

Abstract

Casitas B-lineage lymphoma proto-oncogene-b (Cbl-b) is a RING finger E3 ligase that is responsible for repressing T-cell, natural killer (NK) cell, and B-cell activation. The robust antitumor activity observed in Cbl-b deficient mice arising from elevated T-cell and NK-cell activity justified our discovery effort toward Cbl-b inhibitors that might show therapeutic promise in immuno-oncology, where activation of the immune system can drive the recognition and killing of cancer cells. We undertook a high-throughput screening campaign followed by structure-enabled optimization to develop a novel benzodiazepine series of potent Cbl-b inhibitors. This series displayed nanomolar levels of biochemical potency, as well as potent T-cell activation. The functional activity of this class of Cbl-b inhibitors was further corroborated with ubiquitin-based cellular assays.
Keywords: Cbl-b, T-cell activation, immuno-oncology, benzodiazepines, E3 ligases
One of the major pathways cancer cells utilize to circumvent cell death is to avoid detection by the body’s immune system.1 In order to avoid immune cells such as T-cells or natural killer (NK) cells, cancer cells can display immune checkpoint ligands to bind to immune cells and effectively “turn off” immune response.2 Two of the more understood immune checkpoint binding pairs that have been studied to date are the PD-1/PD-L13,4 and CTLA-4/B74 circuits. In an effort to develop therapeutics to prevent inhibitory signals through these receptors, immune checkpoint inhibitors (ICIs) have been discovered that can bind to the various proteins PD-1, PD-L1, and CTLA-4 for example, which helps activate T-cells for targeted cancer cell killing.5 While this strategy is a valuable therapy platform for various cancer types, several drawbacks to these therapeutics have emerged.6 For one, ICIs are not a general approach for all cancer types, where certain pancreatic and prostate cancers have been shown to be largely resistant to checkpoint-based immunotherapy.7 Additionally, clinical data have shown significant variability in response rate among cancer patients, for reasons that are not fully understood.7,8 Lastly, even among patients who do respond well to ICIs, different resistance mechanisms have been reported after prolonged dosing in some patients.6−8
While there are ongoing efforts toward optimization of these ICIs,9 there is also active research to discover new molecular targets with unique mechanisms to the current ICIs, which could help further drive the immune system toward more efficient elimination of tumor. One such area that has been investigated involves the ubiquitination of proteins by E3 ligases as a novel regulatory mechanism in innate and adaptive immunity.10 Among these E3-ligases, Cbl-b, a member of the Cbl family of RING-type E3 ubiquitin ligases, has demonstrated a key role in regulating effector T-cell function.11 In particular, gene-targeting approaches have shown that Cbl-b negatively regulates T-cell activation.12 It does so through the ubiquitination of proteins downstream of T-cell receptor (TCR) signaling such as ZAP7013 and PLCγ,14,15 thereby targeting them for proteasomal degradation. Cbl-b also regulates T-cell activation through ubiquitination of proteins downstream of CD28 co-stimulation13 The most notable substrate for Cbl-b along this pathway is p85,16 the regulatory subunit of the PI3K protein, which plays a critical role in recruiting and stimulating CD28, a T-cell surface protein.17 Co-stimulatory signals through CD28 lower the threshold for T-cell activation, allowing for a more sensitive immune response.18 As such, evidence for T-cell activation through functional inhibition of Cbl-b has been shown in knockout (KO) mouse models, demonstrating that genetic deletion of Cbl-b resulted in hyperactive T-cells and inhibition of tumor growth.19 Therefore, discovery of small molecule inhibitors of Cbl-b could offer an alternative mechanistic approach to cancer immunotherapy, which could potentially treat patient populations that are nonresponsive to ICI therapies.20,21 In addition, combination of a Cbl-b inhibitor with an ICI could have potential in overcoming resistance mechanisms, which would improve the standard of care for certain types of cancers.20
Cbl-b, like other members of the Cbl family, contains all of the structural features required for E3 ubiquitin ligase activity in a highly conserved N-terminal region.22,23 This region is composed of a substrate binding (TKB, tyrosine kinase-binding) domain and a RING domain, connected by a linker helix region (LHR) (Figure 1). Within the LHR is contained a strictly conserved tyrosine residue (Tyr363) that is considered the master regulator of the protein’s E3 ligase activity.24 In the unphosphorylated state, the LHR binds together the TKB and RING domains in a closed, inactive conformation with Tyr363 buried from solvent at the domain interface.24,25 While in this state, the RING domain of Cbl-b cannot recruit E2 conjugating enzymes, rendering the protein unable to ubiquitinate proteins.23 When Tyr363 is phosphorylated, the LHR no longer can bring together the TKB and RING domains in the closed conformation. Instead, phosphorylated Cbl-b adopts an open, active conformation that allows the RING domain and phospho-Tyr363 to recruit and activate ubiquitin-charged E2s, leading to targeted protein ubiquitination.26,27
Figure 1.
