 0.078 equiv) were added to a dry microwave vial under Ar. Degassed, anhydrous toluene (1 mL) was added, and the resulting suspension was stirred at 70 °C for 16 h. After this time, the reaction mixture was cooled to rt, and filtered through Celite, eluting with toluene, Et2O, and EtOAc. The filtrate was concentrated in vacuo, and then redissolved in EtOAc and satd. NaHCO3(aq). The organic and aqueous components were separated, and the aqueous components were extracted with EtOAc (3 × 10 mL). The combined organic components were dried over Na2SO4, filtered, and concentrated in vacuo. The crude material was purified using silica gel flash column chromatography (0–70% EtOAc/pentane) to give 21 as a yellow solid (40 mg, 48%). Rf 0.15 (50% EtOAc/pentane); [α]25D = +263.8 (c 1.0, CHCl3); mp 114–117 °C (from MeCN); v̅max (thin film)/cm–1 3341 (N–H, w), 2980 (C–H, w), 1711 (C=O, m), 1642 (C=O, m); 1H NMR (500 MHz, CD3CN) δH 8.70 (1H, s), 8.00–7.96 (2H, m), 7.83 (1H, d, J 8.9), 7.63–7.59 (3H, m), 7.56–7.53 (1H, m), 7.38 (1H, d, J 8.1), 7.22 (1H, d, J 2.4), 7.02 (1H, dd, J 8.9, 2.4), 5.13 (1H, d, J 8.0), 4.86–4.74 (1H, m), 4.41 (1H, ddd, J 12.2, 8.0, 4.2), 4.31 (2H, q, J 7.1), 2.72 (1H, ddd, J 12.5, 8.5, 4.2), 2.17 (3H, s), 1.37–1.25 (4H, m), 1.14 (3H, d, J 6.3); 13C NMR (126 MHz, CD3CN) δC 170.0, 166.9, 150.6, 147.5, 147.0, 145.6, 139.3, 138.2, 137.6, 136.9, 130.8, 130.4, 127.9, 127.7, 126.6, 124.5, 123.2, 115.8, 103.6, 61.8, 50.7, 48.6, 41.5, 23.4, 21.7, 14.6; LRMS m/z (ESI+) 486 ([M + H]+, 100%); HRMS m/z (ESI+) [found: 486.1844, C28H28O3N3S, requires [M+H]+ 486.1846]; HPLC (Method 1), retention time = 10.7 min, 96.0%; Chiral HPLC, Chiral AD-H column (50:50 hexane/IPA, 1.0 mL min–1), retention time = 8.0 min (opposite enantiomer, 7.5% UV), retention time = 10.6 (21, 92.5% UV); 85% e.e.
4-{(2S,4R)-1-Acetyl-4-[(1,3-benzothiazol-6-yl)amino]-2-methyl-1,2,3,4-tetrahydroquinolin-6-yl}benzoic acid (14)
Compound 21 (61 mg, 0.126 mmol, 1 equiv) was dissolved in EtOH (0.79 mL) and to this solution, NaOH(aq) (2 M, 0.63 mL, 1.26 mmol, 10 equiv) was added portion wise. The solution was stirred for 1 h 20 min, after which time, HCl(aq) (1 M) was added until a red precipitate formed. The precipitate was redissolved in EtOAc (10 mL) and the solution was diluted with H2O (5 mL). The aqueous and organic components were separated, and the aqueous component was extracted with EtOAc (3 × 10 mL). The combined organic components were dried over Na2SO4, filtered, and concentrated in vacuo. The crude material was purified using silica gel flash column chromatography (reverse phase, 5–31% MeCN/H2O) and lyophilized to give 14 as an orange-brown powder (40 mg, 69%): Rf 0.23 (50% MeCN/H2O); [α]25D = +318.2 (c 1.0, MeOH); mp 168–172 °C (lyophilized from MeCN/H2O); v̅max (thin film)/cm–1 3358 (N–H, br, w), 3074 (O–H, br, w), 1702 (C=O, m), 1636 (N–H, m); 1H NMR (400 MHz, MeOD) δH 8.82 (1H, s), 8.01–7.98 (2H, m), 7.83 (1H, d, J 8.9), 7.65 (1H, dd, J 8.2, 2.2), 7.61–7.57 (3H, m), 7.42 (1H, d, J 8.2), 7.22 (1H, d, J 2.3), 7.07 (1H, dd, J 8.9, 2.3), 4.96–4.83 (1H, m) (partially hidden by H2O peak), 4.41 (1H, dd, J 12.2, 4.2), 2.75 (1H, ddd, J 12.5, 8.5, 4.2), 2.27 (3H, s), 1.44–1.36 (1H, m), 1.20 (3H, d, J 6.3); 13C NMR (151 MHz, MeOD) δC 172.2, 169.8, 151.7, 148.4, 146.4, 146.1, 139.9, 139.2, 137.7, 137.1, 131.4, 131.1, 128.0, 127.8, 126.9, 124.1, 123.8, 116.2, 103.4, 50.9, 49.6, 41.7, 23.0, 21.5; LRMS m/z (ESI+) 458 ([M + H]+, 42%); HRMS m/z (ESI–) [found: 456.1386, C26H23N3O3S, requires [M – H]− 456.1387]; HPLC (Method 1), retention time = 8.2 min, 99.1%; Chiral HPLC, Chiral AD-H column (40:60 hexane:IPA, 1.0 mL min–1), retention time = 5.9 min (opposite enantiomer, 7.8% UV), retention time = 7.8 min (14, 92.2% UV); 84% e.e.
