

Abstract

Inhibition of methionine adenosyltransferase 2A (MAT2A) has received significant interest because of its implication as a synthetic lethal target in methylthioadenosine phosphorylase (MTAP)-deleted cancers. Here, we report the discovery of a series of 3H-pyrido[1,2-c]pyrimidin-3-one derivatives as novel MAT2A inhibitors. The selected compound 30 exhibited high potency for MAT2A inhibition and a favorable pharmacokinetic profile. Furthermore, in an HCT-116 MTAP-deleted xenograft model, compound 30 showed better in vivo potency than current clinical compound AG-270.
Keywords: Allosteric Inhibitor; Methionine Adenosyltransferase 2A (MAT2A); Methylthioadenosine Phosphorylase (MTAP); 5′-Methylthioadenosine (MTA); 3H-Pyrido[1,2-c]pyrimidin-3-one
Methionine adenosyltransferase 2A (MAT2A) is a rate-limiting enzyme in the methionine cycle that synthesizes the cofactor S-adenosylmethionine (SAM) from methionine and adenosine triphosphate (ATP), which plays an important role in cell proliferation and survival.1,2 The methylthioadenosine phosphorylase (MTAP) gene is colocated on chromosome 9p21 with the CDKN2A tumor suppressor, and homozygous codeletion of MTAP and CDKN2A occurs in approximately 15% of all human cancers, which leads to aggressive tumors with poor prognoses.3−6 MTAP is the only enzyme known to catalyze the degradation of 5′-methylthioadenosine (MTA).7 Loss of the function of MTAP results in accumulation of MTA, which inhibits the activity of protein arginine methyltransferase 5 (PRMT5). This inhibition makes the cancer cells vulnerable to further inhibition of the upstream metabolic enzyme MAT2A, which implicates MAT2A as a synthetic lethal target.8 Recent work has demonstrated that depletion of MAT2A with RNA interference leads to a selective antiproliferative effect in MTAP-deficient cancers.9−11 All of these results suggest that targeting MAT2A may provide a novel therapy for MTAP-deleted tumors.
To date, several efforts have been made to develop targeted therapies against MAT2A.11−19 In 2017, Pfizer researchers identified the first allosteric MAT2A inhibitor, PF-9366.15 However, the upregulation of MAT2A gene expression as a feedback mechanism to MAT2A inhibition blunted the cellular potency. To overcome the effect of feedback loop, Agios disclosed their in vivo tool compound AGI-25696 with improved potency relative to PF-9366, and further optimization yielded AG-270 (2), a potent and orally bioavailable MAT2A inhibitor currently in clinical development.16 Subsequently, another publication by Agios described a series of MAT2A inhibitors with differentiated chemical scaffolds. AGI-41998 (3) exhibits good activity and brain penetration.17 Recently, AstraZeneca reported their efforts to the identification of potent MAT2A inhibitors through fragment-based drug discovery:18 the chemical series were similar to the patent compounds of Ideaya Biosciences19 (Figure 1). Crystal structures of MAT2A in complex with compound 4 showed that the quinazolinone formed a bidentate interaction with Arg313 to occupy the lipophilic cryptic pocket and participate in two additional π-stacking interactions with the side chains of Phe20 and Trp274.
Figure 1.

Representative chemical structures of the MAT2A inhibitors.
Following these initial findings, our medicinal strategy was to search for a novel structural motif as the central core, which can maintain the key interaction with MAT2A. Herein, we disclose our discovery of a series of novel MAT2A inhibitors with the 3H-pyrido[1,2-c]pyrimidin-3-one scaffold.
Initially, we synthesized compounds 5 and 6, which contained pyrimidin-2(1H)-one in the bicyclic core structure. MAT2A enzymatic inhibition of compound 5 showed moderate binding potency to the target, and compound 6 with a saturated heterocycle on the left side of the bicycle was completely inactive to MAT2A. Additionally, replacing pyrimidin-2(1H)-one with the piperazin-2-one (7) resulted in a ∼10-fold drop in potency compared with 5. These results indicated that the conjugated π system in the bicyclic core is favorable for the binding with MAT2A. Subsequently, the introduction of 1,8-naphthyridin-2(1H)-one as the core (8) showed comparable potency with compound 5, while compound 9 with 6H-pyrimido[1,6-b]pyridazin-6-one motif was surprisingly inactive in this assay. To our delight, compound 10 containing the 3H-pyrido[1,2-c]pyrimidin-3-one scaffold displayed a significantly increased enzymatic activity with an IC50 of 49 nM (Figure 2).