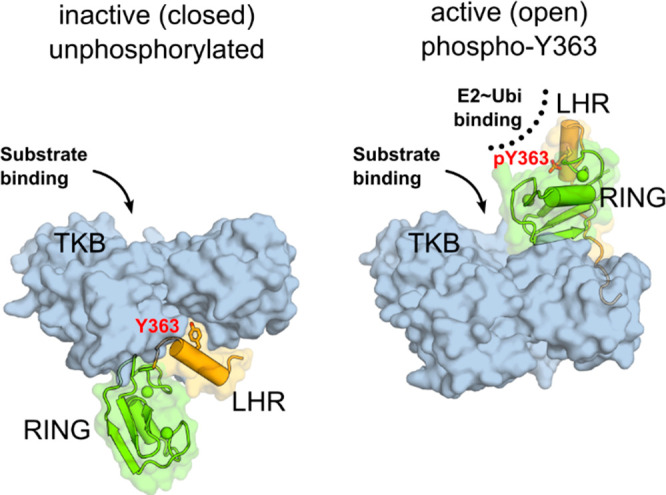
Cbl-b regulation by phosphorylation of Y363. Left: Unphosphorylated, inactive Cbl-b adopts a “closed” conformation with Y363, shown as sticks, buried from solvent. The linker helix region (LHR, orange) and the RING domain (green) are sequestered away from the substrate binding site on the TKB domain (blue surface). Right: Phosphorylation of Y363 induces the active (open) conformation, with the RING domain and phospho-Y363 in the LHR poised to recruit and activate ubiquitin-charged E2 conjugating enzyme (PDB code 3ZNI).28
Given that Cbl-b has both an active and inactive state, there may be multiple inhibitor binding sites that can be targeted, which increases the odds of therapeutic approaches to this target.25 Thus, Cbl-b inhibition becomes an exciting prospect in developing novel chemical matter in immuno-oncology. Nurix first disclosed a Cbl-b inhibitor in 2019 and has since begun clinical trials on NX-160729,30 (structure not disclosed). Since then, others have published patents on similar chemical matter31,32 (Figure 2). Herein, we disclose our efforts toward the development of a novel series of benzodiazepines as Cbl-b inhibitors to enhance antitumor immunity.
Figure 2.

Representative Cbl-b inhibitors from literature.
Our hit-finding approach began with a high-throughput screening (HTS) campaign against Cbl-b. An HTS library consisting of ∼1.8 million compounds was initially screened in a TR-FRET probe displacement assay, followed by orthogonal validation through fluorescence polarization measurements with the same probe. This cascade resulted in 581 confirmed hits, whose binding was further characterized by surface plasmon resonance (SPR). Through these efforts, benzodiazepine (BDZ) compound 1 was identified as a potent hit from this screening cascade with an SPR Kd of 0.40 μM and a TR-FRET IC50 of 0.21 μM for Cbl-b (Table 1, calculated Ki values for all compounds are found in Supporting Information). As compound 1 already displayed good binding affinity for Cbl-b, it provided a strong starting point for further optimization as a novel series of Cbl-b binders.
Table 1. Validation of Initial Hit 1 as a Cbl-b Inhibitor.

IC50 binding values for Cbl-b are based on a TR-FRET probe displacement assay and are the average of at least three determinations.
log D measured via shake-flask method in octanol and water at pH 7.4.
LLE: p(IC50) – log D.
Compound 1 was measured by SPR to have a Kd of 0.40 μM. All SPR measurements are the average of at least two determinations.
In order to determine the binding mode of compound 1, we solved a crystal structure in complex with truncated Cbl-b protein containing the substrate binding (TKB) domain, the linker helix region, and the RING domain (Figure 3A). The structure shows Cbl-b in the inactive, closed conformation, with the compound interacting with the TKB domain and the linker helix region. Although compound 1 is racemic, only the (S)-isomer was found to bind to the protein. A key interaction seen with the benzodiazepine core is a bidentate hydrogen bond (HB) contact between the internal amide of the azepine ring with the backbone of Phe263. Both the rotatable and fused phenyl rings do not seem to be making any notable interactions. Moreover, the fused phenyl ring is solvent exposed, making it an ideal area for modulation of physicochemical properties without affecting the binding potency. The other key interactions are seen with the morpholine ring. The morpholine nitrogen appears to be protonated and participates as a hydrogen bond donor (HBD), interacting with the backbone carbonyl of Ser218. In addition, the morpholine oxygen functions here as a hydrogen bond acceptor (HBA), interacting with the phenol of Tyr260. Interestingly, the morpholine oxygen is also in close proximity (3.8 Å) to the critical Tyr363, which suggested that shortening this distance could lead to a beneficial HB contact.
Figure 3.
