

SUMMARY
Uropathogenic E. coli (UPEC) is a primary organism responsible for urinary tract infections and a common cause of sepsis. Microbially experienced laboratory mice, generated by cohousing with pet store mice, exhibit increased morbidity and mortality to polymicrobial sepsis or lipopolysaccharide challenge. By contrast, cohoused mice display significant resistance, compared with specific pathogen-free mice, to a monomicrobial sepsis model using UPEC. CD115+ monocytes mediate protection in the cohoused mice, as depletion of these cells leads to increased mortality and UPEC pathogen burden. Further study of the cohoused mice reveals increased TNF-α production by monocytes, a skewing toward Ly6ChiCD115+ “classical” monocytes, and enhanced egress of Ly6ChiCD115+ monocytes from the bone marrow. Analysis of cohoused bone marrow also finds increased frequency and number of myeloid multipotent progenitor cells. These results show that a history of microbial exposure impacts innate immunity in mice, which can have important implications for the preclinical study of sepsis.
In brief
Cohousing laboratory mice with pet store mice changes the immune system and alerts responsiveness to future challenges. Martin et al. show that microbial exposure drives changes to the innate immune system, including a skewing toward classical phenotype monocytes driven by enhanced myelopoiesis, resulting in increased resistance to systemic bacterial infection.
Graphical Abstract
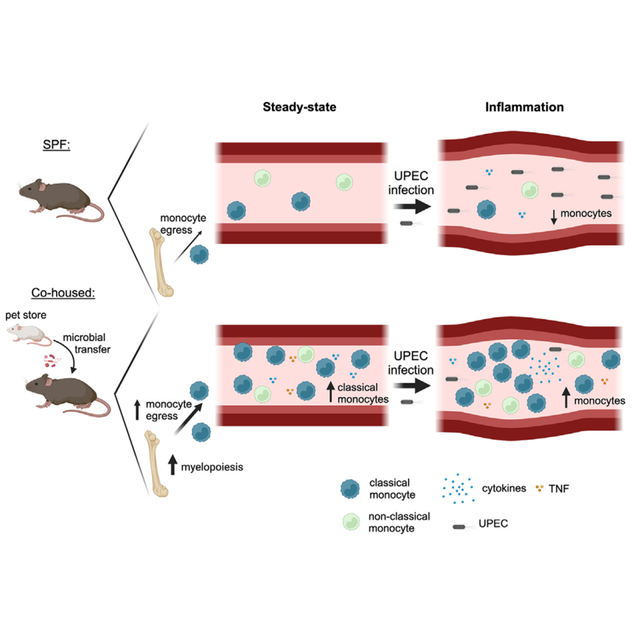
INTRODUCTION
Sepsis is defined as a life-threatening organ dysfunction resulting from a dysregulated host immune response to an infection.1 Each year, ~2 million people in the U.S. experience a septic event, leading to 300,000 deaths.2,3 Bacterial infections are the most common inducers of sepsis, but severe viral/fungal infections or traumatic injury can also progress to sepsis.4,5 The lungs, gastrointestinal tract, kidneys, and bladder are frequent infection sites that can eventually develop into full-blown sepsis.6 For example, if uropathogenic E. coli (UPEC), which causes a majority of urinary tract infections (UTIs), ascends from the bladder to cause pyelonephritis, in severe cases the bacteria can enter the bloodstream causing bacteremia and sepsis.7
Preclinical research using mice has shown that the immune system is involved in the development of pathologies arising due to sepsis and the response to treatments.8 However, knowledge of immune system involvement in the response during a septic event is incomplete, and data from mouse sepsis studies have inconsistently translated to humans. Thus, development and use of experimental models with increased physiological and clinical relevance are critically needed to maintain the robustness of preclinical sepsis research. One important difference between the mice used in almost all preclinical studies and humans is that laboratory mice are typically housed under specific pathogen-free (SPF) conditions, while humans are exposed to a diverse array of commensal and pathogenic microbes (e.g., bacteria, viruses, or fungi) throughout their lives. This microbial exposure, together with vaccinations received over time, shapes the immune system of a person as they age to provide rapid, robust, and long-lasting protection against future infection. However, an immune system primed for a quick and vigorous response can be detrimental to the host in some situations such as sepsis.
The immune system of SPF mice is dominated by naive phenotype immune cells, especially within the T cell compartment, and resembles that of neonatal humans.9,10 Sequential infection of SPF-housed laboratory mice with common experimental pathogens, such as MHV, MCMV, Listeria monocytogenes, LCMV, and influenza A virus,10,11 or cohousing SPF mice with pet store mice carrying multiple pathogenic and commensal bacteria, viruses, and/or fungi9 induces immune system alterations and maturations that more closely resemble the adult human immune system. Such microbial exposure drastically alters the composition and function of the immune system of laboratory mice, which can significantly influence the overall outcome (and survival) to subsequent infection. For example, cohoused (CoH) mice have higher frequencies of phagocytes (Ly6G+ neutrophils and CD64+ monocytes) and Ag-experienced T cells in the circulation and secondary lymphoid organs.12 CoH mice also display qualitatively different T cell-mediated responses and are better able to clear virulent L. monocytogenes infection than SPF mice.9,12
The magnitude of the immune response to a systemic infection plays a major role in both pathogen clearance and host survival.13 One of the canonical features of sepsis is the development of a “cytokine storm.”14 Data from our lab found that CoH mice produce an exaggerated cytokine storm and are more susceptible, compared with SPF mice, to polymicrobial sepsis induced by cecal ligation and puncture (CLP) surgery or cecal slurry injection, or sterile inflammation induced by lipopolysaccharide (LPS) challenge.12 An uncontrolled inflammatory response can result in mortality, but an insufficient response will not clear the infecting pathogen.15 At present, it is unknown how previous microbial exposure influences susceptibility to monomicrobial sepsis or how alterations in innate or adaptive compartments due to microbial exposure determine sepsis outcomes.
As E. coli are a Gram-negative bacteria with abundant LPS in their outer membrane16 and CoH mice are more susceptible to LPS endotoxemia,12 we hypothesized CoH mice would exhibit an enhanced cytokine storm and be more susceptible to sepsis induced by systemic UPEC infection (modeling monomicrobial sepsis). CoH mice did display an increased systemic cytokine response following UPEC infection compared with SPF mice, but contrary to our hypothesis CoH mice were found to be less susceptible to systemic UPEC infection and the UPEC burden in CoH mice was significantly lower in multiple tissues compared with SPF mice 24 h after infection. Interestingly, CD115+ monocytes were crucial for providing CoH mice with enhanced protection against systemic UPEC. These results advance our knowledge of how generalized microbial exposure shapes the phenotype of the innate immune system, in addition to the adaptive immune system, and they further support the use of microbially experienced mice to advance clinically relevant mouse models, including those related to the study of sepsis.
RESULTS
Microbially experienced CoH mice show increased resistance against systemic UPEC infection
Cohousing SPF laboratory mice with microbially experienced pet store mice permits the transfer of diverse microbes that activate and mature the immune system, which can be most easily seen by the increase in memory T cell frequency.9,12 The cohousing model established by Beura et al. combines eight female laboratory mice with one female pet store mouse in a large mouse cage (nine mice total) located in a non-SPF housing facility.17 We and others have used such “dirty” mice to evaluate immune responses in a variety of experimental settings compared with age-matched SPF mice.12,18–23 After 60 days of cohousing, the activation state of circulating CD4 and CD8 T cells in all CoH mice was assessed before use, as a means of confirming microbial exposure. Flow cytometric analysis of peripheral blood leukocytes of female CoH C57Bl/6 (B6) mice revealed increased percentages of CD11a-hiCD49dhi Ag-experienced CD4 T cells24–26 and CD8loCD11ahi Ag-experienced CD8 T cells27 compared with female SPF mice (Figures S1A and S1B). The majority of CD8loCD11ahi Ag-experienced CD8 T cells from CoH mice also had an effector/memory phenotype (i.e., CD62L− and/or KLRG1+). Despite using different pet store mice (often from different local pet stores) for each cohousing cohort, we found consistent increases in CD44hi CD8 T cells in the CoH B6 mice used (Figure S1C), which is similar with recently published data.21
CoH mice exhibit an exacerbated cytokine storm and increased mortality during CLP-induced sepsis and LPS endotoxemia.12 Thus, we were interested to determine whether CoH mice were similarly more susceptible to monomicrobial sepsis caused by systemic infection with Gram-negative UPEC strain UTI89.28 Female SPF and CoH mice were injected intravenously with 4 × 107 CFU UPEC/mouse and survival was monitored. In contrast to other models of sepsis we have used, CoH mice were more resistant to systemic UPEC infection than SPF mice, as 78% of CoH mice survived to day 7 following infection while 94% of SPF mice succumbed to the infection by day 4 (Figure 1A). Pet store mice had similar increased resistance to systemic UPEC infection compared with SPF animals (58% vs. 6%; Figure S1D). We next determined bacterial titers in the blood, spleen, liver, and kidney 24 h after infection to determine the extent to which the increased survival of CoH mice was due to more efficient UPEC clearance. Significantly lower UPEC titers were found in all organs examined from CoH mice compared with SPF mice (Figure 1B), indicating that CoH mice had the capacity to rapidly contain a systemic UPEC infection. We also tested whether CoH mice would have enhanced survival from a different monomicrobial infection. CoH and SPF mice were intravenously infected with virulent L. monocytogenes, modeling listeriosis,29 and survival was monitored over time. Similar to increased resistance to systemic UPEC infection, CoH mice had less mortality than SPF mice after systemic infection with L. monocytogenes (Figure S1E). Collectively, these data suggest that exposure to diverse pathogenic and commensal microbes establishes a state of increased resistance against systemic UPEC infection.
Figure 1. Microbially experienced CoH mice demonstrate increased resistance against systemic UPEC-induced sepsis.
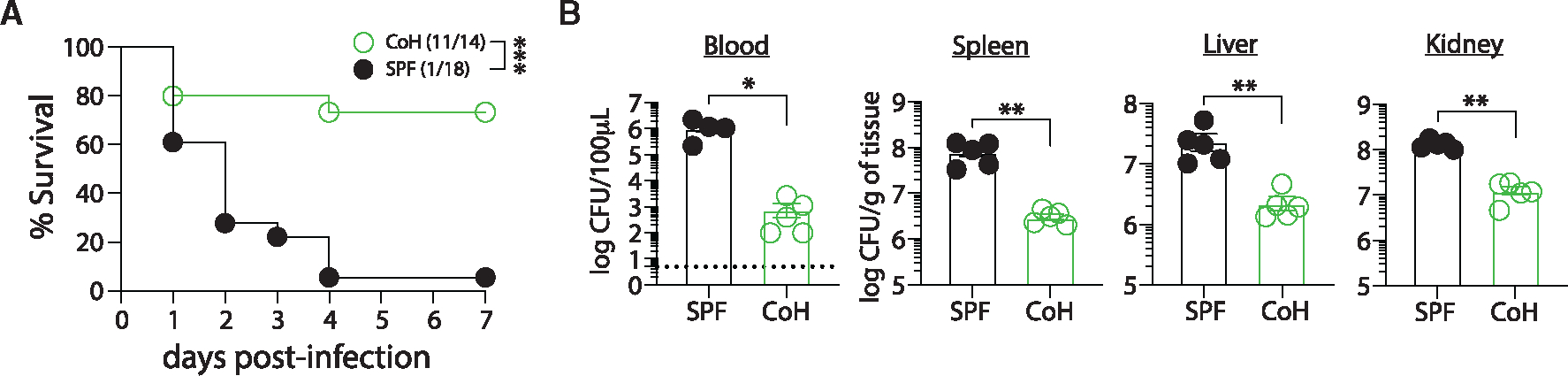
Female SPF and CoH mice were infected with 4 × 107 colony forming units (CFUs) of uropathogenic E. coli (UPEC) intravenously.
(A) Survival at the indicated days post-infection.
(B) Bacterial CFU per 100 μL of blood or gram of spleen, liver, and kidney tissue of SPF and CoH mice 24 h following infection. *p ≤ 0.05, **p ≤ 0.01, and ***p ≤ 0.005 as determined by log rank test in (A) or nonparametric Mann-Whitney test in (B). Data in (A) were combined from two experiments lasting 5–7 days using a total of 14–18 mice per group. Data in (B) are representative from three experiments using 4–5 mice per group, where each symbol represents a mouse and bars indicate means with SEM. Dashed line in (B) indicates limit of detection of the assay.
CoH mice exhibit a heightened inflammatory response and increased number of CD115+ monocytes following systemic UPEC infection
We next wanted to experimentally address if the differences in survival and UPEC clearance between SPF and CoH mice were driven by differences in the immune response following infection. We first examined the magnitude of the inflammatory cytokine/chemokine response in SPF and CoH mice 3 h following UPEC infection, an early time point meant to capture the initial cytokine storm, as well as 24 h after UPEC infection. Consistent with what we saw in other sepsis models, serum concentrations of multiple cytokines and chemokines, including IFN-γ, IL-12p70, IL-6, TNF-α, CCL3, CCL4, and CCL5, were significantly greater in UPEC-infected CoH mice than SPF mice (Figures 2A and 2B). These same cytokines and chemokines were also increased in the serum from uninfected CoH vs. SPF mice (Figures 2A and 2B), which is consistent with our previous findings.12 These data suggest that CoH mice are primed to respond more vigorously and quickly during a systemic UPEC infection.
Figure 2. CoH mice exhibit a heightened inflammatory response following systemic UPEC-induced sepsis.