Ethyl 4-{(2S,4R)-1-Acetyl-2-methyl-4-[(quinolin-6-yl)amino]-1,2,3,4-tetrahydroquinolin-6-yl}benzoate (22)
Compound S7a (56 mg, 0.159 mmol, 1.2 equiv), 6-bromoquinoline (18 μL, 0.132 mmol, 1.0 equiv), CyJohnPhos (5 mg, 0.0143 mmol, 0.11 equiv), Pd(OAc)2 (2 mg, 0.00891 mmol, 0.067 equiv), and NaOtBu (15 mg, 0.156 mmol, 1.2 equiv) were added to a flame-dried microwave vial, and the vial was purged with Ar. Anhydrous, degassed toluene (0.32 mL) was added to the vial, and the resulting solution was stirred at 100 °C for 15 h. After this time, the solution was cooled to rt and filtered through Celite, eluting with toluene, Et2O, and EtOAc. The washings were partially concentrated in vacuo to remove the Et2O, diluted with EtOAc (10 mL), and washed with H2O (20 mL) and then HCl(aq) (1 M, 20 mL). The aqueous components were extracted with EtOAc (3 × 20 mL). The aqueous components were then neutralized with NaHCO3(aq) and further extracted with EtOAc (3 × 40 mL). The combined organic components were washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. The crude material was purified using silica gel flash column chromatography (0–80% EtOAc/petroleum ether, 1% NEt3) to give compound 22 as a yellow solid (19 mg, 30%): Rf 0.16 (60% EtOAc/petroleum ether, 1% NEt3); [α]25D = +405.8 (c 1.0, CHCl3); mp 82–86 °C (from CHCl3); v̅max (thin film)/cm–1 3324 (N–H, m), 2981 (C–H, m), 1712 (C=O, s), 1626 (s), 1608 (s); 1H NMR (400 MHz, CD3CN) δH 8.53 (1H, dd, J 4.2, 1.7), 7.98–7.92 (2H, m), 7.89 (1H, dd, J 8.4, 1.7), 7.84 (1H, d, J 9.1), 7.65–7.57 (3H, m), 7.57–7.53 (1H, m), 7.39 (1H, d, J 8.2), 7.35 (1H, dd, J 9.1, 2.6), 7.24 (1H, dd, J 8.4, 4.2), 6.85 (1H, d, J 2.6), 5.26 (1H, d, J 7.9), 4.90–4.76 (1H, m), 4.49 (1H, ddd, J 12.1, 7.9, 4.1), 4.30 (2H, q, J 7.1), 2.74 (1H, ddd, J 12.5, 8.5, 4.1), 2.19 (3H, s), 1.41–1.29 (1H, m), 1.32 (3H, t, J 7.1), 1.16 (3H, d, J 6.3); 13C NMR (151 MHz, CD3CN) δC 170.1, 166.9, 147.1, 147.0, 145.6, 144.1, 139.1, 138.3, 137.6, 134.5, 131.15, 131.12, 130.8, 130.4, 127.9, 127.7, 126.6, 123.2, 122.5, 122.4, 104.5, 61.8, 50.5, 48.6, 41.4, 23.4, 21.7, 14.6; LRMS m/z (ESI+) 480 ([M+H+]+, 100%); HRMS m/z (ESI+) [found: 480.2278, requires [M + H]+ 480.2282)]; HPLC (Method 1), retention time = 8.6 min, 95.8%; Chiral HPLC, Chiral IG-3 column (40:60 hexane:IPA, 1.0 mL min–1), retention time = 9.8 min (22, 93.1% UV), retention time = 18.4 (opposite enantiomer, 6.9% UV); 86% e.e.
4-{(2S,4R)-1-Acetyl-2-methyl-4-[(quinolin-6-yl)amino]-1,2,3,4-tetrahydroquinolin-6-yl}benzoic Acid (15)
Compound 22 (51 mg, 0.106 mmol, 1 equiv) was dissolved in EtOH (0.66 mL), NaOH(aq) (2 M, 0.53 mL, 1.06 mmol, 10 equiv) was added dropwise, and the resulting solution was left to stir for 2.5 h. After this time, the solution was acidified to approximately pH 5 using HCl(aq) (1 M), and the aqueous solution was extracted with EtOAc (5 × 10 mL). The combined organic components were dried over Na2SO4, filtered, and concentrated in vacuo. The crude material was purified using silica gel flash column chromatography (reverse phase, 5–36% MeCN/H2O) and lyophilized to give 15 as a yellow-green solid (17 mg, 36%, 94.7% purity by HPLC). To achieve a higher purity, 15 was further purified by semipreparative HPLC and lyophilized (>99.9% by HPLC). Rf 0.38 (50% MeCN/H2O; reverse phase); [α]25D = +369.7 (c 0.12, MeOH); mp 194–197 °C (from MeOH); v̅max (thin film)/cm–1 3342 (N–H, br, w), 2970 (O–H, br, w), 1703 (C=O, m), 1625 (N–H, s); 1H NMR (600 MHz, MeOD) δH 8.47 (1H, dd, J 4.3, 1.6), 8.07–8.02 (1H, m), 7.98 (2H, d, J 8.1), 7.83 (1H, d, J 9.1), 7.66 (1H, dd, J 8.2, 2.2), 7.60–7.58 (1H, m), 7.57 (2H, d, J 8.1), 7.47–7.40 (2H, m), 7.32 (1H, dd, J 8.3, 4.3), 6.86 (1H, d, J 2.6), 4.97–4.87 (1H, m), 4.49 (1H, dd, J 12.1, 4.2), 2.79 (1H, ddd, J 12.5, 8.5, 4.2), 2.29 (3H, s), 1.49–1.39 (1H, m), 1.22 (3H, d, J 6.3); 13C NMR (151 MHz, MeOD) δC 172.2, 169.7, 148.3, 146.0, 145.5, 142.2, 139.4, 139.2, 137.7, 137.1, 132.2, 131.4, 131.0, 128.9, 128.1, 127.8, 127.0, 123.8, 123.7, 122.7, 104.2, 50.6, 49.6, 41.6, 23.1, 21.5; LRMS m/z 452 (ESI+) ([M + H]+, 100%); HRMS m/z (ESI+) [Found: 452.1972, C28H26N3O3, requires [M + H]+ 452.1969]; HPLC (Method 1), retention time = 6.8 min, > 99.9%; Chiral HPLC, Chiral IG-3 column (70:30 hexane:IPA, 1.0 mL min–1), retention time = 17.2 min (15, 91.3% UV), retention time = 23.9 (opposite enantiomer, 8.7% UV); 83% e.e.
Biological Methods
Identification of SmBCPs
The identification of S. mansoni BCPs (stably found in both v7 and v10 genome assemblies) was performed using a bioinformatic approach previously described.13 Briefly, a BLASTp search was performed to identify S. mansoni proteins (Smps) sharing high similarity (E value ≤1 × 10–5) to representative H. sapiens BCPs derived from literature data.16,52−55 The results of this method were validated and further expanded using a WormBase ParaSite56-based BioMart search looking for S. mansoni proteins containing annotated Bromo domain (BRD) or BRD associated domains. Interpro, Pfam, PROSITE, and SMART signatures were used for this search.