Figure 2.

Evaluation of the bicyclic core structure and MAT2A IC50.
With the 3H-pyrido[1,2-c]pyrimidin-3-one fragment in hand, we then investigated the structure–activity relationships (SARs) around 10 for potential compounds with better profile. Initial SAR efforts started with modulating the R1 group. As revealed in Table 1, replacing the trifluoromethyl group with a methyl led to an ∼3-fold decrease of potency (11). Introduction of cyclopropyl (12) and difluoromethyl (13) resulted in a negligible impact on potency, which led us to further investigate a more synthetically accessible scaffold.
Table 1. R1 Modifications.

Relative IC50 results were obtained from a single assay run as technical duplicates.
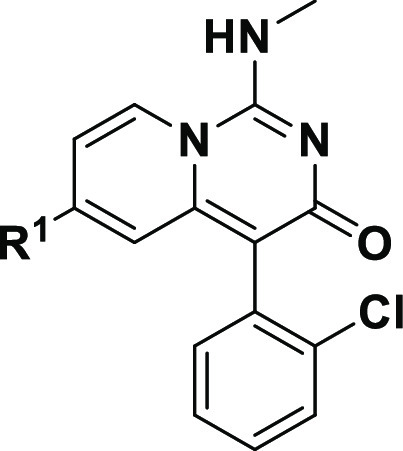
Although 10 was identified as a potent inhibitor of MAT2A, the axial chirality of the biaryl bond becomes a problem. Fast interconversion of the R- and S-atropisomers occurred when we tried to obtain pure isomers via SFC, and the calculated interconversion free energy barrier (ΔG‡) was 23 kcal/mol. Thus, our further investigation focused on exploring the SAR of R2, and the results are summarized in Table 2. Compound 14 bearing a phenyl substituent showed similar potency to 10. Introduction of a bulkier group (15) or a smaller group (16) into the ortho position of the phenyl group led to negligible impact on potency. Bis-ortho substitution (17) also showed similar enzymatic activity to 10. We next turned to probe the effect of the meta substituent. Both the 3-F (18)- and 3-methyl (19)-substituted compound showed comparable potency, while a larger group, such as −Cl (20)- and −CN (21)-substituted compounds, showed decreased enzymatic activity. Additionally, introduction of a methyl group into the para position led to ∼7-fold drop in potency against compound 10.
Table 2. SAR with R2.


Relative IC50 results were obtained from a single assay run as technical duplicates.
Further in vivo investigation of compound 16 and 18 showed that the methylamine group readily undergoes demethylation. We then focused on evaluating the SAR of the R3 group. As shown in Table 3, compound 23 bearing a secondary amine, pyrrolidine (23), was not tolerated. Replacing the methylamine group with isoproylamine (23) showed no obvious change in potency. In an effort to tune the basicity of the NH group, a fluorinated ethylamine group was introduced to give 25, which displayed a 3-fold decrease in potency. Similar enzymatic potency was observed with a variety of analogues, exemplified here by cyclobutylamino (26) and cyclopropylamino (28). Introduction of a hydroxyl group into compound 26 gave 27, which has relatively lower lipophilicity and showed slightly lower potency. These results indicated that bulkier groups were not well accommodated at R3. Interestingly, replacing the 2-fluorophenyl of 28 with 3-fluorophenyl resulted in a comparable activity with compound 18. Previous work has demonstrated that the meta position of the phenyl ring has the potential to form a weak hydrogen bond with the Gly273, which will contribute to the potency.18 We hypothesized that the introduction of an H-bond acceptor to the phenyl group would be favorable for protein binding. Thus, we replaced the phenyl group in compound 28 with pyridyl or introduced a hydroxy group at the meta position of the phenyl ring to generate 30 and 31, respectively. To our delight, both these compounds showed improved potency (3- to 4-fold) relative to compound 28.
Table 3. SAR with R2 and R3.
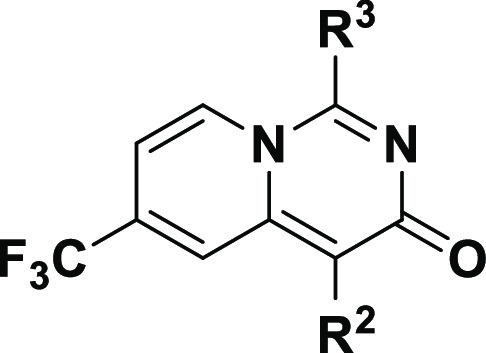

Relative IC50 results were obtained from a single assay run as technical duplicates.
