

Abstract
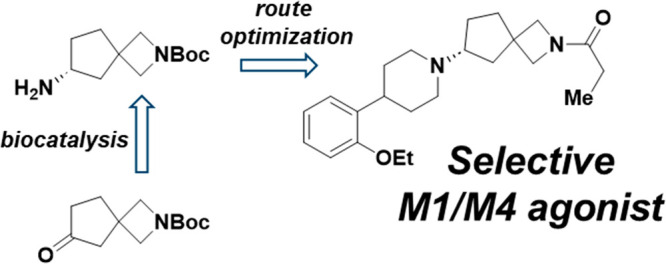
We have developed a chiral route toward the synthesis of muscarinic M4 agonists that was enabled by the biocatalytic synthesis of the key spirocyclic diamine building blocks 10 and 12. Using these bifunctional compounds we were able to optimize a synthetic sequence toward a collection of advanced intermediates for further elaboration. These advanced intermediates were then used as starting points for early medicinal chemistry and the identification of selective M1/M4 agonists.
Keywords: selective CHRM4 agonist, biocatalysis, asymmetric synthesis, muscarinic receptors, bifunctional amine building blocks, aminotransferase
The hallmarks of psychosis, frequent hallucinations and/or delusions, are most commonly associated with schizophrenia, but these symptoms are found in many other psychiatric and neurodegenerative diseases such as unipolar depression and Alzheimer’s disease.1 Current treatments to control psychosis mainly act through the D2 receptor and are associated with poor tolerability and there is consequently a pressing need for novel and mechanistically distinct antipsychotics.2 In this context, the nonselective muscarinic agonist xanomeline reduced the symptoms of psychosis in both schizophrenia3 and dementia.4 However, despite these promising clinical data, the development of xanomeline as a single agent was stopped due to intolerable gastrointestinal side effects caused by the activation of peripheral M2 and M3 receptors.
At the time of the xanomeline clinical trials, it was not clear which muscarinic receptor(s) were responsible for human efficacy. However, later mechanistic studies in transgenic mice determined that the M4 receptor is primarily responsible for the antipsychotic effect of xanomeline.5 These preclinical studies highlight M4 as a compelling, novel target and suggest that a M4 selective agonist would have the antipsychotic efficacy of xanomeline without the associated peripheral side effects of this small molecule. However, the identification of a selective muscarinic agonist has proven challenging due to the high degree of homology in the orthosteric binding pocket of the five muscarinic receptors.6 Nonetheless, we explored the potential of selectively activating M4 because of the promising clinical data for xanomeline and the high unmet need in this therapeutic area, and in this vein, we were able to identify a series of 2-azaspiro[3.4] octane7 and 5-oxa-2-azaspiro[3.4] octane8 spirocycles as M4 agonists. A discussion of the effort leading to the identification of these spirocycles will be detailed elsewhere, while this communication will highlight our efforts to develop a chiral route toward these series as well as some preliminary structure activity relationship (SAR) insights. The starting synthetic route toward our spirocylic M4 agonists is exemplified in Scheme 1. Compounds such as (R)-5 were originally produced racemically starting with an aryl halide such as 1 and vinyl piperidine boronic ester 2. Suzuki coupling followed by hydrogenation and Boc deprotection yielded intermediate 3. Next, piperidine 3 and spirocyclic ketone 4 were combined via reductive amination and subsequent chiral separation yielded (R)-5 and (S)-5. While this racemic route was initially satisfactory, we soon set our sights on a chiral route for several reasons. In order to optimize for muscarinic selectivity, every new analog synthesized had to be chirally separated, which increased the material requirements for compound synthesis as well as slowed the design, make, test, analyze cycle10 with the time required for the supercritical fluid chromatography (SFC) separation of the enantiomers. In addition, the spirocylic building blocks such as ketone 4 are relatively complex and expensive and losing half of the material to a chiral separation was not ideal from a sustainability perspective, especially as we considered potential scale ups for testing in vivo.