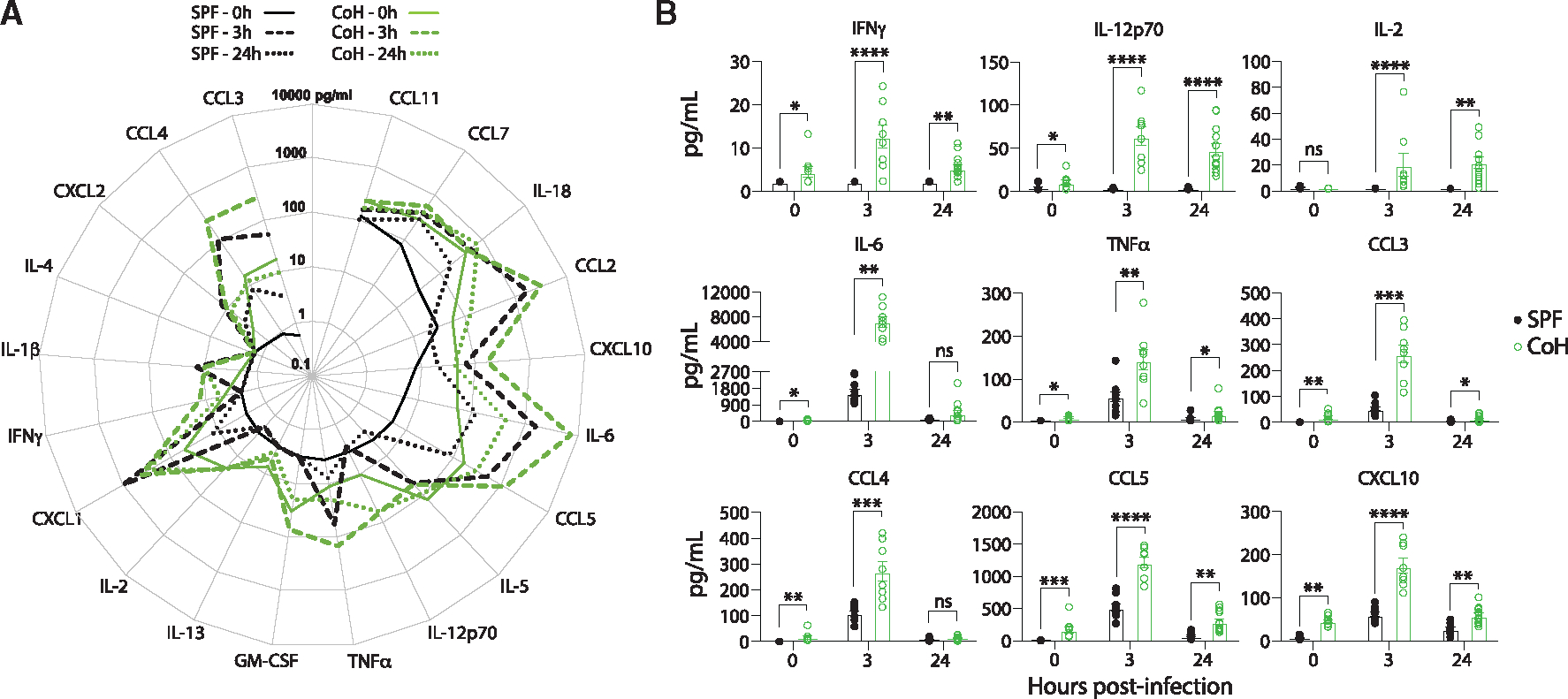
Blood was collected from SPF and CoH mice prior to infection (0 h), and 3 and 24 h after infection with 4 × 107 CFU UPEC i.v. The concentration of 20 cytokines and chemokines in the serum was determined by Luminex.
(A) Radar plot shows the average steady state, 3 and 24 h post-infection serum concentrations (pg/mL) of the indicated cytokines and chemokines.
(B) Amount (pg/mL) of IFN-γ, IL-12p70, IL-2, IL-6, TNF-α, CCL3, CCL4, and CCL5 in serum prior to (0 h), and 3 and 24 h after UPEC infection. For statistical comparisons, SPF mice with undetectable cytokines were given a value of “0.” **p ≤ 0.01, ***p ≤ 0.005, and ****p < 0.0001 as determined by nonparametric Mann-Whitney test. Data in (A) and (B) were combined from two experiments using a total of 7–10 mice per group, where each symbol in (B) represents a mouse and bars indicate means with SEM.
To determine how immune cell numbers in SPF and CoH mice were affected by the systemic UPEC infection, CD4 T cells, CD8 T cells, B cells, NK cells, CD11b+CD115+ monocytes (hereafter referred to as CD115+ monocytes), and neutrophils in the spleens and blood of SPF and CoH mice were quantitated before and 24 h after systemic UPEC infection. Prior to infection, the number of each cell type—except for B cells—was increased in CoH mice (Figures 3A and 3B). Following infection, T cell and B cell numbers were maintained in both SPF and CoH spleens, while NK cell numbers decreased slightly (but significantly) in CoH mice (Figure 3A). Neutrophil numbers increased in both SPF and CoH spleens, which was expected under the early conditions of the infection. The most interesting change in the spleen occurred within the CD115hi monocyte compartment, marked by a significant decrease in SPF mice vs. increase in CoH mice compared with uninfected mice (Figure 3A). In the blood, a significant reduction in number of CD4 T cells, CD8 T cells, B cells, NK cells, neutrophils, and CD115+ monocytes was observed in SPF mice 24 h after UPEC infection compared with uninfected SPF mice (Figure 3B). While a less prominent, but significant, reduction was seen in CoH blood CD4 T cells, CD8 T cells, and B cells 24 h after UPEC infection, the number of CoH NK cells and neutrophils in the blood did not drop (Figure 3B). Most strikingly, unlike the marked reduction of SPF CD115+ monocytes after UPEC infection, blood CD115+ monocytes increased in number in CoH mice (Figure 3B), similar to the increased number of CD115+ monocytes in CoH spleens (Figure 3A). These increases in CD115+ monocytes correlated with the overall survival (Figure 1A) and UPEC CFU numbers in the tissues (Figure 1B). In total, the data in Figures 1, 2, and 3 suggest that the survival and UPEC clearance advantages seen for CoH mice relate to the immune response generated, and the most notable immune cell difference between SPF and CoH was observed within the monocyte compartment.
Figure 3. CoH mice exhibit increased numbers of CD115+ monocytes following systemic UPEC infection.
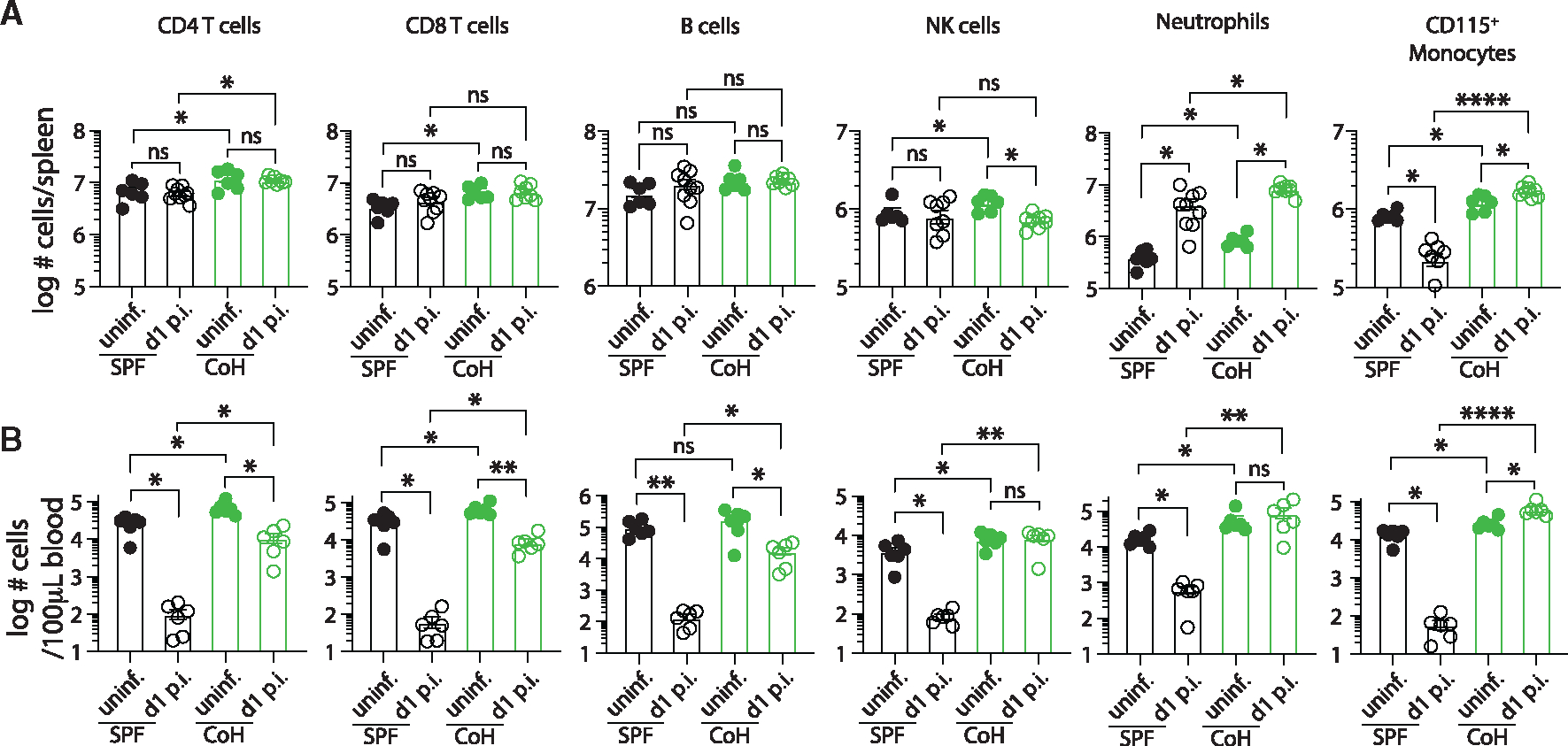
Number of CD4 T cells, CD8 T cells, B cells, NK cells, neutrophils, and CD115+ monocytes in the (A) spleen and (B) blood of SPF and CoH mice before and 24 h after systemic UPEC infection. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.005, and ****p < 0.0001 as determined by Kruskal-Wallis test, with a Dunn’s post hoc test to correct for multiple comparisons. Data in (A) and (B) were combined from two experiments using a total of 6–9 mice per group, where each symbol represents a mouse and bars indicate means with SEM.
CD115+ monocytes from CoH mice are necessary to protect against systemic UPEC infection
We were intrigued by the data showing increased numbers of CD115+ monocytes in the spleens and blood of CoH mice after UPEC infection, leading us to speculate that these cells may be major contributors to the resistance to systemic UPEC infection in CoH mice. Thus, we examined the impact of depleting these cells from CoH mice prior to infection using an anti-mouse CD115 monoclonal antibody (mAb).30,31 We also examined the impact of depleting CD4 T cells or CD8 T cells from the CoH mice because of the skewing of the T cell compartment to Ag-experienced memory cells in CoH mice compared with the predominantly naive T cells in SPF mice. To confirm the efficiency of each mAb used for depletion, we first measured the frequency of CD4 T cells, CD8 T cells, and CD115+ monocytes among CD45+ peripheral blood leukocytes prior to infection. Indeed, the percentages of CD4 T cells, CD8 T cells, and CD115+ monocytes were significantly lower in CoH mice injected with the respective depleting mAb compared with mice that received control IgG (cIgG) (Figures 4A and 4B). Survival of cIgG-treated CoH mice was significantly greater than SPF mice after UPEC infection (Figure 4C), which agrees with the survival differences between SPF and CoH mice shown in Figure 1A. Depletion of either CD4 or CD8 T cells did not influence resistance of CoH mice to infection, suggesting that these populations of cells do not meaningfully contribute to the survival advantage seen in CoH mice following systemic UPEC infection. By contrast, susceptibility of CoH mice to UPEC infection was significantly increased after removal of CD115+ cells (Figure 4C), suggesting that these cells are necessary mediators of the survival advantage for UPEC-infected CoH mice.
Figure 4. CD115+ cells mediate protection in CoH mice against systemic UPEC infection.
