

Abstract
Background:
In addition to its anticoagulant function in downregulating thrombin generation, activated protein C (APC) evokes pleiotropic cytoprotective signaling activities when it binds to endothelial protein C receptor (EPCR) to activate protease-activated receptor 1 (PAR1) in endothelial cells.
Objectives:
To investigate the protective effect of APC in a chlorhexidine gluconate (CG)-induced peritoneal fibrosis model.
Methods:
Peritoneal fibrosis was induced in wild-type and EPCR- and PAR1-deficient mice by daily injection of CG (0.2 mL of 0.1% CG in 15% ethanol and 85% saline) for 21 days with or without concomitant injection of recombinant human APC derivatives (50 μg/kg/bodyweight). Expression of proinflammatory cytokines, profibrotic markers and collagen deposition were analyzed by established methods.
Results:
CG significantly upregulated expression of TGF-β1 in peritoneal tissues that culminated in deposition of excessive extracellular matrix proteins, thickening of the peritoneal membrane and mesothelial-to-mesenchymal transition in the damaged tissues. APC potently inhibited CG-induced peritoneal fibrosis and downregulated expression of proinflammatory cytokines, collagen deposition, Smad3 phosphorylation, and markers of mesothelial-to-mesenchymal transition (α-SMA, vimentin and N-cadherin). APC also inhibited TGF-β1 mediated upregulation of α-SMA, Smad3 and fibronectin in human primary mesothelial cells. Employing signaling-selective and anticoagulant-selective variants of APC and mutant mice deficient for either EPCR or PAR1, we demonstrate that the EPCR-dependent signaling function of APC through PAR1 activation is primarily responsible for its antifibrotic activity in the CG-induced peritoneal fibrosis model.
Conclusions:
APC and signaling-selective variants of APC may have therapeutic potential for preventing/treating pathologies associated with peritoneal fibrosis.
Keywords: Peritoneal fibrosis, TGF-β1, APC, EPCR, PAR1
Introduction
Activated protein C (APC) is the enzymatic product of thrombomodulin (TM)-dependent activation of protein C by thrombin on endothelial cells [1]. It is a vitamin K-dependent serine protease in plasma involved in downregulation of the clotting cascade by inactivating procoagulant cofactors Va and VIIIa by limited proteolysis [2]. In addition to this anticoagulant function, APC also binds to endothelial protein C receptor (EPCR) with high affinity to cleave and activate protease activated receptor 1 (PAR1) to initiate cytoprotective signaling responses in endothelial cells [3-5]. Recently, we constructed signaling-selective (APC-2Cys) and anticoagulant-selective (APC-E170A) mutants of APC [6,7], and demonstrated the cytoprotective signaling function of APC is highly effective in several injury and inflammatory models by preventing injury/inflammation-mediated expression of proinflammatory cytokines by EPCR- and PAR1-dependent inhibition mechanisms [8-10]. The current study was undertaken to investigate the cytoprotective role of APC in preventing peritoneal fibrosis in a chlorhexidine gluconate (CG)-induced peritoneal fibrosis (PF) model. CG-induced PF is an established model that has been extensively used for evaluating and discovery of potential therapeutics for preventing/treating peritonitis and peritoneal dialysis-induced PF [11-14]. Peritoneal dialysis is an alternative method to hemodialysis that is used as a treatment strategy for patients with end-stage kidney disease [15,16]. However, bioincompatible substances in the dialysate and its hyperosmotic and acidic nature cause repeated injury over time to mesothelial cells of the peritoneum, thereby leading to development of fibrosis, thickening of the peritoneal membrane and eventually failure of the filtration [15-17]. It has been hypothesized the pathogenic mechanism involves excessive accumulation of fibrous connective tissue as the result of secretion of significant amounts of growth factors including the profibrotic cytokine TGF-β1 by peritoneal mesothelial and fibroblast cells [12-17]. Dysregulated expression of TGF-β1 by peritoneal cells is thought to cause mesenchymal conversion of mesothelial cells by a process referred to as epithelial-mesenchymal transition (EMT) which is a normal physiological process occurring primarily during embryonic development however its dysregulation can lead to chronic inflammation and fibrosis in affected tissues [15,17].
Here, we set up a CG-induced PF model and evaluated the cytoprotective signaling function of APC in wild-type as well as in EPCR- and PAR1-deficient mice. Results demonstrate APC exerts potent protective effects in preventing PF by inhibiting upregulation of proinflammatory and profibrotic cytokines by EPCR-dependent activation of PAR1 on peritoneal tissues. The signaling-selective variant of APC (APC-2Cys) was highly effective in inhibiting PF in CG-treated mice. Moreover, APC exhibited no cytoprotective effect in mice deficient for either EPCR or PAR1. We further discovered that CG-mediated PF is associated with a dramatic decrease in expression of TM on peritoneal mesothelial cells, suggesting the TM-dependent protein C activation potential of thrombin has also been compromised by peritoneal mesothelial cells of CG-treated mice. APC effectively prevented CG-mediated loss of TM from mesothelial cell surfaces of the peritoneum. APC inhibited TGF-β1 signaling, collagen deposition, and thickening of the submesothelial compact zone in CG-treated mice. APC also inhibited purified TGF-β1-mediated phenotypic changes in human primary mesothelial cells (HPMC) derived from peritoneum. Based on these results, we believe therapeutic potentials of APC derivatives warrant further investigation.
Methods
Materials and reagents
TM antibody was from R&D (Minneapolis, MN, USA). Alexa Fluor 555-conjugated goat anti-rat antibody and Alexa Fluor 568-conjugated goat anti-rabbit antibody were from Thermo Fisher Scientific (Rockford, IL, USA). Antibodies to α-SMA, α2-integrin, vimentin, Snail, Slug, E-cadherin, N-cadherin and pSmad3, fibronectin, and Smad2/3 (#8685) were from Cell Signaling Technology (Beverly, MA, USA). Phycoerythrin (PE)-conjugated TM antibody (#130-113-318) and PE-conjugated mouse isotype control antibody (#130-113-762) were purchased from Miltenyi Biotec, Germany. Lyophilized TGF-β1 (#10804-HNAC) was purchased from Sino Biological (Houston, TX, USA) and dissolved in sterile water to prepare a stock solution of 0.1mg/ml (as per manufacturer instructions). Recombinant human APC derivatives including APC-2Cys, APC-E170A and APC-S195A were prepared as described (6,7). C57BL/6 male and female mice were purchased from Jackson Laboratory. Transgenic of EPCRδ/δ and PAR1−/− male mice have described previously [18-20]. Animals were backcrossed onto an inbred C57BL/6 background. All experiments involving animals were performed according to guidelines for Care and Use of Laboratory Animals approved by Oklahoma Medical Research Foundation.
Animal model
The mouse model of CG-induced PF in 8-weeks old C57BL/6 mice was developed by daily intraperitoneal injection of 0.2mL of 0.1% CG in 15% ethanol and 85% saline for 21 days as described [11-14]. To analyze the protective effect of human APC derivatives (APC-WT, signaling-selective APC-2Cys, anticoagulant-selective APC-E170A and catalytically inactive APC-S195A) in this model, 1h before administration of CG, APC derivatives (50μg/kg/bodyweight, determined to be optimal) were injected daily. Control mice received 0.2mL saline. After 21 days, mice were sacrificed, and the parietal peritoneum was dissected for histological examination.
Mesothelial cell culture
Peritoneal-derived adult human primary mesodermal cells (HPMCs) were obtained from Zenbio Inc. (Durham, NC, USA, cat# MES-F-SL) and cultured according to manufacturer’s manual. The effect of TGF-β1 (2.5ng/mL for 48h) with or without APC and APC-2Cys treatment (50nM) was monitored for morphological changes using inverted microscopy and quantitative Real-time PCR analysis of selected genes of extracellular matrix proteins. The effect of APC derivatives on TGF-α1-mediated nuclear translocation of Smad2/3 and cell surface expression of TM in HPMCs were also analyzed by immunofluorescence and flow cytometry.