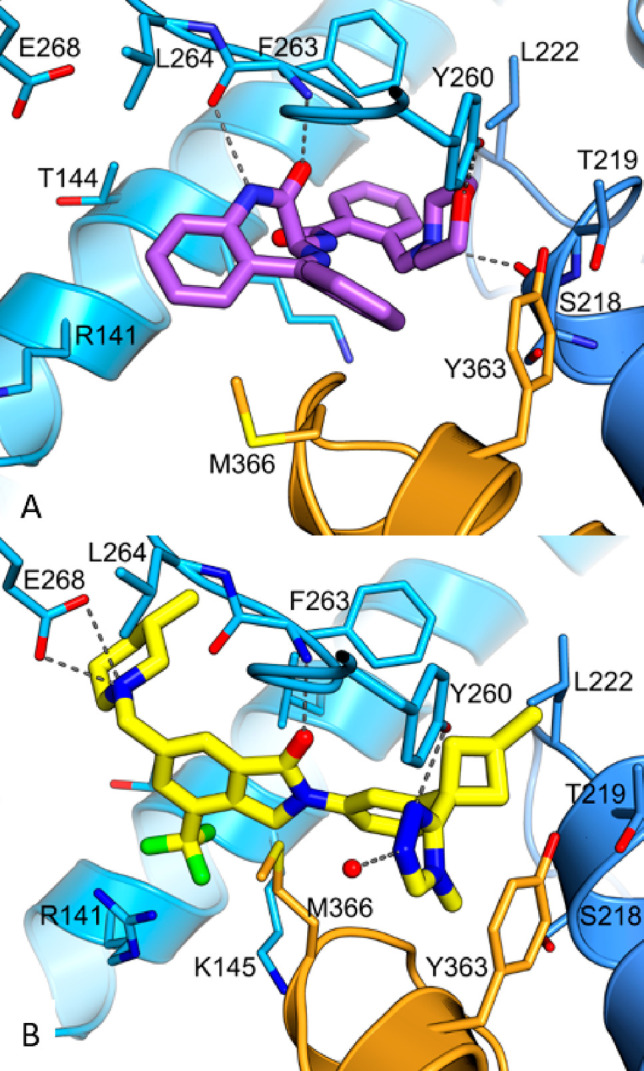
(A) Crystal structure of compound 1 bound to the TKB/RING domain of Cbl-b (PDB code 8QNG). Compound 1 is colored purple, and the protein is in blue, with the linker helix region highlighted in orange. Hydrogen bonds are shown as dashed lines. (B) Crystal structure of Ex23 bound to the TKB/RING domain of Cbl-b (PDB code 8QNH). Ex23 is in yellow, and the protein is in blue, with the linker helix region highlighted in orange. Hydrogen bonds/ion-pair interactions are shown as dashed lines. Ex23 has recently been crystallized in another laboratory to show the same binding mode.34
Seeking to evaluate the binding mode of compound 1 in relation to a known Cbl-b inhibitor, a representative compound, example 23 (Ex23) from patent WO2020264398, was chosen as the exemplar molecule (Figure 2). Ex23 was synthesized and tested internally to give an SPR Kd of 0.003 μM and a TR-FRET IC50 of 0.009 μM for Cbl-b. It was then cocrystallized with the same truncated Cbl-b protein (Figure 3B).33Ex23 also binds in the inactive closed conformation and showcases a HB contact with Tyr260 using a 1,2,4-triazole in similar fashion to the morpholine in compound 1. While it also engages in an HB with Phe263, it is only a single interaction compared to the bidentate interaction seen with compound 1. There are also some clear differences in binding modes between the two compounds, which is noteworthy, as all previously published Cbl-b inhibitors share a high degree of chemical similarity to Ex23. One is the additional ion-pair interaction between the piperidine of Ex23 with a solvent exposed Glu268, which points the chiral methyl group of the piperidine ring into a hydrophobic pocket near the vicinity of Leu264. This may account for the observed boost in potency compared to compound 1. Another major difference seems to come from the positioning of the methionine on the LHR. Compound 1 has a phenyl ring pointed directly out into the LHR, which leads to a conformational shift of Met366 as compared to the structure seen with Ex23, where the Met366 side chain is within ∼4 Å to the isoindolone core. While it is unclear how this conformational change of LHR will affect functional inhibition of Cbl-b, this illustrates clearly that binding to Cbl-b operates in an induced fit model.
Following the analysis of Figure 3A, a small number of compounds were synthesized to answer key questions about the molecule’s binding pose, as shown in the crystal structure. To better understand the effect of BDZ stereochemistry on binding to Cbl-b, both the pure (S)-isomer 2 and the pure (R)-isomer 3 were synthesized via chiral purification of rac-compound 1 (Table 1). The (S)-isomer 2 was shown to be the more potent isomer with a binding IC50 of 94 nM. A crystal structure of 2 was also obtained for absolute stereochemistry, showing the same pose as the crystal structure of compound 1 (see Supporting Information for further details). The (R)-isomer 3 was shown to be a weaker binder at 8 μM, and no crystal structure could be obtained. The observed difference in potency between the two isomers is hypothesized to result from the difficulty of the (R)-isomer morpholine to adopt the desired conformation for the HB to Tyr260. To provide evidence that the morpholine pharmacophore is critical for binding, compound 4 was synthesized, showing that without the oxygen present as an HBA, potency is completely lost. Additionally, shortening the benzyl morpholine to the corresponding aniline derivative 5 was also shown to be inactive, suggesting that precise positioning of the morpholine from the BDZ core is essential for the HB to take place.