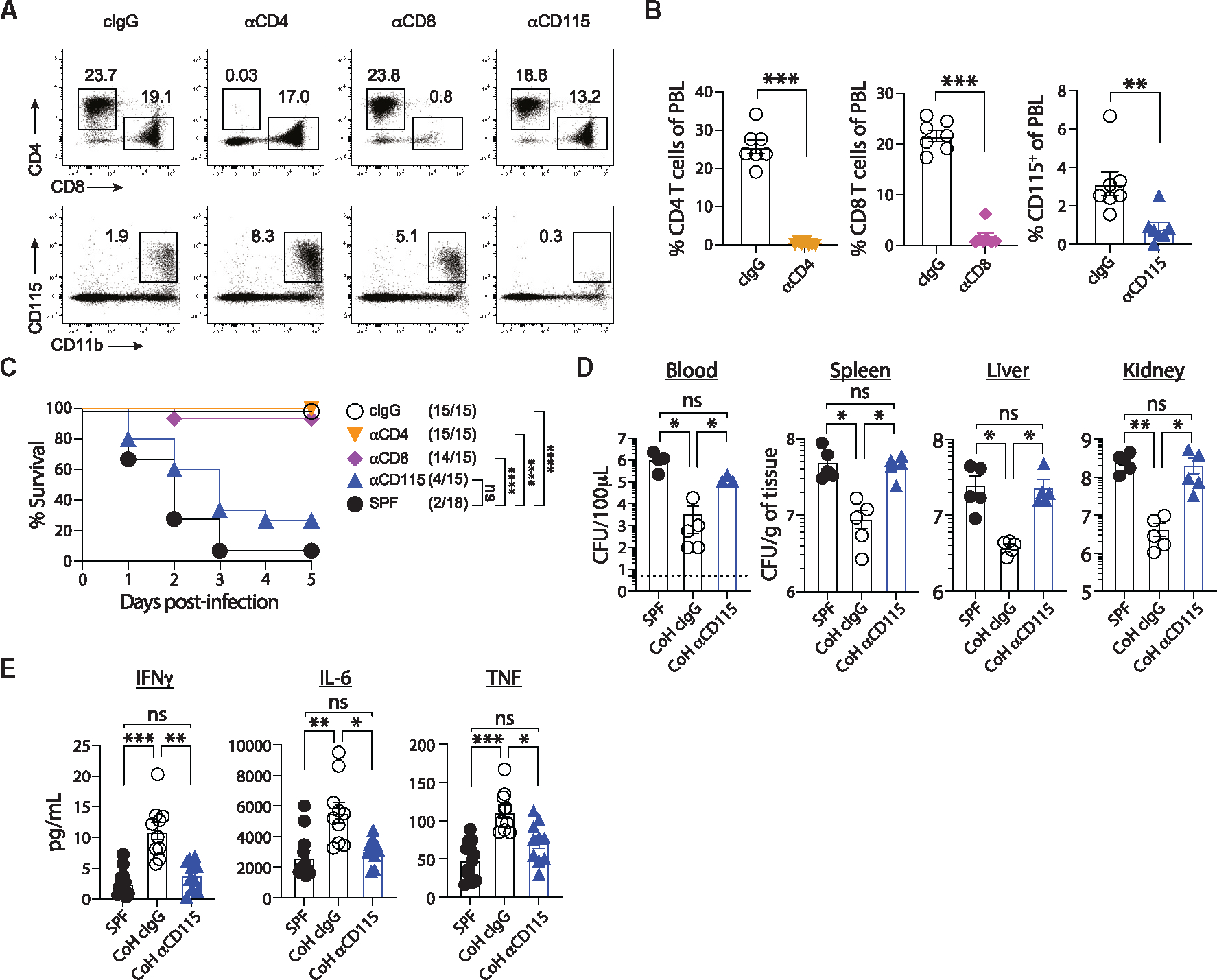
(A–C) CoH mice were injected with control IgG or mAb to deplete CD4 T cells, CD8 T cells, or CD115+ monocytes. (A) Representative dot plots showing detection of CD4 and CD8 T cells (top) and CD115+ monocytes (bottom) in CoH mice after injection with control IgG or anti-CD4, -CD8, or -CD115 depleting mAbs. (B) Percentage of CD4 T cells, CD8 T cells, or CD115+ monocytes among peripheral blood lymphocytes in CoH mice injected with control IgG or anti-CD4, -CD8, or -CD115 depleting mAbs. (C) Survival of SPF mice, CoH mice injected with anti-CD115, anti-CD8, anti-CD4, or control IgG at the indicated days post-infection.
(D) Bacterial CFU per 100 μL of blood or grams of spleen, liver, and kidney 24 h following UPEC infection from SPF mice, CoH mice injected with control IgG, or CoH mice injected with anti-CD115 mAbs.
(E) Serum IFN-γ, IL-6, and TNF-α concentrations from SPF mice, CoH mice injected with control IgG, or CoH mice injected with anti-CD115 mAb 3 h following UPEC infection. ns, not significant, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and ***p ≤ 0.0001, as determined by nonparametric Mann-Whitney test (B), log rank test (C), or Kruskal-Wallis test, with a Dunn’s post hoc test to correct for multiple comparisons (D and E). Data in (A) and (B) are representative from three experiments using 7–8 mice per group, or 4–5 mice per group in (D). Combined data from three experiments using a total of 15–18 mice per group are in (C), and two experiments using a total of 10–12 mice per group are in (E). Each symbol in (B, D, and E) represents a mouse and bars indicate means with SEM. Dashed line in (D) indicates limit of detection of the assay.
To determine the contribution of monocytes in the accelerated UPEC clearance seen in CoH mice, we next depleted CD115+ cells from CoH mice and determined bacterial titers in blood, spleen, liver, and kidney of these mice, as well as SPF mice and cIgG-treated CoH mice, 24 h after infection. While the UPEC burden was significantly lower in cIgG-treated CoH mice compared with SPF mice (which is also consistent with the clearance data shown in Figure 1B), the UPEC burden in CoH mice depleted of CD115+ cells was not statistically different from that seen in SPF mice (Figure 4D). CD115+ mAb depletion also reduced the magnitude of the systemic cytokine response, as the circulating levels of IFN-γ, IL-6, and TNF 3 h after UPEC were reduced to what was measured in SPF mice (Figure 4E), as well as CXCL10, GM-CSF, IL-2, and IL-12p70 (Figure S2). In sum, these data suggest that CD115+ cells from CoH mice are necessary mediators of enhanced protection against systemic UPEC infection and significantly contribute toward the systemic cytokine response.
CoH monocytes/macrophages have enhanced TNF-α production
Since CoH mice had heightened chemokine/cytokine production after UPEC infection compared with SPF mice and that this difference was diminished with depletion of CD115+ cells, we next wanted to assess the functional capacity of CoH vs. SPF monocytes and macrophages (Mφ) to produce TNF-α production, a cytokine known to be important in pathogen clearance.32,33 Because of the rapidity of cytokine production in vivo, a whole blood ex vivo LPS-induced TNF-α production assay, which is used to evaluate innate immune function in sepsis patients,34 was utilized. Higher amounts of TNF-α were detected in samples containing whole blood from CoH mice compared with SPF whole blood (Figure 5A). Similar results were seen when using SPF and CoH splenocytes stimulated in vitro for 4 h with either purified LPS, UPEC or left unstimulated, as CoH splenocyte cultures exhibited greater production of TNF-α and increased frequency and number of TNF-α+ cells (Figure 5B).
Figure 5. CoH monocyte/Mφ produce more TNF-α than SPF monocyte/Mφ during in vitro culture.
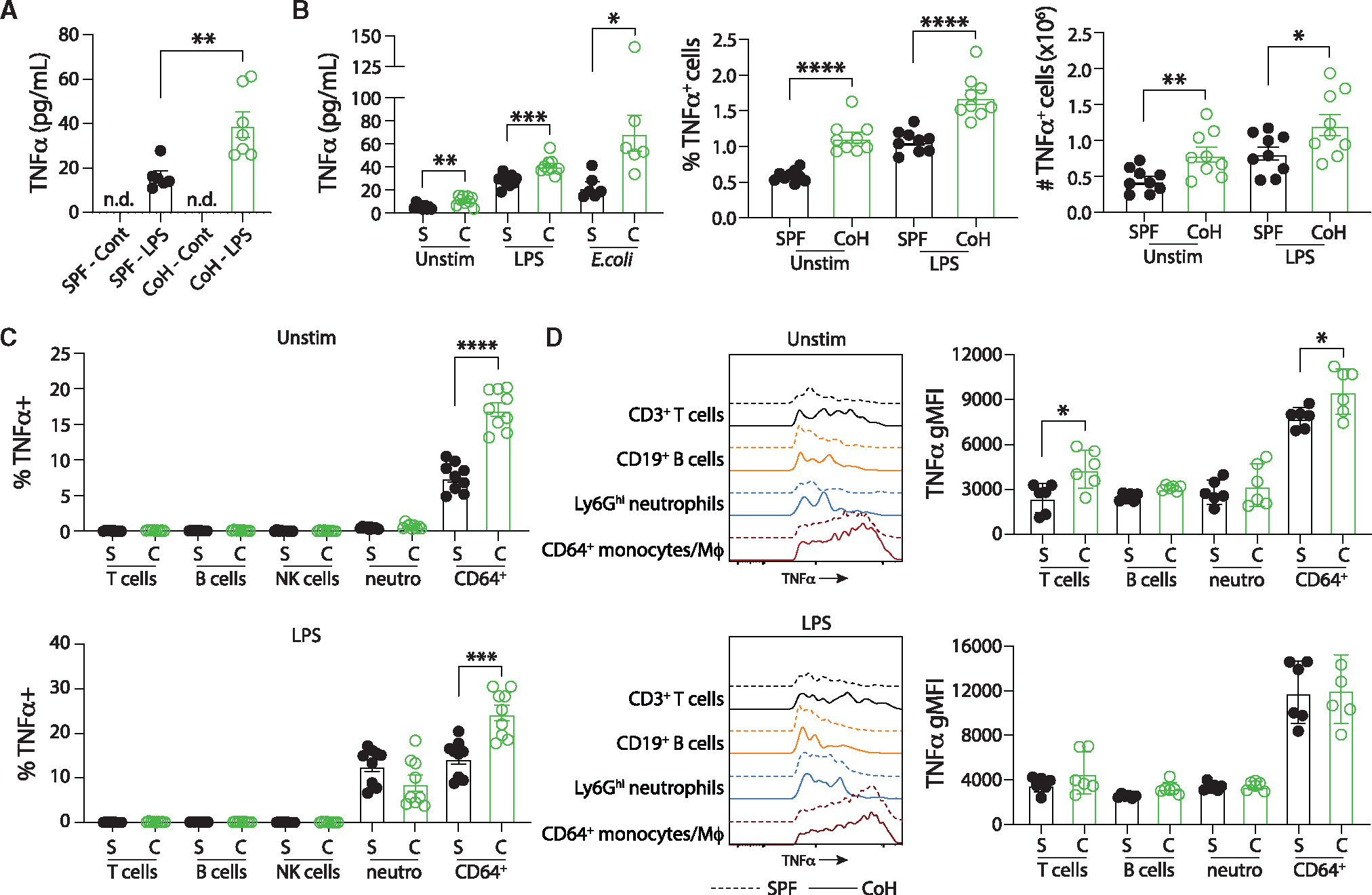
(A) Steady-state innate immune function was measured ex vivo by adding heparinized whole blood to tubes containing RPMI alone (“control” tubes) or RPMI and 0.5 ng/mL LPS and incubated for 4 h at 37°C. The amount of TNF-α in the supernatant was determined by ELISA.
(B) SPF and CoH splenocytes (2 × 106 cells) were incubated alone or with either LPS (100 ng/mL) or UPEC (5:1 UPEC:splenocytes) for 4 h. The amount of TNF-α in the supernatant was then measured by ELISA (left) and the frequency (middle) and number (right) of TNF-α+ cells was determined by flow cytometry.
(C) The frequency of TNF-α+ cells within T cells, B cells, NK cells, neutrophils, and CD64+ monocytes/M4 from the unstimulated and LPS-stimulated splenocyte cultures were also determined by flow cytometry.
(D) Geometric mean fluorescence intensity (gMFI) of TNF-α expression by unstimulated and LPS-stimulated TNF-α+ T cells, B cells, NK cells, neutrophils, and CD64+ monocytes/Mφ was determined by flow cytometry. Representative plots and cumulative data are shown and data in (A–D) were combined from at least two independent experiments using a total of 6–9 mice per group, where each symbol represents a mouse and bars indicate means with SEM. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.005, and ****p < 0.0001 as determined by nonparametric Mann-Whitney test.
CD115 (CSF-1R) is a protein commonly used to identify monocytes (with CD11b) as CD115 expression is highest on this subset, but it is cleaved from the surface of cells when cultured at 37°C and hence unable to be used to identify these cells in in vitro culture studies (Figure S3A).35,36 CD64 can also be used to identify monocytes/Mφ and, while it has less robust expression than CD115 on monocytes (Figure S3B), its expression is not lost with in vitro culture and was a good marker to identify monocytes/Mφ in this setting. Analysis of immune cell subsets within the splenocyte culture determined that CD64+ monocytes/Mφ had the highest percentage of TNF-α-producing cells after 4 h LPS exposure as well as without stimulation (Figures S4A, S4B, and 5C). In addition, when first gating on TNF-α-producing cells and then determining what percentage each immune cell encompasses within that pool, most of the TNF-α-producing cells in both the unstimulated and LPS-stimulated cultures were CD64+ monocytes/Mφ, and this fraction was increased in the CoH samples (Figure S4C). Besides there being more CoH TNF-α-producing CD64+ monocytes/Mφ, TNF-α-producing CoH CD64+ monocytes/Mφ also contained more TNF-α on a per cell basis as TNF-α gMFI was significantly elevated when compared with SPF TNF-α-producing CD64+ monocytes/Mφ without stimulation (Figure 5D). Collectively, these data show that CoH mice exhibit elevated TNF-α production from both total blood cells and splenocytes, and CoH CD64+ monocytes/Mφ produce significantly more TNF-α in the steady state without additional treatment providing additional evidence of the importance of monocytes/Mφ in providing protection against UPEC infection.
CoH mice have an increased frequency of classical Ly6ChiCD115+ monocytes in the steady state, show elevated egress of CD115+ monocytes from the bone marrow, and skew toward myelopoiesis in bone marrow multipotent progenitor subsets
We next examined whether CoH CD115+ monocytes had additional functional differences compared with SPF CD115+ monocytes besides TNF-α production. We conducted bulk RNA sequencing (RNA-seq) of flow sort-purified CD115+ monocytes from the spleens of steady-state SPF and CoH mice (i.e., after 60 days of cohousing with pet store mice and no UPEC infection; Figure S5A). Principal-component analysis of gene expression revealed unique clustering for SPF and CoH CD115+ monocytes (Figure 6A), suggesting that these cells exist in different baseline transcriptional states. Analysis identified 720 genes differentially expressed with a false discovery rate (FDR) < 0.01 among CD115+ monocytes from SPF and CoH mice (Table S1), and differences in gene expression can be seen in volcano plots and heatmaps (Figures 6B and 6C). In addition, GO term analysis identified many differentially regulated functional pathways in SPF and CoH CD115+ monocytes, including phagocytosis and regulation of chemotaxis (Figure 6D).
Figure 6. CoH mice have increased frequency of “classical” Ly6Chi CD115+ monocytes at steady state that produce more TNF-α.