Scheme 1. Synthesizing (R)-5 and (S)-5 through a Racemic Route.

Reagents and conditions: (a) PdCl2(dtbpf), K3PO4, dioxane:water 110 °C, 10 min, 67%; (b) H2, Pt–C, EtOAc, 3 bar, 2.5 h, 80%; (c) TFA, DCM 10 min, quant.; (d) NaBH(OAc)3, DCM, 4 Å molecular sieves, 16 h, 83%; (e) SFC chiral separation.
Toward the development of a chiral route, we determined the stereochemistry of the less active enantiomer to be (S) as shown by the crystal structure in Figure 1 (we used the less active enantiomer for X-ray studies to conserve the more active enantiomer for biological profiling). With these data in hand, we considered potential methods to produce these molecules as single enantiomers and decided to synthesize the spirocycle as a chiral intermediate rather than attempt an asymmetric reductive amination thereby avoiding changes in enantioselectivity based on the spirocylic or piperidine substituents. Conceptually, there are two different synthetic strategies one could pursue in this vein to achieve a stereoselective synthesis. One method would be to synthesize a chiral amine on the spirocycle and use it as a nucleophile in a double substitution reaction to form the central piperidine ring, while an alternative approach would entail the generation of an activated, stereodefined alcohol derivate that could be displaced by an amine nucleophile. While a team at Pfizer chose the latter approach for a related piperazine system,9 we chose the former because we thought that this method would be the least prone to stereochemical erosion in the reaction. To transform this concept into practice, we first used Ellman auxiliary chemistry to synthesize the desired spirocylic amine, but as shown in Scheme 2, this reaction gave an equimolar amount of the two diastereomers. We were able to separate 6 and 7 by SFC to begin exploratory chemistry using these amines, but we required a superior way to access them. We therefore accelerated our investigations into an enzymatic route toward installing the chiral amine, which, from a long-term perspective, was the preferred solution given the sustainability advantages of biocatalysis.11
Figure 1.
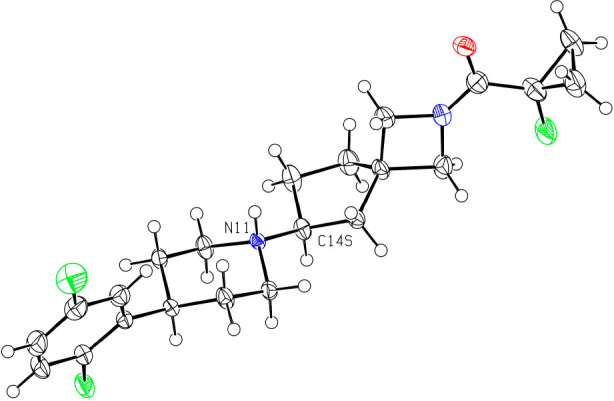
Determination of the desired absolute stereochemistry of the spirocyclic amine of the M4 agonists. The structure of (S)-5 showing the S-configuration at C14. The compound was crystallized as a besylate (protonation site: N11), anion, omitted for clarity. Ellipsoids at 50% probability, radii of hydrogen atoms arbitrary.
Scheme 2. Ellman Auxiliary Does Not Produce the Desired Amine Stereoselectivelya.

Reagents and conditions: (a) Ti(OEt)4, (S)-2-methylpropane-2-sulfinamide, 60 °C, 12 h; NaBH4, −20 → 25 °C, 62%; (b) SFC chiral separation.