After validation of compound 1 was concluded, a strategy was formulated to explore the structure–activity relationship (SAR) of this scaffold (Figure 4).
Figure 4.
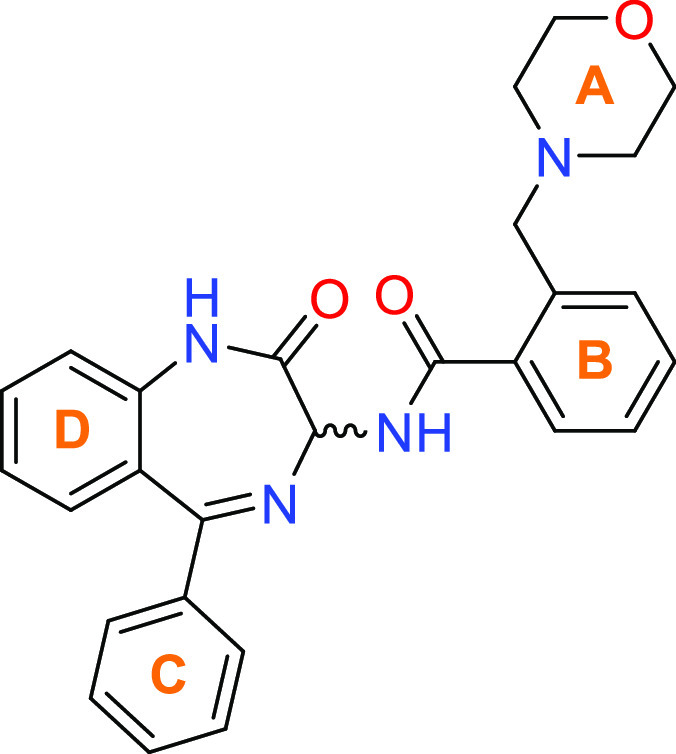
Overview of SAR strategy for benzodiazepines via exploration of A–D rings.
Initial exploration around the A-ring centered mostly around substituted morpholines and morpholine replacements (Table 2). Addition of a methyl group near the morpholine nitrogen, as seen in compounds 6 and 7 led to a reduction in Cbl-b binding affinity compared to initial hit 1. Based on the crystal structure of 1, the introduced methyl group may be sterically disrupting the HB of the morpholine N to Ser218. Addition of a methyl group near the morpholine oxygen, however, afforded a pronounced improvement in binding affinity, with a >6-fold lower IC50 for both compounds 8 and 9, with a modest increase in LLE compared to 1. One explanation for the observed binding improvement could be attributed to the methyl group picking up favorable hydrophobic interactions deeper in the binding pocket. In addition, the methyl group may also be inductively enhancing the HBA ability of the morpholine oxygen. Other methyl substitution patterns, such as those shown in compounds 10 and 11, lose potency with respect to 1, suggesting that the binding space surrounding the morpholine may be limited. Attempts to find other ring types outside of morpholine were less successful, with bicyclic scaffolds 12–14 being the only systems that showed sub-μM binding to Cbl-b.
Table 2. Investigation of A-Ring Variations.

IC50 binding values for Cbl-b are based on a TR-FRET probe displacement assay and are the average of at least three determinations.
log D measured via shake-flask method in octanol and water at pH 7.4.
LLE: p(IC50) – log D.
Exploration of the B- and C-rings was conducted next, where we found that far fewer substitutions were tolerated in these positions (Table 3). With more emphasis on finding substitutions that could maintain potency and lower log D and thus improve LLE, introduction of more polar groups was attempted. Changing the B-ring from a phenyl ring to a pyridine ring shown in compound 15 led to a loss in potency compared to 1, albeit with a reduction in log D. Five-membered rings such as furan 16 and thiophene 17 were tolerated and reduced log D but did not improve potency compared to compound 1. Changes to the C-ring aimed at improving LLE were less tolerated, with only compound 18 showing modest potency at 2.0 μM. Pyridone compounds 19 and 20, while substantially lowering log D and maintaining LLE compared to 8 and 9, lost significant amounts of potency.
Table 3. Investigation of B- and C-Ring Variations.

IC50 binding values for Cbl-b are based on a TR-FRET probe displacement assay and are the average of at least three determinations.
log D measured via shake-flask method in octanol and water at pH 7.4.
LLE: p(IC50) – log D.