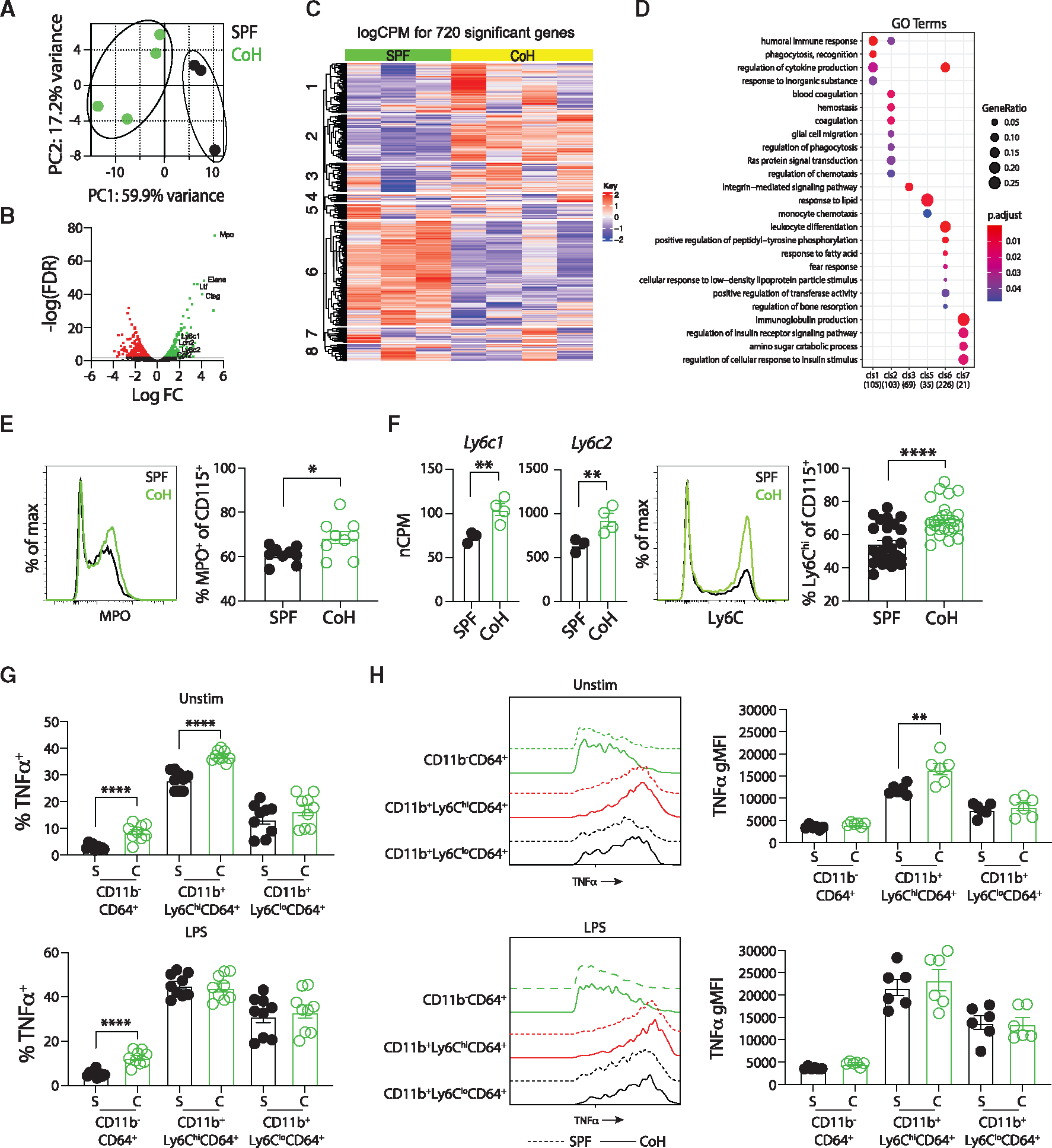
(A–D) CD11b+CD115+ monocytes were flow sort-purified from spleens of steady-state SPF and CoH mice, RNA was isolated, bulk RNA-seq analysis was performed, and comparison of gene expression was conducted.
(A) Principal-component analysis from RNA-seq data showing unique clustering of CD115+ monocytes from steady-state SPF and CoH mice.
(B) Volcano plot of log false discovery rate (FDR, y axis) by log fold change (x axis). Genes with an FDR < 0.01 with increased expression in CD115+ monocytes from spleens of steady-state CoH mice compared with SPF mice are shown in green, while genes with an FDR < 0.01 with decreased expression in CD115+ monocytes from spleens of steady-state CoH mice compared with SPF mice are shown in red.
(C) Heatmap showing relative gene expression in CD115+ monocytes from spleens of steady-state SPF and CoH mice. Seven hundred and twenty genes were differentially expressed with an FDR < 0.01.
(D) GO term analysis for pathways differentially regulated between CD115+ monocytes from spleens of steady-state CoH and SPF mice.
(E) Representative histogram of MPO expression in SPF and CoH CD115+ monocyte/Mφ (left) and frequency of myeloperoxidase (MPO)+ cells among CD115+ monocytes in the spleen as measured by flow cytometry (right).
(F) Number of Ly6c1 and Ly6c2 mRNA transcripts in CD115+ monocytes from steady-state SPF and CoH mice (left), representative histogram of Ly6C protein expression on SPF and CoH CD115+ monocytes (middle), and frequency of Ly6C+ cells among CD115+ monocytes in the spleens of CoH mice (right).
(G) Frequency of TNF-α+ cells within CD11b−CD64+ resident macrophages, Ly6ChiCD64+ monocytes, and Ly6CloCD64+ monocytes in the spleens of SPF and CoH at steady-state and after in vitro LPS stimulation (100 ng/mL).
(H) Geometric mean fluorescence intensity (gMFI) of TNF-α expression by unstimulated and LPS-stimulated TNF-a+CD11b−CD64+ resident macrophages (green), CD11b+Ly6ChiCD64+ classical (red), and CD11b+Ly6CloCD64+ non-classical (black) monocytes was determined by flow cytometry (left). Dashed lines, SPF; solid lines, CoH. TNF-α gMFI within TNF-α+CD11b−CD64+ resident macrophages, Ly6ChiCD64+ classical monocytes, and Ly6CloCD64+ non-classical monocytes in the spleens of SPF and CoH at steady-state and after in vitro LPS stimulation (right). *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.005, and ****p < 0.0001 as determined by nonparametric Mann-Whitney test. Transcript (normalized copies per million [nCPM]) data in (F) were obtained from 3 to 4 mice per group. Flow data in (E) were combined from two experiments using a total of 9 mice per group, in (F) were combined from three experiments using a total of 25–26 mice per group, in (G) were combined from three experiments using a total of 8–9 mice per group, in (H) were combined from two experiments using 3 mice per group. Each symbol in (A and E–H) represents a mouse and bars indicate means with SEM.
Of all the differentially expressed genes in the steady state, transcripts for Mpo were the most increased in CoH CD115+ monocytes (Figure 6B). Mpo encodes for myeloperoxidase (MPO), a peroxidase important for innate microbial defense by catalyzing the formation of reactive oxygen intermediates.37 Importantly, the transcriptional Mpo increase carried over to the protein level as we found there was a higher frequency of CD115+ monocytes from CoH mice expressing MPO based on flow cytometric analysis (Figure 6E). Murine monocytes can be classified into classical (Ly6Chi) or non-classical (Ly6Clo) subsets based on Ly6C expression. Ly6Chi mouse monocytes perform pro-inflammatory functions and are analogous to human CD14+ monocytes.38 Ly6Chi monocytes isolated from the blood express 10-fold higher MPO than Ly6Clo monocytes.39 Mouse Ly6C is comprised of two homologous isoforms encoded by Ly6c1 and Ly6c2.40 Both transcripts were highly expressed in CoH CD115+ monocytes compared with SPF, and we similarly found a significantly higher fraction of the CD115+ monocytes from CoH mice expressed high levels of Ly6C protein (Figure 6F). Examining the CD115+ monocytes for expression of both MPO and Ly6C found that CoH mice had an increased frequency of Ly6ChiMPO+CD115+ monocytes (Figure S5B). Importantly, the expression of MPO on a per cell basis was similar between the SPF and CoH Ly6ChiMPO+CD115+ monocytes (Figure S5C), suggesting that CoH monocytes do not have more MPO on a per cell basis compared with SPF monocytes, but instead have a skewing toward classical Ly6Chi monocytes within the CD115+ compartment. Of the identified top 10 most differentially expressed genes identified by RNA sequencing analysis of blood Ly6Chi vs. Ly6Clo monocytes,41 three of these genes—Rhou, Fn1, and C3—were also upregulated in the CoH CD115+ population analyzed here (Figure S5D), providing additional evidence supporting a skewing of the CoH CD115+ monocyte population toward a predominance of classical monocytes.
We next turned back to the 4 h in vitro splenocyte culture to assess TNF-α production within the CD64+ monocyte/M4 populations. Of all immune cell subsets analyzed, untreated CD11b+Ly6ChiCD64+ classical monocytes had the largest percentage of TNF-α-producing cells and CoH classical monocytes had an elevated percentage (37%) of TNF-α+ cells compared with SPF (28%; Figure 6G). Untreated CoH CD11b+Ly6ChiCD64+ classical monocytes also encompassed a larger percentage of the overall TNF-α-producing subset in the in vitro culture compared with SPF (Figure S4D). In addition, untreated TNF-α-producing CoH CD11b+Ly6ChiCD64+ classical monocytes had a higher TNF-α gMFI compared with TNF-α-producing SPF classical monocytes, suggesting that CoH classical monocytes produce more TNF-α than SPF classical monocytes on a per cell basis (Figure 6H). While CoH CD11b−CD64+ resident Mφ without and with LPS had a heightened percentage of TNF-α-producing cells compared with SPF, the TNF-α gMFI for TNF-α-producing resident Mφ was significantly lower than classical monocytes, suggesting that resident Mφ are not the main contributors of TNF-α production in this system (Figures 6G and 6H). Instead, CoH classical monocytes both had the highest percentage of TNF-α production as well as the highest TNF-α gMFI of TNF-α producers compared with SPF cells, suggesting that CoH classical monocytes are the main drivers of TNF-α production in this context.
Monocyte recruitment and chemotaxis are mediated in part by CCR2, which binds to multiple chemokines including monocyte chemoattractant protein-1 (MCP-1; CCL2) and monocyte chemotactic protein-3 (MCP-3; CCL7). Ccr2 transcript and CCR2 protein expression were increased in CoH CD115+ monocytes (Figure 7A). Moreover, CoH mice had a higher frequency of CCR2+Ly6ChiCD115+ monocytes (Figure 7B), consistent with published literature showing that Ly6Chi classical monocytes have higher CCR2 expression.42 Monocytes that first egress from the bone marrow are phenotypically classical in nature, expressing CCR2 and Ly6C.43 This population of classical monocytes is short-lived and will either infiltrate into tissues or transition into non-classical Ly6Clo monocytes.44 Since we identified an increased frequency of classical monocytes in the periphery of CoH mice, this could be explained by enhanced egress of monocytes from the bone marrow. As part of our evaluation of circulating chemokines, we noted higher concentrations of CCL2 and CCL7 in serum collected from CoH mice at steady state (Figure 7C). When combined with the increased frequency of CCR2+ classical Ly6C+ monocytes, it is tempting to speculate that the increased frequency of Ly6C+CD115+ monocytes in the periphery of CoH mice is due (in part) to the increased expression of CCL2 and CCL7 that call these cells from the bone marrow. To directly test this possibility, we performed a 16 h in vivo BrdU pulse-chase study to measure the egress of CD115+ monocytes from the bone marrow. Monocyte proliferation primarily occurs in the bone marrow, so any BrdU+ monocytes in the blood would be those that were actively proliferating in the bone marrow prior to their recent egress into the periphery. Indeed, there was a significant increase in frequency of Ly6C+CD115+ monocytes in the blood of CoH mice that were BrdU+ (Figure 7D). These data suggest an elevated rate of CD115+ monocyte migration out of the bone marrow of CoH mice.
Figure 7. Increased egress of “classical” Ly6Chi CD115+ monocytes from bone marrow and enhanced myelopoiesis in steady-state CoH mice.
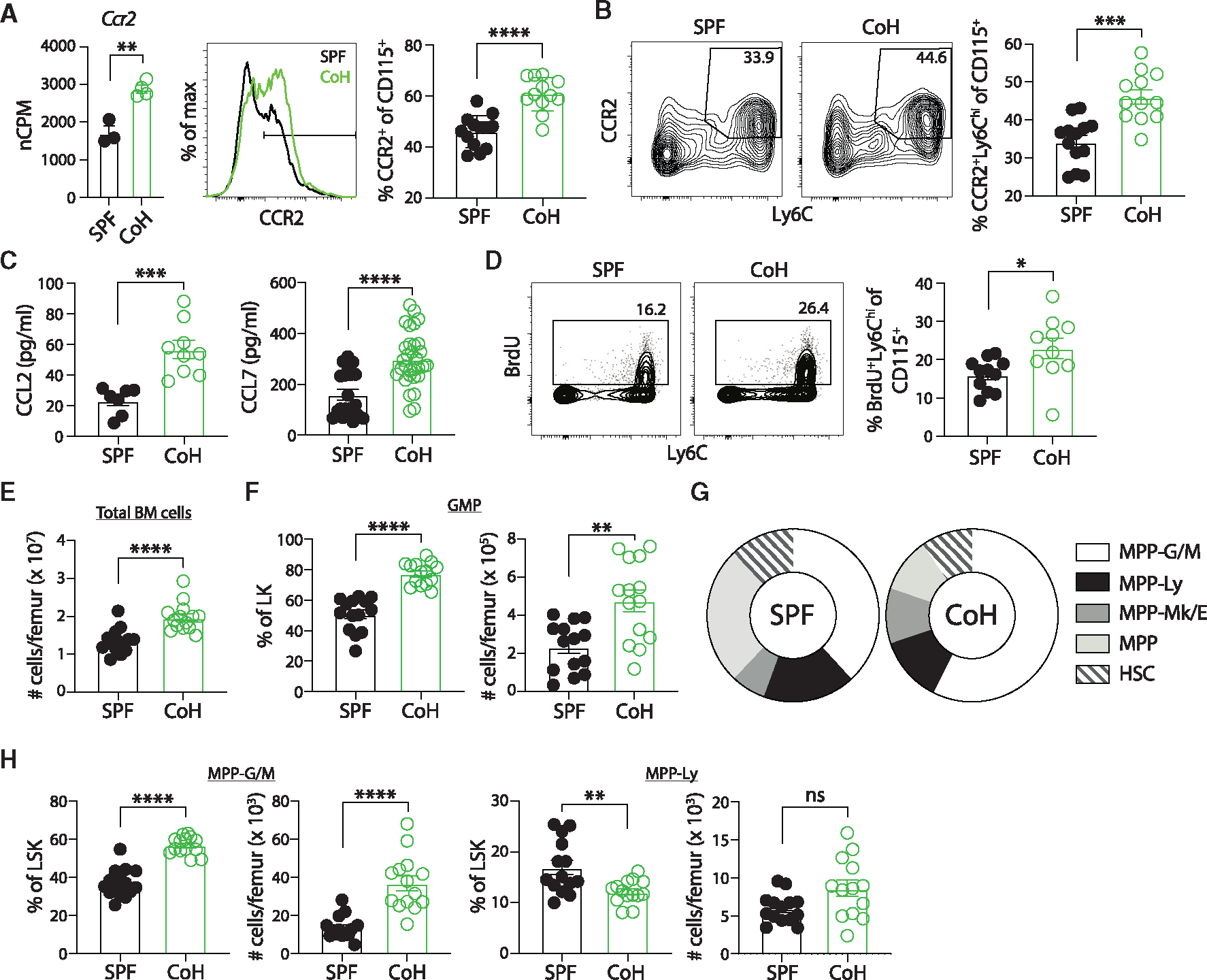
(A) Number of Ccr2 mRNA transcripts in CD115+ monocytes from steady-state SPF and CoH mice (left), representative histogram of CCR2 protein expression on SPF and CoH CD115+ monocytes (middle), and frequency of CCR2+ cells among CD115+ monocytes in the spleens of SPF and CoH mice as measured by flow cytometry (right).