To investigate the potential of enzymatic catalysis to produce the desired enantiopure amine, we screened a set of transaminases against our two spirocycles of interest.12 In addition to cyclopentyl ketone 8, we also examined the reactivity of THF ketone 9 as a related spirocycle. While the Ellman chemistry was performed on the 1-fluorocyclopropyl ketone, we instead performed the transaminase chemistry on the Boc-protected amine to give us flexibility toward the eventual azetidine substituent. A summary of the enzyme screen for spirocycles 8 and 9 is shown in Figure 2. For carbocylic ketone 8 we found the reactivity to be moderate, with conversion rates generally in the 60–80% range and with varying degrees of enantioselectivity. For example, enzyme ATA032 mildly favored the desired enantiomer 10 whereas ATA238 slightly favored the undesired enantiomer 11 but within this screen we were able to identify ATA412 which produced 10 with excellent (>99%) enantioselectivity and acceptable conversion from the ketone (77%). Compared to the carbocycle, the THF spirocycle in general was a much better transaminase substrates with higher observed conversions with the exception of ATA007 that gave poor conversion (38%). Enzymes were found that favored the desired enantiomer 12 with high conversion such as ATA013 (>99%), but this enzyme still gave ∼10% of the undesired enantiomer 13. Compared to ATA013, the conversion of 9 by ATA036 was slightly attenuated (98.6%), but the enantioselectivity toward 12 was excellent (99%). As shown in Figure 2B and 2C, the desired absolute stereochemistry of the two building blocks was confirmed by X-ray crystallography, and so ATA412 and ATA036 were taken forward for further reaction development. The results from the transaminase screen were promising, but the reactions of 8 with ATA412 and 9 with ATA036 had to be optimized to produce sufficient quantities of 10 and 12 to support analog synthesis. As part of the reaction engineering, we investigated the pH, and found the reaction to have higher conversions in the pH range of 8.5–10 with 9.5 being optimal for conversion. Enzyme loading was also examined and while the THF spirocycle 9 required only a 4% (w/w) catalyst loading to achieve >98% conversion, the less reactive cyclopentyl spirocycle 8 could only reach 75% conversion with 10% enzyme loading. Following the optimization of the reaction conditions, both transformations were scaled up to >100 g, which greatly facilitated the medicinal chemistry investigations.
Figure 2.

Enzymatic synthesis of key chiral diamine intermediates. (A) Screen of transaminases identifies promising enzymes for further reaction development. (B) Crystal structure of 10, showing the R-configuration at C2. Compound was crystallized as the hydrochloride and is conformationally disordered over three sites (ratio 0.56:0.24:0.20). Anion and minor conformations are omitted for clarity. Ellipsoids are at 50% probability, and the radii of hydrogen atoms arbitrary. (C) Crystal structure of 12, showing the S-configuration at C2. Compound was crystallized as the hydrochloride. Disordered chloride anion is omitted for clarity. Ellipsoids at 50% probability, radii of hydrogen atoms arbitrary.
With the chiral building blocks in hand, the first route we attempted using these amines is shown in Scheme 3. The key step in this route is a transamination reaction of amine 10 and N,N-dimethyl-4-oxopiperidinium iodide 14 to yield ketone 15.13 Piperidone 15 could then be reacted with organolithiums such as those produced by reacting aryl bromide 16 with n-butyl lithium to afford tertiary alcohol 17.
Scheme 3. First Generation Route Utilizing Chiral Amine 10.

Reagents and conditions: (a) K2CO3, EtOH/H2O, 60 °C, 4 h, 56%; (b) n-BuLi, THF, −78 °C, 45%.
Exposing tertiary alcohols such as 17 to acidic conditions provided the dehydration products, and the resulting olefins could be subsequently hydrogenated toward the final compounds. While this route was successful, the chiral amines were employed in the very first step of the sequence and were carried through each synthetic step, requiring relatively large quantities of the precious chiral spirocycle. In addition, the dehydration step also removed the Boc group, which was not necessarily desired, and both the dehydration and following hydrogenation gave variable yields based on the substituents on the neighboring aryl ring. Since we were actively exploring the SAR of the phenyl ring, this was another significant drawback of this approach.