Classification of SmBCP Domains
The amino acid positions of the catalytic domain of each S. mansoni BCP (Bromo domain - BRD) were extracted from WormBase ParaSite. The architecture of these proteins was then populated with other N-terminal and/or C-terminal domains known from the literature to be associated with the catalytic domain. These additional domains (Figure 1B) were defined as follows: DNA binding homeobox and Different Transcription factors (DDT), Plant homeodomain (PHD), N-terminal Extra Terminal (ET) domain (NET), Transcription adaptor putative zinc finger (TAZ), KID-interacting domain (KIX), CREB-binding protein (CREBBP), ZZ-type zinc finger domain (ZZ), Histone acetyltransferase (HAT), Trp-Asp motif (WD40), Domain of unknown function 3512 (DUF3512), AAA- ATPase - ATPases Associated with various cellular Activities (AAA), Enhanced and polycomb-like (EPL), Pro-Trp-Trp-Pro domain (PWWP), Zinc finger MYND-type (MYND), Transcription initiation factor TFIID subunit 1 (TFIID), Bromo-adjacent homology domain (BAH), High mobility group box (HMG Box), Gln-Leu-Gln (Glutamine-Leucine-Glutamine) domain (QLQ), SNF2-related, N-terminal domain (SNF2-N), Helicase (Hel), Brahma and kismet domain (BRK), Helicase/SANT-associated domain (HAS), B-box-type zinc finger domain (B-BOX), and Forkhead-associated (FHA) domain (FHA). Graphical representation was prepared using Illustrator for Biological Sequences.57
Gene Expression Profiling of SmBCPs
The normalized gene expression values for each smbcp were derived by the RNA-Seq meta database curated by Lu et al.58 and accessible through the ‘schisto_xyz’ search engine (http://schisto.xyz/). This database was obtained by normalizing gene expression values derived RNA-Seq data published by Anderson et al. for egg,59 Wang et al. for miracidia (indicated as Mir in the heat map) and sporocysts (Spo),60 Protasio et al. for cercariae (Cerc), 3h and 24h schistosomula (Som_3h and Som_24h),61 Protasio et al. for 21 day juvenile male and female (Male_21days and Female_21days),62 and Lu et al. for 42 day adult male and female (Male_42days and Female_42days).63 The gene expression values for the 22 BCPs were extracted for each of the 10 life cycle stages and plotted as a heat map in R studio.
Ethics Statement
All mouse procedures performed at Aberystwyth University adhered to the United Kingdom Home Office Animals (Scientific Procedures) Act of 1986 (project license P3B8C46FD) as well as the European Union Animals Directive 2010/63/EU and were approved by Aberystwyth University’s Animal Welfare and Ethical Review Body (AWERB).
Ex Vivo Schistosomula Screening
S. mansoni schistosomula were obtained by mechanical transformation from cercariae as previously described,64 distributed in 384-well tissue culture plates (PerkinElmer, catalogue number 6007460) at 120 parasites/well and dosed with the selected compounds as previously reported.65−68 For this study, each compound (as 10 mM DMSO stock solution) was initially tested at two concentration points (20 and 10 μM final concentration, in duplicates) in at least four independent screens (Z′ scores above 0.35).69 Each screen contained both positive (10 μM auranofin (AUR) in 0.625% DMSO) and negative (0.625% DMSO) controls. For those compounds active at 10 μM (18, 20, 21, and 22), a further 2-fold titration (from 10 to 0.312 μM) was performed to determine the extent of their antischistosomula activity (Figure S48). Following 72 h schistosomula/compound coincubation, the plate was analyzed by the Roboworm platform.70 EC50 values were calculated from the titrated concentrations by nonlinear regression, after log transformation of concentrations and data normalization using GraphPad Prism 7.02.
Ex Vivo Adult Worm Screening
S. mansoni adult worms were recovered by hepatic portal vein perfusion from TO mice (Mus musculus HsdOLa:TO - Tuck Ordinary; Envigo, UK) that were percutaneously infected 7 weeks earlier with 180 cercariae.65,71 Following perfusion, schistosomes were washed and processed as previously described.65,66 Subsequently, schistosome pairs were seeded into 48 well tissue culture plates (1 worm pair/well, in duplicate) and dosed with the compounds (20 μM in 0.2% DMSO). DMSO (0.2%) and praziquantel (10 μM in 0.1% DMSO) were also included as negative and positive controls, respectively. Parasite motility was assessed by a digital image processing-based system (WormassayGP2),66 modified after Wormassay.72In vitro laid eggs (IVLEs) were enumerated and the presence or absence of paired worms was noted.
Ex Vivo Miracidia Screening
S. mansoni miracidia were obtained by hatching of schistosome eggs as previously described.73 The resulting miracidial suspension was collected, washed with Chernin’s balanced salt solution (CBSS),73 and enumerated prior to being used for ex vivo miracidia to sporocyst screens as previously reported.66,73 Each compound was tested at 0.5, 2.0, 5.0, 10, and 25 μM final concentrations (in 1% DMSO). Each treatment was set up in duplicate (15–25 miracidia/well). Each titration was performed in three independent experiments (two technical replicates per data point). Parasites cultured in CBSS (containing 1% DMSO) were included as negative controls. After 48 h incubation at 26 °C in the dark, dead, fully transformed and partially transformed miracidia were enumerated as previously described.74,75
Acknowledgments
M.S. was supported by the Deutsche Forschungsgemeinschaft (SCHI 1408/1-1). D.M. thanks the BBSRC and GSK for studentship support (BB/S507015/1). A.K.N.C. received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreements No 660156. S.J.C. thanks St Hugh’s College, Oxford, for research support. S.J.C. is grateful to Michael and Alice Jung for endowing the Jung Chair in Medicinal Chemistry and Drug Discovery at UCLA, which partially supported this work. K.F.H. and the DLS coauthors thanks the Life Sciences National Research Network Wales for funding. We are grateful to Pearse Solon, under the supervision of Prof. Martin Smith, University of Oxford, for his support in the chiral HPLC analysis of novel compounds. We thank LilyLatimer-Smith for the synthesis of literature intermediates and final compounds, Oliver Stratton and Dr Charles Evans for the synthesis of previously reported compounds, and Dr Corentine Laurin for contributing tool molecules to early studies.