After exploring substitutions for A–C rings, the most potent compounds 8 and 9 were then purified by chiral chromatography to test the more active (S)-isomers of these structures (Table 4). As expected, compounds 21 and 22 both showed equally high levels of potency in the TR-FRET assay, reaching 10 and 13 nM, respectively. In an effort to improve the LLE through lowering of log D, we hypothesized that addition of polar substituents on the D-ring would be a good avenue for modulating log D without harming potency, as the D-ring is largely solvent exposed. Linear and cyclic amides shown in compounds 23 and 24 were well tolerated in the TR-FRET assay, and both led to increases in LLE, especially for compound 24 with a log D of 2.5 and LLE of 5.4. Simple D-ring modification from a phenyl to pyridyl ring shown in compound 25 also maintained potency while improving log D and LLE profiles.
Table 4. Modulation of log D through D-Ring Exploration.

IC50 binding values for Cbl-b are based on a TR-FRET probe displacement assay and are the average of at least three determinations.
log D measured via shake-flask method in octanol and water at pH 7.4.
LLE: p(IC50) – log D.
With a cohort of potent Cbl-b binders in compounds 21–25, functional potency was then tested in a human T-cell IL-2 secretion assay (Figure 5). Interestingly, while all five compounds were similarly potent in the TR-FRET assay, the dose–response curves in the T-cell assay were not similar. All five compounds showed IL-2 production at lower concentrations, although compounds 21 and 22 showed a decrease in IL-2 production at the top concentration (10 μM), leading to bell-shaped curves. Meanwhile compounds 23–25 gave dose–response curves that showed robust IL-2 production. Compounds 23 and 24 showed low-μM EC50 in the T-cell assay, and to our delight, compound 25 showed sub-μM EC50 at 0.41 μM.
Figure 5.

Dose–response curves of selected compounds in a T-cell IL-2 secretion assay. Corresponding T-cell EC50 values are shown on the right as an average of at least three determinations, along with the binding affinity values for off-target CCKA as an average of at least two determinations. *EC50 values calculated are based on fitting to a sigmoidal curve.
Further investigation to better understand why IL-2 production decreased at higher concentrations for compounds 21 and 22 revealed these compounds to be cytotoxic, a finding not observed in cells treated with 23–25. One hypothesis to explain this observation is that off-target activity of compounds 21 and 22 could be leading to T-cell cytotoxicity. As part of our routine safety profiling, compounds 22 and 23 were tested in a panel of 85 distinct in vitro secondary pharmacology targets consisting of enzymes, membrane transporters, G-protein-coupled receptors (GPCRs), kinases, enzymes, nuclear hormone receptors, and ion channels (data generated at CEREP, France), to determine binding affinities for any off-targets. A correlation was found between the functional T-cell activity of these compounds and the binding affinity for the protein cholecystokinin A (CCKA) (Figure 5), leading to compounds 21, 24, and 25 being tested at this focused target. While binding of compounds 23–25 to CCKA showed very weak to no affinity (ranging from 24 to >100 μM), compounds 21 and 22 showed more significant binding affinity at 3.7 and 2.6 μM, respectively. These data accord with reports of CCKA as a regulator in T-cell function, suggesting that agonism of this protein could lead to loss of T-cell function/cell death.35,36 While this is an interesting correlation, further studies would be required to prove causation.
Intrigued by the cellular potency of 25, a cocrystal structure of the compound with Cbl-b was obtained (Figure 6A). Unsurprisingly, compound 25 shares many of the same interactions as compound 1, which are the HB between the morpholine oxygen and Tyr260, as well as the bidentate interaction with the benzodiazepine core amide and Phe263. The fused pyridyl ring designed to modulate physicochemical properties and maintain potency is solvent exposed and binds as expected. Interestingly, a weak HB is also seen between the morpholine oxygen and the critical Tyr363 at 3.5 Å distance compared to 3.8 Å seen in compound 1, which could account for the boost in functional activity. Notably, there was no HB interaction between the morpholine N and Ser218 as seen in Figure 3A. Instead, the morpholine N is observed to be locking the bioactive conformation through an intramolecular hydrogen bond between it and the exocyclic amide NH. Lastly, the chiral methyl group, which confers significant potency in this series, appears to be pointing away from the rest of the molecule, fitting into a hydrophobic portion of the binding pocket.
Figure 6.

(A) Crystal structure of compound 25 bound to the TKB/RING domain of Cbl-b (PDB code 8QNI). Compound 25 is in green, TKB domain in blue, and LHR in orange. Hydrogen bonds are shown as dashed lines; the putative intramolecular hydrogen bond is highlighted in red. (B) Western blot of a dose–response effect of Cbl-b inhibition on phosphorylation of PLCγ1 at Tyr783. Phosphorylated-PLCγ (pPLCγ) increases upon an increased dose of compound 25, in activated human T-cells. The left-most panel shows an unstimulated (UnStim) T-cell as a control, where the total PLCγ (TPLC) is completely unphosphorylated. (C) Inhibition of Cbl-b autoubiquitination via concentration-dependent dosing of compound 25. (D) Inhibition of Cbl-b ubiquitination of ZAP70 via concentration-dependent dosing of compound 25.