Histological evaluation
For evaluation of the cell density and thickness, peritoneal tissues were separated from other tissues and fixed in 10% formalin and immersed in paraffin. Several paraffin sections were made by microtome and stained by Haematoxylin–Eosin (H&E) and Masson Trichrome. The areas of peritoneal fibrosis (PF) were assessed in predetermined fields (200) of the mesothelial compact zone captured by a digital camera and the area stained was analyzed using a Lumina Vision 2.20 (Olympus, USA) in 10 random fields. The number of cells present in the submesothelial compact zone were counted in 10 random fields.
Immunohistochemistry (IHC)
The submesothelial compact zone was analyzed by the IHC for accumulation of collagen I and III, expression of elevated levels of α-smooth muscle actin (α-SMA, as a characteristic marker of myofibroblast), expression of E-cadherin and elevated phosphorylation of Smad3 (as a marker of TGF-β1 signaling pathway). The diaminobenzidine (DAB)-stained positive areas of α-SMA, collagens I, and III were assessed in predetermined fields (×200) of the submesothelial compact zone captured by a digital camera and the stained area was determined by the bio-imaging analysis system (Lumina vision, USA) in 10 random fields. The number of cells positive for pSmad3 (×400) in the submesothelial compact zone was counted in 10 random fields
Immunofluorescence
For immunofluorescence, parietal peritoneum tissues were fixed in 4% paraformaldehyde overnight at 4°C. Tissue samples were washed in PBS, cryoprotected in PBS containing 30% sucrose at 4°C overnight and embedded in 50% tissue freezing medium + 50% OCT compound (Tissue-Tek). All tissues were cryosectioned (10μm sections) with Leica CM3050 Cryomicrotome. Tissue sections were blocked with 1X PBS containing 1% BSA, 5% goat serum and 0.3% Triton-X-100. Samples then incubated with primary antibodies against TM (rat monoclonal) followed by Alexa Fluor 555-conjugated goat anti-rat antibody. Slides were mounted with mounting medium containing DAPI (Vector Laboratories Inc., Burlingame, CA) and images were obtained using a Nikon C2 Confocal Microscope (Melville, NY, USA).
For analysis of smad2/3 nuclear translocation, sub-confluent HPMCs were washed with PBS and fixed with 4% paraformaldehyde for 15min followed by permeabilization with 0.1% Triton X-100/PBS for 10min at room temperature. Cells were then washed with PBS, blocked for 1h with 2% BSA in the same buffer system, and incubated with rabbit monoclonal anti-smad2/3 antibody followed by goat anti-rabbit Alexa Fluor 568-conjugated secondary antibody. The nucleus was stained with Hoechst 33342 and images were captured with a ZOE™ Fluorescent Cell Imager (BioRad Laboratories, CA, USA).
TUNEL assay
The apoptotic effect of CG on the mouse peritoneal tissue with and without APC was analyzed through the TUNEL assay, according to a protocol provided by the manufacturer (Roche, Switzerland).
Flow cytometry
Sub-confluent cultures of HPMCs were treated with and without APC-WT and APC-2Cys for 3h followed by TGF-β1 treatment for 18h. Cells were detached using HBSS containing 10mM EDTA, washed and resuspended in HBSS containing 1% human serum albumin (HSA) and 2mM EDTA. Cells were stained using PE-conjugated anti-TM antibody or PE-conjugated isotype control antibody for 30min on ice. Cell surface levels of TM were detected using FACS Celesta and data was analyzed by FlowJo™ v10 software (BD Biosciences).
Real-time PCR and ELISA
mRNA expression levels of TGF-β1, α-SMA, fibronectin, tPA, PAI-1, α2-integrin, tissue inhibitor of metalloprotease (TIMP)-2, matrix metalloprotease (MMP)-2 and 9, collagen and keratin in peritoneal tissues with or without treatment with APC were determined by quantitative Real-time PCR using the iQ SYBR Green Supermix (Bio-Rad) and a BIO-RAD C1000 Thermal cycle as described. Results were normalized to expression levels of GAPDH and presented as the fold difference relative to the control group employing the 2−ΔCt. Primer pairs for individual genes were synthesized by IDT (Integrated DNA Technologies, Coralville, IA) and presented as Table 1. A commercially available ELISA kit (eBioScience, San Diego, CA) was used to measure the concentration of TGF-β1 in the peritoneal fluid of experimental animals.
Table 1:
Primer sequences employed in Real-Time quantitative PCR assays
| Gene | Primer sequence (5′-3′) |
|---|---|
| TGF-β1 | GAGCCCGAAGCGGACTACTA TGGTTTTCTCATAGATGGCGTTG |
| Keratin-2 | CACGTCTGCGGAGAATGATTT TCCTGTGCTAGTATGTCCAGC |
| TIMP-2 | CTCGCTGTCCCATGATCCC GCCCATTGATGCTCTTCTCTGT |
| α2-Integrin | TGTCTGGCGTATAATGTTGGC TGCTGTACTGAATACCCAAACTG |
| MMP-2 | GGACAAGTGGTCCGCGTAAA CCGACCGTTGAACAGGAAGG |
| MMP-9 | GCGTCGTGATCCCCACTTAC CAGGCCGAATAGGAGCGTC |
| IL-1β | TTCAGGCAGGCAGTATCACTC GAAGGTCCACGGGAAAGACAC |
| TNFα | CAGGCGGTGCCTATGTCTC CGATCACCCCGAAGTTCAGTAG |
| Collagen 1a1 | GCTCCTCTTAGGGGCCACT ATTGGGGACCCTTAGGCCAT |
| Collagen 4a1 | GGCCCCAAAGGTGTTGATG CAGGTAAGCCGTTAAATCCAGG |
| Fibronectin | GCAGTGACCACCATTCCTG GGTAGCCAGTGAGCTGAACAC |
SDS-PAGE and Western-blotting
Tissue lysates were prepared with 1X PBS containing, 0.5% Triton X-100, 0.5% Nonidet P-40, 5 mM EDTA and protease-phosphatase inhibitor cocktail. After clearing the lysate, protein estimation was done by the BCA protein assay kit (Thermo Fisher, Rockford, IL, USA). All lysate samples were boiled in loading buffer with 5% β-mercaptoethanol and resolved on 10% SDS-polyacrylamide gels. Protein samples were transferred to nitrocellulose membrane and incubated with respective antibodies followed by horseradish peroxidase linked anti-rabbit IgG or anti-mouse IgG. Protein bands were detected using an ECL substrate (Thermo Fisher, Rockford, IL, USA).
Statistical analysis
Data are presented as mean ± standard error of mean (SEM) from ≥3 independent experiments. All data were analyzed using analysis of variance (ANOVA) followed by Bonferroni post hoc test using Graph Pad Prism7 (Graph Pad Prism, San Diego, CA). A p value of <0.05 was considered statistically significant.