(B) Representative flow cytometry plots showing expression of CCR2 and Ly6C double-positive cells from CD11b+CD115+ splenocytes (left) and frequency of CCR2+Ly6Chi CD115+ monocytes (right).
(C) Concentration of CCL2 and CCL7 in serum of mice 60 days after cohousing using Luminex.
(D) SPF and CoH mice were injected with 1 mg BrdU i.p. Blood was collected 16 h later, and peripheral blood leukocytes were stained to detect BrdU+ Ly6ChiCD115+ monocytes by flow cytometry.
(E–H) Bone marrow was isolated from steady-state SPF and CoH mice and progenitor populations were analyzed (see Figure S6A for gating strategy). (E) Total number of bone marrow cells per femur of SPF and CoH mice. (F) Absolute number per femur and percentage of granulocyte/monocyte progenitors (GMP) (lineage−Sca1−cKit+CD16+) within the lineage−cKit+ (LK) progenitor population.
(G) Percentage of multipotent progenitor (MMP) populations within the lineage−Sca1+cKit+ (LSK) population. MPP-granulocyte/monocyte (G/M) (Flt3−CD48+CD150−), MPP-lymphocyte (Ly) (Flt3+), MPP-megakaryocyte/erythrocyte (Mk/E) (Flt3−CD48+CD150+), MPP (Flt3−CD48−CD150−), and hematopoietic stem cell (HSC) (Flt3−CD48−CD150+).
(H) Absolute number per femur and percentage of MPP-G/M (left) and MPP-Ly (right) within the LSK population. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.005, and ****p < 0.0001 as determined by nonparametric Mann-Whitney test. Transcript (normalized copies per million [nCPM]) data in (A) were obtained from 3 to 4 mice per group. Flow data in (A) and (B) were combined from three experiments using a total of 12–13 mice per group, (C) were combined from at least two experiments with a total of 7–31 mice per group, (D) were combined from three experiments using a total of 10–11 mice per group, (E–H) were combined from three independent experiments using a total of 14 mice per group. Each symbol represents a mouse and bars indicate means with SEM.
To directly test whether CoH mice have enhanced myelopoiesis, we analyzed bone marrow multipotent progenitor (MPP) populations using the new simplified isolation scheme developed by the International Society for Experimental Hematology community45 (Figure S6A). Steady-state CoH mice had more total bone marrow cells per femur compared with SPF femurs (Figure 7E). In addition, while there was not an increase in the percentage or number of lineage −cKit+ (LK) cells in CoH bone marrow, the percentage and number of granulocyte/monocyte progenitor (GMP) cells within the LK population were significantly increased in CoH bone marrow (Figures 7F and S6B). Analysis of less differentiated lineage −Sca1+cKit+ (LSK) progenitor subsets also showed a skewing toward the MPP-granulocyte/monocyte (MPP-G/M) population and away from the MPP-lymphocyte (MPP-Ly) subset in CoH bone marrow (Figures 7G, 7H, and S6C). Together, these data showing the increased skewing toward both GMP and MPP-G/M progenitor subsets provide further evidence for enhanced myelopoiesis in CoH mice.
Collectively, we show that microbially experienced CoH mice have a heightened inflammatory response and increased resistance against systemic UPEC infection. CoH mice have an increased number of CD115+ monocytes following systemic UPEC infection, and CD115+ cells from CoH mice are necessary to protect against systemic UPEC infection. In addition, CoH mice have an increased frequency of classical Ly6Chi CD115+ monocytes driven by elevated egress from the bone marrow, and this population has heightened TNF-α production at the steady state compared with SPF mice. Bone marrow progenitor analysis confirmed enhanced myelopoiesis in CoH mice. Together these data highlight the importance of CoH classical monocytes in contributing to protection against systemic UPEC infection compared with SPF animals.
DISCUSSION
In this study, we used a laboratory/pet store mouse cohousing system to examine how a history of microbial exposure impacts outcomes after systemic UPEC infection as a model of monomicrobial sepsis. Past studies utilizing CoH mice have shown a history of microbial exposure can either be beneficial or detrimental to the host depending on the infection/disease analyzed.9,10,12 Our data add valuable information to what is known regarding the impact of prior microbial exposure on the response to new pathogens. Whereas other studies have primarily focused on demonstrating how such exposure affects the composition and function of cells within the adaptive immunity arm,9,10,21 the data presented herein show how exposure to a diverse array of pathogenic and commensal microbes can also have profound impact on the functional capacity of innate immune cells. Our data show that CD115+ monocytes of CoH mice were transcriptionally distinct from CD115+ monocytes of SPF mice at steady state (i.e., prior to sepsis induction), and there was a higher frequency of CD115+ monocytes from CoH mice expressing ROS-producing enzyme MPO, Ly6C, and CCR2. In addition, CoH CD64+ monocytes/M4 have a heightened percentage of TNF-α+ cells compared with SPF and comprise the largest subset of cells among the TNF-α-producing splenocytes in the steady state or after LPS stimulation. Within the CD64+ subset, classical monocytes have the greatest production of TNF-α on a per cell basis in the steady state, with CoH classical monocytes producing significantly more TNF-α than their SPF counterparts. TNF-α is key in the handling and clearance of bacterial infections,32,33 leading to TNF-α being the predominant functional cytokine analyzed here. However, it should be noted that other inflammatory cytokines that encompass the septic cytokine storm are likely contributing to the elevated UPEC clearance in CoH mice reported here. Additional studies should be performed to assess the extent to which CD115+ monocytes/Mφ have elevated production of other inflammatory cytokines. These phenotypic features provide evidence that help to explain why CD115+ monocytes/Mφ were key players in providing increased protection for systemic UPEC-infected CoH mice.
Mouse models of sepsis have been vital for identifying key cellular hallmarks, as well as testing novel therapies, to minimize the pathology associated with the cytokine storm and restore immune function during the prolonged state of immunoparalysis related to sepsis. However, recent reports have suggested limited correlation between mouse and human sepsis.46–49 Among the key differences noted was the (nearly) exclusive use of SPF mice in preclinical sepsis research. As such, we were the first group to show that microbially experienced mice displayed an exacerbated response in multiple sepsis models,12 suggesting the perceived differences between mouse and human sepsis data were not because of a species difference but more the result in differences in the baseline status of the mouse immune system. Most studies with microbially experienced laboratory mice, generated by cohousing with non-SPF mice, sequential infection with multiple pathogens, or housing in outdoor enclosures, have focused more on how this exposure impacts adaptive immune cells.9,50–52 By contrast, few experiments or models have examined the impact on general microbial exposure on innate immune cell function. Our findings are consistent with a recent report looking at natural microbial exposure starting at the time of birth, which also showed enhanced hematopoiesis, including myeloid progenitors, in the bone marrow of 3-day-old pups.18 Our data suggest that CD115+ monocytes are important for mediating protection against a monomicrobial systemic E. coli infection as well as for preventing downstream pathologies related to sepsis.
Recently, Chung et al. reported that severe septic patients with initial blood monocyte counts less than 250 cells/μL (compared with 250–500, 500–750, and >750 cells/μL) showed the highest mortality, rate of bacteremia, and organ dysfunction compared with severe septic patients with initial monocytes >250 cells/μL.53 In addition, of patients who also had premorbid differential blood cell counts, there was an increase in absolute monocyte counts from premorbid to sepsis for patients who survived past 28 days. Non-survivors, in contrast, showed a decrease in premorbid to sepsis absolute monocyte counts. These clinical data suggest that the absolute number of monocytes early during a human septic event positively correlates with better outcomes, which is a similar conclusion as to that reported here with increased presence of monocytes positively affecting survival in microbially experienced mice. In addition, classical monocytes account for 85% of the total circulating monocytes in humans during physiological conditions.54 Previous microbial exposure in mice boosts the frequency of classical Ly6Chi monocytes to 75% of total circulating monocytes (vs. 55% in SPF mice), more closely mirroring the monocyte ratio in humans. This is additional evidence to suggest generalized microbial experience in mice brings aspects of the mouse innate immune system closer to humans in physiological conditions. Moreover, these data suggest that addressing inflammatory and other functional differences in SPF vs. CoH monocytes could be instructive for developing therapeutics that target the innate immune system.
Previous work from our lab found that CoH mice are more susceptible, compared with SPF mice, to polymicrobial sepsis (CLP or cecal slurry injection).12 Yet, here we report better survival in CoH vs. SPF mice after monomicrobial UPEC sepsis. The reason for this is not completely clear, but could have to do with the magnitude and diversity of pathogen(s) encountered and whether the pathogen is quickly containable or not. While in both polymicrobial and monomicrobial sepsis models CoH mice have an exacerbated cytokine storm compared with SPF mice, in the context of monomicrobial UPEC infection the CoH mice quickly contained and eliminated the infection (Figure 1), which led to increased survival. In the context of polymicrobial sepsis, the lack of being able to quickly eliminate the pathogen burden due to both pathogen magnitude and diversity could result in prolonged uncontrolled inflammatory response leading to increased death in CoH mice compared with their SPF counterparts. Future experiments are needed to tease apart the differences that lead to worse mortality in some contexts but not others in CoH vs. SPF mice. We detected high UPEC titers in the blood 24 h after infection (Figure 1B), yet multiple reports have described minimal blood titers of Gram-negative bacilli at this time point because the spleen and liver can filter the bacteria rapidly.55–57 However, the high 24 h UPEC blood titers reported herein are consistent with data reported by van Schaik and Abbas.58 It is unclear why some UPEC (or other Gram-negative bacteria) isolates maintain higher blood titers than others after infection, but potential explanations may be due to a specific isolate’s fitness for establishing and maintaining a presence in the blood, impact of inoculum dose, and/or mouse strains used (among other factors).
Our data show that exposure to diverse microbes has significant effects on monocyte gene expression patterns, and several differentially expressed genes were validated to be differentially expressed at the protein level by CoH CD115+ monocytes, which helps to explain the increased resistance to UPEC-induced sepsis. However, the RNA-seq data also pointed to further alterations in genes and pathways that may impact outcomes in other infection/disease models. Of note, pathways that govern inflammatory responses, phagocytosis, and metabolism were differentially regulated between SPF and CoH CD115+ monocytes. Given the importance of monocyte effector functions, including cytokine/chemokine production and phagocytosis to infection clearance,59,60 and the known impacts of metabolism on monocyte functions,61 further examination of the effects of microbial exposure on these pathways and functions will be critical.
Transcriptional and functional changes to monocytes (including alterations to phagocytic abilities, ability to produce ROS, metabolism, and mitochondria) after exposure to microbial components (for example, β-glucan) are hallmarks of trained innate immunity along with enhanced myelopoiesis in bone marrow.62,63 While trained innate immunity has been shown in other systems, it is an understudied aspect of “dirty” mice. Interestingly, our data show enhanced egress of CD115+ monocytes from the CoH bone marrow as well as a skewing toward myeloid MPPs in CoH femurs, which are consistent with hallmarks of trained innate immunity.63 Further studies are needed to discern the relationship between the heightened frequency of classical monocytes and myeloid MPPs identified in CoH mice in this study and trained immunity.
Our data indicate that CD115+ monocytes play a critical role in providing CoH mice protection against E. coli-induced sepsis, but other cell types likely contribute. Adaptive immunity—specifically B and T cell responses following vaccination or infection—is altered in mice with a history of microbial exposure.9,10,21 The number of T cells and B cells in the spleens of SPF and CoH mice did not change following systemic E. coli infection (compared with uninfected mice), and depletion of either CD4 or CD8 T cells from CoH mice did not alter outcomes. Combined with the rapid rates at which UPEC-infected mice die after challenge, whether they are SPF or CoH mice depleted of CD115+ cells, it is unlikely that the primary adaptive immune response meaningfully contributes to protection in CoH mice.64,65 In the innate compartment, neutrophil numbers increased significantly in the spleens of both SPF and CoH mice after infection. Because neutrophil responses can impact monocyte/Mφ function and vice versa,66 it is possible that neutrophils may also make important contributions to protection of CoH mice. Here, we focused on the importance of CD115+ monocytes during the acute cytokine storm phase of sepsis, but it would also be beneficial for future studies to determine the contribution of CD115+ monocytes to sepsis-induced immunoparalysis, a phase that happens much later after a septic event.67
In summary, we have provided further evidence to support the notion that the cohousing mouse model provides valuable insight into understanding the beneficial or detrimental role of the immune system during sepsis. We have added to previous work with CoH mice to show that in addition to impacts on adaptive immunity, cohousing also shapes cells of the innate immune compartment. This work has important implications for understanding how microbial exposure alters immune responses that impact sepsis outcomes and provides a wealth of information on how microbial exposure alters monocyte function that may be important in other disease models.