To better understand the SARs of the compounds in this work, we performed molecular docking20 of compound 30 to MAT2A using a published cocrystal structure of the enzyme with small molecular inhibitor (PDB: 7BHV).18 As shown in Figure 3, 30 occupies the allosteric site with similar binding mode with the original ligand in the cocrystal structure. The pyrimidone core forms two hydrogen bonds with Arg313. The aromatic amino acids in the pocket, Try274, Phe20, Phe333 and Phe18, form π–π interaction with the aromatic rings of 30. It is worth mentioning that the N atom of flanked pyridine contacts the protein through a water molecule, which contributes to the binding affinity. The terminal cyclopropane of 30 enters a small and shallow subpocket formed by Arg313, Phe18, and Gln190. This subpocket, with its amphiphilic nature, can accommodate small substituents with comparable bioactivity but cannot bind well with bulky substituents.
Figure 3.

Predicted binding mode of compound 30 with allosteric site of MAT2A. The binding pocket is shown with electric potential surface, important residues in the binding pocket is shown in sticks. Compound 30 is shown as a red ball and stick. Hydrogen bonds are shown in red dash lines.
On the basis of the above SAR investigations, compounds 30 and 31 were dosed in male mice to determine if the pharmacokinetic (pk) profile supported in vivo use (Table 4). With an intragastric (i.g.) administration of 10 mg/kg, 30 showed a plasma drug exposure (AUC) of 34 009 ng·h/mL. The plasma drug concentration reached a maximum at 0.67 h, and the peak concentration was 16 235 ng/mL. However, with the same oral dosing, compound 31 exhibited significantly lower AUC exposure (8882 ng·h/mL) and Cmax (3957 ng/mL).
Table 4. Pharmacokinetic Properties of 30 and 31a.
| no. | 30 | 31 |
|---|---|---|
| for mice | i.g. | i.g. |
| dose (mg/kg) | 10 | 10 |
| T1/2 | 3.3 | 7.7 |
| Tmax | 0.67 | 0.33 |
| Cmax (ng/mL) | 16 235 | 3957 |
| AUC0-last (ng·h/mL) | 34 009 | 8882 |
The values are presented as mean (n = 3).
Compound 30 was further characterized for cellular assay and absorption, distribution, metabolism, and excretion (ADME) properties in vitro. As shown in Table 5, compound 30 showed potent proliferation inhibition of MTAP-deficient cells. The ADME properties of compound 30 were favorable overall, including high microsomal stability (CLint = 1.4 mL/min/kg) and high free fraction in plasma, and no major drug–drug interaction liability was detected since IC50 against four tested cytochrome P450 (CYP) enzymes was >10 μM.
Table 5. Key In Vitro Parameters of 30.
| parameter | 30 |
|---|---|
| HCT116(MTAP–/−) cell proliferation inhibition IC50 (μM) | 0.273 |
| PPB, human/dog/rat/mouse (% Fu) | 21.6/32.5/32.9/9.1 |
| CLint, (mL/min/kg)a | 1.4 |
| CYP inhibition IC50, 3A4/2C9/2C19/2D6 (μM) | all > 10 |
Intrinsic clearance rate was determined in human liver microsome.
The pk profile of compound 30 was further evaluated in rat and dog; the results are summarized in Table 6. To our delight, compound 30 exhibited a high per os (po) exposure (AUC0-last = 16 244 ng·h/mL) after 10 mg/kg administration in rat. With an oral administration of 3 mg/kg in dog, compound 30 also displayed a favorable plasma drug exposure (9312 ng·h/mL).
Table 6. Pharmacokinetic Properties of 30a.
| no. | 30 | |
|---|---|---|
| species | rat | dog |
| route | po | po |
| dose (mg/kg) | 10 | 3 |
| T1/2 | 3.0 | 2.6 |
| Tmax | 3.3 | 1.3 |
| Cmax (ng/mL) | 2823 | 1677 |
| AUC0-last (ng·h/mL) | 16 244 | 9312 |
The values are presented as mean (n = 3).