Results
APC reduces peritoneal cell density and thickening in CG-induced fibrosis
Intraperitoneal injection of CG has been shown to induce peritoneal fibrosis (PF) by inducing the activation of proinflammatory cytokines including TGF-β1, TNF-α and NF-κB signaling pathways, thereby leading to an increase in cell density and inflammatory thickening of submesothelial peritoneal membrane [11-14]. This model has been extensively used to investigate the pathophysiology of peritonitis and peritoneal dialysis-mediated fibrosis [11-14]. Because of its potent cytoprotective activity, we hypothesized here APC has the potential to prevent PF in this model by inhibiting TGF-β1 and other inflammatory cytokines. Indeed, H&E staining indicated daily intraperitoneal injection of CG for 21 days was associated with induction fibrosis and a dramatic increase in the thickening of the submesothelial compact zone of the peritoneum when compared to the vehicle control mice (Fig. 1A and B). Remarkably, concomitant injection of 50 μg/kg/bodyweight human APC potently inhibited CG-mediated PF, thickening and cellularity of the peritoneum in experimental animals (Fig. 1C, G and H; n=10 male mice in all groups). APC is known to exhibit both anticoagulant and anti-inflammatory activities [1-5]. To understand the mechanism by which APC inhibits the pathogenesis of PF, studies were conducted with two APC variants one of which has near normal anti-inflammatory signaling function (APC-2Cys) but possesses insignificant anticoagulant activity and the other one has normal anticoagulant activity but is defective in its signaling function (APC-E170A) [6-10]. Analysis by H&E staining indicated that, like APC-WT (Fig. 1C), APC-2Cys markedly inhibits thickening and cell density of the peritoneum (Fig. 1D, G, and H). The protective effect, though to a lesser extent, was also observed with the anticoagulant-selective APC-E170A variant (Fig. 1E, G, and H). However, the catalytically inactive APC variant (APC-S195A) exhibited no protective activity (Fig. 1F, G, and H), suggesting an intact active-site pocket for APC is required to exert its protective function.
Figure 1. APC suppresses peritoneal cell density and thickening of peritoneal membrane in CG-injected mice.
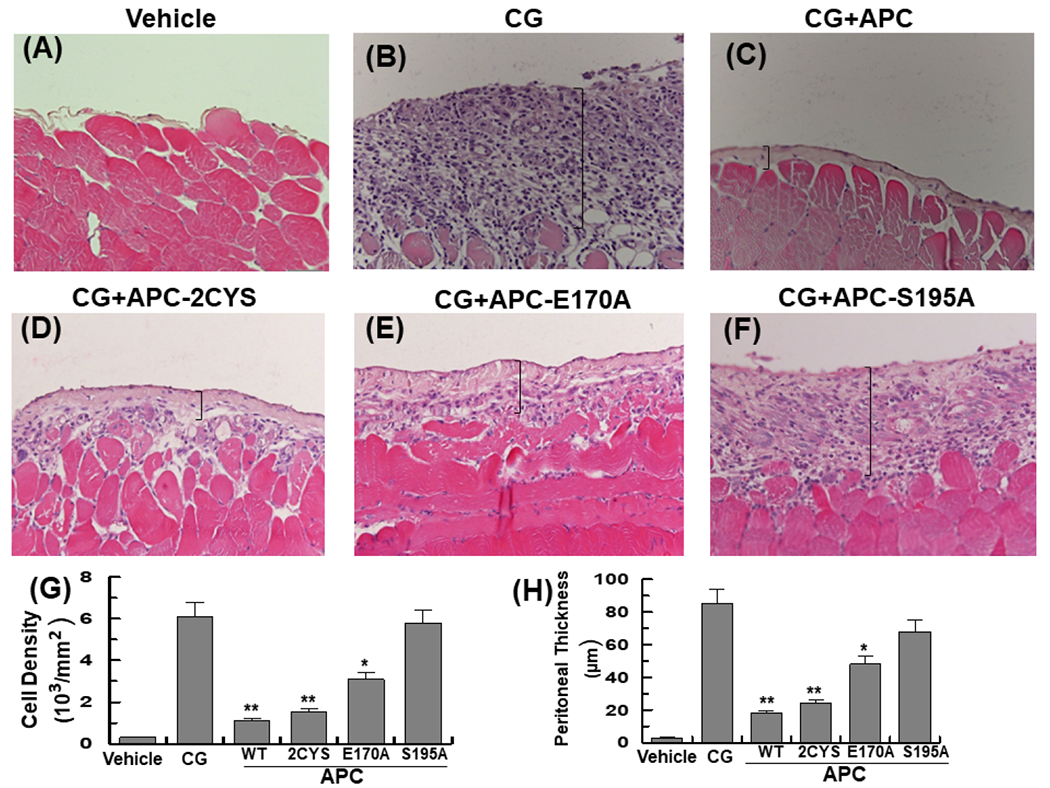
(A-F) Representative light microscopic features of H&E staining of peritoneal tissues on day 21 in control (vehicle, normal saline) mice (A), CG-injected mice treated with the vehicle alone (B), CG-injected mice treated with APC-WT (50 μg/kg/day for all groups) (C), APC-2Cys (D), APC-E170A (E), and APC-S195A (F). Brackets show the thickness of the submesothelial compact zone. (G-H) Quantification of cell density and thickness of the submesothelial compact zone in all groups. The data are shown as mean ± SEM (n= 10), *p< 0.05 and **p<0.01 (APC: 50 ug/kg/day in entire study). Scale bar =5μm. In all figure legends (n) indicates number of animals analyzed.
Analysis by Masson’s trichrome staining indicated the thickened peritoneum in the CG-treated mice contains a significant amount of collagen (Fig. 2A,B) and interestingly, both APC and APC-2Cys effectively inhibited expression and deposition of collagen by peritoneal mesothelial cells (Fig. 2C and D). Like a modest PF inhibitory effect observed by H&E staining, the anticoagulant-selective APC mutant exhibited some protective effect in inhibiting excessive CG-induced collagen deposition in the submesothelial compact zone of the peritoneum (Fig. 2E), however, APC-S195A exhibited no inhibitory function (Fig. 2F). In support of these results, analysis by immunohistochemistry revealed elevated levels of the extracellular matrix proteins collagen I and collagen III in peritoneal tissues of the CG-treated mice which were significantly downregulated in mice treated with APC-WT (Fig. 2G and H).
Figure 2. APC suppresses collagen expression in peritoneal tissues of CG-injected mice.
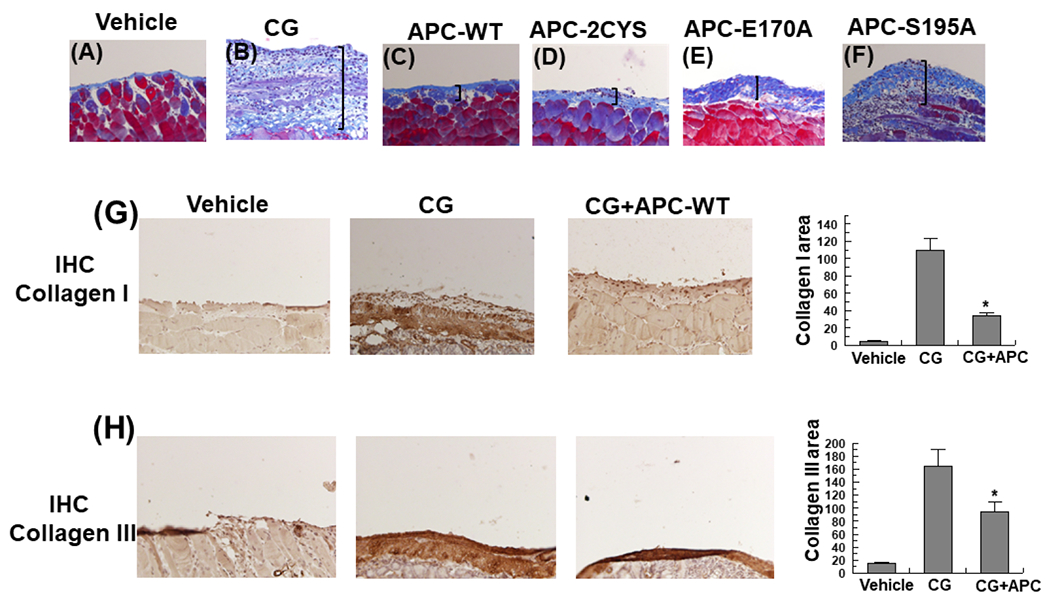
(A-F) Representative light microscopic features of Masson’s trichrome staining of peritoneal tissues on day 21 in control (vehicle) mice (A), CG-injected mice treated with the vehicle (B), CG-injected mice treated with APC-WT (C), APC-2Cys (D), APC-E170A (E) and APC-S195A (F). n= 10 in all group. (G-H) Immunohistochemical analysis of collagen I (G) and collagen III (H) expression in peritoneal tissues on day 21 in vehicle group mice, CG-injected mice treated with the vehicle alone, and CG-injected mice treated with APC-WT. The stained areas for collagens I and III are presented next to each panel on the right. The data are shown as mean ± SEM, n= 5, *p< 0.05 and **p<0.01. Scale bar =50μm.