Desiring a second-generation synthesis to avoid the issues of the piperidone based approach, we next designed a route to form the piperidine ring with the aryl ring preinstalled. Our first attempt at this synthetic plan is shown in Scheme 4 and begins with aldehyde 18, which is exposed to ytterbium(III) chloride and allyl trimethylsilane to generate bis-allyl 19.14 This compound was then subjected to ozonolysis to oxidatively cleave the two vinyl groups, followed by sodium borohydride reduction to yield diol 20. The two alcohols were subsequently activated as tosylates and combined with chiral amine 10 using potassium phosphate as the base to provide bifunctional intermediate 21 which can be further elaborated from either the phenol or the azetidine amine. Using this route, we were able to successfully produce gram quantities of 21, but like our first-generation route, there were some drawbacks to this synthesis. For example, the double allylation reaction is dependent on the electronics of the aryl ring, and when strongly electron withdrawing groups such as trifluoromethoxy are present, the major product is single rather than double allylation. In addition, the allylation required a relatively high loading of ytterbium(III) chloride (25 mol %) increasing the cost and decreasing the sustainability of the synthetic route. Finally, we were also concerned about potential scalability issues with ozonolytic cleavage of the two vinyl moieties. Once the diol was obtained, however, the cyclization to form the piperidine ring worked well, validating the overall synthetic strategy. Thus, for the next-generation route, we searched for an alternative method to access the key diol intermediate 20.
Scheme 4. Next Generation Routes Towards Advanced Bifunctional Intermediates.

Reagents and conditions: (a) YbCl3, MeNO2, allyltrimethylsilane, 25 °C, 72 h, 73%; (b) O3, MeOH, −78 °C 1 h; NaBH4, −78 °C → 23 °C, 16 h, 62%; (c) pTsCl, TEA, 4-DMAP, MeCN, −5 °C → 25 °C, 16 h 80%; (d) 10, K3PO4, MeCN, 90 °C, 16 h, 94%; (e) KOH, [RhCl(COD)]2, dioxane/water, 0 °C → 35 °C, 16 h, 97%; (f) LiAlH4, THF, 0 °C, 2 h, 93%; (g) NaOEt, dimethylmalonate, EtOH, 80 °C, 16 h; 52%; (h) HCl (conc) 120 °C, 16 h, 93%; (i) B2H6, THF, 16 h, 57%.
In this vein, we explored both metal-catalyzed and metal free routes to diol 20. First, we investigated the rhodium catalyzed addition of aryl boronic acids to alpha-beta unsaturated enones because of the reported flexible substrate scope of both reaction components.15−17 Beginning with 2-benzyloxy phenyl boronic acid 22 and alpha-beta unsaturated lactone 23 using cyclooctadiene rhodium chloride dimer as the catalyst we found that this reaction worked very well to provide the phenyl lactone 24 in excellent yield (87%). Reduction of the lactone with lithium aluminum hydride cleanly provided diol 20 which could then intersect with the second-generation route to provide bifunctional intermediates such as 21. Alternatively, to avoid the use of the relatively expensive rhodium catalyst, the previously described alpha-beta unsaturated ester 25(18) was reacted with diethyl malonate to yield triethyl ester 26. Decarboxylation with concentrated hydrochloric acid provided diacid 27 which could then be reduced with diborane to provide diol 28 which was employed in the piperidine formation reaction analogus to diol 20. Overall, the three different paths toward diol 20 shown in Scheme 4 enabled us to access the set of bifunctional building blocks depicted in Table 1. Using these key advanced intermediates, we were able to elaborate from both the azetidine amine and the aryl ring to optimize for muscarinic selectivity and molecular properties.
Table 1. Bifunctional Intermediates Produced Using the Chemistry Shown in Scheme 4.