Additional functional assays were performed on compound 25 to confirm its ability to affect downstream activation of key proteins in the TCR signaling pathway (Figure 6B–D). One of the proteins that Cbl-b functionally inhibits through ubiquitination is PLCγ.14,15 PLCγ is phosphorylated downstream of the T cell receptor and is a key signaling protein that connects the proximal TCR and distal TCR signaling cascades which ultimately drive effector function in T cells.13 Dosing of 25 in activated T-cells led to a concentration-dependent increase in phosphorylated-PLCγ (pPLCγ) compared to the total amount of PLCγ (TPLCγ) by Western blot, demonstrating a clear functional inhibition of Cbl-b (Figure 6B). Ubiquitin transfer FRET assays were also established to probe the functional consequences of compound 25 binding. This was achieved by monitoring the in vitro activation of Cbl-b (pTyr363) and subsequent irreversible transfer of labeled ubiquitin to itself (autoubiquitination) or to downstream substrates like ZAP70. Cbl-b autoubiquitination is a pathway naïve T-cells use to regulate Cbl-b expression via proteasomal degradation, and ZAP70 is a critical kinase in the TCR signaling pathway.13,18 Complete ablation of Cbl-b autoubiquitination (Figure 6C) and ZAP70 substrate ubiquitination (Figure 6D) was observed with increasing concentration of compound 25, providing strong evidence that while the inhibitor is bound to the inactive form of Cbl-b, kinase-dependent activation and subsequent E3 ligase activity cannot occur.
Through our HTS hit finding approach, we were able to discover compound 1, which was an exciting starting point to develop a novel series of benzodiazepines as functional Cbl-b inhibitors. This series is structurally diverse compared to previously reported Cbl-b inhibitors and thus presents a unique binding mode. Guided by crystal structure analysis of our initial hit, we carried out SAR exploration of the four outer rings of the azepine core to design compounds that exhibit potent Cbl-b inhibition in a TR-FRET assay, with demonstrable cellular function in T-cells. This led to the discovery of compound 25, which displayed potent Cbl-b inhibition and strong levels of IL-2 production. Furthermore, functional inhibition of Cbl-b by compound 25 was demonstrated to arrest E3 ligase function on downstream TCR signaling proteins as well as its own autoubiquitination. We believe that compound 25 represents a promising lead compound for further optimization as a functional Cbl-b inhibitor.
Acknowledgments
We gratefully acknowledge William Connors’ early project contributions, as well as James Sheppeck for his manuscript review.
Glossary
Abbreviations
- PD-1
programmed cell death protein 1
- PD-L1
programmed cell death ligand 1
- CTLA-4
cytotoxic T-lymphocyte-associated protein 4
- RING
really interesting new gene
- ZAP70
ζ-chain-associated protein kinase 70
- PLCγ
phospholipase Cγ
- CD28
cluster of differentiation 28
- PI3K
phosphatidylinositol-3-kinase
- Ubi
ubiquitin
- TR-FRET
time-resolved fluorescence resonance energy transfer
- μM
micromolar
- Å
angstrom
- Kd
dissociation constant
- IC50
half maximal inhibitory concentration
- log D
logarithm of the distribution coefficient
- LLE
lipophilic ligand efficiency
- nM
nanomolar
- IL-2
interleukin-2
- GPCR
G-protein-coupled receptor
- EC50
half maximal effective concentration
- Ki
inhibitor constant
- GADPH
glyceraldehyde 3-phosphate dehydrogenase
- UnStim
unstimulated
- ATP
adenosine triphosphate
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.3c00439.
Full details of all materials and methods used in this work including protein production, NMR, X-ray crystallography, biological assays, and chemical synthesis (PDF)
Accession Codes
Atomic coordinates and structure factors have been deposited in the Protein Data Bank with accession codes 8QNG (1), 8QNH (Ex23), and 8QNI (25).