APC inhibits CG-induced TGF-β1 signaling and expression of α-SMA, N-cadherin and vimentin in peritoneal tissues
It is known peritoneal fibrosis including CG-induced PF is associated with induction of the TGF-β1/Smad signaling and expression of α-SMA as a marker of mesothelial to mesenchymal trans-differentiation [12,15]. The protective effect of APC on these cellular alterations was evaluated and results indicated APC effectively inhibits CG-induced phosphorylation Smad3 in peritoneal tissues (Fig. 3A and C). In agreement with this result, the assessment of the peritoneal fluid of CG-treated mice by ELISA indicated the protein level of TGF-β1 has been markedly elevated in experimental animals and both APC-WT and APC-2Cys potently inhibit the expression of this profibrotic cytokine (Fig. 3E and F). Like its inhibitory effect toward TGF-β1, analysis by immunohistochemistry indicated APC also inhibits CG-induced expression α-SMA in peritoneal tissues of the CG-treated mice (Fig. 3B and D).
Figure 3. APC downregulates expression of TGF-β1 and α-SMA and phosphorylation of Smad3 in peritoneal tissues of CG-injected.
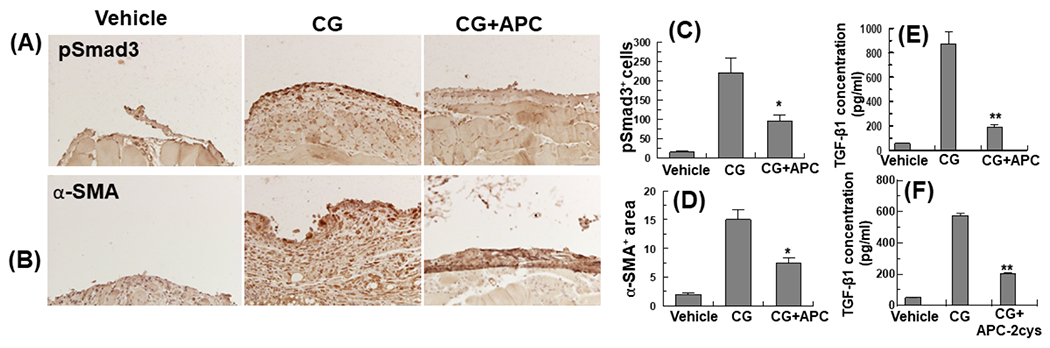
Immunohistochemical analyses of pSmad3 (A) and α-SMA (B) expression in peritoneal tissues on day 21 in vehicle group mice, CG-injected mice treated with the vehicle alone, and CG-injected mice treated with APC-WT. The stained areas encompassing cells positive for pSmad3 (C) and α-SMA (E) are shown next to each panel. (D) The expression of TGF-β1 in the peritoneal fluid was measured by ELISA. The data are shown as mean ± SEM, n= 5, *p<0.05 and **p<0.01. Scale bar =50μm.
Immunoblot analysis of peritoneal tissue lysates indicated expressions of N-cadherin and vimentin are upregulated by CG treatment in mice and both EMT markers were significantly downregulated by APC (Fig. 4A-C).
Figure 4. APC downregulates expression of N-cadherin and vimentin in peritoneal tissues of CG-injected mice and TGF-β1-mediated morphological changes in HPMCs.
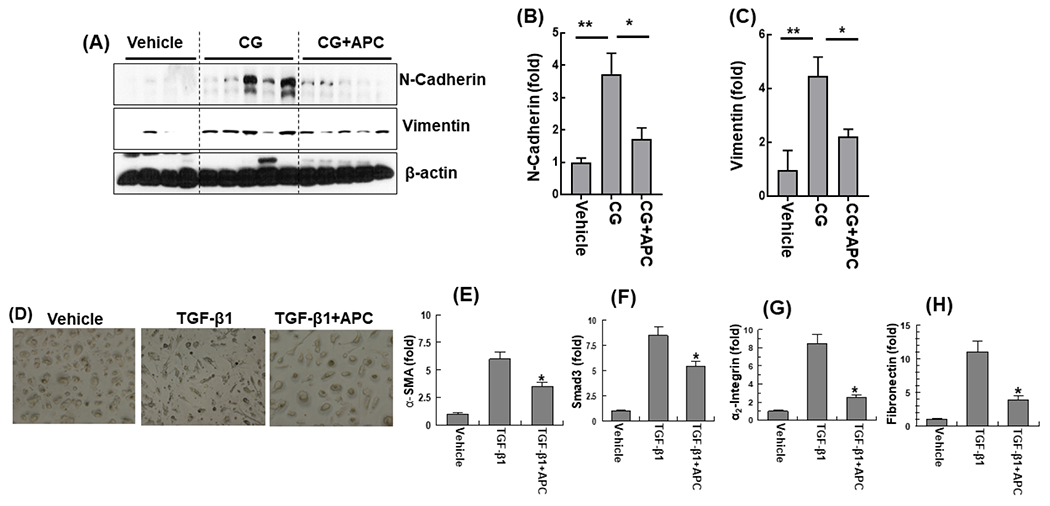
(A) Immunoblot analysis of expression of N-cadherin and vimentin in peritoneal tissues on day 21 in vehicle group mice, CG-injected mice treated with the vehicle alone, and CG-injected mice treated with APC-WT. β-actin was used as loading control. Densitometric analysis of expression of N-cadherin (B) and vimentin (C) in CG-treated mice of panel (A). Data are mean ± SEM, n=5, *p<0.05, **p< 0.01 and ***p< 0.001. (D) Representative photomicrographs untreated and TGF-β1 (48 h) treated HPMCs with or without pretreatment with APC-WT (50 nM, 3h). (E-H) Real-time quantitative PCR of mRNA expression levels in HPMCs in presence to TGF-β1 and with and without pretreatment APC-WT for α-SMA (E), Smad3 (F), α2-integrin (G) and fibronectin (H). The data are shown as mean ± SEM, n= 5, *p<0.05 and **p<0.01.
Morphological analysis and quantitative PCR analysis of gene expression profiles of the TGF-β1-treated HPMCs derived from the omentum, indicated TGF-β1 treatment changes the characteristic cobblestone-like appearances of normal HPMCs to fibroblast-like phenotype after 48h of stimulation and that this phenotype was effectively prevented by pretreatment of cells with APC (Fig. 4D-H). Real-time quantitative PCR analysis revealed TGF-β1 promotes mRNA expression levels of α-SMA, Smad3, α2-integrin and fibronectin and that APC treatment effectively inhibits the elevated expression of these profibrotic mediators in HPMCs (Fig. 4E-H). To determine whether TGF-β1 induces the nuclear translocation of Smad2/3 in HPMCs, sub-confluent HPMCs were treated with the control buffer, APC-WT and APC-2Cys (50nM each) for 3h followed by treatment with or without TGF-β1 for 30min. Immunofluorescence analysis revealed TGF-β1 treatment was associated with translocation of Smad2/3 to the nucleus which was significantly downregulated by both APC-WT and APC-2Cys (Suppl. fig. S1), suggesting the signaling function of APC is responsible for reducing TGF-β1 signaling in mesothelial cells. Consistent with these results, western blot analysis of cell lysates indicated both APC and APC-2Cys downregulate TGF-β1-mediated elevated expression of fibronectin and N-cadherin in HPMCs (Suppl. fig. S2). Analysis by flow cytometry indicated TGF-β1 decreases cell surface expression of TM, but neither APC-WT nor APC-2Cys affected TM expression on cultured cells under these conditions (Suppl. fig. S3).