In vivo efficacy profiling of compound 30 was carried out in an HCT-116 MTAP-deleted xenograft mouse model. After 21 days of po dosing [20 mg/kg, quaque die (qd)], compound 30 induced a continuous decrease in tumor growth (TGI = 60%, Figure 4A), and significant reduction of tumor SAM concentration (79%) was detected at 10 h after the dose of day 21 (Figure 4C). Comparatively, treatment of AG-270 (50 mg/kg, qd)16 resulted in a weaker antitumor response (TGI = 43%). While compound 30 was tolerated over the course of the 21 days of treatment, the mice exhibited a maximum body weight loss of 11% (Figure 4B).
Figure 4.

In vivo efficacy of compound 30 in the HCT-116(MTAP–/−) xenograft tumor model in mice: (A) antitumor response, (B) body weight change, (C) reduction in tumor SAM.
The synthesis of compound 30 is shown in Scheme 1. Commercially available 2-fluoro-3-methylpyridine (30a) was treated with NBS to form 30b. Nucleophilic replacement of bromine with cyano yielded intermediate 30c. Pd-catalyzed coupling between 30c and 2-bromo-4-(trifluoromethyl)pyridine completed the synthesis of 30d. The treatment of intermediate 30d with concentrated sulfuric acid generated 30e. Intermediate 30f was constructed by nucleophilic attack of 30e to isocyanatocyclopropane. Finally, compound 30 was obtained by the I2-mediated cyclization of 30f.
Scheme 1. Synthetic Route of Compound 30.

Reagents and conditions: (i) NBS, benzoyl peroxide, CH3CN, 70 °C, 12 h; (ii) trimethylsilyl cyanide, K2CO3, CH3CN, 30 °C, 8 h; (iii) NIXANTPHOS, Pd(OAc)2, 1,4-dioxane, t-BuOK, 60 °C, 10 h; (iv) concd. H2SO4, 25 °C, 10 h; (v) Cs2CO3, DMF, 25 °C, 12 h; (vi) pyridine, I2, THF, 25 °C, 8 h.
In this work, we identified a fragment, 3H-pyrido[1,2-c]pyrimidin-3-one, from the screen of various bicyclic groups, which led to compound 10 that displayed potent enzymatic inhibition. Through structure–activity relationship exploration around 10, we successfully discovered compound 30 as a potent MAT2A inhibitor. Pharmacokinetic investigation in mice, rat, and dog indicated that 30 exhibited a favorable pharmacokinetic profile with high-plasma drug exposure. Compound 30 also showed favorable drug metabolism and pharmacokinetics (DMPK) properties, including high microsomal stability and low CYP P450 inhibition. Furthermore, compound 30 was identified as a promising in vivo tool that demonstrated reduction in tumor SAM levels and antitumor response in the MTAP-null HCT116 xenograft model.
Glossary
Abbreviations
- CYP
cytochrome P450
- CL
clearance
- PPB
plasma protein binding
- HCT-116
human colorectal carcinoma
- IC50
half-maximal inhibitory concentration
- po
peros
- AUC
area under curve
- TGI
tumor growth inhibition value
- SAR
structure–activity relationship
- NIXANTPHOS
4,6-bis(diphenylphosphino)-10H-phenoxazine
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.3c00488.
Experimental and characterization data for all compounds and biological data (PDF)
Author Contributions
Q.L. and Y.Z. contributed equally to this work. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Roje S. S-Adenosyl-L-methionine: beyond the universal methyl group donor. Phytochemistry 2006, 67, 1686–1698. 10.1016/j.phytochem.2006.04.019. [DOI] [PubMed] [Google Scholar]