Limitations of the study
All experiments were conducted using only female mice, as the cohousing of male mice with pet store mice is not feasible because of fighting restrictions. Cohousing laboratory mice with pet store mice induces a transient systemic inflammation, which peaks 10–14 days after cohousing and then levels out by day 60 of cohousing (but is still elevated compared with SPF mice).22 All our experiments used mice that were CoH at least 60 days. We recognize the possibility that challenging mice with a systemic bacterial infection earlier in the cohousing timeline (e.g., within the first 14 days of cohousing) could yield different outcomes. We also only monitored survival for 7 days after infection. It is possible that more mortality could have been observed (in the CoH mice) if we extended the duration of the experiment, but the significance of the results reported here would be (likely) unchanged. In addition, we used a virulent L. monocytogenes infection to provide further validation that CoH mice handle monomicrobial infections better than SPF. L. monocytogenes is a commonly used Gram-positive experimental pathogen for testing different aspects of immune cell function.68 Even though Listeria sepsis can occur in neonatal and adult humans,69 it is not a common sepsis-inducing pathogen in humans. Additional experiments using other Gram-positive (e.g., Staphylococcus aureus or Streptococcus pyogenes) or Gram-negative (e.g., Klebsiella pneumonia) bacteria that are more frequently associated with sepsis would further strengthen our claims. Finally, our assessment of pathogen burden in the blood and organs was limited to quantitating UPEC, as the UTI89 strain we used is resistant to kanamycin and the agar plates used to quantify bacterial burden contained kanamycin. Cohousing also induces gut microbiota changes,12,22 so we cannot exclude the possibility that some other gut-derived bacteria could be contributing to the phenotype observed in the CoH mice.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Thomas Griffith (tgriffit@umn.edu).
Materials availability
There are no newly generated materials associated with the paper.
Data and code availability
The bulk RNA-seq data generated have been deposited at the National Center for Biotechnology Information Gene Expression Omnibus (NCBI GEO) under accession number GSE237883 and are publicly available as of the date of publication. Any additional details regarding these data will be made available upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Mice
Female C57Bl/6N (B6) mice were purchased from Charles River (Wilmington, MA) at 8–10 weeks of age. Female pet store mice were purchased from local pet stores in the Minneapolis-St. Paul metropolitan area. All mice were housed in AALAC-approved animal facilities at the University of Minnesota (BSL-1/BSL-2 for SPF B6 mice, and BSL-3 for cohoused B6 and pet store mice). SPF B6 and pet store mice were cohoused at a ratio of 8:1, respectively, in large mouse cages for 60 days to facilitate microbe transfer.17 Cohoused and age-matched littermate SPF mice were used for experiments after between 60 and 90 days of cohousing. Experimental procedures were approved by the University of Minnesota Animal Care and Use Committee (protocol #2205–39995A) and performed following the Office of Laboratory Animal Welfare guidelines and PHS policy on Human Cancer and Use of Laboratory Animals.
METHOD DETAILS
Systemic UPEC or virulent L. monocytogenes infection
The RFP+, kanamycin-resistant uropathogenic E. coli UPEC strain UTI89-kan-RFP was used for all infections.70 For bacterial growth prior to infection, a single colony of bacteria grown on LB agar plates containing kanamycin (50 μg/mL) was selected and placed in 10 mL of LB broth containing kanamycin (50 μg/mL) and grown statically overnight at room temperature. The following morning, the optical density (OD600) of the culture was measured and the colony forming units (CFU)/mL were calculated according to the empirically determined formula OD600 0.35 = 2×108 CFU/mL. Bacteria were then prepared at 2×108 CFU/mL in PBS, and 200 μL was injected intravenously (retro-orbital) per mouse for an infectious dose of 4×107 CFU/mouse. Inoculum dosage was confirmed by dilution plating an aliquot of prepared bacteria on LB agar plates containing kanamycin (50 μg/mL). Frozen 1 mL aliquots of virulent L. monocytogenes were thawed and added to 5 mL of sterile tryptic soy broth containing streptomycin (50 μg/mL final concentration). Bacteria was incubated in a bacterial shaker (37°C at 250 RPM) for 2h to reach an OD600 of ~0.06–0.08. The bacterial concentration was calculated according to the empirically determined formula OD600 0.1 = 108 CFU/mL. Bacteria were then prepared at 5×104 CFU/mL in PBS, and 200 μL was injected intravenously (retro-orbital) per mouse for an infectious dose of 104 CFU/mouse. Inoculum dosage was confirmed by dilution plating an aliquot of prepared bacteria on tryptic soy broth agar plates containing streptomycin (50 μg/mL).
Measurement of bacterial clearance
UPEC-infected SPF and CoH mice were euthanized 24 h after infection. 20 μL of whole blood was collected and placed in 180 μL of dH20 containing 0.2% IGEPAL. Spleens, livers, and kidneys were collected and placed in 2 mL of dH20 containing 0.2% IGEPAL and disrupted using a gentleMACS dissociator (Miltenyi). Serial dilutions of samples were made in dH20 containing 0.2% IGEPAL and plated on LB agar plates containing kanamycin (50 μg/mL). Plates were incubated at room temperature for 24 h, and CFU were counted.
Measurement of cytokines and chemokines
Blood was collected from SPF and CoH mice prior to and either 3 h or 24 h post UPEC infection, and serum was separated and collected by centrifugation of samples at 13,000×g for 1 min. Serum cytokines were quantitated by ProcartaPlex immunoassays (ThermoFisher) using a Luminex 200 with Bio-plex Manager Software 5.0. The amount of TNFα present in culture supernatants from in vitro/ex vivo assays was quantitated by ELISA (Biolegend ELISA MAX standard set mouse TNFα; cat. No 430901).
Detection of immune cell subsets using flow cytometry
For detection of peripheral blood leukocytes, blood was collected by retro-orbital puncture and red blood cells were lysed with ACK. For detection of cells in spleens, tissue was processed into single-cell suspension using a gentleMACS dissociator before ACK lysis. Neutrophils and monocytes were identified using the following gating scheme: Cells were first gated based on forward scatter-A and side scatter-A, singlets were gated based on forward scatter-W and side scatter-A, and then neutrophils (Ly6G+CD11b+), non-neutrophils (Ly6G−), and monocytes (CD11b+CD115+) identified. For some samples, cells were also stained with mAb specific for myeloperoxidase or CCR2. B cells, NK cells, CD4 T cells, CD8 T cells, and Ag-experienced CD4 and CD8 T cells were identified using the following gating scheme: Cells were first gated based on forward scatter-A and side scatter-A, singlets were gated based on forward scatter-W and side scatter-A, and then live cells (ghost dye−) were gated. B cells (CD3−CD19+), NK cells (CD3−NK1.1+) and T cells (CD3+NK1.1−) were identified. Among the gated CD4+ and CD8+ T cells, Ag-experienced CD4 T cells (CD11ahiCD49dhi) and Ag-experienced CD8 T cells (CD8aloCD11ahi) were determined. All samples were acquired on Fortessa X20 or LSRFortessa H0081 flow cytometers (BD) and analyzed using FlowJo software.
Cell depletions
To deplete specific cell populations from CoH mice, the following mAb were used: control anti-rat IgG2a – 400 μg/mouse i.p. on days 7, 5, and 2 prior to infection; anti-mouse CSF1R (CD115; AFS98, BioXCell) – 400 μg/mouse i.p. on days 7, 5, and 2 prior to infection; anti-mouse CD4 (GK1.5, BioXCell) – 400 μg/mouse i.p. on days 5 and 2 prior to infection; and anti-mouse CD8β (53–5.8, BioXCell) – 400 μg/mouse i.p. on days 5 and 2 prior to infection. Depletion of CD115+ cells, CD4 T cells, and CD8 T cells in the circulation was assessed by flow cytometry one day prior to infection.
Whole blood ex vivo stimulation
LPS-induced TNFα production by whole blood was determined as described.34 Briefly, 50 μL of heparinized whole blood from SPF and CoH mice was added to tubes containing RPMI alone (control) or RPMI and LPS (phenol-extracted from Salmonella abortus equi (Sigma)). Tubes were incubated at 37°C for 4 h, after which the supernatant was collected and frozen at −80°C until analysis. Tubes were obtained from Dr. Mark Hall (Nationwide Children’s Hospital, Columbus, OH).
In vitro splenocyte culture for TNFα production
Spleens were isolated from untreated CoH and SPF mice, ACK lysed, counted, and resuspended in a final concentration of 2×106 cells/ml in RPMI complete (RPMI-1640 + 10% FBS, 100U/ml penicillin and streptomycin, 1mM sodium pyruvate, 10mM HEPES, 1x non-essential amino acids (Hyclone SH30238.01), and 50μM 2-mercaptoethanol) (for UPEC cultures cell media did not include pen/strep) in either polypropylene tubes or 24-well plates with 100 ng/ml LPS EB (Invivogen Lrl-eblps E. coli 0111:B4), 5:1 MOI UPEC-RFP:splenocytes, or left untreated. Splenocytes were cultured at 37°C for 4 h and then supernatant was collected for TNFα ELISA quantification. For cellular analysis of TNFα production, 5μg/ml brefeldin A was added at 2 h of the total 4 h of in vitro culture. Cells were then collected and blocked with FC receptor block and 1:50 normal mouse serum and normal rat serum for 10 min on ice, 30 min surface stain on ice (Thy1.2, UV blue Live/dead, CD11b, CD45, CD19, Ly6G, Ly6C, CD115, NK1.1, CD64, TNFα), and intracellular TNFα was determined following the BD cytofix/cytoperm kit standard protocol and anti-TNFα antibody. Cells were analyzed by flow cytometry for surface markers distinguishing cellular subsets and intracellular TNFα (see Figures S4A and S4B for gating strategy and representative intracellular TNFα staining).
Cell sorting, RNAseq, and biostatistical analysis
For sorting of CD115+ monocytes, SPF and CoH spleens were collected from uninfected mice. Tissue was homogenized into a single-cell suspension using a gentleMACS dissociator, red blood cells were lysed with ACK, and cells were re-suspended in 1mL of FACS buffer. CD3+ cells were depleted from samples using an EasySep Mouse T cell Isolation kit. Flow through cells were then stained with the following mAbs: ghost dye 510 (live/dead), CD3, Ly6G, CD11b, and CD115. Cells were run and collected on a FACSDiva (BD) (see Figure S5A for sorting gating strategy). The sorted cells were re-suspended in Trizol, RNA was purified, and libraries were prepared and sequenced using Illumina NovSeq platforms (Genewiz, Azenta Life Sciences). Raw data was trimmed and mapped, and differential gene expression was determined (Institute for Health Informatics, University of Minnesota), where additional biostatistical data analysis was also performed. The sequencing reads were mapped to the mouse genome (GRCm38) using Bowtie aligner (bowtie2 version 2.3.4.1) with local mode, -L 22 and -N 1 parameters.71 Reads were assigned to Ensembl gene models (Mus_musculus.GRCm38.87.gtf) with featureCounts of the Subread software package (version 1.5.1).72 The reads count matrices were organized corresponding to experimental design and used for subsequent statistical analysis using the bioconductor package edgeR (version 3.24.3).73,74 The raw reads count table were normalized by using default method in the package prior to generating statistics. Multidimensional scaling was performed with edgeR using the top 500 differentially expressed genes across samples. The regression model in the edgeR package was used for statistical analysis to select the corresponding significant genes. Clustering and gene ontology analysis was performed by using ComplexHeatmap and clusterProfiler (5,6), respectively on genes with an adjusted p value < 0.01.
In vivo BrdU pulse-chase
BrdU (1 mg) was injected into the peritoneal cavity of SPF and CoH mice. Blood was collected 16 h later, and red blood cells were removed by ACK lysis. Peripheral blood leukocytes were stained and fixed by following the protocol provided by the manufacturer (FITC-BrdU kit, BD Pharmingen) and detected by flow cytometry.