APC inhibits CG-induced mRNA transcript levels of inflammatory cytokines in peritoneal tissues
In agreement with the cytoprotective function of APC against CG-induced PF as shown above, real-time quantitative PCR analysis of mRNA levels indicated CG markedly upregulated transcript levels of TGF-β1 (Fig. 5A) IL-1β (Fig. 5B), TNF-α (Fig. 5C), plasminogen activator inhibitor 1 (PAI-1, Fig. 5D), α2-integrin (Fig. 5E), matrix metalloproteinase-2 and 9 (MMP-2 and MMP-9, Fig. 5F and G) in peritoneal tissues of CG-treated mice and APC effectively inhibits mRNA levels of proinflammatory markers. However, APC reversed CG-mediated downregulation of the mRNA level of the metalloproteinase inhibitor-2 (TIMP-2) in peritoneal tissues (Fig. 5H). mRNA expression levels of keratin-2, collagen 1a1 and collagen 1a4 were all significantly elevated in peritoneal tissues of CG-treated animals and APC was highly effective in reducing these transcripts as well (Suppl. fig. S4).
Figure 5. Analysis of mRNA expression levels of peritoneal tissues.
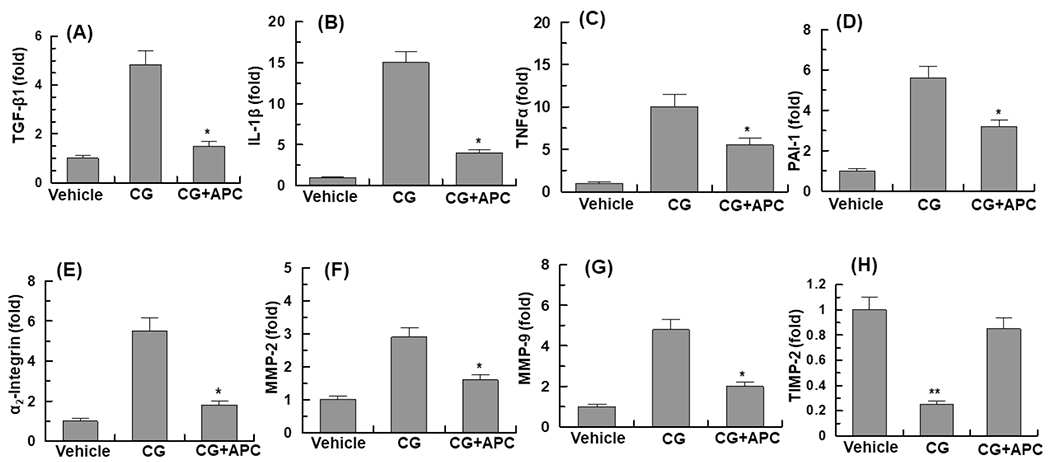
Real-time quantitative PCR was used to analyze the mRNA expression levels in peritoneal tissues of vehicle group, CG-injected treated with vehicle and CG-injected mice treated with APC-WT: (A), TGF-β 1; (B), IL1-β; (C), TNFα; (D), PAI-1; (E), α2-integrin; (F), MMP-2; (G), MMP-9; (H), TIMP-2. The data are shown as mean ± SEM, n=5, *p<0.05 and **p<0.01 as compared to CG-injected vehicle treatment.
Protective effects of APC are mediated through EPCR and PAR1
To determine whether the EPCR-dependent activation of PAR1 by APC accounts for its protective function, the same CG-mediated PF experiments were conducted in genetically altered EPCR deficient (EPCRδ/δ) mice, which express greatly diminished amount of the receptor (<10% of cell-surface expression) and in PAR1−/− knockout mice [18-20]. Analysis of results indicated EPCR is required for the antifibrotic activity of APC as evidenced by inability of APC to inhibit CG-induced increase in the cell density and thickening of the submesothelial compact zone of the peritoneum of EPCRδ/δ mice (Fig. 6A,B). Like its effect in EPCRδ/δ mice, APC exhibited no protective effect against CG-mediated peritoneal fibrosis in PAR1−/− knockout mice (Fig. 6C and D), suggesting mesothelial cells express receptors of APC signaling and that the protective effect of APC is largely mediated through EPCR-dependent activation of PAR1 in peritoneal mesothelial cells. These cells, like endothelial and pleural mesothelial cells [21-23], possess similar phenotypic characteristics with respect to APC signaling, thus the receptor deficiency eliminated barrier protective, anti-inflammatory, and anti-fibrotic functions of APC in both knockout models. A recent study analyzed mechanism of the protective EPCR-PAR1 signalingng and reported expression of EPCR on endothelial cells is downregulated by deletion of PAR1 [24]. Further studies will be required to determine whether this is also for mesothelial cells.
Figure 6. APC cannot inhibit CG-mediated PF in EPCR- and PAR1-deficient mice.
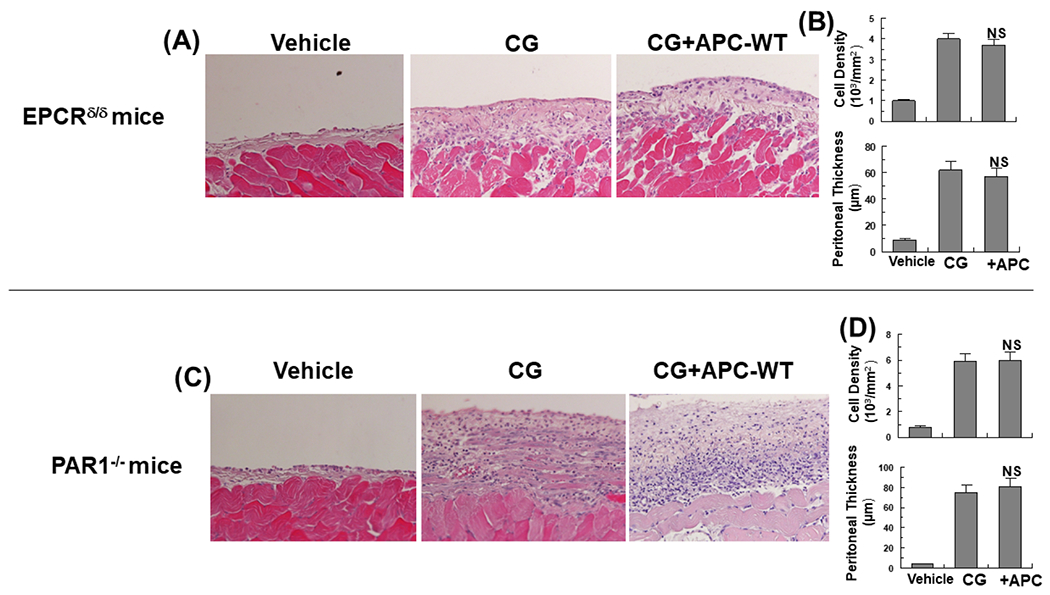
Representative light microscopic features of H&E staining of peritoneal tissues on day 21 in control (vehicle) mice, CG-injected mice treated with the vehicle alone, CG-injected mice treated with APC-WT in the EPCRδ/δ (A) and PAR1−/− (C) mice. Increased cell density and thickness of the submesothelial compact zone are presented for the CG-treated EPCRδ/δ (B) and PAR1−/− (D) mice. The data are shown as mean ± SEM, n= 10, *p<0.05 and **p<0.01. Scale bar =50μm.