Glossary
Abbreviations
- BCPs
bromodomain-containing proteins;
- BRDs
bromodomains;
- Cer
cercaria;
- ΔTm
protein thermal shift;
- HsBRD3/4
human bromodomain containing protein 3/4;
- ITC
isothermal titration calorimetry;
- KAc
acetylated lysine;
- Mir
miracidia;
- MUSCLE
multiple sequence comparison by log-expectation;
- PDB
protein data bank;
- PZQ
praziquantel;
- SmBRD3
Schistosoma mansoni bromodomain containing protein 3;
- SmBRD3(1/2)
the first/second bromodomain of SmBRD3;
- SmCBP1/2
cyclic AMP response element binding protein binding protein1/2 from S. mansoni;
- SmGCN5
general control nondepressible 5 from S. mansoni;
- Som
schistosomula;
- T. cruzi
Trypanosoma cruzi
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.3c01321.
Author Present Address
# Current address: Swansea University Medical School, Grove Building, Swansea University, Swansea, Wales, United Kingdom
Author Contributions
⊥ M.S., D. J.B.M., and G.P. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Geyer K. K.; Hoffmann K. F. Epigenetics: A Key Regulator of Platyhelminth Developmental Biology?. Int. J. Parasitol. 2012, 42, 221–224. 10.1016/j.ijpara.2012.02.003. [DOI] [PubMed] [Google Scholar]
- Cosseau C.; Wolkenhauer O.; Padalino G.; Geyer K. K.; Hoffmann K. F.; Grunau C. (Epi)Genetic Inheritance in Schistosoma Mansoni: A Systems Approach. Trends Parasitol. 2017, 33, 285–294. 10.1016/j.pt.2016.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogg S. J.; Beavis P. A.; Dawson M. A.; Johnstone R. W. Targeting the Epigenetic Regulation of Antitumour Immunity. Nat. Rev. Drug Discovery 2020, 19, 776–800. 10.1038/s41573-020-0077-5. [DOI] [PubMed] [Google Scholar]
- Schiedel M.; Conway S. J. Small Molecules as Tools to Study the Chemical Epigenetics of Lysine Acetylation. Curr. Opin. Chem. Biol. 2018, 45, 166–178. 10.1016/j.cbpa.2018.06.015. [DOI] [PubMed] [Google Scholar]
- Zaware N.; Zhou M.-M. Bromodomain Biology and Drug Discovery. Nat. Struct. Mol. Biol. 2019, 26, 870–879. 10.1038/s41594-019-0309-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiedel M.; Moroglu M.; Ascough D. M. H.; Chamberlain A. E. R.; Kamps J. J. A. G.; Sekirnik A. R.; Conway S. J. Chemical Epigenetics: The Impact of Chemical- and Chemical Biology Techniques on Bromodomain Target Validation. Angew. Chem., Int. Ed. 2019, 58, 17930–17952. 10.1002/anie.201812164. [DOI] [PubMed] [Google Scholar]
- Tallant C.; Bamborough P.; Chung C.; Gamo F. J.; Kirkpatrick R.; Larminie C.; Martín J.; Prinjha R.; Rioja I.; Simola D. F.; Gabarró R.; Calderón F. Expanding Bromodomain Targeting into Neglected Parasitic Diseases. ACS Infect. Dis. 2021, 7, 2953–2958. 10.1021/acsinfecdis.1c00387. [DOI] [PubMed] [Google Scholar]
- Carneiro V. C.; de Abreu da Silva I. C.; Torres E. J. L.; Caby S.; Lancelot J.; Vanderstraete M.; Furdas S. D.; Jung M.; Pierce R. J.; Fantappié M. R.; Knight M. Epigenetic Changes Modulate Schistosome Egg Formation and Are a Novel Target for Reducing Transmission of Schistosomiasis. PLoS Pathog. 2014, 10, e1004116 10.1371/journal.ppat.1004116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins J. N. R.; Collins J. J. Tissue Degeneration Following Loss of Schistosoma Mansoni Cbp1 Is Associated with Increased Stem Cell Proliferation and Parasite Death In Vivo. PLoS Pathog. 2016, 12, e1005963 10.1371/journal.ppat.1005963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Paz C.; Padalino G.; Coghlan A.; Lu Z.; Gradinaru I.; Collins J. N. R.; Berriman M.; Hoffmann K. F.; Collins J. J. Large-Scale RNAi Screening Uncovers Therapeutic Targets in the Parasite Schistosoma Mansoni. Science 2020, 369, 1649–1653. 10.1126/science.abb7699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus D. P.; Dunne D. W.; Sacko M.; Utzinger J.; Vennervald B. J.; Zhou X.-N. Schistosomiasis. Nat. Rev. Dis. Prim. 2018, 4, 13. 10.1038/s41572-018-0013-8. [DOI] [PubMed] [Google Scholar]
- Ending the Neglect to Attain the Sustainable Development Goals: A Road Map for Neglected Tropical Diseases 2021–2030.