Author Contributions
All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Whiteside T. L.; Mandapathil M.; Szczepanski M.; Szajnik M. Mechanisms of tumor escape from the immune system: adenosine-producing Treg, exosomes and tumor-associated TLRs. Bull. Cancer 2011, 98, E25–31. 10.1684/bdc.2010.1294. [DOI] [PubMed] [Google Scholar]
- Hargadon K. M.; Johnson C. E.; Williams C. J. Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int. Immunopharmacol. 2018, 62, 29–39. 10.1016/j.intimp.2018.06.001. [DOI] [PubMed] [Google Scholar]
- Han Y.; Liu D.; Li L. PD-1/PD-L1 pathway: current researches in cancer. Am. J. Cancer Res. 2020, 10, 727–742. [PMC free article] [PubMed] [Google Scholar]
- Seidel J. A.; Otsuka A.; Kabashima K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86. 10.3389/fonc.2018.00086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtukiewicz M. Z.; Rek M. M.; Karpowicz K.; Gorska M.; Politynska B.; Wojtukiewicz A. M.; Moniuszko M.; Radziwon P.; Tucker S. C.; Honn K. V. Inhibitors of immune checkpoints-PD-1, PD-L1, CTLA-4-new opportunities for cancer patients and a new challenge for internists and general practitioners. Cancer Metastasis Rev. 2021, 40, 949–982. 10.1007/s10555-021-09976-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghahremanloo A.; Soltani A.; Modaresi S. M. S.; Hashemy S. I. Recent advances in the clinical development of immune checkpoint blockade therapy. Cell Oncol. (Dordr) 2019, 42, 609–626. 10.1007/s13402-019-00456-w. [DOI] [PubMed] [Google Scholar]
- Li X.; Shao C.; Shi Y.; Han W. Lessons learned from the blockade of immune checkpoints in cancer immunotherapy. J. Hematol. Oncol. 2018, 11, 31. 10.1186/s13045-018-0578-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H.; Lan J.; Yang J. Mechanisms of Resistance to Checkpoint Blockade Therapy. Adv. Exp. Med. Biol. 2020, 1248, 83–117. 10.1007/978-981-15-3266-5_5. [DOI] [PubMed] [Google Scholar]
- Bai R.; Lv Z.; Xu D.; Cui J. Predictive biomarkers for cancer immunotherapy with immune checkpoint inhibitors. Biomarker Res. 2020, 8, 34. 10.1186/s40364-020-00209-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhoj V. G.; Chen Z. J. Ubiquitylation in innate and adaptive immunity. Nature 2009, 458, 430–437. 10.1038/nature07959. [DOI] [PubMed] [Google Scholar]
- Chiang Y. J.; Kole H. K.; Brown K.; Naramura M.; Fukuhara S.; Hu R. J.; Jang I. K.; Gutkind J. S.; Shevach E.; Gu H. Cbl-b regulates the CD28 dependence of T-cell activation. Nature 2000, 403, 216–220. 10.1038/35003235. [DOI] [PubMed] [Google Scholar]
- Paolino M.; Penninger J. M. Cbl-b in T-cell activation. Semin. Immunopathol. 2010, 32, 137–148. 10.1007/s00281-010-0197-9. [DOI] [PubMed] [Google Scholar]
- Shah K.; Al-Haidari A.; Sun J.; Kazi J. U. T cell receptor (TCR) signaling in health and disease. Signal Transduction Target Ther. 2021, 6, 412. 10.1038/s41392-021-00823-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qingjun L.; Zhou H.; Langdon W.; Zhang J. E3 ubiquitin ligase Cbl-b in innate and adaptive immunity. Cell Cycle 2014, 13, 1875–1884. 10.4161/cc.29213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassi I.; Presti R.; Kim S.; Yokoyama W. M.; Gilfillan S.; Colonna M. Phospholipase C-gamma 2 is a critical signaling mediator for murine NK cell activating receptors. J. Immunol. 2005, 175, 749–754. 10.4049/jimmunol.175.2.749. [DOI] [PubMed] [Google Scholar]
- Fang D.; Liu Y. C. Proteolysis-independent regulation of PI3K by Cbl-b-mediated ubiquitination in T cells. Nat. Immunol. 2001, 2, 870–875. 10.1038/ni0901-870. [DOI] [PubMed] [Google Scholar]
- Schmitz M. L. Activation of T cells: releasing the brakes by proteolytic elimination of Cbl-b. Sci. Signaling 2009, 2, pe38 10.1126/scisignal.276pe38. [DOI] [PubMed] [Google Scholar]
- Tang R.; Langdon W. Y.; Zhang J. Regulation of immune responses by E3 ubiquitin ligase Cbl-b. Cell Immunol. 2019, 340, 103878 10.1016/j.cellimm.2018.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz B.; An W.; Mohapatra B.; Zutshi N.; Iseka F.; Storck M. D.; Meza J.; Sheinin Y.; Band V.; Band H. A novel CBL-Bflox/flox mouse model allows tissue-selective fully conditional CBL/CBL-B double-knockout: CD4-Cre mediated CBL/CBL-B deletion occurs in both T-cells and hematopoietic stem cells. Oncotarget 2016, 7, 51107–51123. 10.18632/oncotarget.9812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly R. J.; Scott A. M.; Klein O.; Ernst M. Enhancing therapeutic anti-cancer responses by combining immune checkpoint and tyrosine kinase inhibition. Mol. Cancer 2022, 21, 189. 