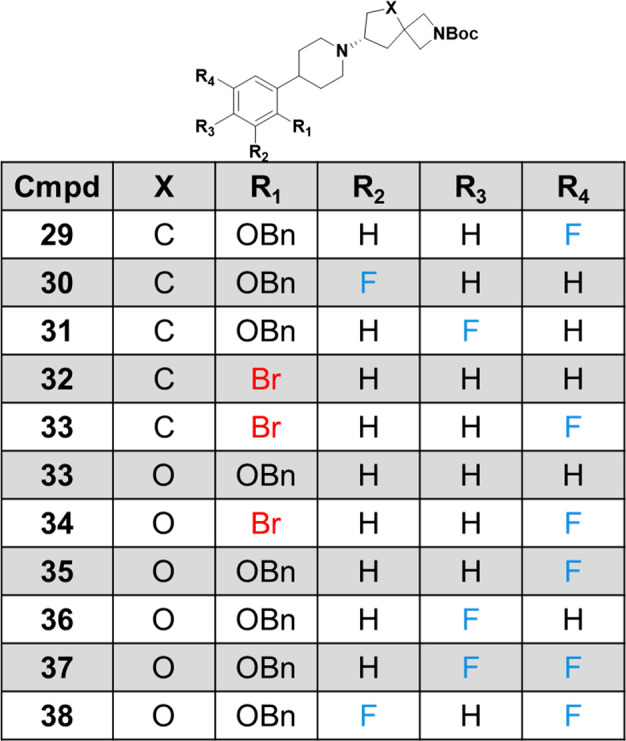
With ready access to the intermediates in Table 1, we began to explore the SAR of the carbocylic and THF spirocycles, and these early investigations are shown in Table 2. First, we found that in addition to the 1-fluorocyclopropyl amide contained in (R)-5, small amides such as methyl (40), ethyl (41), and 3-oxetane amide (42) are also potent on M4 at a similar level to xanomeline (39). Next, because the intolerability of xanomeline is due in part to the activation of M2 and M3, we also profiled the spirocycles against these receptors and found that they generally have greater selectivity than xanomeline, with several of the analogues having no agonist activity on either M2 or M3. Overall, the selectivity toward M1 was more attenuated compared to M2 or M3 though, similar to what was reported for a series of carbamate based M4 agonists.19−21 In addition to these general observations, we found that the identity of the amide affects muscarinic selectivity as shown by methyl amide 40 and ethyl amide 41 which are inactive against M2, compared to 3-oxetane amide 42, which is a weak partial agonist of M2. The effect of the amide on muscarinic selectivity is also demonstrated by methyl amide 40, which has a slight preference for M1 over M4 (19 nM vs 42 nM) that is unique to this amide. Next, we investigated the effect of substituting the phenyl ring with a fluorine, as illustrated with compounds 43–45. When a fluorine is added in the 4-position relative to the piperidine, the partial M2 agonist activity of the 3-oxetane amide is negated as shown by 44 but the activity is not significantly altered otherwise. In contrast, installing a fluorine to the five-position relative to the piperidine increases activity for M1, M2, M4 and M5, though 5-fluorine 45 remains more selective than xanomeline. Finally, we also explored the activity of THF spirocycles 46–48. Comparing the matched pairs for the carbocyclic and THF spirocycles, we found that the THF spirocycles are about 50-fold less active on M4 but that these compounds remain selective for M4 over M2, M3, and M5 with some activity on M1. From this early SAR, we see that the compounds in Table 2 are more selective than the clinically utilized muscarinic agonist xanomeline, and a more comprehensive detailing of the evolution, properties, and muscarinic activity of these two subseries will be described in the future.
Table 2. M4 Activity Data for the Initial Analogs.

Compound 43 is a single enantiomer obtained through a racemic route followed by chiral separation. The data shown are for the more potent enantiomer.