- Padalino G.; Ferla S.; Brancale A.; Chalmers I. W.; Hoffmann K. F. Combining Bioinformatics, Cheminformatics, Functional Genomics and Whole Organism Approaches for Identifying Epigenetic Drug Targets in Schistosoma Mansoni. Int. J. Parasitol.: Drugs Drug Resist. 2018, 8, 559–570. 10.1016/j.ijpddr.2018.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzuk M. M.; McKeown M. R.; Filippakopoulos P.; Li Q.; Ma L.; Agno J. E.; Lemieux M. E.; Picaud S.; Yu R. N.; Qi J.; Knapp S.; Bradner J. E. Small-Molecule Inhibition of BRDT for Male Contraception. Cell 2012, 150, 673–684. 10.1016/j.cell.2012.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan X.; Cheryala N.; Karim R. Md.; Chan A.; Berndt N.; Qi J.; Georg G. I.; Schönbrunn E. Bivalent BET Bromodomain Inhibitors Confer Increased Potency and Selectivity for BRDT via Protein Conformational Plasticity. J. Med. Chem. 2022, 65, 10441–10458. 10.1021/acs.jmedchem.2c00453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P.; Picaud S.; Mangos M.; Keates T.; Lambert J.-P.; Barsyte-Lovejoy D.; Felletar I.; Volkmer R.; Muller S.; Pawson T.; Gingras A.-C.; Arrowsmith C. H.; Knapp S. Histone Recognition and Large-Scale Structural Analysis of the Human Bromodomain Family. Cell 2012, 149, 214–231. 10.1016/j.cell.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciel R. de M.; Dutra D. L.; da S.; Rumjanek F. D.; Juliano L.; Juliano M. A. Fantappié, M. R. Schistosoma Mansoni Histone Acetyltransferase GCN5: Linking Histone Acetylation to Gene Activation. Mol. Biochem. Parasitol. 2004, 133, 131–135. 10.1016/j.molbiopara.2003.09.005. [DOI] [PubMed] [Google Scholar]
- Bertin B.; Oger F.; Cornette J.; Caby S.; Noël C.; Capron M.; Fantappie M. R.; Rumjanek F. D.; Pierce R. J. Schistosoma Mansoni CBP/P300 Has a Conserved Domain Structure and Interacts Functionally with the Nuclear Receptor SmFtz-F1. Mol. Biochem. Parasitol. 2006, 146, 180–191. 10.1016/j.molbiopara.2005.12.006. [DOI] [PubMed] [Google Scholar]
- Fantappíé M. R.; de Oliveira F. M. B.; Santos R. d. M. M. d.; Mansure J. J.; Furtado D. R.; da Silva I. C. d. A.; Rumjanek F. D. Control of Transcription in Schistosoma Mansoni: Chromatin Remodeling and Other Regulatory Elements. Acta Tropica 2008, 108, 186–193. 10.1016/j.actatropica.2007.11.006. [DOI] [PubMed] [Google Scholar]
- Whatley K. C. L.; Padalino G.; Whiteland H.; Geyer K. K.; Hulme B. J.; Chalmers I. W.; Forde-Thomas J.; Ferla S.; Brancale A.; Hoffmann K. F. The Repositioning of Epigenetic Probes/Inhibitors Identifies New Anti-Schistosomal Lead Compounds and Chemotherapeutic Targets. PLoS Neglected Trop. Dis. 2019, 13, e0007693 10.1371/journal.pntd.0007693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper J.; Evans R.; Pritzel A.; Green T.; Figurnov M.; Ronneberger O.; Tunyasuvunakool K.; Bates R.; Žídek A.; Potapenko A.; Bridgland A.; Meyer C.; Kohl S. A. A.; Ballard A. J.; Cowie A.; Romera-Paredes B.; Nikolov S.; Jain R.; Adler J.; Back T.; Petersen S.; Reiman D.; Clancy E.; Zielinski M.; Steinegger M.; Pacholska M.; Berghammer T.; Bodenstein S.; Silver D.; Vinyals O.; Senior A. W.; Kavukcuoglu K.; Kohli P.; Hassabis D. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeira F.; Pearce M.; Tivey A. R. N.; Basutkar P.; Lee J.; Edbali O.; Madhusoodanan N.; Kolesnikov A.; Lopez R. Search and Sequence Analysis Tools Services from EMBL-EBI in 2022. Nucleic Acids Res. 2022, 50, W276–W279. 10.1093/nar/gkac240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn R. D.; Bateman A.; Clements J.; Coggill P.; Eberhardt R. Y.; Eddy S. R.; Heger A.; Hetherington K.; Holm L.; Mistry J.; Sonnhammer E. L. L.; Tate J.; Punta M. Pfam: The Protein Families Database. Nucleic acids research 2014, 42, D222–D230. 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewings D. S.; Rooney T. P. C.; Jennings L. E.; Hay D. A.; Schofield C. J.; Brennan P. E.; Knapp S.; Conway S. J. Progress in the Development and Application of Small Molecule Inhibitors of Bromodomain-Acetyl-Lysine Interactions. J. Med. Chem. 2012, 55, 9393–9413. 10.1021/jm300915b. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P.; Knapp S. The Bromodomain Interaction Module. FEBS letters 2012, 586, 2692–2704. 10.1016/j.febslet.2012.04.045. [DOI] [PubMed] [Google Scholar]
- Chung C.-W.; Coste H.; White J. H.; Mirguet O.; Wilde J.; Gosmini R. L.; Delves C.; Magny S. M.; Woodward R.; Hughes S. A.; Boursier E. V.; Flynn H.; Bouillot A. M.; Bamborough P.; Brusq J.-M. G.; Gellibert F. J.; Jones E. J.; Riou A. M.; Homes P.; Martin S. L.; Uings I. J.; Toum J.; Clément C. A.; Boullay A.-B.; Grimley R. L.; Blandel F. M.; Prinjha R. K.; Lee K.; Kirilovsky J.; Nicodeme E. Discovery and Characterization of Small Molecule Inhibitors of the BET Family Bromodomains. J. Med. Chem. 2011, 54, 3827–3838. 10.1021/jm200108t. [DOI] [PubMed] [Google Scholar]
- Niesen F. H.; Berglund H.; Vedadi M. The Use of Differential Scanning Fluorimetry to Detect Ligand Interactions That Promote Protein Stability. Nat. Protoc. 2007, 2, 2212–2221. 10.1038/nprot.2007.321. [DOI] [PubMed] [Google Scholar]