Protective effect of APC against CG-mediated PF is independent of the gender
Since the studies described above was conducted in male mice, we excluded the gender specificity of the protective effect of APC by reproducing the results in the female mice by employing the same experimental protocols. Like in male mice, intraperitoneal injection of CG in female mice (n=5) resulted in induction of PF, increased cell density, thickening of the submesothelial compact zone and enhanced collagen deposition which, like in male mice, were all reversed by the APC treatment (Fig. 7A,B). Peritoneal tissues were evaluated for TM expression in CG-treated mice. Immunofluorescence analysis indicated mesothelial cells abundantly express TM and CG treatment results in a significant loss of TM from the surface of peritoneal mesothelial cells as well as of vasculature tissues in the peritoneum (Fig. 7C,D). Immunofluorescence analysis further revealed a significant discontinuity in mesothelial layer bordering the abdominal cavity (Fig. 7C,D). Analysis by a TUNEL assay indicated CG-mediated injury is associated with apoptosis of mesothelial cells that is largely inhibited by the APC treatment (Suppl. fig. S5). This was also the case with the male mice where IHC analysis indicated majority of the cell surface TM was lost in peritoneal tissues of CG-treated mice, but a significant number of mesothelial cells in APC-treated group were positive for TM, suggesting TM expressing cells in the peritoneal membrane of CG-treated animals has been dramatically decreased (data not shown). These results implicate activation of the anticoagulant protein C pathway has also been adversely impacted on peritoneal mesothelial cells of the CG-treated animals and thus the dysregulation of the clotting cascade may also contribute to the pathogenesis of PF.
Figure 7. APC attenuates CG-induced PF in female mice.
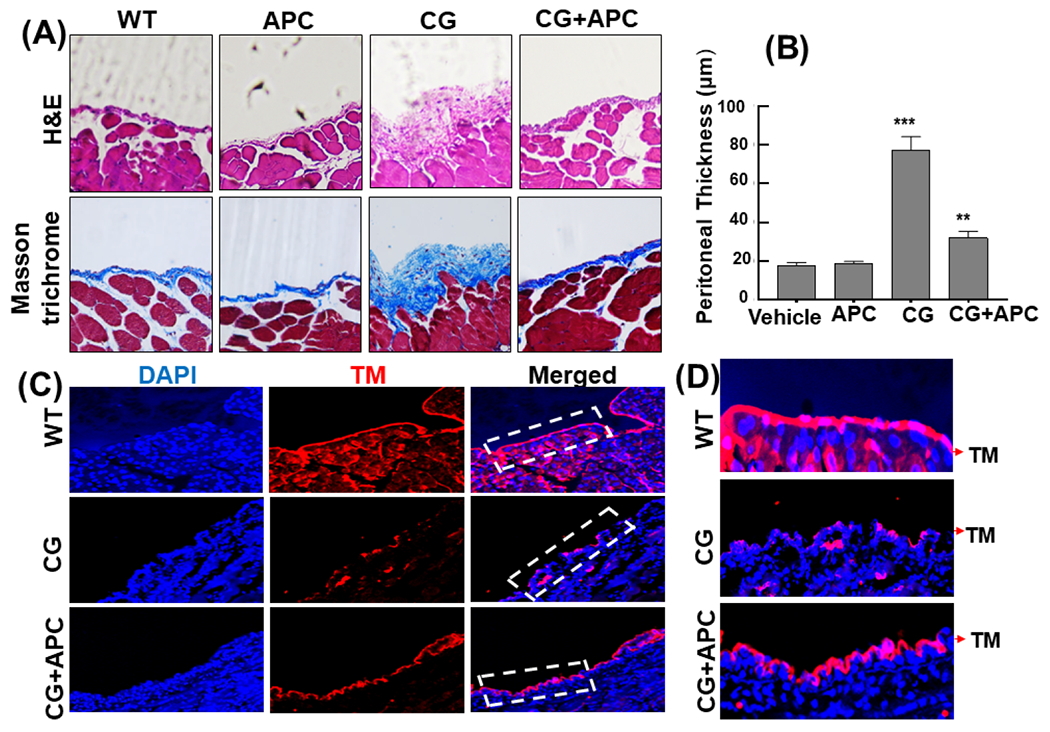
(A) Representative light microscopic features of H&E and Masson’s trichrome staining of peritoneal tissues on day 21 in control (vehicle, normal saline) mice, CG-injected mice treated with the vehicle alone, and CG-injected mice treated with APC-WT (50 μg/kg/day and n=5 for all groups). (B) Thickness of the submesothelial compact zone derived from the H&E staining. Data are mean ± SEM, **p<0.01 and ***p<0.001. (C) Peritoneal membrane cryosections were fixed, permeabilized, and incubated with anti-TM rat monoclonal antibody followed by Alexa Fluor 555-conjugated anti-rat antibody. DAPI was used to stain the nucleus. The magnified images correspond to the regions marked with white dashed boxes. The arrows in white dashed boxes mark the location of mesothelial TM on peritoneal membrane sections of the vehicle control, CG-treated and CG+APC-treated mice. (D) Restoration of APC-WT-mediated expression of TM in the peritoneal tissues of CG-treated mice. Scale bar =50μm.
Discussion
We have demonstrated in this study APC and the signaling-selective APC-2Cys variant are highly effective in preventing CG-induced peritoneal fibrosis (PF) in mice by downregulating proinflammatory cytokines and the profibrotic growth factor TGF-β1 in the peritoneal tissues of the experimental animals. APC effectively inhibited CG-mediated thickening of the submesothelial compact zone and excessive deposition of collagen in peritoneal tissues. The protective activity of APC was primarily mediated through EPCR-dependent activation of PAR1 as evidenced by inability of APC to inhibit CG-induced PF in either EPCR- or PAR1-deficient mice. Further support for this hypothesis was provided by the observation that the signaling-selective APC-2Cys mutant inhibited CG-mediated PF and collagen deposition in peritoneal tissues. The anticoagulant-selective APC-E170E also exhibited some protective effect, though to a lesser extent, in reducing CG-mediated peritoneal pathology, suggesting the activation of both coagulation and inflammatory pathways are involved in the pathogenesis of the disease. The anticoagulant function of APC-E170A is normal, thus it is likely that CG also induces the activation of the coagulation cascade and generation of thrombin, thereby promoting inflammation through activation of PAR1 on injured peritoneal membrane under conditions in which the TM receptor has been lost. The inhibition of thrombin generation by the APC-E70A variant may account for its partial protective effect in this model. The APC-S195A variant exhibited no protective effect, suggesting an intact active-site is required for APC to exert its protective effect against CG-mediated PF in mice.
Because of its clinical importance, understanding the pathogenic mechanism of peritoneal fibrosis has attracted much attention in recent years. It has been established that the activation of TGF-β1/Smad signaling is primarily responsible for the pathogenesis of fibrosis observed in experimental models of CG-mediated PF [11-14]. Previous results have indicated dysregulated TGF-β1 signaling causes a mesenchymal conversion of mesothelial cells by inducing epithelial-to-mesenchymal transition (EMT) in the chronically inflamed tissues of the peritoneum [12,15]. A key pathologic feature of the EMT is the loss of E-cadherin at intercellular junctions that has been shown to correlate with the ability of peritoneal mesothelial cells to adopt a mesenchymal migratory and invasive phenotype [15]. It has been also demonstrated the Snail family of transcription factors, which are upregulated by TGF-β/Smad signaling pathway, play key roles in the progression of the EMT process [15,17,25,26]. Our results suggest another EMT marker, N-cadherin, has been upregulated in CG-induced PF, suggesting E-cadherin to N-cadherin switch, which is a hallmark of EMT in cancer invasion and tumorigenesis [27,28], is also involved in the pathogenesis of CG-induced PF.