To summarize, in an effort to identify selective M4 agonists, we have developed a robust synthetic route to the chiral intermediates shown in Table 1. Beginning with the racemic synthesis depicted in Scheme 1, we determined the absolute stereochemistry of the less active enantiomer through X-ray crystallography, and to improve cycle time and reduce material requirements, we investigated methods to produce the required chiral amines and enable the synthesis of single enantiomers. To this end, we screened a series of transaminases and were able to identify enzymes that could stereoselectively produce desired spirocyclic amines 10 and 12. The reaction screening hits were then optimized for conversion and scaled up to provide the medicinal chemistry team with sufficient material for chiral route development. Using these chiral amines, we first progressed the route shown in Scheme 3 using a transamination reaction to generate piperidone 15. However, this initial route required relatively large quantities of our valuable chiral spirocycles, so we instead designed a strategy to install the amine later in the synthesis to conserve these building blocks. Thus, we established a route utilizing diol 20 that, upon activation as a bis-tosylate, could react with either amine 10 or 12 to form the piperidine ring as demonstrated by 21. This route was not ideal, however, due to limitations of the allylation and ozonolysis reactions, and we therefore developed an alternative route using rhodium catalyzed conjugate addition to yield lactone 24 en route to the key intermediate diol 20. In addition, we developed a metal-free synthesis relying on a conjugate addition-decarboxylation sequence to produce a bis-acid such as 27 as a diol precursor. These routes are a significant improvement over the beginning racemic synthesis and were successfully employed to generate the collection of compounds shown in Table 1, which greatly expedited medicinal chemistry efforts. The early SAR results shown in Table 2 demonstrate that the spirocylic amides gain significant selectivity for M4 compared with the clinical compound xanomeline. In addition, we found that the identity of the amide can affect selectivity, as demonstrated for the moderate M1 selectivity of methyl amide 40 and the weak M2 partial agonism of 3-oxetane amides 42 and 43. Next, we also found that adding a fluorine to the ring can modulate both the activity and selectivity of these compounds. Finally, comparing the carbocylic and THF spirocycles we see that THF spirocycles are less active than the matched carbocyclic analogues. Given that the clinical intolerability of xanomeline as a single agent was due to the activation of the M2 and M3 receptors, the selectivity data for the compounds depicted in Table 2 are compelling. From a strategic perspective, these results clearly demonstrate the value of investing early in the synthesis of chiral intermediates compared to relying on the asymmetric separation of final compounds to enhance compound throughput and improve sustainability by avoiding the synthesis of undesired stereoisomers. Furthermore, the production of 10 and 12 shows the utility of transaminase chemistry to access unique chiral amines that are difficult to access through other means such as Ellman auxiliary chemistry. For these particular bifunctional amines, we predict that 10 and 12 will have broad applications as structurally complex chiral building blocks for drug design.
Acknowledgments
We thank Philippe Piechon, Lauren Connor, and Trixie Wagner for assistance with the small molecule X-ray studies and our Wuxi collaborators for synthetic chemistry support. We also thank Kaila Margrey for careful review of the manuscript.
Glossary
Abbreviations
- SAR
structure activity relationship
- SFC
supercritical fluid chromatography
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.3c00331.
Detailed experimental procedures, analytical data for compounds tested in vitro and small molecule X-ray crystallographic data (PDF)
Author Present Address
# Pharmaron UK Ltd., Innovation Park, West Cl, Hertford Road, Hoddesdon EN11 9FH, United Kingdom
Author Present Address
□ Vertex Pharmaceuticals, 50 Northern Avenue, Boston, MA 02210, United States
Author Present Address
○ Relay Therapeutics, 399 Binney Street, Suite 2, Cambridge, Massachusetts 02142, United States
Author Present Address
△ Karuna Therapeutics, 99 High St Floor 26, Boston, MA 02110
Author Present Address
▽ Theseus Pharmaceuticals, 314 Main St Suite 04–200, Cambridge MA, 02142
Author Present Address
■ Eisai Co Ltd., 35 Cambridgepark Dr, Cambridge, MA 02140
Author Present Address
● Octet Medical 13400 Sabre Springs Parkway, San Diego CA 92128
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare the following competing financial interest(s): At the time of the work, all authors were employees of Novartis AG, which has filed patents on selective M4 agonists for the treatment of psychosis.