Bone marrow progenitor cell analysis
Femurs from SPF and CoH mice were removed and placed in RPMI media. One end of the bones was cut off and placed in a 250 μL Eppendorf tube, with three 25g needle holes poked in the bottom, within a 1.5mL Eppendorf tube. The bones were spun down momentarily at 6000 RPM, ACK lysed for 3 min on ice, and counted. 3×106 bone marrow cells were stained with FLT-3, c-Kit, CD34, CD48, Sca-1, CD16, CD150, B220, CD11c, Ly6G, Ter119, and CD3 to identify bone marrow progenitor populations. See Figure S6A for gating strategy.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed with the GraphPad Prism v10 (GraphPad Software Inc., San Diego, CA) for Mac OS X software package, where the Log Rank (Mantel-Cox) test was used for survival curves. Statistical comparisons of two groups were done using the unpaired nonparametric Mann-Whitney test. Statistical comparisons of more than two groups were done using Kruskal-Wallis tests, where the multiple comparisons were corrected with Dunn’s post hoc test. The calculated p values were corrected for multiple testing using the false discovery rate (FDR) method to determine the FDR-adjusted p value. Statistical details for each experiment can be found in the figure legends.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Anti-mouse B220 (RA3–6B2, APC-eF780) | Invitrogen | Cat# 47–0452-82; RRID:AB_1518810 |
| Anti-mouse CD3 (145–2C11, PerCP-Cy5.5) | BioLegend | Cat# 100328; RRID:AB_893318 |
| Anti-mouse CD3 (UCHT1, APC-eF780) | Invitrogen | Cat# 47–0038-42; RRID:AB_1272042 |
| Anti-mouse CD4 (GK1.5, AF700) | BioLegend | Cat# 100430; RRID:AB_493699 |
| Anti-mouse CD8a (53–6.7, BV605) | BioLegend | Cat# 100744; RRID:AB_2561352 |
| Anti-mouse CD11a (M17/4, PE-Cy7) | BioLegend | Cat# 101122; RRID:AB_2562781 |
| Anti-mouse CD11b (M1/70, BV421) | BioLegend | Cat# 101236; RRID:AB_10897942 |
| Anti-mouse CD11b (M1/70, APC-eF780) | eBioscience | Cat# 47–0112-82; RRID:AB_1603193 |
| Anti-mouse CD11c (N418, APC-eF780) | eBioscience | Cat# 47–0114-82; RRID:AB_1548652 |
| Anti-mouse CD16/32 (S17011E, PE-Cy7) | BioLegend | Cat# 156610; RRID:AB_2800707 |
| Anti-mouse CD16/32 (FC receptor block) | eBioscience | Cat# 14–0161-85; RRID:AB_467134 |
| Anti-mouse CD19 (6D5, BV711) | BioLegend | Cat# 115555; RRID:AB_2565970 |
| Anti-mouse CD19 Biotin (1D3) | Cytek | Cat# 30–0193; RRID:AB_2621641 |
| Anti-mouse CD34 (RAM34, BV711) | BD | Cat# 751621; RRID:AB_2875614 |
| Anti-mouse CD45.2 (104, BV650) | BioLegend | Cat# 109835; RRID:AB_2563065 |
| Anti-mouse CD48/Blast (HM48–1, PE) | BioLegend | Cat# 103405; RRID:AB_313020 |
| Anti-mouse CD49d (R1–2, PE) | BioLegend | Cat# 103608; RRID:AB_313038 |
| Anti-mouse CD64 (X54–5/7.1, PE-Cy7) | BioLegend | Cat# 139314; RRID:AB_2563904 |
| Anti-mouse CD115 (AFS98, PE) | BioLegend | Cat# 135506; RRID:AB_1937253 |
| Anti-mouse CD117/c-kit (ACK2, BV605) | BioLegend | Cat# 135120; RRID:AB_2650925 |
| Anti-mouse CD150/Slam (TC15–12F12.2, APC) | BioLegend | Cat# 115909; RRID:AB_493461 |
| Anti-mouse CCR2 (TG5, APC) | BioLegend | Cat# 150628; RRID:AB_2810415 |
| Anti-mouse FLT-3/CD135 (A2F10, BV421) | BioLegend | Cat# 135314; RRID:AB_2562339 |
| Anti-mouse GR1(RB6–8C5, APC-eF780) | eBioscience | Cat# 47–5931-82; RRID:AB_1518804 |
| Anti-mouse Ly6C (HK1.4, PerCP-Cy5.5) | BioLegend | Cat# 128012; RRID:AB_1659241 |
| Anti-mouse Ly6G (1A8, FITC) | BioLegend | Cat# 127606; RRID:AB_1236494 |
| Anti-mouse Ly6G (RB6–8C5, APC-eF780) | eBioscience | Cat# 47–5931-82; RRID:AB_1518804 |
| Anti-mouse myeloperoxidase (2D4, FITC) | Abcam | Cat# ab90812; RRID:AB_2050025 |
| Anti-mouse NK1.1 (PK136, APC) | Cytek | Cat# 20–5941; RRID:AB_2621611 |
| Anti-mouse NK1.1 (SI7016D, PE-Fire 700) | BioLegend | Cat# 156528; RRID:AB_2910320 |
| Anti-mouse Sca-1/Ly6A (E13–161.7, PE-Dazzle 594) | BioLegend | Cat# 122528; RRID:AB_2687355 |
| Anti-mouse Ter119 (TER-119, APC-eF780) | eBioscience | Cat# 47–5921-82; RRID:AB_1548786 |
| Anti-mouse TNFα (APC, MP6-XT22) | BioLegend | Cat# 506308; RRID:AB_315429 |
| Anti-rat IgG2a (2A3) | BioXCell | Cat# BE0089; RRID:AB_1107769 |
| Anti-mouse CD4 (GK1.5) | BioXCell | Cat# BE0003–1; RRID:AB_1107636 |
| Anti-mouse CD8b (53–5.8) | BioXCell | Cat# BE0223; RRID:AB_2687706 |
| Anti-mouse CSF1R (CD115, AFS98) | BioXCell | Cat# BE0213; Cat# BE0213 |
| Anti-mouse 90.2 (53–2.1, BUV395) | BD Biosciences | Cat# 565257; RRID:AB_2739136 |
| Ghost Dye UV 450 Fixable Viability Dye | Cell Signaling | Cat# 80862 |
| Ghost Dye V510 Fixable Viability Dye | Cell Signaling | Cat# 59863 |
| Ex vivo LPS stimulation solution tubes | Dr. Mark Hall (Nationwide Children’s Hospital, Columbus, OH) | N/A |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| RFP+ kanamycin-resistant uropathogenic E. coli UPEC | Strain UT189-kan-RFP | N/A |
| Virulent Listeria monocytogenes | Dr. Sarah Hamilton-Hart (University of Minnesota, Minneapolis, MN) | N/A |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| LPS-EB (LPS from E. coli 011:B4) | Invivogen | Cat# Tlrl-eblps |
| Brefeldin A | BioLegend | Cat# 420601 |
| Kanamycin | Gold Bio | Cat# K-120–5 |
| IGEPAL | Sigma | Cat# 542334 |
| RPMI-1640 | HyClone | Cat# SH30027.01 |
| Fetal bovine serum | Atlanta Biologicals | Cat#S11550 |
| Penicillin 10,000 U/ml - Streptomycin 10,000 μg/ml | HyClone | Cat#SV30010 |
| Sodium pyruvate 100mM | HyClone | Cat# SH30239.01 |
| HEPES 1M | HyClone | Cat# SH30237.01 |
| MEM Non-Essential Amino Acids 100X | HyClone | Cat# SH30238.01 |
| 2-Mercaptoethanol solution 50 mM | Gibco | 21985–023) |
|
| ||
| Critical commercial assays | ||
|
| ||
| Mouse Custom ProcartaPlex 20-plex (CCL2, CCL3, CCL4, CCL5, CCL7, CCL11, CXCL1, CXCL2, CXCL10, GM-CSF, IFNγ, IL-1β, IL-2, IL-4, IL-5, IL-6, IL-12p70, IL-13, IL-18, TNFα) | Thermo Fisher Scientific | PPX-20 |
| ELISA MAX standard set mouse TNFα | BioLegend | Cat# 430901 |
| Cytofix/cytoperm fixation/permeabilization solution kid | BD Biosciences | Cat# 554714 |
| EasySep Mouse 90.2 positive selection kit II | STEMCELL Technologies | Cat# 18951CA |
| FITC BrdU flow kit | BD Pharmingen | Cat# 559619 |
|
| ||
| Deposited data | ||
|
| ||
| SPF and CoH murine CD115+ monocytes RNA-seq (bulk) | This study | GEO: GSE237883 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: C57BL/6Ncr | NCI Charles River | N/A |
| Mouse: pet store | Minneapolis/St. Paul area Petco stores | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Prism 10 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| FlowJo | BD Biosciences | www.flowjo.com |
| FacsDiva | BD Biosciences | N/A |
| Bio-plex Manager Software 5.0 | Bio Rad | www.bio-rad.com |
| bowtie2 version 2.3.4.1 | John Hopkins Univ | https://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Subread version 1.5.1 | R package | https://subread.sourceforge.net |
| edgeR version 3.24.3 | R package | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| Complexheatmap | R package | https://bioconductor.org/packages/release/bioc/html/ComplexHeatmap.html |
| clusterProfile | R package | https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html |
Highlights.
Monocytes in CoH mice provide enhanced resistance to UPEC infection
CoH circulating monocytes are skewed toward a classical phenotype
CoH mice have enhanced monocyte egress from the BM and elevated BM myelopoiesis
ACKNOWLEDGMENTS
This study was supported by National Institutes of Health grants AI154527 (to T.S.G.), GM140881 (to T.S.G.), GM134880 (to V.P.B.), and AI165553 (to J.W.W.) and a Veterans Administration Merit Review Award (BX001324 to T.S.G.). V.P.B. is a University of Iowa Distinguished Scholar. T.S.G. is the recipient of a Research Career Scientist award (IK6BX006192) from the Department of Veterans Affairs. This work was also supported in part by NIH P30 CA77598, utilizing the Masonic Cancer Center, University of Minnesota, University of Minnesota Flow Cytometry shared resource, the Urology Care Foundation Research Scholar Award Program, and the AUA North Central Section (to M.D.M.). The graphical abstract was created with BioRender.com.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.113345.
REFERENCES
- 1.Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche JD, Coopersmith CM, et al. (2016). The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 315, 801–810. 10.1001/jama.2016.0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rhee C, Dantes R, Epstein L, Murphy DJ, Seymour CW, Iwashyna TJ, Kadri SS, Angus DC, Danner RL, Fiore AE, et al. (2017). Incidence and Trends of Sepsis in US Hospitals Using Clinical vs Claims Data. JAMA 318, 1241–1249. 10.1001/jama.2017.13836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rudd KE, Johnson SC, Agesa KM, Shackelford KA, Tsoi D, Kievlan DR, Colombara DV, Ikuta KS, Kissoon N, Finfer S, et al. (2020). Global, regional, and national sepsis incidence and mortality, 1990–2017: analysis for the Global Burden of Disease Study. Lancet 395, 200–211. 10.1016/S0140-6736(19)32989-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dolin HH, Papadimos TJ, Chen X, and Pan ZK (2019). Characterization of Pathogenic Sepsis Etiologies and Patient Profiles: A Novel Approach to Triage and Treatment. Microbiol. Insights 12, 1178636118825081. 10.1177/1178636118825081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mas-Celis F, Olea-López J, and Parroquin-Maldonado JA (2021). Sepsis in Trauma: A Deadly Complication. Arch. Med. Res. 52, 808–816. 10.1016/j.arcmed.2021.10.007. [DOI] [PubMed] [Google Scholar]
- 6.Vincent JL, Sakr Y, Sprung CL, Ranieri VM, Reinhart K, Gerlach H, Moreno R, Carlet J, Le Gall JR, and Payen D; Sepsis Occurrence in Acutely Ill Patients Investigators (2006). Sepsis in European intensive care units: results of the SOAP study. Crit. Care Med. 34, 344–353. 10.1097/01.ccm.0000194725.48928.3a. [DOI] [PubMed] [Google Scholar]
- 7.Flores-Mireles AL, Walker JN, Caparon M, and Hultgren SJ (2015). Urinary tract infections: epidemiology, mechanisms of infection and treatment options. Nat. Rev. Microbiol. 13, 269–284. 10.1038/nrmicro3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martin MD, Badovinac VP, and Griffith TS (2020). CD4 T Cell Responses and the Sepsis-Induced Immunoparalysis State. Front. Immunol. 11, 1364. 10.3389/fimmu.2020.01364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beura LK, Hamilton SE, Bi K, Schenkel JM, Odumade OA, Casey KA, Thompson EA, Fraser KA, Rosato PC, Filali-Mouhim A, et al. (2016). Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature 532, 512–516. 10.1038/nature17655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reese TA, Bi K, Kambal A, Filali-Mouhim A, Beura LK, Bürger MC, Pulendran B, Sekaly RP, Jameson SC, Masopust D, et al. (2016). Sequential Infection with Common Pathogens Promotes Humanlike Immune Gene Expression and Altered Vaccine Response. Cell Host Microbe 19, 713–719. 10.1016/j.chom.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berton RR, Jensen IJ, Harty JT, Griffith TS, and Badovinac VP (2022). Inflammation Controls Susceptibility of Immune-Experienced Mice to Sepsis. Immunohorizons 6, 528–542. 10.4049/immunohorizons.2200050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huggins MA, Sjaastad FV, Pierson M, Kucaba TA, Swanson W, Staley C, Weingarden AR, Jensen IJ, Danahy DB, Badovinac VP, et al. (2019). Microbial Exposure Enhances Immunity to Pathogens Recognized by TLR2 but Increases Susceptibility to Cytokine Storm through TLR4 Sensitization. Cell Rep. 28, 1729–1743.e5. 10.1016/j.celrep.2019.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moioffer SJ, Danahy DB, van de Wall S, Jensen IJ, Sjaastad FV, Anthony SM, Harty JT, Griffith TS, and Badovinac VP (2021). Severity of Sepsis Determines the Degree of Impairment Observed in Circulatory and Tissue-Resident Memory CD8 T Cell Populations. J. Immunol. 207, 1871–1881. 10.4049/jimmunol.2001142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chousterman BG, Swirski FK, and Weber GF (2017). Cytokine storm and sepsis disease pathogenesis. Semin. Immunopathol. 39, 517–528. 10.1007/s00281-017-0639-8. [DOI] [PubMed] [Google Scholar]
- 15.Hotchkiss RS, and Karl IE (2003). The pathophysiology and treatment of sepsis. N. Engl. J. Med. 348, 138–150. 10.1056/NEJMra021333. [DOI] [PubMed] [Google Scholar]
- 16.Kaper JB, Nataro JP, and Mobley HL (2004). Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2, 123–140. 10.1038/nrmicro818. [DOI] [PubMed] [Google Scholar]