- Hewings D. S.; Wang M.; Philpott M.; Fedorov O.; Uttarkar S.; Filippakopoulos P.; Picaud S.; Vuppusetty C.; Marsden B.; Knapp S.; Conway S. J.; Heightman T. D. 3,5-Dimethylisoxazoles Act as Acetyl-Lysine-Mimetic Bromodomain Ligands. J. Med. Chem. 2011, 54, 6761–6770. 10.1021/jm200640v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewings D. S.; Fedorov O.; Filippakopoulos P.; Martin S.; Picaud S.; Tumber A.; Wells C.; Olcina M. M.; Freeman K.; Gill A.; Ritchie A. J.; Sheppard D. W.; Russell A. J.; Hammond E. M.; Knapp S.; Brennan P. E.; Conway S. J. Optimization of 3,5-Dimethylisoxazole Derivatives as Potent Bromodomain Ligands. J. Med. Chem. 2013, 56, 3217–3227. 10.1021/jm301588r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay D.; Fedorov O.; Filippakopoulos P.; Martin S.; Philpott M.; Picaud S.; Hewings D. S.; Uttakar S.; Heightman T. D.; Conway S. J.; Knapp S.; Brennan P. E. The Design and Synthesis of 5 and 6-Isoxazolylbenzimidazoles as Selective Inhibitors of the BET Bromodomains. MedChemComm 2013, 4, 140–144. 10.1039/C2MD20189E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney T. P. C.; Filippakopoulos P.; Fedorov O.; Picaud S.; Cortopassi W. A.; Hay D. A.; Martin S.; Tumber A.; Rogers C. M.; Philpott M.; Wang M.; Thompson A. L.; Heightman T. D.; Pryde D. C.; Cook A.; Paton R. S.; Muller S.; Knapp S.; Brennan P. E.; Conway S. J. A Series of Potent CREBBP Bromodomain Ligands Reveals an Induced-Fit Pocket Stabilized by a Cation-π Interaction. Angew. Chem., Int. Ed. 2014, 53, 6126–6130. 10.1002/anie.201402750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay D. A.; Fedorov O.; Martin S.; Singleton D. C.; Tallant C.; Wells C.; Picaud S.; Philpott M.; Monteiro O. P.; Rogers C. M.; Conway S. J.; Rooney T. P. C.; Tumber A.; Yapp C.; Filippakopoulos P.; Bunnage M. E.; Muller S.; Knapp S.; Schofield C. J.; Brennan P. E. Discovery and Optimization of Small-Molecule Ligands for the CBP/P300 Bromodomains. J. Am. Chem. Soc. 2014, 136, 9308–9319. 10.1021/ja412434f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekirnik A. R.; Hewings D. S.; Theodoulou N. H.; Jursins L.; Lewendon K. R.; Jennings L. E.; Rooney T. P. C.; Heightman T. D.; Conway S. J. Isoxazole-Derived Amino Acids Are Bromodomain-Binding Acetyl-Lysine Mimics: Incorporation into Histone H4 Peptides and Histone H3. Angew Chem Int Ed 2016, 55 (29), 8353–8357. 10.1002/anie.201602908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings L. E.; Schiedel M.; Hewings D. S.; Picaud S.; Laurin C. M. C.; Bruno P. A.; Bluck J. P.; Scorah A. R.; See L.; Reynolds J. K.; Moroglu M.; Mistry I. N.; Hicks A.; Guzanov P.; Clayton J.; Evans C. N. G.; Stazi G.; Biggin P. C.; Mapp A. K.; Hammond E. M.; Humphreys P. G.; Filippakopoulos P.; Conway S. J. BET Bromodomain Ligands: Probing the WPF Shelf to Improve BRD4 Bromodomain Affinity and Metabolic Stability. Bioorg. Med. Chem. 2018, 26, 2937–2957. 10.1016/j.bmc.2018.05.003. [DOI] [PubMed] [Google Scholar]
- Catalano M.; Moroglu M.; Balbi P.; Mazzieri F.; Clayton J.; Andrews K. H.; Bigatti M.; Scheuermann J.; Conway S. J.; Neri D. Selective Fragments for the CREBBP Bromodomain Identified from an Encoded Self-assembly Chemical Library. Chemmedchem 2020, 15, 1752–1756. 10.1002/cmdc.202000528. [DOI] [PubMed] [Google Scholar]
- Brand M.; Clayton J.; Moroglu M.; Schiedel M.; Picaud S.; Bluck J. P.; Skwarska A.; Bolland H.; Chan A. K. N.; Laurin C. M. C.; Scorah A. R.; See L.; Rooney T. P. C.; Andrews K. H.; Fedorov O.; Perell G.; Kalra P.; Vinh K. B.; Cortopassi W. A.; Heitel P.; Christensen K. E.; Cooper R. I.; Paton R. S.; Pomerantz W. C. K.; Biggin P. C.; Hammond E. M.; Filippakopoulos P.; Conway S. J. Controlling Intramolecular Interactions in the Design of Selective, High-Affinity Ligands for the CREBBP Bromodomain. J. Med. Chem. 2021, 64, 10102–10123. 10.1021/acs.jmedchem.1c00348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurin C. M. C.; Bluck J. P.; Chan A. K. N.; Keller M.; Boczek A.; Scorah A. R.; See K. F. L.; Jennings L. E.; Hewings D. S.; Woodhouse F.; Reynolds J. K.; Schiedel M.; Humphreys P. G.; Biggin P. C.; Conway S. J. Fragment-Based Identification of Ligands for Bromodomain-Containing Factor 3 of Trypanosoma Cruzi. ACS Infect. Dis. 2021, 7, 2238–2249. 10.1021/acsinfecdis.0c00618. [DOI] [PubMed] [Google Scholar]
- Gosmini R.; Nguyen V.-L.; Toum J.; Simon C.; Brusq J.-M. G.; Krysa G.; Mirguet O.; Riou-Eymard A. M.; Boursier E. V.; Trottet L.; Bamborough P.; Clark H.; Chung C.-W.; Cutler L.; Demont E. H.; Kaur R.; Lewis A. J.; Schilling M. B.; Soden P. E.; Taylor S.; Walker A. L.; Walker M. D.; Prinjha R. K.; Nicodeme E. The Discovery of I-BET726 (GSK1324726A), a Potent Tetrahydroquinoline ApoA1 Up-Regulator and Selective BET Bromodomain Inhibitor. J. Med. Chem. 2014, 57, 8111–8131. 10.1021/jm5010539. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P.; Qi J.; Picaud S.; Shen Y.; Smith W. B.; Fedorov O.; Morse E. M.; Keates T.; Hickman T. T.; Felletar I.; Philpott M.; Munro S.; McKeown M. R.; Wang Y.; Christie A. L.; West N.; Cameron M. J.; Schwartz B.; Heightman T. D.; La Thangue N.; French C. A.; Wiest O.; Kung A. L.; Knapp S.; Bradner J. E. Selective Inhibition of BET Bromodomains. Nature 2010, 468 (7327), 1067–1073. 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steegmaier M.; Hoffmann M.; Baum A.; Lénárt P.; Petronczki M.; Krššák M.; Gürtler U.; Garin-Chesa P.; Lieb S.; Quant J.; Grauert M.; Adolf G. R.; Kraut N.; Peters J.-M.; Rettig W. J. BI 2536, a Potent and Selective Inhibitor of Polo-like Kinase 1, Inhibits Tumor Growth In Vivo. Curr. Biol. 2007, 17, 316–322. 10.1016/j.cub.2006.12.037. [DOI] [PubMed] [Google Scholar]