Supplementary Material
References
- Arciniegas D. B. Psychosis. Continuum 2015, 21, 715–736. 10.1212/01.CON.0000466662.89908.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neef J.; Palacios D. S. Progress in mechanistically novel treatments for schizophrenia. RSC Med. Chem. 2021, 12, 1459–1475. 10.1039/D1MD00096A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekhar A.; Potter W. Z.; Lightfoot J.; Lienemann J.; Dubé S.; Mallinckrodt C.; Bymaster F. P.; McKinzie D. L.; Felder C. C. Selective Muscarinic Receptor Agonist Xanomeline as a Novel Treatment Approach for Schizophrenia. Am. J. Psychiatry 2008, 165, 1033–1039. 10.1176/appi.ajp.2008.06091591. [DOI] [PubMed] [Google Scholar]
- Bodick N. C.; Offen W. W.; Levey A. I.; Cutler N. R.; Gauthier S. G.; Satlin A.; Shannon H. E.; Tollefson G. D.; Rasmussen K.; Bymaster F. P.; Hurley D. J.; Potter W. Z.; Paul S. M. Effects of Xanomeline, a Selective Muscarinic Receptor Agonist, on Cognitive Function and Behavioral Symptoms in Alzheimer Disease. Arch Neurol. 1997, 54, 465–473. 10.1001/archneur.1997.00550160091022. [DOI] [PubMed] [Google Scholar]
- Woolley M. L.; Carter H. J.; Gartlon J. E.; Watson J. M.; Dawson L. A. Attenuation of amphetamine-induced activity by the non-selective muscarinic receptor agonist, xanomeline, is absent in muscarinic M4 receptor knockout mice and attenuated in muscarinic M1 receptor knockout mice. Eur. J. Pharmacol. 2009, 603, 147–149. 10.1016/j.ejphar.2008.12.020. [DOI] [PubMed] [Google Scholar]
- Thal D. M.; Sun B.; Feng D.; Nawaratne V.; Leach K.; Felder C. C.; Bures M. G.; Evans D. A.; Weis W. I.; Bachhawat P.; Kobilka T. S.; Sexton P. M.; Kobilka B. K.; Christopoulos A. Crystal Structures of the M1 and M4Muscarinic Acetylcholine Receptors. Nature 2016, 531, 335–340. 10.1038/nature17188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calhoun A.; Chen X.; Gardinier K. M.; Hall E. C.; Jendza K.; Labbé-Giguere N.; Neef J.; Palacios D. S.; Qian M.; Shultz M. D.; Thomson C. G.; Wang K. Y.; Yang Fan. 2-azaspiro[3.4]octane derivatives as M4 agonists. U.S. Patent 2021/0107889, April 15, 2021.
- Calhoun A.; Chen X.; Gardinier K. M.; Hall E. C.; Jendza K.; Labbé-Giguere N.; Neef J.; Palacios D. S.; Qian M.; Shultz M. D.; Thomson C. G.; Wang K. Y.; Yang F.. 2-azaspiro[3.4]octane derivatives as M4 agonists. U.S. Patent 2021/0130365, May 6, 2021.
- Zhang L.; LaChapelle E. A.; Butler C. R.; Kablaoui N. M.; Brodney M. A.; McAllister L. A.; Yang Q.; Helal C. J.; Webb D.. Piperazine azaspiro derivatives. U.S. Patent 2021/0024497, January 28, 2021.