- 17.Pierson M, Merley A, and Hamilton SE (2021). Generating Mice with Diverse Microbial Experience. Curr. Protoc. 1, e53. 10.1002/cpz1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burger S, Stenger T, Pierson M, Sridhar A, Huggins MA, Kucaba TA, Griffith TS, Hamilton SE, and Schuldt NJ (2023). Natural Microbial Exposure from the Earliest Natural Time Point Enhances Immune Development by Expanding Immune Cell Progenitors and Mature Immune Cells. J. Immunol. 210, 1740–1751. 10.4049/jimmunol.2300061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sjaastad FV, Huggins MA, Lucas ED, Skon-Hegg C, Swanson W, Martin MD, Salgado OC, Xu J, Pierson M, Dileepan T, et al. (2022). Reduced T Cell Priming in Microbially Experienced “Dirty” Mice Results from Limited IL-27 Production by XCR1+ Dendritic Cells. J. Immunol. 209, 2149–2159. 10.4049/jimmunol.2200324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stolley JM, Scott MC, Joag V, Dale AJ, Johnston TS, Saavedra F, Gavil NV, Lotfi-Emran S, Soerens AG, Weyu E, et al. (2023). Depleting CD103+ resident memory T cells in vivo reveals immunostimulatory functions in oral mucosa. J. Exp. Med. 220, e20221853. 10.1084/jem.20221853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fiege JK, Block KE, Pierson MJ, Nanda H, Shepherd FK, Mickelson CK, Stolley JM, Matchett WE, Wijeyesinghe S, Meyerholz DK, et al. (2021). Mice with diverse microbial exposure histories as a model for preclinical vaccine testing. Cell Host Microbe 29, 1815–1827.e6. 10.1016/j.chom.2021.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Block KE, Iijima K, Pierson MJ, Walsh DA, Tei R, Kucaba TA, Xu J, Khan MH, Staley C, Griffith TS, et al. (2022). Physiological microbial exposure transiently inhibits mouse lung ILC2 responses to allergens. Nat. Immunol. 23, 1703–1713. 10.1038/s41590-022-01350-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Camell CD, Yousefzadeh MJ, Zhu Y, Prata LGPL, Huggins MA, Pierson M, Zhang L, O’Kelly RD, Pirtskhalava T, Xun P, et al. (2021). Senolytics reduce coronavirus-related mortality in old mice. Science 373, eabe4832. 10.1126/science.abe4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McDermott DS, and Varga SM (2011). Quantifying antigen-specific CD4 T cells during a viral infection: CD4 T cell responses are larger than we think. J. Immunol. 187, 5568–5576. 10.4049/jimmunol.1102104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martin MD, Danahy DB, Hartwig SM, Harty JT, and Badovinac VP (2017). Revealing the Complexity in CD8 T Cell Responses to Infection in Inbred C57B/6 versus Outbred Swiss Mice. Front. Immunol. 8, 1527. 10.3389/fimmu.2017.01527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martin MD, Sompallae R, Winborn CS, Harty JT, and Badovinac VP (2020). Diverse CD8 T Cell Responses to Viral Infection Revealed by the Collaborative Cross. Cell Rep. 31, 107508. 10.1016/j.celrep.2020.03.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rai D, Pham NLL, Harty JT, and Badovinac VP (2009). Tracking the total CD8 T cell response to infection reveals substantial discordance in magnitude and kinetics between inbred and outbred hosts. J. Immunol. 183, 7672–7681. 10.4049/jimmunol.0902874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mulvey MA, Schilling JD, and Hultgren SJ (2001). Establishment of a persistent Escherichia coli reservoir during the acute phase of a bladder infection. Infect. Immun. 69, 4572–4579. 10.1128/IAI.69.7.4572-4579.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Théroude C, Reverte M, Heinonen T, Ciarlo E, Schrijver IT, Antonakos N, Maillard N, Pralong F, Le Roy D, and Roger T (2021). Trained Immunity Confers Prolonged Protection From Listeriosis. Front. Immunol. 12, 723393. 10.3389/fimmu.2021.723393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arnold IC, Mathisen S, Schulthess J, Danne C, Hegazy AN, and Powrie F (2016). CD11c(+) monocyte/macrophages promote chronic Helicobacter hepaticus-induced intestinal inflammation through the production of IL-23. Mucosal Immunol. 9, 352–363. 10.1038/mi.2015.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, Gupta R, Tsai JM, Sinha R, Corey D, et al. (2017). PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 545, 495–499. 10.1038/nature22396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li X, Körner H, and Liu X (2020). Susceptibility to Intracellular Infections: Contributions of TNF to Immune Defense. Front. Microbiol. 11, 1643. 10.3389/fmicb.2020.01643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pfeffer K, Matsuyama T, Kündig TM, Wakeham A, Kishihara K, Shahinian A, Wiegmann K, Ohashi PS, Krönke M, and Mak TW (1993). Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell 73, 457–467. 10.1016/0092-8674(93)90134-c. [DOI] [PubMed] [Google Scholar]
- 34.Muszynski JA, Nofziger R, Moore-Clingenpeel M, Greathouse K, Anglim L, Steele L, Hensley J, Hanson-Huber L, Nateri J, Ramilo O, and Hall MW (2018). Early Immune Function and Duration of Organ Dysfunction in Critically III Children with Sepsis. Am. J. Respir. Crit. Care Med. 198, 361–369. 10.1164/rccm.201710-2006OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Breslin WL, Strohacker K, Carpenter KC, Haviland DL, and McFarlin BK (2013). Mouse blood monocytes: standardizing their identification and analysis using CD115. J. Immunol. Methods 390, 1–8. 10.1016/j.jim.2011.03.005. [DOI] [PubMed] [Google Scholar]
- 36.Yang J, Zhang L, Yu C, Yang XF, and Wang H (2014). Monocyte and macrophage differentiation: circulation inflammatory monocyte as biomarker for inflammatory diseases. Biomark. Res. 2, 1. 10.1186/2050-7771-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frangie C, and Daher J (2022). Role of myeloperoxidase in inflammation and atherosclerosis (Review). Biomed. Rep. 16, 53. 10.3892/br.2022.1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ingersoll MA, Spanbroek R, Lottaz C, Gautier EL, Frankenberger M, Hoffmann R, Lang R, Haniffa M, Collin M, Tacke F, et al. (2010). Comparison of gene expression profiles between human and mouse monocyte subsets. Blood 115, e10–e19. 10.1182/blood-2009-07-235028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Swirski FK, Wildgruber M, Ueno T, Figueiredo JL, Panizzi P, Iwamoto Y, Zhang E, Stone JR, Rodriguez E, Chen JW, et al. (2010). Myeloperoxidase-rich Ly-6C+ myeloid cells infiltrate allografts and contribute to an imaging signature of organ rejection in mice. J. Clin. Invest. 120, 2627–2634. 10.1172/JCI42304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee PY, Wang JX, Parisini E, Dascher CC, and Nigrovic PA (2013). Ly6 family proteins in neutrophil biology. J. Leukoc. Biol. 94, 585–594. 10.1189/jlb.0113014. [DOI] [PubMed] [Google Scholar]
- 41.Yang P, Liu L, Sun L, Fang P, Snyder N, Saredy J, Ji Y, Shen W, Qin X, Wu Q, et al. (2021). Immunological Feature and Transcriptional Signaling of Ly6C Monocyte Subsets From Transcriptome Analysis in Control and Hyperhomocysteinemic Mice. Front. Immunol. 12, 632333. 10.3389/fimmu.2021.632333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guilliams M, Mildner A, and Yona S (2018). Developmental and Functional Heterogeneity of Monocytes. Immunity 49, 595–613. 10.1016/j.immuni.2018.10.005. [DOI] [PubMed] [Google Scholar]
- 43.Serbina NV, and Pamer EG (2006). Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat. Immunol. 7, 311–317. 10.1038/ni1309. [DOI] [PubMed] [Google Scholar]
- 44.Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, et al. (2013). Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38, 79–91. 10.1016/j.immuni.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Challen GA, Pietras EM, Wallscheid NC, and Signer RAJ (2021). Simplified murine multipotent progenitor isolation scheme: Establishing a consensus approach for multipotent progenitor identification. Exp. Hematol. 104, 55–63. 10.1016/j.exphem.2021.09.007. [DOI] [PubMed] [Google Scholar]
- 46.Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, McDonald-Smith GP, Gao H, Hennessy L, et al. (2013). Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc. Natl. Acad. Sci. USA 110, 3507–3512. 10.1073/pnas.1222878110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stortz JA, Raymond SL, Mira JC, Moldawer LL, Mohr AM, and Efron PA (2017). Murine Models of Sepsis and Trauma: Can We Bridge the Gap? ILAR J. 58, 90–105. 10.1093/ilar/ilx007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang N, Lu Y, Zheng J, and Liu X (2022). Of mice and men: Laboratory murine models for recapitulating the immunosuppression of human sepsis. Front. Immunol. 13, 956448. 10.3389/fimmu.2022.956448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Efron PA, Mohr AM, Moore FA, and Moldawer LL (2015). The future of murine sepsis and trauma research models. J. Leukoc. Biol. 98, 945–952. 10.1189/jlb.5MR0315-127R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Japp AS, Hoffmann K, Schlickeiser S, Glauben R, Nikolaou C, Maecker HT, Braun J, Matzmohr N, Sawitzki B, Siegmund B, et al. (2017). Wild immunology assessed by multidimensional mass cytometry. Cytometry A. 91, 85–95. 10.1002/cyto.a.22906. [DOI] [PubMed] [Google Scholar]
- 51.Rosshart SP, Vassallo BG, Angeletti D, Hutchinson DS, Morgan AP, Takeda K, Hickman HD, McCulloch JA, Badger JH, Ajami NJ, et al. (2017). Wild Mouse Gut Microbiota Promotes Host Fitness and Improves Disease Resistance. Cell 171, 1015–1028.e13. 10.1016/j.cell.2017.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rosshart SP, Herz J, Vassallo BG, Hunter A, Wall MK, Badger JH, McCulloch JA, Anastasakis DG, Sarshad AA, Leonardi I, et al. (2019). Laboratory mice born to wild mice have natural microbiota and model human immune responses. Science 365, eaaw4361. 10.1126/science.aaw4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chung H, Lee JH, Jo YH, Hwang JE, and Kim J (2019). Circulating Monocyte Counts and its Impact on Outcomes in Patients With Severe Sepsis Including Septic Shock. Shock 51, 423–429. 10.1097/SHK.0000000000001193. [DOI] [PubMed] [Google Scholar]
- 54.Ożańska A, Szymczak D, and Rybka J (2020). Pattern of human monocyte subpopulations in health and disease. Scand. J. Immunol. 92, e12883. 10.1111/sji.12883. [DOI] [PubMed] [Google Scholar]
- 55.Hullahalli K, and Waldor MK (2021). Pathogen clonal expansion underlies multiorgan dissemination and organ-specific outcomes during murine systemic infection. Elife 10, e70910. 10.7554/eLife.70910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Smith SN, Hagan EC, Lane MC, and Mobley HLT (2010). Dissemination and systemic colonization of uropathogenic Escherichia coli in a murine model of bacteremia. mBio 1, e00262–10. 10.1128/mBio.00262-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Benacerraf B, Sebestyen MM, and Schlossman S (1959). A quantitative study of the kinetics of blood clearance of P32-labelled Escherichia coli and Staphylococci by the reticuloendothelial system. J. Exp. Med. 110, 27–48. 10.1084/jem.110.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.van Schaik SM, and Abbas AK (2007). Role of T cells in a murine model of Escherichia coli sepsis. Eur. J. Immunol. 37, 3101–3110. 10.1002/eji.200737295. [DOI] [PubMed] [Google Scholar]
- 59.Gordon S, Plüddemann A, and Martinez Estrada F (2014). Macrophage heterogeneity in tissues: phenotypic diversity and functions. Immunol. Rev. 262, 36–55. 10.1111/imr.12223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wynn TA, Chawla A, and Pollard JW (2013). Macrophage biology in development, homeostasis and disease. Nature 496, 445–455. 10.1038/nature12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rosenberg G, Riquelme S, Prince A, and Avraham R (2022). Immunometabolic crosstalk during bacterial infection. Nat. Microbiol. 7, 497–507. 10.1038/s41564-022-01080-5. [DOI] [PubMed] [Google Scholar]
- 62.Stothers CL, Burelbach KR, Owen AM, Patil NK, McBride MA, Bohannon JK, Luan L, Hernandez A, Patil TK, Williams DL, and Sherwood ER (2021). beta-Glucan Induces Distinct and Protective Innate Immune Memory in Differentiated Macrophages. J. Immunol. 207, 2785–2798. 10.4049/jimmunol.2100107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mitroulis I, Ruppova K, Wang B, Chen LS, Grzybek M, Grinenko T, Eugster A, Troullinaki M, Palladini A, Kourtzelis I, et al. (2018). Modulation of Myelopoiesis Progenitors Is an Integral Component of Trained Immunity. Cell 172, 147–161.e12. 10.1016/j.cell.2017.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kaech SM, Wherry EJ, and Ahmed R (2002). Effector and memory T-cell differentiation: implications for vaccine development. Nat. Rev. Immunol. 2, 251–262. 10.1038/nri778. [DOI] [PubMed] [Google Scholar]
- 65.Martin MD, Condotta SA, Harty JT, and Badovinac VP (2012). Population dynamics of naive and memory CD8 T cell responses after antigen stimulations in vivo. J. Immunol. 188, 1255–1265. 10.4049/jimmunol.1101579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nathan C (2006). Neutrophils and immunity: challenges and opportunities. Nat. Rev. Immunol. 6, 173–182. 10.1038/nri1785. [DOI] [PubMed] [Google Scholar]
- 67.Silva EE, Skon-Hegg C, Badovinac VP, and Griffith TS (2023). The Calm after the Storm: Implications of Sepsis Immunoparalysis on Host Immunity. J. Immunol. 211, 711–719. 10.4049/jimmunol.2300171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kaufmann SH (1993). Immunity to intracellular bacteria. Annu. Rev. Immunol. 11, 129–163. 10.1146/annurev.iy.11.040193.001021. [DOI] [PubMed] [Google Scholar]
- 69.Schlech WF (2019). Epidemiology and Clinical Manifestations of Listeria monocytogenes Infection. Microbiol. Spectr. 7. 10.1128/microbiolspec.GPP3-0014-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mora-Bau G, Platt AM, van Rooijen N, Randolph GJ, Albert ML, and Ingersoll MA (2015). Macrophages Subvert Adaptive Immunity to Urinary Tract Infection. PLoS Pathog. 11, e1005044. 10.1371/journal.ppat.1005044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 73.McCarthy DJ, Chen Y, and Smyth GK (2012). Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 40, 4288–4297. 10.1093/nar/gks042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The bulk RNA-seq data generated have been deposited at the National Center for Biotechnology Information Gene Expression Omnibus (NCBI GEO) under accession number GSE237883 and are publicly available as of the date of publication. Any additional details regarding these data will be made available upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.