Interestingly, we also found the expression of TM in the peritoneal mesothelial cells has been essentially eliminated in the CG-treated mice, but APC significantly prevented TM loss in peritoneal tissues. TM is abundantly expressed in mesothelial cells [21]. In addition to its cofactor function in promoting the activation of protein C by thrombin, TM has direct anti-inflammatory function and plays a key role in protecting the peritoneal membrane from disruptive effects of proinflammatory cytokines and damage-associated molecular patterns released by dying cells under different pathophysiological conditions [29,30]. Thus, the loss of mesothelial TM can contribute to the pathogenesis of CG-induced PF. The mechanism by which CG induces the loss of TM from mesothelial cells is not known. It has been reported TM is a downstream target of the Snail transcription factor and is downregulated during the EMT [31]. Our results are consistent with the hypothesis that CG-induced TGF-β1/Smad signaling, and induction of the Snail transcription repressor is responsible for suppression of TM expression on mesothelial cells of the peritoneal membrane as well as vascular tissues of the peritoneum. It was interesting to discover that the signaling function of APC markedly inhibited/reversed pathogenic factors of CG-induced PF including EMT as evidenced by inhibiting the elevated expression levels of α-SMA, TGF-β1 and pSmad, collagens, N-cadherin, and vimentin in peritoneal tissues (Fig. 8). In agreement with results in the animal model, a potent protective effect for APC was also observed in the cellular model in which APC reversed the purified TGF-β1-mediated EMT-like phenotypes in HPMCs derived from human peritoneum.
Figure 8. Hypothetical model of the anti-fibrotic activity of APC in CG-induced PF.
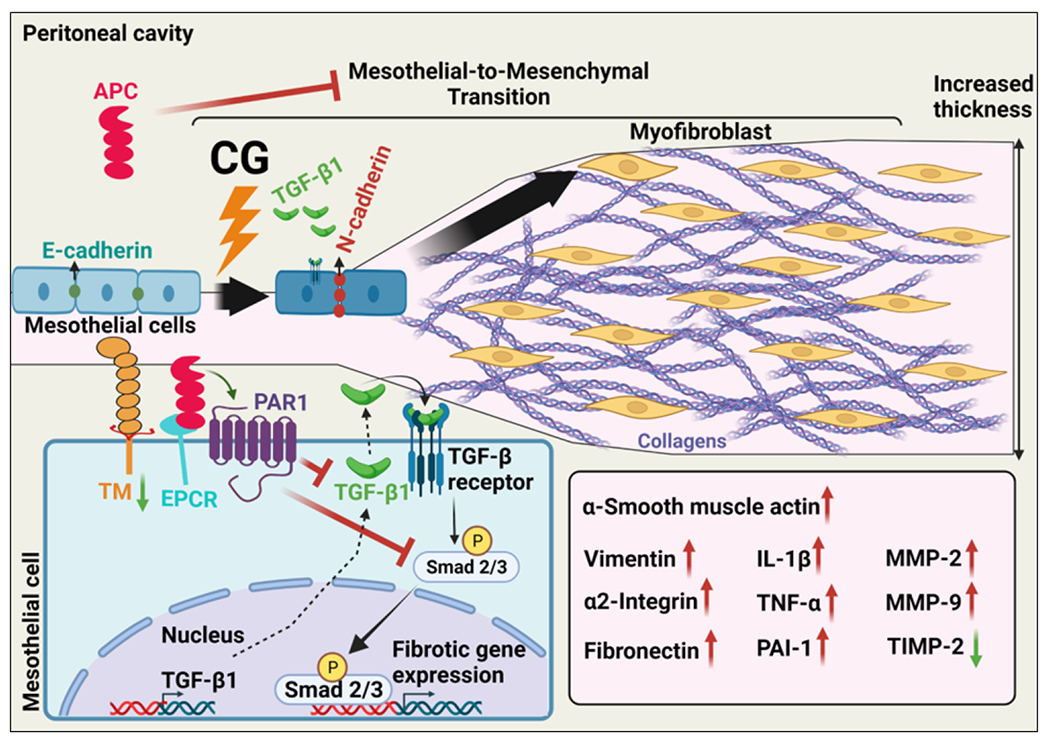
Intraperitoneal administration of CG induces PF by activation of TGF-β1/Smad signaling, thereby causing epithelial-to-mesenchymal transition (EMT) in the chronically inflamed tissues, which leads to increased thickness of the mesothelial compact zone. APC binds to EPCR on mesothelial cells, activating PAR1 and inhibiting CG-induced TGF-β1 signaling and EMT as well as deposition of collagens and elevated expression of α-SMA, vimentin N-cadherin and proinflammatory cytokines.
CG-induced PF has been used as a viable model system for understanding the pathogenic mechanism of PF in peritoneal dialysis, a treatment strategy for patients with end-stage kidney disease [11-14]. However, the unphysiological nature of the dialysate, used in the procedure, causes repeated injury to mesothelial cells of the peritoneum, thereby causing tissue fibrosis, thickening of the peritoneum and eventually failure of the filtration [15]. Similar to CG-induced PF, clinical, translational and cell-based data derived from patients undergoing peritoneal dialysis have indicated the pathogenic mechanism of dialysis-mediated PF involves excessive accumulation of fibrous connective tissue as the result of secretion of a significant amount of growth factors including TGF-β1 by mesothelial and fibroblast cells of the peritoneum that culminates in EMT, loss of E-cadherin, enhanced expression levels of α2-integrin and the transcription repressor Snail [15]. These markers of EMT were all detected in mesothelial cells isolated from effluents in dialysis fluid from patients undergoing continuous ambulatory peritoneal dialysis [15]. Based on remarkable observations of this study that APC through EPCR-dependent activation of PAR1 inhibits all similar pathologies observed in CG-induced PF in mice, we strongly believe the therapeutic potential of APC in dialysis-mediated PF in kidney patients warrants further investigation.
Addendum
H.G. and I.B. designed, performed experiments, and analyzed data. A.R.R. conceived, designed the experiments, analyzed data, and wrote the manuscript. All authors approved the final version of the manuscript.
Supplementary Material
1- Suppl. fig. S1
2- Suppl. fig. S2
3- Suppl. fig. S3
4- Suppl. fig. S4
5- Suppl. fig. S5
Essentials.
In addition to its anticoagulant function, APC possesses cytoprotective activities through EPCR-dependent activation of PAR1
We developed an experimental model of peritoneal fibrosis and evaluated the cytoprotective function of human APC in this model
APC inhibited fibrosis by downregulating TGF-β1 signaling, collagen deposition, and mesothelial-to-mesenchymal transition in peritoneal tissues
Studies with EPCR- and PAR1-deficient mice using APC variants indicate the signaling function of APC is primarily responsible for its anti-fibrotic activity
Acknowledgements
We would like to thank Dr. Hartmut Weiler for the EPCRδ/δ and PAR1−/− knockout mice and Dr. Peyman Dinarvand for breeding the mutant mice and conducting some of the CG-mediated peritoneal fibrosis experiments when he was a postdoctoral fellow at St. Louis University School of Medicine. We also thank Audrey Rezaie for proofreading the manuscript.
Funding Sources
This study was supported by a grant awarded by the National Heart, Lung, and Blood Institute of the National Institutes of Health (HL101917) to ARR.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure of Conflict of Interests
The authors declare no conflict of interests.