- Pendleton K. E.; Chen B.; Liu K.; Hunter O. V.; Xie Y.; Tu B. P.; Conrad N. K. The U6 snRNA m(6)A methyltransferase METTL16 regulates SAM synthetase intron retention. Cell 2017, 169 (5), 824–835. 10.1016/j.cell.2017.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespo I.; Tao H.; Nieto A. B.; Rebelo O.; Domingues P.; Vital A. L.; Patino M. del C.; Barbosa M.; Lopes M. C.; Oliveira C. R.; Orfao A.; Tabernero M. D. Amplified and homozygously deleted genes in glioblastoma: impact on gene expression levels. PLoS One 2012, 7 (9), e46088 10.1371/journal.pone.0046088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell E. L.; Leoni L. M.; Canto M. I.; Forastiere A. A.; Iocobuzio-Donahue C. A.; Wang J. S.; Maitra A.; Montgomery E. Concordant loss of MTAP and p16/CDKN2A expression in gastroesophageal carcinogenesis: evidence of homozygous deletion in esophageal noninvasive precursor lesions and therapeutic implications. Am. J. Surg. Pathol. 2005, 29, 1497–1504. 10.1097/01.pas.0000170349.47680.e8. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Chen Z. H.; Savarese T. M. Codeletion of the genes for p16INK4, methylthioadenosine phosphorylase, interferon-α1, interferon-β1, and other 9p21 markers in human malignant cell lines Cancer Genet. Cytogenet. 1996, 86, 22–28. 10.1016/0165-4608(95)00157-3. [DOI] [PubMed] [Google Scholar]
- Barekatain Y.; Ackroyd J. J.; Yan V. C.; Khadka S.; Wang L.; Chen K. C.; Poral A. H.; Tran T.; Georgiou D. K.; Arthur K.; Lin Y. H.; Satani N.; Ballato E. S.; Behr E. I.; deCarvalho A. C.; Verhaak R. G. W.; de Groot J.; Huse J. T.; Asara J. M.; Kalluri R.; Muller F. L. Homozygous MTAP deletion in primary human glioblastoma is not associated with elevation of methylthioadenosine. Nat. Commun. 2021, 12 (1), 4228. 10.1038/s41467-021-24240-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang P. W.; Jiao J. Y.; Chen Z.; Zhu X. Y.; Cheng C. S. Keep a watchful eye on methionine adenosyltransferases, novel therapeutic opportunities for hepatobiliary and pancreatic tumours. Biochim Biophys Acta Rev. Cancer 2022, 1877 (5), 188793. 10.1016/j.bbcan.2022.188793. [DOI] [PubMed] [Google Scholar]
- Marjon K.; Cameron M. J.; Quang P.; Clasquin M. F.; Mandley E.; Kunii K.; McVay M.; Choe S.; Kernytsky A.; Gross S.; Konteatis Z.; Murtie J.; Blake M. L.; Travins J.; Dorsch M.; Biller S. A.; Marks K. M. MTAP deletions in cancer create vulnerability to targeting of the MAT2A/PRMT5/RIOK1 axis. Cell Rep. 2016, 15, 574–587. 10.1016/j.celrep.2016.03.043. [DOI] [PubMed] [Google Scholar]
- Kalev P.; Hyer M. L.; Gross S.; Konteatis Z.; Chen C. C.; Fletcher M.; Lein M.; Aguado-Fraile E.; Frank V.; Barnett A.; Mandley E.; Goldford J.; Chen Y.; Sellers K.; Hayes S.; Lizotte K.; Quang P.; Tuncay Y.; Clasquin M.; Peters R.; Weier J.; Simone E.; Murtie J.; Liu W.; Nagaraja R.; Dang L.; Sui Z.; Biller S. A.; Travins J.; Marks K. M.; Marjon K. MAT2A inhibition blocks the growth of MTAP-deleted cancer cells by reducing PRMT5-dependent mRNA splicing and inducing DNA damage. Cancer Cell 2021, 39 (2), 209–224. 10.1016/j.ccell.2020.12.010. [DOI] [PubMed] [Google Scholar]
- Kryukov G. V.; Wilson F. H.; Ruth J. R.; Paulk J.; Tsherniak A.; Marlow S. E.; Vazquez F.; Weir B. A.; Fitzgerald M. E.; Tanaka M.; Bielski C. M.; Scott J. M.; Dennis C.; Cowley G. S.; Boehm J. S.; Root D. E.; Golub T. R.; Clish C. B.; Bradner J. E.; Hahn W. C.; Garraway L. A. MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science 2016, 351, 1214–1218. 10.1126/science.aad5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavrakis K. J.; McDonald E. R. 3rd; Schlabach M. R.; Billy E.; Hoffman G. R.; deWeck A.; Ruddy D. A.; Venkatesan K.; Yu J.; McAllister G.; Stump M.; deBeaumont R.; Ho S.; Yue Y.; Liu Y.; Yan-Neale Y.; Yang G.; Lin F.; Yin H.; Gao H.; Kipp D. R.; Zhao S.; McNamara J. T.; Sprague E. R.; Zheng B.; Lin Y.; Cho Y. S.; Gu J.; Crawford K.; Ciccone D.; Vitari A. C.; Lai A.; Capka V.; Hurov K.; Porter J. A.; Tallarico J.; Mickanin C.; Lees E.; Pagliarini R.; Keen N.; Schmelzle T.; Hofmann F.; Stegmeier F.; Sellers W. R. Disordered methionine metabolism in MTAP/CDKN2A-deleted cancers leads to dependence on PRMT5. Science 2016, 351, 1208–1213. 10.1126/science.aad5944. [DOI] [PubMed] [Google Scholar]