10.1186/s12943-022-01656-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z.; McGray A. J. R.; Jiang W.; Lu B.; Kalinski P.; Guo Z. S. Improving cancer immunotherapy by rationally combining oncolytic virus with modulators targeting key signaling pathways. Mol. Cancer 2022, 21, 196. 10.1186/s12943-022-01664-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thien C. B.; Langdon W. Y. c-Cbl and Cbl-b ubiquitin ligases: substrate diversity and the negative regulation of signaling responses. Biochem. J. 2005, 391, 153–166. 10.1042/BJ20050892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buetow L.; Tria G.; Ahmed S. F.; Hock A.; Dou H.; Sibbet G. J.; Svergun D. I.; Huang D. T. Casitas B-lineage lymphoma linker helix mutations found in myeloproliferative neoplasms affect conformation. BMC Biol. 2016, 14, 76. 10.1186/s12915-016-0298-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levkowitz G.; Waterman H.; Ettenberg S. A.; Katz M.; Tsygankov A. Y.; Alroy I.; Lavi S.; Iwai K.; Reiss Y.; Ciechanover A.; Lipkowitz S.; Yarden Y. Ubiquitin ligase activity and tyrosine phosphorylation underlie suppression of growth factor signaling by c-Cbl/Sli-1. Mol. Cell 1999, 4, 1029–1040. 10.1016/S1097-2765(00)80231-2. [DOI] [PubMed] [Google Scholar]
- Kobashigawa Y.; Tomitaka A.; Kumeta H.; Noda N. N.; Yamaguchi M.; Inagaki F. Autoinhibition and phosphorylation-induced activation mechanisms of human cancer and autoimmune disease-related E3 protein Cbl-b. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 20579–20584. 10.1073/pnas.1110712108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng N.; Wang P.; Jeffrey P. D.; Pavletich N. P. Structure of a c-Cbl-UbcH7 complex: RING domain function in ubiquitin-protein ligases. Cell 2000, 102, 533–539. 10.1016/S0092-8674(00)00057-X. [DOI] [PubMed] [Google Scholar]
- Dou H.; Buetow L.; Hock A.; Sibbet G. J.; Vousden K. H.; Huang D. T. Structural basis for autoinhibition and phosphorylation-dependent activation of c-Cbl. Nat. Struct. Mol. Biol. 2012, 19, 184–192. 10.1038/nsmb.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou H.; Buetow L.; Sibbet G. J.; Cameron K.; Huang D. T. Essentiality of a non-RING element in priming donor ubiquitin for catalysis by a monomeric E3. Nat. Struct. Mol. Biol. 2013, 20, 982–986. 10.1038/nsmb.2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barsanti P.; Bence N.; Gosling J.; Saha A.; Taherbhoy A.; Zapf C.; Boyle K.; Cardozo M.; Mihalic J.; Lawrenz M.. Inhibitors of CBL-B and methods of use thereof. WO2019148005A1, 2019.
- Sharp A.; Williams A.; Blagden S. P.; Plummer E. R.; Hochhauser D.; Krebs M.; Pacey S.; Evans T. R. J.; Whelan S.; Nandakumar S.; Rogers S.; Jameson K. L.; Basile F. G.; De Bono J. S.; Arkenau H.-T. A first-in-human phase 1 trial of nx-1607, a first-in-class oral CBL-B inhibitor, in patients with advanced solid tumor malignancies. Journal of Clinical Oncology 2022, 40, TPS2691 10.1200/JCO.2022.40.16_suppl.TPS2691. [DOI] [Google Scholar]
- Sabnis R. W. Novel Lactams as CBL-B Inhibitors for Treating Cancer. ACS Med. Chem. Lett. 2023, 14, 897–898. 10.1021/acsmedchemlett.3c00247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leit de Moradei S. M.; West A. V.; Baker T.; Arregui J. C.; Castagna D.; Greenwood J. R.; Rafi S.; McRobb F.; Zhang Y.. Cbl-b modulators and uses thereof. WO2022217276A1, 2022.
- Sands A. T.; Bence N. F.; Zapf C. W.; Cohen F.; Wang C.; Cummins T.; Tanaka H.; Shunatona H.; Cardozo M.; Weiss D.; Gosling J.. Substituted benzyl-triazole compounds for cbl-b inhibition, and further uses thereof. WO2020264398A1, 2020.
- Kimani S.; Perveen S.; Szewezyk M.; Zeng H.; Dong A.; Li F.; Ghiabi P.; Li Y.; Chau I.; Arrowsmith C.; Barsyte-Lovejoy D.; Santhakumar V.; Vedadi M.; Halabelian L. Probing the mechanism of Cbl-b inhibition by a small-molecule inhibitor. bioRxiv 2023, 10.1101/2023.05.05.539612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. G.; Liu J. X.; Jia X. X.; Geng J.; Yu F.; Cong B. Cholecystokinin octapeptide regulates the differentiation and effector cytokine production of CD4(+) T cells in vitro. Int. Immunopharmacol. 2014, 20, 307–315. 10.1016/j.intimp.2014.03.013. [DOI] [PubMed] [Google Scholar]
- Zhang J. G.; Cong B.; Li Q. X.; Chen H. Y.; Qin J.; Fu L. H. Cholecystokinin octapeptide regulates lipopolysaccharide-activated B cells co-stimulatory molecule expression and cytokines production in vitro. Immunopharmacol. Immunotoxicol 2011, 33, 157–163. 10.3109/08923973.2010.491079. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.