References
- 1.Esmon CT, Owen WG. Identification of an endothelial cell cofactor for thrombin-catalyzed activation of protein C. Proc Natl Acad Sci USA. 1981; 78:2249–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walker FJ, Fay PJ. Regulation of blood coagulation by the protein C system. FASEB J. 1992;6:2561–2567. [DOI] [PubMed] [Google Scholar]
- 3.Riewald M, Petrovan RJ, Donner A, Mueller BM, Ruf W. Activation of endothelial cell protease activated receptor 1 by the protein C pathway. Science. 2002; 296:1880–1882. [DOI] [PubMed] [Google Scholar]
- 4.Mosnier LO, Zlokovic BV, Griffin JH. The cytoprotective protein C pathway. Blood. 2007; 109:3161–3172. [DOI] [PubMed] [Google Scholar]
- 5.Mohan Rao LV, Esmon CT, Pendurthi UR. Endothelial cell protein C receptor: a multiliganded and multifunctional receptor. Blood. 2014; 124:1553–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang L, Bae JS, Manithody C, Rezaie AR. Identification of a specific exosite on activated protein C for interaction with protease-activated receptor 1. J Biol Chem. 2007; 282:25493–25500. [DOI] [PubMed] [Google Scholar]
- 7.Bae JS, Yang L, Manithody C, Rezaie AR. Engineering a disulfide bond to stabilize the calcium-binding loop of activated protein C eliminates its anticoagulant but not its protective signaling properties. J Biol Chem. 2007; 282:9251–9259. [DOI] [PubMed] [Google Scholar]
- 8.Costa R, Morrison A, Wang J, Manithody C, Li J, Rezaie AR. Activated protein C modulates cardiac metabolism and augments autophagy in the ischemic heart. J Thromb Haemost. 2012; 10:1736–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dinarvand P, Hassanian SM, Weiler H, Rezaie AR. Intraperitoneal administration of activated protein C prevents postsurgical adhesion band formation. Blood. 2015; 125:1339–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lattenist L, Jansen MP, Teske G, Claessen N, Meijers JC, Rezaie AR, Esmon CT, Florquin S, Roelofs JJ. Activated protein C protects against renal ischaemia/reperfusion injury, independent of its anticoagulant properties. Thromb Haemost. 2016; 116:124–133. [DOI] [PubMed] [Google Scholar]
- 11.Yokoi H, Kasahara M, Mori K, Ogawa Y, Kuwabara T, Imamaki H, Kawanishi T, Koga K, Ishii A, Kato Y, Mori KP, Toda N, Ohno S, Muramatsu H, Muramatsu T, Sugawara A, Mukoyama M, Nakao K. Pleiotrophin triggers inflammation and increased peritoneal permeability leading to peritoneal fibrosis. Kidney Int. 2012; 81:160–169. [DOI] [PubMed] [Google Scholar]
- 12.Ueno T, Nakashima A, Doi S, Kawamoto T, Honda K, Yokoyama Y, Doi T, Higashi Y, Yorioka N, Kato Y, Kohno N, Masaki T. Mesenchymal stem cells ameliorate experimental peritoneal fibrosis by suppressing inflammation and inhibiting TGF-beta1 signaling. Kidney Int. 2013; 84:297–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lua I, Li Y, Pappoe LS, Asahina K. Myofibroblastic Conversion and Regeneration of Mesothelial Cells in Peritoneal and Liver Fibrosis. Am J Pathol. 2015; 185:3258–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xiong C, Liu N, Fang L, Zhuang S, Yan H. Suramin inhibits the development and progression of peritoneal fibrosis. J Pharmacol Exp Ther. 2014; 351:373–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yanez-Mo M, Lara-Pezzi E, Selgas R, Ramírez-Huesca M, Domínguez-Jiménez C, Jiménez-Heffernan JA, Aguilera A, Sánchez-Tomero JA, Bajo MA, Alvarez V, Castro MA, del Peso G, Cirujeda A, Gamallo C, Sánchez-Madrid F, López-Cabrera M. Peritoneal dialysis and epithelial-to-mesenchymal transition of mesothelial cells. N Engl J Med. 2003; 348:403–413. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Z, Jiang N, Ni Z. Strategies for preventing peritoneal fibrosis in peritoneal dialysis patients: new insights based on peritoneal inflammation and angiogenesis. Front Med. 2017; 11:349–358. [DOI] [PubMed] [Google Scholar]
- 17.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009; 119:1420–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Castellino FJ, Liang Z, Volkir SP, Haalboom E, Martin JA, Sandoval-Cooper MJ, Rosen ED. Mice with a severe deficiency of the endothelial protein C receptor gene develop, survive, and reproduce normally, and do not present with enhanced arterial thrombosis after challenge. Thromb Haemost. 2002; 88:462–472. [PubMed] [Google Scholar]
- 19.Darrow AL, Fung-Leung WP, Ye RD, Santulli RJ, Cheung WM, Derian CK, Burns CL, Damiano BP, Zhou L, Keenan CM, Peterson PA, Andrade-Gordon P. Biological consequences of thrombin receptor deficiency in mice. Thromb Haemost. 1996; 76:860–866. [PubMed] [Google Scholar]
- 20.Connolly AJ, Ishihara H, Kahn ML, Farese RV Jr, Coughlin SR. Role of the thrombin receptor in development and evidence for a second receptor. Nature. 1996; 381:516–519. [DOI] [PubMed] [Google Scholar]
- 21.Verhagen HJ, Heijnen-Snyder GJ, Pronk A, Vroom TM, van Vroonhoven TJ, Eikelboom BC, Sixma JJ, de Groot PG. Thrombomodulin activity on mesothelial cells: perspectives for mesothelial cells as an alternative for endothelial cells for cell seeding on vascular grafts. Br J Haematol. 1996; 95:542–549. [DOI] [PubMed] [Google Scholar]
- 22.Belling F, Ribeiro A, Wörnle M, Ladurner R, Mussack T, Sitter T, Sauter M. PAR-1 mediates the thrombin-induced mesothelial cell overproduction of VEGF and PAI-1. Int J Artif Organs. 2013; 36:97–104. [DOI] [PubMed] [Google Scholar]
- 23.lakhiaev AV, Rezaie AR, Idell S. Thrombomodulin-mediated catabolism of protein C by pleural mesothelial and vascular endothelial cells. Thromb Haemost. 2007; 98:627–634. [PubMed] [Google Scholar]
- 24.Bochenek ML, Gogiraju R, Großmann S, Krug J, Orth J, Reyda S, Georgiadis GS, Spronk HM, Konstantinides S, Münzel T, Griffin JH, Wild P, Espinola-Klein C, Ruf W, Schafer K. EPCR-PAR1 biased signaling regulates perfusion recovery and neovascularization in peripheral ischemia. JCI Insight. 2022;7:e157701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dhasarathy A, Phadke D, Mav D, Shah RR, Wade PA. The transcription factors Snail and Slug activate the transforming growth factor-beta signaling pathway in breast cancer. PLoS One. 2011; 6:e26514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou BP, Deng J, Xia W, Xu J, Li YM, Gunduz M, Hung MC. Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat Cell Biol. 2004; 6:931–940. [DOI] [PubMed] [Google Scholar]
- 27.Loh CY, Chai JY, Tang TF, Wong WF, Sethi G, Shanmugam MK, Chong PP, Looi CY. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells. 2019; 8:1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dong C, Wu Y, Yao J, Wang Y, Yu Y, Rychahou PG, Evers BM, Zhou BP. G9a interacts with Snail and is critical for Snail-mediated E-cadherin repression in human breast cancer. J Clin Invest. 2012; 122:1469–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Conway EM, Van de Wouwer M, Pollefeyt S, Jurk K, Van Aken H, De Vriese A, Weitz JI, Weiler H, Hellings PW, Schaeffer P, Herbert JM, Collen D, Theilmeier G. The lectin-like domain of thrombomodulin confers protection from neutrophil-mediated tissue damage by suppressing adhesion molecule expression via nuclear factor kappaB and mitogen-activated protein kinase pathways. J Exp Med. 2002; 196:565–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ito T, Maruyama I. Thrombomodulin: protectorate God of the vasculature in thrombosis and inflammation. J Thromb Haemost. 2011; 9 (Suppl 1):168–173. [DOI] [PubMed] [Google Scholar]
- 31.Kao YC, Wu LW, Shi CS, Chu CH, Huang CW, Kuo CP, Sheu HM, Shi GY, Wu HL. Downregulation of thrombomodulin, a novel target of Snail, induces tumorigenesis through epithelial-mesenchymal transition. Mol Cell Biol. 2010; 30:4767–4785. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1- Suppl. fig. S1
2- Suppl. fig. S2
3- Suppl. fig. S3
4- Suppl. fig. S4
5- Suppl. fig. S5