- Li C.; Gui G.; Zhang L.; Qin A.; Zhou C.; Zha X. Overview of methionine adenosyltransferase 2A (MAT2A) as an anticancer target: structure, function, and inhibitors. J. Med. Chem. 2022, 65 (14), 9531–9547. 10.1021/acs.jmedchem.2c00395. [DOI] [PubMed] [Google Scholar]
- Atkinson S. J.; Evans L.; Scott J. S. A patent review of MAT2a inhibitors (2018–2021). Expert Opin Ther Pat 2022, 32 (10), 1043–1053. 10.1080/13543776.2022.2119127. [DOI] [PubMed] [Google Scholar]
- Zhang S.; Qing L.; Wang Z.; Zhang Y.; Li Y.; Fang H.; Liu Y.; He H. Design and Structural Optimization of Methionine Adenosyltransferase 2A (MAT2A) Inhibitors with High In Vivo Potency and Oral Bioavailability. J. Med. Chem. 2023, 66 (7), 4849–4867. 10.1021/acs.jmedchem.2c02006. [DOI] [PubMed] [Google Scholar]
- Quinlan C. L.; Kaiser S. E.; Bolanos B.; Nowlin D.; Grantner R.; Karlicek-Bryant S.; Feng J. L.; Jenkinson S.; Freeman-Cook K.; Dann S. G.; Wang X.; Wells P. A.; Fantin V. R.; Stewart A. E.; Grant S. K. Targeting S-adenosylmethionine biosynthesis with a novel allosteric inhibitor of Mat2A. Nat. Chem. Biol. 2017, 13, 785–792. 10.1038/nchembio.2384. [DOI] [PubMed] [Google Scholar]
- Konteatis Z.; Travins J.; Gross S.; Marjon K.; Barnett A.; Mandley E.; Nicolay B.; Nagaraja R.; Chen Y.; Sun Y.; Liu Z.; Yu J.; Ye Z.; Jiang F.; Wei W.; Fang C.; Gao Y.; Kalev P.; Hyer M. L.; DeLaBarre B.; Jin L.; Padyana A. K.; Dang L.; Murtie J.; Biller S. A.; Sui Z.; Marks K. M. Discovery of AG-270, a first-in-class oral MAT2A inhibitor for the treatment of tumors with homozygous MTAP deletion. J. Med. Chem. 2021, 64 (8), 4430–4449. 10.1021/acs.jmedchem.0c01895. [DOI] [PubMed] [Google Scholar]
- Li M.; Konteatis Z.; Nagaraja N.; Chen Y.; Zhou S.; Ma G.; Gross S.; Marjon K.; Hyer M. L.; Mandley E.; Lein M.; Padyana A. K.; Jin L.; Tong S.; Peters R.; Murtie J.; Travins J.; Medeiros M.; Liu P.; Frank V.; Judd E. T.; Biller S. A.; Marks K. M.; Sui Z.; Reznik S. K. Leveraging Structure-based drug design to identify next-generation MAT2A inhibitors, including brain-penetrant and peripherally efficacious leads. J. Med. Chem. 2022, 65 (6), 4600–4615. 10.1021/acs.jmedchem.1c01595. [DOI] [PubMed] [Google Scholar]
- De Fusco C.; Schimpl M.; Borjesson U.; Cheung T.; Collie I.; Evans L.; Narasimhan P.; Stubbs C.; Vazquez-Chantada M.; Wagner D. J.; Grondine M.; Sanders M. G.; Tentarelli S.; Underwood E.; Argyrou A.; Smith J. M.; Lynch J. T.; Chiarparin E.; Robb G.; Bagal S. K.; Scott J. S. Fragment-Based Design of a Potent MAT2a Inhibitor and in Vivo Evaluation in an MTAP Null Xenograft Model. J. Med. Chem. 2021, 64 (10), 6814–6826. 10.1021/acs.jmedchem.1c00067. [DOI] [PubMed] [Google Scholar]
- Alam M.; Cleary L.; Fleury M.; Pei Z.; Steel R.; Sutton J.; Knox J. E.; Newby Z. E. R.. 2-Oxoquinazoline Derivatives as Methionine Adenosyltransferase 2A Inhibitors and Their Preparation. WO2020123395, 2020.
- Glide Schrödinger Release 2022–3; Schrödinger, LLC: New York, NY, 2020.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.