- Brand M.; Measures A. M.; Wilson B. G.; Cortopassi W. A.; Alexander R.; Höss M.; Hewings D. S.; Rooney T. P. C.; Paton R. S.; Conway S. J. Small Molecule Inhibitors of Bromodomain-Acetyl-Lysine Interactions. ACS Chem. Biol. 2015, 10, 22–39. 10.1021/cb500996u. [DOI] [PubMed] [Google Scholar]
- Shadrick W. R.; Slavish P. J.; Chai S. C.; Waddell B.; Connelly M.; Low J. A.; Tallant C.; Young B. M.; Bharatham N.; Knapp S.; Boyd V. A.; Morfouace M.; Roussel M. F.; Chen T.; Lee R. E.; Kiplin Guy R.; Shelat A. A.; Potter P. M. Exploiting a Water Network to Achieve Enthalpy-Driven, Bromodomain-Selective BET Inhibitors. Bioorg. Med. Chem. 2018, 26, 25–36. 10.1016/j.bmc.2017.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padalino G.; Coghlan A.; Pagliuca G.; Forde-Thomas J. E.; Berriman M.; Hoffmann K. F. Using ChEMBL to Complement Schistosome Drug Discovery. Pharmaceutics 2023, 15, 1359. 10.3390/pharmaceutics15051359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulmer G. R.; Miller A. J. M.; Sherden N. H.; Gottlieb H. E.; Nudelman A.; Stoltz B. M.; Bercaw J. E.; Goldberg K. I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. 10.1021/om100106e. [DOI] [Google Scholar]
- Robinson M. W.; Hill A. P.; Readshaw S. A.; Hollerton J. C.; Upton R. J.; Lynn S. M.; Besley S. C.; Boughtflower B. J. Use of Calculated Physicochemical Properties to Enhance Quantitative Response When Using Charged Aerosol Detection. Anal. Chem. 2017, 89, 1772–1777. 10.1021/acs.analchem.6b04060. [DOI] [PubMed] [Google Scholar]
- Valkó K.; Bevan C.; Reynolds D. Chromatographic Hydrophobicity Index by Fast-Gradient RP-HPLC: A High-Throughput Alternative to Log P/Log D. Anal. Chem. 1997, 69, 2022–2029. 10.1021/ac961242d. [DOI] [PubMed] [Google Scholar]
- Bunally S.; Young R. J. The Role and Impact of High Throughput Biomimetic Measurements in Drug Discovery. ADMET DMPK 2018, 6, 74–84. 10.5599/admet.530. [DOI] [Google Scholar]
- Young R. J.; Green D. V. S.; Luscombe C. N.; Hill A. P. Getting Physical in Drug Discovery II: The Impact of Chromatographic Hydrophobicity Measurements and Aromaticity. Drug Discovery Today 2011, 16, 822–830. 10.1016/j.drudis.2011.06.001. [DOI] [PubMed] [Google Scholar]
- Inaloo I. D.; Majnooni S.; Esmaeilpour M. Superparamagnetic Fe3O4 Nanoparticles in a Deep Eutectic Solvent: An Efficient and Recyclable Catalytic System for the Synthesis of Primary Carbamates and Monosubstituted Ureas. Eur. J. Org. Chem. 2018, 2018, 3481–3488. 10.1002/ejoc.201800581. [DOI] [Google Scholar]
- Valenta V.; Bartošová M.; Protiva M. Carbamates and Ureas Derived from 11-Amino-6,11-Dihydrodibenzo[b,e]Thiepin; Synthesis and Pharmacological Screening. Collect. Czech. Chem. Commun. 1980, 45, 517–528. 10.1135/cccc19800517. [DOI] [Google Scholar]
- Furuya T.; Ritter T. Fluorination of Boronic Acids Mediated by Silver(I) Triflate. Org. Lett. 2009, 11, 2860–2863. 10.1021/ol901113t. [DOI] [PubMed] [Google Scholar]
- Furdas S. D.; Carlino L.; Sippl W.; Jung M. Inhibition of Bromodomain-Mediated Protein–Protein Interactions as a Novel Therapeutic Strategy. MedChemComm 2012, 3, 123–134. 10.1039/C1MD00201E. [DOI] [Google Scholar]
- Padmanabhan B.; Mathur S.; Manjula R.; Tripathi S. Bromodomain and Extra-Terminal (BET) Family Proteins: New Therapeutic Targets in Major Diseases. J. Biosci. 2016, 41, 295–311. 10.1007/s12038-016-9600-6. [DOI] [PubMed] [Google Scholar]
- Liu Z.; Wang P.; Chen H.; Wold E. A.; Tian B.; Brasier A. R.; Zhou J. Drug Discovery Targeting Bromodomain-Containing Protein 4. J. Med. Chem. 2017, 60, 4533–4558. 10.1021/acs.jmedchem.6b01761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujisawa T.; Filippakopoulos P. Functions of Bromodomain-Containing Proteins and Their Roles in Homeostasis and Cancer. Nat. Rev. Mol. Cell Biol. 2017, 18, 246–262. 10.1038/nrm.2016.143. [DOI] [PubMed] [Google Scholar]
- Howe K. L.; Bolt B. J.; Shafie M.; Kersey P.; Berriman M. WormBase ParaSite – a Comprehensive Resource for Helminth Genomics. Mol. Biochem. Parasitol. 2017, 215, 2–10. 10.1016/j.molbiopara.2016.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W.; Xie Y.; Ma J.; Luo X.; Nie P.; Zuo Z.; Lahrmann U.; Zhao Q.; Zheng Y.; Zhao Y.; Xue Y.; Ren J. IBS: An Illustrator for the Presentation and Visualization of Biological Sequences. Bioinformatics 2015, 31, 3359–3361. 10.1093/bioinformatics/btv362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z.; Zhang Y.; Berriman M. A Web Portal for Gene Expression across All Life Stages of Schistosoma Mansoni. bioRxiv 2018, 308213. 10.1101/308213. [DOI] [Google Scholar]
- Anderson L.; Amaral M. S.; Beckedorff F.; Silva L. F.; Dazzani B.; Oliveira K. C.; Almeida G. T.; Gomes M. R.; Pires D. S.; Setubal J. C.; DeMarco R.; Verjovski-Almeida S. Schistosoma Mansoni Egg, Adult Male and Female Comparative Gene Expression Analysis and Identification of Novel Genes by RNA-Seq. PLoS Neglected Trop. Dis. 2015, 9, e0004334 10.1371/journal.pntd.0004334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B.; Collins J. J.; Newmark P. A. Functional Genomic Characterization of Neoblast-like Stem Cells in Larval Schistosoma Mansoni. eLife 2013, 2, e00768 10.7554/eLife.00768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protasio A. V.; Tsai I. J.; Babbage A.; Nichol S.; Hunt M.; Aslett M. A.; De Silva N.; Velarde G. S.; Anderson T. J. C.; Clark R. C.; Davidson C.; Dillon