- Andersson S.; Armstrong A.; Björe A.; Bowker S.; Chapman S.; Davies R.; Donald C.; Egner B.; Elebring T.; Holmqvist S.; Inghardt T.; Johannesson P.; Johansson M.; Johnstone C.; Kemmitt P.; Kihlberg J.; Korsgren P.; Lemurell M.; Moore J.; Pettersson J. A.; Pointon H.; Pontén F.; Schofield P.; Selmi N.; Whittamore P. Making medicinal chemistry more effective-application of Lean Sigma to improve processes, speed and quality. Drug Discovery Today 2009, 14, 598–604. 10.1016/j.drudis.2009.03.005. [DOI] [PubMed] [Google Scholar]
- Sheldon R. A.; Woodley J. M. Role of Biocatalysis in Sustainable Chemistry. Chem. Rev. 2018, 118, 801–838. 10.1021/acs.chemrev.7b00203. [DOI] [PubMed] [Google Scholar]
- Gomm A.; O’Reilly E. Transaminases for chiral amine synthesis. Curr. Opin. Chem. Biol. 2018, 43, 106–112. 10.1016/j.cbpa.2017.12.007. [DOI] [PubMed] [Google Scholar]
- Kuehne M. E.; Muth R. S. Total syntheses of yohimbe alkaloids, with stereoselection for the normal, allo, and 3-epiallo series, based on annelations of 4-methoxy-1,2-dihydropyridones. J. Org. Chem. 1991, 56, 2701–2712. 10.1021/jo00008a025. [DOI] [Google Scholar]
- Fang X.; Watkin J. G.; Warner B. P. Ytterbium trichloride-catalyzed allylation of aldehydes with allyltrimethylsilane. Tetrahedron Lett. 2000, 41, 447–449. 10.1016/S0040-4039(99)02090-0. [DOI] [Google Scholar]
- Sakai M.; Hayashi H.; Miyaura N. Rhodium-Catalyzed Conjugate Addition of Aryl- or 1-Alkenylboronic Acids to Enones. Organometallics 1997, 16, 4229–4231. 10.1021/om9705113. [DOI] [Google Scholar]
- Edwards H. J.; Hargrave J. D.; Penrose S. D.; Frost C. G. Synthetic applications of rhodium catalysed conjugate addition. Chem. Soc. Rev. 2010, 39, 2093–2105. 10.1039/b919762c. [DOI] [PubMed] [Google Scholar]
- Tian P.; Dong H.-Q.; Lin G.-Q. Rhodium-Catalyzed Asymmetric Arylation. ACS Catal. 2012, 2, 95–119. 10.1021/cs200562n. [DOI] [Google Scholar]
- Calder E. D. D.; McGonagle F. I.; Harkiss A. H.; McGonagle G. A.; Sutherland A. Preparation of Amino-Substituted Indenes and 1,4-Dihydronaphthalenes Using a One-Pot Multireaction Approach: Total Synthesis of Oxybenzo[c]phenanthridine Alkaloids. J. Org. Chem. 2014, 79, 7633–7648. 10.1021/jo5014492. [DOI] [PubMed] [Google Scholar]
- Sumiyoshi T.; Enomoto T.; Takai K.; Takahashi Y.; Konishi Y.; Uruno Y.; Tojo K.; Suwa A.; Matsuda H.; Nakako T.; Sakai M.; Kitamura A.; Uematsu Y.; Kiyoshi A. Discovery of Novel N-Substituted Oxindoles as Selective M1 and M4 Muscarinic Acetylcholine Receptor Partial Agonists. ACS Med. Chem. Lett. 2013, 4, 244–248. 10.1021/ml300372f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai K.; Inoue Y.; Konishi Y.; Suwa A.; Uruno Y.; Matsuda H.; Nakako T.; Sakai M.; Nishikawa H.; Hashimoto G.; Enomoto T.; Kitamura A.; Uematsu Y.; Kiyoshi A.; Sumiyoshi T. Discovery of N-substituted 7-azaindoline derivatives as potent, orally available M1 and M4 muscarinic acetylcholine receptors selective agonists. Bioorg. Med. Chem. Lett. 2014, 24, 3189–3193. 10.1016/j.bmcl.2014.04.085. [DOI] [PubMed] [Google Scholar]
- Uruno Y.; Konishi Y.; Suwa A.; Takai K.; Tojo K.; Nakako T.; Sakai M.; Enomoto T.; Matsuda H.; Kitamura A.; Sumiyoshi T. Discovery of dihydroquinazolinone derivatives as potent, selective, and CNS-penetrant M1 and M4 muscarinic acetylcholine receptor agonists. Bioorg. Med. Chem. Lett. 2015, 25, 5357–5361. 10.1016/j.bmcl.2015.09.032. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.