G. P.; Holroyd N. E.; LoVerde P. T.; Lloyd C.; McQuillan J.; Oliveira G.; Otto T. D.; Parker-Manuel S. J.; Quail M. A.; Wilson R. A.; Zerlotini A.; Dunne D. W.; Berriman M.; Hoffmann K. F. A Systematically Improved High Quality Genome and Transcriptome of the Human Blood Fluke Schistosoma Mansoni. PLoS Neglected Trop. Dis. 2012, 6, e1455 10.1371/journal.pntd.0001455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protasio A. V.; van Dongen S.; Collins J.; Quintais L.; Ribeiro D. M.; Sessler F.; Hunt M.; Rinaldi G.; Collins J. J.; Enright A. J.; Berriman M.; Brindley P. J. MiR-277/4989 Regulate Transcriptional Landscape during Juvenile to Adult Transition in the Parasitic Helminth Schistosoma Mansoni. PLOS Neglected Trop. Dis. 2017, 11, e0005559 10.1371/journal.pntd.0005559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z.; Sessler F.; Holroyd N.; Hahnel S.; Quack T.; Berriman M.; Grevelding C. G. Schistosome Sex Matters: A Deep View into Gonad-Specific and Pairing-Dependent Transcriptomes Reveals a Complex Gender Interplay. Sci. Rep. 2016, 6, 31150. 10.1038/srep31150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colley D. G.; Wikel S. K. Schistosoma Mansoni: Simplified Method for the Production of Schistosomules. Exp. Parasitol. 1974, 35, 44–51. 10.1016/0014-4894(74)90005-8. [DOI] [PubMed] [Google Scholar]
- Whiteland H. L.; Chakroborty A.; Forde-Thomas J. E.; Crusco A.; Cookson A.; Hollinshead J.; Fenn C. A.; Bartholomew B.; Holdsworth P. A.; Fisher M.; Nash R. J.; Hoffmann K. F. An Abies Procera-Derived Tetracyclic Triterpene Containing a Steroid-like Nucleus Core and a Lactone Side Chain Attenuates in Vitro Survival of Both Fasciola Hepatica and Schistosoma Mansoni. Int. J. Parasitol.: Drugs Drug Resist. 2018, 8, 465–474. 10.1016/j.ijpddr.2018.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padalino G.; El-Sakkary N.; Liu L. J.; Liu C.; Harte D. S. G.; Barnes R. E.; Sayers E.; Forde-Thomas J.; Whiteland H.; Bassetto M.; Ferla S.; Johnson G.; Jones A. T.; Caffrey C. R.; Chalmers I.; Brancale A.; Hoffmann K. F. Anti-Schistosomal Activities of Quinoxaline-Containing Compounds: From Hit Identification to Lead Optimisation. Eur. J. Med. Chem. 2021, 226, 113823 10.1016/j.ejmech.2021.113823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteland H.; Crusco A.; Bloemberg L. W.; Tibble-Howlings J.; Forde-Thomas J.; Coghlan A.; Murphy P. J.; Hoffmann K. F. Quorum Sensing N-Acyl Homoserine Lactones Are a New Class of Anti-Schistosomal. PLoS Neglected Trop. Dis. 2020, 14, e0008630 10.1371/journal.pntd.0008630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partridge F. A.; Bataille C. J. R.; Forman R.; Marriott A. E.; Forde-Thomas J.; Häberli C.; Dinsdale R. L.; O’Sullivan J. D. B.; Willis N. J.; Wynne G. M.; Whiteland H.; Archer J.; Steven A.; Keiser J.; Turner J. D.; Hoffmann K. F.; Taylor M. J.; Else K. J.; Russell A. J.; Sattelle D. B. Structural Requirements for Dihydrobenzoxazepinone Anthelmintics: Actions against Medically Important and Model Parasites: Trichuris Muris, Brugia Malayi, Heligmosomoides Polygyrus, and Schistosoma Mansoni. ACS Infect. Dis. 2021, 7, 1260–1274. 10.1021/acsinfecdis.1c00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.-H.; Chung T. D. Y.; Oldenburg K. R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen. 1999, 4, 67–73. 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- Paveley R. A.; Mansour N. R.; Hallyburton I.; Bleicher L. S.; Benn A. E.; Mikic I.; Guidi A.; Gilbert I. H.; Hopkins A. L.; Bickle Q. D. Whole Organism High-Content Screening by Label-Free, Image-Based Bayesian Classification for Parasitic Diseases. PLoS Neglected Trop. Dis. 2012, 6, e1762 10.1371/journal.pntd.0001762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulme B. J.; Geyer K. K.; Forde-Thomas J. E.; Padalino G.; Phillips D. W.; Ittiprasert W.; Karinshak S. E.; Mann V. H.; Chalmers I. W.; Brindley P. J.; Hokke C. H.; Hoffmann K. F. Schistosoma Mansoni α-N-Acetylgalactosaminidase (SmNAGAL) Regulates Coordinated Parasite Movement and Egg Production. PLoS Pathog. 2022, 18, e1009828 10.1371/journal.ppat.1009828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcellino C.; Gut J.; Lim K. C.; Singh R.; McKerrow J.; Sakanari J. WormAssay: A Novel Computer Application for Whole-Plate Motion-Based Screening of Macroscopic Parasites. PLoS Neglected Trop. Dis. 2012, 6, e1494 10.1371/journal.pntd.0001494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roquis D.; Taudt A.; Geyer K. K.; Padalino G.; Hoffmann K. F.; Holroyd N.; Berriman M.; Aliaga B.; Chaparro C.; Grunau C.; Augusto R. de C. Histone Methylation Changes Are Required for Life Cycle Progression in the Human Parasite Schistosoma Mansoni. PLoS Pathog. 2018, 14, e1007066 10.1371/journal.ppat.1007066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taft A. S.; Norante F. A.; Yoshino T. P. The Identification of Inhibitors of Schistosoma Mansoni Miracidial Transformation by Incorporating a Medium-Throughput Small-Molecule Screen. Exp Parasitol 2010, 125 (2), 84–94. 10.1016/j.exppara.2009.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzi A.; Cosseau C.; Grunau C. Schistosoma Mansoni: Developmental Arrest of Miracidia Treated with Histone Deacetylase Inhibitors. Exp. Parasitol. 2009, 121, 288–291. 10.1016/j.exppara.2008.11.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.